 Hongmei Yang
Hongmei Yang Yue Sun
Yue Sun Qingchao Li
Qingchao Li Fengyan Jin
Fengyan Jin Yun Dai
Yun Dai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 29 March 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.868788
This article is part of the Research Topic Epigenetic Regulation in Cardiovascular Diseases, Volume II View all 6 articles
Emerging research on epigenetics has resulted in many novel discoveries in atherosclerosis (AS), an inflammaging-associated disease characterized by chronic inflammation primarily driven by macrophages. The bulk of evidence has demonstrated the central role of epigenetic machinery in macrophage polarization to pro- (M1-like) or anti-inflammatory (M2-like) phenotype. An increasing number of epigenetic alterations and their modifiers involved in reprogramming macrophages by regulating DNA methylation or histone modifications (e.g., methylation, acetylation, and recently lactylation) have been identified. They may act to determine or skew the direction of macrophage polarization in AS lesions, thereby representing a promising target. Here we describe the current understanding of the epigenetic machinery involving macrophage polarization, to shed light on chronic inflammation-driving onset and progression of inflammaging-associated diseases, using AS as a prototypic example, and discuss the challenge for developing effective therapies targeting the epigenetic modifiers against these diseases, particularly highlighting a potential strategy based on epigenetically-governed repolarization from M1-like to M2-like phenotype.
Atherosclerosis (AS) is known as one of inflammaging-associated diseases (IAADs) that includes more than 100 different diseases involving almost all human organs (1). AS-related acute cardiovascular or cerebrovascular events (e.g., heart attack and ischemic stroke) currently represent the leading causes of death worldwide (2). The common feature of IAADs is sustained inflammation due to impaired transformation from inflammation into resolution, an event driven primarily by the disproportionate polarization of macrophages (3–5). For example, as the most prevalent type of immune cells in AS lesions, macrophages play an essential role in orchestrating the entire process of AS till plaque rupture (4), thus named AS-associated macrophages (ASAM). High plasticity enables macrophages to polarize toward pro- (M1-like) and anti-inflammatory (M2-like) phenotype in response to different environmental cues (6), which is controlled at the transcriptional level via epigenetic modifications (known as marks or codes) of DNA and histones regulated by various epigenetic modifiers (7), including writers that add epigenetic marks onto DNA or histones, erasers that remove epigenetic marks from DNA or histones, and readers that recognize epigenetic marks and facilitate gene transcription. Despite recent discoveries on the essential roles of epigenetics in inflammation and immunity, the functional epigenetic investigation in IAADs is just emerging (8). Here we present the current understanding of the epigenetic mechanisms regulating macrophage polarization and functions, and discuss a novel strategy targeting epigenetic machinery for macrophage repolarization in IAADs such as AS. To avoid the confusion about the terms of various macrophage phenotypes due to the diversity of nomenclature (9), the terms M1 and M2, unless otherwise specified, used in this article only refer to macrophages with pro-inflammatory and anti-inflammatory/pro-resolving properties, respectively.
A widely accepted concept is that M1-like ASAMs play a pro-AS role by initiating and accelerating inflammation; by contrast, M2-like ASAMs act against AS by stopping inflammation and promoting resolution. Moreover, M1-like ASAMs promote necrosis and thin the protective fibrous cap by secreting proteases (e.g., matrix metallopeptidases such as MMP2 and MMP9), leading to plaque rupture and acute thrombosis. M2-like ASAMs remove apoptotic or dead cells through efferocytosis, produce collagen to thicken the fibrous cap, and secrete growth factors to promote tissue repair, thereby facilitating plaque stabilization or even regression (10). Indeed, M1-like ASAMs are often found in the rupture-prone plaques, while M2-like ASAMs usually appear in more stable plaques and away from the lipid core of the lesions (11). AS patients display a heavily-imbalanced ratio of M1-like to M2-like phenotype, which correlates with disease severity (12). The concept that AS is an inflammatory disease has further been supported by the recent identification of various inflammatory (M1-like) phenotypes of ASAMs at single-cell level in murine and human AS plaques (13–15). However, current anti-AS therapies, including cholesterol-lowering agents (e.g., statins), angiotension-converting enzyme inhibitors, β-blockers, and aspirin, have little direct effect on macrophage polarization because they do not specifically target macrophages.
The principle behind the development of an effective anti-AS therapy is to reduce and stabilize AS lesions to prevent disease progression and fatal complications such as myocardial infarction (MI) and stroke. Over the past decades, tremendous efforts have been made in developing anti-inflammatory therapy to reduce inflammation (16), including blocking inflammatory cell recruitment (e.g., by antagonists of chemokine receptors or adhesion molecules) (17), stabilizing plaques (e.g., by inhibitors of MMPs), and neutralizing pro-inflammatory factors (e.g., by monoclonal antibodies against various cytokines or chemokines) (18). However, almost all of them have failed at preclinical or early clinical phases. Notably, a large randomized trial (named CANTOS) involving more than 10,000 patients with previous MI and high C-reactive protein levels has shown that the IL-1β monoclonal antibody canakinumab could reduce C-reactive protein levels and the incidence rate of recurrent cardiovascular events, without affecting the LDL cholesterol level, but no significant difference in all-cause mortality, probably due to increased risk of fatal infection and sepsis (19). Thus, although CANTOS has, for the first time, approved the “inflammatory hypothesis” of AS in a clinical setting, IL-1β seems not an ideal target (20). Of note, these approaches mainly neutralize various pro-inflammatory factors (e.g., cytokines, chemokines) secreted by inflammatory cells (e.g., macrophages) rather than block their production. Another important lesson learned from the previous failures is that an effective anti-inflammatory approach may rely on both inflammation inhibition and resolution promotion, which require not only the termination of inflammatory cell recruitment and activation (reducing M1-like macrophages) but also the removal of apoptotic or dead cells via efferocytosis (increasing M2-like macrophages) (21).
Plaque regression is clinically desirable, which may be achieved by redirecting ASAM polarization from M1-like to M2-like phenotype (10, 22). For example, increasing HDL levels by targeting miR-33 (an intronic microRNA located within the SREBF2 gene that encodes sterol regulatory element binding transcription factor 2) leads to the regression of established plaques in Ldlr−/− mice (23). Silencing of IRF5 (encoding interferon regulatory factor 5) reprograms ASAMs from M1-like to M2-like phenotype and improves post-MI healing by augmenting resolution (24). Whereas the deficiency of efferocytosis due to high levels of the “don’t eat me” signal CD47 is observed in advanced plaques, CD47-blocking antibodies significantly prevent disease progression by restoring the phagocytotic capability of M2-like ASAMs (25). Taken together, these findings strongly argue that the therapeutic strategy promoting phenotypic transition from M1-like to M2-like phenotype may effectively attenuate and even block non-resolving inflammation, delaying or halting AS progression or leading to plaque regression.
Traditionally, macrophages can polarize toward classically activated M1 [e.g., M(IFNγ), M(LPS), and M(LPS + IFNγ)] and alternatively activated M2 [e.g., M(IL-4)] phenotype upon different stimuli (9, 26). While recent application of single-cell RNA sequencing (scRNAseq) has revealed high heterogeneity of ASAMs in AS plaques (13–15), the theme of macrophage polarization seems not to have been changed. However, the classical model for macrophage polarization may be challenged by our genome-wide survey based on a published public available database (27). By comparing the gene expression profiling (GEP) between resting (M0) and M1 [M(LPS + IFNγ)] or M2 [M(IL-4)] macrophages, we have noted some interesting phenomena in gene expression reprogramming during macrophage polarization, including a) that a majority of changes in differentially expressed genes (DEGs, including up- and down-regulated ones) occur during M1 [M(LPS + IFNγ)] polarization (versus M0, particularly involving pro- and anti-inflammatory genes as well as the genes involved in inflammatory pathways), while most of these changes (>80%) are, however, reversed in M2 or M(IL-4) [versus M1 or M(LPS + IFNγ)]; in contrast, much less DEGs (only 16 genes) was observed during M2 or M(IL-4) polarization (versus M0); b) that the DEGs in M1 [M(LPS + IFNγ)] polarization but reversed in M2 [M(IL-4)] are functionally enriched for several key pathways (e.g., proteasome, NF-κB, JAK/STAT, and apoptosis for the up-regulated DEGs; lysosome, oxidative phosphorylation, and PPAR for the down-regulated DEGs); and c) interestingly, these pathways appear to be shared by differentiation of monocytes into macrophages.
Accordingly, we hypothesize an alternative model for macrophage polarization, in which in addition to the classical models (i.e., polarization of M0 to either M1 or M2 upon different stimuli), M1 may be skewed directly to M2 simply by turning off the M1 program, a process termed repolarization, similar to inter-phenotypic transition (or trans-differentiation) in the epigenetic landscape originally described by Waddington and later updated by many others (28). According to this model, non-resolving inflammation in AS lesions might be caused by deficient repolarization of ASAMs (4). Therefore, targeting the machinery that governs the repolarization from M1-like to M2-like phenotype provides a rationale for developing an effective “double-hit” anti-inflammatory therapy, which simultaneously inhibits inflammation (reducing M1-like phenotype) and promotes resolution (increasing M2-like phenotype). In this context, the mechanism driving macrophage repolarization may be associated with histone lactylation, a novel form of epigenetic modification, which occurs in the late stage of M1 [M(LPS + IFNγ)] and is related to M2 gene expression (29). Interestingly, histone lactylation in macrophages is induced primarily by lactate, an “end-product” of glycolysis, which accumulates during M1 polarization due to a metabolic paradigm shift from oxidative phosphorylation to glycolysis (30).
