 Younes Zaid1,2*
Younes Zaid1,2* Yahye Merhi3
Yahye Merhi3
- 1Laboratory of Materials, Nanotechnology and Environment, Faculty of Sciences, Mohammed V University in Rabat, Rabat, Morocco
- 2Immunology and Biodiversity Laboratory, Department of Biology, Faculty of Sciences, Hassan II University, Casablanca, Morocco
- 3Laboratory of Thrombosis and Hemostasis, Montreal Heart Institute, Research Center, The Université de Montréal, Montreal, QC, Canada
In addition to their well-described hemostatic function, platelets are active participants in innate and adaptive immunity. Inflammation and immunity are closely related to changes in platelet reactions and enhanced platelet function in thrombo-inflammation, as well as in microbial and virus infections. A platelet’s immune function is incompletely understood, but an important balance exists between its protective and pathogenic responses and its thrombotic and inflammatory functions. As the mediator of vascular homeostasis, platelets interact with neutrophils, bacteria and virus by expressing specific receptors and releasing granules, transferring RNA, and secreting mitochondria, which controls hemostasis and thrombosis, infection, and innate and adaptive immunity. This review focuses on the involvement of platelets during immuno-thrombosis and thrombo-inflammation.
Introduction
Platelets are small (2–4 μm in diameter) anucleated cells derived from their megakaryocyte’s precursors with 7–10 days lifespan. Nearly one trillion platelets sentinel the blood vessels to monitor and preserve the integrity of the vasculature. There is no nucleus in platelets, but they are prepacked with proteins and various forms of RNA from their precursor cells. When damage to blood vessels occurs, it triggers the formation of a thrombus to stop bleeding (1). The discoid shape changes to a spherical one, resulting in long filopodia that facilitate adhesion. In order for platelets to function, they must involve an array of adhesive and activation receptors, secreted granule reservoirs, and dynamic cytoskeletal proteins (2). Extracellular vesicles (microparticles) can also play a role in the formation of thrombus mediated by platelets, as they provide anionic phospholipids that aid in the coagulation process (3). The role of platelets is not restricted to the hemostatic/thrombotic response, but platelets play a crucial role in inflammatory and immune responses (4–8). Indeed, not only do platelets express an array of molecules serving wound repair, but they also bear immune and inflammatory molecules such as interleukin IL1 (9), and an array of receptors including toll-like-receptors (TLRs), CD154, or CD40L (4), Fc receptor for IgG (FcγRIIA) (10), IgA (FcαRI) and IgE (FcεRI) (11, 12).
In response to harmful pathogens, platelets contribute to the immune system either directly, by producing cytokines and antimicrobial peptides, or indirectly, through interactions with neutrophils, monocytes, lymphocytes, and other cells (13, 14). Immuno-thrombosis may negatively affect hemostatic and immunological processes during a bacterial infection, resulting in adverse clinical outcomes (15). In this review, we will focus on the role of platelets in immuno-thrombosis and thrombo-inflammation.
Platelet-Associated Immunopathology: Immuno-Thrombosis and Thrombo-Inflammation
Mechanisms of Neutrophil Extracellular Trap-Induced Thrombosis
It is unsurprising that platelet-neutrophil interactions are greatly increased during inflammatory responses (16–19). For the most part, soluble mediators initiate these interactions, which directly activate these cells. Platelets and neutrophils co-incubated with septic patient plasma induced platelet adhesion to neutrophils mediated by the TLR4 receptor (20, 21).
Neutrophils are the most abundant subset of leukocytes in arterial thrombi from patients with myocardial infarction (22). Activated neutrophils express adhesion molecules belonging to the selectin and integrin families promoting platelets and the endothelium binding (23). As well, activated platelets express adhesion molecules on their surface membrane, such as P-selectin that mediates binding of platelets to its main receptor on neutrophils, P-selectin-glycoprotein-ligand-1 or PSGL-1 (24). Indeed, neutrophils and activated platelets can recruit each other to inflamed or injured tissues, thereby causing thrombo-inflammation (25). In this regard, we found that platelets can modulate neutrophil adhesion to the injured arterial wall and that both elements influence the degree of post-injury vasoconstriction in in vivo porcine model involving arterial injury by angioplasty (26). More recently, we have revealed that platelet activation and binding to neutrophils enhance the secretion of platelet MMP-2 via an adhesive interaction between P-selectin and PSGL-1, which contributes to increase platelet-neutrophil aggregation (27). Inhibition of platelet-leukocyte binding, using a recombinant PSGL-1 reduced restenosis (28) and prevented in in-stent restenosis via reduction of thrombo-inflammatory reactions (29).
Neutrophil extracellular traps (NETs) are composed of DNA, histones, and antimicrobial peptides, and they are produced as part of an antimicrobial mechanism, which is affected by immune/immune-related cells during NETosis (30). Indeed, NETosis appears to be associated with many inflammatory disorders, including infections, cancers, endothelial dysfunction, atherosclerosis, thrombosis, and ischemia (31, 32).
