
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 11 August 2022
Sec. Cardio-Oncology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.876294
This article is part of the Research Topic Case Reports in Cardio-Oncology: 2022 View all 39 articles
 Lorenzo Bartoli1,2†
Lorenzo Bartoli1,2† Francesco Angeli1,2†Andrea Stefanizzi1,2Michele Fabrizio1,2
Francesco Angeli1,2†Andrea Stefanizzi1,2Michele Fabrizio1,2 Pasquale Paolisso3,4
Pasquale Paolisso3,4 Luca Bergamaschi1,2
Luca Bergamaschi1,2 Alessandro Broccoli2,5
Alessandro Broccoli2,5 Pier Luigi Zinzani2,5
Pier Luigi Zinzani2,5 Nazzareno Galiè1,2
Nazzareno Galiè1,2 Paola Rucci6
Paola Rucci6 Alberto Foà1,2‡
Alberto Foà1,2‡ Carmine Pizzi1,2*‡
Carmine Pizzi1,2*‡Background: Erdheim–Chester disease (ECD) is a rare form of histiocytosis. An increasing number of genetic mutations have been associated with this syndrome, confirming its possible neoplastic origin. Recently, a connection between the BRAF mutational status and a specific phenotype was described; however, no studies have yet evaluated the correlations between other mutations and the clinical features of the disease.
Objectives: This study aims to clarify the association between the clinical phenotype and genetic mutations identified in the neoplastic cell lines of ECD.
Methods: We describe a case of ECD characterized by pericardial involvement and a KRAS mutation shared with chronic myelomonocytic leukemia. Hence, through a meta-analysis of individual participant data of all genetically and clinically described cases of ECD in the literature, we aimed to elucidate the association between its clinical phenotype and baseline genetic mutations.
Results: Of the 760 studies screened, our review included 133 articles published from 2012 to April 2021. We identified 311 ECD patients whose genotype and phenotype were described. We found five main genes (BRAF, KRAS, NRAS, PIK3CA, and MAP2K1) whose mutation was reported at least three times. Mutation of BRAF led to a neurological disease (183 of 273 patients, 67%; p < 0.001); KRAS- and NRAS-mutated patients mainly showed cutaneous (five of six patients, 83.3%, p < 0.004) and pleural (four of nine patients, 44%, p = 0.002) involvement, respectively; PIK3CA was not associated with specific organ involvement; and MAP2K1 mutations caused the disease to primarily involve the peritoneum and retroperitoneum (4 of 11, 36.4%, p = 0.01).
Conclusion: This work implies a possible influence of baseline mutation over the natural history of ECD, underscoring the importance of a thorough genetic analysis in all cases with the ultimate goal of identifying a possible targeted therapy for each patient.
Erdheim–Chester disease (ECD) is a rare non-Langerhans cell histiocytosis (1, 2), with less than 1,000 cases described so far (2). It is known that ECD affects multiple organs with a wide spectrum of clinical presentations, ranging from an asymptomatic to a life-threatening condition. The most common symptom is bone pain (50%) as bilateral symmetric osteosclerosis evaluated by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography/computed tomography (PET/CT) or technetium-99 m (Tc-99m) bone scan is considered a pathognomonic feature, reported by about 80–95% of patients (3, 4). The other organs often infiltrated are the skin (xanthelasmas), central nervous system (CNS; resulting in ataxia, dysarthria, cognitive impairment, headache, peripheral neuropathy, and, sometimes, mood lability), respiratory system, pituitary gland (mostly central diabetes insipidus), retroperitoneum (renal failure requiring dialysis), and retro-orbital tissue (exophthalmos, retro-orbital pain, oculomotor nerve palsy, or vision loss; 2–4). About 50–70% of patients with ECD exhibit cardiovascular involvement. The most common findings include pericardial infiltration and effusion, and right atrioventricular pseudotumor and peri-arterial sheathing, involving mainly the thoracic and abdominal aorta, renal vessels, and coronary arteries (2–4).
Supplementary Box 1 summarizes the multiorgan involvement in ECD and the potential “red flags.” An increasing number of genetic mutations have been associated with ECD. Specifically, recurrent activating mutations involving the mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K)-AKT pathways have been discovered. BRAF V600E is the most frequent mutation encountered (5), although other genes may be affected, such as NRAS, KRAS, PI3KCA, ARAF, MAP2K1, ALK, and NTRK1 (6–9).
These findings suggest that ECD is a clonal neoplastic disorder, and the most common signaling pathways involved could represent the first molecular targets in histiocytic disorders (7, 8, 10–12).
Last, an association between ECD and leukemic/myelodysplastic disorders that share most of the clonal mutations has been described (13), confirming the possible neoplastic origin probably due to a clonal hematopoiesis phenomenon (14–16).
In terms of clinical presentation, an association between the BRAF mutational status and cardiac, as well as neurological, disorders has been reported. Nevertheless, no studies have yet evaluated the correlations between other gene mutations and the specific ECD phenotype.
We report a case of constrictive pericarditis in a man with xanthelasmas and monocytosis; the final diagnosis was ECD associated with chronic myelomonocytic leukemia (CMML) harboring the same clonal mutation in the KRAS gene. Furthermore, we report the results of a meta-analysis of individual participant data (IPD) of all available ECD cases, aiming to elucidate the association between the ECD clinical phenotype and genetic mutations in neoplastic cell lines.
In the early 2000s, a 55-year-old man developed bilateral eyelid and thorax xanthelasmas (Figure 1A). The eyelid lesions relapsed rapidly after two surgical excisions.