Epigenetics is defined as the coding of gene expression in a highly tissue/cell- and context-specific manner via modifications of DNA and histones without altering the DNA sequence itself (8). The transition of cell phenotypes and maintenance of cell identity are controlled by the epigenetic machinery through reprogramming gene expression at the transcriptional level (31). To date, epigenetics has been studied in the context of chromatin modifications in either DNA or histones, and recently in higher-order chromatin structures involving large epigenomic domains named lamina-associated domains (LADs) and large, organized chromatin lysine modifications (LOCKs) (32). Epigenetic modifications, often called codes or marks, include DNA methylation of the nucleotide cytosine (e.g., 5mC and 5hmC, 5fC, 5caC, 3mC, and 6mA) at CpG sites and histone post-translational modifications [PTMs e.g., methylation, acetylation, phosphorylation, ubiquitylation, sumoylation, butyrylation, formylation, propionylation, citrullination, crotonylation, proline isomerization, ADP ribosylation, succinylation, 2-hydroxy isobutylylation, and more recently lactylation (29)] mostly at lysine residues (33). Other epigenetic mechanisms include various RNA modifications (e.g., m6A, m5C, m1A, 2’-O-Me, and Ψ) and non-coding RNAs (e.g., lncRNA, microRNA, and circRNA). DNA methylation is regulated by DNA methyltransferases (e.g., DNMT1, DNMT2, DNMT3A, and DNMT3B) and ten-eleven translocation methylcytosine dioxygenases (e.g., TET1, TET2, and TET3). The most common histone PTMs are lysine methylation and acetylation, which are reciprocally regulated by two classes of histone-modifying enzymes (i.e., “writer” and “eraser”). The writer that adds epigenetic codes to specific residues of histones includes lysine methyltransferase (KMT) and histone acetyltransferase (HAT). The eraser that removes these codes includes lysine demethylase (KDM) and histone deacetylase (HDAC). Histone PTMs result in a “loose” (open) or “tight” (closed) chromatin configuration, called chromatin remodeling, which control the accessibility of transcriptional factors to the promoter or enhancer regions of target genes on DNA, thereby triggering or silencing their expression.
Another category of epigenetic molecules called “reader” that recognizes epigenetic codes and recruit transcription-regulatory factors to target genes includes two families i.e., bromodomain and extraterminal protein (BET) and malignant brain tumor domain protein (MBT) (31). BETs read histone acetylation codes through their distinct bromodomain (BRD) and then recruit positive transcription elongation factor b (P-TEFb, a complex of CDK9 and cyclin T), which in turn phosphorylates the C-terminal domain (CTD) of RNA polymerase II to trigger transcription initiation and elongation (34). MBTs (e.g., MBT, chromodomain, tudor domain) recognize histone methylation codes, but their role in macrophages remains unknown. Taking the advantage of recent advances in the development of specific BRD inhibitors in cancer treatment, these agents may also emerge as a potential therapy for IAADs such as AS.
An additional category of epigenetic molecules called chromatin remodeler (or nucleosome remodeling factor, NURF) includes at least four subfamilies: switch/sucrose non-fermenting (SWI/SNF), imitation switch (ISWI), inositol requiring 80-like (INO80-like) and chromodomain helicase DNA binding (CHD) (35). The remodelers are recruited to their target regions by transcription factors (TFs) or non-coding RNAs and forms the ATP-dependent chromatin remodeling complexes, which facilitate transcription precisely and accurately in time and space, via multiple mechanisms including nucleosome-positioning or nucleosome sliding, creation of a remodeled state for DNA to be more accessible with histones still bound, altering histone–DNA interactions, disassembly of nucleosomes, exchange of histones with variants of different properties, and regulation of higher-order chromatin structures. Although the role of remodelers in macrophages remains unknown, the characterization of their structures may pave an entirely new avenue for drug development to treat various diseases (36, 37).
With further understanding their functions, all epigenetic-regulatory proteins have been re-categorized into three classes, including epigenetic modifiers that directly control DNA methylation (e.g., DNMTs and TETs), histone PTMs (e.g., KMTs and KDMs for methylation and HATs and HDACs for acetylation), or higher-order chromatin structures; epigenetic mediators that serve as the downstream targets of epigenetic modifiers and in turn govern cell plasticity and phenotypes via reprogramming; and epigenetic modulators that regulate the activity or subcellular localization of epigenetic modifiers and bridge (micro)environment with epigenomics (25).
Fast-accumulating evidence indicates the correlation between epigenetic alterations and the risk of AS, including DNA methylation and its regulatory enzymes (e.g., DNMTs and TETs), as well as histone PTMs and their writers and erasers. To date, at least 15 types of histone PTMs and more than 130 sites have been identified, which regulate gene expression via chromatin remodeling (33). Functionally, histone PTMs (particularly methylation and acetylation, mostly involving histones H3 and H4) can be divided into activating and inhibitory types, which either allow or prevent the transcription of target genes during cell phenotype transition like macrophage polarization. In general, histone lysine (K) acetylation often activates gene expression via increasing the accessibility of TFs, which is reciprocally regulated by specific HATs and HDACs. However, histone lysine methylation can be either activating or inhibitory in regulating the transcription of target genes, dependently on which lysine residue(s) is methylated with how many methyl groups, which is reciprocally regulated by KMTs and KDMs. Among numerous epigenetic modifiers identified to regulate histone PTMs, only a small number of them have been identified in the regulation of macrophage polarization and functions thus far.
DNA methylation involves the transfer of a methyl group to the C5 position of the cytosine to form 5mC, an event mediated by DNMTs, which primarily silence the expression of target genes by preventing the binding of TFs to DNA. DNA demethylation is mediated by TETs, which catalyzes oxidation of 5mC to 5hmC, 5fC, and then 5caC, thereby removing this inhibitory epigenetic mark to allow gene transcription. In AS, DNA methylation is specifically associated with disease type and progression, as well as disease onset, vascular events, and plaque stabilization. For example, there is a unique whole-genome landscape of DNA methylation in AS lesions, compared to surrounding normal vessel tissue (38); a disease- or location-specific methylation pattern is associated with MI, rather than ischemic stroke (39); disease progression-specific CpG methylation profiles correlate with the grade of lesions (40); after cerebrovascular events, global demethylation is associated with up-regulation of anti-inflammatory genes and likely contributes to plaque stabilization (41). Moreover, DNA methylation of specific target genes (e.g., TRAF3 that encodes TNF receptor-associated factor 3, PPM1A that encodes protein phosphatase 1A, among many others) is also closely associated with AS or its treatment (42). With rapid advances in the technology of single-cell analysis such as scRNAseq and mass cytometry by the time-of-flight (CyToF) (43, 44), cell type-specific profiles of either global or gene-specific DNA methylomes (e.g., macrophage, endothelial cell/EC, smooth muscle cells/SMC, lymphocyte, etc.) may soon be available for more precisely monitoring the dynamic changes of DNA methylation as well as understanding their clinical significance in AS.
To date, understanding of the functional roles of the epigenetic modifiers involving DNA methylation is relatively limited in AS. Functionally, DNMT3A and DNMT3B catalyze de novo DNA methylation. DNMT3A and TET2 represent the most commonly mutated genes in patients with coronary heart disease carrying clonal hematopoiesis of indeterminate potential (CHIP) (45). Transplantation with bone marrow of Dnmt3a−/− mice significantly increases plaque size of Ldlr−/− mice (46), suggesting the anti-AS role of DNMT3A. However, Dnmt3a expression is suppressed in M2 [M(IL-4)] macrophages, probably via an lncRNA called DNMT3aos located on the antisense strand of Dnmt3a (47). Interestingly, the effect of TET2 resembles that for DNMT3A, although these two epigenetic enzymes have opposite functions in regulating DNA methylation. Partial reconstitution of bone marrow clonal hematopoiesis by transplanting Tet2-mutant cells increases plaque size in Ldlr−/− mice, in association with increased IL-1β production by Tet2-mutant ASAMs via NLRP3 inflammasome (48). TET2 specifically represses IL-6 expression in macrophages, an event for inflammation resolution (48, 49). DNMT1 is a DNA methyltransferase maintaining DNA methylation. Macrophage-specific expression of Dnmt1 promotes AS in Apoe−/− mice, via increasing M1 cytokine production but suppressing the M2 gene expression, in association with promoter methylation and thus down-regulation of PPAR-γ or KLF4 (Kruppel-like factor 4) (50, 51). Following the paradigm shift from profiling epigenomics to functional epigenetics in the epigenetics field (52), the functions of these or other epigenetic enzymes involving DNA methylation will be defined more precisely and thus become a potential targets for anti-inflammatory therapy.
H3K4me3 is one of activating histone PTMs, which is reciprocally regulated by MLL (KMT2A) and KDM5B, respectively. Emerging evidence supports the involvement of H3K4me3 in the regulation of macrophages involving AS. H3K4 methylation stepwisely increases in macrophages during AS progression, in association with disease severity (53). Training monocytes by oxLDL, rather than native LDL, results in up-regulation of multiple pro-inflammatory and pro-AS genes, in association with H3K4me3 on their promoters (Figure 1A), while this event can be abolished via H3K4me3 inhibition by pan-KMT inhibitors (54). MLL is up-regulated in M1 [e.g., M(IFNγ)] macrophages, in association with increased H3K4me3 (Figure 1A); in contrast, its enzyme activity is reduced in M2 [M(IL-4 + IL13)] macrophages (55). Increased H3K4me3 up-regulates the pro-inflammatory genes, which can be prevented by an inhibitor of the MLL-Menin interaction (MI2-2). Consistently, pan-KMT inhibition (e.g., by 5’-methylthioadenosine/MTA) also inhibits LPS-induced expression of M1 genes and LPS + IFNγ-triggered secretions of pro-inflammatory cytokines (56).