Neutrophil extracellular traps contribute to thrombosis through direct and indirect mechanisms. Although the vast majority of studies use NET components rather than intact NETs, the role of intact versus NET component in activating coagulation is controversial (33). In addition to their ability to promote thrombin formation, NETs were also known for providing a scaffold for pro-coagulant molecules such as VWF, fibrinogen, FXII, and tissue factor, as well as pro-coagulant extracellular vesicles TF-bearing EVs for instance (34–38).
Platelets can induce dysregulation of NET, resulting in tissue damage, hypercoagulability, and thrombosis (34). In addition, NETosis is well documented in its role in the pathogenesis of sepsis and ARDS, causing vascular tissue damage and spreading microthrombi that eventually cause multiorgan failure and death (39, 40).
The studies on NETosis provide growing evidence that NETosis is connected to inflammation, atherosclerosis, and atherothrombosis, as well as poor prognoses of ischemic/reperfusion injuries (41).
Mouse studies have examined the effects of NETosis and platelet aggregation on outcome after ischemia/reperfusion (42). Then, Cf-DNAs trigger DNA-platelet and DNA-platelet-granulocyte colonization. This form of NET and platelet aggravation leads to NETosis.
There are no doubts that NETs play a significant role in thrombosis and hemostasis (Figure 1). Both arterial and venous thrombosis are affected by NETs, and NETs are implicated in stenosis that regulate thrombosis in different ways (43, 44). Additionally, preventing the formation of NETs can reduce thrombogenicity, which could be useful in preventing thrombosis (44). A thorough understanding of how new therapeutic options targeting NETs affect thrombosis will require both preclinical and clinical trials.
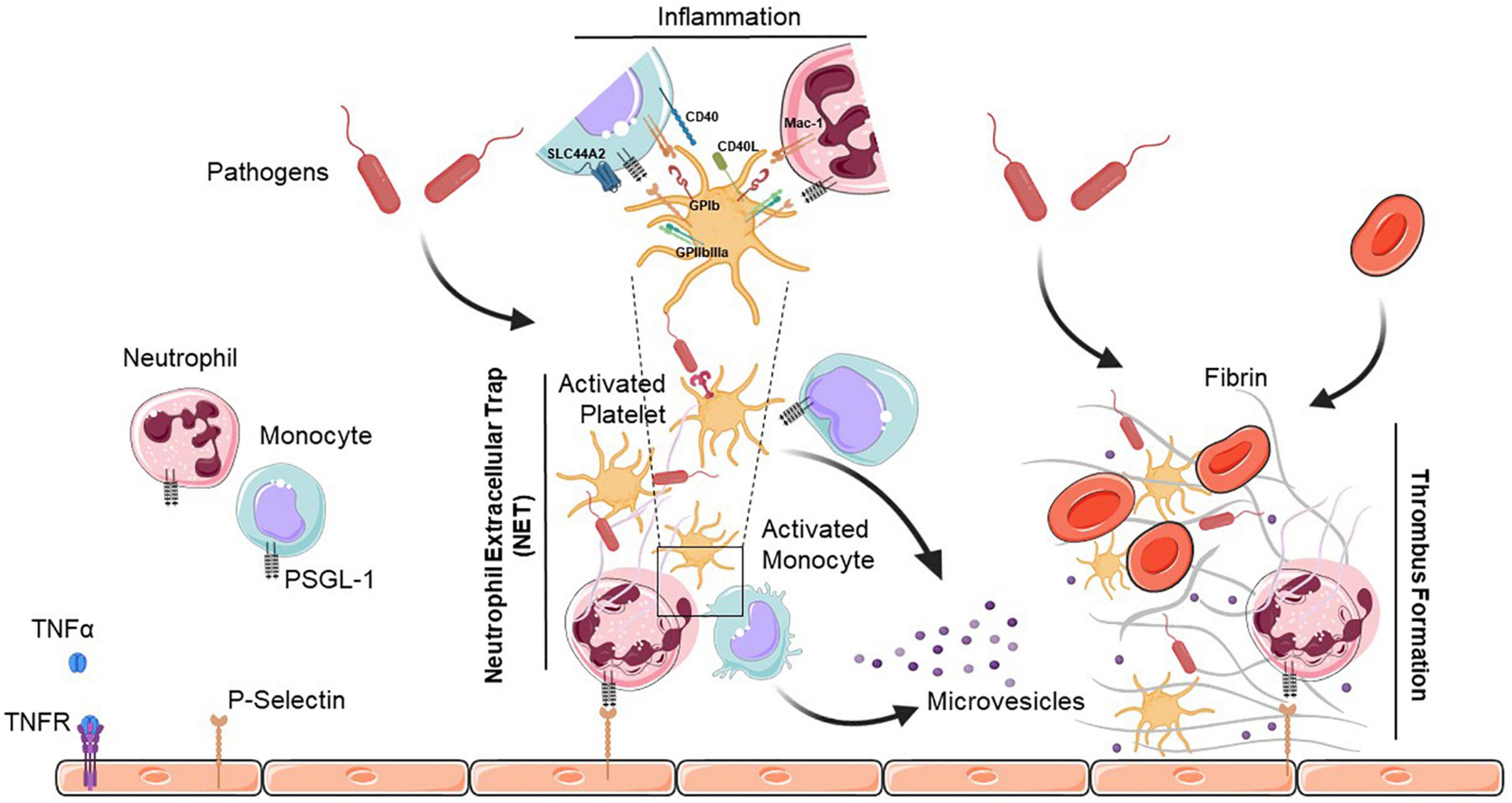
Figure 1. Neutrophil extracellular trap (NET)-mediated platelet thrombosis. In the presence of pathogens, TNFα binds to its receptor (TNFR) to initiate diverse cellular responses, and platelets, neutrophils and monocytes collaborate to form NETs that are highly thrombotic and resistant to tissue plasminogen activator-mediated fibrinolysis. NETs also help to recruit platelets which support the immuno-thrombotic process by promoting fibrin generation along with endothelial cells. Activated platelets release large amounts of pro-inflammatory cytokines in platelet extracellular vesicles. Interactions between platelets and neutrophils or monocytes occur via direct contact between cell surface receptors, or by binding of secreted ligands. PSGL-1, P-selectin glycoprotein ligand-1; TNFα, tumor necrosis factor alpha; TNFR, tumor necrosis factor receptor; GPIb, glycoprotein Ib; SLC44A2, solute carrier family 44 member 2; CD40L, cluster of differentiation 40 lignad; CD40, cluster of differentiation 40; Mac-1, macrophage-1 antigen; GPIIbIIIa, glycoprotein IIb/IIIa.
Platelet Extracellular Vesicles
