
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 04 May 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.868934
This article is part of the Research TopicInflammation and Immunomodulation in Cardiovascular RemodelingView all 10 articles
 Bryce R. Evans1,2†
Bryce R. Evans1,2† Anaïs Yerly1,2†
Anaïs Yerly1,2† Emiel P. C. van der Vorst3,4,5,6Iris Baumgartner1,2Sarah Maike Bernhard1,2
Emiel P. C. van der Vorst3,4,5,6Iris Baumgartner1,2Sarah Maike Bernhard1,2 Marc Schindewolf1,2
Marc Schindewolf1,2 Yvonne Döring1,2,3,4*
Yvonne Döring1,2,3,4*Atherosclerotic vascular disease remains the most common cause of ischemia, myocardial infarction, and stroke. Vascular function is determined by structural and functional properties of the arterial vessel wall, which consists of three layers, namely the adventitia, media, and intima. Key cells in shaping the vascular wall architecture and warranting proper vessel function are vascular smooth muscle cells in the arterial media and endothelial cells lining the intima. Pathological alterations of this vessel wall architecture called vascular remodeling can lead to insufficient vascular function and subsequent ischemia and organ damage. One major pathomechanism driving this detrimental vascular remodeling is atherosclerosis, which is initiated by endothelial dysfunction allowing the accumulation of intimal lipids and leukocytes. Inflammatory mediators such as cytokines, chemokines, and modified lipids further drive vascular remodeling ultimately leading to thrombus formation and/or vessel occlusion which can cause major cardiovascular events. Although it is clear that vascular wall remodeling is an elementary mechanism of atherosclerotic vascular disease, the diverse underlying pathomechanisms and its consequences are still insufficiently understood.
Inflammatory mediators such as chemokines and cytokines are quickly released by a multitude of cell types during inflammation and trauma and thereby orchestrate a vital immune response and vascular remodeling. These interactions can occur in either an autocrine or paracrine fashion resulting in structural and functional changes of the vascular wall. However, during chronic inflammatory conditions these mediators can also cause tissue damage or (irreversible) tissue remodeling, a term referring to structural and functional changes of the arterial vessel wall (1). A chronic disease that is characterized by such arterial vessel remodeling is atherosclerotic vascular disease, which is the most common cause of cardiovascular disease (CVD) (2). Mechanisms involved in atherosclerotic arterial remodeling include hyperplasia of the arterial intima and media, changes in vascular collagen and elastin, endothelial dysfunction, and arterial calcification. Identifying the cross talk between cells of the vasculature which maintain or disrupt vascular homeostasis and result in vascular remodeling may offer strategic insight for CVD prevention.
Atherosclerosis is initiated by endothelial dysfunction allowing lipids to accumulate in the intima, which subsequently results in intimal inflammation driving (persistent) vascular changes (3). Various inflammatory mediators comprising of modified lipids, such as oxidized low-density lipoprotein (oxLDL), but also chemokines, cytokines and lipid mediators further foster the endothelial dysfunction and increase vascular permeability. This increased permeability further stimulates the influx and accumulation of lipids and immune cells in the intimal layer of the vascular wall, resulting in a vicious circle (3). The immune cell infiltration into the vessel wall is further increased by the upregulation of adhesion molecules on the endothelium, stimulated by the inflammatory environment. This further induces the arrest of monocytes and other leukocytes onto the vessel wall which subsequently transmigrate into the intima (4, 5). Infiltrated monocytes subsequently differentiate into macrophages which will engulf excess lipids and develop into lipid laden foam cells. Due to the excess uptake of lipids, these foam cells will eventually undergo apoptosis and necrosis, resulting in the formation of a necrotic core in atherosclerotic lesions (6). Accumulation of such necrotic debris leads to the continued release of toxic and pro-inflammatory stimuli in the intima, further promoting inflammation, remodeling and vulnerability of the plaque.
Key cells in shaping the vascular architecture are vascular smooth muscle cells (VSMCs), which are normally present in the arterial media and express a range of “SMC markers” including smooth muscle cell myosin heavy chain (MYH11), smooth muscle cell actin (SM-α), smoothelin and others. These VSMCs are considered to have a contractile phenotype, which is important to maintain the vascular tone. However, during atherosclerosis formation a phenotype switch is induced by inflammatory mediators resulting in the transition from a contractile phenotype to a synthetic phenotype. Additionally, VSMCs will proliferate and migrate from the media into the intima where they will produce extracellular matrix (ECM) to form a fibrous cap and stabilize the atherosclerotic lesions. Moreover, VSMCs with a synthetic phenotype adopt macrophage-like characteristics and can also develop into SMC-derived foam cells (2). Additionally and especially in later stages of lesion development, synthetic VSMCs also produce and secrete matrix metalloproteinases (MMPs) resulting in greater proteolytic activity toward elastin and collagen, which destabilizes the plaque and increases the risk of plaque rupture and thrombus formation (7).
This review aims to draw attention to the main inflammatory mediators involved in vascular remodeling seen in atherosclerosis.
Chemokines are a family of chemoattractant cytokines secreted by various cells, which play a vital role in cell migration from the bloodstream into tissues. They induce cell movement in response to and toward a chemokine gradient also referred to as chemotaxis (8). In addition, chemokines play an important role in various cellular functions including proliferation, survival and differentiation (9). Chemokines can be classified into four structural subfamilies, CC, CXC, CX3C and C based on the location of the key cysteine residues in the disulfide bonding which are either juxtaposed (CC) or separated by 1 or 3 amino acids (CXC and CX3C) respectively (10). Chemokines initiate cellular responses through interaction with seven-transmembrane G-protein coupled receptors (GPCRs), more specifically classical chemokine receptors, or with atypical chemokine receptors (ACKRs), which do not signal through G-proteins (11).
CC chemokines have at least 27 distinct members reported in mammals, called CC chemokine ligands (CCL) and are typically responsible for the induction of leukocyte migration (10, 11). Currently, 17 different CXC chemokines have been described in mammals, which can be further subdivided into two subcategories, based on the presence or absence a specific amino acid sequence of glutamic acid-leucine-arginine immediately before the first cysteine of the CXC motif (12). The subgroup of CXC chemokines with this sequence specifically induce the migration of neutrophils, while the subgroup without this sequence typically attract lymphocytes. More unique is CX3CL1, which possesses three amino acids between the two cysteines and is also termed CX3C chemokine or fractalkine (12) and the two C chemokines XCL1 (lymphotactin-α) and XCL2 (lymphotactin-β) (12).
The remainder of this chapter will focus on individual chemokines that have been shown to be involved in vascular remodeling during inflammation with a particular focus on atherosclerosis.
CCL2 (Table 1) also known as monocyte chemoattractant protein-1 is a member of the CC chemokine subfamily and exhibits potent chemotactic activity toward monocytes and T lymphocytes (36). Jones et al. (37) demonstrated that activated VSMCs isolated from mice secreted more CCL2 and CXCL1 and in vivo knockout of Protease-Activated Receptor 2 (PAR2) in vascular cells reduced their expression of CCL2/CXCL1 and resulted in a reduction of macrophage content in atherosclerotic lesions compared to the control animals. Additional findings demonstrated increased plaques stability, increased smooth muscle actin alpha 2 ACTA-2, collagen content and reduced interleukin-1 (IL)-1 and tumor necrosis factor (TNF)-α. This suggests that CCL2 acts on macrophage chemotaxis into the lesion in a paracrine fashion (Figure 2). Furthermore, CCL2 may interact on VSMC via an autocrine mechanism to stimulate the phenotypic changes toward a synthetic phenotype. Besides promoting the transmigration of circulating monocytes, CCL2 also promotes cytokine production and adhesion molecule expression on monocytes (38). CCL2 expression is induced by inflammatory cytokines, growth factors, or complement factors in monocytes, ECs, and VSMCs (39, 40). Furthermore, CCL2 seems to be an important chemokine in the development of atherosclerosis, since its expression has been detected in atherosclerotic lesions but not in vessels obtained from healthy individuals (38, 41) and in patients with MI (36, 42). Enhanced CCL2 within the lesion is correlated with histopathologic, molecular, and clinical hallmarks of plaque vulnerability, clearly suggesting that this chemokine also plays a role in advanced stages of lesion development (43). Interestingly, circulating CCL2 levels also correlate with subclinical atherosclerosis disease severity in postmenopausal women and may act as a potential early biomarker in this population (44). Further studies have demonstrated that circulating myeloid cells deposit CCL2 on the arterial endothelium to enhance monocyte recruitment and thereby drive atherogenesis (15).
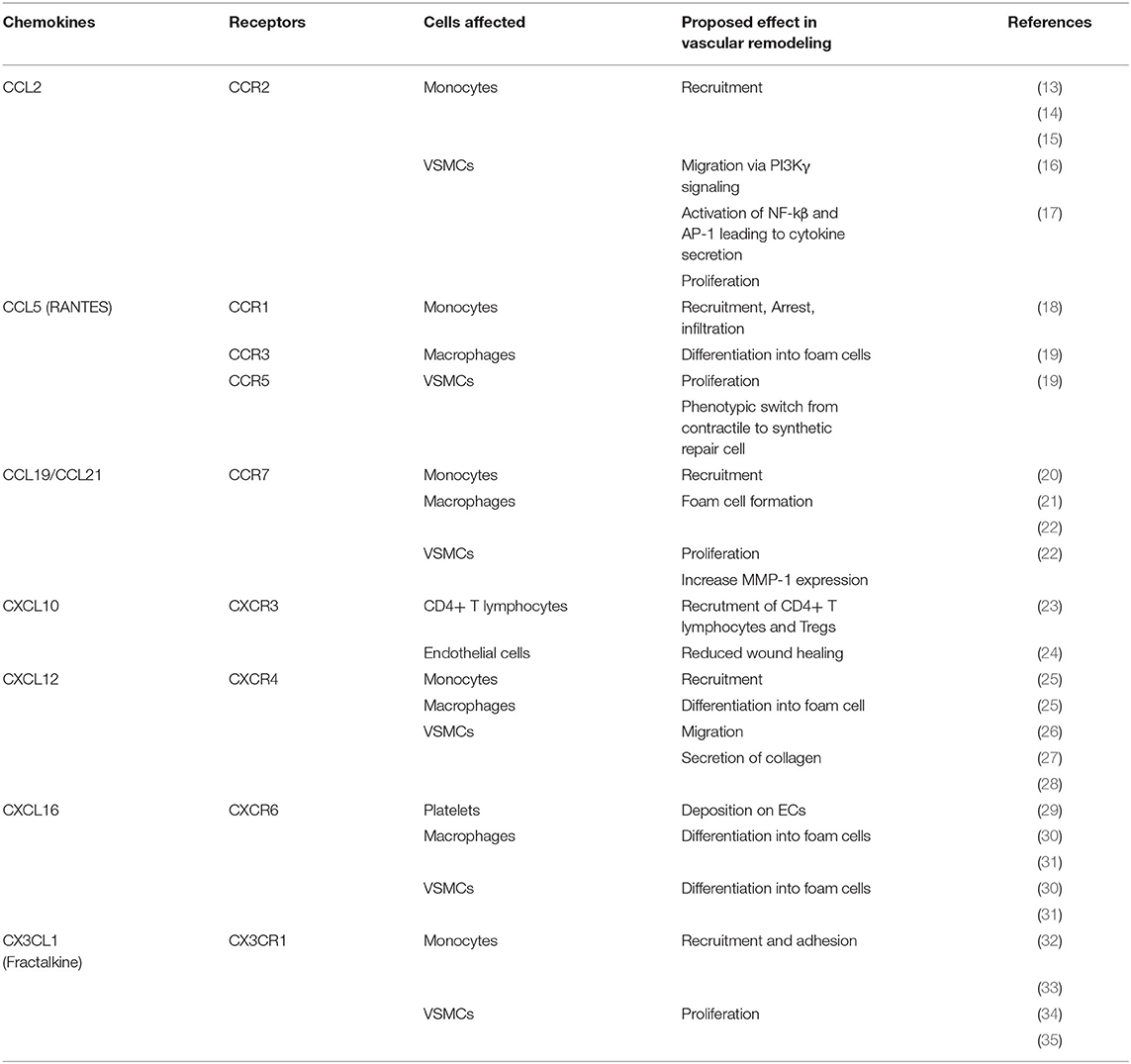
Table 1. Overview of chemokines involved in atherosclerosis remodeling and their physiological effect.
CCL2 is also involved in vascular remodeling as it has been shown to stimulate the binding activity of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) to activator protein-1 (AP-1) in cultured human VSMCs (HVSMCs) grown from unused portions of saphenous veins harvested during coronary artery bypass surgery (17). In addition, CCL2 was reported to induce the proliferation and IL-6 secretion from HVSMCs in vitro (17). Furthermore, recombinant CCL2 stimulated HVSMC proliferation in vitro via AP-1, which was inhibited by mitogen-activated protein kinase (MEK)-1 and MEK2 inhibitor treatment (17). This suggests that CCL2 induces differential activation of NFκB and AP-1 leading to cytokine secretion and proliferation in human VSMCs. Moreover, pharmacological phosphatidylinositol 3-kinase gamma (PI3Kγ) inhibition, inactivation of PI3Kγ, as well as genetic deletion of PI3Kγ in mice was used to study the role of CCL2 on the platelet-derived growth factor (PDGF) signaling pathway and migration processes in primary aortic VSMCs (16). A wound healing assay illustrated that CCL2 is crucial for VSMC migration via PI3Kγ signaling as blocking the CCL2/CCR2 pathway or the inhibition of PI3Kγ reduced PDGF-stimulated aortic VSMC migration by 50% (16). Furthermore, a low-density lipoprotein receptor knock-out (Ldlr−/−) mouse model fed a Western diet (WD) for 8-weeks treated with a PI3Kγ inhibitor showed decreased atherosclerotic lesion size and increased plaque collagen content (45). Finally, VSMCs isolated from PI3Kγ-deficient mice (PI3Kγ−/−), or mice expressing an inactive PI3Kγ, termed PI3KγKD/KD, showed reduced migration when compared to the control cells in response to CCL2 and PDGF (16). Combined these results demonstrate that CCL2 plays a role in lesion formation by promoting VSMC migration in a PI3Kγ dependent manner but can also result in decreased plaque stability by reducing the collagen formation.
Another study demonstrated that a CCL2 competitor (PA508) reduced inflammatory monocyte recruitment, limited neointimal hyperplasia, and attenuated myocardial ischemia/reperfusion injury in an Apolipoprotein E knock-out (Apoe−/−) mouse model, highlighting the potential of PA508 as novel therapeutic approach to treat MI (13). However, the study did not examine mice on a WD to investigate the therapeutic potential of PA508 on atherosclerosis development. Nevertheless, another study treating Apoe−/− mice, fed with a WD for 6-weeks, daily with a CCR2 antagonist (INCB3344) revealed reduced circulating CCR2+ monocytes and diminished atherosclerotic plaques in both the carotid artery and the aortic root (14), proving the therapeutic potential of CCL2/CCR2 targeting for atherogenesis.
Overall, as CCL2 has been shown to be overexpressed in atherosclerotic lesions it is likely that it plays a prominent role in vascular remodeling of VSMCs in atherosclerosis (Figure 1). CCL2 may stabilize the plaque by fostering migration of medial VSMCs into the intima and increase their proliferation while in parallel, recruitment of arterial leukocytes into the lesion triggers atherogenesis (15). Furthermore, therapeutic strategies, such as the PI3Kγ inhibition or CCL2/CCR2 pathway inhibition reveal a decrease in lesion size in mice due to the reduced VSMC migration (45) (Table 2). Nevertheless, further research is necessary to demonstrate whether the putative beneficial effects of CCL2 on VSMCs, e.g., increased proliferation and subsequent increase of plaque stability, outbalance its pro-atherogenic effects on leukocyte recruitment.
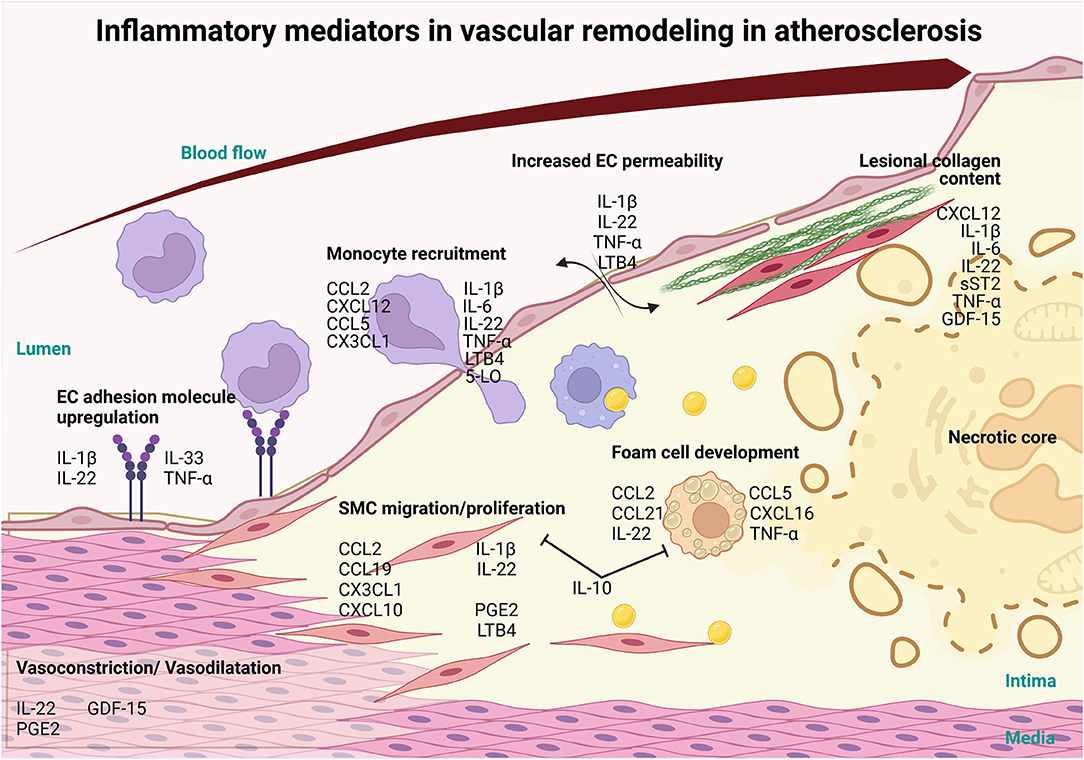
Figure 1. Involvement of inflammatory mediators in vascular remodeling in atherosclerosis. Inflammatory mediators, such as interlukin-1β (IL-1β), IL-22, IL-33 and tumor necrosis factor-α (TNFα) cytokines can influence the progression of atherosclerosis and CVD via the activation of the endothelium resulting in the upregulation of adhesion molecules. Furthermore, these mediators increase vascular permeability, through IL-1β, IL-22, TNFα and LTB4 and along with the adhesion molecule upregulation allows for the infiltration of monocytes and other immune cells recruited via chemokine ligand 2 (CCL2), C-X-C Motif Chemokine Ligand 12 (CXCL12), CCL5, CX3CL1, IL-1β, IL-6, IL-22, TNF-α Leukotriene B4 (LTB4), 5-LO. Mediators like CCL2, CCL19, CXCL10, CX3CL1, IL-1β, IL-22, Prostaglandin E2 (PGE2) and LTB4 also induce the migration and proliferation of VSMCs into the intima and affect the production of collagen, which, in turn, modulates plaque stability. Foam cell formation is initiated by CCL2, CCL5, CCL21, CXCL16, IL-22 and TNFα and exhausted foam cells undergoing apoptosis and necrosis to establish the necrotic core of the lesion. On the other hand, IL-10, a potent anti-inflammatory cytokine, prevents the formation of foam cells and SMC migration and proliferation. As the lesion grows, blood vessel lumen is narrowing eventually causing vessel occlusion which may lead to major adverse cardiovascular complications. Inflammatory mediators CXCL12, IL-1β, IL-6, IL-22, soluble suppression of tumorigenesis-2 (sST2), TNFα and Growth/Differentiation Factor-15 (GDF-15) also play a role in the stability of the lesion by controlling collagen in the fibrous cap. In addition, mediators like LL-22, GDF-15 and PGE2 regulate vasoconstriction and vasodilation of the arteries thereby controlling blood pressure and ensuring proper vascular function (this figure was made with Biorender.com).