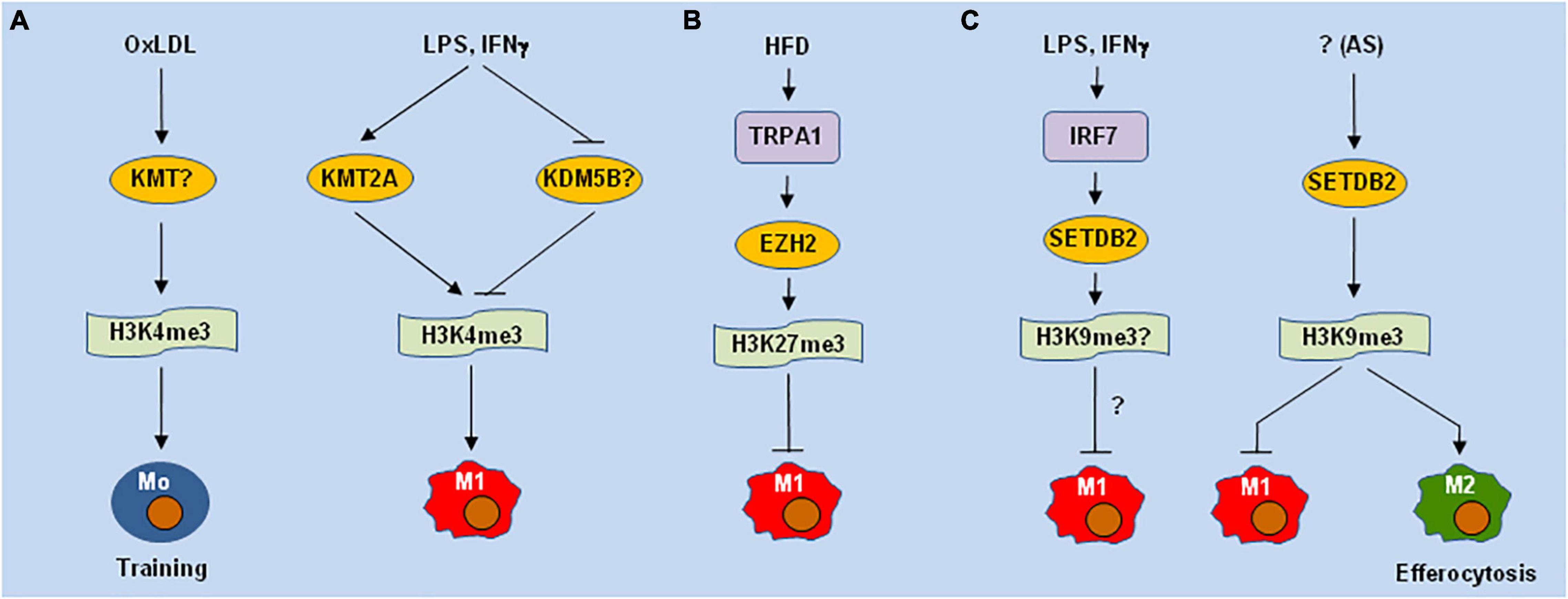
Figure 1. Different lysine methyltransferases may play opposite roles of in macrophage polarization and functions. (A) H3K4me3 mediated by lysine methyltransferases (KMTs e.g., KMT2A/MLL) is involved in oxLDL-induced training immunity of monocytes (Mo) in atherosclerosis (AS) and LPS-induced M1 polarization. (B) High-fat diet (HFD) induces the expression of TRPA1 (a calcium permeable non-selective cation channel), which stabilizes EZH2 (PRC2 or KMT6) to inhibit M1 polarization via H3K27me3. (C) LPS and IFNs up-regulates SETDB2 via IRF7, while deletion of SETDB2 in hematopoietic cells promotes M1-like polarization and impairs efferocytosis, a function of M2 macrophages, via H3K9me3 in AS lesions. oxLDL, oxidized low-density lipoprotein; LPS, lipopolysaccharide; TRPA1, transient receptor potential ankyrin 1; EZH2, enhancer of zeste homolog 2; IFN, interferon; IRF7, interferon regulatory factor 7; SETDB2, SET domain bifurcated histone lysine methyltransferase 2.
H3K9me3 and H3K27me3 represent two inhibitory PTMs. Both of these PTMs are markedly increased in ASAMs and lymphocytes in AS plaques, but they are undetectable in healthy vessel tissues (57). However, these PTMs markedly decrease during disease progression (53), while they do not correlate with the expression of their KMTs, including PRC2 (EZH2 or KMT6, which inhibits M1-like polarization in AS lesions (58)) (Figure 1B), the H3K27me3 methyltransferase G9a (KMT1C), and the H3K9me3 methyltransferases SETDB1 (KMT1E) or SUV39H1/2 (KMT1A/B). SETDB2, a member of the KMT1 family that methylates H3K9, is up-regulated in M1 [M(LPS)] but not M2 [M(IL-4)] macrophages (59). While SETDB2 is highly expressed in murine AS lesions, its genetic deletion in hematopoietic cells promotes inflammation and accelerates AS in Ldlr−/− mice, in association with enhanced expression of pro-inflammatory genes but attenuated efferocytosis in CD45+ ASAMs (Figure 1C).
Jumonji domain-containing 3 (JMJD3) (KDM6B), which specifically demethylates H3K27me3, is involved in the reprogramming of M1 [M(LPS)] polarization (60) (Figure 2A). More than 70% of LPS-inducible genes are JMJD3 targets in macrophages. However, although Jmjd3 deletion increases H3K27me3, but does not markedly affect most of those LPS-induced genes, suggesting that JMJD3 only acts to fine-tune the transcriptional program for inflammatory gene expression in LPS-activated macrophages and this action is independent of its H3K27me3 demethylase function. Although the enzymatic activity of JMJD3 seems unable to determine the direction of macrophage polarization by itself, it works together with the H3K9me2/H3K27me2/H4K20me1-specific demethylase KIAA1718 and certain transcription elongation-regulated proteins to demethylate H3K27me3 of pro-inflammatory genes. In contrast, down-regulation of either JMJD3 or KIAA1718 attenuates mRNA elongation of these genes (61). In this case, JMJD3 may require collaboration with an additional epigenetic modifier (e.g., KIAA1718) to act as an H3K27me3 demethylase in M1 reprogramming, at least in response to LPS. Serum amyloid A (SAA), another pro-inflammatory factor involved in AS, induces JMJD3 expression, in association with reduced H3K27me3 in macrophages (62). Unlike LPS-induced genes, SAA-induced expression of pro-inflammatory genes can, however, be blocked by Jmjd3 knockdown or inactivation, via restoration of H3K27me3, suggesting that the JMJD3 functions may vary in a stimulus-specific manner. Considering JMJD3 and H3K27me3 as a potential target in inflammation (63), the selective inhibitors of H3K27-specific demethylases have been developed as a novel anti-inflammatory therapy. For example, GSK-J1 (an inhibitor of JMJD3 and UTX, both belonging to the KDM6 subfamily), which specifically binds to the catalytic pocket of JMJD3 to inhibit its demethylase activity, sharply inhibits pro-inflammatory gene expression in macrophages, via increasing H3K27me3 (64).
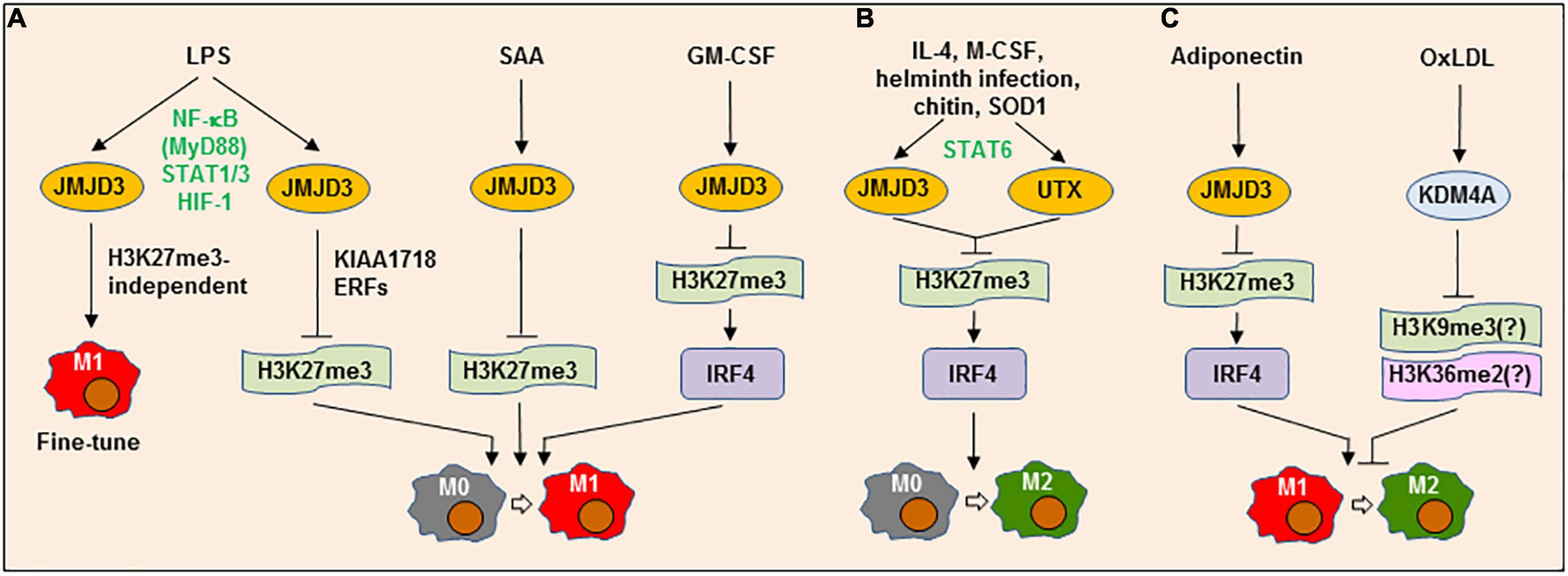
Figure 2. Lysine demethylases can play dual roles in both M1 and M2 polarization or M1→M2 repolarization. (A) LPS up-regulates the lysine demethylase (KDM) jumonji domain-containing (JMJD3) (KDM6B) via a process involving multiple transcription factors (e.g., NF-κB/MyD88, STAT1/3, and HIF-1), which in turn acts to either fine-tune M1 phenotype via an H3K27me3-independent process or promote M1 polarization, which requires another KDM KIAA1718 and elongation-regulatory factors (ERFs). Similarly, JMJD3 is involved in M1-like polarization induced by serum amyloid A (SAA) or granulocyte-macrophage colony-stimulating factor (GM-CSF) by up-regulating IRF4 via H3K27me3. (B) Alternatively, multiple factors (e.g., IL-4, M-CSF, helminth infection, chitin, and SOD1) can induce JMJD3 and UTX (KDM6A) expression via STAT6, which in turn mediate M2 polarization by up-regulating IRF4 via H3K27me3. (C) JMJD3 may promote adiponectin-triggered repolarization from M1-like to M2-like phenotype, likely via the similar mechanism involving H3K27me3-depdendent IRF4 expression. On the contrary, oxLDL induces the expression of KDM4A (JMJD2A), another member of the JMJD demethylase family, which blocks the repolarization from M1-like to M2-like phenotype, an event that can be restored by KDM4A deficiency or inhibition. LPS, lipopolysaccharide; JMJD3, Jumonji domain-containing protein 3; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; STAT, signal transducer and activator of transcription; HIF-1, hypoxia-inducible factor 1; IRF4, interferon regulatory factor 4; IL-4, interleukin-4; M-CSF, macrophage colony-stimulating factor; SOD1, superoxide dismutase 1; oxLDL, oxidized low-density lipoprotein; KDM4A, lysine demethylase 4A.
