 Sachiko Nishimoto
Sachiko Nishimoto Masataka Sata
Masataka Sata Daiju Fukuda
Daiju Fukuda
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 13 September 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.881181
This article is part of the Research Topic Insights in Atherosclerosis and Vascular Medicine: 2022 View all 23 articles
In lifestyle-related diseases, such as cardiovascular, metabolic, respiratory, and kidney diseases, chronic inflammation plays a causal role in their pathogenesis; however, underlying mechanisms of sterile chronic inflammation are not well-understood. Previous studies have confirmed the damage of cells in these organs in the presence of various risk factors such as diabetes, dyslipidemia, and cigarette smoking, releasing various endogenous ligands for pattern recognition receptors. These studies suggested that nucleic acids released from damaged tissues accumulate in these tissues, acting as an endogenous ligand. Undamaged DNA is an integral factor for the sustenance of life, whereas, DNA fragments, especially those from pathogens, are potent activators of the inflammatory response. Recent studies have indicated that inflammatory responses such as the production of type I interferon (IFN) induced by DNA-sensing mechanisms which contributes to self-defense system in innate immunity participates in the progression of inflammatory diseases by the recognition of nucleic acids derived from the host, including mitochondrial DNA (mtDNA). The body possesses several types of DNA sensors. Toll-like receptor 9 (TLR9) recognizes DNA fragments in the endosomes. In addition, the binding of DNA fragments in the cytosol activates cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS), resulting in the synthesis of the second messenger cyclic GMP-AMP (cGAMP). The binding of cGAMP to stimulator of interferon genes (STING) activates NF-κB and TBK-1 signaling and consequently the production of many inflammatory cytokines including IFNs. Numerous previous studies have demonstrated the role of DNA sensors in self-defense through the recognition of DNA fragments derived from pathogens. Beyond the canonical role of TLR9 and cGAS-STING, this review describes the role of these DNA-sensing mechanism in the inflammatory responses caused by endogenous DNA fragments, and in the pathogenesis of lifestyle-related diseases.
An organism needs to efficiently detect and resolve continual pathogenic attacks to maintain host-survival and homeostasis. The innate immune system protects the host from pathogenic infection by employing pattern recognition receptors (PRRs), which recognize pathogen-associated molecular patterns (PAMPs) and coordinate appropriate host defense mechanisms. PRRs include Toll-like receptors (TLRs), retinoic acid-inducible gene I-like receptors (RLRs), and NOD-like receptors (NLRs). After binding to their respective ligands, these receptors are activated, which results in the release of cytokines and chemokines. These first immune responses recruit antigen-presenting cells and leukocytes at the site of infection and induce subsequent adaptive immunity.
Toll-like receptors, which are the most familiar PRRs, are evolutionarily conserved and recognize various components came from bacteria, fungi, and viruses. There are 10 members of the TLR family in humans. Classically, most TLRs are categorized into two sub-groups. The first comprises TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11 which are primarily expressed on the cell surface, and their function is to recognize the components of microbial membranes (1). The other sub-group is composed of TLR3, TLR7, TLR8, and TLR9. These TLRs are expressed intracellularly in vesicles [e.g., lysosomes, endosomes, and the endoplasmic reticulum (ER)] and recognize microbial nucleic acids (2–5). In the past, numerous studies have examined the downstream signaling related to TLRs and demonstrated that it requires the recruitment of several adaptor proteins, which lead to the activation of the nuclear factor-kappa B (NF-κB) and interferon (IFN) regulatory factor (IRF) pathways, accelerating inflammatory responses (2). In addition to TLRs which recognize nucleic acid, cytoplasmic DNA sensors have been known. In particular, stimulator of interferon genes (STING) which recognizes second messenger cyclic guanosine monophosphateâ adenosine monophosphate (GMP-AMP) (cGAMP) generated from cyclic GMP-AMP synthase (cGAS) activated by DNA fragments in the cytosol have been well-studied (6–9). RNA sensors in the cytosol, such as RLRs also have been known (10, 11). Numerous studies have reported that multiple pathways related to inflammation, such as IRFs, NF-κB, and inflammasomes, are activated after these DNA sensors bind to their ligands (12).
Non-communicable diseases (NCDs) are a major contributor to the global burden of disease and account for up to 72% of worldwide deaths (13). Chronic low-grade inflammation, characterized by persistent elevated concentrations of circulating pro-inflammatory cytokines, has been associated with the development of both age and diet-related NCDs, including obesity, cardiometabolic diseases, respiratory and auto-immune diseases, and many cancers (14–16). Recent studies have demonstrated that PRR signaling contributes not only to innate immune responses but also to the pathogenesis of various inflammatory diseases. Especially, the TLR9 signaling and STING signaling have attracted much attention, because emerging evidence suggested their roles in the pathogenesis of lifestyle-related diseases. Lifestyle-related diseases are a group of diseases that onset and progression closely link with lifestyle and behavior factor(s), such as dietary habits, physical activities, rest, smoking, alcohol consumption, etc. Especially cardiovascular diseases, metabolic disorders, respiratory diseases including chronic obstructive pulmonary disease (COPD), chronic kidney diseases (CKD) are focused as major lifestyle-related diseases, that are a health threat to humans in recent decades (17–19). Beyond the canonical role of TLR9 and cGAS-STING in antimicrobial and antiviral immunity, the functional roles of TLR9 and cGAS-STING to lifestyle-related diseases has emerged from recent expanding evidence. This review briefly summarizes the role of TLR9 and STING signaling in the pathogenesis of inflammation caused by self-derived DNA fragments. This review also highlights the roles of the DNA sensing system in the pathophysiology of lifestyle-related diseases and discusses its potential as a therapeutic target for these diseases.
Sterile chronic inflammation is recognized as a shared mechanism of vascular diseases and metabolic diseases; however, the molecular mechanisms of sterile chronic inflammation remain a major medical problem that is yet to be solved. Though the mechanisms which cause DNA damage is multifactorial (20), DNA damage has been reported to play a crucial role in the development of these diseases (21, 22). Previous studies demonstrated that higher oxidative stress (23) and lower oxygen pressure (24) related to pathologic condition in unhealthy lifestyles and metabolic risk factors cause the deterioration of the cells in the vascular system and tissue of the metabolic organs. Subsequently, damaged genomic DNA and mitochondrial DNA (mtDNA) (25–29) are released and/or accumulated within the body (14, 30–32). We previously reported the accumulation of DNA fragments in macrophages, which infiltrate into atherosclerotic lesions and adipose tissue by using immune-electron microscopy and inflammatory activation of macrophages by DNA fragments (17–19). These results suggested that pro-inflammatory activation of macrophages by DNA damage play a key role in the pathophysiology of cardiometabolic diseases (33).
In developed countries, chronic kidney disease (CKD) is the most commonly attribute to diabetes and hypertension. The progression of CKD is associated with adverse clinical outcomes, including end-stage renal disease (ESRD), cardiovascular disease, and increased mortality (34–36). In the kidney, tubular cells contain enriched mitochondria to prepare for higher energy consumption. Recent studies highlight a pathogenic role of mitochondrial damage in the development of kidney disease. In fact, several kidney diseases such as diabetic nephropathy, tubulo-nephritis, and CKD show elevated mtDNA levels not only in the plasma but also in the urine (37–39). A recent study showed that urinary mtDNA levels have no significant association with the rate of worsening of renal function in non-diabetic CKD, although the levels correlate with baseline renal function, proteinuria, and the severity of histological damage (40).
Chronic obstructive pulmonary disease is a respiratory disorder characterized by irreversible limited expiratory airflow and abnormal inflammation. Etiologically, cigarette smoking (CS) is a major risk factor for COPD (41). Here, the impaired function of alveolar macrophages is a notable factor (42). CS-induced abnormal inflammatory responses amplify protease expression and oxidative stress, which accelerate COPD pathogenesis (43, 44). These processes further damage lung cells, including epithelial, vascular, and inflammatory cells, thus altering the lung microenvironment and enhancing the release of endogenous ligands. In fact, previous in vitro and in vivo studies have demonstrated that CS increased mtDNA damage (45, 46).
Deoxyribonucleic acid damage is also a potential marker of inflammatory diseases. The presence of extracellular DNA, which is named as cell-free DNA (cfDNA), has been known for a long time (47). Furthermore, recent studies have reported positive correlations between circulating cfDNA levels and the disease condition such as traumas (48–50), sepsis (51), cancer (52) or inflammatory diseases including autoimmune diseases (53–57), ESRD (58, 59), and neurodegenerative diseases (60). Recent clinical studies also have shown positive correlations between plasma cfDNA levels and the development of cardiometabolic disorders in humans. A clinical study that used coronary computed tomographic (CT) angiography demonstrated that patients with severe coronary artery disease had significantly higher levels of plasma double-stranded DNA and nucleosomes than those in the control group (61). Our previous study that used optical coherence tomography also revealed a positive correlation between cfDNA levels in the target artery and the inflammatory features of plaque in the target lesion of patients with acute myocardial infarction (17). In addition, we demonstrated that obese individuals presented higher cfDNA levels in the plasma and that the severity of abdominal adiposity and insulin resistance positively correlated with plasma levels of cfDNA (33). Similarly, several studies have reported that CS triggered DNA damage, releasing self-derived DNA into the plasma and alveolar space (62, 63). Thus, DNA damage and cfDNA has drawn increasing attention as a causal factor which initiates and accelerates vascular and metabolic diseases (20, 32).