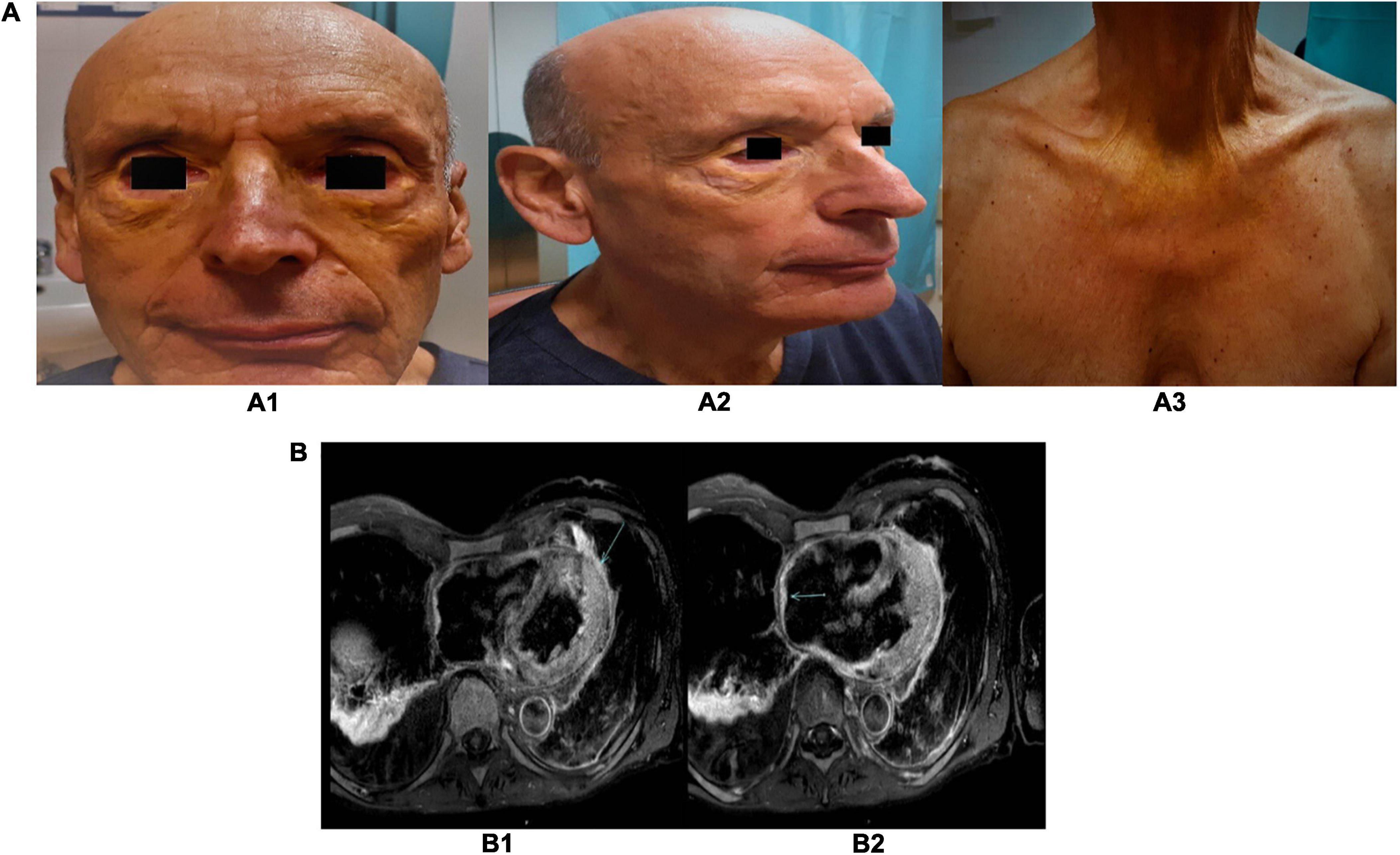
Figure 1. (A) Pictures of the xanthelasmas of the eyelid and thorax. Several yellowish plaque/papules on both upper and lower eyelids, cheeks (A1,A2), and anterior region of the thorax (A3). (B) Cardiac magnetic resonance images. Post-gadolinium T2-weighted sequences, showing a gadolinium-enhanced pericardial nodule (B1) and the pseudotumor of the right atrial roof (B2).
In 2011, blood tests revealed monocytosis, and in the following years, splenomegaly was detected. No further diagnostic or therapeutic approaches were applied due to the lack of systemic and specific organ symptoms, as well as blood test alterations.
In 2017, the patient underwent left pleurodesis due to left pleuritis after a series of ineffective conservative treatments. Bioptic and cytologic materials revealed non-specific inflammation.
The patient remained asymptomatic until October 2019, when he developed fatigue and breathlessness. An electrocardiogram demonstrated sinus tachycardia. Right pleural effusion was detected on chest X-ray. Echocardiography showed findings consistent with constrictive pericarditis; therefore, he was admitted to our cardiology unit.
Blood tests confirmed monocytosis with dysmorphic, broken, and vacuolated monocytes on a blood smear. In addition, anemia and elevated C-reactive protein were observed. Notably, despite the history of xanthelasmas, cholesterol levels were low without lipid-lowering therapy.
A chest CT scan detected nodular pleuro-pericardial lesions. Right heart catheterization and cardiac magnetic resonance (MR; Supplementary Video 1) confirmed the diagnosis of constrictive pericarditis. Furthermore, cardiac MR revealed right atrial posterior wall and pericardial thickening. It also confirmed nodular pericardial and pleural lesions showing hyperemia and edema in T2-weighted sequences (Figure 1B).
Given the multiorgan involvement, including the right atrium and pericardium, which are reported to be commonly affected in cardiac malignancies (17), we hypothesized a hematologic systemic disease. Specifically, the presence of xanthelasma, pleuro-pericardial involvement, and right atrial posterior wall thickening raised the suspicion of ECD.