On the other hand, IL-4 also induces JMJD3 expression and thus activates M2 genes by reducing H3K27me2/3 at their promoters (65), suggesting a role of JMJD3 in M2 [e.g., M(IL-4)] polarization (Figure 2B). JMJD3 also mediates M2-like polarization in response to M-CSF, but is not involved in M1-like polarization induced by GM-CSF. In this context, helminth infection or chitin fails to induce M2-like polarization in Jmjd3−/− mice (66). Although Jmjd3 deficiency results in a global increase in H3K27me3 at the promoters of numerous genes, only a small number of them are specifically affected by Jmjd3 deletion. In the latter, IRF4 (interferon regulatory factor 4) represents one downstream target responsible for M2-like polarization, expression of which is associated with the demethylase activity of JMJD3. While Irf4 knockout copies the phenotype of Jmjd3 deletion, restoration of IRF4 expression can rescue the M2-like polarization that is impaired in Jmjd3−/− macrophages. However, GM-CSF also induces IRF4 expression via enhancing JMJD3 demethylase activity, leading to the production of pro-inflammatory CCL17 in a murine model of arthritis, an event blocked by GSK-J1 (67). In addition, the JMJD3-IRF4 axis may also contribute to repolarization of M1-like to M2-like phenotype mediated by adiponectin (68) (Figure 2C). Therefore, JMJD3 and its downstream targets such as IRF4 play dual functions context-specifically in both M1 and M2 polarization.
JMJD3 expression is regulated at transcriptional level by a number of TFs. LPS induces JMJD3 expression via a MyD88-dependent activation of the NF-κB pathway in ECs (69) or the activation of STAT1 and STAT3. For the latter, knockdown of both Stat1 and Stat3, rather than either of them, inhibits the expression of JMJD3 and pro-inflammatory genes in microglia (70). As one of the HIF-1-dependent genes (71), JMJD3 expression also involves the HIF pathway in response to LPS (72). Whereas IL-4 induces JMJD3 expression via STAT6 activation in macrophages, Stat6 knockout only prevents JMJD3 expression induced by IL-4, but not LPS (65). Moreover, Stat6 deficiency prevents H3K27me3 demethylation and M2 gene expression in response to IL-4, while has no effect on the pro-inflammatory effect of LPS. Similar phenomenon has also been found in SOD1-induced M2-like polarization (73). Thus, although the roles of JMJD3 and its downstream targets (e.g., IRF4) may not be specific to M1 or M2 polarization, the upstream signals (e.g., diverse TFs) of JMJD3 seem to determine the direction of JMJD3-mediated macrophage polarization (74). Of note, myeloid Jmjd3 deficiency leads to progression of AS lesions in Ldlr−/− mice, suggesting an anti-AS role of JMJD3 in ASAMs (75). However, it remains unclear whether JMJD3 acts to promote the polarization of ASAMs toward an anti-inflammatory (M2-like) phenotype in this setting.
KDM4A [JMJD2A or jumonji C-domain-containing histone demethylase 3A (JHDM3A)] demethylates H3K9me3, H3K36me2, and H1.4K26me3. We have identified KDM4A as another member of the JMJD demethylase family that regulates M1-like polarization, at least in response to oxidized low-density lipoprotein (oxLDL) (76) (Figure 2C). Exposure to oxLDL results in KDM4A up-regulation, accompanied by the expression of multiple M1 genes. While KDM4A knockdown prevents oxLDL-induced M1-like polarization, it instead promotes the expression of M2 genes. As an oxygen sensor, hypoxia increases the protein level of KDM4A, but not its mRNA level (77), by preventing its degradation via the SCF-containing ubiquitin ligase complex (78). In contrast, other JMJD family members (e.g., JMJD3) are primarily regulated at the transcriptional level via HIF-1α activation (79). However, although oxLDL also activates the NF-κB and HIF pathways (80), KDM4A expression seems to be independent of the NF-κB and HIF-1 pathways in macrophages exposed to oxLDL (76). Therefore, KDM4A inhibition may skew ASAM polarization straight from M1-like to M2-like phenotype (i.e., repolarization), thus serving as a potential target for the development of anti-inflammatory therapy against AS.
Traditionally, the epigenetic modifications were considered to act only temporarily due to their reversible features and thus not to contribute to immune memory. However, this concept has been challenged by recent discoveries that immune cells (e.g., macrophages) can memorize the cellular states and perturbations (e.g., environmental stimuli) without changing the DNA sequence. Such an epigenetic memory is able to burst a rapid and robust inflammatory response once cells encounter the same or even different stimuli (8). For example, Western diets can cause NLRP3-dependent immune memory in monocytes/macrophages, which contributes to chronic and stepwisely worsening inflammation in AS plaques (81). Later, such phenomenon has been attributed to trained immunity (e.g., by oxLDL) via histone epigenetic modifications (e.g., H3K4me1 and H3K4me3), which could be prevented by the inhibitors of HMTs (54, 82, 83). Of note, trained immunity driven by epigenetic reprogramming is closely associated with metabolic rewiring from oxidative phosphorylation to glycolysis (84–86), a well-documented hallmark of M1 macrophages. However, which HMT(s) or KDM(s) are responsible for trained immunity remains to be defined.
Histone acetylations are generally activating PTMs that promote the expression of target genes. To date, only a few HATs have been identified to be involved in macrophage polarization. EP300 (KAT3B) binds to c-Myc via protein arginine methyltransferase 1 (PRMT1) and is recruited to the promoters of target genes, resulting in M2 gene expression (87). Another HAT CBP (KAT3A) mediates transcriptional activation of IFNβ via increasing H3K56-Ac (88). However, a novel HAT named MOF (KAT8) promotes TNF-α/NF-κB-mediated expression of pro-inflammatory genes via increasing H4K16-Ac (89).
HDAC3, a class I HDAC, deacetylates H4K9-Ac and H4K14-Ac, which is involved in the regulation of both M1 and M2 polarization. In HDAC3-deficient macrophages, LPS fails to activate nearly half of the inflammatory genes, suggesting the role of HDAC3 in M1 polarization (90) (Figure 3A). While there are near 700 genomic regions are hyperacetylated at histone H4 in Hdac3−/− macrophages, the number of hyperacetylated regions are tripled after LPS stimulation. In contrast, a large number of regions display H4 hypoacetylated in both untreated or LPS-treated Hdac3−/− macrophages, in which the recognition motifs for the IRF family and STAT1 are mostly enriched. Moreover, while IRF3 (interferon regulatory factor 3) directly controls Ifnb1 transcription, the pro-inflammatory IFNβ-STAT1 axis is, however, impaired in Hdac3−/− macrophages exposed to LPS, in association with up-regulation of PTGS1 (encodes COX-1). During NLRP3 inflammasome activation, HDAC3 translocates to mitochondria and thus restricts fatty acid oxidation (FAO) by deacetylating the non-histone protein HADHA (mitochondrial trifunctional protein subunit α) at K303, which reduces its FAO enzyme activity and promotes IL-1β production by shaping mitochondrial adaptation (91). Thus, HDAC3 seems to trigger M1 gene expression indirectly via diverse mechanisms.