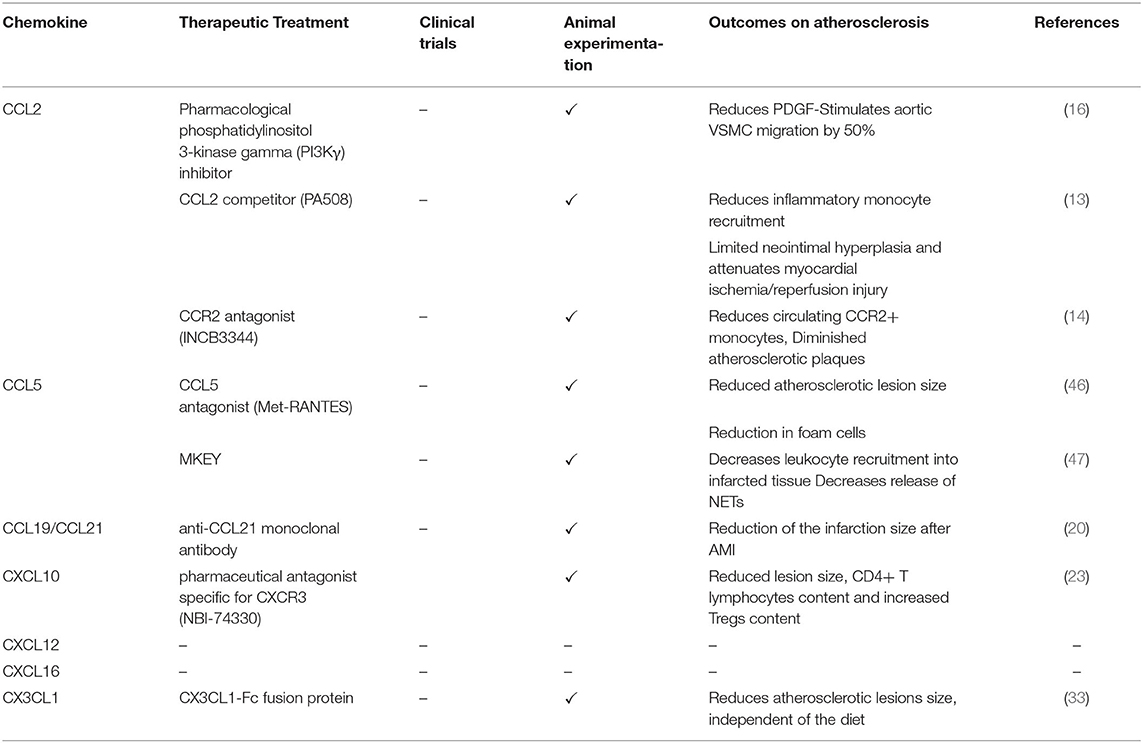
Table 2. Targeting chemokines as therapeutic treatments in vascular remodeling and CVD.
CCL5, a chemokine which is also known as RANTES, can bind to a plethora of receptors including CCR1, CCR3 and CCR5 (Table 1) (48). Platelet-derived deposition of CCL5 on the activated endothelium results in monocyte arrest (49), which appears to be dependent on P-selectin (50). Wire-injury induced neointima formation in the carotid artery of Apoe−/− mice showed that the systemic inhibition of CCL5 or P-selectin deficiency hindered neointima formation as well as monocyte infiltration (50). Furthermore, it could be demonstrated that CCL5 mRNA and protein levels were upregulated in the aortic intima of Ldlr−/− mice fed a WD for 3 weeks and a function-blocking antibody to CCR5 significantly reduced monocyte recruitment into the lesions (18). Further investigations using a bone marrow transplantation (BMT) experiment demonstrated that hematopoietic CCL5 regulates monocyte recruitment and accumulation of macrophages in the lesions after 3 weeks of cholesterol-rich diet feeding of Ldlr−/− mice. However, after 6 weeks of cholesterol-rich diet, CCL5 only plays a minor role in the recruitment of monocytes into the lesions suggesting that CCL5 only plays a role in early stages of lesion formation and subsequent vascular remodeling (18).
Similar results were seen in a 2-week WD Ldlr−/− mouse model where mice were treated with a CCL5 antagonist (Met-RANTES). Treated animals had reduced atherosclerotic lesion size and a reduction in relative lesional foam cell content compared to the control group (46). Thus, CCL5 mediates vascular remodeling in atherogenesis via monocyte arrest and infiltration into the lesion and facilitates the differentiation and development of monocytes into foam cells, although the exact underlying mechanisms behind this CCL5-induced foam cell formation remain to be elucidated. Another study, using CCL5−/−CCR5–/− mice on a 12-week WD, showed significantly reduced expression of the synthetic markers osteopontin and proliferating cell nuclear antigen (PCNA), while the expression of the contractile VSMC marker SMα was increased in the thoracoabdominal aorta compared to the control group (19). Furthermore, in vitro culturing of human aortic SMCs (HASMCs) with palmitic acid resulted in an increased expression of proliferative and synthetic phenotype markers while inhibition of CCR5 using the antagonist maraviroc or RNA interference prevented HASMC proliferation and synthetic phenotype formation (19). Macrophages from stroke patients, exhibit an increased expression of CCL5 which signals through CCR5 on VSMCs driving their proliferation and dedifferentiation, suggesting a paracrine relationship between these macrophages and vascular VSMCs [(19, 51), p. 153]. These data suggest that CCL5 induces VSMC proliferation and phenotypic switching from a contractile to synthetic phenotype via CCR5 (Figure 2). The latter would argue for a pro-atherosclerotic role of CCL5 and induction of a synthetic VSMC phenotype promoting vascular remodeling. However, Lin et al. did not backcross the mice on an atherosclerotic background such as Apoe−/− or Ldlr−/−, making it impossible to determine whether CCL5 also promotes a synthetic VSMC phenotype during atherogenesis.
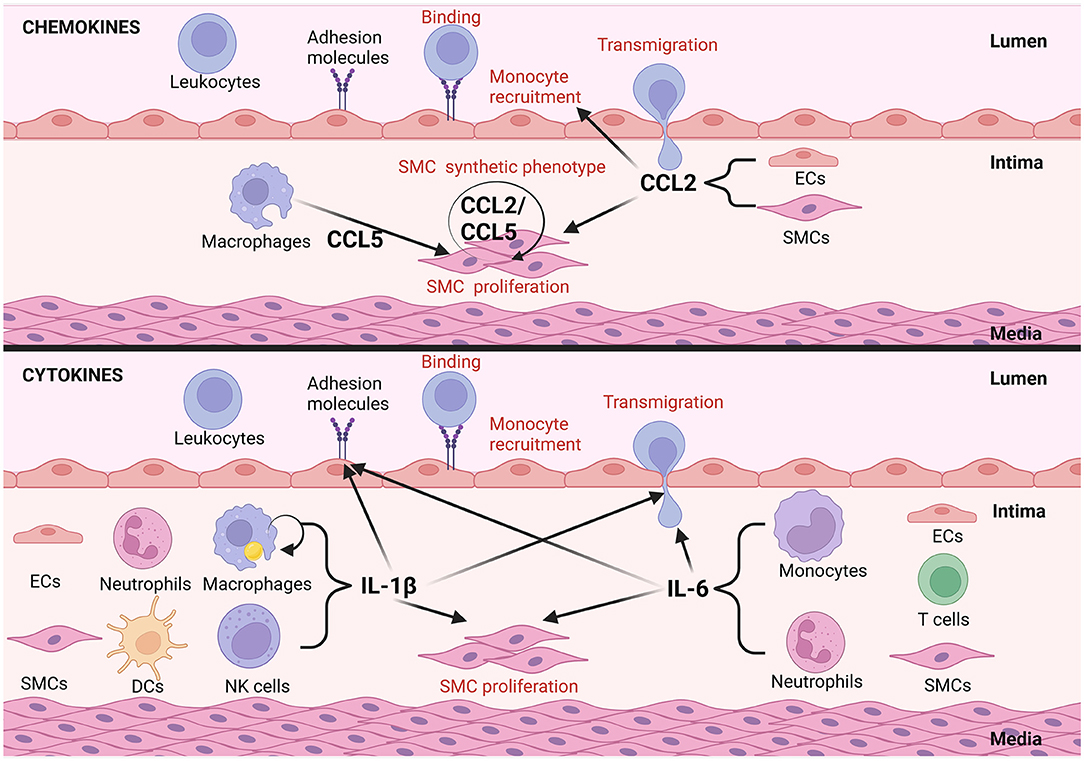
Figure 2. Examples of autocrine/paracrine interactions of inflammatory mediators in vascular remodeling in atherosclerosis. Inflammatory mediators can act in an autocrine manor, CCL2 and CCL5 for example activate VSMCs to undergo phenotypical changes in atherosclerosis. EC and VSMC derived CCL2 can further foster cross-talk between them promoting remodeling. CCL2 from ECs triggers synthetic differentiation of VSMCs and CCL2 from VSMCs promotes monocyte recruitment. Alternatively, mediators like interlukin-1β, expressed by dendritic cells (DCs) and natural killer (NKs) cells promote the upregulation of adhesion molecule expression by ECs, transmigration of circulating leukocytes and VSMC proliferation in a paracrine fashion. IL-1β in addition can act upon macrophages to promote IL-1β secretion in an autocrine manner. IL-6 secreted by ECs, monocytes, T cells, neutrophils and VSMCs promotes adhesion molecule expression by ECs as well as transmigration of circulating immune cells into the atherosclerotic plaque (this figure was made with Biorender.com).
CCL5 can also heterodimerize with CXCL4, another platelet-derived chemokine deposited onto vascular endothelium (52). Enhanced CXCL4 presence in human plasma correlates with the severity of human atherosclerotic disease and the platelet specific deletion of CXCL4 decreases atherosclerotic lesions in mice (53, 54). Furthermore, interfering with this heterodimer via the use of the cyclic peptide MKEY resulted in decreased leukocyte recruitment and release of NETs (47). All in all, these studies suggest that CCL5 at least exerts part of its atherogenic effects by heterodimerizing with CXCL4.
CD146, a cell adhesion molecule expressed on arterial endothelium was also implicated in CCL5-mediated changes to the vascular wall. CD146−/−Apoe−/− mice fed a WD for 24 weeks displayed lesions with a greater neutrophil and macrophage content correlating with the upregulation of CCL5 secretion when compared to the control group (55). Furthermore, neutrophil recruitment was increased in CD146-deficient mice 12 h after thioglycolate-induced peritonitis, whereby this increased recruitment correlated with the enhanced CCL5 secretion in the peritoneal cavity. The same study showed that mice treated with maraviroc between week 12 to 24 of WD feeding showed smaller atherosclerotic lesions and reduced neutrophilia in CD146−/−Apoe−/− mice to the same level found in Apoe−/− mice (55). These data suggest that CCL5 plays a prominent role in neutrophil recruitment and foam cell formation in atherosclerotic lesions and identifies CD146 agonists as potential therapeutic targets to lower CCL5 levels (Figure 1). Hence, increased levels of CCL5 may act as a biomarker for the severity of atherosclerosis and may even be a potent therapeutic target for SMC proliferation and phenotype switching (Table 2).
Chemokines CCL19 and CCL21 and their receptor CCR7 (Table 1) are associated with the modulation of inflammatory responses in lymphoid and nonlymphoid tissues, including atherosclerotic lesions (56–58). Elevated levels of circulating CCL19 and CCL21 can be found in patients with unstable angina pectoris compared to controls (57) and enhanced levels of these chemokines were also observed in patients with carotid atherosclerosis, both systemically (CCL21) and within the lesion (CCL19 and CCL21) (22).
In a mouse model studying MI, the inhibition of CCL21 via intravenous injection of anti-CCL21 monoclonal antibodies led to the reduction of the infarction size after acute MI (20). Anti-CCL21 monoclonal antibody treatment further resulting in reduced MMP-9 and total collagen content in the myocardium. CCL21 was also shown to increase the binding of acetylated LDL (ac-LDL) to macrophages, inducing the up-regulation of LDL receptor-1 (LOX-1), a scavenger receptor of ox-LDL which has been shown to drive foam cell formation (21, 22). Furthermore, the lipid droplet marker adipose differentiation-related protein (ADRP) was upregulated in CCL21-treated macrophages indicating that CCL21 induces foam cell formation (59).
CCL19 has been shown to induce proliferation of VSMCs and results in increased MMP-1 serum levels (22), which positively correlates with total plaque burden (60). Using a BMT experiment of either CCL19−/− or CCL21−/− bone marrow into Ldlr−/− mice it was further demonstrated that CCL21 directs leukocyte homing into atherosclerotic lesions, whereas CCL19 induces the activation of leukocytes, lipid uptake by macrophages and foam-cell formation (61). Thus, CCL19 is involved in vascular remodeling in atherosclerosis, by inducing VSMC proliferation and enhancing protein expression of MMP-1 in VSMCs, which acts to destabilize the plaques, while both CCL19 and CCL21 foster macrophage foam cell formation (Figure 1). Blocking CCL19 and/or CCL21 may therefore be a potent therapeutic target to decrease the lipid content in lesions and increase plaque stability (Table 2).
EC, VSMCs, and macrophages all express CXCL10 during atherosclerosis while its receptor CXCR3 is mainly expressed on CD4+ T cells (Table 1) (62). Evidence from Apoe−/− Cxcl10−/− mice on a WD for 6 and 12 weeks demonstrated attenuated lesions, reduced CD4+ T cell content and increased regulatory T cells (Tregs) markers within the lesions, suggesting an increase in Tregs within the lesion of Apoe−/− Cxcl10−/− compared to the control (63). Furthermore, Ldlr−/− mice fed a 2-week WD before a collar placement and a further 8 week WD with daily treatments of a pharmaceutical antagonist specific for CXCR3 (NBI-74330) demonstrated a reduced lesion size in the aorta and aortic root compared to the control (23). Further investigation showed reduced CD4+ T cells in the lesion and greater expression of genes associated with greater presence of Tregs in the plaques from mice treated with NBI-74330. This suggests that CXCL10 plays a prominent role in vascular remodeling and may be a potential therapeutic target (Table 2). Further experiments were conducted in Apoe−/− mice fed a WD for 2 weeks followed by induction of an unstable plaque with a flow-altering device around the carotid artery. These animals were further fed a WD for 9 weeks and treated with a bioactivity-neutralizing monoclonal CXCL10 antibody (MAB466) (64). The CXCL10 antibody treatment resulted in a more stable lesion phenotype with increased VSMCs content compared to untreated controls. Hence, CXCL10 may also influence VSMC behavior affecting plaque stability, potentially making CXCL10 an attractive therapeutic target for vascular remodeling (65). Adding to this, in PI3KγKD/KD mice treated with the CXCL10 antibody (MAB466) demonstrated that CXCL10 production by VSMCs inhibited endothelial healing in a wire induced scratch model (24). This suggests that CXCL10 also has a paracrine relationship with endothelial healing and may play a role in maintaining endothelial dysfunction in atherosclerosis. However, this would need to be further investigated in an atherosclerotic mouse model on a WD. Furthermore, studies using an in vitro model in which human endothelial cells (SGHEC-7) were co-cultured with the human VSMC cell line SGHVSMC-9 demonstrated that CXCL10 expression contributes to remodeling by altering the motility and differentiation of the VSMCs (66). Future studies could investigate the role of CXCL10 on the phenotypic switching of VSMCs within atherosclerosis.
The chemokine CXCL12 (Table 1) is highly expressed in human atherosclerotic lesions (32, 67) and genome-wide association studies noted that the genomic locus 10q11, hosting the CXCL12 gene and the intergenic single nucleotide polymorphism (SNP) rs2802492 located near CXCL12, are independently associated with CXCL12 plasma levels and coronary artery disease (CAD) risk (68, 69). Furthermore, a causative detrimental role of enhanced CXCL12 titers in CAD was described (70) and may also account for CVD in general, rendering CXCL12 a useful biomarker for CVD risk. In line with this, mouse studies could show that EC-specific CXCL12 knock out in Apoe−/−mice fed a WD for 12 weeks reduced the lesion area in the thoracoabdominal aorta and the aortic arch as well as CXCL12 plasma levels compared to control mice (69). Furthermore, a positive correlation between CXCL12 plasma levels and lesion area was seen in the control but not in EC-CXCL12 deficient mice. Combined, these observations suggest that EC-derived CXCL12 promotes atherosclerosis and vascular remodeling (69).
In vitro assays exploring the effects of ox-LDL treatment demonstrated an increase in CXCL12 protein and mRNA expression compared to untreated controls for both THP-1 cells (human monocyte cell line derived from a patient with acute monocytic leukemia) and HASMCs (71). Furthermore, Gao et al. (25) showed that CXCL12-treated THP-1 cells expressed less ATP Binding Cassette Subfamily A Member 1 (ABCA1) and subsequently resulted in a reduced cholesterol efflux capacity toward apolipoprotein A1 (ApoA-I). The same study also revealed that systemic overexpression of CXCL12 in Apoe−/− mice, using a lentivirus, resulted in increased lesion formation as well as macrophage accumulation in the plaques. A more detailed analysis demonstrated decreased ABCA1 mRNA expression in lesional macrophages in Apoe−/− mice which overexpressed CXCL12 supporting the observations made in vitro (25). Together, these data suggest that CXCL12 promotes the formation of macrophage-derived foam cells in atherosclerotic lesions via inhibition of ABCA1 expression. The potential of CXCL12 to promote foam cell formation by VSMCs or any other analysis of VSMCs was not investigated in the in vivo model in this study and should therefore be a subject for future research.
In contrast to the above described observations, CXCL12 is upregulated in the artery via lysophosphatidic acid in response to vessel injury, and the increase in circulating CXCL12 induces the migration of a subtype of Sca-1+ Lin− smooth muscle progenitor cells (SPCs) (26, 27). Intravenous injection of CXCL12 in Apoe−/− mice fed a WD resulted in increased plaque stabilization, characterized by increased collagen content and a thicker fibrous cap, via the accumulation of SPCs (28). However, this study analyzed ligated carotid arteries, which display significant phenotypical differences in their lesion composition compared to ≪native≫ atherosclerotic lesions.Contrary, presence of CXCL12 does also induce recruitment of endothelial cell progenitors and fosters neovascularization in MI (72). While neovascularlarization in the context of MI seems beneficial another recent study has shown that active non-canonical NF-κB signaling in microvessels of carotid atherosclerotic lesions together with enhanced CXCL12 expresssion and neovascularization may induce plaque instability (73).
Overall, CXCL12 plays a critical role in the development of vascular remodeling as demonstrated by EC-derived CXCL12 mediating atheroprogression or the accumulation of VSMCs in the intima in response to increased CXCL12 titers (Figure 1) (26, 27, 69). Taken together, CXCL12 may serve as biomarker for CAD risk (70) and seems to drive macrophage foam cell formation and lesion growth. On the other hand, animal studies also suggest that it increases lesion stability by recruitment of SPCs and infiltration of medial VSMCs. Hence, therapeutical targeting of CXCL12 should only be cell specific and considered with great care.
CXCL16 is an atypical chemokine containing a mucin-like stalk, transmembrane and cytoplasmic domains, which are not found in other CXC chemokines (74). CXCL16 has two distinct forms, the membrane-bound form which promotes the firm adhesion of cells expressing the receptor CXCR6 and the soluble form, generated by proteolytic cleavage of membrane-bound CXCL16, which acts as a chemoattractant for CXCR6+ cells (74). CXCL16 (Table 1) is expressed on stimulated ECs and VSMCs, macrophages, dendritic cells (DC) and platelets, whereas its receptor CXCR6 is expressed on memory and effector T cells, natural killer (NK) cells and NK T cells and is also found on plasma B cells (29, 75). CXCL16 is expressed in human atherosclerotic plaques and lesion severity is correlated with increased CXCL16 levels, as shown in human carotid endarterectomy specimens (76). It could be demonstrated that CXCL16 expression in the plaque enhanced platelet deposition on the endothelium (29). Furthermore, in vitro studies using human umbilical vein endothelial cells (HUVECs) established that the immobilization of CXCL16 promoted CXCR6-dependent platelet adhesion to the endothelium during physiologic flow and at low shear rates (77). This data implies that CXCL16 may play a role in vascular inflammation and thrombo-occlusive diseases. However, future investigations need to assess this relationship also on the more pathologically relevant arterial endothelium to understand platelet adhesion in an atherosclerotic model. Zhao et al. demonstrated that during ischemia reperfusion injury, cardiac EC-derived CXCL16 recruits CD11b+Ly6Chigh inflammatory cells and facilitates the release of tumor necrosis factor α (TNFα) (interferon) IFN-γ and interleukin (IL)-17 in the heart. In line with this, silencing of CXCL16 by applying a specific shRNA reduced cardiac apoptosis, inflammation and dysfunction in ischemia reperfusion induced mice (78).
