 Andrew Owen
Andrew Owen Jaimin M. Patel1,2
Jaimin M. Patel1,2 Mansoor N. Bangash
Mansoor N. Bangash
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 15 July 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.854421
This article is part of the Research Topic Cardiovascular Involvement During Sepsis: Therapeutic and prognostic consequences View all 7 articles
Prolonged critical care stays commonly follow trauma, severe burn injury, sepsis, ARDS, and complications of major surgery. Although patients leave critical care following homeostatic recovery, significant additional diseases affect these patients during and beyond the convalescent phase. New cardiovascular and renal disease is commonly seen and roughly one third of all deaths in the year following discharge from critical care may come from this cluster of diseases. During prolonged critical care stays, the immunometabolic, inflammatory and neurohumoral response to severe illness in conjunction with resuscitative treatments primes the immune system and parenchymal tissues to develop a long-lived pro-inflammatory and immunosenescent state. This state is perpetuated by persistent Toll-like receptor signaling, free radical mediated isolevuglandin protein adduct formation and presentation by antigen presenting cells, abnormal circulating HDL and LDL isoforms, redox and metabolite mediated epigenetic reprogramming of the innate immune arm (trained immunity), and the development of immunosenescence through T-cell exhaustion/anergy through epigenetic modification of the T-cell genome. Under this state, tissue remodeling in the vascular, cardiac, and renal parenchymal beds occurs through the activation of pro-fibrotic cellular signaling pathways, causing vascular dysfunction and atherosclerosis, adverse cardiac remodeling and dysfunction, and proteinuria and accelerated chronic kidney disease.
The global burden of critical illness is difficult to quantify due to many factors but even prior to the Coronavirus Disease 2019 (COVID-19) pandemic it was likely to be substantial and broad reaching (1). Traditionally the focus of large multi-center randomized controlled trials in critical illness has been on interventions aimed at decreasing inpatient mortality, but the spotlight is increasingly turning to post-critical illness survivorship and quality of life (2–4). Several recent studies highlight that even in previously healthy individuals a period of critical illness is associated with the onset of long-term chronic health problems. In this review we critically appraise the literature surrounding post critical illness cardiovascular disease and explore the potential mechanisms.
Admission to critical care usually marks a patient's most severe lifetime episode of acute illness. Two fundamental lessons can be learned from critical illness survivor studies—the effects of critical illness are not short-lived or trivial, and the severity of these long-term effects relate to the severity and duration of the acute illness. For example, 3-year mortality in survivors of critical illness is elevated compared to non-critically ill patients, and these elevations in mortality are considerably increased by higher critical illness severity (5). Similar associations between illness severity, duration and one-year mortality are also found in ARDS when examining survivor data (6–8). Although co-morbidity and advanced age deleteriously synergise with acute illness severity, studies attempting to correct for this still show an elevated subsequent mortality associated with sepsis and acute respiratory failure diagnoses (8–11). The recent COVID-19 pandemic has served to emphasize these matters though intensive care unit (ICU) follow-up has naturally been shorter in this disease than for other critical illnesses (12).
Examination of the temporal patterns of mortality in ICU survivors (compared to hospital controls) shows a rapid cumulative excess mortality over the first 6-months following discharge, which slows in its accrual rate over the next 2.5 years of follow-up (5). One study found that over 35% of ICU survivor deaths within 1-year of ICU admission relate to cardiovascular, renal and cerebrovascular diseases and interestingly this cluster of diseases was ahead of malignancy as the leading cause of post-ICU survivor deaths (13). Other studies also point to elevated rates of cardiovascular and renal diseases and associate them with blood-stream markers of inflammation, showing an accelerated mortality which gradually settles with time (8, 14).
The question arises as to whether this is a generalizable phenomenon or whether there are only specific subsets of the critically ill to whom this applies. Data from previously young healthy adults who have survived severe traumatic injuries show an increased rate of new onset cardiovascular disease (15). Furthermore, there is a dose-response relationship between severity of injury (by injury severity score) and cumulative incidences of hypertension, coronary artery disease, diabetes mellitus, and chronic kidney disease (CKD). These very same diseases are also seen in adult burn patients (16, 17). Trauma and burns, as with many other critical illnesses, are often associated with acute kidney injury (AKI). Data from a retrospective study in Denmark demonstrated a one-year mortality rate of over 50% following dialysis-requiring AKI (though not strictly isolated to the critically ill), and a 1-year end-stage renal failure rate of over 5% (18). Similarly, follow-up of a large randomized controlled trial of differing dialysis intensities for AKI in critical care also showed that the overall 1-year mortality rate following AKI is over 50% with the majority of deaths occurring in the first 90 days, but with a significant and ongoing mortality over several years (19). The aforementioned study found an association between the severity of critical illness (determined by APACHE III score) and the risk of long-term mortality and also showed the association of AKI survival with high rates of cardiovascular disease, proteinuria and CKD. There are obvious parallels with the findings of studies in trauma and burns where injury severity correlated with subsequent cardiovascular morbidity (15). Sepsis studies show similar trends of elevated cardiovascular risk in survivors, as do studies of viral [e.g., influenza and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)] and bacterial respiratory infection (20–26). Interestingly, the elevated cardiovascular risk seen in sepsis survivors is similar to non-infected critically ill controls in some studies, suggesting that this phenomenon is either not infection specific, or that non-infection diagnostic categories fail to adequately capture same-stay inpatient infection/sepsis events (20).
Were this phenomenon universal, one would also expect it to be recapitulated in the pediatric setting. Pediatric critical care survivor data suffers from problems of a relative sparsity coupled with inclusion of large numbers of patients with confounding inherited or congenital co-morbidities. Furthermore, pediatric survivorship is usually coupled with continued physical growth and maturation which has the potential to offset pathological adversity. Nevertheless, there are data that supports a generalisability of the phenomenon seen in critically ill adults. Children who have suffered AKI (whether in ICU or not) show a similar pattern of progression to proteinuria, CKD and end-stage renal disease later in adulthood (27, 28). In the pediatric burn population, studies show increased rates of type 2 diabetes mellitus (T2DM), cardiovascular disease and significant myocardial and circulatory impairments in survivors (17, 29, 30). Irrespective of age, surviving a wide variety of severe critical illnesses results in an accelerated onset of chronic disease, particularly that which relates to the cardiovascular and renal systems.
The constellation of cardiovascular diseases comprising hyperlipidaemia, essential hypertension, coronary heart disease, cerebrovascular disease, heart failure, and CKD have substantial clinical overlap (31–35). Although it was over a century ago that Virchow suggested involvement of the immune system in atherosclerosis based on post-mortem studies, the understanding of the pathogenesis of atherosclerosis came to center on abnormalities of lipid homeostasis (36). Hypertension research only became possible after the introduction of routine and non-invasive blood pressure measurement, and associations between hypertension and cardiovascular diseases such as atherosclerosis, heart failure, or stroke soon became apparent (37). Abnormalities of salt and water homeostasis and autonomic regulation as drivers of hypertension were considered important, implicating renal, humoral, and neural mechanisms alongside abnormalities in lipid metabolism as drivers of cardiovascular disease. Other common findings that were considered important but assumed to have secondary roles in the pathophysiology of cardiovascular disease related to the immune infiltrates that Virchow and others had observed, subsequent oxidative and free-radical mediated damage, microvascular dysfunction with blunted endothelial-dependent vasodilatory responses, extracellular matrix remodeling and fibrosis, and platelet activation. Many of these secondary findings are also seen in T2DM, though the underlying driver there is believed to be pathologically elevated plasma glucose levels and tissue insulin resistance.
