 Xuan Xiao
Xuan Xiao Yonghong Luo
Yonghong Luo Daoquan Peng
Daoquan Peng- Department of Cardiovascular Medicine, The Second Xiangya Hospital of Central South University, Changsha, China
Glucose and cholesterol engage in almost all human physiological activities. As the primary energy substance, glucose can be assimilated and converted into diverse essential substances, including cholesterol. Cholesterol is mainly derived from de novo biosynthesis and the intestinal absorption of diets. It is evidenced that glucose/insulin promotes cholesterol biosynthesis and uptake, which have been targeted by several drugs for lipid-lowering, e.g., bempedoic acid, statins, ezetimibe, and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. Inversely, these lipid-lowering drugs may also interfere with glucose metabolism. This review would briefly summarize the mechanisms of glucose/insulin-stimulated cholesterol biosynthesis and uptake, and discuss the effect and mechanisms of lipid-lowering drugs and genetic mutations on glucose homeostasis, aiming to help better understand the intricate relationship between glucose and cholesterol metabolism.
Introduction
As indispensable nutrients, glucose and cholesterol are of prime importance in maintaining human physiological activities. In normal physiological state, hepatic gluconeogenesis and glycogenolysis maintain the normal blood glucose level for continuous consumption for energy during fasting. Glycemic hormones, including glucagon, epinephrine, glucocorticoids, and asprosin, activate a series of signal pathways of hepatic gluconeogenesis and glycogenolysis (1).Glucagon is of principal significance in endogenous glucose production among these glycemic hormones. Postprandially, the elevated blood glucose level incites insulin secretion to stimulate peripheral uptake of blood glucose, promote hepatic glycogen synthesis, and repress gluconeogenesis, thereby maintaining a normal blood glucose level (1). Since the blood insulin level increases simultaneously with blood glucose after feeding, it's difficult to distinguish the effects of insulin and glucose in vivo. Thus, in the following review, we may not specify the effect of glucose or insulin in some cases. However, in in vitro studies (e.g., hepatocytes), when using glucose as a stimulator, it is mostly the effects of glucose but not insulin. Insulin may inhibit gluconeogenesis via multiple way, such as downregulating the expression of gluconeogenesis genes, suppressing the secretion of glucagon, reducing white adipose tissue lipolysis, and cutting down skeletal muscle proteolysis (1). Generally, glucose obtained from diets, gluconeogenesis, and glycogenolysis can be decreased by experiencing aerobic oxidation for energy or converting into energy storage substances (such as glycogen and lipids) with the assistance of insulin. However, individuals with type 1 diabetes mellitus (T1DM) or type 2 diabetes mellitus (T2DM) exhibit hyperglucagonemia and hyperglycemia because of insufficient insulin secretion or insulin resistance. Besides, patients with T2DM are also characterized by defected hepatic glucose uptake and enhanced hepatic gluconeogenesis, which collectively accelerate hepatic glucose production (2, 3). Cholesterol is one of the principal lipids of cell membranes in eukaryotic cells, and the content of cholesterol influences cell membranes' physical properties and functions (1). According to current knowledge, there are two pathways for a human to acquire cholesterol: absorbing cholesterol directly from diets and synthesizing de novo based on acetyl-CoA, an intermediate product of glycolysis, β-oxidation of fatty acids and catabolism of amino acids (4, 5). To date, it has been proved that glucose can not only provide raw materials for cholesterol synthesis but also serve as a regulator of cholesterol biosynthesis enzymes and cholesterol uptake (as shown in Figure 1). Clinical trials indicated that the antidiabetic drug metformin reduced the blood cholesterol level in both diabetic and non-diabetic individuals (ClinicalTrials.gov Identifier: NCT01483560) (6, 7). On the other hand, cholesterol may impact glucose homeostasis, as dyslipidemic subjects treated with statin tend to develop new-onset diabetes (NOD) (8, 9). The intricate crosstalk between glucose/insulin and cholesterol has not been sufficiently discussed. This review aims to discuss the relevant roles of glucose/insulin in the biosynthesis and uptake of cholesterol based on the updated findings. Meanwhile, we will briefly summarize the relevant roles of current cholesterol-lowering drugs and cholesterol metabolism-related gene mutations in glucose regulation (as shown in Figure 2).
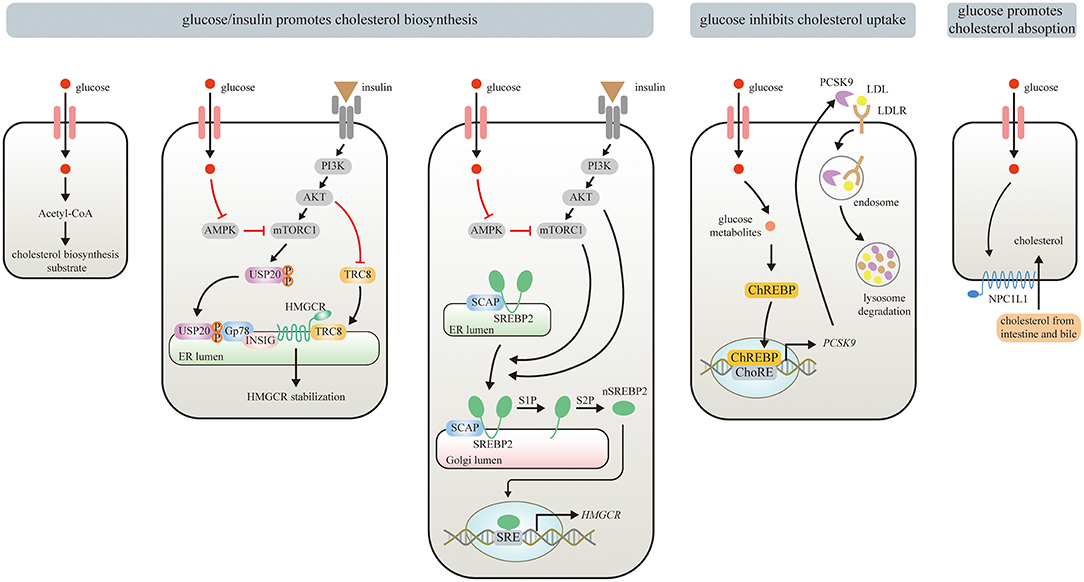
Figure 1. Effects of Glucose/insulin on cholesterol metabolism. Glucose provides Acetyl-CoA for cholesterol biosynthesis. Glucose/insulin also enhances cholesterol biosynthesis by stabilizing HMGCR and increasing HMGCR expression. Glucose or insulin activates mTORC1 by repressing AMPK or stimulating insulin-mediated PI3K/AKT signaling pathway, respectively. The USP20 phosphorylated by mTORC1 prevents HMGCR from being degraded by GP78. PI3K/Akt signaling pathway also stabilizes HMGCR via inhibiting the recruitment of E3 ligase TRC8. The upregulated mTORC1 can promote the translocation of SREBP2 from ER to the Golgi apparatus, where nSREBP2 is produced sequentially by the S1P and S2P. nSREBP2 translocates into the nucleus and binds to SRE sequences to stimulate HMGCR expression. Meanwhile, PI3K/Akt signaling pathway upregulates HMGCR via promoting SREBP–SCAP complex to migrate into the Golgi. Besides, glucose and its metabolites inhibit cholesterol uptake by activating ChREBP, which enters the nucleus to augment human PCSK9 expression, thereby increasing PCSK9-induced LDLR degradation. Moreover, elevated circulating glucose levels can enhance enterocyte NPC1L1 expression via some unknown mechanisms, thereby strengthening intestinal absorption of cholesterol.
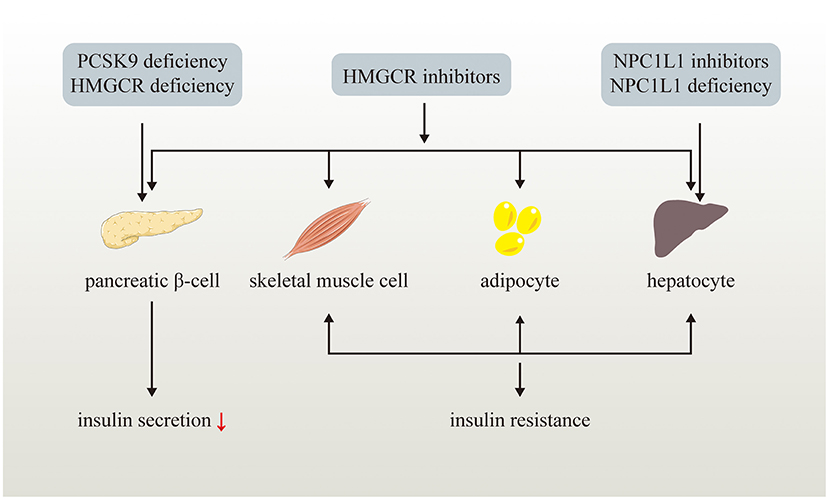
Figure 2. Effects of LDL-C lowering drugs or genetic variants on glucose metabolism. LDL-C lowering drugs or genetic variants disturb glucose homeostasis via multiple ways. Genetic PCSK9 deficiency impairs pancreatic β-cell insulin secretion while NPC1L1 inhibitors and genetic NPC1L1 deficiency lead to insulin resistance of hepatocyte. HMGCR inhibitors and genetic HMGCR deficiency impairs pancreatic β-cell insulin secretion and induce insulin resistance of skeletal muscle cell, adipocyte, and hepatocyte.