CXCL16 may also play a role in vascular remodeling through alteration of foam cell formation as its expression has been found to be upregulated in lipid-laden intimal macrophages and VSMCs (30). Moreover, CXCL16 may also directly contribute to foam cell formation as it is known to be a scavenger receptor for phosphatidylserine and oxLDL and its expression is upregulated in lipid-laden intimal macrophages and VSMCs via an autocrine mechanism (30). Growing evidence highlights the potential for VSMCs to develop into foam cells (71) and, like macrophages, VSMCs also express CXCL16 in response to IFN-γ stimulation, which also correlates with an increased uptake of oxLDL by VSMCs (31). Therefore, it can be surmised that CXCL16 facilitates the development of macrophage and VSMC foam cells (Figure 1). However, to our best knowledge so far no therapeutic treatment against CXCL16 has been investigated in the context of atherosclerosis and chronic vascular inflammation opening a potential interesting novel avenue of future research.
CX3CL1 also known as fractalkine is also a chemokine which can be present as a membrane-bound or soluble form (79). Both forms activate the chemokine receptor CX3CR1 (Table 1), where the transmembrane form induces integrin-independent leukocyte adhesion and the soluble form is a chemoattractant for leukocytes (80). During atherosclerosis, monocytes expressing CX3CR1 bind and adhere to the endothelium which express the membrane-bound form of CX3CL1 (81). CD16+CX3CR1HIGH monocytes then activate endothelial signal transducer and activator of transcription 1 (STAT1), NFκB and p65 phosphorylation to upregulate proliferation and expression of CX3CL1, IL-1β, Intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) by ECs (82, 83). Hence, the CX3CL1-CX3CR1 axis enhances a pro-atherosclerotic EC phenotype via the upregulation of adhesion molecules and inflammatory mediators. In this context, in vitro studies have shown that CX3CL1 is upregulated on activated VSMCs and triggers monocyte adhesion to VSMCs (32). Further work has demonstrated that Cx3cr1−/− animals—post femoral arterial injury induced via an angioplasty guide wire—were protected against intimal hyperplasia due to decreased monocyte trafficking to the lesion compared to the control group (34). In addition, CX3CR1 deficiency resulted in decreased VSMC proliferation and intimal accumulation, which is either directly or indirectly a result of defective monocyte infiltration (34). The relationship between CX3CL1-CX3CR1 and inhibited VSMC proliferation and intimal accumulation in vascular remodeling still needs to be elucidated in detail. One study has demonstrated that a mononuclear CX3CR1+ cell population residing in murine bone marrow provides a source of SPCs after wire-induced vascular injury, which differentiate into VSMCs within the neointimal plaque (35). Furthermore, BMT of CX3CR1 deficient bone marrow into C57BL6/J mice demonstrated that Cx3cr1 expression is essential for VSMC differentiation from SPCs in the vascular wall (35). Thus, CX3CL1 may play a prominent role in intimal hyperplasia promoting atherosclerosis through increased VSMC proliferation and monocyte trafficking. However, it would be worthwhile to perform similar studies using either an Apoe−/−or a Ldlr−/− mouse fed a WD to provide results that truly reflect CVD triggered mechanisms.
Another recent study treated Ldlr−/− mice with a CX3CL1-Fc fusion protein inhibiting the CX3CR1-CX3CL1 interaction. This fusion protein treatment significantly reduced atherosclerotic lesion size, independent of WD diet feeding and reduced M1-like macrophage and T cell accumulation in the aortic wall (33). Thus CX3CL1-Fc could be a potent therapeutic option interfering with vascular remodeling and atherosclerosis (Table 2). Along this line, another study implemented the use of a DNA vaccine, a vector that contains genes encoding a single-chain antibody specific for the mouse dendritic cell (DC) antigen DEC205 and the receptor CX3CR1 (DEC-CX3CR1) and a non-DC DNA vaccine (Con-CX3CR1). Both vectors were injected into Apoe−/−mice fed a normal chow diet (84). DEC-CX3CR1 vaccinated mice demonstrated a significantly reduced atherosclerotic plaque size compared to both Con-CX3CR1 vaccinated and unvaccinated mice (84). Furthermore, DEC-CX3CR1 mice showed reduced monocyte infiltration and lipid deposition in the lesions compared to unvaccinated mice, although the lesional macrophages still possessed an M1 phenotype. Not only does this further support the role of the CX3CL1-CX3CR1 axis in vascular remodeling (Figure 1), but it also emphasizes the need to conduct more studies on DNA vaccination (Table 2) as an effective therapeutic strategy against atherosclerosis and vascular remodeling. Overall, CX3CL1 plays an important role in M1 macrophage differentiation and the migration/proliferation of VSMC resulting in vascular changes (Figure 1).
Cytokines are molecules that are secreted by immune cells and other specific cell types that modulate the inflammatory immune response and mediate cell-cell communication. Cytokines are subdivided into different classes: TNFs, IFNs, ILs, transforming growth factors (TGFs) including growth/differentiation growth factors (GDF), colony-stimulating factors (CSFs) and chemokines as detailed before. In atherosclerosis, pro-inflammatory cytokines play an important role in the initiation and progression of the disease and in the instigation of endothelial dysfunction, upregulation of adhesion molecules and promotion of immune cell migration as well as their infiltration into the lesion. All these different factors lead to arterial remodeling and subsequent changes in vascular function (85). The most important cytokines that are known to contribute to atherosclerosis lesion remodeling are IL-1, IL-6, IL-10, IL-22, IL-33, TNF-α and GDF-15 which will be discussed below in greater detail:
IL-1 was one of the first cytokines to be discovered and it is divided into two related but functionally distinct isoforms: IL-1α and IL-1β. Here, the main focus will be on IL-1β as this is a potent driver of the inflammatory response in atherosclerosis and vascular inflammation. IL-1β is synthesized by many cells including neutrophils, NK cells, DCs, ECs macrophages, monocytes and SMCs (Figure 2). In atherosclerosis, its synthesis is triggered by the uptake of cholesterol crystals by macrophages activating the NLR family pyrin domain containing 3 (NLRP3) inflammasome or by the binding of IL-1 family members to their receptor IL-1R resulting in a positive autocrine inflammatory feedback loop (86). In addition to cholesterol, monocytic inflammasomes and the production of IL-1β can be activated by dying VSMC and in turn promotes VSMC proliferation as shown in vitro and in vivo in C57BL/6 mice of vein graft injury (87). IL-1β itself induces the upregulation of adhesion molecules such as ICAM1 and VCAM1 as well as the monocyte chemoattractant chemokine CCL2 (please see section CCL2) on ECs promoting leukocyte recruitment into the atherosclerotic plaque (Figure 1; Table 3) (88, 115, 116). Pro-inflammatory cytokine IL-1β has also an important role in cross-talk between ECs and the underlying VSMCs. Specific secretion of IL-1β by SMC induces E-selectin expression by ECs (117). In turn, IL-1β secretion by activated ECs promotes VSMC proliferative, synthetic and macrophage-like phenotypes (118). The barrier function of ECs is crucial to maintain vascular wall homeostasis. However, in atherosclerotic conditions, dyslipidemia and proinflammatory cytokines such as IL-1β promote excessive adhesion molecule expression by ECs leading to endothelial dysfunction and increased permeability of the EC barrier due to the disruption of intercellular junctions, resulting in increased leukocyte infiltration and vascular remodeling (Figures 1, 2; Table 3) (90). In addition, IL-1β promotes VSMC proliferation by stimulation of autocrine production of PDGF (Figure 1) and the production and release of the pro-inflammatory cytokine IL-6 (Table 3) (92, 93). IL-1β-induced EC dysfunction is also promoted by plasma trimethylamine-N-oxide (TMAO). TMAO is an oxidation product of the liver that is made from compounds synthetized by intestinal bacteria and an elevated concentration of TMAO increases monocyte mobilization and activation leading to low-grade inflammation (119). In line with this, high plasma levels of TMAO are associated with atherosclerosis and increased risk of CVD. In this context Boini et al. (119) showed that TMAO increases the assembly and activation of the NLRP3 inflammasome leading to an increased production of IL-1β in carotid arteries of WT mice with partially ligated carotid artery. In addition, in vitro experiments indicate that TMAO treatment induces NLRP3-dependent endothelial hyperpermeability by decreasing zonula occludens-1 (ZO-1), a tight junction protein responsible for junction integrity, expression in mouse carotid arterial endothelial cells (CAECs) (119). Therefore, targeting TMAO may help to reduce adverse remodeling in atherosclerosis by preventing EC leakage and infiltration of inflammatory cells as well as reducing IL-1β driven inflammation (119). IL-1β also increases the expression of dipeptidyl peptidase 4 (DPP4), which is a transmembrane protein expressed on ECs that is involved in glucose metabolism and cardiometabolic disease (120). Recent studies showed that DPP4 inhibition decreases atherosclerotic plaque burden. For example, Meng et al. investigated the atheroprotective role of the DPP4 inhibitor trelagliptin on HAECs exposed to IL-1β in vitro, showing that trelagliptin treatment had a strong inhibitory effect on the expression of adhesion molecules and pro-inflammatory chemokines and cytokines that orchestrate monocyte adhesion on the endothelium (89). Mechanistically, trelagliptin inhibits IL-1β induced NFκB transcription factor activation which subsequently prevents the transcription of the monocyte chemoattractant chemokines CCL2, CXCL1 as well as the pro-inflammatory cytokine IL-6 and the adhesion molecules ICAM1 and VCAM1 (mRNA and protein levels) (Table 3) (89).
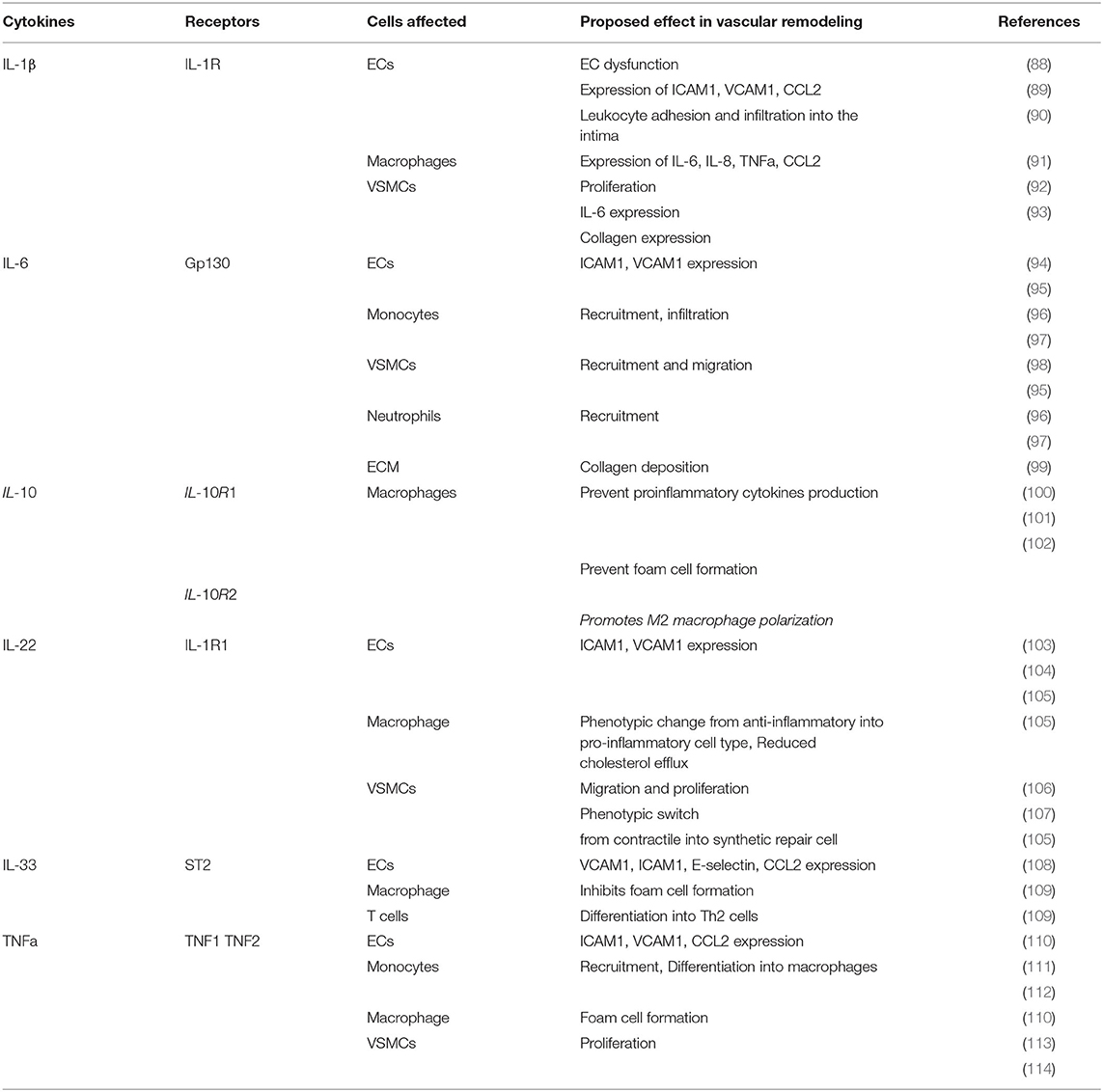
Table 3. Overview of cytokines involved in atherosclerosis remodeling and their physiological effect.
Results from studies that focus on IL-β antibody blocking or knock out in vivo are less straightforward. Earlier work in Apoe−/−mice in which the anti-IL-1β antibody XMA052 MG1K was injected twice weekly during 16 weeks of WD showed a decrease of aortic lesion area of 37, 22 and 29% with 0.1, 1.0, 10 mg/kg XMA052 MG1K, respectively, compared to IgG injected control Apoe−/− mice (91). In vitro experiments performed in the same study revealed reduced release of other pro-inflammatory cytokines such as IL-6, IL-8, TNF-α and CCL2 from cultured macrophages and reduced release of the proteolytic enzymes MMP-3 and MMP-9 from ECs and VSMCs, after XOMA 052, a human engineered IgG2 anti-IL-1β antibody, treatment (Tables 3, 4) (91). However, MMP-3 and MMP-9 expression were not affected in vivo, and no difference was observed in the plaque collagen content suggesting that plaque stability is not modified by this treatment (91). Yet, when anti-IL-1β treatment was tested to investigate its role in established atherosclerotic lesions, it led to adverse remodeling. Cell-specific effects of the IL-1β antibody (mouse monoclonal antibody, Novartis, 10 mg/kg) were tested by Gomez et al. on VSMC lineage tracing Apoe−/− mice where the fate and migration of SMCs can be monitored during the development of atherosclerosis by using an inducible Cre-flox system to label MYH11+ SMC specific YFP expression (Apoe−/−Myh11-CreERT2R26R-YFP). The anti-IL-1β monoclonal antibody treatment did not lead to any differences in the aortic plaque size compared to IgG treated control animals after 18 weeks WD. In addition, anti-IL-1β treated mice showed thinner fibrous caps characterized by a 30% decrease of collagen, a 40% decrease of VSMC content, though a surprising 50% increase of lesional macrophages within the fibrous cap (Table 4) (121). These results indicate that anti-IL-1β treatment has a detrimental effect on fibrous cap remodeling promoting plaque instability.
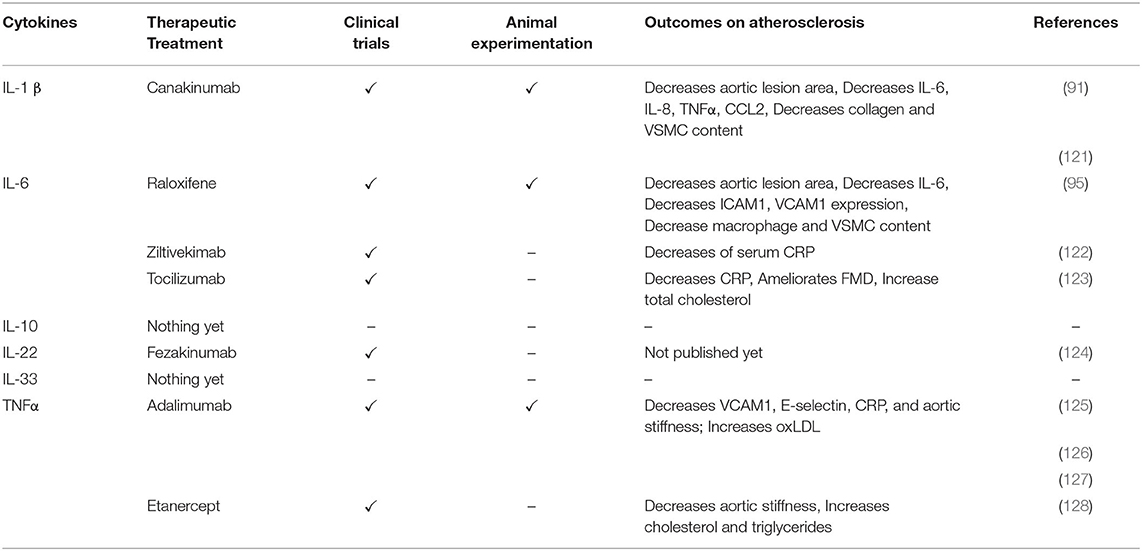
Table 4. Targeting cytokines as therapeutic treatments in vascular remodeling and CVD.
Nevertheless, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) was the first big scale clinical trial confirming the potential of anti-inflammatory IL-1β therapy in CVD. In the CANTOS trial, anti-IL-1β (canakinumab) treatment resulted in a decrease of inflammatory markers in the plasma of patients such as high-sensitivity C-reactive protein (hsCRP) (26–41%) and IL-6 (6–25%) (129–131).
The receptor for IL-1β, IL-1β receptor type 1 (IL-1R1), was also described to reduce atherosclerotic plaque development (132, 133). However and in sharp contrast, lack of the IL-1β receptor antagonist (IL-1ra) promotes atherosclerotic plaque formation (134). More recent studies further support this controversial role of IL-1R1 and IL-1ra in atherosclerosis development and more precisely in atherosclerotic plaque remodeling.
IL-1R1 deletion has a dual role in plaque stability and outward vessel remodeling as shown in a female mouse model lacking IL-1R1 (IL-1R1−/−Apoe−/−) and fed a WD for 30 weeks. These mice showed reduced aortic root lesion size as well as a 50% reduction of the lumen area of the brachiocephalic artery when compared to control IL-1R1+/+Apoe−/− mice. However, these reductions were accompanied by enhanced features of plaque instability, characterized by decreased VSMCs and collagen content. MMP-3 expression in the brachiocephalic artery was also significantly lower in IL-1R1−/−Apoe−/− animals which may explain the reduced collagen content in the plaques (135). In line with this, IL-1Ra-deficient mice exhibited considerable inflammation in the aorta accompanied by an increased macrophage content in the adventitia and destruction of the elastic lamina, which is normally important to maintain arterial wall stability prevent intima lesion progression (136, 137). Another study also investigated the role of IL-1Ra in atherosclerosis lesion development by making use of IL-1Ra+/+ and IL-1Ra+/− mice (IL-1Ra−/− mice were excluded from this study due to reduced weight gain and differential cholesterol metabolism) on Apoe−/− background. Heterozygous and control animals were fed normal chow diet for 16 weeks and IL-1Ra+/−Apoe−/− mice showed a 30% increase of atherosclerotic lesion size compared to IL-1Ra+/+Apoe−/− mice (134). Interestingly, after 32 weeks of chow diet feeding the lesion size was similar in both groups, but macrophage accumulation in the lesions was reduced in IL-1Ra+/−Apoe−/− animals compared to control. VSMCs in the same lesions showed a 15% decrease compared to control IL-1Ra+/+/Apoe−/−animals (134). These findings suggest that IL1-Ra plays an important role in the development of atherosclerotic lesions and also suggests that it impacts plaque composition, modulation and stability (Figure 1).
Taken together, IL-1β is detrimental and promotes early-stage atherosclerosis development but on the other hand animal models suggest that it is beneficial in an advanced stage of the disease to maintain plaque stability and avoid major cardiovascular complications. Plaque stability also seems to be affected by interfering with IL1-R1 or IL-1RA. Nevertheless, it is important to keep in mind that research on mice and in vitro assays have certain limitations with regards to the translation to human pathologies. Therefore, efficacy and safety of the CANTOS trial and its promising beneficial results in patients with adverse cardiovascular events still hold true even if research on IL-1β on atherosclerosis in mice and in vitro findings are sometimes conflictual meaning that the results are not always consistent from one study to another. In addition, one should keep in mind that Apoe−/− mice are severely hypercholesterolemic which does not fully translate with the CANTOS patients who were under statin treatment. Moreover, canakinumab in CANTOS was only administered quarterly and not weekly which may explain why no effects on lesion stability were reported yet. The latter also underlines the importance of careful timing of treatment regimens.