To investigate the possible bone involvement, we performed a bilateral femoral X-ray and MR scan.
Subsequently, a whole-body 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) bone scintigraphy and 18F-FDG PET/TC were performed to evaluate disease extension and to guide a subsequent biopsy.
We performed blood tests to exclude endocrinopathy, namely, adrenal and pituitary involvement. The CNS was evaluated by a head MR scan. To demonstrate pulmonary, perivascular, and perinephric infiltrates, CT scans of the neck, chest, abdomen, and pelvis were acquired.
The presence of peripheral monocytes and splenomegaly led us to perform a bone marrow biopsy. To detect the typical components of histiocytic infiltrates, tissue was taken from the xanthelasmas of upper lateral and lower medial left eyelids, and a bone biopsy from regions with maximum osteosclerosis on the previous femoral MR.
Moreover, immunohistochemistry (IHC) was performed to evaluate the presence of the typical BRAF V600E mutation. Targeted capture next-generation sequencing (NGS) was executed to identify ECD-related mutations in the skin biopsy, blood plasma, and bone marrow. The panels used for genetic analysis, the genes specifically evaluated by these techniques, and methods used are reported in the section “Supplementary material.”
Last, we performed a meta-analysis of IPD of all ECD cases in the literature. Design and reporting were carried out according to PRISMA-IPD Statement (18) (Supplementary Tables 1, 2). Our methods are reported in the Supplementary material.
Bilateral femoral X-ray and MR revealed osteosclerosis. In addition, MR images showed a symmetrical increase in the red bone marrow. On the other hand, both whole-body 99mTc-DPD scintigraphy and 18F- FDG PET/CT were negative for bone tracer uptake. 18F-FDG PET/CT showed bone marrow glucose uptake consistent with a myeloproliferative disease and there was a mild uptake by the known pleural and pericardial nodular lesions (19).
The head MR scan excluded CNS, pituitary, or retro-orbital involvement. The CT scans of the neck, chest, abdomen, and pelvis only showed marked splenomegaly, without any further sign of organ involvement. The hypothalamic–pituitary–adrenal axis and thyroid and sex hormones were within the normal range.
A right femur biopsy did not show any relevant histiocytic and/or inflammatory component. Conversely, the skin specimen presented diffuse histiocytic infiltrates, with histopathologic and immunohistochemical features, consistent with ECD (CD 68+, CD163+, S100-, and CD1a-; Supplementary Figures 1A, A1, A2, and A3).
The bone marrow biopsy showed a monocytic component (CD14+, CD68PGM1+, and CD163+) compatible with CMML, a rare myeloproliferative disorder (Supplementary Figures 1B, B1, B2, and B3).
Next-generation sequencing analysis of the skin biopsy, blood, and bone marrow, and immunofluorescence of the skin biopsy excluded the presence of typical BRAFV600E mutation but showed a clonal mutation involving KRAS gene (p. Gly12Asp, c.35G > A9), shared by both histiocytes and leukemic cells (Supplementary Figures 1A, A4, 1B, B4). Moreover, adjunctive mutations were reported on the peripheral blood and bone marrow, namely, ASXL1 (p. Leu815Pro), SETBP1 (p.Val1101Ile a), EZH2 (p.Asp185His), and TP53 (p.Pro72Arg).
The final diagnosis was ECD and concomitant chronic myelomonocytic leukemia with a shared clonal mutation.
A treatment with corticosteroid was started, and clinical conditions rapidly improved. A chest X-ray at 3 months showed the resolution of pleural effusion. Blood tests indicated a progressive increase in the blood cell count with no signs of inflammation markers. Cutaneous lesions showed a progressive reduction, leading to complete disappearance.
At the 6-month follow-up, the patient was completely asymptomatic. A cardiac MR scan demonstrated resolution of pericardial constriction (Supplementary Video 1), and neither hyperemia nor edema was observed at T2-weighted images. Similarly, a contrast-enhanced chest CT scan demonstrated complete resolution of pleuro-pericardial effusion and nodular lesions.
Of 760 studies screened, our review of the literature selected 133 articles. The first article included was published in September 2012, and it was the first molecularly characterized case report of ECD (20). In the end, we identified 311 ECD patients whose genotype and phenotype were described (flow diagram of IPD search). The major traits of ECD are summarized in Figure 2, which illustrates the main clinical and radiographic findings of ECD (Figure 2A), the clinical features of specific subgroups (Figure 2B), and the influences of the main mutation over clinical phenotypes (Figure 2C).
