 Rishika Banydeen
Rishika Banydeen Astrid Monfort2,3†
Astrid Monfort2,3† Remi Neviere
Remi Neviere
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 06 June 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.898033
Cardiac amyloidosis (CA) is a myocardial disease characterized by extracellular amyloid infiltration throughout the heart, resulting in increased myocardial stiffness, and restrictive heart wall chamber behavior. Its diagnosis among patients hospitalized for cardiovascular diseases is becoming increasingly frequent, suggesting improved disease awareness, and higher diagnostic capacities. One predominant functional manifestation of patients with CA is exercise intolerance, objectified by reduced peak oxygen uptake (VO2 peak), and assessed by metabolic cart during cardiopulmonary exercise testing (CPET). Hemodynamic adaptation to exercise in patients with CA is characterized by low myocardial contractile reserve and impaired myocardial efficiency. Rapid shallow breathing and hyperventilation, in the absence of ventilatory limitation, are also typically observed in response to exercise. Ventilatory inefficiency is further suggested by an increased VE-VCO2 slope, which has been attributed to excessive sympathoexcitation and a high physiological dead space (VD/VT) ratio during exercise. Growing evidence now suggests that, in addition to well-established biomarker risk models, a reduced VO2 peak is potentially a strong and independent predictive factor of adverse patient outcomes, both for monoclonal immunoglobulin light chain (AL) or transthyretin (ATTR) CA. Besides generating prognostic information, CPET can be used for the evaluation of the impact of therapeutic interventions in patients with CA.
Cardiac amyloidosis (CA) is a myocardial disease characterized by extracellular amyloid infiltration throughout the heart (1, 2). CA diagnosis among patients hospitalized for cardiovascular diseases is increasingly frequent, suggesting improved disease awareness and higher diagnostic capacities. Till now, only clinical and biomarker scoring systems have been accurately used to stratify disease severity in patients with CA (1, 2). In addition to these well-established risk models, it has been recently described that reduced peak aerobic capacity (VO2 peak) can help identify patients with poor prognoses (3–6).
The aim of the present review, addressed to clinicians and cardiologists, is to describe the utility of cardiopulmonary exercise testing (CPET) as an aid to patient orientation and clinical decision-making in CA management. Indeed, as patients with CA experience progressive exercise intolerance over their disease course, they will be increasingly referred for CPET evaluation. We outline here the role of CPET in assessing the functional status and prognosis in patients with CA, with a particular focus on ventilatory inefficiency.
Cardiac amyloidosis is a restrictive cardiomyopathy caused by the extracellular deposition of proteins in the myocardium (1, 2, 7, 8). The unstable structure of proteins causes them to misfold, aggregate, and deposit as amyloid fibrils. CA is caused mainly by misfolded monoclonal immunoglobulin light chains (AL) resulting from the abnormal clonal proliferation of plasma cells, or by misfolded transthyretin (TTR), a liver-synthesized protein (previously called pre-albumin) involved in hormone thyroxine and retinol protein binding and blood transportation. TTR can either be inherited as an autosomal dominant trait caused by pathogenic variants in the TTR gene (TTRv), or by the deposition of wild-type TTR protein (TTRwt) (7, 8). Cardiac involvement in systemic amyloidosis is suspected using electrocardiography, transthoracic echocardiography, as well as cardiac magnetic resonance and nuclear imaging. Confirmation of cardiac amyloid deposition is achieved through tissue biopsy, using thioflavin dyes (most commonly Congo red), displaying birefringence under cross-polarized light and conferring the characteristic apple-green color to amyloid deposits (1, 7, 8). Cardiac biomarkers are also helpful for risk stratification and staging of patients with AL and ATTR systemic amyloidosis (1, 2). Overall, CA is associated with a poor prognosis due to a high risk of sudden death, congestive heart failure, conductive disorders, and arrhythmias. One of the most frequent functional manifestations in patients with CA is exercise intolerance, which can be objectively evaluated by reduced exercise aerobic capacity.
The link between impaired exercise capacity and central hemodynamic alterations has been extensively studied in patients with CA (9–12). Typically, CA is predominantly considered a diastolic disease caused by increased myocardial stiffness and restrictive heart wall chamber behavior (9). Recent studies have also underlined a profound lack of myocardial contractile reserve and coronary flow velocity reserve (10–12). A blunt increase in stroke volume during exercise has been attributed to this decreased contractile reserve, rather than changes in preload and afterload (10). Impaired systolic myocardial performance during exercise has been further attributed to abnormal myocardial external efficiency, i.e., the ratio of left ventricle external stroke work to myocardial oxygen consumption (12). Impaired myocardial efficiency suggests that the coupling of myocardial oxidative metabolism to contractile work deteriorates, possibly due to structural amyloid damage to the heart, as well as cardiac cell toxicity in the case of AL amyloidosis (13, 14). Direct toxic effects of ATTR amyloid on cardiomyocytes have also been reported, such as oxidative stress, altered calcium cycling, and apoptotic pathways (15).