In view of the critical role for platelets in thrombosis, studying platelet function may provide novel biomarkers for arterial thrombosis (45). There are numerous platelet function tests available in clinical practice, but most commonly, aggregometry-based tests are carried out (46). The aggregometry method, however, provides information on platelet functionality in the presence of exogenous and soluble agonists, which does not represent in vivo platelet activation. Thus, so far results from platelet function tests in vitro have limited value as biomarkers of arterial thrombosis.
A biomolecule or metabolite associated with activated platelets may be able to provide information about arterial thrombosis (47). Platelet-derived extracellular vesicles (PEV) are among these biomarkers, covering platelet microparticles/microvesicles and exosomes (48).
A number of benefits attributed to platelets are likely mediated by platelet-derived extracellular vesicles (PEVs), which are small vesicles released from activated platelets. Indeed, the release of platelet α-granules have been considered as small vesicles, as revealed by electron microscopic analyses (49). Also known as microparticles, this release of vesicles from the platelet plasma membrane occurs as a result of the extrusion of the platelet cytomembrane structures (50). Later studies using electron microscopy confirmed the presence of the two types of PEVs: small vesicles with diameters of 80–200 nm, and larger vesicles with diameters of 400–600 nm, which retained procoagulant activity mediated by factor V-like activity and tissue factor (51).
Platelet-derived extracellular vesicle can serve as biomarkers for autoimmune diseases, cancer, cardiovascular diseases, and infectious diseases (52–55). Rheumatoid arthritis patients have PEV detected in their synovial fluid (56), and increased levels of circulating PEV correlate with disease activity (57). Additionally, in murine models of atherosclerosis and autoimmune arthritis, PEV concentrations have been found to be increased in lymph (52, 58). As a result of systemic lupus erythematosus (SLE), PEV levels in blood can be increased. Higher levels of PEV have been associated with declining kidney function (59). Recently, an increase in PEV circulating in blood of COVID-19 patients has been observed (55, 60, 61).
Aside from transporting and producing inflammation-promoting mediators such as prostaglandins and leukotrienes, the PEV is also the transporter and producer of many lipid mediators (62).
Receptors in Platelet–Immune Cell Interactions
Platelets enhance leukocyte recruitment by expressing an arsenal of complement system receptors including cC1qR, gC1qR, C3aR, and C5aR, and storing complement proteins and regulators such as C3 and factor H. Platelets stimulate the classical and alternative pathways of complement, causing the accumulation of opsonin C3b as well as the release of anaphylatoxins C3a and C5a, which chemoattract innate immune cells (63, 64).
Among innate immune system receptors, TLRs are pattern recognition receptors that link various pathogen-shared molecules, including lipopolysaccharide (LPS), lipoproteins, and other bacterial wall constituents (65). As a consequence of expressing TLRs involved in the innate immunity response, platelets may contribute to the immune response and to infections by secreting a number of inflammatory mediators and pro-inflammatory factors (5, 66, 67).