Therefore, investigating pro-inflammatory roles of endogenous DNA fragments released from the host and the mechanisms by which endogenous DNA fragments accelerates inflammation associated with lifestyle-related diseases have become a research topic of great interest.
Toll-like receptor 9 is a well-studied DNA-sensing TLR. It recognizes unmethylated CpG motif-containing DNA fragments and induces innate immune response (64). After binding with ligands, TLR9 activates inflammatory pathways such as myeloid differentiation primary response 88 (MyD88)–IRF7 pathway and MyD88–NF-κB pathway, resulted in the production of type I IFN and inflammatory cytokines (Figure 1; 45, 65–67).
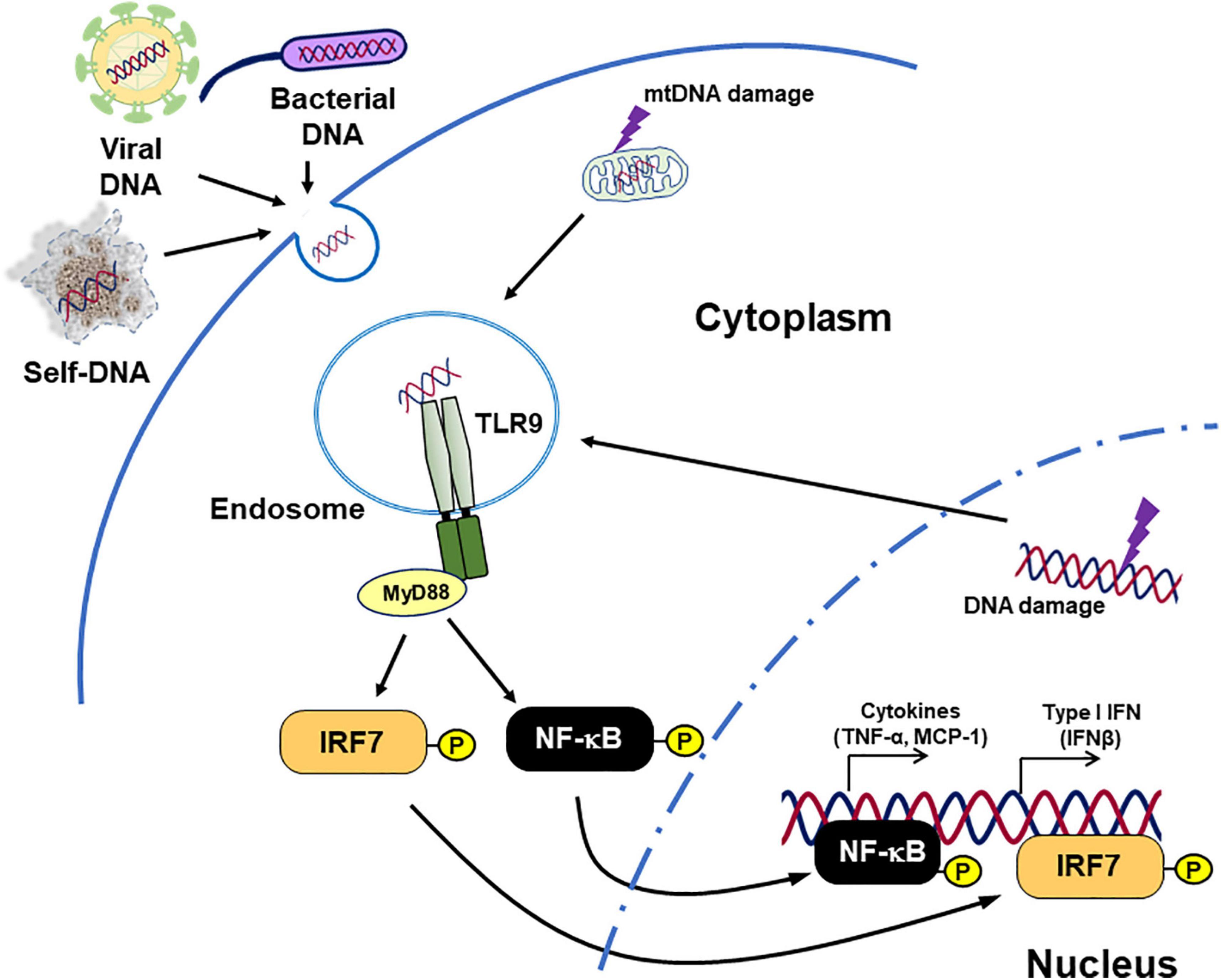
Figure 1. Inflammation caused by TLR9. TLR9 binds not only DNA originating from bacteria and viruses but also endogenous DNA and triggers signaling cascades related to pro-inflammatory responses. Diseases such as cancer, chronic infection, and lifestyle-related diseases can modulate TLR9 expression. In addition, tissue damage caused by these diseases increases the release of TLR9 agonists. This figure is reproduced from Fukuda et al. (80) with modification.
The cGAS-STING pathway is originally known as cytosolic DNA sensor machinery which recognizes pathogen-derived DNA, thus regulating the innate immune response (6, 68–70). STING ligates with cGAMP which is generated by cGAS activated with DNA fragments presented in the cytoplasm. Subsequently, STING activates NF-κB and IRF3, inducing IFNs and other pro-inflammatory cytokines (Figure 2; 7–9, 68, 71, 72).
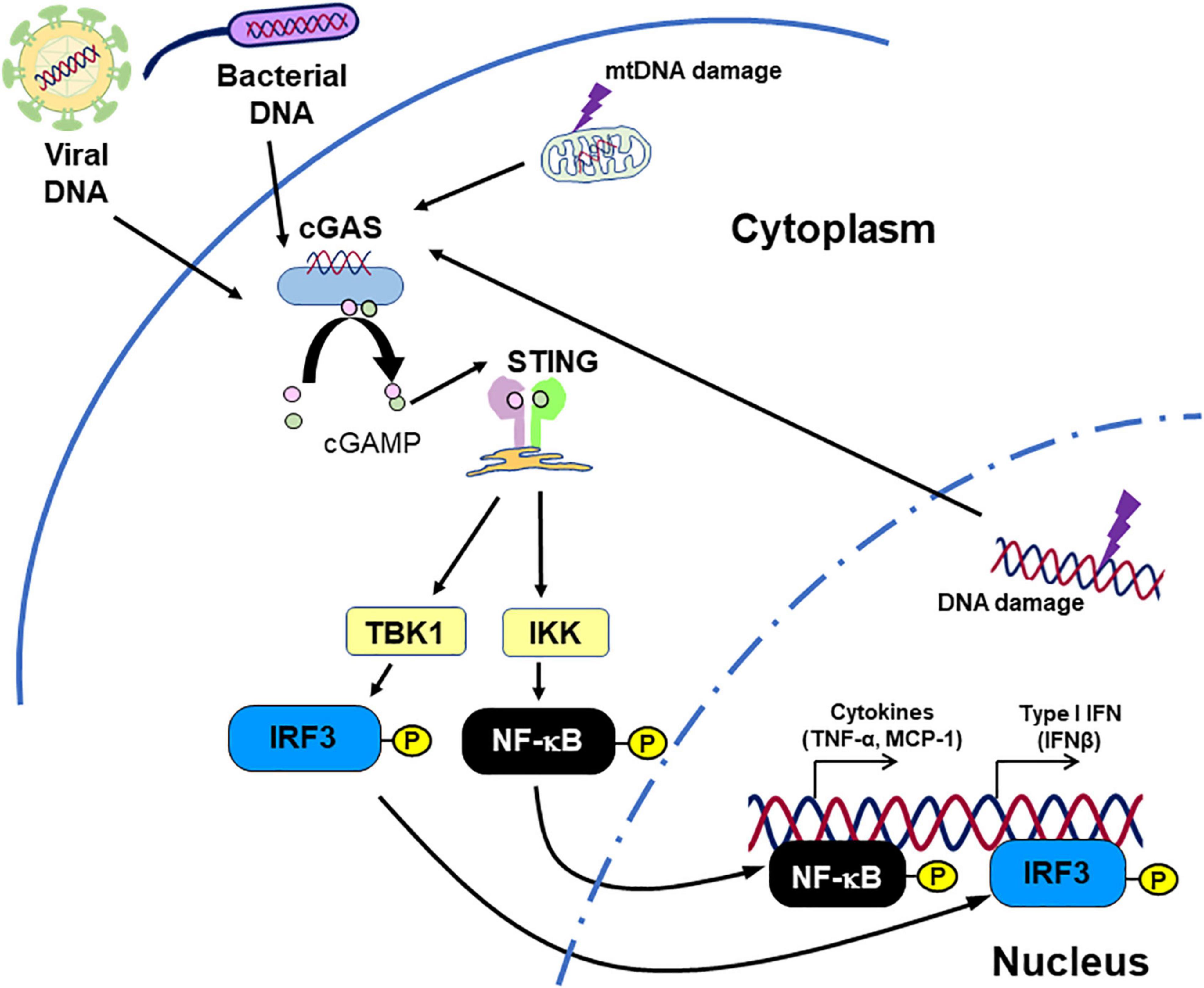
Figure 2. Inflammation caused by STING. STING recognizes cGAMP, which originates from DNA fragments. Not only DNA fragments derived from pathogens, but also endogenous DNA fragments can activate STING signaling, promoting inflammatory responses. This figure is reproduced from Fukuda et al. (80) with modification.