IL-6 is secreted by various cells including epithelial cells, VSMCs, vascular ECs, monocytes, and T cells (Figure 2) (138) and promotes the synthesis of acute phase proteins such as CRP, amyloid A, β2 protein, hemopexin and haptoglobin. Both CRP and amyloid A are biomarkers of chronic inflammation and their levels predict cardiovascular risk (97, 139). In addition, IL-6 shows chemotactic activity for monocytes and neutrophils and promotes the expression of adhesion molecules (Table 3) (94) and chemokines leading to vascular remodeling by the induction of leukocyte adhesion and infiltration into the intima (Figures 1, 2; Table 3) (96, 97). More precisely, paracrine secretion of IL-6 by VSMC induces expression of the adhesion molecule E-selectin on ECs (117). In addition, in response to IL-6 signaling ECs and SMC also start to secrete IL-6 (140). TNFα activated ECs also induce SMC production of IL-6 (141).
Circulating IL-6 is a biomarker for CVD such as acute coronary syndrome with atherosclerosis (142). A study on carotid atherosclerosis showed that blood levels of IL-6 and TNFα were higher in patients with carotid atherosclerosis compared to control subjects and the cytokine levels increased with increasing amounts of carotid atherosclerosis stenosis (143). IL-6 was also shown to promote migration and viability of macrophages and VSMCs in vitro and regulate ECM deposition and reorganization (Table 3) (95, 98). One mechanism of IL-6 driven SMC migration is made via MyD88 and TRIF action and activation of p38 MAPK and ERK1/2 signaling) (144). Schieffer et al. investigated the role of IL-6 deficiency on atherosclerosis development. In this study, IL-6 deficient Apoe−/− mice (Apoe−/−IL-6−/−) were fed with chow diet for 53 ± 4 weeks and showed a significant decrease in aortic transcript and protein levels of MMP-9, tissue inhibitor of metalloproteinase-1, collagen I and V and lysyl oxidase (145), an important protein for the formation and repair of the ECM, compared to Apoe−/− control mice (146). Additionally, Apoe−/−IL-6–/− mice exhibited a decrease in macrophage and leukocyte infiltration into the lesions, although atherosclerotic lesion formation was enhanced in Apoe −/−IL-6–/− compared to Apoe−/− animals (28.1 vs. 14.9% respectively). The latter may be explained by the observed increase of total cholesterol, LDL, and very-low-density lipoprotein VLDL in IL-6 deficient animals and a decrease of plasma IL-10, compared to control mice (146).
Interestingly, a recent study showed an atheroprotective effect of raloxifene treatment in male Apoe−/− mice fed a WD for 12 weeks. Raloxifene is a drug that is prescribed to post menopause women to prevent osteoporosis and is able to inhibit IL-6 binding to the IL-6 receptor subunit GP130, thereby prohibiting downstream signaling pathways such as STAT3. In this study, it was observed that daily administration of 5 mg/kg raloxifene reduced the area of atherosclerotic lesion size in the aorta and aortic root in Apoe−/− mice compared with untreated control Apoe−/− mice (95). In addition, raloxifene significantly decreased the expression of IL-6, ICAM1, and VCAM1 in the aortic vascular ECs and reduced the lesional macrophage and VSMC content in these aortic lesions. Mechanistically it could be demonstrated that raloxifene decreases atherosclerosis by preventing IL-6-induced phosphorylation of STAT3 and inhibiting the IL-6/GP130 interaction (Tables 3, 4) (95).
IL-6 secretion by aortic perivascular adipose tissue (PVAT) also promotes aortic stiffness and remodeling in Ldlr−/− mice compared to WT C57BL/6 mice fed with chow diet (99). Aortic stiffness, mediated by changes in the ECM protein expression, is caused by an increase in cross-linking of collagen (147). Consistent with other studies, Du et al. (99) also observed an increase in IL-6-ediated collagen type I expression in the aorta of Ldlr−/− mice (Table 3). These findings suggest that IL-6 may play an important role in inducing changes in the ECM by increasing collagen type I expression and thereby promoting subsequent arterial stiffness and thus vascular remodeling (Figure 1).
RESCUE, a trial to evaluate the reduction in inflammation in patients with advanced chronic renal disease utilizing antibody-mediated IL-6 inhibition investigated the effect of ziltivekimab in patients with high cardiovascular risk (122). Patients in this study had elevated serum CRP levels and chronic kidney disease and were randomly allocated into groups with differential monthly subcutaneous administrations of ziltivekimab (total of 7.5 mg, 15 mg or 30 mg) or placebo. After 12 weeks of treatment, CPR levels decreased by 77% in the 7.5 mg group and by 92% in the 30 mg group. Biomarkers of systemic inflammation and thrombosis, relevant to atherosclerosis, were also reduced in a dose-dependent manner, while no severe side effects were detected. Due to the COVID-19 pandemic, the study unfortunately had to be terminated before the planned secondary 24-week endpoint to avoid exogenous causes of increased CPR levels and thereby bias the interpretation of the results. However, it is planned to do a large-scale cardiovascular outcome trial on the same population to investigate the effect of ziltivekimab on the recurrence rate of vascular events (Table 4) (122). Another clinical study investigated the efficiency of an anti-IL-6 treatment, tocilizumab, on endothelial function in high-risk CVD patients with rheumatoid arthritis (RA). Tocilizumab is a humanized monoclonal antibody that targets the soluble and membrane-bound IL-6 receptor. The study consisted of three groups, whereby 18 patients received tocilizumab (8 mg/kg IV) every 4 weeks, 24 patients underwent anti-TNFα treatment (methotrexate 15–25 mg/week or leflunomide 20 mg/d) and 18 control patients who were treated with synthetic disease-modifying antirheumatic drugs (123). Endothelial function was evaluated by flow-mediated dilation (FMD) measurements before and after 16 weeks of starting the different treatments. As expected, patients from the tocilizumab treated group showed the most striking reduction in mean CRP levels compared to the other treatments. Furthermore, the mean FMD only significantly improved in the tocilizumab group, increasing from 3.43 to 4.96%. On the other hand, anti-IL-6 treatment greatly enhanced the atherogenic lipid profile and increased total cholesterol levels (Table 4) (123). Long-term treatment with tocilizumab was also addressed in a pilot study with 16 female RA patients receiving monthly injections with tocilizumab (8 mg/kg IV) and endothelial function was again measured by FMD. The endothelial function significantly improved after 6 months of treatment compared to 16 non-RA control subjects (148). As underlined by various clinical trials, tocilizumab worsens the lipid profile, total cholesterol burden, LDL, and triglyceride profile. However, it does also improve endothelial function and therefore reduces vascular remodeling. Therefore, to minimize the detrimental effects of elevated lipids, it would be ideal to for example combine tocilizumab with statin treatment.
IL- 10 is an anti-inflammatory cytokine produced mainly by macrophages and T cells. IL-10 signaling is mediated through a two-receptor complex named IL-10 receptor 1 (IL-10R1) and IL-10R2. The receptor complex is constitutively expressed on numerous hematopoietic and non-hematopoietic cells such as epithelial cells and fibroblast and is upregulated upon activation (149). In atherosclerosis, IL-10 prevents remodeling by inhibiting macrophage activation and their proinflammatory cytokine production including TNFα, IL-1β, IL-6, IL-8, granulocyte stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF), foam cell formation, MMP expression, VSMC proliferation and therefore helps to reduce atherosclerotic plaque formation (Figure 1; Table 3) (101, 150).
In contrast, IL-10 deficiency in Apoe−/− after 16 weeks on chow diet leads to greater atherosclerosis plaque formation, an increase in blood cholesterol (e.g., LDL), an increase in Th1 cell response in the lesion as well as greater tissue factor activities, systemic coagulation and vascular thrombosis compared to control (151). In APOE*3-Leiden mice under cholesterol-enriched high-fat diet with cuff-induced neointima formation in the femoral artery, IL-10 deficiency led to increased neointima formation. On the other hand, in the same mouse model, IL-10 overexpression by means of single intramuscular injection of IL-10 resulted in a 45% decrease of neointima surface (152). However, Ldlr−/− mice transplanted with marrow cells from IL-10 transgenic male mice on a C57BL/6J background and fed chow diet for 4 weeks prevents the formation of advanced lesions, shifts Th1 cells toward Th2 phenotype and decreases IFN-γ levels in the lesion (153). Interestingly, IL-10 overexpression leads to increased modified LDL uptake by macrophages, although it also stimulates its efflux and thus prevents foam cell formation (Figure 1) (100). Overall, IL-10 diminishes lesion burden by decreasing proinflammatory cytokine levels and promoting an anti-inflammatory environment in the lesion with M2 polarized macrophages as well as Th2 cells and prevents foam cell formation (100, 154, 155). IL-10 also inhibits VSMCs activation in vitro and in vivo. IL-10 treatment of LPS-stimulated rat SMCs resulted in a decreased NF-κB activation, IL-6 secretion as well as reduced SMC migration and proliferation (Figure 1). In line with these observations, in a rat model of intimal hyperplasia, IL-10 treatment led to reduced SMC proliferation and intimal growth 14 days after balloon abrasion of the aorta compared with saline-injected control animals (150).
Furthermore, Jung et al. investigated more in detail the protective cellular and molecular mechanisms of IL-10 that prevent adverse MI LV remodeling. The authors show that IL-10 promotes M2 macrophage polarization in vitro and in turn the M2 secretome induces cardiac fibroblast activation, proliferation, migration and α-SMA expression. In addition, the same study showed that IL-10 treatment of fibroblasts reduces the ratio of collagen I to III secretion 7 days post-MI leading therefore to decrease fibrosis formation (Table 3) (102). These observations could mean that IL-10 levels may be implicated in plaque stability.
Systemic treatment with IL-10 is not ideal as it inhibits inflammation even when it is needed to fight pathogens. Indeed, long-term treatment of IL-10 increases the prevalence of intracellular infection such as Chlamydia and Listeria (156). In this regard, a study has tried cell-specific systems to avoid these off-target side effects. Exosomes loaded with IL-10 mRNA were engineered to target inflamed macrophages in the atherosclerotic lesion. These exosomes were used to treat Apoe−/− mice under 8 weeks high fat diet and resulted in decreased atherosclerotic plaque formation compared to PBS receiving control or exosome treatment (157).
Treatments and clinical trials are currently undergoing to explore the anti-inflammatory properties of IL-10 in various chronic diseases including RA, multiple sclerosis, allergies and inflammatory intestinal disease among others. However, IL-10 therapy for atherosclerosis and its effect on vascular remodeling remains to be investigated.
IL-22 is a member of the IL-10 cytokine family and is secreted by both innate and adaptive immune cells such as activated T cells especially T helper (Th) 22 cells and Th17 cells, NK cells, neutrophils, fibroblasts, and macrophages. IL-22 is involved in many cellular processes including lipid metabolism regulation, maintenance of bacterial homeostasis in the intestine and tissue regeneration (158).
The function of IL-22 in atherosclerosis is largely unknown, although evidence suggests that IL-22 is involved in vascular remodeling by promoting pro-inflammatory chemokines and antimicrobial peptide secretion as well as increasing VSMC migration and proliferation (Figure 1) (159, 160). In addition, IL-22 regulates adhesion molecule expression by ECs such as ICAM1 and VCAM1 as well as the production of chemokine ligands that have been implicated in adhesion, migration and recruitment of monocytes in atherogenesis (Figure 1) (103–105) (Table 3). The receptor of IL-22, being IL-22R1, is widely expressed on VSMCs, macrophages and ECs and mediates enhanced proliferation and migration through NFκB-, STAT3-, MAPK- and ERK1/2-dependent pathways in VSMCs (Figure 1). Furthermore, paracrine IL-22 promotes macrophage differentiation from anti-inflammatory into a pro-inflammatory phenotype and impairs the cholesterol efflux capacity of the cells, thereby promoting foam cell formation (Figure 1) (161, 162). Rattik et al. demonstrated that IL-22-deficient Apoe−/− mice (IL-22−/− Apoe−/−) fed a WD for 14 weeks had a significant reduction of atherosclerotic plaque size in both the aortic root and aorta, compared to control Apoe−/− mice. In addition, IL-22−/− Apoe−/−mice depicted reduced collagen content but increased expression of genes associated with VSMC contraction, namely α-actin, vinculin and caldesmon (Table 3) (106). The same study also explored the role of IL-22 in tissue repair mediated by arterial VSMCs in a carotid artery injury model in C57BL/6 mice. Here, an increased expression of IL-22 on VSMCs could be observed in the injured compared with non-injured arteries (106). These results suggest that IL-22 plays a key role in atherosclerotic plaque formation via the stimulation of dedifferentiation of contractile VSMCs toward synthetic repair cells resulting in plaque growth and that IL-22 is involved in plaque stability by thickening the fibrous cap, rendering it more stable by promoting lesional collagen content (Figure 1). IL-22 is therefore a double-edged sword. On the one hand, IL-22 worsens atherosclerosis by promoting inflammation, dysregulating macrophage cholesterol metabolism leading to more foam cell formation and promoting VSMCs proliferation and fibrous cap thickening (Table 3) (105). While on the other hand, IL-22 also leads to more stable plaques by increasing VSMC proliferation and migration into the intima forming a thick fibrous cap which decreases the risk of rupture and acute cardiovascular events.
Besides the above described protective effect of IL-22 it was also demonstrated that this cytokine can prevent atherosclerosis by promoting the expression of antimicrobial peptides which limit the spread of proatherogenic bacteria such as Enterobacteriaceae (Klebsiella sp), Prevotellaceae (Prevotella copri), Lachnospiraceae, Clostridiaceae and Ruminococcaceae. Additionally, a BMT of IL-22−/− BM into Ldlr−/− mice fed with WD for 16-weeks revealed larger atherosclerotic lesion sizes in hematopoietic IL-22-deficient Ldlr−/− mice compared to the control group (158). In these mice the aortic plaque was characterized by an increase of T cells and myeloid cell content as well as enhanced expression of aortic and intestinal proinflammatory cytokines like IL-1β, TNFα, CCL2 and CCL5. Intestinal gene expression of anti-microbial peptides C-type regenerating islet derived-3 (Reg3)-b and Reg3-g were also significantly reduced in hematopoietic IL-22 deficient Ldlr−/− mice compared to controls and whole metagenome shotgun sequencing analysis of cecal luminal microbiota revealed an increase of the above mentioned pro-inflammatory and proatherogenic bacterial species in absence of IL-22 (158).
However, in sharp contrast, more recently Shi et al. (107) underlined the pro-atherogenic effects of IL-22 produced by Th22 cells. After 12 weeks of WD feeding while Apoe−/− mice were treated with intraperitoneal injections of 2 μg recombinant mouse IL-22 (rIL-22) three times a week, it could be shown that IL-22 treatment resulted in larger atherosclerotic plaque sizes in the aortic root and the aorta as well as an increase in lesional macrophages. Th22, Th17 cells as well as DCs, collagen and serum IL-6 levels were also enhanced compared to PBS-treated Apoe−/− control mice. In addition, SMC α-actin was reduced in mice undergoing rIL-22 treatment. These observations were abolished when mice were treated three times a week with 20 μg anti-IL-22 monoclonal antibody (IL-22 mAb), proving the causal role of IL-22 in the observed effects. The same study also showed that BM-derived DCs from Apoe−/− mice treated with 100 ng/mL rIL-22 followed by stimulation with oxLDL displayed enhanced maturation properties and were able to induce differentiation and proliferation of naïve CD4+T cells into Th17 cells (107). The authors concluded that IL-22 secretion by Th22 aggravates atherosclerosis by promoting T cells (Th17), DCs and macrophage infiltration in the plaque and by inducing the dedifferentiation of contractile SMCs into synthetic SMCs (Table 3).
Human studies have also observed a correlation between circulating IL-22 levels and atherosclerosis. 45 patients with carotid artery disease were selected and were classified as symptomatic or asymptomatic based on the presence or absence of cerebrovascular symptoms. Immunostaining of plaques revealed a 7.15-fold higher IL-22 occurrence in symptomatic patients compared to the asymptomatic ones (104). Moreover, significant higher plasma levels of IL-22 were measured in patients with acute MI compared to healthy controls (163). Pre-clinical and clinical studies testing fezakinumab, a human anti-IL-22 mAb, and mAbs targeting the IL-22 receptor are still ongoing in patients with severe inflammatory diseases such as psoriasis, atopic dermatitis and rheumatoid arthritis. The limited results that are already published suggest no adverse safety concerns and <50% skin improvement, based on Percentage Change in the Scoring of Atopic Dermatitis (SCORAD) scores in atopic dermatitis patients (Table 4) (124, 164). Since atherosclerosis is also an inflammatory disease, interfering with IL-22 or IL-22 receptor may also represent a promising therapeutic target for CVD, although more elaborate research is needed to pinpoint the exact mechanisms of action.
IL-33, the most recently discovered member of the IL-1 cytokine family may also play a crucial role in vascular remodeling. This cytokine is expressed by ECs, VSMCs, epithelial cells and immune cells such as macrophages and T cells. For example, IL-33 is released from injured or necrotic ECs and acts as an alarmin leading to pro-inflammatory responses, both from innate and adaptive immune cells (165, 166). IL-33 binds to IL-1 receptor like protein (IL-1RL1 or also known as ST2). ST2 has two forms, a transmembrane (ST2L) and a soluble (sST2) one, which compete with each other for IL-33 binding. ST2L is expressed by various immune cells such as macrophages, T cells (predominantly Th2), mast cells, and innate lymphoid cells (167). Elevated levels of IL-33 and its soluble receptor sST2 were observed in patients suffering from pathologies such as diabetes, obesity, CAD, stroke and atherosclerosis (168).
IL-33 promotes the expression and secretion of various pro-inflammatory cytokines, adhesion molecules, proteolytic and coagulation factors. Investigations of IL-33 in atherosclerosis revealed that this cytokine stimulates the expression of the endothelial adhesion molecules VCAM1, ICAM1 and E-selectin as well as the expression of CCL2 in HUVECs in a concentration-dependent manner resulting in increased leukocyte adhesion (Figure 1; Table 3) (108). On the other hand, by binding to the ST2L, IL-33 can also influence the phenotype and function of macrophages and T cells (167). Furthermore, Miller et al. (109) found that IL-33 reduces atherosclerotic lesion size in the aortic sinus of 6-week-old male Apoe−/− mice fed with WD for 12 weeks while treated twice weekly with IL-33 injections (1 μg/injection) compared to the PBS-injected control group. In the same study, it could be demonstrated using in vitro serum assays that the atheroprotective effect of IL-33 was due to IL-33-mediated polarization of T cells into Th2 cells, inhibition of foam cell formation and an increase of antibody production targeting oxidized LDL. Nevertheless, plaque stability from the treated and untreated group remained unchanged, characterized by similar VSMC and collagen content, suggesting that IL-33 treatment decreases atherosclerosis plaque size without affecting plaque stability (Table 3) (109).
However, also some controversial results regarding the effects of IL-33 were found in human and rat studies. In humans, circulating IL-33 levels were found to correlate with vulnerable and high-risk plaques in 191 patients with carotid artery atherosclerosis. Analysis of carotid endarterectomies (CEA) from these patients also revealed an increased plaque expression of IL-33 and its receptor ST2 as well as enhanced IL-33 serum levels in CEA patients with vulnerable plaques compared with CEA patient with stable plaques (166). In contrast to what was observed in humans, obese rats showed increased sST2 levels in the aorta which correlated with an increase in the production of collagen, fibronectin and profibrotic molecules enhancing ECM formation and vascular fibrosis (Figure 1) (169). IL-33 seems to have differential effects on the stability of the atherosclerotic plaque depending on the species, in humans it was associated with vulnerable plaques however in rats, IL-33 seems to increase plaque stability.
In addition, it was found that certain genetic polymorphisms in the IL-33 locus resulted in deferential atherosclerosis development in patients with RA. From 576 RA patients carrying the mutant TT genotype of IL-33 rs3939286 polymorphism had a significantly lower carotid intima-media thickness (cIMT) evaluated by carotid ultrasound, compared to the wild-type CC genotype. The heterozygous CT genotype had an intermediate cIMT value. Combined, these results suggest a potential protective effect of the IL-33 rs3939286 T allele in atherosclerosis development by decreasing IL-33 expression (170).