Recent studies found significantly elevated levels of CXCL1, CXCL8, CXCL12, CCL2, CCL3, CCL5, EGF, VEGF, and PDGF-AB/BB in plasma and BAL fluid of patients with severe COVID-19, all of which can be found in platelet granules, indicating that platelets may play a role in hyperinflammation observed in patients with ARDS (68, 69). These factors mediate the inflammatory response by acting synergistically, through both autocrine and paracrine pathways.
There is a wealth of evidence suggesting that platelets modulate both the innate and adaptive immune systems. Platelets and platelet-derived microparticles are active in killing foreign pathogens in addition to directing a cascade of proteases and inhibitors to sites of vascular injury and inflammation (70). As well, secreted chemokines and cytokines (RANTES, IL-1β, and MCP-1) and platelet-expressed P-selectin promote leukocyte recruitment, adhesion, and transmigration at sites of vascular injury (Figure 1).
A CD40L-expressing platelet is capable of not only increasing inflammatory responses in the endothelium, but also triggering antigen-presenting cells (dendritic cells and macrophages), resulting in enhanced antigen delivery to T lymphocytes. As demonstrated in CD40L deficient mice infected with viruses, CD40L on activated platelets also facilitates B cell differentiation and Ig class switching (71, 72). Also, patients with immune thrombocytopenia have reduced levels of regulatory T (TReg) cells, and therapeutic increases in platelet counts restore TReg cell numbers and functions in such individuals (73–75). The effect of TGFβ secreted by platelets in TReg formation is unclear, but since differentiation of TReg cells requires TGFβ, platelets may contribute to TReg formation.
Platelets in Bacterial and Viral Infections
In Bacterial Infections
A wide variety of bacteria can interact with platelets, including the Staphylococci family, Neisseria gonorrhea, Porphyromonas gingivalis, and Helicobacter pylori (76–78). GPIIb/IIIa, FcγRIIa, and IgG receptors on platelets help them adhere and aggregate around bacteria, as well as fibrinogen and fibronectin (79, 80). TLR1, 2, 4, 6, and 9 within platelets enable them to bind bacteria, which will either cause platelets to secrete thrombocidins (antibacterial proteins within platelet α-granule, including thrombocidin 1 and 2) or aggregate around bacteria to trap them for elimination by phagocytes (20, 81–84). Upon interacting with bacteria, platelets release antimicrobial compounds that are contained within their granules. Alpha toxin released by S. Aureus mediates the release of β-defensin from platelets, which is responsible for NET formation (85). Platelets binding to Gram-negative bacteria releases a PF4 with exposed heparin-like epitopes, increasing antibody binding to the surface of bacteria and possibly leading to neutrophil opsonization and phagocytosis (86, 87). Some Gram-negative bacteria, such as Yersinia pestis, are insensitive to mammalian TLR4s, making this mechanism crucial (88, 89). Based on in vivo experiments with another Gram-negative bacteria, Porphyromonas gingivalis, it has been shown that during infection, platelets interact with neutrophils forming heterotypic aggregates in a TLR2-dependant fashion and that TLR2 promotes the aggregation of platelets (90). These study findings indicate that platelets can trigger platelet thrombotic pathway and/or inflammatory pathway activation upon recognizing bacterial components (90).
In Viral Infections
On the other hand, platelets interact with various types of viruses and their phenotype may vary depending on the type of viral infection (91). Thrombocytopenia and even thrombosis can accompany viral infections (92). Viruses can be divided into those that have either DNA or RNA genomes. Furthermore, RNA viruses can be divided into double-stranded and single-stranded viruses. Several DNA viruses, such as the herpes simplex virus type 1 (HSV1), cytomegalovirus (CMV) and vaccinia, have been identified associated with platelets. However, it is unknown if these viruses can be internalized by platelets (93–95). In contrast, RNA viruses such as HIV, hepatitis C virus (HCV), dengue, influenza, CVB, and EMCV have smaller sizes and are easily internalized by platelets (96–101).
There was recently evidence that SARS-CoV-2, a single-stranded RNA virus, can increase platelet activity and formation of platelet-monocyte aggregates facilitated by TF expression on monocytes (55, 102–105). Koupenova et al. (106) reported that SARS-CoV-2 promotes programmed cell death in platelets in another aspect of platelet-SARS-CoV-2 interaction. According to this study, RNA sequence analysis for SARS-CoV-2 shown by ARTIC v3 sequencing, transmission electron microscopy and immunofluorescence showed that SARS-CoV-2 virions internalized when attached to microparticles. As a consequence of such internalization, apoptosis, necroptosis, and EV release occur, which contribute to impaired immunity and thrombosis (106).
Several studies suggest that platelets may associate with SARS-CoV-2 RNA molecules, and that this event may be more likely to occur in older patients (55, 105, 107). However, Bury et al. did not detect viral RNA in platelets from COVID-19 patients (108). The disparity could be due to the size of the cohort which is significantly larger in the studies that detected traces of viral RNA in platelets of some COVID-19 patients.