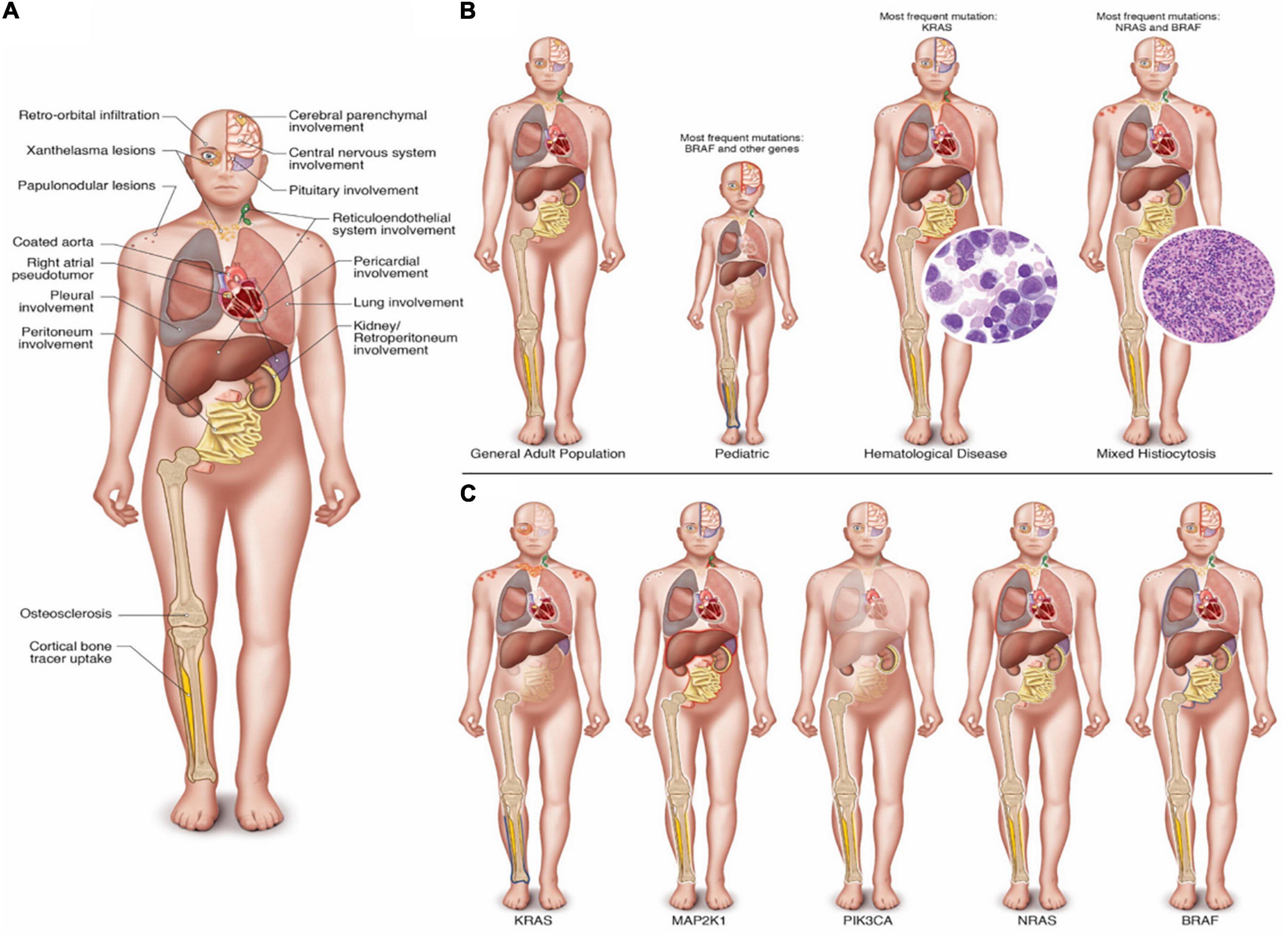
Figure 2. (Central Illustration): (A) Main clinical and radiographic findings in ECD. (B) Clinical and radiographic findings in specific subgroups of ECD. Characteristics of the general adult population, the pediatric population, patients with associated hematologic disorder, in mixed histiocytosis and the respective most frequently associated genetic mutations reported in the literature. (C) Clinical and radiographic findings of ECD in patients with mutation of KRAS, MAP2K1, PIK3CA, NRAS, and BRAF. Note: In (B,C), clinical and radiological features of the specific subgroups were depicted transparent if they were never reported in the literature. Instead, they were reported with a blue halo if there was a statistically significant low frequency involvement; a white halo if there was a normal frequency involvement; and finally with a red halo if there was a statistically significant common involvement in the respective group.