Role of Glucose in Cholesterol Metabolism
Glucose and Cholesterol Biosynthesis
Glucose Derived Acetyl-CoA Is the Material of Cholesterol Synthesis
Acetyl-CoA, one of the vital metabolites of glucose, serves as the direct raw material for endogenous cholesterol synthesis. Generally, acetyl-CoA deriving from glucose metabolism participates in various metabolic pathways as a substrate, such as in the tricarboxylic acid (TCA) cycle, in the acetylation reaction, and in the synthesis of ketone bodies, fatty acids, and cholesterol. The TCA cycle is initiated with the condensation of acetyl-CoA with oxaloacetate by citrate synthase, followed by the generation of citrate, the first intermediate product of the TCA cycle.
ATP-citrate lyase (ACL), which can convert citrate into oxaloacetate and acetyl-CoA, is proposed as a new target to reduce cholesterol synthesis (10). Bempedoic acid (ETC-1002) has been considered as a first-class lipid-lowering drug for its ability to inhibit the expression of ACL. Clinic trial disclosed that patients with hypercholesterolemia treated with bempedoic acid exhibit a significant reduction of low-density lipoprotein cholesterol (LDL-C) compared with placebo or standard treatment (ClinicalTrials.gov Identifier: NCT02988115) (11, 12). The beneficial effect was also observed in those who lack ample response to maximally tolerated lipid-lowering therapies (ClinicalTrials.gov Identifier: NCT02991118) (13). Meta-analyses of randomized controlled trials have shown that bempedoic acid treatment resulted in a decreased incidence of NOD (12, 14).
Glucose/Insulin Regulates HMGCR
It has been known since the 1970s that cholesterol biosynthesis is induced by feeding but suppressed by fasting, which is closely correlated with the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (HMGCR), the pivotal enzyme of cholesterol biosynthesis (15). The increased circulating glucose and insulin levels are the most remarkable changes after feeding, indicating that glucose/insulin may account for the change of HMGCR activity. It is found that the elevated glucose levels tend to downregulate the expression of adenosine monophosphate-activated protein kinase (AMPK) by lowering adenosine monophosphate (AMP)/ adenosine triphosphate (ATP) and adenosine diphosphate (ADP)/ATP ratios (16). The clinical data indicated that the first-line hypoglycemic drug, metformin, which can repress gluconeogenesis in hepatocytes via obstructing a mitochondrial redox shuttle and reduce net glucose uptake from diets by motivating anaerobic glucose metabolism of enterocytes, tended to increase AMPK and reduce serum LDL cholesterol and total cholesterol (17, 18). For example, treatment with 2.55 g/d metformin for 28 weeks reduced the plasma level of LDL-C by 14 mg/dL in 31 non-diabetic but morbidly obese individuals (7). A recent observational study including 912 participants indicated that treatment with 0.1 g/d metformin for 7 years was correlated with a 11.83 mg/dL reduction of LDL-C levels (17). Another study further revealed that plasma LDL-C levels were reduced by 16.79 mg/dL after treated with 2 g/d metformin added to titrated insulin therapy for 3 years in diabetic participants (ClinicalTrials.gov Identifier: NCT01483560) (6).
As an energy-sensing enzyme, AMPK is activated by an elevation in AMP/ATP and ADP/ATP ratios. AMPK activation tends to strengthen catabolism (e.g., glycolysis and fatty acid oxidation) but weaken anabolism (e.g., gluconeogenesis and cholesterol synthesis) (16, 19). Interestingly, it is shown that mammalian AMPK can be also restrained directly by extracellular glucose and intracellular fructose-1,6-bisphosphate in unchanged cellular energy conditions (20). The activated AMPK triggered by energy stress is likely to antagonize the biosynthetic process of cholesterol by suppressing the expression of the mammalian target of rapamycin complex 1 (mTORC1), a crucial nutrient sensor, which participates in the activation of HMGCR on the endoplasmic reticulum (ER) (21). It was reported that the repression of mTORC1 by AMPK was related to the upregulation of mTORC1 inhibitor, TSC2 gene (22). The interaction between circulating insulin and insulin receptor (INSR) phosphorylates insulin receptor substrates (IRSs), which can also enhance mTORC1 expression by initiating phosphoinositide 3-kinase (PI3K)/Akt (namely protein kinase B) signaling pathway (23). Furthermore, activation of PI3K/Akt signaling pathway caused by insulin can stabilize HMGCR via inhibiting the recruitment of E3 ligase TRC8, which can also supply an explanation for glucose/insulin-induced cholesterol synthesis (24).
Recently, Song et al. discovered that feeding would stabilize HMGCR via facilitating mTORC1 to phosphorylate Ser132 and Ser134 of the deubiquitylase ubiquitin-specific peptidase 20 (USP20), thereby protecting HMGCR from being degraded (25). Conversely, AMPK activation by fasting and metformin was likely to decrease the mTORC1 level, thereby accelerating HMGCR ubiquitination impelled by the E3 ubiquitin-protein ligase GP78 (25). Furthermore, USP20 knockout or administration of GSK2643943A, a specific USP20 inhibitor, distinctly lessened cholesterol biosynthesis after feeding compared with controls (25). Hence, it is proposed that the application of USP20 inhibitors may offer a new insight to lower cholesterol levels in hyperlipidemia (25).
Glucose/Insulin and SREBP2
Sterol regulatory element-binding proteins (SREBPs), a member of the membrane-bound transcription factors family, have received wide attention due to their role in regulating the synthesis of unsaturated fatty acids, cholesterol, and triglycerides (26). Three isoforms of SREBPs, namely SREBP1a, SREBP1c, and SREBP2, are encoded by SREBP1 gene and SREBP2 gene, respectively. SREBP1a and SREBP1c account for the activation of genes involved in fatty acids and triglyceride synthesis, such as fatty acid synthase (27), while SREBP2 promotes the transcription of enzymes that participated in cholesterol synthesis and uptake, including HMGCR, HMG-CoA synthase, and low-density lipoprotein receptor (LDLR) (27). When cholesterol is excess, SREBP2 is bound to SREBP cleavage-activating protein (SCAP) in ER. When the ER is deprived of cholesterol, the SREBP2-SCAP complex is transported to Golgi and SREBP2 is cleaved by two Golgi proteases (the Site-1 protease (S1P) and S2P) sequentially to release the active nuclear SREBP2 (nSREBP2). The nSREBP2 will be translocated to the nucleus and bind to nuclear sterol regulatory element (SRE) sequences, initiating the transcription of downstream genes (28).
Increased circulating glucose promotes insulin secretion after feeding. PI3K/Akt signaling pathway activated by insulin drives the movement of the SREBP–SCAP complex to the Golgi through regulating a series of classic signaling pathways, including glycogen synthase kinase-3β (GSK3β), and cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB)-regulated transcription coactivator 2 (CRTC2) (27, 29, 30). Moreover, the high-glucose conditions can also enhance the stability of SCAP by directly stimulating the N-glycosylation of SCAP, facilitating the relocation of the SREBP-SCAP complex to the Golgi (31). It has been found that the mTORC1 upregulated by PI3K/Akt signaling pathway can decrease the content of cholesterol in ER by prohibiting membrane-derived cholesterol from arriving lysosomes, thereby actuating the translocation of SREBP2 from ER to the Golgi apparatus and activating cholesterol synthesis (28).
AMPK downregulation caused by increased glucose levels also prevents CRTC2 from phosphorylation, then the dephosphorylated mTORC2 is transported into the nucleus where mTORC2 enhances the transcription of gluconeogenic genes and SREBP2 (32, 33). Recently, a newly synthesized compound, Kanglexin, blockades SREBP2 signal pathway by activating AMPK, thus having the potential to lower blood cholesterol and treat atherosclerosis (34). The above evidence further elucidates the possibility to target glucose pathways to inhibit cholesterol biosynthesis and indicates that the phosphorylation of CRTC2 caused by AMPK may provide a new target to alleviate dyslipidemia, insulin resistance, and atherosclerosis.