Conclusion
Anucleated megakaryocyte-derived platelets play an important role in hemostasis and thrombosis. Platelets serve as additional mediators of inflammation beyond hemostasis and contribute to several aspects of immune response, including priming of other immune cells and integration of extrinsic immunological stimuli. Indeed, platelets initiate innate immunity as well as adaptive immunity, which is beneficial for host defenses at certain stages of infection. The functions of platelets are thus diversified and rooted firmly in their interactions with vascular and circulating immune cells. Ultimately, uncontrolled endothelial damage and inflammation caused by viral infection progression can result in enhanced platelet reactions which amplify thrombosis and inflammation leading to higher cardiovascular, cerebral and lung pathologic events.
Author Contributions
YZ and YM wrote and edited the manuscript, read, and agreed to the published version of the manuscript.
Funding
The authors acknowledge support from the Fonds de Recherche de l’Institut de Cardiologie de Montréal (FRICM) (Montreal, QC, Canada).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. (2007) 357:2482–94. doi: 10.1056/NEJMra071014
2. El-Kadiry AE, Merhi Y. The role of the proteasome in platelet function. Int J Mol Sci. (2021) 22:3999. doi: 10.3390/ijms22083999
3. Ridger VC, Boulanger CM, Angelillo-Scherrer A, Badimon L, Blanc-Brude O, Bochaton-Piallat ML, et al. Microvesicles in vascular homeostasis and diseases. position paper of the European society of cardiology (ESC) working group on atherosclerosis and vascular biology. Thromb Haemost. (2017) 117:1296–316. doi: 10.1160/TH16-12-0943
4. Semple JW, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
5. Kapur R, Zufferey A, Boilard E, Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol. (2015) 194:5579–87.
6. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. (2014) 123:2759–67. doi: 10.1182/blood-2013-11-462432
7. Allaoui A, Khawaja AA, Badad O, Naciri M, Lordkipanidze M, Guessous F, et al. Platelet function in viral immunity and SARS-CoV-2 infection. Semin Thromb Hemost. (2021) 47:419–26. doi: 10.1055/s-0041-1726033
8. Limami Y, Senhaji N, Zaid N, Khalki L, Naya A, Hajjaj-Hassouni N, et al. PKC-delta-dependent pathways contribute to the exacerbation of the platelet activity in Crohn’s disease. Semin Thromb Hemost. (2021) 48:246–50. doi: 10.1055/s-0041-1736571
9. Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. (2001) 154:485–90. doi: 10.1083/jcb.200105058
10. Karas SP, Rosse WF, Kurlander RJ. Characterization of the IgG-Fc receptor on human platelets. Blood. (1982) 60:1277–82.
11. Idzko M, Pitchford S, Page C. Role of platelets in allergic airway inflammation. J Allergy Clin Immunol. (2015) 135:1416–23. doi: 10.1016/j.jaci.2015.04.028
12. Arman M, Krauel K. Human platelet IgG Fc receptor FcgammaRIIA in immunity and thrombosis. J. Thromb Haemost. (2015) 13:893–908. doi: 10.1111/jth.12905
13. Auerbach DJ, Lin Y, Miao H, Cimbro R, Difiore MJ, Gianolini ME, et al. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc Natl Acad Sci USA. (2012) 109:9569–74. doi: 10.1073/pnas.1207314109
14. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. (2015) 114:449–58. doi: 10.1160/TH14-12-1067
15. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
16. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. (1999) 93:876–85.
17. Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood. (2000) 95:1158–66.