Cardiopulmonary exercise testing with gas exchange determination not only enables peak oxygen uptake (VO2 peak) to be determined, but also sheds light upon the mechanisms of exercise curtailment, such as cardiovascular, ventilatory, and metabolic/muscular limitations (16). Peak exercise aerobic capacity (VO2 peak) is defined as the maximum ability of the cardiovascular system to deliver oxygen (O2) to exercising skeletal muscle, along with the ability of the skeletal muscle to maximally extract O2 from the blood (16, 17). As a result, exercise tolerance is determined by adequate cardiovascular performance, pulmonary gas exchange, and skeletal muscle metabolism. Key parameters estimated during CPET include oxygen uptake (VO2), pulmonary elimination of carbon dioxide (VCO2), minute ventilation (VE), as well as changes in cardiovascular parameters, such as heart rate and systemic arterial pressure rise (16, 17). Exercise intolerance in patients with CA was first demonstrated in case reports, describing symptoms of exertional syncope (18). Since then, severely reduced aerobic capacity (VO2 peak) in CA has been objectively demonstrated using the 6-minute walk test and CPET (3–6, 10, 12, 19–23). Table 1 summarizes the main CPET characteristics of patients with CA, which include reduced VO2 peak (expressed as low absolute peak values and anaerobic threshold VO2), increased VE-VCO2 slope, and episodes of oscillatory ventilation (EOV). The ventilatory response to exercise in these patients is further characterized by hyperventilation with rapid shallow breathing, in the absence of ventilatory limitation as evidenced by a normal ventilatory reserve (Table 1). A common additional feature of patients with CA is chronotropic incompetence, defined as insufficient cardio-acceleration (typically less than 80–85%) at peak exercise (4, 24). While the increase in normal stroke volume during exercise is lost in most patients with CA, cardiac output rises are primarily dependent on heart rate elevation. Thus, chronotropic incompetence may be considered a critical factor limiting exercise capacity in these patients. However, before concluding that a patient has chronotropic incompetence, it is important to consider the level of achieved effort and reasons for effort termination. Low exercise intensity will equal low metabolic stress and insufficient heart rate rise without true chronotropic incompetence. Chronotropic incompetence can be objectively demonstrated by the calculation of the slope between heart rate rise and VO2 increase during exercise. Using this metabolic-chronotropic relationship approach, no evidence of chronotropic incompetence was found in patients with ATTR CA (22).
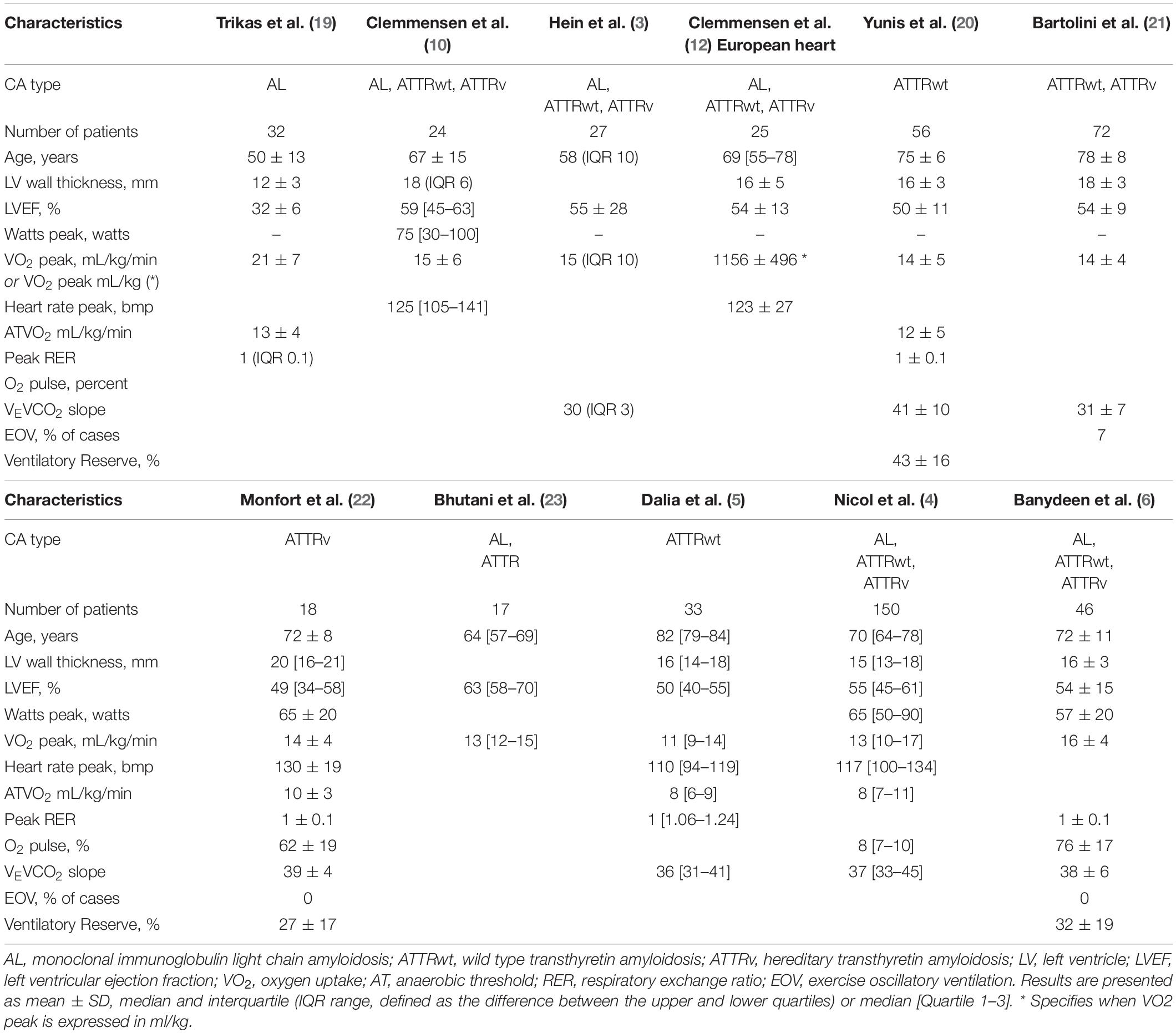
Table 1. Main characteristics of cardiopulmonary exercise testing (CPET) in patients with cardiac amyloidosis (CA).