Although inflammation has long been recognized as playing a potential causative role in cardiovascular disease (particularly atherosclerosis), significant doubt existed whether detected inflammation was a result or cause of atherosclerotic disease (38, 39). Although acute inflammation can be shown to associate with elevated cardiovascular risk in humans, definitive proof that inflammation is an independent mechanistic driver of human cardiovascular disease was until recently lacking. Clinical studies suggest that modifying inflammation may hold cardiovascular benefit (40, 41), while some animal studies have also confirmed accelerated cardiovascular pathology following inflammatory insults (42, 43). It is only recently that the role of inflammation as a therapeutic target for combating cardiovascular disease has been confirmed by the CANTOS trial, where interleukin (IL)-1β antagonism showed a reduction in composite cardiovascular outcomes in patients with established coronary artery disease independent of plasma low density lipoprotein (LDL) cholesterol (44–46). While this is an exciting development, the mortality benefits of therapy in this setting overrode, but nonetheless, were accompanied by an increased likelihood of death due to infection, a particularly important finding for survivors of sepsis and critical illnesses who have a high rate of infection-related hospital & ICU readmissions (47).
Given that inflammation is an aetiological factor in chronic cardiovascular disease, we hypothesize that critical illness is a phase of hyperinflammation that can drive de novo or accelerate pre-existing cardiovascular morbidity and mortality. Critical illness is undoubtedly associated with inflammation and survivor chronic illness and mortality, particularly of cardiovascular origin (14). However, it is not immediately obvious how relatively brief acute illnesses that have lasted only days to weeks and that end in a repair and resolution phase of illness can induce a step change in cardiovascular risk. In this phase the levels of many pro-inflammatory cytokines have decreased, patients are often normotensive (and may have spent more time during their illnesses with lower than normal blood pressures), with low levels of total cholesterol, and may have low circulating levels of cortisol and aldosterone. Patients also display functional immunosuppression (48), and so this constellation of features would appear to run counter to the traditional and inflammatory models of cardiovascular disease (see Table 1). However, by turning to modern insights in to the pathogenesis of cardiovascular disease this apparent paradox is somewhat resolved.
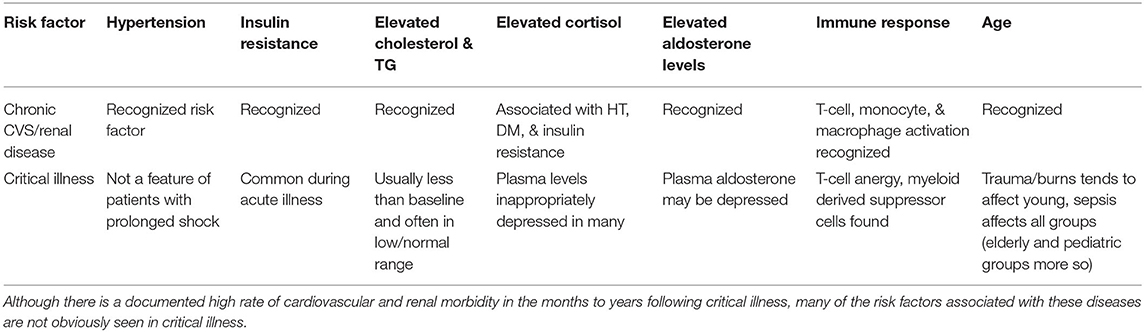
Table 1. The paradox of a lack of traditional risk factors for renal and cardiovascular disease during critical illness.
Considering that plasma endotoxin levels associate with incident diabetes and cardiovascular disease in the community setting, there is at least at the epidemiological level a strong association between innate immune activation and the development of metabolic and cardiovascular diseases (49–51). One mechanism of this linkage is suggested to be through molecular pattern receptor activation triggering low grade inflammation and cytokine mediated insulin resistance (52). There is no reason to suspect such phenomena are isolated to chronic disease and do not occur alongside or as a consequence of acute inflammatory events such as those which cause critical illness:
The acute phase of critical illness is undoubtedly characterized by tremendous immune activation and consequential free radical mediated damage (53, 54). Myeloperoxidase (MPO) is an important enzyme in the process of oxidative damage, it is the most abundant enzyme found in neutrophils, and is capable of inducing microvascular dysfunction by decreasing endothelial nitric oxide bioavailability, both independently and when complexed with high density lipoprotein (HDL) (55, 56). Furthermore, under various conditions, including pre-clinical models of sepsis, the free-radical mediated damage by MPO and other enzymes such as NADPH Oxidase (NOX) results in the formation of highly reactive isolevuglandins (γ-ketoaldehydes formed from the isoprostane pathway) that rapidly modify cellular proteins and activate pro-inflammatory pathways in tissue antigen presenting cells (57–60). Isolevuglandin protein adducts, found at elevated levels in patients with atherosclerosis and end-stage renal disease, are presented by dendritic cells bringing about monocyte recruitment to the endothelium, linking free-radical mediated damage with the activation and further recruitment of immune cells to the vasculature (61). Furthermore, these protein adducts modify LDL resulting in the uptake of LDL by receptors for oxidized LDL on macrophages, and they modify HDL, resulting, resulting in dysfunctional circulating HDL (62). Although leucocyte-endothelial interactions necessarily increase at sites of infection or tissue injury through mechanisms that are independent of this specific pathway, isoprostanes have also been detected at elevated levels in patients with sepsis (63). This suggests that this pathway, which is of importance in hypertension and atherosclerosis, could be of importance in human post critical illness cardiovascular pathogenesis.
HDL is considered anti-inflammatory, atheroprotective, and inhibitory of microvascular dysfunction by virtue of its myriad effects on inflammatory signaling and adhesion molecule upregulation, foam cell cholesterol efflux, and inhibitory effects on LDL oxidation and Toll-like receptors (TLRs) (64). It serves as a carrier molecule for transferring endotoxins such as lipopolysaccharide (LPS) and lipoteichoic acid (LTA) from the bloodstream to the liver where they can be rapidly cleared and detoxified by enzymes such as acyloxyacyl hydrolase, but HDL also carries other molecules such as the enzyme symmetric dimethyl arginase (SDMA) which limits nitric oxide bioavailability in the circulation (65, 66). It has been shown in some patient groups that share clinical overlap with critical illness survivors (e.g., those with CKD and T2DM) that HDL is abnormally modified by isoprostanes as described above, or by the presence of SDMA, in both cases inducing endothelial dysfunction and atherosclerosis via the activation of macrophage TLRs and inhibition of nitric oxide (67–69). One study showed fifty percent of 28-day sepsis survivors have SDMA levels of 0.82 μmol.l−1 or more, which is a level where population nomograms show severe HDL impairment (70, 71). The amount of circulating functional HDL is therefore also likely low in a substantial number of sepsis, and possibly other previously hyperinflamed, ICU survivors. This is not to implicate these pathways as being central drivers of the severe microvascular dysfunction which occurs during critical illnesses such as sepsis (though it may partly explain why patients with pre-existing cardiovascular diseases have worse basal microvascular function than others). Instead we propose that these pathways become relevant for ongoing vascular dysfunction after the critical phase of illnesses have subsided.