Glucose and Cholesterol Uptake
Glucose Regulates NPC1L1
The absorption of cholesterol in diets depends on Niemann-Pick type C1-like 1 (NPC1L1) protein on the apical membrane of enterocytes, which transports the cholesterol from the intestinal lumen to enterocytes (35, 36). NPC1L1 expressed in hepatocytes contributes to uptake of biliary cholesterol back to liver (37, 38). To some extent, NPC1L1 may play a role in preventing excessive excretion of cholesterol mediated by ATP-binding cassette transporter G (ABCG)5/8 heterodimer in hepatocytes and enterocytes. It has been reported that consumption of food with higher carbohydrate tends to incur higher postprandial chylomicrons (39). Several in vitro studies revealed that the promoter activity, mRNA levels, and protein expression of NPC1L1 in human intestinal Caco2 cells was remarkably reduced when the medium was deficient in glucose, and the promoter activity of NPC1L1 could be restored by replenishing glucose (40–42). The basolateral site of Caco-2/15 cells is responsible for sensing high glucose concentration (41), which means that the expression of enterocyte NPC1L1 may be stimulated by elevated circulating glucose levels. Unfortunately, the detailed mechanism involved in this process remains to be investigated.
Glucose Regulates PCSK9
PCSK9 is a plasma enzyme mainly secreted by hepatocytes but also presents in a relatively lower level in extrahepatic tissues, including the brain and the pancreas (43, 44). Recent research reveals that the presence of PCSK9 protein cannot be detected in the plasma of liver-selective PCSK9 knockout mice, indicating that the liver might be the only source of circulating PCSK9 (44). The circulating PCSK9 increases the circulating LDL-C level by promoting LDLR degradation (45). PCSK9 binds to LDLR on the plasma membrane of hepatocytes and the PCSK9-LDLR complex is then delivered to lysosomal for degradation, leading to the depletion of LDLR and subsequently elevated plasma level of LDL-C (46). Individuals with obesity and T2DM are more likely to display a higher level of PCSK9 compared with controls (47, 48). This phenomenon is probably related to the transcription of PCSK9 activated by SREBP2 due to a SRE contained in the promoter region of the PCSK9 gene (49). Interestingly, administration of metformin in patients with T2DM who had received statin treatment for more than 3 months avoided the statins-caused increase of circulating PCSK9 level in contrast with the controls without metformin treatment (50). Metformin is primarily known for inhibiting hepatic gluconeogenesis by directly restricting intracellular glucose metabolites' production (51, 52). Our latest finding reveals that the expression of the carbohydrate-responsive element-binding protein (ChREBP), a glucose sensor responsive to increased glucose and its metabolites, is repressed due to the metformin-induced reduction of intracellular glucose and its metabolites in human hepatocytes (53). ChREBP upregulates the transcription genes related to glycolysis and de novo lipogenesis. Previous studies have proved that the genetic deletion of ChREBP reduced levels of circulating cholesterol and LDL-C in humans and mice (54–56). We found that ChREBP activated by increased intracellular glucose and metabolites translocated to the nucleus, where it bound to carbohydrate response element (ChoRE) in the PCSK9 promoter and inducing PCSK9 transcription, eventually decreasing LDLR and elevating plasma LDL-C levels (53). Both the nuclear translocation of ChREBP and the expression of PCSK9 were notably restricted under lower intracellular glucose states triggered by metformin or glucose deprivation, but were reversed by replenishing glucose (53). Although it has been proposed that metformin may directly activate AMPK and subsequently repress fatty acid desaturase (FADS) to reduce the production of endogenous arachidonic acid, thus indirectly contributing to the recycling of LDLR via enhancing membrane fluidity (17), we observed that PCSK9 downregulation induced by metformin is unrelated to activation of AMPK and SREBP2 pathway since we did not observed changes in SREBP2 and PCSK9 expression after treated with metformin and AMPK agonists (53). The findings indicate that ChREBP has the potential to serve as a new target for hepatic PCSK9 suppression to treat dyslipidemia.
Cholesterol-Lowering Drugs and Glucose Metabolism
Statins
Statins are the first-line cholesterol-lowering drugs based on their ability to inhibit HMGCR (57, 58). Statins reduce intracellular cholesterol and incite SREBP2, upregulating LDLR and LDL-C uptake, thus reducing circulating LDL-C. Administration of statins in diabetic patients apparently decreased the occurrence of atherosclerotic cardiovascular disease, such as myocardial infarction (59, 60). However, increasing findings indicate that statins treatment is correlated to elevated occurrence of NOD (61, 62). Several observational studies and meta-analyses of randomized controlled trials demonstrated that statin therapy yields side effects on glucose metabolism, increasing the NOD risk by around 12% (63–66). Although the definite mechanisms behind statins-induced NOD are still uncertain, it is disclosed that statins may indirectly promote NOD by inciting pancreatic β-cells dysfunction and insulin resistance (61, 62).
Statins and Pancreatic β-Cells Dysfunction
Pancreatic β-cells are the only cell population that recognizes increased plasma glucose (>100 mg/dL) and secrets insulin, which is of paramount importance in controlling glucose homeostasis for its unique hypoglycemic effects, including the promotion of glycogen synthesis, glucose transport mediated by glucose transporter 4 (GLUT-4), glucose oxidation in peripheral tissues, and the suppression of glycogenolysis and gluconeogenesis.
Pancreatic β-cells dysfunction caused by statins is characterized by decreased insulin secretion. The latest findings reveal that pancreatic β-cells of HMGCR knockout mice are accompanied by severe hyperglycemia due to compromised insulin secretion and impaired pancreatic β-cell proliferation (67). The mevalonate pathway initiated by HMGCR produces isoprenoid, a kind of intermediate metabolite which contributes to insulin granule exocytosis via enhancing the posttranslational modification of small G proteins (sGPs), such as Rab5a (62). Some sGPs function as activators of mTOR, which upregulates some key pancreatic transcription factors, such as v-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), thereby retaining mature β-cell functional mass (68). Statins lead to isoprenoid deficiency and disturbed protein prenylation, thus impairing insulin secretion (68). Supplementation of geranylgeranyl pyrophosphate, one of the intermediates in the mevalonate pathway, significantly reverses MIN6 cells (a mouse pancreatic β-cell line) function damaged by atorvastatin (68). Hence, targeting the mevalonate pathway may also provide a new strategy for avoiding statin-induced hyperglycemia.
GLUT-2 and ion channels may also partially account for the correlation between statins and NOD. GLUT-2 transports glucose from extracellular space to cytoplasm, increasing cytosolic ATP/ADP ratio as a result of augmented glycolysis in β-cells (69). The high level of ATP is prone to stimulate instant calcium influx by blocking K+ -ATP channels and opening voltage-gated Ca2+ channel (VGCC), thereby causing exocytosis of insulin secretory vesicles (70, 71). Glucose-induced insulin secretion is decreased by repression of P2X and P2Y purinergic receptors (72, 73). It is also found that ATP and ADP present in the insulin exocytosis granules, enabling them to activate β-cell purinergic P2 receptors by serving as autocrine activators (74).
GLUT-2 expression in pancreatic β-cells is inversely proportional to the dosage of atorvastatin and pravastatin (75). Further study indicates that statins impair GLUT-2 expression, thus obstructing glucose uptake of β-cells (76). A study using MIN6 cells implied that statins deceased GLUT-2 expression by reducing the generation of ATP (77). Furthermore, it is reported that statins directly suppress VGCC expression in β-cells, eventually leading to decreased insulin secretion (78, 79).
Some findings indicate that statins promote β-cells apoptosis via prompting cytochrome c expulsion from mitochondria, providing another explanation for pancreatic β-cells deprivation and development of NOD by statins (80). Statins suppress mitochondrial complex II and III activity and reduce mitochondrial membrane potential (80, 81), which incites mitochondrial oxidative stress, eventually decreasing the synthesis of ATP and then inhibiting insulin secretion (82). A further study unveils that simvastatin may also restrain K+-ATP channels function directly independent of mitochondria (83).
Stains may also reduce insulin secretion by downregulating G protein-coupled receptor 40 (GPR40) and glucagon-like peptide 1 (GLP-1) receptor (84). GPR40 elevates intracellular free calcium concentration level via lessening the voltage-gated K+ current (85). The activated GLP-1 receptor facilitates insulin secretion by inciting adenylate cyclase, which accelerates the transformation of ATP to cAMP (86). The downstream molecules of cAMP, the cAMP-dependent protein kinase (PKA) and Epac (exchange protein activated by cyclic-AMP) 2, stimulate inositol 1,4,5-triphosphate (IP3) receptor on the ER and lead to the release of Ca2+ from ER, thus intensifying insulin secretion (86, 87). GPR40 and GLP-1 suppressed by statins reopen K+-ATP channels and decrease intracellular Ca2+, further hindering insulin secretion (84).