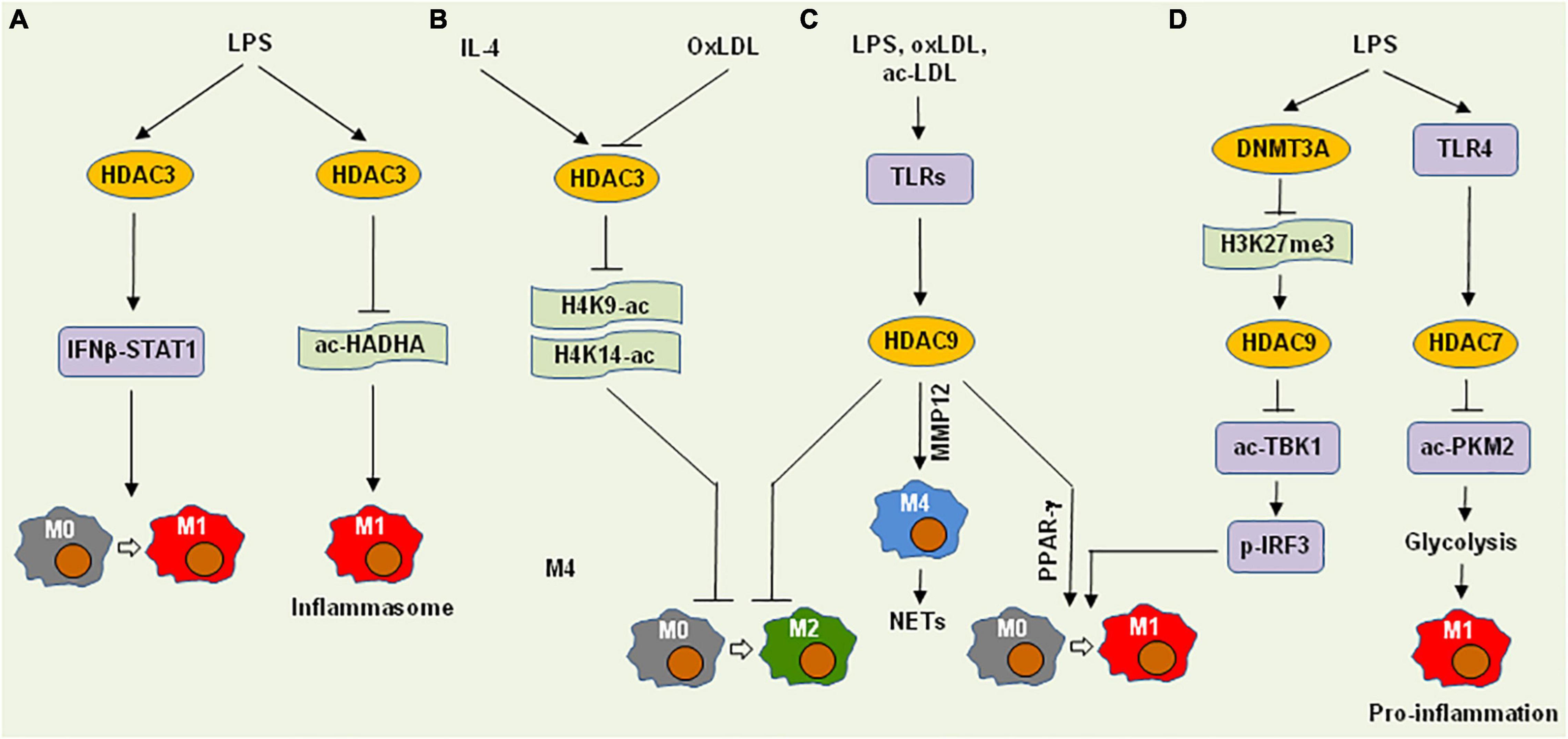
Figure 3. Histone deacetylases promote M1 polarization and pro-inflammatory functions of macrophages via deacetylation of histones or non-histone proteins. (A) HDAC3 mediates LPS-induced M1 polarization and inflammasome activation via the IFNβ-STAT1 signaling cascade and HADHA deacetylation that restricts fatty acid oxidation, respectively. (B) On the other hand, HDAC3 can block M2 polarization involving either IL-4 or oxLDL via deacetylation of H4K9-ac and H4K14-ac. (C) Multiple pro-inflammatory factors (e.g., LPS, oxLDL, and ac-LDL) induce the expression of HDAC9 via toll-like receptors (TLR), which in turn inhibits M2 polarization, promotes M1 polarization via PPARγ, or induces M4 macrophages (in conjunction with MMP12) that recruit neutrophils to form neutrophil extracellular traps (NETs). (D) LPS induces HDAC9 expression via DNMT3A-associated repression of H3K27me3, thus triggering M1 polarization via deacetylation of TBK1 that phosphorylates IRF3. Alternatively, LPS up-regulates HDAC7 via TLR4, which deacetylates PKM2 to promote glycolysis, thereby promoting pro-inflammatory function of M1 macrophages. HDAC, histone deacetylase; IFNβ, interferon β; STAT1, signal transducer and activator of transcription 1; HADHA, hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha; oxLDL, oxidized low-density lipoprotein; ac-LDL, acetylated low-density lipoprotein; MMP12, matrix metallopeptidase 12; PPARγ, peroxisome proliferator-activated receptor gamma; DNMT3A, DNA methyltransferase 3A; TBK1, TANK-binding kinase 1; PKM2, pyruvate kinase muscle isozyme 2; ac-, acetylation; p-, phosphorylation.
HDAC3 is also involved in the regulation of M2 polarization (Figure 3B). In this case, Hdac3−/− macrophages display an M2-like phenotype analogous to IL-4-induced alternative activation, in association with increased H3K9-Ac and H3K14-Ac (92). Interestingly, although most up-regulated genes in Hdac3−/− macrophages can be induced by both IL-4 and LPS in wild-type macrophages, a large number of the down-regulated genes in Hdac3−/− macrophages are up-regulated only by LPS in wild-type macrophages. Thus, HDAC3 acts as a suppressor of M2 polarization via its deacetylase activity even in the absence of pro-inflammatory stimuli. After exposure to oxLDL, Hdac3−/− macrophages secrete more TGF-β, an M2 cytokine, than wild-type counterparts, in association with increased H3K9/14-Ac at the Tgfb locus. This suggests that HDAC3 directly binds to the regions near the Tgfb promoter and inhibit its expression. Taken together, HDAC3 activation may skew the direction of macrophage polarization from M1-like to M2-like phenotype.
HDAC3 expression is associated with plaque vulnerability in human AS (93), suggesting its pro-AS role. Moreover, conditional knockout of Hdac3 in macrophages transforms their phenotype to an anti-AS fibrotic phenotype, leading to increased collagen deposition and thus more stable plaques in Ldlr−/− mice. Interestingly, while HDAC3 overexpression in ECs promotes endothelial-to-mesenchymal transition (EndMT), HDAC3 inhibition reduces AS lesions in Ldlr−/− mice, further supporting the pro-AS role of HDAC3 (94). On the contrary, up-regulation of HDAC3 in ECs may also inhibit inflammation and AS (95). Nevertheless, whether the functions of HDAC3 described above in the regulation of macrophage polarization would be applied in AS remains uncertain.
During the differentiation of monocytes to macrophages, HDAC9 (a class IIa HDAC) are up-regulated (96), which consists of two isoforms with or without HDAC domain, respectively. The latter, named HDAC-related protein (HDRP) or MEF2-interacting transcription repressor protein (MITR), is a truncated form that lacks deacetylase activity, which functions to recruit other HDACs (e.g., HDAC1 or HDAC3). Although HDAC9 is highly expressed in macrophages, it can be further induced by LPS (via toll-like receptor, TLR), oxLDL, and acetylated LDL (Figure 3C). High levels of HDAC9 in macrophages is maintained by DNMT3A via H3K27me3 repression at it distal promoter region, while HDAC9 in turn binds to TBK1 to enhance its K241 deacetylation and kinase activity (97) (Figure 3D). In this context, Dnmt3a deficiency selectively impairs the expression of type I interferons (e.g., IFN-α and IFN-β) induced by LPS, due to inhibition of TBK1-mediated IRF3 phosphorylation. Therefore, HDAC9 could execute its pro-inflammatory functions via diverse mechanisms involving multiple upstream and downstream pathways. As relatively high basal levels of HDAC9 seems to be required for macrophages, it would be quite challenging to target only the inducible part of HDAC9 to inhibit inflammation triggered by (micro) environmental stimuli.
A genome-wide association study (GWAS) has unveiled a SNP within the HDAC9 gene, which is significantly associated with the risk of large vessel stroke (98). Another genome-wide association meta-analysis has identified two genetic loci (HDAC9 and RAP1GAP) associated with aortic calcification, an independent predictor for the risk of cardiovascular events (99). These observations in the large population-based cohorts provide a strong link between HDAC9 and AS. In this context, Hdac9 deficiency inhibits M1 gene expression via up-regulating PPAR-γ, while promotes M2-like polarization of macrophages and expression of the ATP-binding cassette transporter ABCA1 and ABCG1 via increasing H3K9-ac at their promoters in Ldlr−/− mice (96). In AS plaques, HDAC9 is associated with MMP12 expression in the regions clustered with inflammatory genes in ASAMs. The expression of both Mmp12 and Hdac9 is associated with a unique subtype of macrophages named M4 (100), an inflammatory phenotype that recruits neutrophils to form neutrophil extracellular traps (NETs) in response to oxLDL (101). In Apoe−/− mice, Hdac9 deficiency confers plaque stability via an alternative mechanism involving deacetylation of the non-histone protein IKKα and IKKβ, which leads to the activation of the NF-κB pathway in both macrophages and ECs to drive inflammatory response (102). Of note, a specific inhibitor of the class IIa HDACs (including HDAC9) limits this pro-inflammatory response and attenuates lesion formation. Thus, due to its dual roles in pro-inflammatory response and plaque vulnerability, HDAC9 is considered as a very promising target for the treatment of AS-related diseases.
HDAC7, another class IIa HDAC, is structurally similar to HDAC9. Unlike the latter, HDAC7, however, has minimal deacetylase activity, while often binds to HDAC3 to suppress gene expression. In pre-B cells, HDAC7 inhibits pro-inflammatory genes essential for macrophage functions, while HDAC7 is specifically down-regulated to release this brake during transdifferentiation of pre-B cells into macrophages (103). In differentiated macrophages, HDAC7 promotes the TLR4-induced expression of a subset of pro-inflammatory genes (Figure 3D), an event prevented by a selective inhibitor of the class IIa HDACs (104). In macrophages, HDAC7 binds to and deacetylates PKM2 (pyruvate kinase muscle isozyme 2) at K433, therefore increasing LPS-induced inflammatory responses via promoting glycolysis (105), suggesting a role of HDAC7 in immunometabolism (106). In this case, while the role of HDAC7 in M1 polarization remains uncertain, it appears to be required for maintaining the pro-inflammatory property of M1-like macrophages. Thus, targeting HDAC7 could suppress the pro-inflammatory functions of macrophages, but not reprogramming for M1 polarization. While the role of HDAC7 remains uncertain in AS, it has been demonstrated that HDAC7 plays an important role in the maintenance of vascular integrity by repressing MMP10 expression in ECs (107). The latter would be a major concern for targeting HDAC7 in AS.