In patients with heart failure, VO2 measured at peak exercise is the most important predictor of prognosis (25, 26). A VO2 peak of less than 12–14 ml/kg/min is associated with a poor outcome. Of note, the predictive value of the VO2 peak is accurate only when exercise capacity is limited by heart failure, which means that factors prematurely terminating the test must be excluded. Such factors include peripheral muscular deconditioning, peripheral artery disease, myocardial ischemia, or low patient motivation. Besides VO2 peak, parameters derived from the ventilatory response are also useful to determine risk in patients with heart failure. The ventilatory equivalent for carbon dioxide (VE/VCO2 ratio) and ventilatory efficiency (VE-VCO2 slope) are well-established indicators of prognosis (25–27). In patients with CA, growing evidence suggests that VO2 peak, along with usual risk scores, are strong and independent predictive factors, independently of CA etiology (AL or ATTR) (3–6, 10, 12, 19–23) (Figure 1). Overall, these findings are fairly consistent with the large body of evidence upholding the added value of VO2 peak in terms of efficacious risk stratification and outcome prediction in patients with heart failure. This constitutes a strong ground to believe that future multicenter studies will soon be confirming the prognostic capacity of CPET in patients with ATTR CA.
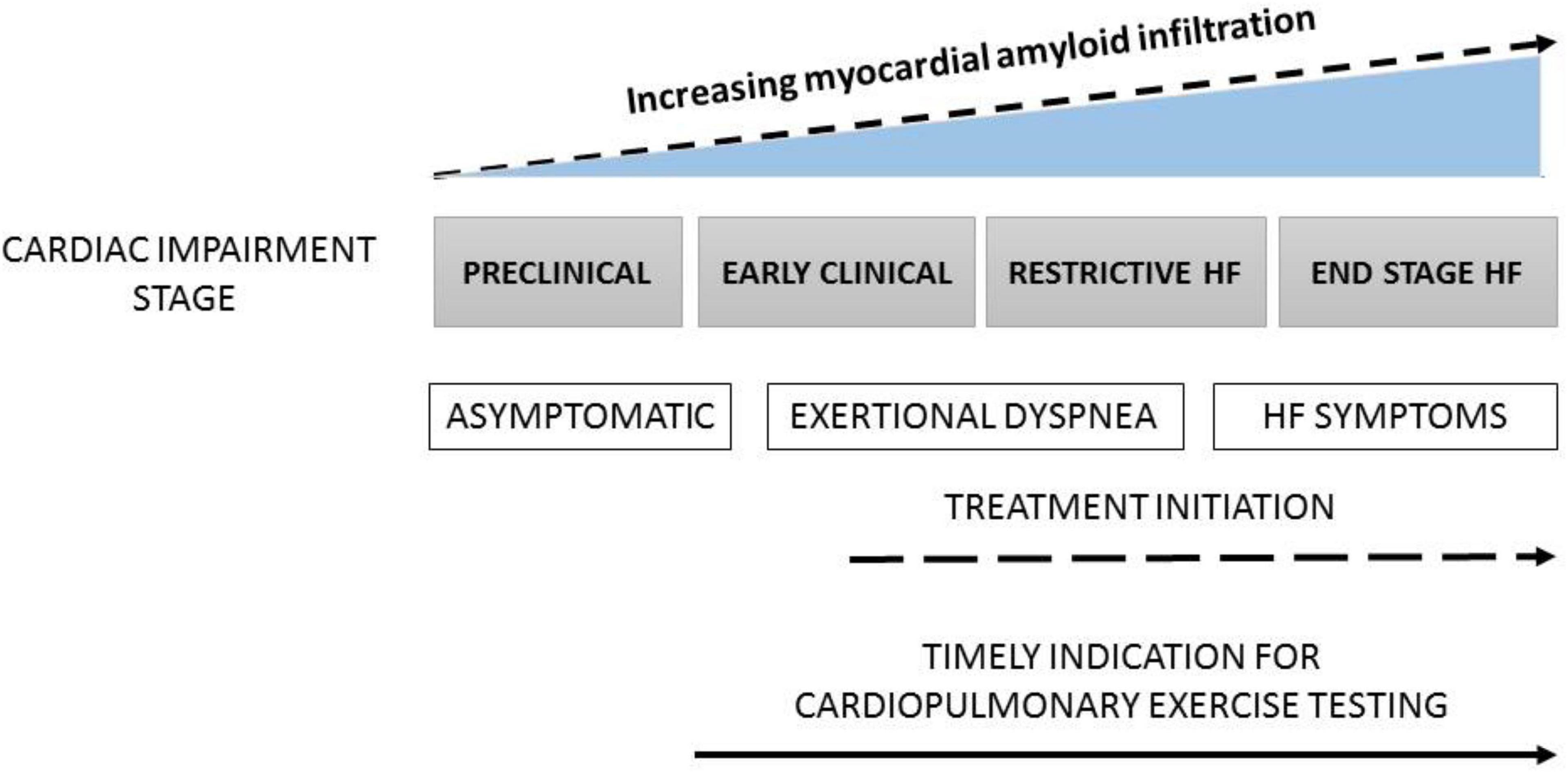
Figure 1. The progression of CA from asymptomatic stage to end stage heart failure (HF) and timing for the use of cardiopulmonary exercise testing (CPET).