Due to lifestyle changes, the prevalence of obesity is increasing worldwide. This condition has close link with multiple metabolic abnormalities, including insulin resistance, and hepatic steatosis. Chronic sterile inflammation in metabolic organs plays a central role in the pathology of obesity and its associated complications. The mechanisms by which obesity promotes inflammation in metabolic organs are still undefined. However, obesity disturb the balance of the hypertrophy and the proliferation of adipocytes, angiogenesis, and the role of immune cells (24, 73, 74) because of higher oxidative stress, lower oxygen pressure (23), and excess inflammation, initiating cellular degeneration in adipose tissue (75–77). Here, local and/or systemic adipocyte-derived factors are suggested to participate in multiple pathways which accelerate inflammation within adipose tissue (77). Thus, many studies have been investigating the contribution of TLRs (78, 79). Recent studies, including our own, have demonstrated that among adipocyte-derived factors, self-derived DNA fragments released from metabolic organs promote chronic sterile inflammation by acting as endogenous ligands for DNA sensors.
We previously demonstrated that obesity caused by a high fat diet (HFD)-feeding increased plasma single-stranded DNA (ssDNA) levels in mice. Similarly, patients with visceral obesity, determined by CT, showed higher plasma ssDNA levels compared with the non-obese control. Furthermore, we reported that plasma ssDNA levels have a positive correlation with insulin resistance as determined by HOMA-IR, in humans (33). We also investigated the role of TLR9 in adipose tissue inflammation as it is one of the major receptors of nucleic acids (2). At first, we confirmed that HFD-induced obesity promotes TLR9 expression in the visceral fat. To explore the role of TLR9 in obese condition, we employed diet-induced obesity mouse model. TLR9 deficiency decreases macrophage accumulation into the visceral fat along with the reduction of inflammation in the adipose tissue and the inhibition of the development of obesity-induced insulin resistance (33). Similarly, pharmacological blocking of TR9 with iODN2088, a specific inhibitory oligonucleotide for TLR9, in HFD-fed wild-type mice suppressed inflammation in adipose tissue and ameliorated insulin sensitivity. In contrast, restoration of TLR9 only in BM aggravated insulin resistance in HFD-fed TLR9 deficient mice. In agreement with in vivo studies, our in vitro studies revealed that DNA fragments released from obese adipocytes partially promoted pro-inflammatory activation of macrophages via TLR9 signaling. Our results indicate that obesity overstimulates the innate immune system by increasing both ligand and receptor levels. In tandem, these results suggest a link between TLR9 and obesity-associated insulin resistance. Simultaneously, these results suggested that cfDNA-TLR9 signaling can be a potential therapeutic target for insulin resistance in obese subjects. Other TLR9 ligands, such as HMGB1, might also participate in TLR9 activation in obese subjects (81). However, a previous study reported opposite results. In that study, the authors demonstrated the protective role of TLR9 in the development of insulin resistance by showing the exacerbation of insulin resistance and pro-inflammatory activation of macrophages in TLR9 deficient mice (82). Further studies are needed to explore the function of TLR9 in the pathogenesis of obesity-induced insulin resistance.
Recent evidence has also demonstrated the contribution of TLR9 signaling to the pathogenesis of non-alcoholic fatty liver disease (NAFLD) (27). A preclinical study using a mouse model reported that TLR9 signaling activated by mtDNA accelerates the progression of hepatocyte damage and liver fibrosis, and that a TLR7/9 antagonist ameliorated the development of hepatic steatosis. As with animal studies, clinical studies have revealed that patients with non-alcoholic steatohepatitis (NASH) exhibits higher mtDNA levels compared to the controls (83, 84). Metabolic stresses caused by fat accumulation in the liver are thought to induce hepatocyte damage, leading to the release and accumulation of endogenous DNA fragments. DNA fragment-induced TLR9 activation might be an important driver of inflammatory responses in this widespread liver disease. Thus, TLR9 activation caused by self-derived DNA may be one of the molecular mechanisms of NAFLD.
In addition to TLR9, the expression of signaling molecules related to STING pathway is enhanced in metabolic organs in obese mouse models (85–87). For instance, the activation of cGAS– cGAMP–STING pathway by mtDNA derived from adipose tissues promoted chronic sterile inflammation in adipose tissue and contributed to the development of insulin resistance (87, 88). The activation of STING signaling in pancreatic β-cells was also documented in genetically obese mice such as db/db mice, suggesting that STING signaling is involved in the pathophysiology of type 2 diabetes (T2D) which is characterized by dysfunction of pancreatic β-cells (89).
Resembling TLR9, a number of studies have reported the role of STING signaling in the disease processes in NAFLD (90–92). The excess of fat accumulation in the liver triggers mitochondria dysfunction and mtDNA damage in hepatocytes (93). The activation of STING induced by these mtDNA fragments increases production of type I IFN (90, 91), accelerating oxidative stress and inflammation as hepatic diseases develop (94). In fact, genetically deficiency of STING ameliorated non-alcoholic steatohepatitis and insulin resistance in wild-type mice (90, 91). Here, Kupffer cells, a type of macrophage in the liver, are suggested to play a pivotal role. Kupffer cells increase the expression of inflammatory molecules such as TNF-α and interleukin-6 in the response to released mtDNA from hepatocytes, which accelerates disease processes (91). Furthermore, some clinical studies have shown potential contribution of STING signaling in patients with NAFLD by linking the release of mtDNA and the progression inflammation and fibrosis in the liver (92, 95).
Lifestyle and dietary changes tend to enlarge the number of patients with obesity and its complications worldwide. This could result in more attention being directed toward the role of TLR9 and STING signaling in the development of metabolic diseases, such as insulin resistance and hepatic diseases and advances being made in new therapeutic strategies. To explore the function of DNA sensors in the pathogenesis of metabolic diseases further studies are warranted.
Chronic inflammation in the vascular system initiates impairment of endothelial function, accelerating atherogenic process (96). Efficient intervention to risk factors such as dyslipidemia, T2D, and hypertension decreases cardiovascular events, although significant residual risk is still concerned (97). This also indicates that molecular and cellular mechanisms of atherogenesis are not completely understood.
Accumulating evidence demonstrates the participation of innate immune system in the process of vascular inflammation despite its multifactorial etiology (98). A variety of cells in arterial lesions, including endothelial cells, macrophages, and dendritic cells express PRRs, including TLRs (99–102). TLR9 plays a crucial role in atherosclerosis development. Several studies have demonstrated that the stimulation of TLR9 signaling accelerates pro-inflammatory activation of macrophages and dendritic cells (103–105). In addition, we found that ODN1826, a TLR9 agonist, partially through p38 MAPK signaling, increased the expression of pro-inflammatory molecules in apolipoprotein E-deficient (ApoE KO) macrophages (17). Previous studies have revealed the damage of vascular cells in atherosclerotic lesions (106–108), suggesting the release of various endogenous ligands for TLRs (109). In our in vivo study, genetic deletion of TLR9 suppressed atherogenesis in ApoE KO mice which received angiotensin II infusion (17). The blockade of TLR9 with the administration of iODN2088, an inhibitory oligodeoxynucleotide specific to TLR9, reduced atherosclerotic lesion development when compared to the control group in the same mouse model. Both genetical and pharmacological TLR9 blocking also abated the inflammatory features of atherosclerotic plaques at both the RNA and protein levels, while restoration of TLR9 in the bone marrow exacerbated atherogenesis in TLR9-deficient ApoE KO mice. These findings suggest that TLR9 has pro-atherogenic roles (17). Similarly, another study reported pharmacological blockade of TLR9 by IRS869 mitigated atherosclerotic lesion development and shifted macrophage polarization to the anti-inflammatory M2 phenotype (110). Pro-atherogenic properties of TLR9 was also reported by showing impaired reendothelialization and advanced atherosclerotic plaques in ApoE KO mice which received the administration of TLR9 agonist (111). Furthermore, we demonstrated that TLR9 contributes to the impairment of blood flow recovery in the ischemic limb by using a hind-limb ischemia model (18). TNF-α released from accumulated macrophages via TLR9 signaling played an important role. All these studies suggest that TLR9 activation enhances inflammatory responses and accelerates the development of vascular diseases.
Several groups have reported incongruous results by reporting that TLR9 has anti-atherogenic effects (112–114). Koulis et al. (114) demonstrated anti-atherogenic role of TLR9 by showing that TLR9-deficient ApoE KO mice exhibited increased inflammation in the plaque along with an increase in blood lipid levels. They also reported that the administration of CpG-ODN1668, a TLR9 agonist, suppressed atherosclerotic lesion development in ApoE KO mice. Thus, the role of TLR9 which have been reported is discrepant. Interestingly, a previous study reported conflicting roles of TLR9 activation depending on the concentration of its ligand (115). Therefore, the difference between the mouse model and experimental methods might result in the variance in the levels of ligands, which explains the discrepancy reported in previous studies. Further investigations are needed to clarify the effects of TLR9 on atherosclerosis.