18. Kasper B, Brandt E, Brandau S, Petersen F. Platelet factor 4 (CXC chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J Immunol. (2007) 179:2584–91. doi: 10.4049/jimmunol.179.4.2584
19. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. (2005) 105:924–30. doi: 10.1182/blood-2004-06-2475
20. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
21. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. (2016) 99:153–62. doi: 10.1189/jlb.3A0415-161R
22. Novotny J, Chandraratne S, Weinberger T, Philippi V, Stark K, Ehrlich A, et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS One. (2018) 13:e0190728. doi: 10.1371/journal.pone.0190728
23. Swystun LL, Liaw PC. The role of leukocytes in thrombosis. Blood. (2016) 128:753–62. doi: 10.1182/blood-2016-05-718114
24. Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. (2002) 196:887–96. doi: 10.1084/jem.20012044
25. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
26. Merhi Y, Provost P, Chauvet P, Theoret JF, Phillips ML, Latour JG. Selectin blockade reduces neutrophil interaction with platelets at the site of deep arterial injury by angioplasty in pigs. Arterioscler Thromb Vasc Biol. (1999) 19:372–7. doi: 10.1161/01.atv.19.2.372
27. Abou-Saleh H, Theoret JF, Yacoub D, Merhi Y. Neutrophil P-selectin-glycoprotein-ligand-1 binding to platelet P-selectin enhances metalloproteinase 2 secretion and platelet-neutrophil aggregation. Thromb Haemost. (2005) 94:1230–5. doi: 10.1160/TH05-05-0344
28. Bienvenu JG, Tanguay JF, Theoret JF, Kumar A, Schaub RG, Merhi Y. Recombinant soluble p-selectin glycoprotein ligand-1-Ig reduces restenosis through inhibition of platelet-neutrophil adhesion after double angioplasty in swine. Circulation. (2001) 103:1128–34. doi: 10.1161/01.cir.103.8.1128
29. Tanguay JF, Geoffroy P, Sirois MG, Libersan D, Kumar A, Schaub RG, et al. Prevention of in-stent restenosis via reduction of thrombo-inflammatory reactions with recombinant P-selectin glycoprotein ligand-1. Thromb Haemost. (2004) 91:1186–93. doi: 10.1160/TH03-11-0701
30. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
31. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. (2011) 18:581–8. doi: 10.1038/cdd.2011.1
32. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
33. Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: back in the thrombosis spotlight. Blood. (2019) 133:2186–97. doi: 10.1182/blood-2018-10-862243
34. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
35. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Massberg, Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
36. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. (2015) 36:1405–14. doi: 10.1093/eurheartj/ehv007
37. Wang Y, Luo L, Braun OO, Westman J, Madhi R, Herwald H, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. (2018) 8:4020. doi: 10.1038/s41598-018-22156-5
38. Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, et al. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J Thromb Haemost. (2015) 13:1310–9. doi: 10.1111/jth.13002
39. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One. (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
40. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. (2018) 3:e98178. doi: 10.1172/jci.insight.98178
41. Qi H, Yang S, Zhang L. Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis. Front Immunol. (2017) 8:928. doi: 10.3389/fimmu.2017.00928
42. Shrestha B, Ito T, Kakuuchi M, Totoki T, Nagasato T, Yamamoto M, et al. Recombinant thrombomodulin suppresses histone-induced neutrophil extracellular trap formation. Front Immunol. (2019) 10:2535. doi: 10.3389/fimmu.2019.02535
43. Kopytek M, Kolasa-Trela R, Zabczyk M, Undas A, Natorska J. NETosis is associated with the severity of aortic stenosis: links with inflammation. Int J Cardiol. (2019) 286:121–6. doi: 10.1016/j.ijcard.2019.03.047
44. Laridan E, Martinod K, De Meyer SF. Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost. (2019) 45:86–93. doi: 10.1055/s-0038-1677040
45. Jurk K. Analysis of platelet function and dysfunction. Hamostaseologie. (2015) 35:60–72. doi: 10.5482/HAMO-14-09-0047
46. Kim MH, Choi SY, An SY, Serebruany V. Validation of three platelet function tests for bleeding risk stratification during dual antiplatelet therapy following coronary interventions. Clin Cardiol. (2016) 39:385–90. doi: 10.1002/clc.22540
47. Ferroni P, Riondino S, Vazzana N, Santoro N, Guadagni F, Davi G. Biomarkers of platelet activation in acute coronary syndromes. Thromb Haemost. (2012) 108:1109–23. doi: 10.1160/TH12-08-0550
48. van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. (2012) 64:676–705. doi: 10.1124/pr.112.005983
49. Webber AJ, Johnson SA. Platelet participation in blood coagulation aspects of hemostasis. Am J Pathol. (1970) 60:19–42.
50. Crawford N. The presence of contractile proteins in platelet microparticles isolated from human and animal platelet-free plasma. Br J Haematol. (1971) 21:53–69. doi: 10.1111/j.1365-2141.1971.tb03416.x
51. Sandberg H, Bode AP, Dombrose FA, Hoechli M, Lentz BR. Expression of coagulant activity in human platelets: release of membranous vesicles providing platelet factor 1 and platelet factor 3. Thromb Res. (1985) 39:63–79. doi: 10.1016/0049-3848(85)90122-7
52. Tessandier N, Melki I, Cloutier N, Allaeys I, Miszta A, Tan S, et al. Platelets disseminate extracellular vesicles in lymph in rheumatoid arthritis. Arterioscler Thromb Vasc Biol. (2020) 40:929–42. doi: 10.1161/ATVBAHA.119.313698
53. Gitz E, Pollitt AY, Gitz-Francois JJ, Alshehri O, Mori J, Montague S, et al. CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood. (2014) 124:2262–70. doi: 10.1182/blood-2014-05-572818
54. Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, et al. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood. (2014) 123:2854–63. doi: 10.1182/blood-2013-07-515536
55. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. (2020) 127:1404–18. doi: 10.1161/CIRCRESAHA.120.317703
56. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. (2010) 327:580–3. doi: 10.1126/science.1181928
57. Knijff-Dutmer EA, Koerts J, Nieuwland R, Kalsbeek-Batenburg EM, van de Laar MA. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum. (2002) 46:1498–503. doi: 10.1002/art.10312
58. Milasan A, Tessandier N, Tan S, Brisson A, Boilard E, Martel C. Extracellular vesicles are present in mouse lymph and their level differs in atherosclerosis. J Extracell Vesicles. (2016) 5:31427. doi: 10.3402/jev.v5.31427
59. Mobarrez F, Vikerfors A, Gustafsson JT, Gunnarsson I, Zickert A, Larsson A, et al. Microparticles in the blood of patients with systemic lupus erythematosus (SLE): phenotypic characterization and clinical associations. Sci Rep. (2016) 6:36025. doi: 10.1038/srep36025
60. Guervilly C, Bonifay A, Burtey S, Sabatier F, Cauchois R, Abdili E, et al. Dissemination of extreme levels of extracellular vesicles: tissue factor activity in patients with severe COVID-19. Blood Adv. (2021) 5:628–34. doi: 10.1182/bloodadvances.2020003308
61. Cappellano G, Raineri D, Rolla R, Giordano M, Puricelli C, Vilardo B, et al. Circulating platelet-derived extracellular vesicles are a hallmark of Sars-Cov-2 infection. Cells. (2021) 10:10.3390/cells10010085.