The findings involving the effects of HDAC inhibition on inflammation remains controversial thus far. The pan-HDAC inhibitor SAHA (vorinostat) reduces immune cell infiltration and inflammation in AS plaques of hypercholesterolemic Apoe−/− mice (108), while another pan-HDAC inhibitor TSA increased macrophage infiltration in AS lesions, in association with increased histone H4 acetylation (109). In macrophages, TSA impairs the expression of most M1 markers and cytokines induced by LPS or IFNγ + LPS, but also inhibits the expression of M2 markers induced by IL-4 (56). In the latter case, TSA down-regulates both basal and IL-4-induced expression of Arg1 via an HDAC3-independent process (92). MS-275 (entinostat), a selective inhibitor of class I HDACs (e.g., HDAC1 and HDAC3), increases the basal level of the M2 marker Arg1, similar to the phenotype of Hdac3 deletion, while does not affect IL-4-induced Arg1 expression.
Numerous non-histone proteins also serve as substrates of deacetylation by HDACs (110), including various TFs (e.g., RelA/p65, p53, STAT1). For example, the activation of the NF-κB pathway represents a major signal required by M1 [M(IFNγ)] polarization and functions, while NF-κB inhibition can promote M2 [M(IL-4 + IL-13)] polarization (111). Inhibition of class I HDACs (e.g., HDAC3) lead to persistent activation of the NF-κB pathway via preventing deacetylation of RelA (p65) (112–114), which may induce the IKK-dependent expression of pro-inflammatory CXCL8 (IL-8) (115) or up-regulate CD47 to impair efferocytosis by M2-like macrophages (25, 116). Similarly, acetylation of STAT6 also suppresses M2 [M(IL-4)] polarization (117). Therefore, owe to such diversity of HDACs’ substrates, HDAC inhibition may have either anti- or pro-inflammatory activities, dependently upon which class or individual HDAC(s) are targeted more specifically as well as which substrate(s) are involved more preferentially (118).
Although multiple clinical trials have recently shown promising benefits of anti-inflammatory agents or immunotherapies in AS-related cardiovascular diseases, several major challenges for targeting inflammation in AS remains to be addressed to achieve successful clinical translation of novel targets as well as their targeted agents or therapies (119). Of note, a paradigm shift of AS from an “inflammatory” disease to an “epigenetic” disorder is emerging (120). Fast-increasing evidence supports that the epigenetic machinery plays a central role in the regulation of inflammation. It has been widely accepted that high phenotypic plasticity and functional diversity of macrophages stem from their flexibility in reprogramming gene expression at the transcriptional level, a process primarily orchestrated by the epigenetic machinery (121). Among numerous DNA (as well as RNA) or histone epigenetic modifications and their epigenetic modifiers (including writer, eraser, and reader), an increasing number of them have been demonstrated to be associated with macrophage polarization and functions in inflammation or AS. Theoretically, they and many more candidates could be considered a potential therapeutic target for the treatment of AS. However, only a few therapeutic agents targeting these epigenetic modifications or their modifiers have been investigated thus far in the animal models of AS. Therefore, it remains to be defined whether this approach would be effective and safe in the treatment of patients with AS-related diseases. But, a caution should be taken in the development of their inhibitors as anti-inflammatory therapeutics, due to their diverse functions in macrophages as well as their “off-target” effects (e.g., those involving non-histone proteins). Moreover, there are extensive cross-talks between different epigenetic mechanisms (e.g., DNA methylation and histone PTMs), epigenetic regulation and TFs, or epigenetic reprogramming and metabolic rewiring in the regulation of macrophage polarization and functions (31, 122–125), which make targeting the epigenetic machinery even more challenging. To deal with this challenge, it is necessary to better understand the epigenetic mechanisms underlying macrophage-mediated non-resolving inflammation that drives AS (126). To this end, in addition to the traditional models for M1 or M2 polarization from M0 (resting) macrophages, which have been widely used to develop anti-inflammatory therapy, our observations from the GEP analysis raise a potential alternative model, in which M1 macrophages might be directly skewed to M2 phenotypes (repolarization) via epigenetic reprogramming of gene expression or silencing at the transcriptional level. Of note, this model has been supported by accumulating evidence from recent studies. For example, it has been found that histone lactylation might mediate the repolarization from M1 to M2 via a transition from glycolysis to oxidative phosphorylation (29, 30). Inhibition of NO production leads to repolarization of M1 to M2 via restoration of mitochondrial function that is impaired in M1, thus inhibiting AS (127). However, it is worth mentioning that M2 macrophages may have certain pro-AS properties. For example, CD163+ (M2-like) macrophages could promote intraplaque angiogenesis, vascular permeability, and leukocyte infiltration, leading to AS progression (128, 129). Nevertheless, the model proposed here could be useful to discover the epigenetic modifiers specific for governing the repolarization of macrophage from pro- (M1-like) to anti-inflammatory (M2-like) phenotype as novel therapeutic targets, which would hopefully change the game in the development of effective and safe anti-inflammatory therapy for IAADs like AS.
YD and FJ conceptualized, wrote, edited, and revised the manuscript. YD and HY gathered and analyzed the literature, and prepared the figures. YS and QL contributed to literature search and collection. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Nos. 81471165, 81670190, 81671108, 81670189, and 81870160), the Natural Science Foundation of the Jilin Province (Nos. 20190201042JC and 20190201163JC), Science and Technology Development Program of the Jilin Province (No. 20210509010RQ), and Interdisciplinary Integration and Innovation Project of JLU (Jilin University).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Oishi Y, Manabe I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech Dis. (2016) 2:16018. doi: 10.1038/npjamd.2016.18
2. Institute for Health Metrics and Evaluation [IHME].Findings From the Global Burden of Disease Study 2017. Seattle, WA: IHME (2018).
3. Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, et al. Macrophage polarization in chronic inflammatory diseases: killers or builders? J Immunol Res. (2018) 2018:8917804. doi: 10.1155/2018/8917804
4. Bäck M, Yurdagul A Jr., Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
5. Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight. (2020) 5:e134796. doi: 10.1172/jci.insight.134796
6. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. (2014) 159:1327–40. doi: 10.1016/j.cell.2014.11.023
7. Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martín D, et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell. (2016) 167:1398–414. doi: 10.1016/j.cell.2016.10.026
8. Khyzha N, Alizada A, Wilson MD, Fish JE. Epigenetics of atherosclerosis: Emerging mechanisms and methods. Trends Mol Med. (2017) 23:332–47. doi: 10.1016/j.molmed.2017.02.004
9. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
10. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40:20–33. doi: 10.1161/atvbaha.119.312802
11. Stoger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EAL, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. (2012) 225:461–8. doi: 10.1016/j.atherosclerosis.2012.09.013
12. Fadini GP, Simoni F, Cappellari R, Vitturi N, Galasso S, de Kreutzenberg SV, et al. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis. (2014) 237:805–8. doi: 10.1016/j.atherosclerosis.2014.10.106
13. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
14. Da LJ, Nishi H, Poles J, Niu X, Mccauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. (2019) 4:e124574. doi: 10.1172/jci.insight.124574
15. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E-AD, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
16. Charo IF, Taub R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov. (2011) 10:365–76. doi: 10.1038/nrd3444
17. Schall TJ, Proudfoot AEI. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. (2011) 11:355–63. doi: 10.1038/nri2972
18. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. (2011) 473:317–25. doi: 10.1038/nature10146
19. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31.