Microvascular dysfunction has been found in a number of different types of critical illnesses and is associated with endothelial damage and disruption of the endothelial glycocalyx (72). Due to the inability to measure basal microvascular health before admission (except in elective surgical patients), no studies have specifically looked at the rate of normalization of microvascular abnormalities in individuals after critical illnesses, which makes it difficult to assess the potential importance of these pathways in convalescence. However, one study in sepsis assessed biomarkers of endothelial health from ICU admission to 1-year out. This study showed that in 14–38% of surviving patients, levels of all measured soluble endothelial adhesion molecules and markers of coagulation activation [e.g., plasminogen activator inhibitor (PAI) −1] remained elevated above normal from 90 days after admission until a year out (73). Many of these patients have persistently elevated levels of IL-6, inducing both the endothelial release of PAI-1 and the upregulation of endothelial adhesion molecules, suggesting that the ongoing presence of inflammation and circulating cytokines such as IL-6 sustains endothelial activation (74–76). Furthermore microalbuminuria, a condition that is highly associated with systemic microvascular dysfunction, is particularly common after admission to ICU in those with severe kidney injuries (19, 77). Therefore, abnormalities in endothelial health and microvascular function probably persist after severe critical illnesses for a significant period of time in a substantial proportion of ICU survivors. As endothelial dysfunction is also involved in the initiation and progression of atherosclerotic disease its persistence is likely to contribute to ongoing cardiovascular disease. This paradigm of a seamless progression of endothelial dysfunction from one disease state to another is also supported by conserved inflammatory signaling pathways that induce endothelial activation both in peritonitis and atherosclerosis (78, 79). Furthermore, recent findings that chronic microvascular rarefaction precedes tissue aging suggests that critical illness could induce a step-change toward chronic organ dysfunction and tissue aging at least through the premature induction of chronic vascular dysfunction (80).
The fourth universal definition of myocardial infarction provides a framework for understanding increases in the cardiac injury biomarker Troponin (cTn), —essentially dichotomising to acute myocardial infarction of a variety of ischaemic subtypes and causes external to it (non-ischaemic injuries) (81). Although non-ischaemic myocardial injuries do not involve clinically detectable ischaemia, they can be contributed to by processes that can cause ischaemic injuries and myocardial infarction (e.g., anemia, sustained and severe tachycardia); correctly differentiating non-ischaemic from ischaemic injuries is therefore difficult but improved by rigorous clinical ischaemia assessment (82). Increases in cardiac injury biomarkers such as cTn are common during critical illnesses and are not always accompanied by overt myocardial ischaemia, dysfunction or shock. However, myocardial injury in critical illness is independently associated with adverse acute disease outcomes (83, 84). Outside of ischaemia organ-specific extra-cardiac processes such as pulmonary embolism, hyperadrenergic-associated myocardial injuries in a variety of brain injuries, and polytrauma with myocardial contusion can contribute to cTn release (85–87). Notwithstanding that, there are substantial numbers of critically ill patients who see increases in cTn without fitting in to the aforementioned etiologies. These patients may appear completely stable, such as the routinely observed post-operative major surgical patient with a peri-operative myocardial injury, or alternatively may have severe shock and multiple-organ failure due to a variety of potential causes.
Elevated cTn levels in this context are not dependent on a prior history of ischaemic heart disease or congestive heart failure, though levels tend to be higher in those with such a history (88). Similarly while renal impairment can somewhat affect the circulating levels of cTn through decreased elimination, it is erroneous to dismiss new or large increases in cTn to renal dysfunction alone—cTn clearance is less reliant on renal clearance as blood concentrations increase following its release in to the circulation (89, 90). Furthermore, the persistently raised cTn that is commonly seen in CKD outside of critical illness is also likely to be minimally contributed to by renal impairment per se, implying ongoing myocardial injury instead (91). However, the presence of shock, commonly associated with myocardial dysfunction and usually requiring vasoactive/inotropic drugs, is associated with a higher degree of cTn release than non-shocked states (88, 92). Similarly higher degrees of ARDS severity associate with higher levels of cTn, and also associate with worse endothelial function (93). Other observational studies in mixed cohorts of critically ill suggest associations of cTn with IL-6 and inflammatory state, which is further supported by peri-operative studies (94, 95). Sympathoadrenal hyperactivation in the context of inflammation and tissue injury, proposed to induce shock-induced-endotheliopathy (SHINE), is a potentially contributory mechanism to cTn release, though clearly sympathetic activation is not unique to shock and occurs to a greater or lesser degree in all critically ill patients so that it could partially account for cTn elevation outside of shock states too (96–98). Our findings of an independent association of both heart rate and markers of vasoplegia with hyperdynamic left ventricular function (where cTn release is higher than any other echocardiographic phenotype of septic shock) lends further support to the SHINE concept (99).
In line with this angiography studies in patients with post-operative ACS demonstrate little in the way of obstructive coronary artery disease or plaque thrombosis, but instead show a high prevalence of Takotsubo's cardiomyopathy, a catecholamine-mediated condition (100, 101). Similarly a study in critically ill medical patients also showed Takotsubo-type left ventricular apical ballooning in approximately one third of patients (102). Critical illness related myocardial injury may therefore occur through a combination of sympathoadrenal and cytokine induced cardiomyocyte dysfunction and death—these can drive immunogenic and destructive regulated cell death processes within the tissue microenvironment, such as necroptosis and pyroptosis (103, 104). These latter processes are preceded by and result in the further release of large amounts IL-1β and IL-6. Although some histological studies suggest cell death is infrequent in septic myocardial injury, cardiomyocyte death can be driven by very brief ischaemia that does not produce overt infarction (105). This can drive inflammatory death in macrophages in the cardiac microenvironment through cytosolic nucleic acid detection and the triggering of type 1 interferon responses, implying a self-reinforcing loop of cardiomyocyte death and inflammation (106). However, sources of nucleic acids beyond those in necrosing cardiomyocytes also exist, such as circulating mitochondrial and cell free DNA in trauma, and cytosolic DNA due to sub-lethal cell stress (107, 108). The cardiovascular dysfunction that features as part of shock in critical illness is associated with leukocyte infiltration of the myocardium, myocardial oedema, electrophysiological and cardiomyocyte contractile dysfunction (109). These abnormalities are thought to have their origins in enhanced adrenergic signaling with increased oxidative and nitrosative cell stress, abnormalities of mitochondrial structure and function, abnormalities of calcium sparking, and also possibly through alteration of cardiac macrophage behaviors which are normally involved in co-ordinate action potential propagation through Connexin-43 mediated interactions with cardiomyocytes (109–111). Pre-clinical studies have shown disruption of cardiac gap junctions and Connexin-43 in both septic and traumatic cardiac dysfunction, suggesting this may be involved in arrhythmogenesis, such as atrial fibrillation (AF) (112, 113). The finding that β-adrenoceptor provoked S-nitrosylation of Connexin-43 is arrhythmogenic in pre-clinical models of inherited cardiomyopathies is also notable in this regard (114).