62. Boilard E. Extracellular vesicles and their content in bioactive lipid mediators: more than a sack of microRNA. J Lipid Res. (2018) 59:2037–46. doi: 10.1194/jlr.R084640
63. Peerschke EI, Yin W, Ghebrehiwet B. Platelet mediated complement activation. Adv Exp Med Biol. (2008) 632:81–91. doi: 10.1007/978-0-387-78952-1_7
64. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. (2005) 201:871–9. doi: 10.1084/jem.20041497
65. Janeway CA Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol. (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
66. Ebermeyer T, Cognasse F, Berthelot P, Mismetti P, Garraud O, Hamzeh-Cognasse H. Platelet innate immune receptors and TLRs: a double-edged sword. Int J Mol Sci. (2021) 22:7894. doi: 10.3390/ijms22157894
67. Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, et al. Platelets endocytose viral particles and are activated via TLR (toll-like receptor) signaling. Arterioscler Thromb Vasc Biol. (2020) 40:1635–50. doi: 10.1161/ATVBAHA.120.314180
68. Zaid Y, Dore E, Dubuc I, Archambault AS, Flamand O, Laviolette M, et al. Chemokines and eicosanoids fuel the hyperinflammation within the lungs of patients with severe COVID-19. J Allergy Clin Immunol. (2021) 148:368.e–80.e. doi: 10.1016/j.jaci.2021.05.032
69. Archambault AS, Zaid Y, Rakotoarivelo V, Turcotte C, Dore E, Dubuc I, et al. High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. FASEB J. (2021) 35:e21666. doi: 10.1096/fj.202100540R
70. Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. (2010) 47:2170–5. doi: 10.1016/j.molimm.2010.05.009
71. Elzey BD, Schmidt NW, Crist SA, Kresowik TP, Harty JT, Nieswandt B, et al. Platelet-derived CD154 enables T-cell priming and protection against Listeria monocytogenes challenge. Blood. (2008) 111:3684–91. doi: 10.1182/blood-2007-05-091728
72. Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive immunity. Cell Immunol. (2005) 238:1–9. doi: 10.1016/j.cellimm.2005.12.005
73. Ling Y, Cao X, Yu Z, Ruan C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. Eur J Haematol. (2007) 79:310–6. doi: 10.1111/j.1600-0609.2007.00917.x
74. Liu B, Zhao H, Poon MC, Han Z, Gu D, Xu M, et al. Abnormality of CD4(+)CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. Eur J Haematol. (2007) 78:139–43. doi: 10.1111/j.1600-0609.2006.00780.x
75. Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood. (2008) 112:1147–50. doi: 10.1182/blood-2007-12-129262
76. Yeaman MR. Platelets in defense against bacterial pathogens. Cell Mol Life Sci. (2010) 67:525–44. doi: 10.1007/s00018-009-0210-4
77. Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol. (2006) 4:445–57. doi: 10.1038/nrmicro1425
78. Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections - complex interactions with bacteria. Front Immunol. (2015) 6:82. doi: 10.3389/fimmu.2015.00082
79. Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, et al. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol Microbiol. (2006) 59:212–30. doi: 10.1111/j.1365-2958.2005.04922.x
80. Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, et al. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol. (2005) 57:804–18. doi: 10.1111/j.1365-2958.2005.04731.x
81. Beaulieu LM, Freedman JE. The role of inflammation in regulating platelet production and function: toll-like receptors in platelets and megakaryocytes. Thromb Res. (2010) 125:205–9. doi: 10.1016/j.thromres.2009.11.004
82. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
83. Yeaman MR, Puentes SM, Norman DC, Bayer AS. Partial characterization and staphylocidal activity of thrombin-induced platelet microbicidal protein. Infect Immun. (1992) 60:1202–9. doi: 10.1128/iai.60.3.1202-1209.1992
84. Krijgsveld J, Zaat SA, Meeldijk J, van Veelen PA, Fang G, Poolman B, et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem. (2000) 275:20374–81. doi: 10.1074/jbc.275.27.20374
85. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