We identified five main genes (BRAF, KRAS, NRAS, PIK3CA, and MAP2K1) whose mutation was reported at least three times in the literature. We merged all the others (Kit, JAK2, PDGFRA, ARAF, CSF1R, and FLT3-ITD) in the same group “other genes.”
BRAF was the most frequently mutated gene (Table 1). Its mutation is linked to neurological disease (67.9%), with one-third of the patients showing pituitary involvement, while in less than 10%, the pleura and peritoneum were affected (Tables 2, 3, and Figure 2C).
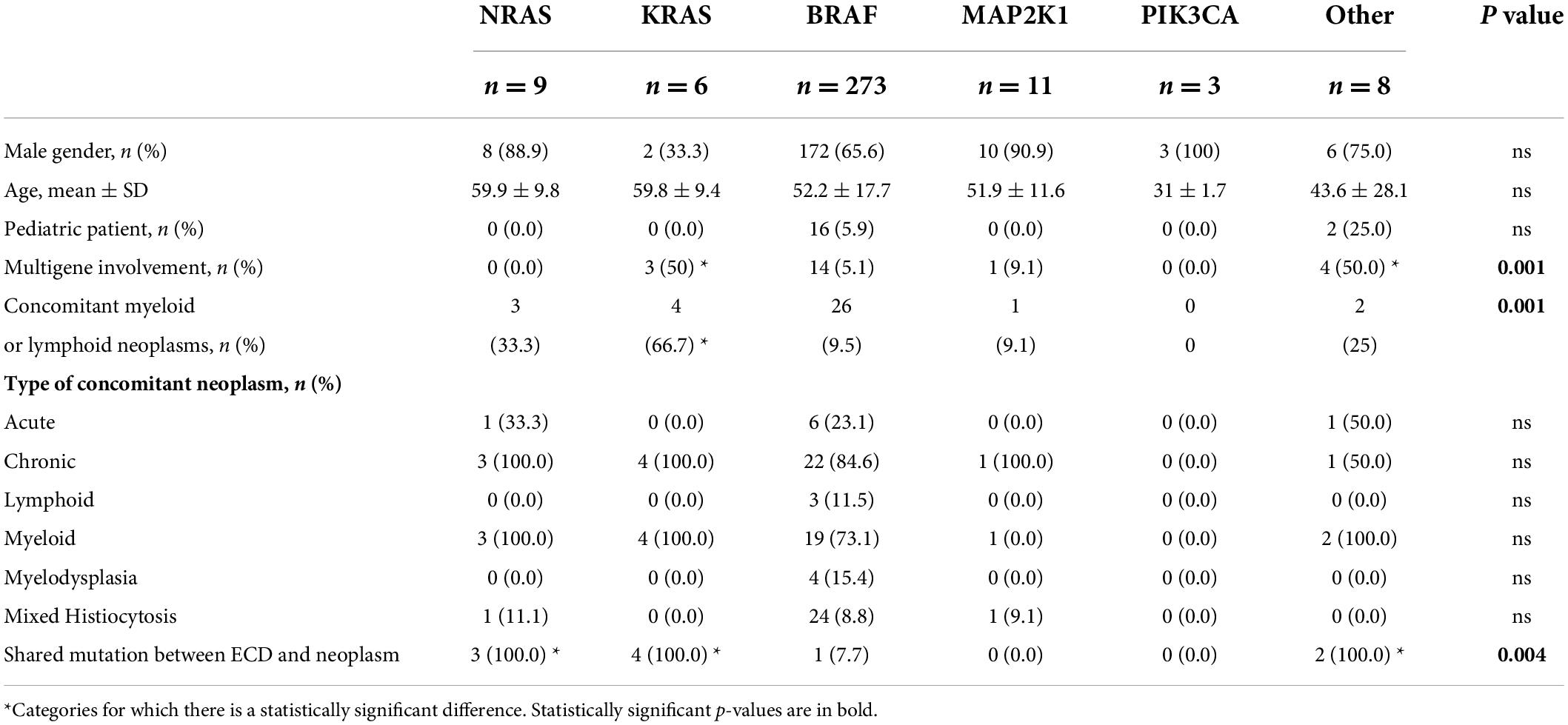
Table 1. General characteristics of Erdheim–Chester patients according to different genotypes.
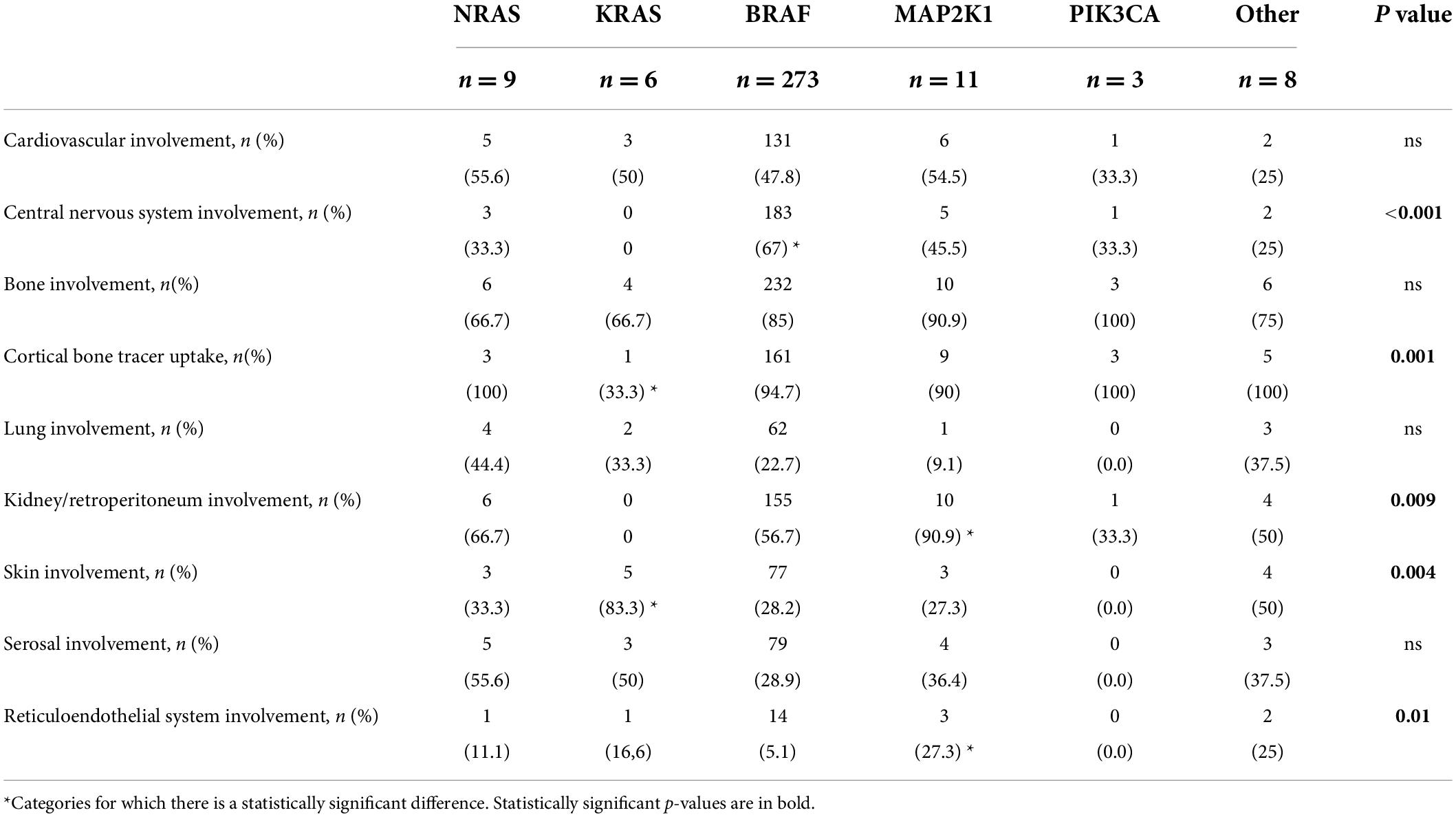
Table 2. Clinical characteristics of Erdheim–Chester patients according to different genotypes.