A number of previous studies have investigated the role of cGAS-STING pathway as a major cytosolic DNA sensor and demonstrated its activation in response not only to pathogen-derived DNA but also endogenous DNA (6, 68–70). Amongst others, we have described the role of STING in the pathogenesis of atherosclerosis. To explore direct evidence of the contribution of STING signaling in vascular inflammation and subsequent atherogenesis, we first attempted to detect the presence of DNA damage in mouse atherosclerotic lesions by using WTD-fed ApoE KO mice, one of the widely used hypercholesterolemic mouse models, because released endogenous DNA initiates the production of STING ligands (19). The results of western blotting and immune-electron microscopy demonstrated the expression of γH2AX, a DNA damage marker, and the accumulation of DNA fragments in macrophages, respectively. Furthermore, we demonstrated the presence of cGAMP, a direct agonist of STING, in the atherosclerotic aorta of this mouse model using liquid chromatograph–mass spectrometry. Therefore, we deleted STING in ApoE KO mice to investigate its role in atherogenesis. Genetic deletion of STING decreased atherogenesis and attenuated the inflammatory features of the vasculature. Pharmacological blocking of STING using C-176 also decreased atherogenesis in the aorta compared to that in control groups. In contrast, its BM-specific expression promotes atherosclerotic lesion progression in ApoE KO mice. These results suggest causal roles of STING in the pathogenesis of atherosclerosis development. Additionally, a recent study showed that the genetic deficiency of IRF3, which is an adaptor molecule in downstream of STING signaling, attenuated the progression and the vulnerability of atherosclerotic plaques in ApoE KO mice (116). Similarly, administration of IFN-β, which is a downstream molecule of STING signaling, promotes atherogenesis in hypercholesterolemic mouse models (117). In our study, STING deficiency reduced the expression of IFN-β in the aorta of ApoE KO mice (19), which is consistent with these results. We further demonstrated that both cGAMP and mtDNA promoted the expression of inflammatory molecules, such as IFN-β, in both mouse and human macrophages (19). In addition, we demonstrated the expression of STING and cGAMP in atherosclerotic plaques collected by carotid endarterectomy, the levels of which were significantly higher in atherosclerotic lesions than control samples purchased from a tissue bank (19).
Recently, one study reported causal role of STING signaling in the pathogenesis of aortic disease (118). Genetic deletion of STING significantly decreased the aortic diameter, dissection, and aortic aneurysm formation in a mouse model. The underlying mechanisms included damage and release of DNA fragments from smooth muscle cells, and activation of macrophages by these DNA fragments. They also showed the inhibitory effects of C-176, a specific STING inhibitor, in their mouse model, suggesting the contribution of STING signaling to aortic aneurysm formation. The results of these studies suggest that the activation of STING signaling promotes vascular inflammation and that it could be a potential therapeutic focus for vascular diseases.
Contribution of STING to the development of vascular diseases has been also suggested in humans. The gain-of-function mutation in STING is reported to have close link with vasculopathy observed in STING-associated vasculopathy with onset in infancy (SAVI), which is a rare familial autoinflammatory disease (119–121). Enhanced IFN-β transcription in peripheral blood mononuclear cells in SAVI patients is thought to be one of the mechanisms involved (115). In contrast, one study reported protective effect of single-nucleotide polymorphism R293Q on STING on cardiovascular disease associated with obesity (122, 123). Evidence regarding the contribution of cGAS–cGAMP–STING signaling to vascular diseases is still limited; however, the results of recent studies suggest that STING signaling contributes to the pathogenesis of vascular diseases.
Thus, the role thought to be played by TLR9 and cGAS–cGAMP–STING signaling is expanding to vascular diseases in addition to the innate immune system. Further investigation of the role of these signaling in the development of vascular diseases would improve the understanding of the pathogenesis of atherosclerosis and might stimulate the development of new therapeutic approaches.
The roles of TLRs in inflammation observed in kidney diseases have been established in both animal models and patients. Several studies have confirmed the association between the TLR9 gene and CKD (124, 125). A human study demonstrated that patients with ESRD showed significant upregulation of TLR2 and TLR4, but not TLR7 or TLR9, in monocytes (126). On the other hand, polymorphisms of TLR9 have been confirmed to be associated with CKD in the Han Chinese population (124). A following study showed that the -1237T/C SNP of the TLR9 gene is significantly associated with ESRD in this population and that -1237T/C may be involved in the development of ESRD through transcriptional modulation of TLR9 (125). Therefore, TLR9 may play a critical role in the development of CKD.
Chronic kidney diseases is manifested by chronic inflammation, with continuous, unsuccessful injury-repair cycles and following fibrosis. These processes involve the activation of macrophages (127). Previous studies have reported the leading role of chronic inflammation and macrophage polarization in the progression of CKD. An animal study demonstrated that systemic exposure to CpG-DNA 1668, one of the TLR9 ligand, increases CD11b + /Ly6Chi macrophages and induces classically activated renal M1 macrophages that enhance intrarenal inflammation and disease progression of Alport nephropathy and other types of chronic kidney diseases (128). Activation of TLR9 induces accumulation of M1 macrophages and increased expression of pro-inflammatory cytokines in the renal interstitial compartment (129). Several studies also have explored the role of TLR9 using experimental acute kidney injury (AKI). Previous studies indicated that TLR9 does not contribute to the development of ischemic AKI by showing that TLR9 deficient mice were not protected against ischemic AKI (130, 131). In contrast, Han et al. reported that renal proximal tubular TLR9 activation exacerbates ischemic AKI by accelerating renal tubular inflammation, apoptosis as well as necrosis via NF-κB and caspase activation after ischemia-reperfusion injury, which leads to the development of AKI (132–134). The fibrosis of the kidney is another feature of AKI. One animal study using a mouse AKI-CKD transition model demonstrated that attenuation of CKD in the TLR9 deficient mice mainly relies on the effects of TLR9 on macrophages (129). In this study, TLR9 deficiency decreases the number of leukocyte and macrophage in the kidney following ischemia-reperfusion injury.
Chronic kidney diseases is associated with accelerated atherosclerosis progression and high incidence of cardiovascular events (135–138). An animal study using 5/6 nephrectomy or unilateral nephrectomy in ApoE KO mice demonstrated that CKD markedly accelerates atherogenesis in ApoE KO mice (136). They suggested that the CKD models which employed ApoE KO mouse are a useful tool to explore the mechanisms of uremic atherosclerosis. Due to a typical oxidative stress, CKD has emerged as a particularly strong risk factor for CVD (135). Thrombotic events are more likely to occur in patients with CKD, as well as in ApoE KO mice with CKD (138), suggesting that the plaques in CKD possess vulnerable features. A recent study using ApoE KO mice demonstrated that induction of CKD increases the release of mtDNA because of oxidative stress-induced mitochondrial damage, which activates the cGAS-STING pathway and subsequently induces type 1 IFN response in vascular smooth muscle cells (138). Interestingly, a recent study which used diabetic mouse models such as db/db mice and KKAy mice has reported that self-DNA-activated cGAS-STING pathway can be a new mechanism causing inflammation in the kidneys in diabetic condition (139). Alleviating type 1 IFN via cGAS-STING pathway may become a potential treatment strategy against diabetic kidneys as well as CKD-associated cardiovascular diseases.
These studies demonstrate that activation of the DNA sensors contributes to the development of kidney diseases, suggesting potential new therapeutic targets for preventing the progression of AKI, CKD, and diabetic kidney diseases. However, how DNA sensors affects CKD progression remains unclear. Further studies are needed.
Chronic obstructive pulmonary disease is a respiratory disorder characterized by irreversible limited expiratory airflow and aberrant inflammation. Inflammation plays a pivotal role in the pathogenesis of COPD (140, 141). The underlying molecular mechanisms are not completely understood; however, recent evidence has suggested that the innate immune system partially contributes to its pathogenesis (124, 142). Previous studies have determined the expression and function of TLRs in the development of COPD (143, 144).
The expression of TLR9 in the lungs (145) and its contribution to the pathogenesis of COPD have been reported (146). Foronjy et al. (147) demonstrated that genetic deletion of TLR9 prevents the development of CS-induced COPD in mice. The authors reported that in addition to inflammatory cells, epithelial cells play a pivotal role in COPD development. This is logical because the epithelium of the airways acts as the first line of defense against pathogens through the process of using a variety of receptors, such as TLRs. In immune responses, reactive-oxygen species (ROS) production is beneficial in the process of self-defense (148), and its accumulation is evident in patients with COPD (149). The overproduction of ROS is intended to abolish invading pathogens, meanwhile it can induce unwanted cellular damage. Excess ROS can directly initiate an inflammatory response and negatively affect tissue function and cellular structure (150). A previous study reported that ROS-induced mtDNA release stimulates immune cells because of TLR9 activation (151). The role and mechanism of activation of TLR9 in the development of COPD remain unclear; however, recent studies have reported that TLR9 polymorphisms are associated with both lung dysfunction and COPD (152, 153). Further study is required to understand the pathophysiology of COPD and develop novel therapeutic strategies that target TLR9.
Some viral-related innate immune mediators, such as RIG1, MDA5, LGP2, STING, and DAI are expressed in the lung tissue and bronchi of patients with COPD (154). Host-derived DNA fragments may act as pro-inflammatory signals for pulmonary inflammation (155). Recent data indicated the role of DAMPs in COPD (156) and many studies have attempted to reveal the relationship between STING and its ligand in the development of this particular disease (157). Nascimento et al. (62) showed that mouse CS-exposure promotes self-DNA release, which correlates with a neutrophil influx into the bronchoalveolar space via STING signaling. In a mouse model, acute CS exposure increased the self-DNA content in the alveolar space and accelerated the inflammatory response through the cGAS-STING pathway (62). Deslee et al. (158) demonstrated the nucleic-acid oxidation in alveolar fibroblasts of patients with severe emphysema. Similarly, CS exposure to mice accumulated nucleic acid oxidation in alveolar fibroblasts time dependently. DNase I treatment also reduced CS-induced lung inflammation (159). On the other hand, several previous studies have suggested that the suppression of cGAS-STING pathway and lower IFN levels are associated with a poor immune response to pathogens in patients with COPD (160, 161). These impeded STING-related immune responses may cause immune compromise in patients. Therefore, targeting the DNA-sensing mechanism such as STING is a double-edged sword. Further studies are needed to establish potential therapeutic strategies in lung diseases.