86. Krauel K, Weber C, Brandt S, Zahringer U, Mamat U, Greinacher A, et al. Platelet factor 4 binding to lipid A of Gram-negative bacteria exposes PF4/heparin-like epitopes. Blood. (2012) 120:3345–52. doi: 10.1182/blood-2012-06-434985
87. Krauel K, Potschke C, Weber C, Kessler W, Furll B, Ittermann T, et al. Platelet factor 4 binds to bacteria, [corrected] inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood. (2011) 117:1370–8. doi: 10.1182/blood-2010-08-301424
88. Dziarski R. Deadly plague versus mild-mannered TLR4. Nat Immunol. (2006) 7:1017–9. doi: 10.1038/ni1006-1017
89. Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. (2006) 7:1066–73. doi: 10.1038/ni1386
90. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. (2009) 104:346–54. doi: 10.1161/CIRCRESAHA.108.185785
91. Zaid Y, Guessous F, Puhm F, Elhamdani W, Chentoufi L, Morris AC, et al. Platelet reactivity to thrombin differs between patients with COVID-19 and those with ARDS unrelated to COVID-19. Blood Adv. (2021) 5:635–9. doi: 10.1182/bloodadvances.2020003513
92. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
93. Assinger A, Kral JB, Yaiw KC, Schrottmaier WC, Kurzejamska E, Wang Y, et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler Thromb Vasc Biol. (2014) 34:801–9. doi: 10.1161/ATVBAHA.114.303287
94. Forghani B, Schmidt NJ. Association of herpes simplex virus with platelets of experimentally infected mice. Arch Virol. (1983) 76:269–74. doi: 10.1007/BF01311111
95. Bik T, Sarov I, Livne A. Interaction between vaccinia virus and human blood platelets. Blood. (1982) 59:482–7.
96. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. (2014) 124:791–802. doi: 10.1182/blood-2013-11-536003
97. Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. (2002) 99:4021–9. doi: 10.1182/blood-2001-12-0191
98. Danon D, Jerushalmy Z, De Vries A. Incorporation of influenza virus in human blood platelets in vitro. Electron microscopical observation. Virology. (1959) 9:719–22. doi: 10.1016/0042-6822(59)90168-0
99. de Almeida AJ, Campos-de-Magalhaes M, Brandao-Mello CE, de Oliveira RV, do Espirito-Santo MP, Yoshida CF, et al. Detection of hepatitis C virus in platelets: evaluating its relationship to antiviral therapy outcome. Hepatogastroenterology. (2009) 56:429–36.
100. Noisakran S, Gibbons RV, Songprakhon P, Jairungsri A, Ajariyakhajorn C, Nisalak A, et al. Detection of dengue virus in platelets isolated from dengue patients. Southeast Asian J Trop Med Public Health. (2009) 40:253–62.
101. Negrotto S, Jaquenod de Giusti C, Rivadeneyra L, Ure AE, Mena HA, Schattner M, et al. Platelets interact with Coxsackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. J Thromb Haemost. (2015) 13:271–82. doi: 10.1111/jth.12782
102. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13:120. doi: 10.1186/s13045-020-00954-7
103. Shen S, Zhang J, Fang Y, Lu S, Wu J, Zheng X, et al. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J Hematol Oncol. (2021) 14:72. doi: 10.1186/s13045-021-01082-6
104. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. (2020) 136:1330–41. doi: 10.1182/blood.2020007252
105. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136:1317–29. doi: 10.1182/blood.2020007214
106. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res. (2021) 129:631–46. doi: 10.1161/CIRCRESAHA.121.319117
107. Zaid Y, Guessous F. The ongoing enigma of SARS-CoV-2 and platelet interaction. Res Pract Thromb Haemost. (2022) 6:e12642. doi: 10.1002/rth2.12642
Keywords: platelets, immuno-thrombosis, thrombo-inflammation, NETosis, extracellular vesicles (EVs)
Citation: Zaid Y and Merhi Y (2022) Implication of Platelets in Immuno-Thrombosis and Thrombo-Inflammation. Front. Cardiovasc. Med. 9:863846. doi: 10.3389/fcvm.2022.863846
Received: 27 January 2022; Accepted: 04 March 2022;
Published: 25 March 2022.
Edited by:
Robert Campbell, The University of Utah, United StatesReviewed by:
Frederik Denorme, The University of Utah, United StatesNirav Dhanesha, The University of Iowa, United States
Copyright © 2022 Zaid and Merhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Younes Zaid, eS56YWlkQHVtNXIuYWMubWE=