However, there are also studies showing that atherosclerosis severity was not affected by IL-33/ST2 signaling. For example, there were no significant differences in atherosclerotic lesion area between IL33−/−Apoe−/−, ST2−/−Apoe−/− or Apoe−/− mice fed with 10-weeks-high cholesterol diet (171).
In conclusion, research on IL-33 and its multiple controversial effects on atherosclerosis are not yet fully elucidated and need to be better understood. A potential explanation for the controversial results are differences in tissue specific expression patterns, the applied model (e.g., deficiency vs. receptor blocking) or the prescribed medication, which should be investigated and compared more closely in future studies.
TNFα is a pro-inflammatory cytokine that is expressed mainly by macrophages, DCs and T cells and activates various pathways including cell survival, apoptosis, necrosis, migration, proliferation, barrier disruption, cell adhesion and actin cytoskeleton modification (172). It has two receptors TNFR1 and TNFR2, although several studies showed that the majority of TNFα signaling is mediated through TNFR1 (173–175). TNFα modulates vascular remodeling by increasing EC permeability, up-regulation of endothelial adhesion molecules leading to monocyte adhesion to the endothelium, matrix degradation and VSMCs proliferation in the intima (Figure 1; Table 3) (111, 113, 176, 177). TNFα can act both in a paracrine or autocrine manner. For example, TNFα-activated ECs stimulate VSMC expression of the pro-inflammatory cytokine IL-6 but also vascular endothelial growth factor (VEGF) and further TNFα expression (141). Furthermore, TNFα secreted by ECs leads to VSMC proliferation as well as promoting synthetic and macrophage-like phenotype differentiation (118).
Therefore, TNFα is a key atherosclerotic cytokine in atherosclerosis (178) and its genetic deletion in atherogenic Apoe−/− mice fed 10 weeks of WD was found to decrease atherosclerotic lesion size by 50% compared to control Apoe−/− animals (112). In the same study a BMT of 10-week-old Apoe−/− mice with age-matched Apoe−/−TNFα−/− BM resulted in a 83% reduction of lesion size after 25 weeks of WD in mice with hematopoietic TNFα-deficiency (112). Unexpectedly, no differences in lipid burden, VCAM1 expression, macrophage, B cell and T cell numbers in the circulation could be observed comparing TNFα-deficient mice with control animals. These results suggest that hematopoietic TNFα may not be involved in monocyte mobilization, since adhesion molecules as well as macrophage content in the plaques were similar in both groups (Table 3) (112). Another study using Apoe−/−TNFα−/− mice found a decreased expression of the adhesion molecules VCAM1 and ICAM1 and the cytokine CCL2 in TNFα-deficient mice on chow diet compared to Apoe−/− control mice (Table 3) (110). The same study also described an reduced capacity of macrophages to phagocytose LDL particles, thereby promoting foam cell formation as well as an decreased expression level of the scavenger receptor class A in TNFα-deficient Apoe−/− mice compared to Apoe−/− controls (110) (Figure 1; Table 3). Taken together, it seems that somatic TNFα deficiency has a greater impact on atherosclerosis development as it decreases endothelial adhesion molecule expression as well as foam cell formation compared to hematopoietic TNF-α deficiency.
Anti-TNFα therapies in RA patients are also known to decrease the level of serum chemerin, an adipokine that has an important role in CVD, although the relationship of chemerin and TNFα in remodeling remains unknown (179). Nevertheless, vascular remodeling seems to be affected by chemerin levels as shown in vitro, where chemerin deficiency decreases VSMCs proliferation and in vivo where it decreases neointima hyperplasia after angioplasty (180). Lack of chemerin also leads to the reduction of pro-inflammatory cytokines, like TNFα, in the serum suggesting a feedback loop between the two (179). Another component that modulates TNFα-induced remodeling is the erythropoietin-producing human hepatocellular receptor (EphA), which is a receptor tyrosine kinase that mediates cell-adhesion and leukocyte homing in atherosclerosis by promoting ICAM1 and VCAM1 expression on ECs (181). EphA2-deficient Apoe−/− mice which were fed a WD for 12 weeks developed smaller innominate artery and carotid sinus plaque size compared to Apoe−/−control animals (182). Furthermore, in vitro knockout of EphA2 in HAECs revealed that TNFα treatment is unable to induce monocyte adhesion to ECs lacking EphA2, suggesting that the TNFα-induced expression of adhesion molecules is inhibited upon EphA2 deficiency (182).
The effect of TNFα on endothelial function may also be modulated by food consumption. For example, aged garlic extract (AGE) and its sulfur-containing constituents improve the endothelial barrier function elicited by TNFα through stimulation of anti-inflammatory, anti-oxidative and anti-hypersensitive pathways in humans thereby also preventing CVD development including atherosclerosis. Active substances in AGE consisting of S-1-prpenylcysteine (S1PC) particularly interfere with TNFα-induced hyperpermeability of the endothelium (183). Therefore, adverse remodeling in atherosclerosis may also be reduced by food supplements.
Furthermore, tongxinluo (TXL), a traditional Chinese medicine product has anti-inflammatory as well as vasoprotective properties. In C57BL/6 mice subjected to carotid artery ligation, TXL treatment significantly reduced neointima hyperplasia in a dose-dependent manner by inhibiting macrophage infiltration as well as VSMCs proliferation in the intima of the artery, compared to untreated control mice (114). Further analysis revealed that the protective effects of TXL in hyperplasia are due to the inhibition of TNFα-induced miRNA-155 expression, a generally pro-inflammatory acting miRNA (114).
Nevertheless, until recently the therapeutic potential of TNFα blockage in atherosclerosis by pharmacological inhibitors such as monoclonal antibodies remained unknown. Oberoi et al. tested weekly injections of mouse-specific anti-TNFα monoclonal antibody CNTO5048 (12 mg/kg) in 10-week-old Ldlr−/− mice fed with high fat, high cholesterol diet for either 6 or 12 weeks. Plasma inflammatory markers such as IL-6, CCL2 and TNFα were significantly decreased in mice receiving CNTO5048 after 12 weeks of treatment compared to control animals injected with IgG antibody (184). However, no differences were observed in the 6 weeks group, although mRNA expression levels of IL-6, CXCL1 and ICAM1 in vascular tissue of the aortic arch were increased after both 6- and 12-weeks treatment. On the other hand, plasma lipid profiles revealed a significant increase in VLDL cholesterol and triglycerides due to CNTO5048 treatment. Moreover, atherosclerotic plaque burden was also augmented in CNTO5048 mice. Detailed examination of the plaque composition revealed a reduction of intimal VSMC infiltration and lower collagen type I deposition within the atherosclerotic plaque in the CNTO5048-treated group which is associated with plaque instability. These counter-intuitive results revealing reduced systemic inflammation though enlarged lesion growth are most likely due to the enhanced cholesterol levels observed in the treated group. Yet, analysis of genes regulating lipid and cholesterol metabolism, such as Apob, Mttp or Apoa5 in the liver, did not reveal any differences between the two groups (184). In contrast, adalimumab, a human-specific anti-TNFα monoclonal antibody binding and blocking both soluble and membrane bound TNFα, administered to Ldlr−/− mice fed 10-weeks of WD, revealed reduced atherosclerotic lesion size by 52% in the anti-TNFα treated mice (2.2 mg/kg twice weekly via intraperitoneal injection) compared to IgG controls, while the cholesterol and triglyceride levels did not change between the groups (185). These differential results may be due to the different antibodies that were applied, since the mouse model and length of WD feeding were comparable between the two studies (184, 185).
Various clinical trials have already been conducted to evaluate the effect of anti-TNFα treatment in patients with CVD. For example, recent findings reveal a decrease in cardiovascular biomarkers, such as soluble VCAM1, in patients after treatment with adalimumab. Psoriasis is well known to increase CVD risk as well as sharing different biomarkers and pathophysiological mechanisms such as systemic inflammation and endothelial dysfunction with CVD, suggesting that this pathology might benefit from similar therapeutic approaches (186, 187). Therefore, Zdanowska et al. conducted a study using 34 patients with psoriasis and 8 healthy volunteers between 30 and 73 years old, which were treated with an initial dose of 80 mg and then 40 mg of adalimumab every 2 weeks. After 12 weeks of treatment, soluble VCAM1 serum levels were significantly decreased comparing psoriasis patients with controls, while E-selectin was not affected (Table 4) (125). On the other hand, E-selectin levels were decreased in a bi-yearly study examining the effect of adalimumab in 17 patients with psoriasis compared with 24 healthy age-, gender- and BMI-matched volunteers with the same treatment regimen. Serum levels of E-selectin as well as plasma levels of IL-22 were significantly decreased compared to baseline, both after 12 and 24 weeks of treatment (126). Plasma CPR levels also decreased but only reached a significant difference compared to baseline after 24 weeks of treatment. Notably and in sharp contrast to the previously described beneficial outcomes, circulating oxLDL levels increased during the 2 years follow-up (Table 4) (126). The atheroprotective effects of adalimumab may therefore be a cumulative effect from a repression of inflammation and reduction of cholesterol uptake by macrophages leading to reduced foam cell formation. However, since cholesterol uptake by macrophage is decreased, it causes an augmentation of circulating cholesterol, which is a side-effect that could be solved with a combinational treatment with statins.
In addition, adalinumab decreases aortic stiffness as tested in 18 RA patients receiving either a monotherapy of subcutaneous adalinumab (40 mg/ 2 weeks) or a combination with disease modifying antirheumatic drugs in comparison to control patients using methotrexate (MTX). After 3 months of therapy, adalinumab treatment significantly decreased the aortic stiffness indices as measured by carotid-femoral pulse wave velocity (cfPWV) from 8.18 m/s to 7.01 m/s while no difference was observed in patients using MTX. On the other hand, there was no significant difference in the disease augmentation index (AIx), a systemic arterial stiffness parameter (188), or other cardiovascular risk factors after the adalinumab treatment (Table 4) (127). Besides adalinumab, also another TNFα inhibitor called etanercept is used as therapy for patients with various chronic inflammatory diseases such as RA. Based on a recent meta-analysis, both treatments significantly decreased vascular remodeling by limiting aortic stiffness and wave reflection in RA patients, independently of the treatment duration (duration varied from 6 to 56 weeks) (Table 4) (128). However, in all the studies mentioned above, TNFα treatment led to enhanced cholesterol and triglyceride titers (Table 4).
In conclusion, TNFα therapy holds great promise in reducing CVD and benefitting cardiovascular patients by decreasing atherosclerotic plaque size as well as decreasing several pro-inflammatory markers and ameliorating aortic stiffness. However, the long-term effects of anti-TFNα treatment and augmentation of cholesterol levels need to be critically considered when judging on the superiority of the treatment for CVD patients in light of the unfavorable lipid profile (128). However, based on clinical studies, the increase in lipid profiles in patients undergoing TNF-α treatment could be reduced by combining it with statin treatment (189).
In addition to chemokines and cytokines also other factors influence vascular function and composition. Among them are for example growth factors, prostaglandins and leukotrienes which will be summarized with respect to their role in vascular remodeling in the following paragraphs.
Growth differentiation factor-15 (GDF-15), also known as macrophage inhibitory cytokine-1, is a member of the TGF-β family which is induced under stress conditions such as oxidative stress, inflammation, ischemia, or mechanical stretch (190). GDF-15 is known to be expressed in cardiomyocytes, especially in response to MI, but also by adipocytes, macrophages, ECs, and VSMCs from both healthy and injured tissues (191). A receptor for GDF-15 has been identified called glial cell-derived neurotrophic factor (GDNF) family receptor α-like (GFRAL). However, this receptor is mainly expressed on human brain stem cells and based on the various physiological effects of GDF-15 throughout the body it is hypothesized that other receptors should exist which are still to be discovered (192). Inflammatory proteins such as IL-1β, TNFα, IL-2 and macrophage stimulating factor (MCSF)-1, all of which are upregulated in atherosclerosis, induce the expression of GDF-15 (192). In line with this, an increase in circulating GDF-15 protein was recently associated with an elevated risk of adverse events in patients suffering from acute coronary syndrome, chronic kidney disease or heart failure (193–197). For example, the study by Gohar et al. (194), demonstrated that high plasma levels of GDF-15 in women with carotid atherosclerotic disease are predictive for secondary cardiovascular events. However, the effect of GDF-15 on cardiac remodeling is still poorly understood. One study from Xu et al. (198) showed that GDF-15 blocks norepinephrine-induced myocardial hypertrophy through a novel pathway involving the inhibition of epidermal growth factor receptor (EGFR) transactivation. In line with this observation, a recent overview from Wesseling et al. (199) emphasizes the role of GDF-15 in endothelial dysfunction, hypertrophy, and fibrosis (Table 3). GDF-15 was also associated with an elevated risk of adverse events in patients suffering from coronary syndrome, chronic kidney disease or heart failure (199). Heart failure involves cardiac remodeling following tissue injury which is caused by inflammation, volume, or pressure overload (199).
Growing evidence indicates that GDF-15 has detrimental effects on endothelial function by causing an increase in adhesion molecule expression and influencing the balance between vasoconstriction and vasodilatation (Figure 1). Indeed, GDF-15 leads to endothelial dysfunction by reducing vascular contraction and relaxation, which ultimately leads to an impaired cardiac function (Table 3) (190, 200). In addition, GDF-15 is implicated in cardiac hypertrophy which is described as an increased heart size and insufficient cardiac output (199). The exact role of GDF-15 on cardiomyocytes is still not fully understood but it seems that GDF-15 has both pro-hypertrophic and anti-hypertrophic effects depending on environmental cues. For example, GDF-15 promotes cardiac hypertrophy by protecting cardiomyocytes from apoptotic stimuli (201), but in constrast GDF-15 also seems to reduce myocardial hypertrophy by inhibiting the transactivation of EGFR (Table 3) (198). Further research also suggests that the anti-hypertrophic effects of GDF-15 on cardiomyocytes may be due to GDF-15-induced activation of small mother against decapentaplegic (SMAD) 2/3 proteins (202). GDF-15 also promotes cardiac fibrosis in heart failure and increases collagen turnover in MI as well as collagen deposition in atherosclerotic plaques (Figure 1; Table 3) (203–205).
In addition, elevated circulating GDF-15 levels are suggested as a potential biomarker for cardiovascular risk and outcome as it is directly linked to atherosclerosis progression. GDF-15 promotes plaque vulnerability through pro-inflammatory and angiogenic effects, especially in the early stages of the disease (205, 206). Furthermore, GDF-15 has been linked to cardiovascular event prediction in general and identification of high-risk patients (205, 206). Therefore, monitoring and targeting GDF-15 in CVD may improve early diagnosis and treatment strategies (206).
Prostaglandins are a group of lipids synthesized at sites of tissue damage or infection and play an important role in resolving injury and illness by regulating inflammation, blood flow and the formation of blood clots (207). The production of prostaglandins is initiated by cyclooxygenase (COX) which induces the production of thromboxane A2 (TXA2) and prostaglandins, such as prostaglandin (PG)-D2, PG-I2, PG-E2 and PG-F2α (208). TNFα induces the expression of the prostaglandin endoperoxide synthase COX2 in a variety of cell types including ECs and VSMCs (186, 209, 210). PGs have also been found to induce diverse effects on VSMCs. For example, PG-D2 dictates the balance of VSMC proliferation and apoptosis and PG-E2 causes VSMCs to contract by inhibiting the potassium current (211, 212). PG-E2 also regulates VSMC tone through the prostaglandin E 1 (EP-1) and EP-3 receptors. Furthermore, EP-1 and EP-3 activation mediates intracellular Ca2+ pathway activation and the reduction of cAMP induced vasoconstriction (213). In sharp contrast, PG-E2 stimulation of EP-2 and EP-4 increases cAMP promoting vasodilation (214). Moreover, stimulating contractile VSMCs with PG-D2 activates the ERK pathway resulting in PG-D2 dependent VSMC phenotypic switching (215, 216). In CVD animal models, e.g. in microsomal prostaglandin E2 synthase-1−/− (mPGES1−/−) Ldlr−/−mice on a 3- and 6-month WD, PGES1-deficiency, resulting in reduced circulating PG-E2 levels and resulted inreduced plaque burden and foam cell accumulation without affecting the blood pressure (217). PG-D2 is also induced by TNFα and, in turn, propagates the dedifferentiation of contractile VSMCs into a synthetic phenotype via the upregulation of proliferator-activated receptor (PPAR) (218). Furthermore, the loss of TNFα or the inhibition of COX2 repressed the induction of intimal thickening in mice (218). The same study also found that TNFα stimulates the activity of COX2 and thereby enhances COX2 expression in contractile VSMCs, while inhibition of COX2 suppressed TNFα-induced contractile VSMC phenotypic switching and neointima formation (218). Taken together, PGs have a diverse role in vascular remodeling (Figure 1), from regulating vasoconstriction and vasodilation to VSMC proliferation and apoptosis. Yet, specifically in atherosclerosis PGs like PG-E2 can dictate lesion size (217) and PG-D2 plays an important role in the phenotypic switching of VSMCs to their pro-atherosclerotic synthetic phenotype (218). However, it is difficult to suggest PGs as a potential therapeutic target as COX2 selective inhibitors are associated with increased atherothrombotic risk (219). Therefore, further research is needed to better understand the role of PGs in vascular remodeling before developing specific treatment approaches.
Leukotrienes (LTs) are biologically active molecules produced by leukocytes, mastocytoma cells and macrophages in response to immunological and nonimmunological stimuli. LTs are well known as allergic, acute and chronic inflammatory mediators and are involved in several inflammatory conditions such as human arthritis, asthma, allograft rejection and atherosclerosis (220, 221). These inflammatory LTs originate from the 5-lipoxygenase (5-LO) pathway of arachidonic acid metabolism and, together with the 5-LO-activating protein (FLAP), catalyze the arachidonic acid metabolism from membrane phospholipids resulting in the formation of LT-A4, which is an unstable precursor leukotriene. 5-LO can further be metabolized into LT-B4 or form cysteinyl-leukotrienes like LT-C4, -D4 and -E4 after conjugation with glutathione (222, 223). Macrophages are the main producers of 5-LO and a correlation has been found between macrophage content of 5-LO and 5-LO localized in DCs, foams cells, mast cells and neutrophils and atherosclerotic plaque size in humans (224). In addition, clinical, population genetic, cell biological and mouse studies have all linked the 5-LO pathway to atherogenesis and arterial wall remodeling (223). In atherosclerosis, LTs promote the migration and accumulation of inflammatory cells into the intima of the vascular wall resulting in the initiation and progression of the disease (Figure 1). In addition, the inflammatory response is enhanced by the activation of leukotriene B4 receptors 1 (BLT1) and 2 (BLT2) as well as the cysteinyl-leukotrienes receptors (CysL)-T1 and CysLT2.These receptors are expressed on immune cells and vascular cells which are associated with atherogenesis such as ECs, VSMCs and monocytes/macrophages and their activation through leukotriene binding lead to structural alterations of the vascular wall and thus vascular remodeling (222, 223).
Direct effects of LTs on blood vessels include left ventricular contractility modifications, blood pressure regulation, coronary artery contraction and leukocyte recruitment into the perivascular space (225–228). In vitro studies revealed that LT treatment of ECs enhances the surface expression of P-selectin, secretion of von Willebrand factor and stimulates the synthesis of platelet activation factors and promotes VSMCs proliferation (229–231). LT-B4 is expressed on diverse cell types and for example neutrophils, eosinophils, VSMCs and monocytes all respond to LT-B4-dependent cell migration and recruitment to sites of inflammation (Figure 1) (220, 228, 232). In addition, the LT-B4-BLT1 pathway mediates VSMC recruitment and proliferation in the atherosclerotic plaque leading to intima hyperplasia as shown in rats treated with the BLT receptor antagonist BIIL 284 (10 mg/kg, once daily for 14 days) after balloon-induced injury of the carotid artery (233). Overall, it is clear that LT-B4 plays an important role in leukocyte attraction and adhesion to the vascular endothelium and the proliferation and migration of VSMCs (Figure 1) (231, 234).
As 5-LO is the regulator of the production of LT-B4 and cysteinyl leukotrienes, it is an interesting therapeutic target. 5-LO inhibition with intake of 0,1% BHB-TZD (5-(3,5-di-tert-butyl-4-hydroxybenzylidene)thiazolidin-2,4-dione) mixed in food was described to prevent plaque progression in atherogenic Ldlr−/− mice fed a WD for 8 weeks (235). Moreover, 5-LO inhibition with licofelone in rabbits significantly decreased the femoral artery intima/media ratio as well as macrophage infiltration in the neointima, CCL2 expression and the activation of NFκB in the vascular lesion (236). Therefore, licofelone seems to diminish neointima formation following arterial wall injury and reduces inflammatory cell recruitment and adhesion into the arterial wall, thereby reducing atherosclerosis vascular remodeling.