Statins and Insulin Resistance
The hypoglycemic effect exerted by insulin is initiated by the combination of insulin to INSR, thereby triggering the insulin signaling, including the phosphorylation of IRSs and then the activation of various kinases (such as Akt, hepatic p70 S6 kinase (S6K1), and mTOR) (88, 89). The activated Akt stimulates glycogen synthesis by repressing glycogen synthase kinase and accelerating glucose uptake via promoting GLUT-4 translocation to the plasma membrane of skeletal muscle cells and adipose tissue (9, 90). Insulin resistance, which is defined as loss of appropriate response to ordinary circulating insulin levels in insulin-targeted cells, such as hepatocytes, adipocytes, and skeletal muscle cells, is one of the pivotal causes of T2DM (91). Stains promote NOD not only by impairing pancreatic β-cells' function, but also by inducing insulin resistance.
As a vital digestive organ, the liver serves as a sensitive sensor of insulin to maintain glucose homeostasis. Insulin controls multiple hepatic metabolic pathways, such as glucose output and lipid synthesis. To date, increasing findings denote that statins therapy correlates with the aggravation of glycemic control in the liver (76). Statins stimulate hepatic gluconeogenesis by activating the key gluconeogenic genes, phosphoenolpyruvate carboxykinase 1 (PEPCK1) and glucose-6-phosphatase (G6Pase) genes (92). The pregnane X receptor (PXR) is a nuclear receptor and exert multiple functions in mediating hepatic lipid and glucose metabolism (93). Stains stimulate PXR, which prompts serum/glucocorticoid regulated kinase 2 (SGK2) dephosphorylation by the protein phosphatase 2CA (PP2CA) (92). Then, PXR and the dephosphorylated SGK2 located in the cytoplasm simultaneously transfer into the nucleus and interact with the nuclear retinoid X receptor (RXR), thereby upregulating the expression of PEPCK1 and G6Pase (92). In contrast, a different study indicates that atorvastatin increases serum glucose level by activating PXR to hamper the expression of GLUT-2 and glucokinase, rather than PXR/SGK2-mediated signaling pathway (76).
As an energy storage organ, adipose tissue also participates in statins-induced NOD as a result of the weakened insulin signal transduction process. Statins treatment is associated with decreased expression of GLUT-4 in adipocytes (94, 95). The further study indicates that statins reduce GLUT-4 translocation to the plasma membrane via inhibiting isoprenoid synthesis (95), which is indispensable for functions of Rab-4 and RhoA, two proteins facilitating GLUT-4 translocation (96). Statins also disturb the function of caveolae, where GLUT-4 inserts in the plasma membrane after being activated by insulin (97). INSR is extremely abundant in adipocyte caveolae (98, 99), which means that caveolae is required for correct insulin signaling in adipocytes. Cholesterol is essential for maintaining the characteristic shape of caveolae (100). Therefore, statins-induced cholesterol insufficiency may disrupt caveolar formation, further interrupting insulin signaling.
Skeletal muscle consumes most of the circulating glucose (~75%), and damaged glucose uptake by skeletal muscle results in T2DM (101). Therefore, statins-induced NOD may partially depend on skeletal muscle despite unclear mechanisms. Similar to adipocytes, skeletal muscle cells uptake glucose primarily via GLUT-4, and the insulin signaling may also be harmed by statins, resulting in elevated plasma glucose levels (102). It is recently found that the total expression of GLUT-4 protein in C2C12 myotubes is unaffected despite reduced GLUT-4 membrane translocation after atorvastatin treatment (103). Furthermore, simvastatin-related INSR and mTORC2 dysfunction may weaken Akt activation and disturb the phosphorylation of GSK3β in C2C12 myotubes, thus inhibiting GLUT-4 translocation (104). It is also proposed that simvastatin may incur insulin resistance in skeletal muscle by increasing fatty acid production. Simvastatin leads to acetyl CoA accumulation due to HMGCR suppression in L6 myotubes. The excess acetyl CoA acts as a precursor to enhance fatty acid synthesis, which further restrain glucose uptake by disrupting GLUT-4 translocation (105, 106). Besides interfering GLUT-4, simvastatin inhibits IR/IRS-1/Akt signaling cascade and dysregulates glycogen synthesis in skeletal muscle cells (107).
Mechanisms behind statins-induced NOD are not completely understood. Unrevealing more mechanisms may help to prevent the generation of NOD by statins.
Ezetimibe
Ezetimibe is the only inhibitor of NPC1L1 used in the clinic to lower blood cholesterol by hindering cholesterol uptake from diet (108). Adding ezetimibe to statin therapy further reduces the plasma LDL-C both in diabetics and nondiabetics when compared with statin monotherapy (ClinicalTrials.gov Identifier: NCT00202878) (109). Long-term combination therapy with ezetimibe and acarbose improved insulin sensitivity in a high-fat diet-induced non-alcoholic fatty liver disease (NAFLD) mouse model by upregulating the mRNA expression of peroxisome proliferators-activated receptor-alpha (PPAR-α) 1 and microsomal triglyceride transfer protein (MTP) in hepatocytes (110). Besides decreasing LDL-C, ezetimibe ameliorates metabolic syndrome and reduces visceral fat (111, 112).
Interestingly, hepatic NPC1L1 overexpression inhibits hepatic gluconeogenesis and ameliorates glucose metabolism in diabetic mouse models via repressing forkhead box O 1 (FoxO1) and reducing G6Pase and PEPCK expression (113). It is reasonable to estimate that NPC1L1 suppression by ezetimibe may impair hepatic glucose metabolism and increase the risk of diabetes. However, we still lack experimental evidence to confirm the association between ezetimibe and NOD, and the underlying mechanisms remain to be investigated.
PCSK9 Inhibitors
PCSK9 inhibitors, including anti-PCSK9 monoclonal antibodies and anti-PCSK9 vaccines (114–116), lower LDL-C by cutting down PCSK9-mediated LDLR degradation and promote hepatic LDL uptake from circulation. PCSK9 inhibitors further decrease blood cholesterol in individuals with statin tolerance (117, 118), which is partially due to elevated PCSK9 expression by statins-induced SREBP-2 activation (48, 49). Up to now, only two anti-PCSK9 monoclonal antibodies, namely evolocumab and alirocumab, have received approval on the hypercholesterolemia treatment from the United States Food and Drug Administration and the European Union (117). Numerous anti-PCSK9 vaccines are still in preclinical or clinical phases to confirm their safety and efficacy (115).
Appropriate PCSK9 expression is beneficial to maintain intracellular cholesterol homeostasis by restricting LDLR level, thereby avoiding excessive cholesterol accumulation in β-cell (44). Excess cholesterol in pancreatic β-cells undermines glucose-stimulated insulin secretion by disturbing the function of organelles, GLUT-2 and K+ -ATP channels, resulting in hyperglycemia (119). However, unlike statins, observational studies and meta-analyses show that alirocumab or evolocumab does not lead to NOD or aggravate preexisting diabetes mellitus (120–122). The latest findings reveal that it is the local rather than circulating PCSK9 accounts for the upregulated expression of LDLR in pancreatic β-cells, consequently incurring cholesterol overload and β-cells dysfunction (44). PCSK9 existing in pancreatic islets is derived from pancreatic δ-cell (44), which implies that the expression of PCSK9 and LDLR in the pancreas can be exempt from changes in circulating PCSK9. Both alirocumab and evolocumab primarily target liver-derived circulating PCSK9, thereby exerting finite influence on β-cells dysfunction and NOD (44). Besides, PCSK9 deficiency does not harm insulin signaling in hepatocytes and skeletal muscle cells (44). Hence, PCSK9 inhibitors may manage hypercholesterolemia without disturbing glucose metabolism.
Cholesterol-Lowering Gene Variants and Nod
HMG-CoA Reductatse Gene and NOD
Mendelian Randomization (MR), which utilizes genetic mutations as an instrumental variable for studying exposure factors, has emerged as a popular approach to mimic the association of exposure factors with the corresponding disease. MR approach is less susceptible to multiple confounding factors and may supply rational evidence of causation. Researchers have used this approach to explore the relationship between statins therapy and the incidence of diabetes (123, 124). A large genetic analysis based on 2,23,463 subjects showed that the amount of rs17238484-G allele, an HMGCR genetic variant used to imitate HMGCR inhibition by statins, is positively related to the degree of body weight gain and the risk of developing NOD (124). It is found that each supplementary rs17238484-G allele is correlated to a statistically significant odds ratio (OR) of 1.02 for T2DM (124). Meanwhile, genetic analysis of randomized trials including 12,9170 individuals observed a statistically significant OR 1.12 for statins-induced NOD at a mean follow-up of 4.2 years (124). Hence, the application of HMGCR gene variants proves that statin-induced HMGCR inhibition might explain the occurrence of NOD.