In patients with known heart failure, ventilatory inefficiency, suggested by an increased VE-VCO2 slope, characterizes those with more severe heart disease and is considered an independent marker of event-free survival (28, 29). Interestingly, one of the most striking findings in patients with CA is an increased VE-VCO2 slope (Table 1). Of note, the latter is observed even in the absence of severe myocardial dysfunction, as suggested by the mild reduction in left ventricular ejection fraction (Table 1).
The physiological determinants and underlying major mechanisms of ventilatory efficiency in patients with chronic heart failure and CA are presented next (Figure 2). The ramp with which ventilation rises with increased CO2 production, i.e., VE-VCO2 slope, reflects ventilatory efficiency (30, 31). Increased VE-VCO2 slope identifies an abnormal ventilatory response to exercise and has been consistently used as a prognostic marker in several chronic cardiopulmonary diseases, independently of other CPET-derived variables, such as VO2 peak. VE-VCO2 slope is calculated off-line after exercise using a linear regression function, and is established by the formula: , where the factor 863 accounts for corrections related to body temperature, ambient pressure, water vapor saturated conditions (BTPS), VCO2 is the rate of CO2 pulmonary elimination, PaCO2 is arterial CO2 partial pressure, and VD/VT is the physiological dead space ratio (30, 31). This formula predicts that VE-VCO2 slope is determined by two factors: (1) the direction and magnitude of PaCO2 change and (2) the fraction of the tidal volume that goes to dead space, i.e., the physiological dead space ratio (VD/VT). Therefore, VE-VCO2 slope increases if VD/VT ratio remains elevated during exercise, or if PaCO2 is reduced.
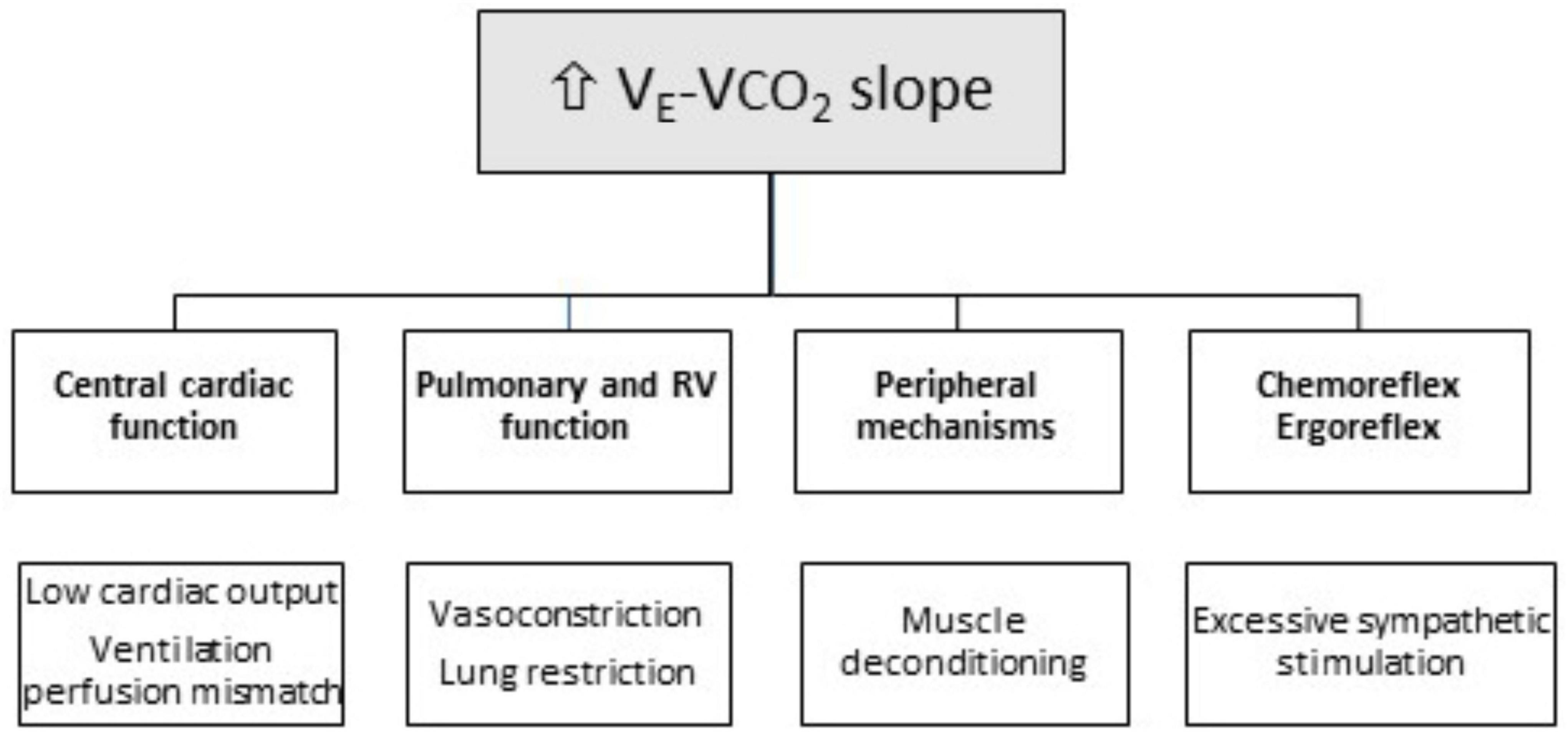
Figure 2. Mechanisms of ventilatory inefficiency (increased VE-VCO2 slope) in patients with cardiac amyloidosis (CA). RV, Right Ventricle.