Targeting LT receptors may represent an additional putative therapeutic target for the treatment of atherosclerosis and for preventing intimal hyperplasia after angioplasty. The Carotid Atherosclerosis Progression Study (CAPS) investigated 8 genetic polymorphisms associated with the leukotriene pathway and early atherosclerosis and remodeling in 969 patients. However, no significant effect of these polymorphisms was observed on atherosclerosis and remodeling risk based on carotid intima-media thickness (237). This result may be explained by the fact that most patients only showed signs of early atherosclerosis, while there was insufficient plaque advancement and stenosis to demonstrate associations with advanced atherosclerosis. Another randomly sampled cohort of 470 healthy, middle-aged women and men from the Los Angeles Atherosclerosis Study (LAAS) investigated the association between 5-LO gene promoter polymorphism, dietary arachidonic acid intake and the effect on atherosclerosis. An increase in intima-media thickness and atherosclerotic plaques could be observed in patients carrying two variant alleles of the 5-lipoxygenase compared with patients with the common allele. In addition, among the persons with the two variant alleles, the ones ingesting more arachidonic acid had significantly elevated intima-media thickness compared to patients with a marine n-3 fatty acid rich diet. These findings suggest a diet-gene interaction effect which impacts the development of atherosclerosis (238).
An ongoing clinical study (started in May 2020, estimated end in December 2023) is examining the role of a cyteinyl leukotriene antagonist in atherosclerosis (NCT04277702). They aim to study its effect on lower limb artery re-occlusion rate in 200 patients with peripheral artery disease after endovascular treatment. Results will further demonstrate whether LTs are a promising therapeutic option.
Taken together many mediators exert divers and, in some cases, even opposing functions in atherosclerotic vascular remodeling. Future studies are needed to demonstrate whether the effects of CCL2, CCL5, CCL19, CXCL12, CXCL16 and CX3CL1 on VSMCs like increased proliferation and subsequent increase of plaque stability but also induction of phenotypic switching balance out their more pro-atherogenic effects on leukocyte recruitment and lesional foam cell formation. Hence, while considering the therapeutic targeting potential of chemokines in general one should keep in mind that chemokine blocking could reduce plaque size at costs of reduced lesion stability. Still, some chemokines may be useful as biomarkers since augmented levels of CCL5 or CXCL12 correlate with the severity of atherosclerosis or CAD respectively.
Cytokines like IL1-β, IL-6, IL-22, TNFα and GDF-15 seem to particularly foster EC activation and drive early atherogenesis while the effects of IL-33, PGs and LTs are less clear. In addition, IL1-β and IL-22 are also involved in VSMC proliferation and phenotypic switching, but animal models also suggest that IL-1β is beneficial in an advanced stage of the disease by maintaining plaque stability. Caution is also warranted when blocking IL-6 and TNFα. Blocking of those two cytokines enhances total cholesterol burden, LDL, and triglyceride profiles despite improving endothelial function and reducing vascular remodeling. Therefore, to minimize the detrimental effects of elevated lipids, it is important to combine IL-6 and TNFα blocking with lipid lowering strategies like statin treatment. On the other hand, fostering beneficial effects of IL-10 for example to inhibit phenotypic switching of VSMCs could also be a promising therapeutic approach to delay vascular wall remodeling.
Overall, it is important keep in mind that tissue specific expression patterns, the applied model, the prescribed medication and deficiency vs. receptor blocking all differentially impact on well-orchestrated immune functions and, in addition, time and spatial resolution significantly contribute to the results summarized above.
BE, AY, and YD performed literature research, drafted the manuscript, and made the figures. IB, SB, EV, and MS wrote the manuscript and provided corrections. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the Swiss National Foundation (SNF) Project IDs 310030_197655 and 4078P0_198297 to YD and CRSII5_193694 to IB as well as by the Swiss Heart Foundation, Project ID FF20099 to YD, a grant from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University and NWO-ZonMw Veni (91619053) to EV.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. (2017) 13:368–80. doi: 10.1038/nrneph.2017.51
2. Van Varik BJ, Rennenberg RJ, Reutelingsperger CP, Kroon AA, De Leeuw PW, Schurgers LJ. Mechanisms of arterial remodeling: lessons from genetic diseases. Front Genet. (2012) 3:290. doi: 10.3389/fgene.2012.00290
3. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Discov. (2021) 20:589–610. doi: 10.1038/s41573-021-00198-1
4. Barthwal MK, Anzinger JJ, Xu Q, Bohnacker T, Wymann MP, Kruth HS. Fluid-phase pinocytosis of native low density lipoprotein promotes murine M-CSF differentiated macrophage foam cell formation. PLoS ONE. (2013) 8:e58054. doi: 10.1371/journal.pone.0058054
5. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. (2015) 107:321–30. doi: 10.1093/cvr/cvv147
6. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
7. Airhart N, Brownstein BH, Cobb JP, Schierding W, Arif B, Ennis TL, et al. Smooth muscle cells from abdominal aortic aneurysms are unique and can independently and synergistically degrade insoluble elastin. J Vasc Surg. (2014). 60:1033–41; discussion 1041–1032. doi: 10.1016/j.jvs.2013.07.097
8. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. (2018) 285:2944–71. doi: 10.1111/febs.14466
9. Raman D, Sobolik-Delmaire T, Richmond A. Chemokines in health and disease. Exp Cell Res. (2011) 317:575–89. doi: 10.1016/j.yexcr.2011.01.005
10. Miller MC, Mayo KH. Chemokines from a structural perspective. Int J Mol Sci. (2017) 18:2088. doi: 10.3390/ijms18102088
11. Gencer S, Van Der Vorst EPC, Aslani M, Weber C, Döring Y, Duchene J. Atypical chemokine receptors in cardiovascular disease. Thromb Haemost. (2019) 119:534–41. doi: 10.1055/s-0038-1676988
12. Laing KJ, Secombes CJ. Chemokines. Dev Comp Immunol. (2004) 28:443–60. doi: 10.1016/j.dci.2003.09.006
13. Liehn EA, Piccinini AM, Koenen RR, Soehnlein O, Adage T, Fatu R, et al. A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol. (2010) 56:1847–57. doi: 10.1016/j.jacc.2010.04.066
14. Bot I, Ortiz Zacarías NV, De Witte WE, De Vries H, Van Santbrink PJ, Van Der Velden D, et al. A novel CCR2 antagonist inhibits atherogenesis in apoE deficient mice by achieving high receptor occupancy. Sci Rep. (2017) 7:52. doi: 10.1038/s41598-017-00104-z
15. Winter C, Silvestre-Roig C, Ortega-Gomez A, Lemnitzer P, Poelman H, Schumski A, et al. Chrono-pharmacological Targeting of the CCL2-CCR2 Axis Ameliorates Atherosclerosis. Cell Metab. (2018) 28:e175–82. doi: 10.1016/j.cmet.2018.05.002
16. Fougerat A, Smirnova NF, Gayral S, Malet N, Hirsch E, Wymann MP, et al. Key role of PI3Kgamma in monocyte chemotactic protein-1-mediated amplification of PDGF-induced aortic smooth muscle cell migration. Br J Pharmacol. (2012) 166:1643–53. doi: 10.1111/j.1476-5381.2012.01866.x
17. Viedt C, Vogel J, Athanasiou T, Shen W, Orth SR, Kubler W, et al. Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-kappaB and activator protein-1. Arterioscler Thromb Vasc Biol. (2002) 22:914–20. doi: 10.1161/01.ATV.0000019009.73586.7F
18. Jongstra-Bilen J, Tai K, Althagafi MG, Siu A, Scipione CA, Karim S, et al. Role of myeloid-derived chemokine CCL5/RANTES at an early stage of atherosclerosis. J Mol Cell Cardiol. (2021) 156:69–78. doi: 10.1016/j.yjmcc.2021.03.010
19. Lin CS, Hsieh PS, Hwang LL, Lee YH, Tsai SH, Tu YC, et al. The CCL5/CCR5 Axis promotes vascular smooth muscle cell proliferation and atherogenic phenotype switching. Cell Physiol Biochem. (2018) 47:707–20. doi: 10.1159/000490024
20. Jiang Y, Bai J, Tang L, Zhang P, Pu J. Anti-CCL21 antibody attenuates infarct size and improves cardiac remodeling after myocardial infarction. Cell Physiol Biochem. (2015) 37:979–90. doi: 10.1159/000430224
21. Stein S, Lohmann C, Schafer N, Hofmann J, Rohrer L, Besler C, et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. (2010) 31:2301–9. doi: 10.1093/eurheartj/ehq107
22. Halvorsen B, Dahl TB, Smedbakken LM, Singh A, Michelsen AE, Skjelland M, et al. Increased levels of CCR7 ligands in carotid atherosclerosis: different effects in macrophages and smooth muscle cells. Cardiovasc Res. (2014) 102:148–56. doi: 10.1093/cvr/cvu036
23. Van Wanrooij EJ, De Jager SC, Van Es T, De Vos P, Birch HL, Owen DA, et al. CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:251–7. doi: 10.1161/ATVBAHA.107.147827
24. Lupieri A, Smirnova NF, Solinhac R, Malet N, Benamar M, Saoudi A, et al. Smooth muscle cells-derived CXCL10 prevents endothelial healing through PI3Kgamma-dependent T cells response. Cardiovasc Res. (2020) 116:438–49. doi: 10.1093/cvr/cvz122
25. Gao JH, He LH, Yu XH, Zhao ZW, Wang G, Zou J, et al. CXCL12 promotes atherosclerosis by downregulating ABCA1 expression via the CXCR4/GSK3β/β-catenin(T120)/TCF21 pathway. J Lipid Res. (2019) 60:2020–33. doi: 10.1194/jlr.RA119000100
26. Schober A, Knarren S, Lietz M, Lin EA, Weber C. Crucial role of stromal cell-derived factor-1alpha in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation. (2003) 108:2491–7. doi: 10.1161/01.CIR.0000099508.76665.9A
27. Subramanian P, Karshovska E, Reinhard P, Megens RT, Zhou Z, Akhtar S, et al. Lysophosphatidic acid receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in neointima formation. Circ Res. (2010) 107:96–105. doi: 10.1161/CIRCRESAHA.109.212647
28. Akhtar S, Gremse F, Kiessling F, Weber C, Schober A. CXCL12 promotes the stabilization of atherosclerotic lesions mediated by smooth muscle progenitor cells in Apoe-deficient mice. Arterioscler Thromb Vasc Biol. (2013) 33:679–86. doi: 10.1161/ATVBAHA.112.301162
29. Borst O, Munzer P, Gatidis S, Schmidt EM, Schonberger T, Schmid E, et al. The inflammatory chemokine CXC motif ligand 16 triggers platelet activation and adhesion via CXC motif receptor 6-dependent phosphatidylinositide 3-kinase/Akt signaling. Circ Res. (2012) 111:1297–307. doi: 10.1161/CIRCRESAHA.112.276444
30. Minami M, Kume N, Shimaoka T, Kataoka H, Hayashida K, Akiyama Y, et al. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. (2001) 21:1796–800. doi: 10.1161/hq1001.096652
31. Wagsater D, Olofsson PS, Norgren L, Stenberg B, Sirsjo A. The chemokine and scavenger receptor CXCL16/SR-PSOX is expressed in human vascular smooth muscle cells and is induced by interferon gamma. Biochem Biophys Res Commun. (2004) 325:1187–93. doi: 10.1016/j.bbrc.2004.10.160
32. Zeiffer U, Schober A, Lietz M, Liehn EA, Erl W, Emans N, et al. Neointimal smooth muscle cells display a proinflammatory phenotype resulting in increased leukocyte recruitment mediated by P-selectin and chemokines. Circ Res. (2004) 94:776–84. doi: 10.1161/01.RES.0000121105.72718.5C
33. Riopel M, Vassallo M, Ehinger E, Pattison J, Bowden K, Winkels H, et al. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol Metab. (2019) 20:89–101. doi: 10.1016/j.molmet.2018.11.011
34. Liu P, Patil S, Rojas M, Fong AM, Smyth SS, Patel DD. CX3CR1 deficiency confers protection from intimal hyperplasia after arterial injury. Arterioscler Thromb Vasc Biol. (2006) 26:2056–62. doi: 10.1161/01.ATV.0000234947.47788.8c
35. Kumar AH, Metharom P, Schmeckpeper J, Weiss S, Martin K, Caplice NM. Bone marrow-derived CX3CR1 progenitors contribute to neointimal smooth muscle cells via fractalkine CX3CR1 interaction. FASEB J. (2010) 24:81–92. doi: 10.1096/fj.09-132225
36. Rollins BJ. Monocyte chemoattractant protein 1: a potential regulator of monocyte recruitment in inflammatory disease. Mol Med Today. (1996) 2:198–204. doi: 10.1016/1357-4310(96)88772-7
37. Jones SM, Mann A, Conrad K, Saum K, Hall DE, Mckinney LM, et al. PAR2 (protease-activated receptor 2) deficiency attenuates atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2018) 38:1271–82. doi: 10.1161/ATVBAHA.117.310082
38. Terkeltaub R, Boisvert WA, Curtiss LK. Chemokines and atherosclerosis. Curr Opin Lipidol. (1998) 9:397–405. doi: 10.1097/00041433-199810000-00003
39. Taubman MB, Rollins BJ, Poon M, Marmur J, Green RS, Berk BC, et al. JE mRNA accumulates rapidly in aortic injury and in platelet-derived growth factor-stimulated vascular smooth muscle cells. Circ Res. (1992) 70:314–25. doi: 10.1161/01.RES.70.2.314
40. Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. (1994) 153:2052–63.
41. Yla-Herttuala S, Lipton BA, Rosenfeld ME, Sarkioja T, Yoshimura T, Leonard EJ, et al. Expression of monocyte chemoattractant protein 1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci USA. (1991) 88:5252–6. doi: 10.1073/pnas.88.12.5252
42. Matsumori A, Furukawa Y, Hashimoto T, Yoshida A, Ono K, Shioi T, et al. Plasma levels of the monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 are elevated in patients with acute myocardial infarction. J Mol Cell Cardiol. (1997) 29:419–23. doi: 10.1006/jmcc.1996.0285
43. Georgakis MK, Van Der Laan SW, Asare Y, Mekke JM, Haitjema S, Schoneveld AH, et al. Monocyte-chemoattractant protein-1 levels in human atherosclerotic lesions associate with plaque vulnerability. Arterioscler Thromb Vasc Biol. (2021) 41:2038–48. doi: 10.1161/ATVBAHA.121.316091
44. Basurto L, Gregory MA, Hernandez SB, Sanchez-Huerta L, Martinez AD, Manuel-Apolinar L, et al. Monocyte chemoattractant protein-1 (MCP-1) and fibroblast growth factor-21 (FGF-21) as biomarkers of subclinical atherosclerosis in women. Exp Gerontol. (2019) 124:110624. doi: 10.1016/j.exger.2019.05.013
45. Fougerat A, Gayral S, Gourdy P, Schambourg A, Ruckle T, Schwarz MK, et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation. (2008) 117:1310–7. doi: 10.1161/CIRCULATIONAHA.107.720466
46. Veillard NR, Kwak B, Pelli G, Mulhaupt F, James RW, Proudfoot AE, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. (2004) 94:253–61. doi: 10.1161/01.RES.0000109793.17591.4E
47. Vajen T, Koenen RR, Werner I, Staudt M, Projahn D, Curaj A, et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci Rep. (2018) 8:10647. doi: 10.1038/s41598-018-29026-0
48. Marques RE, Guabiraba R, Russo RC, Teixeira MM. Targeting CCL5 in inflammation. Expert Opin Ther Targets. (2013) 17:1439–60. doi: 10.1517/14728222.2013.837886
49. Von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. (2001) 103:1772–7. doi: 10.1161/01.CIR.103.13.1772
50. Schober A, Manka D, Von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. (2002) 106:1523–9. doi: 10.1161/01.CIR.0000028590.02477.6F
51. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
52. Fox JM, Kausar F, Day A, Osborne M, Hussain K, Mueller A, et al. CXCL4/Platelet Factor 4 is an agonist of CCR1 and drives human monocyte migration. Sci Rep. (2018) 8:9466. doi: 10.1038/s41598-018-27710-9
53. Pitsilos S, Hunt J, Mohler ER, Prabhakar AM, Poncz M, Dawicki J, et al. Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemost. (2003) 90:1112–20. doi: 10.1160/TH03-02-0069
54. Sachais BS, Turrentine T, Dawicki Mckenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE−/− mice. Thromb Haemost. (2007) 98:1108–13. doi: 10.1160/TH07-04-0271
55. Blin MG, Bachelier R, Fallague K, Moussouni K, Aurrand-Lions M, Fernandez S, et al. CD146 deficiency promotes plaque formation in a mouse model of atherosclerosis by enhancing RANTES secretion and leukocyte recruitment. J Mol Cell Cardiol. (2019) 130:76–87. doi: 10.1016/j.yjmcc.2019.03.017
56. Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. (1998) 391:799–803. doi: 10.1038/35876
57. Damas JK, Smith C, Oie E, Fevang B, Halvorsen B, Waehre T, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol. (2007) 27:614–20. doi: 10.1161/01.ATV.0000255581.38523.7c
58. Yan Y, Chen R, Wang X, Hu K, Huang L, Lu M, et al. CCL19 and CCR7 expression, signaling pathways, and adjuvant functions in viral infection and prevention. Front Cell Dev Biol. (2019) 7:212. doi: 10.3389/fcell.2019.00212
59. Zhou F, Pan Y, Huang Z, Jia Y, Zhao X, Chen Y, et al. Visfatin induces cholesterol accumulation in macrophages through up-regulation of scavenger receptor-A and CD36. Cell Stress Chaperones. (2013) 18:643–52. doi: 10.1007/s12192-013-0417-z
60. Lehrke M, Greif M, Broedl UC, Lebherz C, Laubender RP, Becker A, et al. MMP-1 serum levels predict coronary atherosclerosis in humans. Cardiovasc Diabetol. (2009) 8:50. doi: 10.1186/1475-2840-8-50
61. Akhavanpoor M, Gleissner CA, Gorbatsch S, Doesch AO, Akhavanpoor H, Wangler S, et al. CCL19 and CCL21 modulate the inflammatory milieu in atherosclerotic lesions. Drug Des Devel Ther. (2014) 8:2359–71. doi: 10.2147/DDDT.S72394
62. Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, et al. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. (1999) 104:1041–50. doi: 10.1172/JCI6993
63. Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, Ahluwalia N, et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. (2006) 113:2301–12. doi: 10.1161/CIRCULATIONAHA.105.605121
64. Segers D, Lipton JA, Leenen PJ, Cheng C, Tempel D, Pasterkamp G, et al. Atherosclerotic plaque stability is affected by the chemokine CXCL10 in both mice and humans. Int J Inflam. (2011) 2011:936109. doi: 10.4061/2011/936109
65. Niki T, Soeki T, Yamaguchi K, Taketani Y, Yagi S, Iwase T, et al. Elevated concentration of interferon-inducible protein of 10 kD (IP-10) is associated with coronary atherosclerosis. Int Heart J. (2015) 56:269–72. doi: 10.1536/ihj.14-300
66. Wallace AE, Cartwright JE, Begum R, Laing K, Thilaganathan B, Whitley GS. Trophoblast-induced changes in C-x-C motif chemokine 10 expression contribute to vascular smooth muscle cell dedifferentiation during spiral artery remodeling. Arterioscler Thromb Vasc Biol. (2013) 33:e93–e101. doi: 10.1161/ATVBAHA.112.300354
67. Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P, Luster AD. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res. (2000) 86:131–8. doi: 10.1161/01.RES.86.2.131
68. Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. (2011) 43:333–8. doi: 10.1038/ng.784
69. Doring Y, Van Der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, et al. CXCL12 derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation. (2019) 139:1338–40. doi: 10.1161/CIRCULATIONAHA.118.037953
70. Sjaarda J, Gerstein H, Chong M, Yusuf S, Meyre D, Anand SS, et al. Blood CSF1 and CXCL12 as causal mediators of coronary artery disease. J Am Coll Cardiol. (2018) 72:300–10. doi: 10.1016/j.jacc.2018.04.067
71. Li L, Du Z, Rong B, Zhao D, Wang A, Xu Y, et al. Foam cells promote atherosclerosis progression by releasing CXCL12. Biosci Rep. (2020) 40:BSR20193267. doi: 10.1042/BSR20193267
72. Saxena A, Fish JE, White MD, Yu S, Smyth JW, Shaw RM, et al. Stromal cell-derived factor-1alpha is cardioprotective after myocardial infarction. Circulation. (2008) 117:2224–31. doi: 10.1161/CIRCULATIONAHA.107.694992
73. Maracle CX, Agca R, Helder B, Meeuwsen J a.L., Niessen HWM, et al. Noncanonical NF-kappaB signaling in microvessels of atherosclerotic lesions is associated with inflammation, atheromatous plaque morphology and myocardial infarction. Atherosclerosis. (2018) 270:33–41. doi: 10.1016/j.atherosclerosis.2018.01.032
74. Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol. (2000) 1:298–304. doi: 10.1038/79738
75. Ludwig A, Weber C. Transmembrane chemokines: versatile 'special agents' in vascular inflammation. Thromb Haemost. (2007) 97:694–703. doi: 10.1160/TH07-01-0035
76. Korbecki J, Bajdak-Rusinek K, Kupnicka P, Kapczuk P, Simińska D, Chlubek D, et al. The role of CXCL16 in the pathogenesis of cancer and other diseases. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22073490
77. Meyer Dos Santos S, Blankenbach K, Scholich K, Dorr A, Monsefi N, Keese M, et al. Platelets from flowing blood attach to the inflammatory chemokine CXCL16 expressed in the endothelium of the human vessel wall. Thromb Haemost. (2015) 114:297–312. doi: 10.1160/TH14-11-0911
78. Zhao G, Zhang H, Zhu S, Wang S, Zhu K, Zhao Y, et al. Interleukin-18 accelerates cardiac inflammation and dysfunction during ischemia/reperfusion injury by transcriptional activation of CXCL16. Cell Signal. (2021) 87:110141. doi: 10.1016/j.cellsig.2021.110141
79. Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. (1997) 385:640–4. doi: 10.1038/385640a0
80. Saederup N, Chan L, Lira SA, Charo IF. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2−/− mice: evidence for independent chemokine functions in atherogenesis. Circulation. (2008) 117:1642–8. doi: 10.1161/CIRCULATIONAHA.107.743872
81. Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. (2003) 197:1701–7. doi: 10.1084/jem.20022156
82. Li H, Zhang HM, Fan LJ, Li HH, Peng ZT, Li JP, et al. STAT3/miR-15a-5p/CX3CL1 Loop Regulates Proliferation and Migration of Vascular Endothelial Cells in Atherosclerosis. Int J Med Sci. (2021) 18:964–74. doi: 10.7150/ijms.49460
83. Roy-Chowdhury E, Brauns N, Helmke A, Nordlohne J, Bräsen JH, Schmitz J, et al. Human CD16+ monocytes promote a pro-atherosclerotic endothelial cell phenotype via CX3CR1-CX3CL1 interaction. Cardiovasc Res. (2021) 117:1510–22. doi: 10.1093/cvr/cvaa234
84. Zhou JJ, Wang YM, Lee VWS, Zhang GY, Medbury H, Williams H, et al. DEC205-DC targeted DNA vaccine against CX3CR1 protects against atherogenesis in mice. PLoS One. (2018) 13:e0195657. doi: 10.1371/journal.pone.0195657
85. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. (2009) 78:539–52. doi: 10.1016/j.bcp.2009.04.029
86. Mai W, Liao Y. Targeting IL-1beta in the treatment of atherosclerosis. Front Immunol. (2020) 11:589654. doi: 10.3389/fimmu.2020.589654
87. Li P, Li YL, Li ZY, Wu YN, Zhang CC, A X, et al. Cross talk between vascular smooth muscle cells and monocytes through interleukin- 1beta/interleukin-18 signaling promotes vein graft thickening. Arterioscler Thromb Vasc Biol. (2014) 34:2001–11. doi: 10.1161/ATVBAHA.113.303145
88. Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MAJr. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am J Pathol. (1985) 121:394–403.