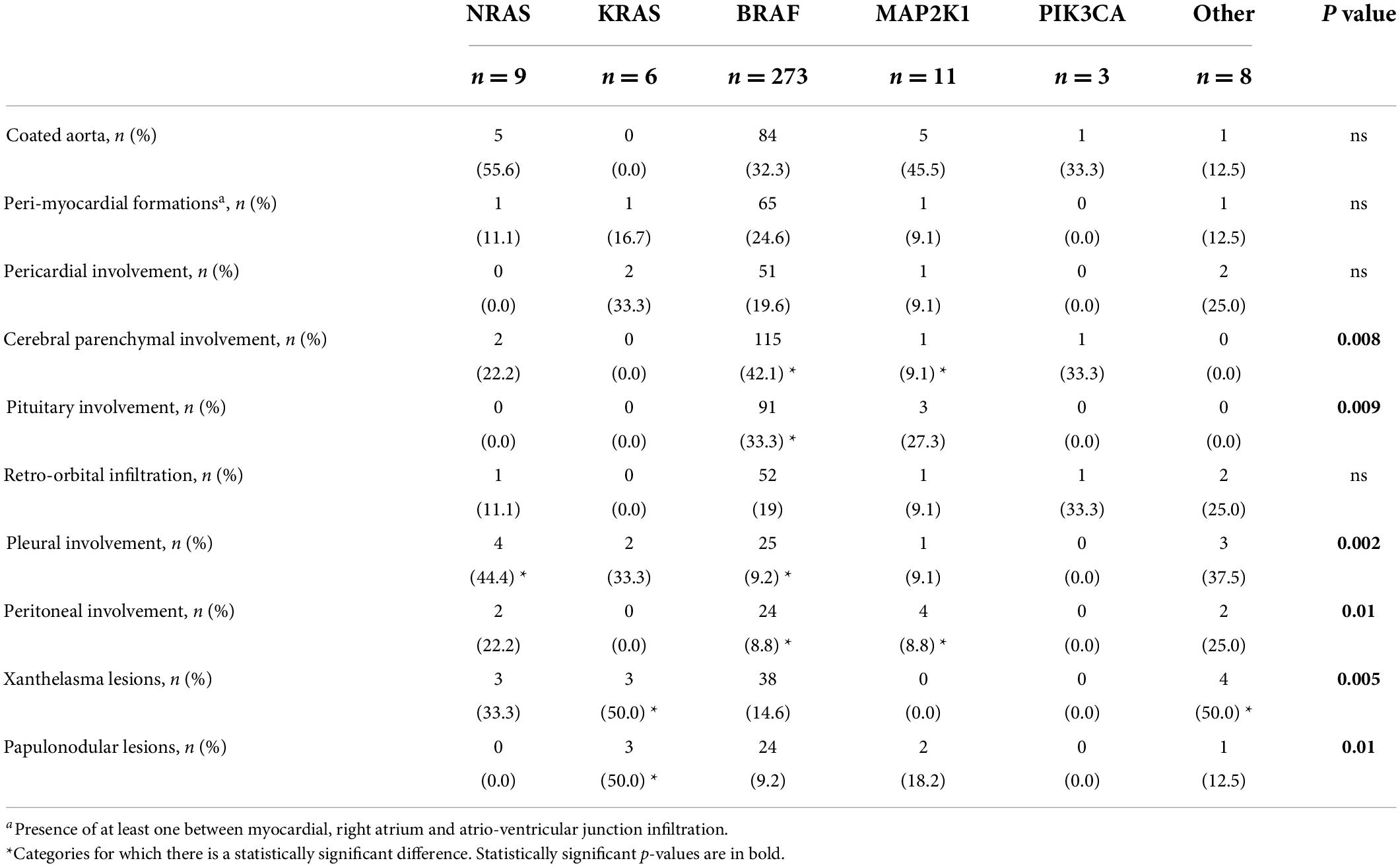
Table 3. Specific cardiac and non-cardiac involvements of Erdheim–Chester patients according to different genotypes.
KRAS mutation is closely related to the skin (both xanthelasma-like and nodular lesions). Furthermore, there was a lower incidence of bone tracer uptake at PET-FDG and bone scan (in case of bone involvement) and a higher frequency of concomitant hematologic neoplasia. The CNS, retroperitoneum, and mesentery were never affected. Finally, patients with KRAS-mutated ECD frequently showed multigene involvement (Tables 1–3 and Figure 2C). These features were also observed in our case report.
Erdheim–Chester disease patients carrying NRAS mutations did not present pericardial or pituitary disease but had a high frequency of pleural involvement (Tables 2, 3, and Figure 2C).
Patients with MAP2K1-mutated ECD showed more frequent abdominal involvement, with both peritoneal and retroperitoneal fibrosis and/or hairy kidney features. Moreover, they tended to show reticuloendothelial system involvement, and they never had xanthelasma lesions (Tables 2, 3, and Figure 2C).
PIK3CA mutation affected young patients without a specific organ involvement. Notably, the lung, skin, serosa, pituitary gland, and reticuloendothelial system were never affected (Tables 1–3 and Figure 2C).
Furthermore, we noticed peculiar characteristics of three different populations: pediatric individuals, patients with concomitant hematologic disease, and subjects with mixed histiocytosis (defined as the simultaneous presence of another histiocytic disease).
In pediatric patients, the most frequently mutated gene was BRAF. These patients frequently exhibited CNS involvement with a lower incidence of serosal and bone disease (with lower tracer uptake at PET-FDG or bone scan), while cardiovascular involvement and abdominal involvement were never observed (Supplementary Table 3 and Figure 2B).