Indeed, in patients with heart failure, VD/VT ratio typically remains high during exercise due to increased heterogeneity of pulmonary ventilation-to-perfusion (VA/Q) ratios and/or blunted tidal volume increase VT (30, 31) (Figure 2). In these patients, true VA/Q mismatch is uncommon, in the absence of coexisting lung disease. As such, while ventilation increases during exercise, cardiac output does not proportionally rise, which tends to augment the VA/Q ratio, resulting in a higher VD/VT ratio and an increased VE-VCO2 slope. Similarly, patients with chronic heart failure often display a reduced VT (volume of air moved into or out of the lungs during a normal breath) during exercise, which may also contribute to a higher VD/VT ratio and increased VE-VCO2 slope (30, 31).
As for PaCO2, the other factor influencing VE-VCO2 slope, its reduction during exercise results in a rise in VE-VCO2 slope (30, 31). The point at which PaCO2 is set is determined by factors, such as metabolic acidosis, hypoxemia, baroreceptors in the pulmonary vasculature, and sympathetic nervous system hyperactivity. The PaCO2 set-point is driven down with excessive ventilatory stimulation from sensitized peripheral chemoreceptors and baroreceptors, or by ergoreceptors in the skeletal muscle. In patients with chronic heart failure, sympathetic activity is overstimulated and ventilatory responses to hypercapnia and hypoxia are exacerbated (32) (Figure 2). Furthermore, PaCO2 either remains stable or modestly declines from rest to peak exercise (31, 32). However, due to chemoreceptor hypersensitivity, even minor PaCO2 changes can elicit excessive hyperventilation and VE-VCO2 slope elevation (31, 32).
Mechanisms leading to VE-VCO2 slope rise in patients with CA (Figure 2) have not been extensively studied. Autonomic dysfunction, along with sympathoexcitation and abnormal ventilatory patterns, may explain VE-VCO2 slope increase in patients with CA (33, 34). Amyloid disorders preferentially involve small unmyelinated nerve fibers, i.e., thinly myelinated Aδ and unmyelinated C fibers, and manifest with sensory loss mainly affecting pain and temperature sensation, as well as autonomic dysfunction (35, 36). The latter, a distinctive feature of patients with CA, is characterized by exaggerated sympathoexcitation and attenuated parasympathetic tone (37, 38). Cardiac dysautonomia manifests with altered heart rate variability and blood pressure regulation, arrhythmias or heart block, and autonomic (sympathetic) denervation (33, 34, 37–39). It is plausible to consider that sympathoexcitation may enhance the ventilatory response of both peripheral and central chemoreflexes, thus leading to enhanced ventilation and an increased VE-VCO2 slope (31, 32).
In patients with CA, right ventricular dysfunction is also becoming increasingly recognized. It is a well-known mechanism that results in an increased VD/VT ratio and VE-VCO2 slope. Impaired pulmonary blood flow due to right ventricular dysfunction increases VD/VT ratio, as regions of ventilated lung remain under perfused at rest and during exercise. A significant correlation between right ventricular dysfunction and VE-VCO2 slope has been reported in patients with CA, in accordance with similar findings in patients with chronic heart failure, in whom an increased VE-VCO2 slope also results from advanced right-sided heart dysfunction and impaired pulmonary hemodynamics.
Another mechanism that may contribute to increased VD/VT ratio and VE-VCO2 slope in patients with CA is the absence of tidal volume VT increase during exercise (32). While ventilatory reserve at peak exercise in patients with CA is typically unaffected, typical manifestations are rapid shallow breathing patterns characterized by low VT and increased breathing frequency (6, 22). The inability to increase O2 delivery to respiratory muscles, translating into reduced respiratory muscle strength, and enhanced central and peripheral chemoreflexes, may explain the absence of tidal volume VT rise during exercise (31, 32). Accordingly, we have shown that patients with CA commonly display a restrictive lung disease characterized by reduced lung volumes (6, 22). In this setting, reduced lung compliance, along with increased elastic loading on respiratory muscles, lung elastic recoil, and stimulated thoracic mechanoreceptors, limit the rise in VT below 50–60% of maximal lung vital capacity, leading to a characteristic shallow breathing pattern with a high respiratory frequency (40).
Treatments for CA vary depending on the type of amyloid. They include heart failure supportive therapy, therapies inhibiting the production of amyloid precursor proteins, and novel strategies inhibiting amyloid fibril formation, amyloid deposits, or stabilizing precursor proteins. In AL amyloidosis, the aim of treatment is to eradicate the underlying plasma cell clone that produces excess light chains (41). Chemotherapy regimens and autologous stem cell transplantation have been adapted from treatment of multiple myeloma, a related plasma cell dyscrasia. Well-established risk stratification dictates treatment strategy for patients with AL (42). Until recently, treatment of TTR CA was aimed at managing symptoms and disease-related complications. Novel therapies for the treatment of TTRwt and TTRv CA have since emerged. Over the past decade, different molecules targeting specific steps of the amyloidogenic cascade have been evaluated as potential drug candidates contributing to modified exercise tolerance in patients with TTR CA (43) (Table 2). Tafamidis, which binds to TTR and prevents tetramer dissociation and amyloidogenesis, has been shown to lower all-cause mortality at 30 months, as well as the deterioration rate during the 6-min walk test (44). Further investigations in patients with TTR CA have confirmed the beneficial effects of tafamidis on exercise capacity, associated with improved cardiac function and a reduction in myocardial amyloid deposition (45). Small interfering RNA or antisense oligonucleotide technologies are also highly effective for the blockade of TTR liver expression. While not a primary objective of randomized controlled studies (Table 2), the analysis of cardiac parameters in hereditary CA cohorts has suggested a beneficial effect on the progression of cardiomyopathy. Indeed, preliminary data suggest that inotersen, a second-generation antisense oligonucleotide targeting TTR messenger RNA, can improve exercise tolerance and reduce left ventricular mass over a 3-year treatment period (46). Large clinical trials involving RNA-targeting and gene editing therapies are currently underway and will probably confirm the beneficial effects of these novel treatments on exercise tolerance in patients with TTR CA (43).