89. Meng J, Zhang W, Wang C, Xiong S, Wang Q, Li H, et al. The dipeptidyl peptidase (DPP)-4 inhibitor trelagliptin inhibits IL-1beta-induced endothelial inflammation and monocytes attachment. Int Immunopharmacol. (2020) 89:106996. doi: 10.1016/j.intimp.2020.106996
90. Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial barrier and its abnormalities in cardiovascular disease. Front Physiol. (2015) 6:365. doi: 10.3389/fphys.2015.00365
91. Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. (2011) 216:313–20. doi: 10.1016/j.atherosclerosis.2011.02.026
92. Libby P, Warner SJ, Friedman GB. Interleukin 1: a mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J Clin Invest. (1988) 81:487–98. doi: 10.1172/JCI113346
93. Loppnow H, Libby P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J Clin Invest. (1990) 85:731–8. doi: 10.1172/JCI114498
94. Didion SP. Cellular and oxidative mechanisms associated with interleukin-6 signaling in the vasculature. Int J Mol Sci. (2017) 18:2563. doi: 10.3390/ijms18122563
95. Luo P, Shi W, Wang Y, Ma H, Liu T, Yan D, et al. Raloxifene inhibits IL-6/STAT3 signaling pathway and protects against high-fat-induced atherosclerosis in ApoE(−/−) mice. Life Sci. (2020) 261:118304. doi: 10.1016/j.lfs.2020.118304
96. Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. (2003) 24:25–9. doi: 10.1016/S1471-4906(02)00013-3
97. Madan M, Bishayi B, Hoge M, Amar S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis. (2008) 197:504–14. doi: 10.1016/j.atherosclerosis.2007.02.023
98. Ju X, Ijaz T, Sun H, Lejeune W, Vargas G, Shilagard T, et al. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J Am Heart Assoc. (2014) 3:e000476. doi: 10.1161/JAHA.113.000476
99. Du B, Ouyang A, Eng JS, Fleenor BS. Aortic perivascular adipose-derived interleukin-6 contributes to arterial stiffness in low-density lipoprotein receptor deficient mice. Am J Physiol Heart Circ Physiol. (2015) 308:H1382–1390. doi: 10.1152/ajpheart.00712.2014
100. Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. (2010) 24:2869–80. doi: 10.1096/fj.09-148155
101. Han X, Boisvert WA. Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function. Thromb Haemost. (2015) 113:505–12. doi: 10.1160/TH14-06-0509
102. Jung M, Ma Y, Iyer RP, Deleon-Pennell KY, Yabluchanskiy A, Garrett MR, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. (2017) 112:33. doi: 10.1007/s00395-017-0622-5
103. Van Hoeven V, Munneke JM, Cornelissen AS, Omar SZ, Spruit MJ, Kleijer M, et al. Mesenchymal stromal cells stimulate the proliferation and IL-22 production of group 3 innate lymphoid cells. J Immunol. (2018) 201:1165–73. doi: 10.4049/jimmunol.1700901
104. Xia Q, Xiang X, Patel S, Puranik R, Xie Q, Bao S. Characterisation of IL-22 and interferon-gamma-inducible chemokines in human carotid plaque. Int J Cardiol. (2012) 154:187–9. doi: 10.1016/j.ijcard.2011.10.093
105. Luo JW, Hu Y, Liu J, Yang H, Huang P. Interleukin-22: a potential therapeutic target in atherosclerosis. Mol Med. (2021) 27:88. doi: 10.1186/s10020-021-00353-9
106. Rattik S, Hultman K, Rauch U, Soderberg I, Sundius L, Ljungcrantz I, et al. IL-22 affects smooth muscle cell phenotype and plaque formation in apolipoprotein E knockout mice. Atherosclerosis. (2015) 242:506–14. doi: 10.1016/j.atherosclerosis.2015.08.006
107. Shi L, Ji Q, Liu L, Shi Y, Lu Z, Ye J, et al. IL-22 produced by Th22 cells aggravates atherosclerosis development in ApoE(−/−) mice by enhancing DC-induced Th17 cell proliferation. J Cell Mol Med. (2020) 24:3064–78. doi: 10.1111/jcmm.14967
108. Demyanets S, Konya V, Kastl SP, Kaun C, Rauscher S, Niessner A, et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2011) 31:2080–9. doi: 10.1161/ATVBAHA.111.231431
109. Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. (2008) 205:339–46. doi: 10.1084/jem.20071868
110. Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, et al. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. (2005) 180:11–7. doi: 10.1016/j.atherosclerosis.2004.11.016
111. Carlos TM, Schwartz BR, Kovach NL, Yee E, Rosa M, Osborn L, et al. Vascular cell adhesion molecule-1 mediates lymphocyte adherence to cytokine-activated cultured human endothelial cells. Blood. (1990) 76:965–70. doi: 10.1182/blood.V76.5.965.965
112. Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. (2004) 24:2137–42. doi: 10.1161/01.ATV.0000143933.20616.1b
113. Davis R, Pillai S, Lawrence N, Sebti S, Chellappan SP. TNF-alpha-mediated proliferation of vascular smooth muscle cells involves Raf-1-mediated inactivation of Rb and transcription of E2F1-regulated genes. Cell Cycle. (2012) 11:109–18. doi: 10.4161/cc.11.1.18473
114. Zhang RN, Zheng B, Li LM, Zhang J, Zhang XH, Wen JK. Tongxinluo inhibits vascular inflammation and neointimal hyperplasia through blockade of the positive feedback loop between miR-155 and TNF-alpha. Am J Physiol Heart Circ Physiol. (2014) 307:H552–562. doi: 10.1152/ajpheart.00936.2013
115. Zapolska-Downar D, Naruszewicz M. Propionate reduces the cytokine-induced VCAM-1 and ICAM-1 expression by inhibiting nuclear factor-kappa B (NF-kappaB) activation. J Physiol Pharmacol. (2009) 60:123–31.
116. Shen CK, Huang BR, Yeh WL, Chen CW, Liu YS, Lai SW, et al. Regulatory effects of IL-1beta in the interaction of GBM and tumor-associated monocyte through VCAM-1 and ICAM-1. Eur J Pharmacol. (2021) 905:174216. doi: 10.1016/j.ejphar.2021.174216
117. Chiu JJ, Chen LJ, Lee CI, Lee PL, Lee DY, Tsai MC, et al. Mechanisms of induction of endothelial cell E-selectin expression by smooth muscle cells and its inhibition by shear stress. Blood. (2007) 110:519–28. doi: 10.1182/blood-2006-08-040097
118. Sorokin V, Vickneson K, Kofidis T, Woo CC, Lin XY, Foo R, et al. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front Immunol. (2020) 11:599415. doi: 10.3389/fimmu.2020.599415
119. Boini KM, Hussain T, Li PL, Koka S. Trimethylamine-N-oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell Physiol Biochem. (2017) 44:152–62. doi: 10.1159/000484623
120. Cao F, Wu K, Zhu YZ, Bao ZW. Roles and mechanisms of dipeptidyl peptidase 4 inhibitors in vascular aging. Front Endocrinol. (2021) 12:731273. doi: 10.3389/fendo.2021.731273
121. Gomez D, Baylis RA, Durgin BG, Newman A, Alencar GF, et al. (2018). Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med 24, 1418–1429. doi: 10.1038/s41591-018-0124-5
122. Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. (2021) 397:2060–9. doi: 10.1016/S0140-6736(21)00520-1
123. Bacchiega BC, Bacchiega AB, Usnayo MJ, Bedirian R, Singh G, Pinheiro GD. Interleukin 6 inhibition and coronary artery disease in a high-risk population: a prospective community-based clinical study. J Am Heart Assoc. (2017) 6:e005038. doi: 10.1161/JAHA.116.005038
124. Guttman-Yassky E, Brunner PM, Neumann AU, Khattri S, Pavel AB, Malik K, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. (2018) 78:872–81 e876. doi: 10.1016/j.jaad.2018.01.016
125. Zdanowska N, Owczarczyk-Saczonek A, Czerwinska J, Nowakowski JJ, Kozera-Zywczyk A, Owczarek W, et al. Methotrexate and adalimumab decrease the serum levels of cardiovascular disease biomarkers (VCAM-1 and E-Selectin) in plaque psoriasis. Medicina (Kaunas). (2020) 56:473. doi: 10.3390/medicina56090473
126. Gkalpakiotis S, Arenbergerova M, Gkalpakioti P, Potockova J, Arenberger P, Kraml P. Long-term impact of adalimumab therapy on biomarkers of systemic inflammation in psoriasis: Results of a 2 year study. Dermatol Ther. (2020) 33:e14110. doi: 10.1111/dth.14110
127. Vassilopoulos D, Gravos A, Vlachopoulos C, Kandili A, Ioakeimidis N, Pectasides D, et al. Adalimumab decreases aortic stiffness independently of its effect in disease activity in patients with rheumatoid arthritis. Clin Rheumatol. (2015) 34:359–64. doi: 10.1007/s10067-014-2718-8
128. Vlachopoulos C, Gravos A, Georgiopoulos G, Terentes-Printzios D, Ioakeimidis N, Vassilopoulos D, et al. The effect of TNF-a antagonists on aortic stiffness and wave reflections: a meta-analysis. Clin Rheumatol. (2018) 37:515–26. doi: 10.1007/s10067-017-3657-y
129. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. (2017) 70:2278–89. doi: 10.1016/j.jacc.2017.09.028
130. Ridker PM, Macfadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
131. Hassan M. CANTOS: A breakthrough that proves the inflammatory hypothesis of atherosclerosis. Glob Cardiol Sci Pract. (2018) 2018:2. doi: 10.21542/gcsp.2018.2
132. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. (2003) 23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3
133. Chi H, Messas E, Levine RA, Graves DT, Amar S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmacotherapeutic implications. Circulation. (2004) 110:1678–85. doi: 10.1161/01.CIR.0000142085.39015.31
134. Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M, et al. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2004) 24:1068–73. doi: 10.1161/01.ATV.0000127025.48140.a3
135. Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, et al. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest. (2012) 122:70–9. doi: 10.1172/JCI43713
136. Aiello VD, Gutierrez PS, Chaves MJ, Lopes AA, Higuchi ML, Ramires JA. Morphology of the internal elastic lamina in arteries from pulmonary hypertensive patients: a confocal laser microscopy study. Mod Pathol. (2003) 16:411–6. doi: 10.1097/01.MP.0000067685.57858.D7
137. Merhi-Soussi F, Kwak BR, Magne D, Chadjichristos C, Berti M, Pelli G, et al. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc Res. (2005) 66:583–93. doi: 10.1016/j.cardiores.2005.01.008
138. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
139. Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, et al. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J Lipid Res. (2015) 56:286–93. doi: 10.1194/jlr.M054015
140. West NR. Coordination of Immune-Stroma Crosstalk by IL-6 Family Cytokines. Front Immunol. (2019) 10:1093. doi: 10.3389/fimmu.2019.01093
141. Chu F, Wang M, Ma H, Zhu J. Simvastatin modulates interaction between vascular smooth muscle cell/macrophage and TNF-alpha-activated endothelial cell. J Cardiovasc Pharmacol. (2018) 71:268–74. doi: 10.1097/FJC.0000000000000567
142. Gager GM, Biesinger B, Hofer F, Winter MP, Hengstenberg C, Jilma B, et al. Interleukin-6 level is a powerful predictor of long-term cardiovascular mortality in patients with acute coronary syndrome. Vascul Pharmacol. (2020) 135:106806. doi: 10.1016/j.vph.2020.106806
143. Huang P, He XY, Xu M. The role of miRNA-146a and proinflammatory cytokines in carotid atherosclerosis. Biomed Res Int. (2020) 2020:6657734. doi: 10.1155/2020/6657734
144. Lee GL, Wu JY, Tsai CS, Lin CY, Tsai YT, Lin CS, et al. TLR4-activated MAPK-IL-6 axis regulates vascular smooth muscle cell function. Int J Mol Sci. (2016) 17:1394. doi: 10.3390/ijms17091394
145. Kumari S, Panda TK, Pradhan T. Lysyl oxidase: its diversity in health and diseases. Indian J Clin Biochem. (2017) 32:134–41. doi: 10.1007/s12291-016-0576-7
146. Schieffer B, Selle T, Hilfiker A, Hilfiker-Kleiner D, Grote K, Tietge UJ, et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation. (2004) 110:3493–500. doi: 10.1161/01.CIR.0000148135.08582.97
147. Pierce GL. Mechanisms and Subclinical Consequences of Aortic Stiffness. Hypertension. (2017) 70:848–53. doi: 10.1161/HYPERTENSIONAHA.117.08933
148. Protogerou AD, Zampeli E, Fragiadaki K, Stamatelopoulos K, Papamichael C, Sfikakis PP. A pilot study of endothelial dysfunction and aortic stiffness after interleukin-6 receptor inhibition in rheumatoid arthritis. Atherosclerosis. (2011) 219:734–6. doi: 10.1016/j.atherosclerosis.2011.09.015
149. Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. (2014) 122:177–210. doi: 10.1016/B978-0-12-800267-4.00005-5
150. Mazighi M, Pelle A, Gonzalez W, Mtairag El M, Philippe M, Henin D, et al. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am J Physiol Heart Circ Physiol. (2004) 287:H866–871. doi: 10.1152/ajpheart.00918.2003
151. Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. (2003) 9:10–7. doi: 10.1007/BF03402102
152. Eefting D, Schepers A, De Vries MR, Pires NM, Grimbergen JM, Lagerweij T, et al. The effect of interleukin-10 knock-out and overexpression on neointima formation in hypercholesterolemic APOE*3-Leiden mice. Atherosclerosis. (2007) 193:335–42. doi: 10.1016/j.atherosclerosis.2006.09.032
153. Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ Res. (2002) 90:1064–71. doi: 10.1161/01.RES.0000018941.10726.FA
154. Namiki M, Kawashima S, Yamashita T, Ozaki M, Sakoda T, Inoue N, et al. Intramuscular gene transfer of interleukin-10 cDNA reduces atherosclerosis in apolipoprotein E-knockout mice. Atherosclerosis. (2004) 172:21–9. doi: 10.1016/j.atherosclerosis.2003.08.032
155. Zimmerman MA, Reznikov LL, Raeburn CD, Selzman CH. Interleukin-10 attenuates the response to vascular injury. J Surg Res. (2004) 121:206–13. doi: 10.1016/j.jss.2004.03.025
156. Terkeltaub RA. IL-10: an “immunologic scalpel” for atherosclerosis? Arterioscler Thromb Vasc Biol. (1999) 19:2823–5. doi: 10.1161/01.ATV.19.12.2823
157. Bu T, Li Z, Hou Y, Sun W, Zhang R, Zhao L, et al. Exosome-mediated delivery of inflammation-responsive Il-10 mRNA for controlled atherosclerosis treatment. Theranostics. (2021) 11:9988–10000. doi: 10.7150/thno.64229
158. Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, Mcculloch JA, Thovarai V, et al. (2018). An interleukin-23-interleukin-22 axis regulates intestinal microbial homeostasis to protect from diet-induced atherosclerosis. Immunity 49 943–957 e949. doi: 10.1016/j.immuni.2018.09.011
159. Che Y, Su Z, Xia L. Effects of IL-22 on cardiovascular diseases. Int Immunopharmacol. (2020) 81:106277. doi: 10.1016/j.intimp.2020.106277
160. Xu S, Zhang J, Liu J, Ye J, Xu Y, Wang Z, et al. The role of interleukin-10 family members in cardiovascular diseases. Int Immunopharmacol. (2021) 94:107475. doi: 10.1016/j.intimp.2021.107475
161. Chellan B, Yan L, Sontag TJ, Reardon CA, Hofmann Bowman MA. IL-22 is induced by S100/calgranulin and impairs cholesterol efflux in macrophages by downregulating ABCG1. J Lipid Res. (2014) 55:443–54. doi: 10.1194/jlr.M044305
162. Hou Y, Zhu L, Tian H, Sun HX, Wang R, Zhang L, et al. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. (2018) 9:1027–38. doi: 10.1007/s13238-018-0505-z
163. Zhang L, Wang T, Wang XQ, Du RZ, Zhang KN, Liu XG, et al. Elevated frequencies of circulating Th22 cell in addition to Th17 cell and Th17/Th1 cell in patients with acute coronary syndrome. PLoS ONE. (2013) 8:e71466. doi: 10.1371/journal.pone.0071466
164. Kragstrup TW, Andersen T, Heftdal LD, Hvid M, Gerwien J, Sivakumar P, et al. The IL-20 cytokine family in rheumatoid arthritis and spondyloarthritis. Front Immunol. (2018) 9:2226. doi: 10.3389/fimmu.2018.02226
165. Aimo A, Migliorini P, Vergaro G, Franzini M, Passino C, Maisel A, et al. The IL-33/ST2 pathway, inflammation and atherosclerosis: trigger and target? Int J Cardiol. (2018) 267:188–92. doi: 10.1016/j.ijcard.2018.05.056
166. Stankovic M, Ljujic B, Babic S, Maravic-Stojkovic V, Mitrovic S, Arsenijevic N, et al. IL-33/IL-33R in various types of carotid artery atherosclerotic lesions. Cytokine. (2019) 120:242–50. doi: 10.1016/j.cyto.2019.05.010
167. Lu J, Kang J, Zhang C, Zhang X. The role of IL-33/ST2L signals in the immune cells. Immunol Lett. (2015) 164:11–7. doi: 10.1016/j.imlet.2015.01.008
168. Altara R, Ghali R, Mallat Z, Cataliotti A, Booz GW, Zouein FA. Conflicting vascular and metabolic impact of the IL-33/sST2 axis. Cardiovasc Res. (2018) 114:1578–94. doi: 10.1093/cvr/cvy166
169. Martinez-Martinez E, Miana M, Jurado-Lopez R, Rousseau E, Rossignol P, Zannad F, et al. A role for soluble ST2 in vascular remodeling associated with obesity in rats. PLoS One. (2013) 8:e79176. doi: 10.1371/journal.pone.0079176
170. Lopez-Mejias R, Genre F, Remuzgo-Martinez S, Robustillo-Villarino M, Garcia-Bermudez M, Llorca J, et al. Protective role of the interleukin 33 rs3939286 gene polymorphism in the development of subclinical atherosclerosis in rheumatoid arthritis patients. PLoS ONE. (2015) 10:e0143153. doi: 10.1371/journal.pone.0143153
171. Martin P, Palmer G, Rodriguez E, Woldt E, Mean I, James RW, et al. Atherosclerosis severity is not affected by a deficiency in IL-33/ST2 signaling. Immun Inflamm Dis. (2015) 3:239–46. doi: 10.1002/iid3.62
172. Full LE, Monaco C. Targeting inflammation as a therapeutic strategy in accelerated atherosclerosis in rheumatoid arthritis. Cardiovasc Ther. (2011) 29:231–42. doi: 10.1111/j.1755-5922.2010.00159.x
173. Zhang L, Peppel K, Sivashanmugam P, Orman ES, Brian L, Exum ST, et al. Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler Thromb Vasc Biol. (2007) 27:1087–94. doi: 10.1161/01.ATV.0000261548.49790.63
174. Xanthoulea S, Thelen M, Pottgens C, Gijbels MJ, Lutgens E, De Winther MP. Absence of p55 TNF receptor reduces atherosclerosis, but has no major effect on angiotensin II induced aneurysms in LDL receptor deficient mice. PLoS ONE. (2009) 4:e6113. doi: 10.1371/journal.pone.0006113
175. Kim CW, Oh ET, Park HJ. A strategy to prevent atherosclerosis via TNF receptor regulation. FASEB J. (2021) 35:e21391. doi: 10.1096/fj.202000764R
176. Goerdt S, Zwadlo G, Schlegel R, Hagemeier HH, Sorg C. Characterization and expression kinetics of an endothelial cell activation antigen present in vivo only in acute inflammatory tissues. Exp Cell Biol. (1987) 55:117–26. doi: 10.1159/000163407
177. Brett J, Gerlach H, Nawroth P, Steinberg S, Godman G, Stern D. Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J Exp Med. (1989) 169:1977–91. doi: 10.1084/jem.169.6.1977
178. Tousoulis D, Oikonomou E, Economou EK, Crea F, Kaski JC. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J. (2016) 37:1723–32. doi: 10.1093/eurheartj/ehv759
179. Liu H, Xiong W, Luo Y, Chen H, He Y, Cao Y, et al. Adipokine chemerin stimulates progression of atherosclerosis in ApoE(–/–) mice. Biomed Res Int. (2019) 2019:7157865. doi: 10.1155/2019/7157865
180. Xiong W, Luo Y, Wu L, Liu F, Liu H, Li J, et al. Chemerin stimulates vascular smooth muscle cell proliferation and carotid neointimal hyperplasia by activating mitogen-activated protein kinase signaling. PLoS ONE. (2016) 11:e0165305. doi: 10.1371/journal.pone.0165305
181. London M, Gallo E. The EphA2 and cancer connection: potential for immune-based interventions. Mol Biol Rep. (2020) 47:8037–48. doi: 10.1007/s11033-020-05767-y
182. Finney AC, Funk SD, Green JM, Yurdagul AJr, Rana MA, Pistorius R, et al. EphA2 expression regulates inflammation and fibroproliferative remodeling in atherosclerosis. Circulation. (2017) 136:566–82. doi: 10.1161/CIRCULATIONAHA.116.026644
183. Kunimura K, Miki S, Takashima M, Suzuki JI. S-1-propenylcysteine improves TNF-alpha-induced vascular endothelial barrier dysfunction by suppressing the GEF-H1/RhoA/Rac pathway. Cell Commun Signal. (2021) 19:17. doi: 10.1186/s12964-020-00692-w
184. Oberoi R, Vlacil AK, Schuett J, Schosser F, Schuett H, Tietge UJF, et al. Anti-tumor necrosis factor-alpha therapy increases plaque burden in a mouse model of experimental atherosclerosis. Atherosclerosis. (2018) 277:80–9. doi: 10.1016/j.atherosclerosis.2018.08.030
185. Zhu L, Giunzioni I, Tavori H, Covarrubias R, Ding L, Zhang Y, et al. Loss of macrophage low-density lipoprotein receptor-related protein 1 confers resistance to the antiatherogenic effects of tumor necrosis factor-alpha inhibition. Arterioscler Thromb Vasc Biol. (2016) 36:1483–95. doi: 10.1161/ATVBAHA.116.307736
186. Kim YA, Kim JY, Kim MR, Hwang KJ, Chang DY, Jeon MK. Tumor necrosis factor-alpha-induced cyclooxygenase-2 overexpression in eutopic endometrium of women with endometriosis by stromal cell culture through nuclear factor-kappaB activation. J Reprod Med. (2009) 54:625–30.