Patients with an associated hematologic neoplasia are older, with a higher frequency of pulmonary, reticuloendothelial, and serosal manifestations, while CNS involvement is sporadic. Notably, subjects with KRAS or NRAS-mutated ECD frequently shared the genetic mutation between histiocytes and hematologic neoplasia (Supplementary Table 4 and Figure 2B).
Finally, patients with mixed histiocytosis showed clinical features similar to those of the conventional ECD cohort with, a higher incidence of skin involvement, such as papulonodular lesions (Supplementary Table 5 and Figure 2B).
Our clinical case demonstrated that targeted therapy is driven by a precise diagnosis.
The goal of precision medicine is “to provide the best available care for each individual,” (21) and an accurate diagnostic approach is crucial to allow the best therapeutic option. This concept is pivotal in entities with a heterogeneous clinical presentation, such as ECD.
In fact, our clinical case differs from the classical presentation of this disease. The patient developed bilateral eyelid and thorax xanthelasmas, pleuritis, and constrictive pericarditis without typical ECD findings, such as tracer uptake on the bone scan or FDG-PET, CNS involvement, coated aorta, and the hairy kidney/retroperitoneal fibrosis, which usually raise the suspicion of ECD.
Furthermore, the hematologic neoplasm and the presence of histiocytes and leukemic cells of KRAS mutation were confounding factors in the diagnostic process.
Precision medicine means to match clinical, diagnostic, and genetic features of a disease. Therefore, to further promote the use of precision medicine and considering the growing body of evidence that ECD is a clonal hematopoietic disorder (3), we hypothesized that extremely variable clinical features may relate to the primitive mutations that generate neoplastic germlines. These germlines give birth to both histiocytic and, if present, hematologic neoplastic cells, and the baseline genetic mutations could greatly influence the clinical phenotype and the natural history of the disease.
To test this hypothesis, we performed an individual meta-analysis of all case reports and case series of patients with a diagnosis of ECD and known mutational status. The results of our work, which is the most comprehensive to date, support our hypothesis since each mutation demonstrated a trend toward organ-specific involvement.
Not surprisingly, the clinical features of our case matched with the results of our analysis since they could be explained by the KRAS mutated status and the concomitant hematologic neoplasm.
Our results, which help in the diagnostic process and promote a thorough genetic analysis of ECD, may have also a positive therapeutic and prognostic impact following a precision model of therapy for Mendelian disease, focused on treating the underlying mechanism using a genetic therapy.
This approach is increasingly used and continuously developing: BRAF and MEK/ERK inhibitors have been already used extensively in ECD (7, 8, 10–12), while others have been used in sporadic case reports. Moreover, a new molecule targeting KRAS has been successfully administrated for the first time in a phase I trial in lung cancer (4, 22) and has recently been approved by the Food and Drug Administration for therapeutic use (23). Our work suggests a possible influence of baseline mutation over the natural history of the disease, underscoring the importance of a thorough genetic analysis in all ECD cases with the ultimate goal of identifying a targeted therapy for each patient.
Our sample size is small (311 patients), except for BRAF, which had a small number of cases for other mutations. This is due to the rarity of this syndrome. Also, there is lack of systematic or thorough genetic analysis in all cases found in the literature, so a high number of patients could not be included in our study owing to the absence of a known mutation.
On the contrary, some studies were excluded because they did not provide the clinical phenotype of the study population, or because a clear link between the genotype and phenotype of individual patients was not always possible (i.e., aggregate data).
Since our meta-analysis focused only on those cases whose both genotype and clinical phenotype were available, confounding and selection bias cannot be excluded.
Finally, there was a risk of including low-quality articles or studies with heterogenous data. Nevertheless, data from individual patients retrieved from different articles were harmonized for statistical analysis, and the methodological quality of the studies was assessed using the method proposed by Murad et al. (see Supplementary material). Thus, of the 131 studies included, 106 and 27 showed good and moderate overall quality, respectively. No study revealed a low methodological quality.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/s for the publication of this case report.
LBa and FA contributed to the conception or design of the work and drafted the manuscript. AS, MF, PP, LBe, and PR contributed to the acquisition, analysis, or interpretation of data for the work. AB, PZ, NG, AF, and CP critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
PP was supported by a research grant from the CardioPaTh Ph.D. Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.876294/full#supplementary-material
Supplementary Figure 1 | (A) Biopsy of the eyelid. Images of hematoxylin-eosin stain (A1), CD 68 (A2), and CD163 (A3) immunohistochemistry. The illustrations show foamy, CD68+ and CD163+ histiocytes, typical of Erdheim-Chester disease. In the last image (A4), demonstration of a KRAS exon 2 mutation (p.Gly12Asp) by NGS (S5 Gene Studio) in the specimen. (B) Biopsy of the bone marrow (BM). Images of hematoxylin-eosin (B1), and May–Grünwald (B2) stain and CD14 immunohistochemistry (B3). The illustrations show granulocytic proliferation in an hypercellular bone marrow, with monocytes (CD14+), erythroid precursors and abundant hypolobulated and small megakaryocytes, suggestive of chronic myelomonocytic leukemia. The last image (B4) shows the detection by NGS (S5 Gene Studio) in both BM and peripheral blood of the same KRAS mutation already detected in the eyelid specimen.
Supplementary Video 1 | In the video frames real T1 cine sequences showing evident respiratory shift of the interventricular septum due to enhanced ventricular interdependence, which completely resolves at follow-up.
1. Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. (2014) 124:483–92. doi: 10.1182/blood-2014-03-561381
2. Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH, et al. Erdheim-Chester disease: Consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. (2020) 135:1929–45. doi: 10.1182/blood.2019003507
3. Estrada-Veras JI, O’Brien KJ, Boyd LC, Dave RH, Durham B, Xi L, et al. The clinical spectrum of Erdheim-Chester disease: An observational cohort study. Blood Adv. (2017) 1:357–66. doi: 10.1182/bloodadvances.2016001784
4. Cohen-Aubart F, Emile JF, Carrat F, Helias-Rodzewicz Z, Taly V, Charlotte F, et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am J Hematol. (2018) 93:E114–7. doi: 10.1002/ajh.25055
5. Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-langerhans cell histiocytoses. Blood. (2012) 120:2700–3. doi: 10.1182/blood-2012-05-430140
6. Emile JF, Diamond EL, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. (2014) 124:3016–9. doi: 10.1182/blood-2014-04-570937
7. Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. (2016) 6:154–65. doi: 10.1158/2159-8290.CD-15-0913
8. Durham BH, Lopez Rodrigo E, Picarsic J, Abramson D, Rotemberg V, De Munck S, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. (2019) 25:1839–42. doi: 10.1038/s41591-019-0653-6
9. Tzankov A, Kremer M, Leguit R, Orazi A, van der Walt J, Gianelli U, et al. Histiocytic cell neoplasms involving the bone marrow: Summary of the workshop cases submitted to the 18th meeting of the European association for haematopathology (EAHP) organized by the European bone marrow working group, Basel 2016. Ann Hematol. (2018) 97:2117–28. doi: 10.1007/s00277-018-3436-0
10. Haroche J, Amoura Z, Charlotte F, Salvatierra J, Wechsler B, Graux C, et al. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim-Chester histiocytosis. Blood. (2008) 111:5413–5. doi: 10.1182/blood-2008-03-148304
11. Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, et al. Vemurafenib for BRAF V600-mutant Erdheim– Chester disease and langerhans cell histiocytosis: Analysis of data from the histology– independent, phase 2, open-label VE-BASKET study. JAMA Oncol. (2018) 4:384–8. doi: 10.1001/jamaoncol.2017.5029
12. Bhatia A, Ulaner G, Rampal R, Hyman DM, Abdel-Wahab O, Durham BH, et al. Single-agent dabrafenib for BRAFV600E-mutated histiocytosis. Haematologica. (2018) 103:e177–80. doi: 10.3324/haematol.2017.185298
13. Papo M, Diamond EL, Cohen-Aubart F, Emile JF, Roos-Weil D, Gupta N, et al. High prevalence of myeloid neoplasms in adults with non-langerhans cell histiocytosis. Blood. (2017) 130:1007–13. doi: 10.1182/blood-2017-01-761718
14. Milne P, Bigley V, Bacon CM, Néel A, McGovern N, Bomken S, et al. Hematopoietic origin of langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. (2017) 130:167–75. doi: 10.1182/blood-2016-12-757823
15. Durham BH, Roos-Weil D, Baillou C, Cohen-Aubart F, Yoshimi A, Miyara M, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. (2017) 130:176–80. doi: 10.1182/blood-2016-12-757377
16. Cohen Aubart F, Roos-Weil D, Armand M, Marceau-Renaut A, Emile JF, Duployez N, et al. High frequency of clonal hematopoiesis in Erdheim-Chester disease. Blood. (2021) 137:485–92. doi: 10.1182/blood.2020005101
17. Foà A, Paolisso P, Bergamaschi L, Rucci P, Di Marco L, Pacini D, et al. Clues and pitfalls in the diagnostic approach to cardiac masses: Are pseudo-tumours truly benign? Eur J Prev Cardiol. (2021) 29:zwab032. doi: 10.1093/eurjpc/zwab032
18. Stewart LA, Clarke M, Rovers M, Riley RD, Simmonds M, Stewart G, et al. Preferred reporting items for systematic review and meta-analyses of individual participant data: The PRISMA-IPD statement. JAMA. (2015) 313:1657–65. doi: 10.1001/jama.2015.3656
19. D’Angelo EC, Paolisso P, Vitale G, Foà A, Bergamaschi L, Magnani I, et al. Diagnostic accuracy of cardiac computed tomography and 18-F fluorodeoxyglucose positron emission tomography in cardiac masses. JACC Cardiovasc Imaging. (2020) 13:2400–11. doi: 10.1016/j.jcmg.2020.03.021
20. Blombery P, Wong SQ, Lade S, Prince HM. Erdheim-Chester disease harboring the BRAF V600E mutation. J Clin Oncol. (2012) 30:e331–2. doi: 10.1200/JCO.2012.43.2260
21. National Research Council. Toward precision medicine: Building a knowledge network for biomedical research and a new taxonomy of disease. Washington, DC: The National Academies Press (2011).
22. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS G12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
Keywords: Erdheim-Chester disease, constrictive pericarditis, genotype, phenotype, precision medicine, case-report
Citation: Bartoli L, Angeli F, Stefanizzi A, Fabrizio M, Paolisso P, Bergamaschi L, Broccoli A, Zinzani PL, Galiè N, Rucci P, Foà A and Pizzi C (2022) Genetics and clinical phenotype of Erdheim–Chester disease: A case report of constrictive pericarditis and a systematic review of the literature. Front. Cardiovasc. Med. 9:876294. doi: 10.3389/fcvm.2022.876294
Received: 15 February 2022; Accepted: 18 July 2022;
Published: 11 August 2022.
Edited by:
Reto Asmis, Wake Forest School of Medicine, United StatesCopyright © 2022 Bartoli, Angeli, Stefanizzi, Fabrizio, Paolisso, Bergamaschi, Broccoli, Zinzani, Galiè, Rucci, Foà and Pizzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmine Pizzi, Y2FybWluZS5waXp6aUB1bmliby5pdA==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.