Table 2. Main studies evaluating treatment for transthyretin (TTR) CA with functional end-points.
Cardiac involvement is the major negative prognostic factor, as deaths are mainly due to heart failure or arrhythmias associated with amyloidosis. Diagnostic strategies for symptomatic CA, presenting with typical morphological features, such as increased ventricular wall thickness, are well established. On the other hand, detection of subclinical CA is much more limited. At a time when effective therapies suppressing or delaying CA progression are available, the early detection of non-clinically overt forms of the disease is more than ever warranted. A large number of extra-cardiac symptoms, called “red flags,” can direct toward CA investigation and detection in its sub-clinical phase. Indeed, from a patient’s perspective, many other extra-cardiac symptoms are typically reported, such as breathlessness, tingling sensation, pain, difficulty in walking, and weight loss.
In the same line of thought, CPET, while not a diagnostic tool, can be extremely useful for the detection of early stages of cardiovascular and lung diseases, as well as for the evaluation of response to therapies (Figure 1). Indeed, it is today quite established that CPET can non-invasively identify early hemodynamic and metabolic alterations in patients with various cardiomyopathies, long before the latter become clinically overt and cardiac structural changes are already in place. In CA, CPET should be used to detect early functional involvement of the disease.
Patients with CA display exertional dyspnea and exercise intolerance, evidenced mainly by poor aerobic capacity (reduced VO2 peak) and ventilatory inefficiency (increased VE-VCO2 slope). Both impaired VO2 peak and increased VE-VCO2 slope, two key parameters estimated during CPET, have been associated with poor outcome, both in monoclonal immunoglobulin light chain (AL) and transthyretin (ATTR) CA. These findings quite strongly advocate for the consideration of CPET, alongside traditional clinical and biological risk models at CA diagnosis, in order to optimize patient risk stratification. Ongoing large multicenter studies are currently evaluating the value of CPET in the prognostic assessment of patients with systemic amyloidosis. In time, CPET should become an integral part of CA management, contributing toward the assessment of the impact of novel therapies on the functional capacity and quality of life of patients with CA.
AM and RB collected the literature data. JI and RN analyzed and interpreted the literature data. RN, RB, and AM wrote the manuscript. All authors contributed, read, and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Our thanks go to the medical staff who contributed toward our study.
1. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. (2016) 387:2641–54. doi: 10.1016/S0140-6736(15)01274-X
2. Joury A, Faaborg-Andersen C, Quintana RA, daSilva-deAbreu A, Nativi-Nicolau J. Diagnostic tools for cardiac amyloidosis: a pragmatic comparison of pathology, imaging and laboratories. Curr Probl Cardiol. (2022) 101106.
3. Hein S, Aus Dem Siepen F, Bauer R, Katus HA, Kristen AV. Peak V’O2 is an independent predictor of survival in patients with cardiac amyloidosis. Amyloid. (2018) 25:167–73. doi: 10.1080/13506129.2018.1496077
4. Nicol M, Deney A, Lairez O, Vergaro G, Emdin M, Carecci A, et al. Prognostic value of cardiopulmonary exercise testing in cardiac amyloidosis. Eur J Heart Fail. (2021) 23:231–9. doi: 10.1002/ejhf.2016
5. Dalia T, Acharya P, Chan WC, Sauer AJ, Weidling R, Fritzlen J, et al. Prognostic role of cardiopulmonary exercise testing in wild-type transthyretin amyloid cardiomyopathy patients treated with tafamidis. J Card Fail. (2021) 27:1285–9. doi: 10.1016/j.cardfail.2021.06.022
6. Banydeen R, Vergaro G, Deney A, Monfort A, Emdin M, Lairez O, et al. Restrictive spirometry pattern and abnormal cardiopulmonary response to exercise in transthyretin cardiac amyloidosis. Eur Respir J. (2022) 59:2102838. doi: 10.1183/13993003.02838-2021
7. Griffin JM, Rosenblum H, Maurer MS. Pathophysiology and therapeutic approaches to cardiac amyloidosis. Circ Res. (2021) 128:1554–75. doi: 10.1161/CIRCRESAHA.121.318187
8. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. (2019) 12:e006075. doi: 10.1161/CIRCHEARTFAILURE.119.006075
9. Razvi Y, Patel RK, Fontana M, Gillmore JD. Cardiac amyloidosis: a review of current imaging techniques. Front Cardiovasc Med. (2021) 8:751293. doi: 10.3389/fcvm.2021.751293