187. Raaby L, Ahlehoff O, De Thurah A. Psoriasis and cardiovascular events: updating the evidence. Arch Dermatol Res. (2017) 309:225–8. doi: 10.1007/s00403-016-1712-1
188. Janner JH, Godtfredsen NS, Ladelund S, Vestbo J, Prescott E. The association between aortic augmentation index and cardiovascular risk factors in a large unselected population. J Hum Hypertens. (2012) 26:476–84. doi: 10.1038/jhh.2011.59
189. Hassan S, Milman U, Feld J, Eder L, Lavi I, Cohen S, et al. Effects of anti-TNF-alpha treatment on lipid profile in rheumatic diseases: an analytical cohort study. Arthritis Res Ther. (2016) 18:261. doi: 10.1186/s13075-016-1148-1
190. Rochette L, Dogon G, Zeller M, Cottin Y, Vergely C. GDF15 and cardiac cells: current concepts and new insights. Int J Mol Sci. (2021) 22:8889. doi: 10.3390/ijms22168889
191. Adela R, Banerjee SK. GDF-15 as a target and biomarker for diabetes and cardiovascular diseases: a translational prospective. J Diabetes Res. (2015) 2015:490842. doi: 10.1155/2015/490842
192. Wischhusen J, Melero I, Fridman WH. Growth/differentiation factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint. Front Immunol. (2020) 11:951. doi: 10.3389/fimmu.2020.00951
193. Dominguez-Rodriguez A, Abreu-Gonzalez P, Hernandez-Baldomero IF, Avanzas P, Bosa-Ojeda F. Change in growth differentiation factor 15, but not C-reactive protein, independently predicts major cardiac events in patients with non-ST elevation acute coronary syndrome. Mediators Inflamm. (2014) 2014:929536. doi: 10.1155/2014/929536
194. Gohar A, Goncalves I, Vrijenhoek J, Haitjema S, Van Koeverden I, Nilsson J, et al. Circulating GDF-15 levels predict future secondary manifestations of cardiovascular disease explicitly in women but not men with atherosclerosis. Int J Cardiol. (2017) 241:430–6. doi: 10.1016/j.ijcard.2017.03.101
195. Nair V, Robinson-Cohen C, Smith MR, Bellovich KA, Bhat ZY, Bobadilla M, et al. Growth differentiation factor-15 and risk of CKD progression. J Am Soc Nephrol. (2017) 28:2233–40. doi: 10.1681/ASN.2016080919
196. Liu JX, Li YP, Liu BH, Zhao XJ, Zhang ZY, Wang JD, et al. Repeated measurement of growth-differentiation factor-15 in Chinese Han patients with post-myocardial infarction chronic heart failure. J Geriatr Cardiol. (2018) 15:618–27. doi: 10.11909/j.issn.1671-5411.2018.10.002
197. Mirna M, Rohm I, Jirak P, Wernly B, Baz L, Paar V, et al. Analysis of novel cardiovascular biomarkers in patients with pulmonary hypertension (PH). Heart Lung Circ. (2020) 29:337–44. doi: 10.1016/j.hlc.2019.03.004
198. Xu XY, Nie Y, Wang FF, Bai Y, Lv ZZ, Zhang YY, et al. Growth differentiation factor (GDF)-15 blocks norepinephrine-induced myocardial hypertrophy via a novel pathway involving inhibition of epidermal growth factor receptor transactivation. J Biol Chem. (2014) 289:10084–94. doi: 10.1074/jbc.M113.516278
199. Wesseling M, De Poel JHC, De Jager SCA. Growth differentiation factor 15 in adverse cardiac remodelling: from biomarker to causal player. ESC Heart Fail. (2020) 7:1488–501. doi: 10.1002/ehf2.12728
200. Mazagova M, Buikema H, Landheer SW, Vavrinec P, Buiten A, Henning RH, et al. Growth differentiation factor 15 impairs aortic contractile and relaxing function through altered caveolar signaling of the endothelium. Am J Physiol Heart Circ Physiol. (2013) 304:H709–718. doi: 10.1152/ajpheart.00543.2012
201. Heger J, Schiegnitz E, Von Waldthausen D, Anwar MM, Piper HM, Euler G. Growth differentiation factor 15 acts anti-apoptotic and pro-hypertrophic in adult cardiomyocytes. J Cell Physiol. (2010) 224:120–6. doi: 10.1002/jcp.22102
202. Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, et al. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. (2006) 98:342–50. doi: 10.1161/01.RES.0000202804.84885.d0
203. De Jager SC, Bermudez B, Bot I, Koenen RR, Bot M, Kavelaars A, et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med. (2011) 208:217–25. doi: 10.1084/jem.20100370
204. Lok SI, Winkens B, Goldschmeding R, Van Geffen AJ, Nous FM, Van Kuik J, et al. Circulating growth differentiation factor-15 correlates with myocardial fibrosis in patients with non-ischaemic dilated cardiomyopathy and decreases rapidly after left ventricular assist device support. Eur J Heart Fail. (2012) 14:1249–56. doi: 10.1093/eurjhf/hfs120
205. Wang FF, Chen BX, Yu HY, Mi L, Li ZJ, Gao W. Correlation between growth differentiation factor-15 and collagen metabolism indicators in patients with myocardial infarction and heart failure. J Geriatr Cardiol. (2016) 13:88–93. doi: 10.11909/j.issn.1671-5411.2016.01.002
206. Wang J, Wei L, Yang X, Zhong J. Roles of growth differentiation factor 15 in atherosclerosis and coronary artery disease. J Am Heart Assoc. (2019) 8:e012826. doi: 10.1161/JAHA.119.012826
207. Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:986–1000. doi: 10.1161/ATVBAHA.110.207449
208. Bozza PT, Bakker-Abreu I, Navarro-Xavier RA, Bandeira-Melo C. Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot Essent Fatty Acids. (2011) 85:205–13. doi: 10.1016/j.plefa.2011.04.020
209. Itatsu K, Sasaki M, Yamaguchi J, Ohira S, Ishikawa A, Ikeda H, et al. Cyclooxygenase-2 is involved in the up-regulation of matrix metalloproteinase-9 in cholangiocarcinoma induced by tumor necrosis factor-alpha. Am J Pathol. (2009) 174:829–41. doi: 10.2353/ajpath.2009.080012
210. Battula S, Hao S, Pedraza PL, Stier CT, Ferreri NR. Tumor necrosis factor-alpha induces renal cyclooxygenase-2 expression in response to hypercalcemia. Prostaglandins Other Lipid Mediat. (2012) 99:45–50. doi: 10.1016/j.prostaglandins.2012.07.001
211. Ren J, Karpinski E, Benishin CG. Prostaglandin E2 contracts vascular smooth muscle and inhibits potassium currents in vascular smooth muscle cells of rat tail artery. J Pharmacol Exp Ther. (1995) 275:710–9.
212. Ragolia L, Palaia T, Paric E, Maesaka JK. Prostaglandin D2 synthase inhibits the exaggerated growth phenotype of spontaneously hypertensive rat vascular smooth muscle cells. J Biol Chem. (2003) 278:22175–81. doi: 10.1074/jbc.M302769200
213. An S, Yang J, So SW, Zeng L, Goetzl EJ. Isoforms of the EP3 subtype of human prostaglandin E2 receptor transduce both intracellular calcium and cAMP signals. Biochemistry. (1994) 33:14496–502. doi: 10.1021/bi00252a016
214. Foudi N, Gomez I, Benyahia C, Longrois D, Norel X. Prostaglandin E2 receptor subtypes in human blood and vascular cells. Eur J Pharmacol. (2012) 695:1–6. doi: 10.1016/j.ejphar.2012.08.009
215. Hayashi K, Takahashi M, Kimura K, Nishida W, Saga H, Sobue K. Changes in the balance of phosphoinositide 3-kinase/protein kinase B (Akt) and the mitogen-activated protein kinases (ERK/p38MAPK) determine a phenotype of visceral and vascular smooth muscle cells. J Cell Biol. (1999) 145:727–40. doi: 10.1083/jcb.145.4.727
216. Yoshida T, Gan Q, Shang Y, Owens GK. Platelet-derived growth factor-BB represses smooth muscle cell marker genes via changes in binding of MKL factors and histone deacetylases to their promoters. Am J Physiol Cell Physiol. (2007) 292:C886–895. doi: 10.1152/ajpcell.00449.2006
217. Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, Fitzgerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. (2006) 103:14507–12. doi: 10.1073/pnas.0606586103
218. Lee HS, Yun SJ, Ha JM, Jin SY, Ha HK, Song SH, et al. Prostaglandin D2 stimulates phenotypic changes in vascular smooth muscle cells. Exp Mol Med. (2019) 51:1–10. doi: 10.1038/s12276-019-0299-y
219. Mcgettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. (2011) 8:e1001098. doi: 10.1371/journal.pmed.1001098
220. Friedrich EB, Tager AM, Liu E, Pettersson A, Owman C, Munn L, et al. Mechanisms of leukotriene B4–triggered monocyte adhesion. Arterioscler Thromb Vasc Biol. (2003) 23:1761–7. doi: 10.1161/01.ATV.0000092941.77774.3C
221. Yokomizo T, Nakamura M, Shimizu T. Leukotriene receptors as potential therapeutic targets. J Clin Invest. (2018) 128:2691–701. doi: 10.1172/JCI97946
222. Back M, Hansson GK. Leukotriene receptors in atherosclerosis. Ann Med. (2006) 38:493–502. doi: 10.1080/07853890600982737
223. Colazzo F, Gelosa P, Tremoli E, Sironi L, Castiglioni L. Role of the cysteinyl leukotrienes in the pathogenesis and progression of cardiovascular diseases. Mediators Inflamm. (2017) 2017:2432958. doi: 10.1155/2017/2432958
224. Spanbroek R, Grabner R, Lotzer K, Hildner M, Urbach A, Ruhling K, et al. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc Natl Acad Sci USA. (2003) 100:1238–43. doi: 10.1073/pnas.242716099
225. Dahlen SE, Bjork J, Hedqvist P, Arfors KE, Hammarstrom S, Lindgren JA, et al. Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: in vivo effects with relevance to the acute inflammatory response. Proc Natl Acad Sci USA. (1981) 78:3887–91. doi: 10.1073/pnas.78.6.3887
226. Michelassi F, Landa L, Hill RD, Lowenstein E, Watkins WD, Petkau AJ, et al. Leukotriene D4: a potent coronary artery vasoconstrictor associated with impaired ventricular contraction. Science. (1982) 217:841–3. doi: 10.1126/science.6808665
227. Allen S, Dashwood M, Morrison K, Yacoub M. Differential leukotriene constrictor responses in human atherosclerotic coronary arteries. Circulation. (1998) 97:2406–13. doi: 10.1161/01.CIR.97.24.2406
228. Subramanian BC, Majumdar R, Parent CA. The role of the LTB4-BLT1 axis in chemotactic gradient sensing and directed leukocyte migration. Semin Immunol. (2017) 33:16–29. doi: 10.1016/j.smim.2017.07.002
229. Datta YH, Romano M, Jacobson BC, Golan DE, Serhan CN, Ewenstein BM. Peptido-leukotrienes are potent agonists of von Willebrand factor secretion and P-selectin surface expression in human umbilical vein endothelial cells. Circulation. (1995) 92:3304–11. doi: 10.1161/01.CIR.92.11.3304
230. Pedersen KE, Bochner BS, Undem BJ. Cysteinyl leukotrienes induce P-selectin expression in human endothelial cells via a non-CysLT1 receptor-mediated mechanism. J Pharmacol Exp Ther. (1997) 281:655–62.
231. Kaetsu Y, Yamamoto Y, Sugihara S, Matsuura T, Igawa G, Matsubara K, et al. Role of cysteinyl leukotrienes in the proliferation and the migration of murine vascular smooth muscle cells in vivo and in vitro. Cardiovasc Res. (2007) 76:160–6. doi: 10.1016/j.cardiores.2007.05.018
232. Heller EA, Liu E, Tager AM, Sinha S, Roberts JD, Koehn SL, et al. Inhibition of atherogenesis in BLT1-deficient mice reveals a role for LTB4 and BLT1 in smooth muscle cell recruitment. Circulation. (2005) 112:578–86. doi: 10.1161/CIRCULATIONAHA.105.545616
233. Back M, Bu DX, Branstrom R, Sheikine Y, Yan ZQ, Hansson GK. Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci USA. (2005) 102:17501–6. doi: 10.1073/pnas.0505845102
234. Aiello RJ, Bourassa PA, Lindsey S, Weng W, Freeman A, Showell HJ. Leukotriene B4 receptor antagonism reduces monocytic foam cells in mice. Arterioscler Thromb Vasc Biol. (2002) 22:443–9. doi: 10.1161/hq0302.105593
235. Choi JH, Jeon HJ, Park JG, Sonn SK, Lee MR, Lee MN, et al. Anti-atherogenic effect of BHB-TZD having inhibitory activities on cyclooxygenase and 5-lipoxygenase in hyperlipidemic mice. Atherosclerosis. (2010) 212:146–52. doi: 10.1016/j.atherosclerosis.2010.05.003
236. Vidal C, Gomez-Hernandez A, Sanchez-Galan E, Gonzalez A, Ortega L, Gomez-Gerique JA, et al. Licofelone, a balanced inhibitor of cyclooxygenase and 5-lipoxygenase, reduces inflammation in a rabbit model of atherosclerosis. J Pharmacol Exp Ther. (2007) 320:108–16. doi: 10.1124/jpet.106.110361
237. Bevan S, Lorenz MW, Sitzer M, Markus HS. Genetic variation in the leukotriene pathway and carotid intima-media thickness: a 2-stage replication study. Stroke. (2009) 40:696–701. doi: 10.1161/STROKEAHA.108.525733
Keywords: atherosclerosis, remodeling, inflammatory mediator, chemokines, cytokines
Citation: Evans BR, Yerly A, van der Vorst EPC, Baumgartner I, Bernhard SM, Schindewolf M and Döring Y (2022) Inflammatory Mediators in Atherosclerotic Vascular Remodeling. Front. Cardiovasc. Med. 9:868934. doi: 10.3389/fcvm.2022.868934
Received: 03 February 2022; Accepted: 11 April 2022;
Published: 04 May 2022.
Edited by:
Paul H. A. Quax, Leiden University, NetherlandsReviewed by:
Teresa Padro, Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau, SpainCopyright © 2022 Evans, Yerly, van der Vorst, Baumgartner, Bernhard, Schindewolf and Döring. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yvonne Döring, eXZvbm5lLmRvZXJpbmdAaW5zZWwuY2g=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.