New-onset AF occurs in roughly one in five critically ill patients, predominantly in a paroxysmal fashion, and is associated with the risk of in-hospital death (115, 116). It occurs sub-clinically in a high proportion of patients and therefore may affect patients without being detected. One follow-up study of critical illness survivors demonstrated an association of ICU-acquired AF with 1-year post-discharge mortality, while the MANAGE trial which investigated the pharmacological management of peri-operative elevated cTn in those having major non-cardiac surgery (a population which is highly prevalent in surgical ICUs) showed improved outcomes in patients randomized to the anti-coagulant dabigatran (116, 117). In a pre-planned secondary analysis of MANAGE the reduction in the composite primary outcome (a composite of vascular mortality and non-fatal myocardial infarction, non-haemorrhagic stroke, peripheral arterial thrombosis, amputation, and symptomatic venous thromboembolism) was seen to be significantly reduced only for non-haemorrhagic stroke, a diagnostic category with strong aetiological association with AF. Furthermore, when looking to survivors of sepsis, those with new onset AF during their acute illness were confirmed in another study to have higher risks of post-critical illness mortality, stroke and also recurrent AF and heart failure than those without (118). New onset AF may then associate with AF in convalescence, the importance of this being that AF is a risk factor for both stroke and heart failure. Given a link between (post-operative) IL-1β release, abnormal cardiomyocyte calcium handling and AF has already been established, it may be that residual inflammation and metabolic dysfunction are targetable drivers of post-ICU AF (119, 120).
Prolonged exposure of the injured myocardium to catecholamines is associated with fibrotic change (121). Studies in patients with Takotsubo's cardiomyopathy show (iron-laden) monocyte/macrophage infiltrates in the myocardium which would seem to be akin to monocytic infiltrates seen in sepsis (122). Pre-clinical studies also support the role of iron-processing macrophages in septic cardiac dysfunction through the enhancement of nitric oxide production (which leads to a vicious cycle of increased catecholamine dosing through reduced adrenoceptor sensitivity), and is also in keeping with studies in human heart failure (123–126). Although Takotsubo studies show echocardiography normalizes some months after the initial event just as echocardiographic abnormalities normalize soon after sepsis, there is still associated residual poor functional ability, ongoing systemic inflammation, magnetic resonance imaging evidence of diastolic dysfunction and a persistently abnormal energetic status common to heart failure (122, 127). Remarkably heart failure, particularly that with preserved ejection fraction (HFpEF), commonly occurs after critical illness in groups without a prior cardiac history (e.g., after severe pediatric burn) suggesting that the acute intracardiac nitrosative stress that is involved in acute cardiac dysfunction remains latent and plays an important aetiological role in HFpEF pathogenesis (29, 126). It is also notable that the recently devised HFpEF score marks elevated body mass index (BMI) and AF as high scoring risk factors for the likelihood of HFpEF—both of these risk factors are prevalent in survivors of critical illness (128). Thus, patients surviving severe critical illnesses with myocardial injury and prolonged periods of high catecholamine exposure (not necessarily exogenous) are likely to develop myocardial inflammatory infiltrates which in the context of ongoing inflammation, neurohumoral activation [including components of the renin-angiotensin-aldosterone system (RAAS)] and ongoing preferential adipose deposition, could develop in to latent heart failure through immune-mediated mechanisms that stimulate myofibroblast proliferation (124).
The immune response to AKI is associated with increased levels of pro-inflammatory cytokines and immune cell secreted Galectin-3, the latter being implicated in myocardial inflammation and fibrosis in acute and chronic settings (129, 130). Sympathoadrenal and neurohumoral activation occur in the acute phase of severe critical illness, as exemplified by sepsis (131, 132). Galectin-3 expression and secretion is increased in response to the terminal RAAS mineralocorticoid hormone aldosterone, salt intake, and beta-adrenoceptor agonism (133–135). However, these sympathoadrenal and neurohumoral changes are also important in driving chronic disease through oxidative damage. Renal nerve derived noradrenaline and circulating Angiotensin II [Ang II; tightly linked to NOX via the type 1 angiotensin II (AT1) receptor] are recognized vasoconstrictive mediators of chronic hypertension, and have also been shown to induce the formation of renal dendritic cell isolevuglandin protein adducts (136). Antigen presentation by these dendritic cells activates and drives renal T-cell infiltration with attendant renal pro-inflammatory cytokine release and altered renal histology (136, 137). AKI studies also show the macrophage chemokine CCL-14 as a highly predictive biomarker of persistent (>72 h) high grade AKI (138). As higher grade AKI and acute kidney disease (AKD; AKI of 7 days or more duration) is predictive of CKD progression this suggests renal macrophages are involved in imperfect repair processes that cause renal fibrosis (139). This is consistent with data from animal studies of AKI showing that renally secreted transforming growth factor (TGF)-β and damage associated molecular patterns (DAMPs) draw in macrophage and T-cell populations which drive renal fibrotic repair programs as part of a cycle of ongoing tubular cell death and macrophage recruitment and activation (140, 141). One study showed that renal mesenchymal cells such as fibroblasts and pericytes are the main cell population that become activated myofibroblasts, driving extracellular matrix deposition and renal fibrosis in response to TGF-β and platelet-derived growth factor (PDGF) signaling (142). Although this study didn't implicate immune cell infiltration as a major component of CKD progression, the model proposed is nonetheless in keeping with one which involves parenchymal and macrophage derived PDGF and TGF-β as initiators of chronic fibrosis in the early stages of injury and is also similar to fibrotic circuits found in the heart (124, 143). As Galectin-3 stabilizes cell surface cytokine receptors and inhibits T-cell apoptosis it may hinder inflammation resolution across the cardiovascular and renal systems, thus skewing toward maladaptive repair and chronicity following AKI (144).
Importantly, although AKI is likely a multi-faceted process and varies in its pathophysiology depending upon the nature of the initiating injury (e.g., sepsis vs. polytrauma), the transition to fibrotic repair and CKD is transcriptionally preserved among different types of CKD pathophysiology and implicates the immune system, renal parenchymal hypoxia, and pro-fibrotic growth factor signaling molecules such as TGF-β (145–147). The early understanding of idiopathic hypertension as being secondary to changes in the kidneys may therefore partly hold true for post critical-illness induced hypertension, potentially aligns renal microvascular dysfunction and microalbuminuria, and might also explain the beneficial effects on mortality of post-AKI Angiotensin Converting Enzyme inhibitors (ACEi) in observational studies (77, 148, 149). The interaction of vascular, renal and heart diseases as is currently understood for chronic disease therefore also apply in the setting of acute illness and its repair phase, though the specifics of the interaction after critical illnesses are unique to acute injury and repair programs, probably occur at a more rapid pace than occur naturally in aging, but clearly only become a concern for patients that can survive the resuscitative phases of illness (150, 151). Furthermore, the persistence of CKD progression may contribute, alongside inflammatory programs, to sarcopenia and frailty in convalescence (152).