The detection of exogenous DNA is the most fundamental function of the innate immune system and is the first line of self-defense in the human body (67). This system promotes inflammation against endogenous DNA fragments, as well as exogenous DNA, under certain circumstances. The underlying mechanism by which DNA-sensing mechanisms cause an unwanted immune response to host-derived DNA remains completely unknown. In this review, we synthesized findings from a very large and rapidly growing body of research investigating associations between DNA-sensing mechanism and the development of lifestyle-related diseases (80). The evidence suggests that host-derived DNA fragment including mtDNA potently works as a stimulator of TLR9 and/or cGAS-STING pathway under a certain circumstance, accelerating chronic inflammation (Figure 3). Although there are increasing number of studies, remaining discrepancies of the results may attribute to the study design such as experiment duration, types of agonists and antagonists, and animal background. Furthermore, methods of preparing DNA sample from blood or culture medium and quantification of the amount, sequences of DNA fragments which have higher potential as exogenous ligands for DNA sensors, and the origin of DNA fragments should be established to elucidate the importance of this system as a therapeutic target in the future.
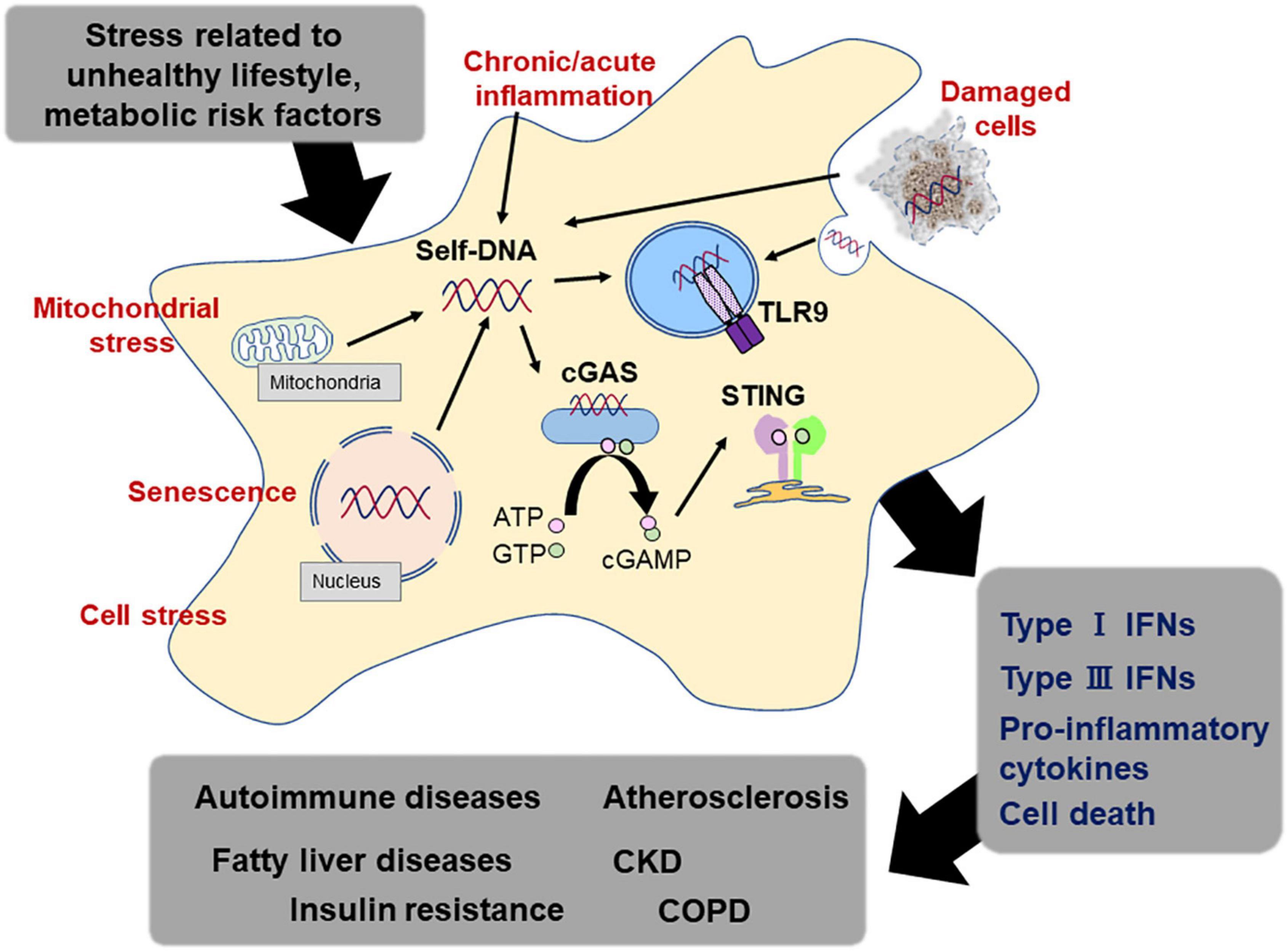
Figure 3. DNA sensing system and lifestyle-related diseases. Endogenous DNA fragments activate the DNA sensing mechanism, which participates in the activation of the immune response. Immune response caused by endogenous DNA fragments via DNA sensors such as TLR9 and STING accelerates sterile inflammation, leading to the development of lifestyle-related diseases, such as atherosclerosis, metabolic diseases, and pulmonary diseases.
As previous studies have demonstrated, risk factors, such as obesity, T2D, and dyslipidemia, induce tissue damage, which suggests the release of exogenous ligands, including nucleic acids. Therefore, controlling these risk factors by using the combination of medical treatment and a healthy lifestyle is indispensable in regulating inflammation caused by DNA-sensing mechanisms.
From the view of drug development, identifying the crosstalk between TLR9, cGAS-STING, and other types of DNA sensors of specific cell type in each stage of diseases will also help developing effective treatment in the future. Indeed, several animal studies have suggested that the administration of inhibitors for DNA sensors including TLR9 and STING attenuates the development of several lifestyle-related diseases such as insulin resistance, hepatic diseases, and atherosclerosis. However, DNA-sensing mechanism basically functions as self-defense. Further studies are needed to establish therapeutic strategies targeting this system. In addition, several studies have mentioned the crosstalk between TLR9 and cGAS-STING in response to DNA damage rerated with infectious diseases and some pathological conditions. In a mouse model of malarial infection, cGAS-STING pathway has been shown to induce suppressor of cytokine signaling (SOCS)1/3 to downregulate TLR9 signaling (162, 163). Deb et al. reported that triggering of the cGAS-STING pathway in plasmacytoid dendritic cells can induce expression of SOCS molecules, leading to inhibit the TLR9 pathway-mediated IFN production (162). In contrast, a synergistic role of TLR9 and STING by has also been reported in animal models such as acute peripheral tissue trauma models or other chemically induced lung injury (164, 165). Here, mtDNA activates neutrophils through both cGAS-STING and TLR9 pathways and leads to an increase in the production of neutrophil elastase and extracellular neutrophil-derived DNA in neutrophil extracellular traps, resulting in acceleration of subsequent sterile inflammation. At in vitro level, a study using cell lines such as human monocyte and human pDC has shown that a particular type of ODN can induce a strong cGAS-STING-dependent IFN response (166), suggesting that we need careful interpretation of the results derived from in vitro experiments which employed CpG-ODNs for distinguishing between TLR9- and cGAS-dependent effects. Until now, the number of studies which examined the crosstalk between multiple signal pathways related to innate immune systems in lifestyle-related diseases. Therefore, further detailed studies about the differences in the temporal and spatial expression and function of these innate immune signals in lifestyle-related diseases will help us to clarify the pathogenic roles of these DNA sensors in these diseases and provide evidence for further prevention and therapeutics.
In summary, DNA sensors, such as TLR9 and cGAS-STING, participate in the development of lifestyle-related diseases such as vascular, metabolic, kidney, and pulmonary diseases. These findings also highlight the potential benefits of transitioning to a healthy lifestyle to decrease disease risk. Further studies are required to understand the stimuli and mechanisms by which one of the most essential immune systems can play a harmful role, and to establish well-tolerated methods targeting DNA-sensing mechanism for these lifestyle-related diseases.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This work was partially supported by JSPS Kakenhi Grants (Number 20K19695 to SN, Number 19K08584 to DF, and Number 19H03654 to MS), Bristol-Myers Squibb Research Grants (DF), The 2022 Osaka Metropolitan University Strategic Research Promotion Project (Priority Research) (DF), The Uehara Memorial Foundation (DF), Takeda Science Foundation (MS), and the Vehicle Racing Commemorative Foundation (MS). The funders had no role in the study design, data collection and analysis, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84.
3. Yamamoto M, Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. (2010) 2010:240365.
4. Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol. (2012) 3:138. doi: 10.3389/fphys.2012.00138
8. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. (2014) 54:289–96.
9. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. (2016) 17:1142–9.