10. Clemmensen TS, Mølgaard H, Sörensen J, Eiskjaer H, Andersen NF, Mellemkjaer S, et al. Inotropic myocardial reserve deficiency is the predominant feature of exercise haemodynamics in cardiac amyloidosis. Eur J Heart Fail. (2017) 19:1457–65. doi: 10.1002/ejhf.899
11. Clemmensen TS, Eiskjær H, Mølgaard H, Larsen AH, Soerensen J, Andersen NF, et al. Abnormal coronary flow velocity reserve and decreased myocardial contractile reserve are main factors in relation to physical exercise capacity in cardiac amyloidosis. J Am Soc Echocardiogr. (2018) 31:71–8. doi: 10.1016/j.echo.2017.09.007
12. Clemmensen TS, Soerensen J, Hansson NH, Tolbod LP, Harms HJ, Eiskjær H, et al. Myocardial oxygen consumption and efficiency in patients with cardiac amyloidosis. J Am Heart Assoc. (2018) 7:e009974. doi: 10.1161/JAHA.118.009974
13. Koike H, Katsuno M. The ultrastructure of tissue damage by amyloid fibrils. Molecules. (2021) 26:4611. doi: 10.3390/molecules26154611
14. Aimo A, Buda G, Fontana M, Barison A, Vergaro G, Emdin M, et al. Therapies for cardiac light chain amyloidosis: an update. Int J Cardiol. (2018) 271:152–60. doi: 10.1016/j.ijcard.2018.05.018
15. Saito Y, Nakamura K, Ito H. Molecular mechanisms of cardiac amyloidosis. Int J Mol Sci. (2021) 23:25. doi: 10.3390/ijms23010025
16. Glaab T, Taube C. Practical guide to cardiopulmonary exercise testing in adults. Respir Res. (2022) 23:9. doi: 10.1186/s12931-021-01895-6
17. American Thoracic Society; American College of Chest Physicians. ATS/ACCP statement on cardiopulmonary exercise testing. Erratum in: Am J Respir Crit Care Med. (2003). Am J Respir Crit Care Med. (2003) 167:211–77. doi: 10.1164/rccm.167.2.211.1451-2
18. Fujii B, Matsuda Y, Ohno H, Hamada Y, Takashiba K, Ebihara H, et al. A case of cardiac amyloidosis presenting with symptoms of exertional syncope. Clin Cardiol. (1991) 14:267–8. doi: 10.1002/clc.4960140317
19. Trikas A, Rallidis L, Hawkins P, Oakley CM, Nihoyannopoulos P. Comparison of usefulness between exercise capacity and echocardiographic indexes of left ventricular function in cardiac amyloidosis. Am J Cardiol. (1999) 84:1049–54. doi: 10.1016/s0002-9149(99)00497-x
20. Yunis A, Doros G, Luptak I, Connors LH, Sam F. Use of ventilatory efficiency slope as a marker for increased mortality in wild-type transthyretin cardiac amyloidosis. Am J Cardiol. (2019) 124:122–30. doi: 10.1016/j.amjcard.2019.03.035
21. Bartolini S, Baldasseroni S, Fattirolli F, Silverii MV, Piccioli L, Perfetto F, et al. Poor right ventricular function is associated with impaired exercise capacity and ventilatory efficiency in transthyretin cardiac amyloid patients. Intern Emerg Med. (2021) 16:653–60. doi: 10.1007/s11739-020-02474-1
22. Monfort A, Banydeen R, Demoniere F, Courty B, Codiat R, Neviere R, et al. Restrictive cardiac phenotype as primary cause of impaired aerobic capacity in Afro-Caribbean patients with val122ile variant transthyretin amyloid cardiomyopathy. Amyloid. (2020) 27:145–52. doi: 10.1080/13506129.2020.1722098
23. Bhutani D, Pan S, Latif F, Goldsmith RL, Saith SE, Mapara MY, et al. Cardiopulmonary exercise testing in patients with cardiac amyloidosis. Clin Lymphoma Myeloma Leuk. (2021) 21:545–8. doi: 10.1016/j.clml.2021.03.015
24. Nativi-Nicolau J, Stehlik J, Al-Dulaimi R, Rodriguez C, Jaramillo J, Conte J, et al. Chronotropic incompetence and autonomic dysfunction as mechanisms of dyspnoea in patients with late stage cardiac amyloidosis. Amyloid. (2019) 26(Supp. 1):134–5. doi: 10.1080/13506129.2019.1582024
25. Cipriano G Jr., Cipriano VT, da Silva VZ, Cipriano GF, Chiappa GR, de Lima AC, et al. Aerobic exercise effect on prognostic markers for systolic heart failure patients: a systematic review and meta-analysis. Heart Fail Rev. (2014) 19:655–67. doi: 10.1007/s10741-013-9407-6
26. Malhotra R, Bakken K, D’Elia E, Lewis GD. Cardiopulmonary exercise testing in heart failure. JACC Heart Fail. (2016) 4:607–16. doi: 10.1016/j.jchf.2016.03.022
27. Myers J, Arena R, Cahalin LP, Labate V, Guazzi M. Cardiopulmonary exercise testing in heart failure. Curr Probl Cardiol. (2015) 40:322–72. doi: 10.1016/j.cpcardiol.2015.01.009
28. Arena R, Myers J, Guazzi M. The clinical and research applications of aerobic capacity and ventilatory efficiency in heart failure: an evidence-based review. Heart Fail Rev. (2008) 13:245–69. doi: 10.1007/s10741-007-9067-5