Amongst the most obvious metabolic changes that occur in critical illnesses are early increases in circulating levels of lactate and glucose, but there are other changes which are just as obvious but perhaps less appreciated. The onset of illness leads to sickness behavior favoring the diversion of energy utilization—a reduction in physical activity is partly possible due to a cytokine mediated induction of lethargy and fatigue and also a reduction in appetite that neutralizes the urge to seek food. Changes in appetite are partly regulated by the IL-1β induced hormone growth differentiation factor (GDF)-15 which signals via central nervous system glial cell derived neurotrophic factor (GDNF) family receptors α-like (GFRAL) receptors to induce anorexia (153). The energy furnished by sickness behavior and tissue breakdown is diverted toward powering the febrile response, increased cardiac work, and the systemic response required for tissue repair and immunity. For example, pre-clinical studies suggest that the GDF-15-GFRAL signaling pathway induces centrally driven sympathetic output to the liver which supports hepatic triglyceride output. This may provide an energy substrate for the heart and other tissues despite an IL-1β mediated downregulation of VLDL receptors in the septic heart, thus enhancing tolerance of cardiac injury in the face of sepsis (154–156). Notably, GDF-15 signaling induces muscle breakdown in studies inside and outside of ICU, thus contributing another systemically available fuel source in illness (157–159). Furthermore, the hormone is also seen as a biomarker of mitochondrial health and cellular stress and senescence as it increases with increasing cellular and organismal dysfunction (160). Whether the linkage between high GDF-15 levels and poor ICU outcomes is related to its contribution to cachexia remains unknown, but levels of this hormone remain elevated in ICU survivors for at least 1 week after ICU discharge so that potentially undesirable effects of GDF-15 could persist in convalescence (161).
Concurrent decreased levels of LDL cholesterol and increased levels of triglycerides and free fatty acids (FFAs) have been observed in critically ill patients, and may occur partly due to increased hepatic triglyceride output, early catecholamine driven increases in adipose tissue lipoprotein lipase and hormone sensitive lipase activity, and additional factors that take precedence at different times such as dynamic reductions in non-adipose lipoprotein lipase activity (162–164). Both obesity and critical illness see high levels of macrophage and other immune cell infiltrates in adipose tissue which contribute to adipose-related inflammation and insulin resistance in the outpatient setting; the role of these immune infiltrates beyond possibly serving as immune memory is unknown in critical illness convalescence (162, 165). However, the serine-phosphorylation of insulin-receptor-substrate-1 by cytokines and TLR4 agonists such as FFAs means that convalescent obesity and adverse convalescence metabolic profiles in addition to residual inflammation can be expected to perpetuate insulin resistance (166, 167). As the normal action of insulin is to facilitate glucose uptake via glucose transporter type 4 (GLUT4) channel expression in muscle, and reduce glucose output from the liver through the inhibition of both glycogenolysis and gluconeogenesis, the combination of enhanced hepatic glucose release and the inhibition of muscle glucose uptake leads to relative or overt hyperglycaemia. This acute stress hyperglycaemia has been shown to be associated with increased rates of post-critical illness DM (168). Although it is likely that post-critical illness DM is a latent disease revealed by acute illness for many (169), evidence of increased rates of insulin resistance and metabolic syndrome that relate to the magnitude of traumatic injuries in UK veterans suggests this is not necessarily the case (170). Furthermore, hyperglycaemia and insulin resistance, which can continue alongside low grade inflammation for months beyond the acute phase of critical illness in the absence of overt DM, are known to contribute to sustained vascular dysfunction via endothelial glycocalyx disruption, the inhibition of endothelial nitric oxide mediated vasodilatation and aggressive pro-atherosclerotic myeloid behaviors (171–174). In this context the emergence of GDF-15 as a cardiovascular and all-cause mortality risk biomarker outside of ICU is notable.
Acute critical illness related weight loss involves a combination of adipose, muscle and skeletal compartment biomass depletion. Muscle mass is lost partly due to immobilization but in common with lipolysis, proteolysis also occurs due to the rewiring of systemic metabolism during critical illness, furnishing substrate particularly to the starved tricarboxylic acid cycle (175, 176). Brief reductions in cellular adenosine triphosphate (ATP) levels, which can occur due to many factors such as hypoxia, the activation of glycolysis and closely related β2-adrenergic activation of the Na+/K+-ATPase, result in a decrease in cellular energy charge which can be compensated for by the conversion of 2 molecules of adenosine diphosphate (ADP) to one molecule each of ATP and AMP. The generated adenosine monophosphate (AMP) in turn activates the cellular energy node 5′ AMP kinase (AMPK) which alters metabolic flux to generate more ATP by favoring catabolism over anabolism. Thus, the activation of fat and protein catabolism is intimately linked with the aforementioned alterations in lipid metabolism and insulin resistance, and is tied to glycolysis activation and the attempted restoration of cellular energy charge. Although glycolysis generates pyruvate which usually enters the tricarboxylic acid cycle (TCA), studies from critically ill patients show alterations in pyruvate dehydrogenase activity and defects in fatty acid beta-oxidation, associated with the development of lipid droplets in immune cells and multiple different non-adipose tissue beds (177–179). The biochemical bottleneck downstream of pyruvate and around long chain fatty acid metabolism coupled with the ability to utilize lactate to feed the TCA cycle is possibly why septic cardiac dysfunction is associated with an increased reliance on lactate as an energy substrate and a reduced use of FFA and ketones (180, 181).