20. Harrington RA. Targeting inflammation in coronary artery disease. N Engl J Med. (2017) 377:1–2. doi: 10.1155/2020/4641013
21. Puleston DJ, Pearce EL. Appetite for arginine: metabolic control of macrophage hunger. Cell Metab. (2020) 31:441–2. doi: 10.1016/j.cmet.2020.02.005
22. Sharma M, Schlegel MP, Afonso MS, Brown EJ, Karishma Rahman K, Weinstock A, et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res. (2020) 127:335–53. doi: 10.1161/CIRCRESAHA.119.316461
23. Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. (2011) 121:2921–31. doi: 10.1172/JCI57275
24. Courties G, Heidt T, Sebas M, Jeon D, Truelove J, Tricot B, et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. (2014) 63:1556–66. doi: 10.1016/j.jacc.2013.11.023
25. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller C, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. (2016) 536:86–90. doi: 10.1038/nature18935
27. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. (2006) 177:7303–11. doi: 10.4049/jimmunol.177.10.7303
28. Shakiba N, Chunhe Li C, Garcia-Ojalvo J, Cho K-H, Patil K, Walczak A, et al. How can Waddington-like landscapes facilitate insights beyond developmental biology? Cell Systems. (2022) 13:4–9. doi: 10.1016/j.cels.2021.12.003
29. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
30. Izzo LT, Wellen KE. Histone lactylation links metabolism and gene regulation. Nature. (2019) 574:492–3. doi: 10.1038/d41586-019-03122-1
31. Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, Sabò A, et al. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross talks. Nat Immunol. (2017) 18:530–40. doi: 10.1038/ni.3710
32. Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. (2018) 378:1323–34. doi: 10.1056/NEJMra1402513
33. Stillman B. Histone modifications: insights into their influence on gene expression. Cell. (2018) 175:6–9. doi: 10.1016/j.cell.2018.08.032
34. Dai Y, Jin F, Wu W, Kumar SK. Cell cycle regulation and hematologic malignancies. Blood Sci. (2019) 1:34–43. doi: 10.1097/bs9.0000000000000009
35. Giles KA, Gould CM, Du Q, Du Q, Skvortsova K, Song JZ, et al. Integrated epigenomic analysis stratifies chromatin remodellers into distinct functional groups. Epigenetics Chromatin. (2019) 12:12. doi: 10.1186/s13072-019-0258-9
36. He S, Wu Z, Tian Y, Yu Y, Yu J, Wang X, et al. Structure of nucleosome-bound human BAF complex. Science. (2020) 367:875–81. doi: 10.1126/science.aaz9761
37. Han Y, Reyes AA, Malik S, He Y. Cryo-EM structure of SWI/SNF complex bound to a nucleosome. Nature. (2020) 579:452–5. doi: 10.1038/s41586-020-2087-1
38. Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, et al. DNA methylation map of human atherosclerosis. Cir Cardiovas Genet. (2014) 7:692–700. doi: 10.1161/CIRCGENETICS.113.000441
39. Rask-Andersen M, Martinsson D, Ahsan M, Enroth S, Ek WE, Gyllensten U, et al. Epigenome-wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction. Hum Mol Genet. (2016) 25:4739–48. doi: 10.1093/hmg/ddw302
40. del Pilar Valencia-Morales M, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, et al. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genomics. (2015) 8:7. doi: 10.1186/s12920-015-0085-1
41. Zaina S, Gonçalves I, Carmona FJ, Gomez A, Heyn H, Mollet IG, et al. DNA methylation dynamics in human carotid plaques after cerebrovascular events. Arterioscler Thromb Vasc Biol. (2015) 35:1835–42. doi: 10.1161/ATVBAHA.115.305630
42. Gallego-Fabrega C, Carrera C, Reny JL, Fontana P, Slowik A, Pera J, et al. TRAF3 epigenetic regulation is associated with vascular recurrence in patients with ischemic stroke. Stroke. (2016) 47:1180–6. doi: 10.1161/STROKEAHA.115.012237
43. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh H, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Cir Res. (2018) 122:1675–88. doi: 10.1161/CIRCRESAHA.117.312513
44. Cole JE, Park I, Ahern DJ, Kassiteridi CD, Danso Abeam DD, Goddard ME, et al. Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc Res. (2018) 114:1360–71. doi: 10.1093/cvr/cvy109
45. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
46. Rauch PJ, Silver AJ, Gopakumar J, McConkey M, Sinha E, Fefer M, et al. Loss-of-function mutations in Dnmt3a and Tet2 lead to accelerated atherosclerosis and convergent macrophage phenotypes in mice. Blood. (2018) 132:745. doi: 10.1182/blood-2018-99-118288
47. Li X, Zhang Y, Pei W, Zhang M, Yang H, Zhong M, et al. LncRNA Dnmt3aos regulates Dnmt3a expression leading to aberrant DNA methylation in macrophage polarization. FASEB J. (2020) 34:5077–91. doi: 10.1096/fj.201902379R
48. Fuster JJ, MacLauchlan S. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. (2017) 355:842–7. doi: 10.1126/science.aag1381
49. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. (2015) 525:389–93. doi: 10.1038/nature15252
50. Yu J, Qiu Y, Yang J, Bian S, Chen G, Deng M, et al. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci Rep. (2016) 6:30053. doi: 10.1038/srep30053
51. Tang RZ, Zhu JJ, Yang FF, Zhang YP, Xie SA, Liu YF, et al. DNA methyltransferase 1 and Krüppel-like factor 4 axis regulates macrophage inflammation and atherosclerosis. J Mol Cell Cardiol. (2019) 128:11–24. doi: 10.1016/j.yjmcc.2019.01.009
52. Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. (2017) 18:51–66. doi: 10.1038/nrg.2016.138
53. Greissel A, Culmes M, Burgkart R, Zimmermann A, Eckstein H-H, Zernecke A, et al. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovas Pathol. (2016) 25:79–86. doi: 10.1016/j.carpath.2015.11.001
54. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea M, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
55. Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PloS One. (2013) 8:e78045. doi: 10.1371/journal.pone.0078045
56. Van den Bossche J, Neele AE, Hoeksema MA, de Heij F, Boshuizen MCS, van der Velden S, et al. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. (2014) 455:396–402. doi: 10.1016/j.bbrc.2014.11.029
57. Wierda RJ, Rietveld IM, van Eggermond MCJA, Belien JAM, van Zwet EW, Lindeman JHN, et al. Global histone H3 lysine 27 triple methylation levels are reduced invessels with advanced atherosclerotic plaques. Life Sci. (2015) 129:3–9. doi: 10.1016/j.lfs.2014.10.010
58. Wang Q, Chen K, Zhang F, Peng K, Wang Z, Yang D, et al. TRPA1 regulates macrophages phenotype plasticity and atherosclerosis progression. Atherosclerosis. (2020) 301:44–53. doi: 10.1016/j.atherosclerosis.2020.04.004
59. Zhang X, Sun J, Canfrán-Duque A, Aryal B, Tellides G, Chang YJ, et al. Deficiency of histone lysine methyltransferase SETDB2 in hematopoietic cells promotes vascular inflammation and accelerates atherosclerosis. JCI Insight. (2021) 6:e147984. doi: 10.1172/jci.insight.147984
60. De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. (2009) 28:3341–52. doi: 10.1038/emboj.2009.271
61. Chen S, Ma J, Wu F, Ma H, Xu W, Lv R, et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Gene Dev. (2012) 26:1364–75. doi: 10.1101/gad.186056.111
62. Yan Q, Sun L, Zhu Z, Wang L, Li S, Ye RD. Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene expression in serum amyloid A-stimulated macrophages. Cell Signal. (2014) 26:1783–91. doi: 10.1016/j.cellsig.2014.03.025
63. Burchfield JS, Li Q, Wang HY, Wang R-F. JMJD3 as an epigenetic regulator in development and disease. Biochem Cell Biol. (2015) 67:148–57. doi: 10.1016/j.biocel.2015.07.006
64. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. (2012) 488:404–8. doi: 10.1038/nature11262
65. Hsu AT, Lupancu TJ, Lee MC, Fleetwood AJ, Cook AD, Hamilton JA, et al. Epigenetic and transcriptional regulation of IL4-induced CCL17 production in human monocytes and murine macrophages. J Biol Chem. (2018) 293:11415–23. doi: 10.1074/jbc.RA118.002416
66. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. (2010) 11:936–44. doi: 10.1038/ni.1920
67. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew H-W, Chang MWN, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. (2016) 126:3453–66. doi: 10.1172/JCI87828
68. Xuan D, Han Q, Tu Q, Zhang L, Yu L, Murry D, et al. Epigenetic modulation in periodontitis: Interaction of adiponectin and JMJD3-IRF4 axis in macrophages. J Cell Physiol. (2016) 231:1090–6. doi: 10.1002/jcp.25201
69. Yu S, Chen X, Xiu M, He F, Xing J, Min D, et al. The regulation of Jmjd3 upon the expression of NF-κB downstream inflammatory genes in LPS activated vascular endothelial cell. Biochem Biophys Res Commun. (2017) 485:62–8. doi: 10.1016/j.bbrc.2017.02.020
70. Przanowski P, Dabrowski M, Ellert-Miklaszewska A, Kloss M, Mieczkowski J, Kaza B, et al. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J Mol Med. (2014) 92:239–54. doi: 10.1007/s00109-013-1090-5
71. Lee HY, Choi K, Oh H, Park YK, Park H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol Cells. (2014) 37:43–50. doi: 10.14348/molcells.2014.2250
72. Lin N, Simon MC. Hypoxia-inducible factors: key regulators of myeloid cells during inflammation. J Clin Invest. (2016) 126:3661. doi: 10.1172/JCI84426
73. He C, Larson-Casey JL, Gu L, Ryan AJ, Murthy S, Carter AB. Cu, Zn-superoxide dismutase-mediated redox regulation of Jumonji domain containing 3 modulates macrophage polarization and pulmonary fibrosis. Am J Respiratory Cell Mol Biol. (2016) 55:58–71. doi: 10.1165/rcmb.2015-0183OC
74. Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Histone demethylase Jumonji D3 (JMJD3/KDM6B) at the nexus of epigenetic regulation of inflammation and the aging process. J Mol Med. (2014) 92:1035–43. doi: 10.1007/s00109-014-1182-x
75. Neele AE, Gijbels MJJ, van der Velden S, Hoeksema MA, Boshuizen MCS, Prange KHM, et al. Myeloid Kdm6b deficiency results in advanced atherosclerosis. Atherosclerosis. (2018) 275:156–65. doi: 10.1016/j.atherosclerosis.2018.05.052
76. Wang X, Wang S, Yao G, Yu D, Chen K, Tong Q, et al. Identification of the histone lysine demethylase KDM4A/JMJD2A as a novel epigenetic target in M1 macrophage polarization induced by oxidized LDL. Oncotarget. (2017) 8: 114442–56. doi: 10.18632/oncotarget.17748