NPC1L1 Gene and NOD
Similar to HMGCR alleles, LDL-C-lowering NPC1L1 alleles are also utilized as genetic alternatives to mimic ezetimibe efficacy (125). A genetic meta-analysis of 50 775 T2DM individuals and 270 269 controls observed that per genetically foreseen 1 mmol/L decrease in LDL-C by NPC1L1 variants is associated with a significant OR of 2.42 for developing T2DM (125). Although the cholesterol-lowering effect of ezetimibe has been widely accepted, the application of ezetimibe is also likely to augment the risk of T2DM based on this genetic study (125).
PCSK9 Gene and NOD
PCSK9 genetic variants can be divided into gain of function (GOF) mutations and loss of function (LOF) mutations according to their effects on circulating LDL clearance (126, 127). Considering the interaction between LDLR and PCSK9, flow cytometry analyses detect the expression level of LDLR in HEK293 cells transfected with PCSK9 variants, which may be an effective and reliable way to distinguish these two distinct types of PCSK9 variants (128). PCSK9-GOF variants tend to increase LDLR degradation in multiple cells, followed by the high level of plasma LDL-C (129). On the contrary, PCSK9-LOF mutations are more likely to increase LDLR expression in various cells, including pancreatic β-cell, which contributes to LDL-C removal from circulation but enhances cholesterol accumulation in pancreatic β-cell. Excess cholesterol accumulation results in β-cell dysfunction, promoting the development of hypoinsulinemic hyperglycemia and impaired glucose tolerance (119). Several clinical and experimental studies unveiled that individuals with PCSK9-LOF variants tended to have higher circulating glucose levels and elevated incidence of T2DM despite lower LDL-C levels (125, 130). Familial hypercholesterolemia is mainly caused by LDLR-LOF or PCSK9-GOF. The probability of patients with familial hypercholesterolemia developing into T2DM is much lower than their unaffected relatives (131), which indirectly means that PCSK9-GOF variants might be associated with NOD. However, it should be noted that the effect of PCSK9 genetic variants on T2DM risk is different from alirocumab and evolocumab, which mainly target liver-derived circulating PCSK9 rather than systemic PCSK9.
Conclusion
Glucose/insulin promotes cholesterol biosynthesis and cholesterol uptake, which indicates that drugs targeting lowering glucose may help to control hypercholesterolemia. On the contrary, cholesterol-lowering drugs or genetic variants could impair glucose homeostasis and lead to diabetes by decreasing pancreatic β-cell insulin secretion or inducing insulin resistance of skeletal muscle cells, adipocytes, or hepatocytes. Understanding the crosstalk between glucose metabolism and cholesterol metabolism, such as glucose-ChREBP/HMGCR- cholesterol pathway, may help to locate safe therapeutic targets for controlling both glucose and cholesterol dysregulation.
Author Contributions
DP was the originator and supervisor of the project. DP and YL conducted elaborate polishment on the article. XX collected and analyzed relevant literature, then completed the writing of the first draft of the article. All authors read and agree with the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by grants from National Natural Science Foundation of China (No.81870336 to DP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen L, Chen XW, Huang X, Song BL, Wang Y, Wang Y. Regulation of glucose and lipid metabolism in health and disease. Sci China Life Sic. (2019) 62:1420–58. doi: 10.1007/s11427-019-1563-3
2. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. (2017) 13:572–87. doi: 10.1038/nrendo.2017.80
3. Iozzo P, Hallsten K, Oikonen V, Virtanen KA, Kemppainen J, Solin O, et al. Insulin-Mediated hepatic glucose uptake is impaired in type 2 diabetes: evidence for a relationship with glycemic control. J Clin Endocrinol Metab. (2003) 88:2055–60. doi: 10.1210/jc.2002-021446
4. Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl coa links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. (2015) 160:745–58. doi: 10.1016/j.cell.2015.01.012
5. Sharabi K, Tavares CD, Rines AK, Puigserver P. Molecular pathophysiology of hepatic glucose production. Mol Aspects Med. (2015) 46:21–33. doi: 10.1016/j.mam.2015.09.003
6. Petrie JR, Chaturvedi N, Ford I, Brouwers M, Greenlaw N, Tillin T, et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (removal): a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. (2017) 5:597–609. doi: 10.1016/S2213-8587(17)30194-8
7. Glueck CJ, Fontaine RN, Wang P, Subbiah MT, Weber K, Illig E, et al. Metformin reduces weight, centripetal obesity, insulin, leptin, and low-density lipoprotein cholesterol in nondiabetic, morbidly obese subjects with body mass index greater than 30. Metabolism. (2001) 50:856–61. doi: 10.1053/meta.2001.24192
8. Corrao G, Ibrahim B, Nicotra F, Soranna D, Merlino L, Catapano AL, et al. Statins and the risk of diabetes: evidence from a large population-based cohort study. Diabetes Care. (2014) 37:2225–32. doi: 10.2337/dc13-2215
9. Galicia-Garcia U, Jebari S, Larrea-Sebal A, Uribe KB, Siddiqi H, Ostolaza H, et al. Statin treatment-induced development of type 2 diabetes: from clinical evidence to mechanistic insights. Int J Mol Sci. (2020) 21:4725. doi: 10.3390/ijms21134725
10. Pinkosky SL, Newton RS, Day EA, Ford RJ, Lhotak S, Austin RC, et al. Liver-Specific Atp-Citrate lyase inhibition by bempedoic acid decreases Ldl-C and attenuates atherosclerosis. Nat Commun. (2016) 7:13457. doi: 10.1038/ncomms13457
11. Laufs U, Banach M, Mancini GBJ, Gaudet D, Bloedon LT, Sterling LR, et al. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia and statin intolerance. J Am Heart Assoc. (2019) 8:e011662. doi: 10.1161/JAHA.118.011662
12. Di Minno A, Lupoli R, Calcaterra I, Poggio P, Forte F, Spadarella G, et al. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia: systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc. (2020) 9:e016262. doi: 10.1161/JAHA.119.016262
13. Goldberg AC, Leiter LA, Stroes ESG, Baum SJ, Hanselman JC, Bloedon LT, et al. Effect of bempedoic acid vs. placebo added to maximally tolerated statins on low-density lipoprotein cholesterol in patients at high risk for cardiovascular disease: the clear wisdom randomized clinical trial. Jama. (2019) 322:1780–8. doi: 10.1001/jama.2019.16585
14. Wang X, Zhang Y, Tan H, Wang P, Zha X, Chong W, et al. Efficacy and safety of bempedoic acid for prevention of cardiovascular events and diabetes: a systematic review and meta-analysis. Cardiovasc Diabetol. (2020) 19:1–9. doi: 10.1186/s12933-020-01101-9
15. Edwards PA, Muroya H, Gould RG. In Vivo demonstration of the circadian thythm of cholesterol biosynthesis in the liver and intestine of the rat. J Lipid Res. (1972) 13:396–401. doi: 10.1016/S0022-2275(20)39403-7
16. Salt IP, Johnson G, Ashcroft SJ, Hardie DG. Amp-Activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J. (1998) 335:533–9. doi: 10.1042/bj3350533
17. Xu T, Brandmaier S, Messias AC, Herder C, Draisma HH, Demirkan A, et al. Effects of metformin on metabolite profiles and Ldl cholesterol in patients with type 2 diabetes. Diabetes Care. (2015) 38:1858–67. doi: 10.2337/dci15-0022
18. Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. (2014) 510:542–6. doi: 10.1038/nature13270
19. Carling D. Ampk signalling in health and disease. Curr Opin Cell Biol. (2017) 45:31–7. doi: 10.1016/j.ceb.2017.01.005
20. Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by Ampk. Nature. (2017) 548:112–6. doi: 10.1038/nature23275
21. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. Ampk phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
22. Inoki K, Zhu T, Guan KL. Tsc2 mediates cellular energy response to control cell growth and survival. Cell. (2003) 115:577–90. doi: 10.1016/S0092-8674(03)00929-2
23. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: Mtorc1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. (2010) 107:3441–6. doi: 10.1073/pnas.0914798107
24. Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA. Sterol-Induced degradation of Hmg Coa reductase depends on interplay of two insigs and two ubiquitin ligases, Gp78 and Trc8. Proc Natl Acad Sci U S A. (2011) 108:20503–8. doi: 10.1073/pnas.1112831108
25. Lu X-Y, Shi X-J, Hu A, Wang J-Q, Ding Y, Jiang W, et al. Feeding induces cholesterol biosynthesis via the Mtorc1–Usp20–Hmgcr axis. Nature. (2020) 588:479–84. doi: 10.1038/s41586-020-2928-y
26. Horton JD, Goldstein JL, Brown MS. Srebps: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI0215593
27. DeBose-Boyd RA, Ye J. Srebps in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci. (2018) 43:358–68. doi: 10.1016/j.tibs.2018.01.005
28. Eid W, Dauner K, Courtney KC, Gagnon A, Parks RJ, Sorisky A, et al. Mtorc1 activates Srebp-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Nat Acad Sci. (2017) 114:7999–8004. doi: 10.1073/pnas.1705304114
29. Shimano H, Sato R. Srebp-Regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
30. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme Pck1 phosphorylates Insig1/2 for lipogenesis. Nature. (2020) 580:530–5. doi: 10.1038/s41586-020-2183-2
31. Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, et al. Glucose-Mediated N-Glycosylation of scap is essential for Srebp-1 activation and tumor growth. Cancer Cell. (2015) 28:569–81. doi: 10.1016/j.ccell.2015.09.021
32. Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, et al. The creb coactivator Torc2 is a key regulator of fasting glucose metabolism. Nature. (2005) 437:1109–11. doi: 10.1038/nature03967
33. Li Y, Song Y, Zhao M, Guo Y, Yu C, Chen W, et al. A novel role for Crtc2 in hepatic cholesterol synthesis through Srebp-2. Hepatology (Baltimore, Md). (2017) 66:481–97. doi: 10.1002/hep.29206
34. Li X, Hu X, Pan T, Dong L, Ding L, Wang Z, et al. Kanglexin, a new anthraquinone compound, attenuates lipid accumulation by activating the Ampk/Srebp-2/Pcsk9/Ldlr signalling pathway. Biomed Pharmacother. (2021) 133:110802. doi: 10.1016/j.biopha.2020.110802
35. Wang DQ. Regulation of intestinal cholesterol absorption. Annu Rev Physiol. (2007) 69:221–48. doi: 10.1146/annurev.physiol.69.031905.160725
36. Wang LJ, Song BL. Niemann-Pick C1-Like 1 and cholesterol uptake. Biochim Biophys Acta. (2012) 1821:964–72. doi: 10.1016/j.bbalip.2012.03.004
37. Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, et al. Hepatic Niemann-Pick C1-Like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest. (2007) 117:1968–78. doi: 10.1172/JCI30060
38. Wang LJ, Wang J, Li N, Ge L, Li BL, Song BL. Molecular characterization of the Npc1l1 variants identified from cholesterol low absorbers. J Biol Chem. (2011) 286:7397–408. doi: 10.1074/jbc.M110.178368
39. Harbis A, Defoort C, Narbonne H, Juhel C, Senft M, Latgé C, et al. Acute hyperinsulinism modulates plasma apolipoprotein B-48 Triglyceride-Rich lipoproteins in healthy subjects during the postprandial period. Diabetes. (2001) 50:462–9. doi: 10.2337/diabetes.50.2.462
40. Villa-Rodriguez JA, Kerimi A, Tumova S, Williamson G. Inhibition of intestinal glucose transport by polyphenols: a mechanism for indirect attenuation of cholesterol absorption? Food Funct. (2019) 10:3127–34. doi: 10.1039/C9FO00810A
41. Grenier E, Mailhot G, Dion D, Ravid Z, Spahis S, Bendayan M, et al. Role of the apical and basolateral domains of the enterocyte in the regulation of cholesterol transport by a high glucose concentration. Biochem Cell Biol. (2013) 91:476–86. doi: 10.1139/bcb-2013-0053
42. Malhotra P, Boddy CS, Soni V, Saksena S, Dudeja PK, Gill RK, et al. D-Glucose modulates intestinal Niemann-Pick C1-Like 1 (Npc1l1) gene expression via transcriptional regulation. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G203–10. doi: 10.1152/ajpgi.00288.2012
43. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (Narc-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. (2003) 100:928–33. doi: 10.1073/pnas.0335507100
44. Da Dalt L, Ruscica M, Bonacina F, Balzarotti G, Dhyani A, Di Cairano E, et al. Pcsk9 deficiency reduces insulin secretion and promotes glucose intolerance: the role of the low-density lipoprotein receptor. Eur Heart J. (2019) 40:357–68. doi: 10.1093/eurheartj/ehy357
45. Schulz R, Schlüter KD. Pcsk9 Targets important for lipid metabolism. Clin Res Cardiol Suppl. (2017) 12:2–11. doi: 10.1007/s11789-017-0085-0
46. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, et al. Secreted Pcsk9 decreases the number of Ldl receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. (2006) 116:2995–3005. doi: 10.1172/JCI29383
47. Silbernagel G, Steiner LK, Hollstein T, Fauler G, Scharnagl H, Stojakovic T, et al. The interrelations between Pcsk9 metabolism and cholesterol synthesis and absorption. J Lipid Res. (2019) 60:161–7. doi: 10.1194/jlr.P088583
48. Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, et al. Statins upregulate Pcsk9, the gene encoding the proprotein convertase neural apoptosis-regulated Convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. (2004) 24:1454–9. doi: 10.1161/01.ATV.0000134621.14315.43
49. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-Dependent regulation of proprotein convertase Subtilisin/Kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. (2008) 49:399–409. doi: 10.1194/jlr.M700443-JLR200
50. Shek AB, Alieva RB, Kurbanov RD, Hoshimov SU, Nizamov UI, Ziyaeva AV. Can metformin stabilize Pcsk9 level in stable coronary artery disease patients treated with statins? Arch Med Sci Atheroscler Dis. (2019) 4:e144–50. doi: 10.5114/amsad.2019.86752
51. Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M, Zeqiraj E, et al. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat Med. (2018) 24:1395–406. doi: 10.1038/s41591-018-0159-7
52. Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang X-M, Zhang D, et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in Vivo. Nat Med. (2018) 24:1384–94. doi: 10.1038/s41591-018-0125-4
53. Hu D, Guo Y, Wu R, Shao T, Long J, Yu B, et al. New insight into metformin-induced cholesterol-lowering effect crosstalk between glucose and cholesterol homeostasis via chrebp (Carbohydrate-Responsive Element-Binding Protein)-mediated Pcsk9 (Proprotein Convertase Subtilisin/Kexin Type 9) Regulation. Arterioscler Thromb Vasc Biol. (2021) 41:e208–e23. doi: 10.1161/ATVBAHA.120.315708
54. Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, et al. Liver-Specific inhibition of Chrebp improves hepatic steatosis and insulin resistance in Ob/Ob Mice. Diabetes. (2006) 55:2159–70. doi: 10.2337/db06-0200
55. Wu CY, Tso SC, Chuang JL, Gui WJ, Lou M, Sharma G, et al. Targeting hepatic pyruvate Dehydrogenase Kinases restores insulin signaling and mitmgates Chrebp-mediated lipogenesis in diet-induced obese Mice. Molecular metabolism. (2018) 12:12–24. doi: 10.1016/j.molmet.2018.03.014
56. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (Chrebp) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. (2004) 101:7281–6. doi: 10.1073/pnas.0401516101
57. van Stee MF, de Graaf AA, Groen AK. Actions of metformin and statins on lipid and glucose metabolism and possible benefit of combination therapy. Cardiovasc Diabetol. (2018) 17:94. doi: 10.1186/s12933-018-0738-4
58. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from Plaques to Genes to statins. Cell. (2015) 161:161–72. doi: 10.1016/j.cell.2015.01.036
59. Mortensen MB, Kulenovic I, Falk E. Statin use and cardiovascular risk factors in diabetic patients developing a first myocardial infarction. Cardiovasc Diabetol. (2016) 15:81. doi: 10.1186/s12933-016-0400-y