29. Guazzi M. Abnormalities in cardiopulmonary exercise testing ventilatory parameters in heart failure: pathophysiology and clinical usefulness. Curr Heart Fail Rep. (2014) 11:80–7. doi: 10.1007/s11897-013-0183-3
30. Phillips DB, Collins SÉ, Stickland MK. Measurement and interpretation of exercise ventilatory efficiency. Front Physiol. (2020) 11:659. doi: 10.3389/fphys.2020.00659
31. Weatherald J, Sattler C, Garcia G, Laveneziana P. Ventilatory response to exercise in cardiopulmonary disease: the role of chemosensitivity and dead space. Eur Respir J. (2018) 51:1700860. doi: 10.1183/13993003.00860-2017
32. Woods PR, Olson TP, Frantz RP, Johnson BD. Causes of breathing inefficiency during exercise in heart failure. J Card Fail. (2010) 16:835–42. doi: 10.1016/j.cardfail.2010.05.003
33. Algalarrondo V, Antonini T, Théaudin M, Chemla D, Benmalek A, Lacroix C, et al. Cardiac dysautonomia predicts long-term survival in hereditary transthyretin amyloidosis after liver transplantation. JACC Cardiovasc Imaging. (2016) 9:1432–41. doi: 10.1016/j.jcmg.2016.07.008
34. Goldstein DS. Cardiac dysautonomia and survival in hereditary transthyretin amyloidosis. JACC Cardiovasc Imaging. (2016) 9:1442–5. doi: 10.1016/j.jcmg.2016.08.003
35. Asiri MMH, Engelsman S, Eijkelkamp N, Höppener JWM. Amyloid proteins and peripheral neuropathy. Cells. (2020) 9:1553. doi: 10.3390/cells9061553
36. Kaku M, Berk JL. Neuropathy associated with systemic amyloidosis. Semin Neurol. (2019) 39:578–88. doi: 10.1055/s-0039-1688994
37. Gonzalez-Duarte A. Autonomic involvement in hereditary transthyretin amyloidosis (hATTR amyloidosis). Clin Auton Res. (2019) 29:245–51. doi: 10.1007/s10286-018-0514-2
38. Koike H, Nakamura T, Nishi R, Ikeda S, Kawagashira Y, Iijima M, et al. Cardiac and peripheral vasomotor autonomic functions in hereditary transthyretin amyloidosis with non-Val30Met mutation. Amyloid. (2019) 26(Supp. 1):13–4. doi: 10.1080/13506129.2019.1582023
39. Damy T, Kristen AV, Suhr OB, Maurer MS, Planté-Bordeneuve V, Yu CR, et al. THAOS Investigators. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J. (2019) 43:391–400. doi: 10.1093/eurheartj/ehz173
40. Gille T, Laveneziana P. Cardiopulmonary exercise testing in interstitial lung diseases and the value of ventilatory efficiency. Eur Respir Rev. (2021) 30:200355. doi: 10.1183/16000617.0355-2020
41. Stelmach-Gołdyś A, Zaborek-Łyczba M, Łyczba J, Garus B, Pasiarski M, Mertowska P, et al. Physiology, diagnosis and treatment of cardiac light chain amyloidosis. J Clin Med. (2022) 11:911. doi: 10.3390/jcm11040911
42. Sidana S, Milani P, Binder M, Basset M, Tandon N, Foli A, et al. A validated composite organ and hematologic response model for early assessment of treatment outcomes in light chain amyloidosis. Blood Cancer J. (2020) 10:41. doi: 10.1038/s41408-020-0306-5
43. Aimo A, Castiglione V, Rapezzi C, Franzini M, Panichella G, Vergaro G, et al. RNA-targeting and gene editing therapies for transthyretin amyloidosis. Nat Rev Cardiol. (2022). doi: 10.1038/s41569-022-00683-z
44. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. (2018) 379:1007–16. doi: 10.1056/NEJMoa1805689
45. Rettl R, Mann C, Duca F, Dachs TM, Binder C, Ligios LC, et al. Tafamidis treatment delays structural and functional changes of the left ventricle in patients with transthyretin amyloid cardiomyopathy. Eur Heart J Cardiovasc Imaging. (2021) jeab226. doi: 10.1093/ehjci/jeab226
46. Dasgupta NR, Rissing SM, Smith J, Jung J, Benson MD. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid. (2020) 27:52–8. doi: 10.1080/13506129.2019.1685487
47. Adams D, Polydefkis M, González-Duarte A, Wixner J, Kristen AV, Schmidt HH, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. (2021) 20(1):49–59. doi: 10.1016/S1474-4422(20)30368-9
Keywords: cardiac amyloidosis, cardiopulmonary exercise testing, oxygen uptake, ventilatory efficiency, transthyretin (ATTR) amyloidosis, AL amyloidosis
Citation: Banydeen R, Monfort A, Inamo J and Neviere R (2022) Diagnostic and Prognostic Values of Cardiopulmonary Exercise Testing in Cardiac Amyloidosis. Front. Cardiovasc. Med. 9:898033. doi: 10.3389/fcvm.2022.898033
Received: 16 March 2022; Accepted: 09 May 2022;
Published: 06 June 2022.
Edited by:
Gian Marco Rosa, San Martino Hospital (IRCCS), ItalyReviewed by:
Shinichi Okuda, Yamaguchi Prefectural Grand Medical Center, JapanCopyright © 2022 Banydeen, Monfort, Inamo and Neviere. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Remi Neviere, cmVtaS5uZXZpZXJlQGNodS1tYXJ0aW5pcXVlLmZy
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.