77. Black JC, Atabakhsh E, Kim J, Biette KM, Van Rechem C, Ladd B, et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. (2015) 29:1018–31. doi: 10.1101/gad.259796.115
78. Tan MK, Lim HJ, Harper JW. SCF(FBXO22) regulates histoneH3 lysine 9 and 36 methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol Cell Biol. (2011) 31:3687–99. doi: 10.1128/MCB.05746-11
79. Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. (2018) 27:281–98. doi: 10.1016/j.cmet.2017.10.005
80. Jin F, Wang X, Wu J, Dai Y. Targeting the NF-κB-dependent HIF-1β pathway reprograms macrophage polarization induced by oxidized LDL. Blood. (2017) 130:993. doi: 10.1182/blood.v130.suppl_1.993.993
81. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75. doi: 10.1016/j.cell.2017.12.013
82. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
83. Leentjens J, Bekkering S, Joosten LAB, Netea MG, Burgner DP, Riksen NP. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ Res. (2018) 122:664–9. doi: 10.1161/CIRCRESAHA.117.312465
84. Stienstra R, Netea-Maier RT, Riksen NP, Joosten LAB, Netea MG. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. (2017) 26:142–56. doi: 10.1016/j.cmet.2017.06.001
85. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
86. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135.–146. doi: 10.1016/j.cell.2017.11.025
87. Tikhanovich I, Zhao J, Bridges B, Kumer S, Roberts B, Weinman SA. Arginine methylation regulates c-Myc-dependent transcription by altering promoter recruitment of the acetyltransferase p300. J Biol Chem. (2017) 292:13333–44. doi: 10.1074/jbc.M117.797928
88. Liu Z, Yang L, Sun Y, Xie X, Huang J. ASF1a enhances antiviral immune response by associating with CBP to mediate acetylation of H3K56 at the Ifnb promoter. Mol Immunol. (2016) 78:57–64. doi: 10.1016/j.molimm.2016.08.008
89. denDekker AD, Davis FM, Joshi AD, Wolf SJ, Allen R, Lipinski J, et al. TNF-α regulates diabetic macrophage function through the histone acetyltransferase MOF. JCI Insight. (2020) 5:e132306. doi: 10.1172/jci.insight.132306
90. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci USA. (2012) 109:E2865–74. doi: 10.1073/pnas.1121131109
91. Chi Z, Chen S, Xu T, Zhen W, Yu W, Jiang D, et al. Histone deacetylase 3 couples mitochondria to drive IL-1β-dependent inflammation by configuring fatty acid oxidation. Mol Cell. (2020) 80:43.e–58.e. doi: 10.1016/j.molcel.2020.08.015
92. Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. (2011) 25:2480–8. doi: 10.1101/gad.175950.111
93. Hoeksema MA, Gijbels MJJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. (2014) 6:1124–32. doi: 10.15252/emmm.201404170
94. Chen L, Shang C, Wang B, Wang G, Jin Z, Yao F, et al. HDAC3 inhibitor suppresses endothelial-to-mesenchymal transition via modulating inflammatory response in atherosclerosis. Biochem Pharmacol. (2021) 192:114716. doi: 10.1016/j.bcp.2021.114716
95. Wang J, Xu X, Li P, Zhang B, Zhang J. HDAC3 protects against atherosclerosis through inhibition of inflammation via the microRNA-19b/PPARγ/NF-κB axis. Atherosclerosis. (2021) 323:1–12. doi: 10.1016/j.atherosclerosis.2021.02.013
96. Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol. (2014) 34:1871–9. doi: 10.1161/ATVBAHA.114.303393
97. Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat Immunol. (2016) 17:806–15. doi: 10.1038/ni.3464
98. Bellenguez C, Bevan S, Gschwendtner A, Spencer CCC, Burgess AI, Pirinen M, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. (2012) 44:328–33. doi: 10.1038/ng.1081
99. Malhotra R, Mauer AC, Cardenas CLL, Guo X, Yao J, Zhang X, et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. (2019) 51:1580–7. doi: 10.1038/s41588-019-0514-8
100. Oksala NKJ, Seppälä I, Rahikainen R, Mäkelä K-M, Raitoharju E, Illig T, et al. Synergistic expression of histone deacetylase 9 and matrix metalloproteinase 12 in M4 macrophages in advanced carotid plaques. Eur J Vasc Endovasc Surg. (2017) 53:632–40. doi: 10.1016/j.ejvs.2017.02.014
101. Maretti-Mira AC, Golden-Mason L, Salomon MP, Kaplan MJ, Rosen HR. Cholesterol-induced M4-like macrophages recruit neutrophils and induce NETosis. Front Immunol. (2021) 12:671073. doi: 10.3389/fimmu.2021.671073
102. Asare Y, Campbell-James TA, Bokov Y, Yu LL, Prestel M, Bounkari OE, et al. Histone deacetylase 9 activates IKK to regulate atherosclerotic plaque vulnerability. Circ Res. (2020) 127:811–23. doi: 10.1161/CIRCRESAHA.120.316743
103. Barneda-Zahonero B, Roma’n-Gonza’ lez L, Collazo O, Rafati H, Islam ABMMK, Bussmann LH, et al. HDAC7 is a repressor of myeloid genes whose downregulation is required for transdifferentiation of pre-B cells into macrophages. PLoS Genet. (2013) 9:e1003503. doi: 10.1371/journal.pgen.1003503
104. Shakespear MR, Hohenhaus DM, Kelly GM, Kamal NA, Gupta P, Labzin LI, et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J Biol Chem. (2013) 288:25362–74. doi: 10.1074/jbc.M113.496281
105. Das Gupta K, Shakespear MR, Curson JEB, Murthy AMV, Iyer A, Hodson MP, et al. Class IIa histone deacetylases drive Toll-like receptor-inducible glycolysis and macrophage inflammatory responses via pyruvate kinase M2. Cell Rep. (2020) 30:2712–28. doi: 10.1016/j.celrep.2020.02.007
106. Tabas I, Bornfeldt KE. Intracellular and intercellular aspects of macrophage immunometabolism in atherosclerosis. Circ Res. (2020) 126:1209–27. doi: 10.1161/CIRCRESAHA.119.315939
107. Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. (2006) 126:321–34. doi: 10.1016/j.cell.2006.05.040
108. Manea SA, Vlad ML, Fenyo IM, Lazar A-G, Raicu M, Muresian H, et al. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. (2020) 28:101338. doi: 10.1016/j.redox.2019.101338
109. Choi JH, Nam KH, Kim J, Baek MW, Park JE, Park HY, et al. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2005) 25:2404–9. doi: 10.1161/01.ATV.0000184758.07257.88
110. Dai Y, Bose P, Grant S. Chapter 16 – cell-cycle regulation and hematologic disorders. 9th ed. In: K Kaushansky, et al. editors. Williams Hematology. McGraw Hill, publisher. (2015).
111. Haydar D, Cory TJ, Birket SE, Murphy BS, Pennypacker KR, Sinai AP, et al. Azithromycin polarizes macrophages to an M2 phenotype via inhibition of the STAT1 and NF-κB signaling pathways. J Immunol. (2019) 203:1021–30. doi: 10.4049/jimmunol.1801228
112. Dai Y, Rahmani M, Grant S. An intact NF-κB pathway is required for histone deacetylase inhibitor-induced G1 arrest and maturation in U937 human myeloid leukemia cells. Cell Cycle. (2003) 2:467–72.
113. Dai Y, Rahmani M, Dent P, Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-κB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. (2005) 25:5429–44. doi: 10.1128/MCB.25.13.5429-5444.2005
114. Dai Y, Chen S, Wang L, Pei XY, Funk VL, Kramer LB, et al. Disruption of IκB kinase (IKK)-mediated RelA serine 536 phosphorylation sensitizes human multiple myeloma cells to histone deacetylase (HDAC) inhibitors. J Biol Chem. (2011) 286:34036–50. doi: 10.1074/jbc.M111.284216
115. Gatla HR, Zou Y, Uddin MM, Singha B, Bu P, Vancura A, et al. Histone deacetylase (HDAC) inhibition induces IkB kinase (IKK)-dependent interleukin-8/CXCL8 expression in ovarian cancer cells. J Biol Chem. (2017) 292:5043–54. doi: 10.1074/jbc.M116.771014
116. Flores AM, Hosseini-Nassab N, Jarr KU, Ye J, Zhu X, Wirka R, et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat Nanotechnol. (2020) 15:154–61. doi: 10.1038/s41565-019-0619-3
117. Yu T, Gan S, Zhu Q, Dai D, Li N, Wang H, et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat Commun. (2019) 10:4353. doi: 10.1038/s41467-019-12384-2
118. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
119. Engelen SE, Robinson AJB, Zurke Y-X, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol. (2022) 31:1–21. doi: 10.1038/s41569-021-00668-4
120. Xu S, Pelisek J, Jin ZG. Atherosclerosis is an epigenetic disease. Trends Endocrinol Metabol. (2018) 29:739–42. doi: 10.1016/j.tem.2018.04.007
121. Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol. (2013) 34:216–23. doi: 10.1016/j.it.2012.11.001
122. Kuznetsova T, Prange KHM, Glass CK, de Winther MPJ. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat Rev Cardiol. (2020) 17:216–28. doi: 10.1038/s41569-019-0265-3
123. Schito L, Rey S. Cell-autonomous metabolic reprogramming in hypoxia. Trends Cell Biol. (2018) 28:128–42. doi: 10.1016/j.tcb.2017.10.006
124. Ivashkiv LB. The hypoxia-lactate axis tempers inflammation. Nat Rev Immunol. (2020) 20:85–6. doi: 10.1038/s41577-019-0259-8
125. Shakespear MR, Iyer A, Cheng CY, Gupta KD, Singhal A, Fairlie DP, et al. Lysine deacetylases and regulated glycolysis in macrophages. Trends Immunol. (2018) 39:473–88. doi: 10.1016/j.it.2018.02.009
126. Kasikara C, Doran AC, Cai B, Tabas I. The role of non-resolving inflammation in atherosclerosis. J Clin Invest. (2018) 128:2713–23. doi: 10.1172/JCI97950
127. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
128. Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V, et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest. (2018) 128:1106–24. doi: 10.1172/JCI93025
Keywords: macrophage, polarization, epigenetic, reprogramming, chronic inflammation, atherosclerosis, therapeutic target
Citation: Yang H, Sun Y, Li Q, Jin F and Dai Y (2022) Diverse Epigenetic Regulations of Macrophages in Atherosclerosis. Front. Cardiovasc. Med. 9:868788. doi: 10.3389/fcvm.2022.868788
Received: 03 February 2022; Accepted: 04 March 2022;
Published: 29 March 2022.
Edited by:
Emiel Van Der Vorst, Institute for Molecular Cardiovascular Research (IMCAR), GermanyReviewed by:
Marten A. Hoeksema, Amsterdam UMC, NetherlandsCopyright © 2022 Yang, Sun, Li, Jin and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengyan Jin, ZmVuZ3lhbmppbkBqbHUuZWR1LmNu, orcid.org/0000-0003-2288-556X; Yun Dai, ZGFpeXVuQGpsdS5lZHUuY24=, ZGFpeXVuNjJAaG90bWFpbC5jb20=, orcid.org/0000-0003-2285-0818
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.