11. Schlee M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology. (2013) 218:1322–35. doi: 10.1016/j.imbio.2013.06.007
12. Okude H, Ori D, Kawai T. Signaling through nucleic acid sensors and their roles in inflammatory diseases. Front Immunol. (2020) 11:625833. doi: 10.3389/fimmu.2020.625833
13. Global Burden of Disease 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. (2017) 390:1151–210. doi: 10.1016/S0140-6736(17)32152-9
14. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85.
15. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation-nature’s way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front Med. (2018) 5:316. doi: 10.3389/fmed.2018.00316
16. Calder PC, Bosco N, Bourdet-Sicard R, Capuron L, Delzenne N, Doré J, et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res Rev. (2017) 40:95–119. doi: 10.1016/j.arr.2017.09.001
17. Fukuda D, Nishimoto S, Aini K, Tanaka A, Nishiguchi T, Kim-Kaneyama JR, et al. Toll-like receptor 9 plays a pivotal role in angiotensin ii-induced atherosclerosis. J Am Heart Assoc. (2019) 8:e010860. doi: 10.1161/JAHA.118.010860
18. Nishimoto S, Aini K, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, et al. Activation of toll-like receptor 9 impairs blood flow recovery after hind-limb ischemia. Front Cardiovasc Med. (2018) 5:144. doi: 10.3389/fcvm.2018.00144
19. Pham PT, Fukuda D, Nishimoto S, Kim-Kaneyama JR, Lei XF, Takahashi Y, et al. a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur Heart J. (2021) 42:4336–48. doi: 10.1093/eurheartj/ehab249
20. Shah NR, Mahmoudi M. The role of DNA damage and repair in atherosclerosis: a review. J Mol Cell Cardiol. (2015) 86:147–57.
21. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, et al. Mitochondrial integrity and function in atherogenesis. Circulation. (2002) 106:544–9.
22. Włodarczyk M, Nowicka G. Obesity, DNA damage, and development of obesity-related diseases. Int J Mol Sci. (2019) 20:1146.
23. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. (2004) 114:1752–61.
24. Sung HK, Doh KO, Son JE, Park JG, Bae Y, Choi S, et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. (2013) 17:61–72.
25. Fetterman JL, Holbrook M, Westbrook DG, Brown JA, Feeley KP, Bretón-Romero R, et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc Diabetol. (2016) 15:53.
26. Kawane K, Tanaka H, Kitahara Y, Shimaoka S, Nagata S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc Natl Acad Sci USA. (2010) 107:19432–7. doi: 10.1073/pnas.1010603107
27. Saito Y, Hikita H, Nozaki Y, Kai Y, Makino Y, Nakabori T, et al. DNase II activated by the mitochondrial apoptotic pathway regulates RIP1-dependent non-apoptotic hepatocyte death via the TLR9/IFN-β signaling pathway. Cell Death Differ. (2019) 26:470–86. doi: 10.1038/s41418-018-0131-6
28. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. (2018) 9:1249. doi: 10.1038/s41467-018-03555-8
29. Zhang B, Davidson MM, Hei TK. Mitochondria regulate DNA damage and genomic instability induced by high LET radiation. Life Sci Space Res. (2014) 1:80–8.
31. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. doi: 10.1038/nature10992
32. Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. (2014) 20:967–77.
33. Nishimoto S, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Murata C, et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci Adv. (2016) 2:e1501332. doi: 10.1126/sciadv.1501332
34. Matsushita K, Coresh J, Sang Y, Chalmers J, Fox C, Guallar E, et al. Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: a collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. (2015) 3:514–25.
35. Astor BC, Matsushita K, Gansevoort RT, van der Velde M, Woodward M, Levey AS, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int. (2011) 79:1331–40.
36. van der Velde M, Matsushita K, Coresh J, Astor BC, Woodward M, Levey A, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int. (2011) 79:1341–52.
37. Whitaker RM, Stallons LJ, Kneff JE, Alge JL, Harmon JL, Rahn JJ, et al. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney Int. (2015) 88:1336–44.
38. Wei PZ, Kwan BC, Chow KM, Cheng PM, Luk CC, Li PK, et al. Urinary mitochondrial DNA level is an indicator of intra-renal mitochondrial depletion and renal scarring in diabetic nephropathy. Nephrol Dial Transplant. (2018) 33:784–8. doi: 10.1093/ndt/gfx339
39. Chang CC, Chiu PF, Wu CL, Kuo CL, Huang CS, Liu CS, et al. Urinary cell-free mitochondrial and nuclear deoxyribonucleic acid correlates with the prognosis of chronic kidney diseases. BMC Nephrol. (2019) 20:391. doi: 10.1186/s12882-019-1549-x
40. Wei Z, Kwan BC-H, Chow KM, Cheng PM-S, Luk CC-W, Lai K-B, et al. Urinary mitochondrial DNA level as a biomarker of tissue injury in non-diabetic chronic kidney diseases. BMC Nephrol. (2018) 19:367. doi: 10.1186/s12882-018-1178-9
41. Salvi S. Tobacco smoking and environmental risk factors for chronic obstructive pulmonary disease. Clin Chest Med. (2014) 35:17–27.
42. Sethi S, Mallia P, Johnston SL. New paradigms in the pathogenesis of chronic obstructive pulmonary disease II. Proc Am Thorac Soc. (2009) 6:532–4.
44. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. (2008) 359:2355–65.
45. Kaisho T, Tanaka T. Turning NF-kappaB and IRFs on and off in DC. Trends Immunol. (2008) 29:329–36. doi: 10.1016/j.it.2008.03.005
46. Yao H, Yang SR, Kode A, Rajendrasozhan S, Caito S, Adenuga D, et al. Redox regulation of lung inflammation: role of NADPH oxidase and NF-kappaB signalling. Biochem Soc Trans. (2007) 35:1151–5.
47. Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost. (2015) 13:S82–91. doi: 10.1111/jth.12977
48. Simmons JD, Lee Y-L, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. (2013) 258:591–8. doi: 10.1097/SLA.0b013e3182a4ea46
49. Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PLoS One. (2013) 8:e72834. doi: 10.1371/journal.pone.0072834
50. Aswani A, Manson J, Itagaki K, Chiazza F, Collino M, Wupeng WL, et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front Immunol. (2018) 9:891. doi: 10.3389/fimmu.2018.00891
51. Yamanouchi S, Kudo D, Yamada M, Miyagawa N, Furukawa H, Kushimoto S. Plasma mitochondrial DNA levels in patients with trauma and severe sepsis: time course and the association with clinical status. J Crit Care. (2013) 28:1027–31. doi: 10.1016/j.jcrc.2013.05.006
52. Singel KL, Grzankowski KS, Khan A, Grimm MJ, D’Auria AC, Morrell K, et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br J Cancer. (2019) 120:207–17. doi: 10.1038/s41416-018-0339-8
53. Mosca M, Giuliano T, Cuomo G, Doveri M, Tani C, Curcio M, et al. Cell-free DNA in the plasma of patients with systemic sclerosis. Clin Rheumatol. (2009) 28:1437–40.
54. Arneth B. Systemic lupus erythematosus and DNA degradation and elimination defects. Front Immunol. (2019) 10:1697. doi: 10.3389/fimmu.2019.01697
55. Duvvuri B, Lood C. Cell-Free DNA as a biomarker in autoimmune rheumatic diseases. Front Immunol. (2019) 10:502. doi: 10.3389/fimmu.2019.00502
56. Rykova E, Sizikov A, Roggenbuck D, Antonenko O, Bryzgalov L, Morozkin E, et al. Circulating DNA in rheumatoid arthritis: pathological changes and association with clinically used serological markers. Arthritis Res Ther. (2017) 19:85. doi: 10.1186/s13075-017-1295-z
57. Swarup V, Rajeswari MR. Circulating (cell-free) nucleic acids–a promising, non-invasive tool for early detection of several human diseases. FEBS Lett. (2007) 581:795–9. doi: 10.1016/j.febslet.2007.01.051
58. Atamaniuk J, Kopecky C, Skoupy S, Säemann MD, Weichhart T. Apoptotic cell-free DNA promotes inflammation in haemodialysis patients. Nephrol Dial Transplant. (2012) 27:902–5. doi: 10.1093/ndt/gfr695
59. Coimbra S, Rocha S, Nascimento H, Valente MJ, Catarino C, Rocha-Pereira P, et al. Cell-free DNA as a marker for the outcome of end-stage renal disease patients on haemodialysis. Clin Kidney J. (2021) 14:1371–8.
60. Wilkins HM, Weidling IW, Ji Y, Swerdlow RH. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front Immunol. (2017) 8:508. doi: 10.3389/fimmu.2017.00508
61. Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. (2013) 33:2032–40. doi: 10.1161/ATVBAHA.113.301627
62. Nascimento M, Gombault A, Lacerda-Queiroz N, Panek C, Savigny F, Sbeity M, et al. Self-DNA release and STING-dependent sensing drives inflammation to cigarette smoke in mice. Sci Rep. (2019) 9:14848. doi: 10.1038/s41598-019-51427-y
63. Giordano L, Gregory AD, Pérez Verdaguer M, Ware SA, Harvey H, DeVallance E, et al. Extracellular release of mitochondrial DNA: triggered by cigarette smoke and detected in COPD. Cells. (2022) 11:369. doi: 10.3390/cells11030369
64. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. Toll-like receptor recognizes bacterial DNA. Nature. (2000) 408:740–5.
65. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. (2004) 5:190–8. doi: 10.1038/ni1028
66. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
67. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801.
68. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8.
69. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. (2008) 29:538–50.
70. Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. (2011) 79:688–94. doi: 10.1128/IAI.00999-10
71. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. (2013) 339:826–30.
72. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
74. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. (2008) 8:923–34.
75. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW II, DeFuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. (2007) 56:2910–8.
76. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. (2008) 49:1562–8. doi: 10.1194/jlr.M800019-JLR200
77. Rigamonti A, Brennand K, Lau F, Cowan CA. Rapid cellular turnover in adipose tissue. PLoS One. (2011) 6:e17637. doi: 10.1371/journal.pone.0017637
78. Fresno M, Alvarez R, Cuesta N. Toll-like receptors, inflammation, metabolism and obesity. Arch Physiol Biochem. (2011) 117:151–64.
80. Fukuda D, Pham PT, Sata M Emerging roles of the innate immune system regulated by DNA sensors in the development of vascular and metabolic diseases. J Atheroscler Thromb. (2022) 29:297–307.
81. Guzmán-Ruiz R, Ortega F, Rodríguez A, Vázquez-Martínez R, Díaz-Ruiz A, Garcia-Navarro S, et al. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. Int J Obes. (2014) 38:1545–54. doi: 10.1038/ijo.2014.36
82. Hong CP, Yun CH, Lee GW, Park A, Kim YM, Jang MH. TLR9 regulates adipose tissue inflammation and obesity-related metabolic disorders. Obesity. (2015) 23:2199–206. doi: 10.1002/oby.21215
83. Kamfar S, Alavian SM, Houshmand M, Yadegarazari R, Seifi Zarei B, Khalaj A, et al. Liver mitochondrial DNA copy number and deletion levels may contribute to nonalcoholic fatty liver disease susceptibility. Hepat Mon. (2016) 16:e40774. doi: 10.5812/hepatmon.40774
84. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. (2016) 126:859–64. doi: 10.1172/JCI83885
85. Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases TBK1 and IKK-ε improves obesity-related metabolic dysfunctions in mice. Nat Med. (2013) 19:313–21. doi: 10.1038/nm.3082
86. Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. (2018) 172:731–43.e12. doi: 10.1016/j.cell.2018.01.007
87. Bai J, Cervantes C, Liu J, He S, Zhou H, Zhang B, et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc Natl Acad Sci USA. (2017) 114:12196–201. doi: 10.1073/pnas.1708744114
88. Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, et al. IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol. (2017) 37:920–9.
89. Hu HQ, Qiao JT, Liu FQ, Wang JB, Sha S, He Q, et al. The STING-IRF3 pathway is involved in lipotoxic injury of pancreatic β cells in type 2 diabetes. Mol Cell Endocrinol. (2020) 518:110890. doi: 10.1016/j.mce.2020.110890
90. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, et al. Expression of STING Is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. (2018) 155:1971–84.e4. doi: 10.1053/j.gastro.2018.09.010
91. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING- mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. (2019) 129:546–55. doi: 10.1172/JCI121842
92. Wang X, Rao H, Zhao J, Wee A, Li X, Fei R, et al. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab Invest. (2020) 100:542–52. doi: 10.1038/s41374-019-0342-6
93. Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. (2006) 6:1–28. doi: 10.1016/j.mito.2005.10.004
94. Zhai Y, Qiao B, Gao F, Shen X, Vardanian A, Busuttil RW, et al. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. (2008) 47:199–206.
95. Akazawa Y, Nakashima R, Matsuda K, Okamaoto K, Hirano R, Kawasaki H, et al. Detection of DNA damage response in nonalcoholic fatty liver disease via p53-binding protein 1 nuclear expression. Mod Pathol. (2019) 32:997–1007. doi: 10.1038/s41379-019-0218-8
97. Aday AW, Ridker PM. Targeting residual inflammatory risk: a shifting paradigm for atherosclerotic disease. Front Cardiovasc Med. (2019) 6:16. doi: 10.3389/fcvm.2019.00016
98. Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. (2002) 91:281–91.
99. Roshan MH, Tambo A, Pace NP. The Role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. Int J Inflamm. (2016) 2016:1532832.
100. Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. (2001) 104:3103–8. doi: 10.1161/hc5001.100631
101. Scholtes VP, Versteeg D, de Vries JP, Hoefer IE, Schoneveld AH, Stella PR, et al. Toll-like receptor 2 and 4 stimulation elicits an enhanced inflammatory response in human obese patients with atherosclerosis. Clin Sci. (2011) 121:205–14. doi: 10.1042/CS20100601
102. Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. (2013) 191:4337–47. doi: 10.4049/jimmunol.1300298
103. Sorrentino R, Morello S, Chen S, Bonavita E, Pinto A. The activation of liver X receptors inhibits toll-like receptor-9-induced foam cell formation. J Cell Physiol. (2010) 223:158–67. doi: 10.1002/jcp.22022
104. Lee JG, Lim EJ, Park DW, Lee SH, Kim JR, Baek SH. A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell Signal. (2008) 20:2266–75. doi: 10.1016/j.cellsig.2008.08.022
105. Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation. (2006) 114:2482–9. doi: 10.1161/CIRCULATIONAHA.106.642801
106. Littlewood TD, Bennett MR. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol. (2003) 14:469–75.
107. Isner JM, Kearney M, Bortman S, Passeri J. Apoptosis in human atherosclerosis and restenosis. Circulation. (1995) 91:2703–11.
108. Martinet W, Schrijvers DM, De Meyer GR. Necrotic cell death in atherosclerosis. Basic Res Cardiol. (2011) 106:749–60.
109. Zheng Y, Gardner SE, Clarke MC. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2011) 31:2781–6.
110. Ma C, Ouyang Q, Huang Z, Chen X, Lin Y, Hu W, et al. Toll-like receptor 9 inactivation alleviated atherosclerotic progression and inhibited macrophage polarized to M1 phenotype in ApoE-/- mice. Dis Mark. (2015) 2015:909572. doi: 10.1155/2015/909572
111. Krogmann AO, Lüsebrink E, Steinmetz M, Asdonk T, Lahrmann C, Lütjohann D, et al. Proinflammatory stimulation of toll-like receptor 9 with high dose CpG ODN 1826 impairs endothelial regeneration and promotes atherosclerosis in mice. PLoS One. (2016) 11:e0146326. doi: 10.1371/journal.pone.0146326
112. Waibler Z, Anzaghe M, Konur A, Akira S, Müller W, Kalinke U. Excessive CpG 1668 stimulation triggers IL-10 production by cDC that inhibits IFN-alpha responses by pDC. Eur J Immunol. (2008) 38:3127–37. doi: 10.1002/eji.200838184
113. Bouaziz JD, Calbo S, Maho-Vaillant M, Saussine A, Bagot M, Bensussan A, et al. 10 produced by activated human B cells regulates CD4(+) T-cell activation in vitro. Eur J Immunol. (2010) 40:2686–91. doi: 10.1002/eji.201040673
114. Koulis C, Chen YC, Hausding C, Ahrens I, Kyaw TS, Tay C, et al. Protective role for Toll-like receptor-9 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2014) 34:516–25.
115. Wu J, Cui H, Dick AD, Liu L. TLR9 agonist regulates angiogenesis and inhibits corneal neovascularization. Am J Pathol. (2014) 184:1900–10. doi: 10.1016/j.ajpath.2014.03.001
116. Liu H, Cheng WL, Jiang X, Wang PX, Fang C, Zhu XY, et al. Ablation of interferon regulatory factor 3 protects against atherosclerosis in apolipoprotein E-deficient mice. Hypertension. (2017) 69:510–20. doi: 10.1161/HYPERTENSIONAHA.116.08395
117. Goossens P, Gijbels MJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, et al. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. (2010) 12:142–53. doi: 10.1016/j.cmet.2010.06.008
118. Luo W, Wang Y, Zhang L, Ren P, Zhang C, Li Y, et al. Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation. (2020) 141:42–66. doi: 10.1161/CIRCULATIONAHA.119.041460
119. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. (2014) 124:5516–20. doi: 10.1172/JCI79100
120. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
121. Munoz J, Rodière M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, et al. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol. (2015) 151:872–7. doi: 10.1001/jamadermatol.2015.0251
122. Hamann L, Ruiz-Moreno JS, Szwed M, Mossakowska M, Lundvall L, Schumann RR, et al. STING SNP R293Q is associated with a decreased risk of aging-related diseases. Gerontology. (2019) 65:145–54.
123. Hamann L, Szwed M, Mossakowska M, Chudek J, Puzianowska-Kuznicka M. First evidence for STING SNP R293Q being protective regarding obesity-associated cardiovascular disease in age-advanced subjects - a cohort study. Immun Ageing. (2020) 17:7. doi: 10.1186/s12979-020-00176-y
124. Lu KC, Yang HY, Lin YF, Kao SY, Lai CH, Chu CM, et al. The T-1237C polymorphism of the Toll-like receptor-9 gene is associated with chronic kidney disease in a Han Chinese population. Tohoku J Exp Med. (2011) 225:109–16. doi: 10.1620/tjem.225.109
125. Yang HY, Lu KC, Lee HS, Huang SM, Lin YF, Wu CC, et al. Role of the functional Toll-Like receptor-9 promoter polymorphism (-1237T/C) in increased risk of end-stage renal disease: a case-control study. PLoS One. (2013) 8:e58444. doi: 10.1371/journal.pone.0058444
126. Gollapudi P, Yoon JW, Gollapudi S, Pahl MV, Vaziri ND. Leukocyte toll-like receptor expression in end-stage kidney disease. Am J Nephrol. (2010) 31:247–54.