60. Bibbins-Domingo K, Grossman DC, Curry SJ, Davidson KW, Epling JW. Jr., García FAR, et al. Statin use for the primary prevention of cardiovascular disease in adults: Us preventive services task force recommendation statement. Jama. (2016) 316:1997–2007. doi: 10.1001/jama.2016.15450
61. Agarwala A, Kulkarni S, Maddox T. The association of statin therapy with incident diabetes: evidence, mechanisms, and recommendations. Curr Cardiol Rep. (2018) 20:50. doi: 10.1007/s11886-018-0995-6
62. Betteridge DJ, Carmena R. The diabetogenic action of statins - mechanisms and clinical implications. Nat Rev Endocrinol. (2016) 12:99–110. doi: 10.1038/nrendo.2015.194
63. Kohli P, Waters DD, Nemr R, Arsenault BJ, Messig M, DeMicco DA, et al. Risk of new-onset diabetes and cardiovascular risk reduction from high-dose statin therapy in pre-diabetics and non-pre-diabetics: an analysis from Tnt and ideal. J Am Coll Cardiol. (2015) 65:402–4. doi: 10.1016/j.jacc.2014.10.053
64. Thakker D, Nair S, Pagada A, Jamdade V, Malik A. Statin use and the risk of developing diabetes: a network meta-analysis. Pharmacoepidemiol Drug Saf. (2016) 25:1131–49. doi: 10.1002/pds.4020
65. Casula M, Mozzanica F, Scotti L, Tragni E, Pirillo A, Corrao G, et al. Statin use and risk of new-onset diabetes: a meta-analysis of observational studies. NMCD. (2017) 27:396–406. doi: 10.1016/j.numecd.2017.03.001
66. Wang S, Cai R, Yuan Y, Varghese Z, Moorhead J, Ruan XZ. Association between reductions in low-density lipoprotein cholesterol with statin therapy and the risk of new-onset diabetes: a meta-analysis. Sci Rep. (2017) 7:39982. doi: 10.1038/srep39982
67. Takei S, Nagashima S, Takei A, Yamamuro D, Wakabayashi T, Murakami A, et al. B-Cell–Specific deletion of Hmg-Coa (3-Hydroxy-3-Methylglutaryl-Coenzyme a) reductase causes overt diabetes due to reduction of B-Cell mass and impaired insulin secretion. Diabetes. (2020) 69:2352–63. doi: 10.2337/db19-0996
68. Shen L, Gu Y, Qiu Y, Cheng T, Nie A, Cui C, et al. Atorvastatin targets the islet mevalonate pathway to dysregulate Mtor signaling and reduce B-Cell functional mass. Diabetes. (2020) 69:48–59. doi: 10.2337/db19-0178
69. Nolan CJ, Prentki M. The islet beta-cell: fuel responsive and vulnerable. Trends Endocrinol Metab. (2008) 19:285–91. doi: 10.1016/j.tem.2008.07.006
70. MacDonald PE, Wheeler MB. Voltage-Dependent K(+) channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia. (2003) 46:1046–62. doi: 10.1007/s00125-003-1159-8
71. Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. (2013) 75:155–79. doi: 10.1146/annurev-physiol-030212-183754
72. Geisler JC, Corbin KL Li Q, Feranchak AP, Nunemaker CS Li C. Vesicular nucleotide transporter-mediated Atp release regulates insulin secretion. Endocrinology. (2013) 154:675–84. doi: 10.1210/en.2012-1818
73. Khan S, Yan-Do R, Duong E, Wu X, Bautista A, Cheley S, et al. Autocrine activation of P2y1 receptors couples Ca (2+) influx to Ca (2+) release in human pancreatic beta cells. Diabetologia. (2014) 57:2535–45. doi: 10.1007/s00125-014-3368-8
74. Bertrand G, Chapal J, Loubatieres-Mariani MM. Potentiating synergism between adenosine diphosphate or triphosphate and acetylcholine on insulin secretion. Am J Physiol. (1986) 251:E416–21. doi: 10.1152/ajpendo.1986.251.4.E416
75. Zhao W, Zhao SP. Different effects of statins on induction of diabetes mellitus: an experimental study. Drug Des Devel Ther. (2015) 9:6211–23. doi: 10.2147/DDDT.S87979
76. Ling Z, Shu N, Xu P, Wang F, Zhong Z, Sun B, et al. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem Pharmacol. (2016) 100:98–111. doi: 10.1016/j.bcp.2015.11.023
77. Zhou J, Li W, Xie Q, Hou Y, Zhan S, Yang X, et al. Effects of simvastatin on glucose metabolism in mouse Min6 cells. J Diabetes Res. (2014) 2014:376570. doi: 10.1155/2014/376570
78. Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol. (1999) 126:1205–13. doi: 10.1038/sj.bjp.0702397
79. Xia F, Xie L, Mihic A, Gao X, Chen Y, Gaisano HY, et al. Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage-gated calcium channel function in pancreatic beta-cells. Endocrinology. (2008) 149:5136–45. doi: 10.1210/en.2008-0161
80. Sadighara M, Amirsheardost Z, Minaiyan M, Hajhashemi V, Naserzadeh P, Salimi A, et al. Toxicity of atorvastatin on pancreas mitochondria: a justification for increased risk of diabetes mellitus. Basic Clin Pharmacol Toxicol. (2017) 120:131–7. doi: 10.1111/bcpt.12656
81. Schirris TJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BG, et al. Statin-Induced myopathy is associated with mitochondrial complex Iii inhibition. Cell Metab. (2015) 22:399–407. doi: 10.1016/j.cmet.2015.08.002
82. Urbano F, Bugliani M. Atorvastatin but not pravastatin impairs mitochondrial function in human pancreatic islets and rat B-Cells. direct effect oxidative stress. Sci Rep. (2017) 7:11863. doi: 10.1038/s41598-017-11070-x
83. Curry L, Almukhtar H, Alahmed J, Roberts R, Smith PA. Simvastatin inhibits L-type Ca2+-channel activity through impairment of mitochondrial function. Toxicol Sci. (2019) 169:543–52. doi: 10.1093/toxsci/kfz068
84. Yaluri N, Modi S, López Rodríguez M, Stančáková A, Kuusisto J, Kokkola T, et al. Simvastatin impairs insulin secretion by multiple mechanisms in min6 cells. PLoS ONE. (2015) 10:e0142902. doi: 10.1371/journal.pone.0142902
85. Feng DD, Luo Z, Roh SG, Hernandez M, Tawadros N, Keating DJ, et al. Reduction in voltage-gated K+ currents in primary cultured rat pancreatic beta-cells by linoleic acids. Endocrinol. (2006) 147:674–82. doi: 10.1210/en.2005-0225
86. Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. (2007) 113:546–93. doi: 10.1016/j.pharmthera.2006.11.007
87. Sugawara K, Shibasaki T, Takahashi H, Seino S. Structure and functional roles of Epac2 (Rapgef4). Gene. (2016) 575:577–83. doi: 10.1016/j.gene.2015.09.029
88. Lizcano JM, Alessi DR. The insulin signalling pathway. Current biology: CB. (2002) 12:R236–8. doi: 10.1016/S0960-9822(02)00777-7
89. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins Irs1 and Irs2. Diabetologia. (2012) 55:2565–82. doi: 10.1007/s00125-012-2644-8
90. Huang S, Czech MP. The Glut4 glucose transporter. Cell Metab. (2007) 5:237–52. doi: 10.1016/j.cmet.2007.03.006
91. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. (2016) 126:12–22. doi: 10.1172/JCI77812
92. Gotoh S, Negishi M. Statin-Activated nuclear receptor Pxr promotes Sgk2 dephosphorylation by scaffolding Pp2c to induce hepatic gluconeogenesis. Sci Rep. (2015) 5:14076. doi: 10.1038/srep14076
93. Hakkola J, Rysä J, Hukkanen J. Regulation of hepatic energy metabolism by the nuclear receptor Pxr. Biochim Biophys Acta. (2016) 1859:1072–82. doi: 10.1016/j.bbagrm.2016.03.012
94. Moraes-Vieira PM, Saghatelian A, Kahn BB. Glut4 expression in adipocytes regulates de novo lipogenesis and levels of a novel class of lipids with antidiabetic and anti-inflammatory effects. Diabetes. (2016) 65:1808–15. doi: 10.2337/db16-0221
95. Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (Slc2a4): implications in glycaemic control. Diabetologia. (2006) 49:1881–92. doi: 10.1007/s00125-006-0269-5
96. Takaguri A, Satoh K, Itagaki M, Tokumitsu Y, Ichihara K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3t3l1 adipocytes. J Pharmacol Sci. (2008) 107:80–9. doi: 10.1254/jphs.FP0072403
97. Gustavsson J, Parpal S, Strålfors P. Insulin-Stimulated glucose uptake involves the transition of glucose transporters to a caveolae-rich fraction within the plasma membrane: implications for type Ii diabetes. Mol Med. (1996) 2:367–72. doi: 10.1007/BF03401634
98. Gustavsson J, Parpal S, Karlsson M, Ramsing C, Thorn H, Borg M, et al. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. (1999) 13:1961–71. doi: 10.1096/fasebj.13.14.1961
99. Sekimoto J, Kabayama K, Gohara K, Inokuchi J. Dissociation of the insulin receptor from caveolae during Tnfα-Induced insulin resistance and its recovery by D-Pdmp. FEBS Lett. (2012) 586:191–5. doi: 10.1016/j.febslet.2011.12.019