Lipid droplets ordinarily harbor contact sites for mitochondria for the co-ordinate running of cellular metabolism, but in the presence of intracellular bacterial infection and presumably pathogen associated molecular pattern (PAMP) sensing, mitochondrial-lipid droplet contacts decrease, altering the flux of lipid metabolism while improving innate immunity through the secretion of antimicrobial lipoproteins (182). In keeping with their important role in immunity, lipid droplets also serve as platforms for nucleic acid detection, enhancing type 1 antiviral interferon responses and are also formed in response to endoplasmic reticulum stress and other forms of cellular stress (183). Therefore, these changes in cellular lipid content could be seen partly as an evolutionary conserved innate immune response to perceived pathogen threat and also a defensive sequestration of backed up cytotoxic lipids such as diacylglycerol. However, they come at a cost of an obligate deterioration in mitochondrial substrate processing when the number of contact sites decrease. Defects of mitochondrial fatty acid delivery and oxidation with subsequent lipid droplet accumulation that occur in the context of insulin resistance superficially appear similar to an accelerated from of metabolic syndrome associated fatty liver disease (MAFLD) and DM. However, acute phase metabolic and organ dysfunction abnormalities in critical illnesses appear to share greater overlap with those responsible for acute fatty liver of pregnancy, such as inherited defects in long chain 3-hydroxyacyl coenzyme A dehydrogenase (LCAD) and carnitine palmitoyl-transferase 1, both enzymes under the regulatory control of peroxisome proliferator-activated receptor (PPAR) pathways. Liver biopsy samples from acute fatty liver of pregnancy patients and from those with liver failure due to mitochondrial damaging therapeutics all converge histologically or biochemically on lipotoxicity and impairment of mitochondrial function at the level of beta oxidation, though the clinical context is clearly liver specific (184). In the case of non-liver critical illnesses such as sepsis, post-mortem histology reveals hepatic steatosis in roughly three quarters of patients (185). Furthermore, the same PPAR-α regulated defects in lipid handling and beta oxidation have been found in small cohorts of critically ill patients and a much larger cohort of stable cirrhotics with multi-organ failure (177–179, 186). This suggests that impaired beta oxidation occurs in the liver and also in a systemic manner in multiple organ failure and that the metabolic abnormalities that cause steatosis and an acute liver failure in AFLP merely signpost how the same process is likely to occur in other organs in critical illnesses (185, 187). In support of this abundant lipid droplets and impairments in fatty acid oxidation have also been found in the septic heart, acutely injured kidney and the skeletal muscle of critically ill patients, while reduced expression of PPAR-α has also been found in patients with poor sepsis outcomes (188–191). Findings of beta oxidation impairment associating with CKD stage, and lipid droplet accumulation in diabetic nephropathy highlight that these acute changes of metabolism in critical illness might lead to ongoing mitochondrial and chronic organ dysfunction in the context of residual inflammation and insulin resistance through lipotoxicity (192, 193). The importance of these changes and how they are also linked with acute changes in mitochondrial responses to stress and late immune and organ dysfunction are discussed further below.
Critical illness occurs when detected threat or injury, synergises with the dynamic host response to it, so that critical illness necessarily is associated with substantial activation of any of a number of pattern recognition receptors such as TLRs. The nucleotide-binding domain leucine-rich repeat and pyrin domain containing receptor 3 (NLRP3) inflammasome operates as a threat-detecting intracellular platform that is required for the formation of IL-1β from its precursor pro-IL-1β, and is instrumental in its cellular release through gasdermin membrane pores in response to detected DAMPs, PAMPs and harmful alterations in the cellular interior milieu, such as alterations in redox status and mitochondrial dysfunction (194). In keeping with this it has been suggested that familial Mediterranean fever, an autoinflammatory condition known to associate with cardiovascular disease and high IL-1β secretion through pyrin overactivity, may have emerged through selection pressures for enhanced resistance to epidemic infections such as Yersinia pestis (195, 196). Importantly, the induction of NLRP3 assembly and pro-IL-1β (and pro-IL-18) production is dependent on a priming signal which in modern times might originate from persistent low grade systemic inflammation as occurs in obesity or aging, or indeed following a first-hit acute inflammatory insult such as surgery/trauma or early infection. Secretion of the pro-inflammatory cytokine IL-1β (and its IL-1 superfamily member IL-18) is thus intricately linked to cellular stress and regulated, necrosis-like cell death (pyroptosis) which may be partly responsible for myocardial injuries that are seen post-surgically and in association with sepsis and other critical illnesses (197, 198). Although this implicates this pathway in pyroptosis, other forms of regulated cell-death (e.g., necroptosis) also implicated in cardiomyocyte death share a high degrees of cross-talk with it and may also be up or down-regulated (199). NLRP3 activation in cardiomyocytes has been shown in pre-clinical models to trigger spontaneous AF, and is upregulated in the atrial cardiomyocytes of patients with post-operative AF, implying that increased activation of this signaling pathway acutely and during convalescence may be an important contributing factor to paroxysmal AF and its consequences (120, 200).
IL-1β signals via its cytokine receptor IL-1R and induces the production of IL-6, which leads to C-reactive protein (CRP) production by the liver. In the aftermath of critical illness, residual inflammatory activity (measured by high sensitivity (hs)CRP) is very common and is associated with higher rates of metabolic syndrome and an elevated risk of death from cardiovascular causes (8, 73, 170). Other studies suggest both IL-1β and IL-6 levels remain elevated in over 30% of ICU survivors at 3-months while CRP is detectable in at least 60%, with correlation between functional outcomes and inflammatory state—this is partially in keeping with pre-clinical studies that suggest IL-1β activity is part of a disease tolerance programme that comes at the cost of fibrosis, cachexia and of course, cardiovascular disease risk (201, 202). As IL-1R mediates mtDNA release and a nucleic acid sensing (antiviral) response which should trigger cellular resilience through tolerance (203, 204), the involvement of persistent inflammation through IL-1β in chronicity may be a function of a dysregulated genetic tolerance programme that leads to a vicious cycle of chronic infection, inflammation and fibrosis (205). In addition to the primary findings of CANTOS where IL-1β repression resulted in a reduction in cardiovascular morbidity and mortality, residual inflammation after treatment with canakinumab (assessed through IL-6 and IL-18 tertiles) also associated with incident cardiovascular diseases (206). Furthermore, genetically determined decreases in IL-6 signaling reduce the likelihood of cardiovascular events in patients with IL-1β related cardiovascular risk factors, but not in those without (207). It therefore seems likely that IL-1β, IL-6 and possibly CRP activity after critical illness perpetuate vascular dysfunction and cardiovascular disease just as they do in the outpatient setting, and this is likely a consequence of ongoing processes linked to cell stress, inflammatory cell death and the engagement of a disease tolerance programme. Furthermore, pro-fibrotic AngII has also been shown to activate the NLRP3 inflammasome via AT1 receptor ligation, downstream NOX activation, with attendant vascular remodeling and renal inflammation (208, 209). Similarly, β1-adrenoceptor activation can also trigger myocardial NLRP3, contributing to immune cell ingress and myocardial fibrosis (210). In this regard it is notable that murine models of sepsis and acute inflammation show significant acceleration of the atherosclerotic process, with increased vascular macrophage infiltration seen in the recovery stage correlating with acute stage upregulation of inflammatory cytokines such as IL-6, and adhesion molecules in vasculature (42, 43). This finding in mice is further corroborated by epidemiological human data showing increased rates of cardiovascular pathology for many years following sepsis (23).