127. Engel JE, Chade AR. Macrophage polarization in chronic kidney disease: a balancing act between renal recovery and decline? Am J Physiol Renal Physiol. (2019) 317:F1409–13. doi: 10.1152/ajprenal.00380.2019
128. Ryu M, Kulkarni OP, Radomska E, Miosge N, Gross O, Anders HJ. Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-α-mediated podocyte loss. Kidney Int. (2011) 79:189–98. doi: 10.1038/ki.2010.373
129. Zheng H, Zhang Y, Li L, Zhang R, Luo Z, Yang Z, et al. Depletion of toll-like receptor-9 attenuates renal tubulointerstitial fibrosis after ischemia-reperfusion injury. Front Cell Dev Biol. (2021) 9:641527. doi: 10.3389/fcell.2021.641527
130. Li X, Yun Z, Tan Z, Li S, Wang D, Ma K, et al. The role of Toll-like receptor (TLR) 2 and 9 in renal ischemia and reperfusion injury. Urology. (2013) 81:1379.e15–20.
131. Bakker PJ, Scantlebery AM, Butter LM, Claessen N, Teske GJ, van der Poll T, et al. TLR9 mediates remote liver injury following severe renal ischemia reperfusion. PLoS One. (2015) 10:e0137511. doi: 10.1371/journal.pone.0137511
132. Han SJ, Li H, Kim M, Shlomchik MJ, Lee HT. Kidney proximal tubular TLR9 exacerbates ischemic acute kidney injury. J Immunol. (2018) 201:1073–85.
133. Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. (2015) 11:88–101.
134. Forni LG, Darmon M, Ostermann M, Oudemans-van Straaten HM, Pettilä V, Prowle JR, et al. Renal recovery after acute kidney injury. Intensive Care Med. (2017) 43:855–66.
135. Drüeke TB, Massy ZA. Atherosclerosis in CKD: differences from the general population. Nat Rev Nephrol. (2010) 6:723–35.
136. Bro S, Bentzon JF, Falk E, Andersen CB, Olgaard K, Nielsen LB. Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice. J Am Soc Nephrol. (2003) 14:2466–74. doi: 10.1097/01.asn.0000088024.72216.2e
137. Yang K, Du C, Wang X, Li F, Xu Y, Wang S, et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood. (2017) 129:2667–79. doi: 10.1182/blood-2016-10-744060
138. Bi X, Du C, Wang X, Wang XY, Han W, Wang Y, et al. Mitochondrial damage-induced innate immune activation in vascular smooth muscle cells promotes chronic kidney disease-associated plaque vulnerability. Adv Sci. (2021) 8:2002738. doi: 10.1002/advs.202002738
139. Myakala K, Jones BA, Wang XX, Levi M. Sacubitril/valsartan treatment has differential effects in modulating diabetic kidney disease in db/db mice and KKAy mice compared with valsartan treatment. Am J Physiol Renal Physiol. (2021) 320:F1133–51. doi: 10.1152/ajprenal.00614.2020
140. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. (2014) 35:71–86.
141. Di Stefano A, Caramori G, Ricciardolo FL, Capelli A, Adcock IM, Donner CF. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy. (2004) 34:1156–67.
142. Holt PG, Strickland DH, Wikström ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. (2008) 8:142–52.
143. Doz E, Noulin N, Boichot E, Guénon I, Fick L, Le Bert M, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. (2008) 180:1169–78.
144. Geraghty P, Dabo AJ, D’Armiento J. TLR4 protein contributes to cigarette smoke-induced matrix metalloproteinase-1 (MMP-1) expression in chronic obstructive pulmonary disease. J Biol Chem. (2011) 286:30211–8. doi: 10.1074/jbc.M111.238824
145. Schneberger D, Caldwell S, Kanthan R, Singh B. Expression of Toll-like receptor 9 in mouse and human lungs. J Anat. (2013) 222:495–503.
146. Nadigel J, Préfontaine D, Baglole CJ, Maltais F, Bourbeau J, Eidelman DH, et al. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8(+) T cells in chronic obstructive pulmonary disease. Respir Res. (2011) 12:149. doi: 10.1186/1465-9921-12-149
147. Foronjy RF, Salathe MA, Dabo AJ, Baumlin N, Cummins N, Eden E, et al. TLR9 expression is required for the development of cigarette smoke-induced emphysema in mice. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L154–66. doi: 10.1152/ajplung.00073.2016
148. Song JH, Ahn JH, Kim SR, Cho S, Hong EH, Kwon BE, et al. Manassantin B shows antiviral activity against coxsackievirus B3 infection by activation of the STING/TBK-1/IRF3 signalling pathway. Sci Rep. (2019) 9:9413. doi: 10.1038/s41598-019-45868-8
149. Zuo L, Hallman AH, Yousif MK, Chien MT. Oxidative stress, respiratory muscle dysfunction, and potential therapeutics in chronic obstructive pulmonary disease. Front Biol. (2012) 7:506–13. doi: 10.1007/s11515-012-1251-x
150. Zuo L, Zhou T, Pannell BK, Ziegler AC, Best TM. Biological and physiological role of reactive oxygen species–the good, the bad and the ugly. Acta Physiol. (2015) 214:329–48. doi: 10.1111/apha.12515
151. Lai JH, Wu DW, Wu CH, Hung LF, Huang CY, Ka SM, et al. Mitochondrial CMPK2 mediates immunomodulatory and antiviral activities through IFN-dependent and IFN-independent pathways. iScience. (2021) 24:102498. doi: 10.1016/j.isci.2021.102498
152. Berenson CS, Kruzel RL, Wrona CT, Mammen MJ, Sethi S. Impaired innate COPD alveolar macrophage responses and toll-like receptor-9 polymorphisms. PLoS One. (2015) 10:e0134209. doi: 10.1371/journal.pone.0134209
153. Pabst S, Bradler O, Gillissen A, Nickenig G, Skowasch D, Grohe C. Toll-like receptor-9 polymorphisms in sarcoidosis and chronic obstructive pulmonary disease. Adv Exp Med Biol. (2013) 756:239–45. doi: 10.1007/978-94-007-4549-0_30
154. D’Anna SE, Maniscalco M, Carriero V, Gnemmi I, Caramori G, Nucera F, et al. Evaluation of innate immune mediators related to respiratory viruses in the lung of stable COPD patients. J Clin Med. (2020) 9:1807. doi: 10.3390/jcm9061807
155. Pouwels SD, Faiz A, den Boef LE, Gras R, van den Berge M, Boezen HM, et al. Genetic variance is associated with susceptibility for cigarette smoke-induced DAMP release in mice. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L559–80. doi: 10.1152/ajplung.00466.2016
156. Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. (2014) 7:215–26.
157. Yu L, Liu P. Cytosolic DNA sensing by cGAS: regulation, function, and human diseases. Signal Transduct Target Ther. (2021) 6:170.
158. Deslee G, Adair-Kirk TL, Betsuyaku T, Woods JC, Moore CH, Gierada DS, et al. Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am J Respir Cell Mol Biol. (2010) 43:576–84.
159. King PT, Sharma R, O’Sullivan KM, Callaghan J, Dousha L, Thomas B, et al. Deoxyribonuclease 1 reduces pathogenic effects of cigarette smoke exposure in the lung. Sci Rep. (2017) 7:12128. doi: 10.1038/s41598-017-12474-5
160. García-Valero J, Olloquequi J, Montes JF, Rodríguez E, Martín-Satué M, Texidó L, et al. Deficient pulmonary IFN-β expression in COPD patients. PLoS One. (2019) 14:e0217803. doi: 10.1371/journal.pone.0217803
161. Qin H, Huang G, Gao F, Huang B, Wang D, Hu X, et al. Diminished stimulator of interferon genes production with cigarette smoke-exposure contributes to weakened anti-adenovirus vectors response and destruction of lung in chronic obstructive pulmonary disease model. Exp Cell Res. (2019) 384:111545.
162. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J. Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity. (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
163. Deb P, Dai J, Singh S, Kalyoussef E, Fitzgerald-Bocarsly P Triggering of the cGAS-STING pathway in human plasmacytoid dendritic cells inhibits TLR9-mediated IFN production. J Immunol. (2020) 205:223–36.
164. Temizoz B, Kuroda E, Ohata K, Jounai N, Ozasa K, Kobiyama K, et al. TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. Eur J Immunol. (2015) 45:1159–69. doi: 10.1002/eji.201445132
165. Liu L, Mao Y, Xu B, Zhang X, Fang C, Ma Y. Induction of neutrophil extracellular traps during tissue injury: involvement of STING and Toll-like receptor 9 pathways. Cell Prolif. (2019) 52:e12579–12579.
Keywords: DNA-sensing mechanism, chronic inflammation, atherosclerosis, metabolic diseases, COPD, TLR9, STING, CKD
Citation: Nishimoto S, Sata M and Fukuda D (2022) Expanding role of deoxyribonucleic acid-sensing mechanism in the development of lifestyle-related diseases. Front. Cardiovasc. Med. 9:881181. doi: 10.3389/fcvm.2022.881181
Received: 22 February 2022; Accepted: 15 August 2022;
Published: 13 September 2022.
Edited by:
Tatsuya Iso, Gunma University of Health and Welfare, JapanCopyright © 2022 Nishimoto, Sata and Fukuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daiju Fukuda, ZGFpanUuZnVrdWRhQG9tdS5hYy5qcA==, ZGFpanUuZnVrdWRhQHRva3VzaGltYS11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.