100. Breen MR, Camps M, Carvalho-Simoes F, Zorzano A, Pilch PF. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of Cavin-2 in a switch-like fashion. PloS ONE. (2012) 7:e34516. doi: 10.1371/journal.pone.0034516
101. Carnagarin R, Dharmarajan AM, Dass CR. Molecular aspects of glucose homeostasis in skeletal muscle–a focus on the molecular mechanisms of insulin resistance. Mol Cell Endocrinol. (2015) 417:52–62. doi: 10.1016/j.mce.2015.09.004
102. Bradley H, Shaw CS, Worthington PL, Shepherd SO, Cocks M, Wagenmakers AJ. Quantitative immunofluorescence microscopy of subcellular Glut4 distribution in human skeletal muscle: effects of endurance and sprint interval training. Physiol Rep. (2014) 2:e12085. doi: 10.14814/phy2.12085
103. Sun B, Zhong Z, Wang F, Xu J, Xu F, Kong W, et al. Atorvastatin impaired glucose metabolism in C2c12 cells partly via inhibiting cholesterol-dependent glucose transporter 4 translocation. Biochem Pharmacol. (2018) 150:108–19. doi: 10.1016/j.bcp.2018.01.021
104. Sanvee GM, Panajatovic MV, Bouitbir J, Krähenbühl S. Mechanisms of insulin resistance by simvastatin in C2c12 myotubes and in mouse skeletal muscle. Biochem Pharmacol. (2019) 164:23–33. doi: 10.1016/j.bcp.2019.02.025
105. Kain V, Kapadia B, Misra P, Saxena U. Simvastatin may induce insulin resistance through a novel fatty acid mediated cholesterol independent mechanism. Sci Rep. (2015) 5:13823. doi: 10.1038/srep13823
106. Roden M. How Free fatty acids inhibit glucose utilization in human skeletal Muscle. News Physiol Sci. (2004) 19:92–6. doi: 10.1152/nips.01459.2003
107. Yaluri N, Modi S, Kokkola T. Simvastatin induces insulin resistance in L6 skeletal muscle myotubes by suppressing insulin signaling, Glut4 expression and Gsk-3β phosphorylation. Biochem Biophys Res Commun. (2016) 480:194–200. doi: 10.1016/j.bbrc.2016.10.026
108. Pirillo A, Catapano AL, Norata GD. Niemann-Pick C1-Like 1 (Npc1l1) inhibition and cardiovascular diseases. Curr Med Chem. (2016) 23:983–99. doi: 10.2174/0929867323666160229114111
109. Giugliano RP, Cannon CP, Blazing MA, Nicolau JC, Corbalán R, Špinar J, et al. Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and safety in patients with vs. without diabetes mellitus: results from improve-it (improved reduction of outcomes: vytorin efficacy international trial). Circulation. (2018) 137:1571–82. doi: 10.1161/CIRCULATIONAHA.117.030950
110. Nozaki Y, Fujita K, Yoneda M, Wada K, Shinohara Y, Takahashi H, et al. Long-Term combination therapy of ezetimibe and acarbose for non-alcoholic fatty liver disease. J Hepatol. (2009) 51:548–56. doi: 10.1016/j.jhep.2009.05.017
111. Yagi S, Akaike M, Aihara K, Iwase T, Ishikawa K, Yoshida S, et al. Ezetimibe Ameliorates metabolic disorders and microalbuminuria in patients with hypercholesterolemia. J Atheroscler Thromb. (2010) 17:173–80. doi: 10.5551/jat.2378
112. Takase H, Dohi Y, Okado T, Hashimoto T, Goto Y, Kimura G. Effects of ezetimibe on visceral fat in the metabolic syndrome: a randomised controlled study. Eur J Clin Invest. (2012) 42:1287–94. doi: 10.1111/eci.12000
113. Kurano M, Hara M, Satoh H, Tsukamoto K. Hepatic Npc1l1 overexpression ameliorates glucose metabolism in diabetic Mice via suppression of gluconeogenesis. Metabolism. (2015) 64:588–96. doi: 10.1016/j.metabol.2015.01.011
114. Momtazi-Borojeni AA, Jaafari MR, Badiee A, Sahebkar A. Long-Term generation of Antipcsk9 antibody using a nanoliposome-based vaccine delivery system. Atherosclerosis. (2019) 283:69–78. doi: 10.1016/j.atherosclerosis.2019.02.001
115. Momtazi-Borojeni AA, Jaafari MR, Badiee A, Banach M, Sahebkar A. Therapeutic effect of nanoliposomal Pcsk9 vaccine in a mouse model of atherosclerosis. BMC Med. (2019) 17:1–15. doi: 10.1186/s12916-019-1457-8
116. Pan Y, Zhou Y, Wu H, Chen X, Hu X, Zhang H, et al. A therapeutic peptide vaccine against Pcsk9. Sci Rep. (2017) 7:1–10. doi: 10.1038/s41598-017-13069-w
117. Chen B, Shi X, Cui Y, Hou A, Zhao P. A review of Pcsk9 inhibitors and their effects on cardiovascular diseases. Curr Top Med Chem. (2019) 19:1790–817. doi: 10.2174/1568026619666190809094203
118. Visseren FLJ, Mach F, Smulders YM, Carballo D, Koskinas KC, Bäck M, et al. 2021 Esc guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. (2021) 42:3227–337. doi: 10.1093/eurheartj/ehab484
119. Perego C, Da Dalt L, Pirillo A, Galli A, Catapano AL, Norata GD. Cholesterol metabolism, pancreatic B-Cell function and diabetes. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2149–56. doi: 10.1016/j.bbadis.2019.04.012
120. Karatasakis A, Danek BA, Karacsonyi J, Rangan BV, Roesle MK, Knickelbine T, et al. Effect of Pcsk9 inhibitors on clinical outcomes in patients with hypercholesterolemia: a meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. (2017) 6:e006910. doi: 10.1161/JAHA.117.006910
121. Colhoun HM, Ginsberg HN, Robinson JG, Leiter LA, Müller-Wieland D, Henry RR, et al. No effect of Pcsk9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 Odyssey phase 3 studies. Eur Heart J. (2016) 37:2981–9. doi: 10.1093/eurheartj/ehw292
122. de Carvalho LSF, Campos AM, Sposito AC. Proprotein convertase Subtilisin/Kexin Type 9 (Pcsk9) inhibitors and incident type 2 diabetes: a systematic review and meta-analysis with over 96,000 patient-years. Diabetes care. (2018) 41:364–7. doi: 10.2337/dc17-1464
123. Liu G, Shi M, Mosley JD, Weng C, Zhang Y, Lee MTM, et al. A mendelian randomization approach using 3-Hmg-Coenzyme-a reductase gene variation to evaluate the association of statin-induced low-density lipoprotein cholesterol lowering with noncardiovascular disease phenotypes. JAMA Network Open. (2021) 4:e2112820. doi: 10.1001/jamanetworkopen.2021.12820
124. Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, et al. Hmg-Coenzyme a reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet (London, England). (2015) 385:351–61. doi: 10.1016/j.atherosclerosis.2015.04.076
125. Lotta LA, Sharp SJ, Burgess S, Perry JRB, Stewart ID, Willems SM, et al. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: a meta-analysis. Jama. (2016) 316:1383–91.
126. Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in Pcsk9 cause autosomal dominant hypercholesterolemia. Nat Genet. (2003) 34:154–6. doi: 10.1038/ng1161
127. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low Ldl cholesterol in individuals of African descent resulting from frequent nonsense mutations in Pcsk9. Nat Genet. (2005) 37:161–5. doi: 10.1038/ng1509
128. Uribe KB, Chemello K, Larrea-Sebal A, Benito-Vicente A, Galicia-Garcia U, Bourane S, et al. A systematic approach to assess the activity and classification of Pcsk9 variants. Int J Mol Sci. (2021) 22:13602. doi: 10.3390/ijms222413602
129. Maxwell KN, Breslow JL. Adenoviral-Mediated expression of Pcsk9 in Mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. (2004) 101:7100–5. doi: 10.1073/pnas.0402133101
130. Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst-Hunter Z, Lyall DM, et al. Pcsk9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes endocrinol. (2017) 5:97–105. doi: 10.1016/s2213-8587(16)30396-5
Keywords: glucose/insulin, cholesterol, statins, ezetimibe, PCSK9 inhibitors
Citation: Xiao X, Luo Y and Peng D (2022) Updated Understanding of the Crosstalk Between Glucose/Insulin and Cholesterol Metabolism. Front. Cardiovasc. Med. 9:879355. doi: 10.3389/fcvm.2022.879355
Received: 19 February 2022; Accepted: 07 April 2022;
Published: 29 April 2022.
Edited by:
Xuewei Zhu, Wake Forest Baptist Medical Center, United StatesReviewed by:
Lin Jia, The University of Texas at Dallas, United StatesJenny E. Kanter, University of Washington, United States
Copyright © 2022 Xiao, Luo and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daoquan Peng, cGVuZ2RxQGNzdS5lZHUuY24=