The recruitment of innate immune cells to the vascular and renal compartments is a phenomenon that is well-recognized in atherosclerosis, AKI and CKD (211, 212). However, many studies in humans and animals have also supported the concept of T-cell and macrophage involvement in the pathogenesis of hypertensive vascular dysfunction (213). CD4+ (Th1 & Th17), CD8+, and γδT-cells have been shown to be important in driving hypertensive changes while regulatory T lymphocytes (Treg)and myeloid derived suppressor cells (MDSC) ameliorate hypertension related vascular dysfunction. Renal infiltrates of cytotoxic CD8+ T lymphocytes are also associated with vascular dysfunction and hypertension. Furthermore, it would appear that the CD8+ T-cells that are found in hypertensives have a greater degree of immunosenescence than age and sex-matched controls, and as such they produce greater quantities of Interferon (IFN)-γ, tumor necrosis factor (TNF)-β, granzyme B, and perforin (214). Studies of the immune landscape of symptomatic and asymptomatic atherosclerotic plaques also show large numbers of T-cells that are primed for granzyme and perforin secretion (212). These observations are important for understanding how cardiovascular and renal diseases might continue to progress in the immediate post-critical illness period. Immunosenescence is a characteristic of critical illness survivorship, and the abnormalities in the immune system with persistent inflammation take more than a year to return to normal levels (48, 215). T-cell Programmed Death (PD)-1 receptor positivity is a mark of exhaustion/anergy is both common in vulnerable atherosclerotic plaques and is a result of persistent inflammatory and hypoxic stimuli, such as that occurring during critical illness and infections (212, 216). Furthermore, findings that immunosenescence alone can drive tissue aging (217), and that biological age can be accurately predicted by both cardiovascular biomarker scores and an associated immune-aging score based on various markers of immunosenescence, such as T-cell PD-1 receptor positivity, lends further support to the hypothesis that the immunesenescence that follows critical illness results in a drive toward both cardiovascular disease and aging (218, 219). It is also notable that rates of viral reactivation in critical illness are high, though it is unclear whether re-activation of cytomegalovirus (CMV) and Epstein Barr Virus (EBV) exacerbate the T-cell immune dysfunction or are its epiphenomena (48, 220, 221). There is evidence that CMV infection is associated with hypertension and vascular dysfunction, and that viral persistence is associated with alterations in T-cell distributions and function similar to those found in persistent critical illness (222–225). Importantly, suppression of viral reactivation in critical care is safe and feasible, but so far has not shown significant mortality benefits, suggesting that latent processes driving functional immunosuppression are of greater importance than CMV disease in driving mortality and morbidity (226).
The traditional model of the innate immune system describes a system without any retained memory function. This concept has radically changed following the recognition that epigenetic reprogramming of myeloid precursors occurs after innate immune stimulation, resulting in a shift in innate immune responses to either trained or tolerant states (227). The proposed mechanisms for this reprogramming are sophisticated and relate immunometabolic changes that occur in infection with those that are seen in vascular dysfunction and atherosclerosis. These immunometabolic changes are intricately tied to the redox state of the cell (Figure 1).
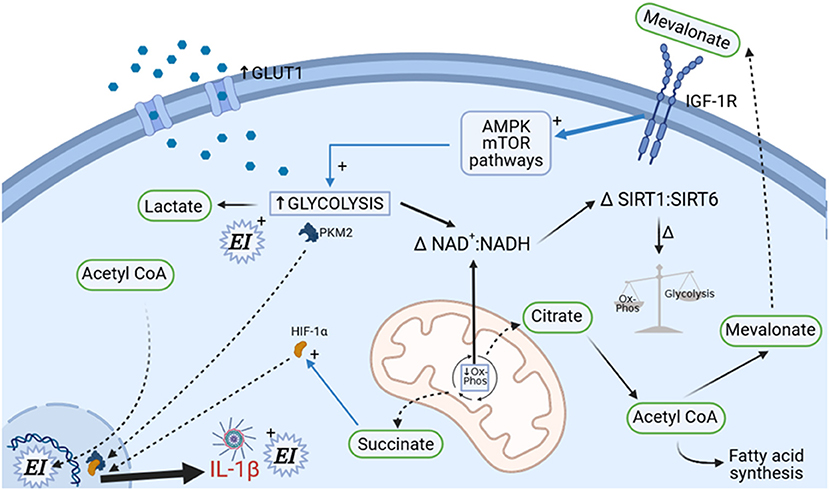
Figure 1. Immunometabolic determination of immune cell polarization. A complex set of metabolic changes occur under cell stress, involving upregulation of GLUT1 transporters, upregulation of cholesterol biosynthesis, decrease in mitochondrial oxidative phosphorylation (causing increased succinate formation), and upregulation of glycolytic enzyme activity. Cellular redox status (reflected in NAD+/NADH+ ratio) is consequently altered. These changes determine future immune cell behavior through alterations in epigenetic imprinting (EI), but also determine current immune cell cytokine responses via HIF-1α. The involvement of mitochondria (center in picture) is fundamental to this process; alterations in fusion/fission status under the control of mTOR, PGC1α, and Drp1 control immune cell functional state and might also directly contribute to organ dysfunction (not shown).
The metabolic response to stress and infection involves altered cellular patterns of glycolysis and oxidative phosphorylation along with alterations in the NAD+/NADH balance of the cell. The glycaemic response to sepsis is well-recognized and has been clearly linked to upregulation of glycolytic pathways in immune cells with involvement of cellular energy sensors such as mechanistic target of rapamycin (mTOR) and AMPK (228, 229). Enhanced glycolysis triggers alterations in the TCA cycle resulting in mitochondrial succinate generation and also driving nuclear translocation of the glycolytic enzyme pyruvate kinase M2 (PKM2). Nuclear PKM2 dimerises and forms a complex with the transcription factor hypoxia inducible factor (HIF)-1α (which is activated by succinate), resulting in acceleration of the Warburg effect, greater lactate production, and pro-inflammatory cytokine generation in macrophages. This HIF-1α related phenomenon occurs in both the atherosclerotic and septic settings and may also mediate enhanced neutrophil mobilization from the bone marrow through lactate (230–235). These metabolic changes are associated with increased intracellular concentrations of mevalonate, a product of the cholesterol biosynthesis pathway. Mevalonate signals in an autocrine and paracrine fashion via insulin-like growth factor-1 (IGF-1) which further enhances glycolysis via sensitization of mTOR and AMPK. Crucially, mevalonate also translocates to the cell nucleus where it is responsible for epigenetic modification of the monocyte and macrophage genome such that host defense molecules and cytokine response (particularly IL-1β) is more vigorous on subsequent exposure to stressors. Statins inhibit mevalonate production and in addition to their effect on circulating cholesterol also stabilize atherosclerotic plaques through a reduction in monocyte and plaque macrophage trained immunity and hyperinflammation (234, 236). Whether their effects on monocyte and macrophage immunometabolism is an important mechanism in their potential action in hyperinflammatory ARDS is unclear at this time (237).
The macrophage phenotype of primed glycolysis (as opposed to mitochondrial dependent oxidative-phosphorylation) coupled with appropriate histone modifications on the macrophage genome and a tendency to IL-1β production has been found in the cells of vulnerable atherosclerotic plaques in several studies—the so-called M1 phenotype (238). Furthermore, in addition to LPS/TLR4 and β-d-Glucan/Dectin-1 signaling (sepsis activated pathways), oxidized LDL, aldosterone and catecholamines have also been shown to trigger trained immunity (227, 239, 240). In order for trained immunity to persist, it must be imprinted in the bone marrow niche. This training process occurs in haematopoietic progenitor cells causing myeloid precursors to be released into the circulation primed in this state (241). In summary, an epigenetic training of monocytes, macrophages and T-cells (see below) which is associated with atherosclerosis and vascular dysfunction follows on from the altered redox status and metabolite profile that itself results from the cellular stress of critical illness and infection (242).
When Ox-phos is interrupted, such as in ischaemia, the TCA intermediate succinate accumulates (243). In stressed cardiomyocytes succinate signals via its receptor GPR91, leading to mitochondrial fission and dysfunction, and cardiomyocyte apoptosis (244). This has been shown to be an important mechanism in myocardial post-ischaemia-reperfusion dysfunction, but whether this also is true for septic cardiac dysfunction remains unknown. Mitochondrial fission involving succinate, Dynamin related protein-1 (Drp1) and the mTOR regulated transcription factor PPAR-γ co-activator 1 (PGC1α), is part of a pattern of metabolic change occurring under cell stress that is required to drive the switch from resting state to the pro-inflammatory phenotype, thus linking altered mitochondrial metabolism to cell stress/death and pro-inflammatory IL-1β secretion (245–248). On the other hand, fused mitochondria show more efficient oxidative phosphorylation and associate with the endoplasmic reticulum (notably, the site of lipid droplet formation) where they sequester calcium. This is associated with the alternately activated (M2) macrophage phenotype and Th2 responses (249–251).
However, the epigenetic imprinting of T-cells relates to their anergic/exhausted status too, so that epigenetic reprogramming of immune cells as previously described is not solely an innate immune process, nor one which always enhances an inflammatory immune response (252). Indeed the late phase of acute illness which is characterized by T-cell anergy and PD-1 upregulation is also accompanied by a switch of leukocytes back from a glycolytic pro-inflammatory state to an immunotolerant mode where ox-phos is also inhibited (this is as opposed to an ordered return to ox-phos with preserved glycolysis) (253, 254). This may provide a metabolic explanation for the increased propensity to infections seen in late stage critical illness—a broad dysfunction of immune cell metabolism means cells are sluggish in responding to a secondary or residual stimulus due to a switch at the cellular level. This presumably costs the host less energy than continual crudescence, but possibly represents a high risk strategy of immune tolerance that may also result in reduced pathogen clearance (255). This is somewhat in keeping with ideas regarding the two strategies by which organisms handle infection, resistance and tolerance. Resistance refers to host strategies that ensure survival by actively reducing pathogen burden, whereas tolerance programs improve host survival at any given level of pathogen load through resilience. Although complimentary, these strategies may dynamically change in dominance depending on the stage of illness, the switch of strategy being determined by energetic considerations that have been selected through evolution (256, 257). Studies show that reductions in complex 1 activity of the electron transport chain in mitochondria, whether through hormetic processes or ribosomally targeted drugs (258, 259), results in changes in mitochondrial substrate handling that increase disease tolerance. In the case of exposure to LPS, ox-phos and fatty oxidation decrease with subsequent reductions in pro-inflammatory epigenetic imprinting through histone acetylation and a concurrent switch from pro-inflammatory M1 to tolerant M2 macrophages. These metabolic changes are in keeping with a reduction in mitochondrial oxygen consumption that is under the control of aforementioned mTOR and PGC-1α (247).
This immunoparesis would tend to decrease the risk of autoimmunity to neoantigens following severe acute inflammation—the evidence that autoimmunity can occur in response to anti-PD-1 therapy in cancer, supports this response representing a deleterious but programmed protective mechanism (255, 260). The importance of epigenetic mechanisms linked to cell metabolism is further demonstrated by the fact that this bi-directional, NAD+ dependent switch of ox-phos to glycolysis, and the switch to T-cell anergy, is mediated by histone modifying sirtuin enzymes at the epigenetic level (261, 262). It is then tempting to speculate that in those critically ill patients with the worst outcomes (death or chronicity) an early pro-inflammatory stage could result in decreases in cellular NAD+ and a subsequent failure of SIRT3 to prevent LCAD and glycolytic enzyme hyperacetylation—this would not only result in impaired fatty acid oxidation and subsequent cellular lipid accumulation, but also the acetylation of numerous mitochondrial proteins and glycolytic enzymes with subsequent immunoparesis and chronic organ dysfunction, as seen in sepsis and CKD (177, 190, 193, 253, 263).
Critical illness presents multiple stresses on the bone marrow. Immobility, inactivity and sedentarism are associated with prolonged increases in bone marrow fat content, which leads to increased leptin signaling and inflammatory monocytic output (264, 265). Furthermore, there is sustained cytokine, glucocorticoid and sympathetic nervous system activity that places additional replicative pressure on this niche with a resultant myeloid skewing to its output (266, 267). Dysglycaemia, which commonly accompanies the insulin resistance of severe illness and continues in convalescence, also leads to increased myelopoiesis with inflammatory monocytosis and neutrophilia, at least one mechanism being via S100A8/A9-mediated activation of the receptor for advanced glycation end-products (174). Sleep, which normally places a brake on bone marrow replication rates, is severely disrupted during severe critical illnesses and remains disrupted for a long time after and this will further compound bone marrow replicative pressure in convalescence (268, 269). The combined effect of these disruptions during critical illness convalescence would be to result in a sustained increase in myeloid progenitor replication rates, a myeloid skewed output with an inflammatory monocyte and neutrophil cohort, and the propagation of atherosclerotic disease and myocardial injuries.
The burden of somatic mutations in the human body is greater than previously appreciated (270). Physico-chemical attack on the genome, for example by ultraviolet radiation and cigarette smoking, results in DNA damage and consequent repair mechanisms which are imperfect, thus resulting in mutation. This also occurs under other circumstances where free-radical mediated damage is increased, such as under conditions of severe inflammation as seen in critical care (271, 272). Emergency myelopoiesis is a feature of the acute response to inflammation and is dependent on epigenetic modifying enzymes such as Tet2 (273). However, inflammation favors not only myelopoiesis and increased rates of somatic mutation, but also increases the probability of myeloid clonal survival (274). A specific form of myeloid clonal survival with a blood variant fraction of 2% or more [termed clonal haematopoiesis (CH) of indeterminate potential (CHIP)] is very strongly associated with driver mutations in genes for epigenetic regulators such as TET2, DNMT3A, ASXL1, and also in genes that code for signaling molecules such as JAK2 and TP53 (275). CHIP is also associated with age, being highly prevalent above the age of 60 years, coupled with an increased incidence of cardiovascular disease (276). These clonal changes are of indeterminate potential as they are preleukaemic, though only a small proportion of those with CHIP go on to develop myelodysplastic syndromes and acute myeloid leukaemias. These preleukaemic changes are driven by TLR2-sensed microbial signals as may be found in critically ill patients with infections or with gut barrier dysfunction (277). Studies support a bi-directionality of causation between inflammation (vascular or otherwise) and CHIP, and as CH exacerbates insulin resistance in preclinical models (278, 279), the hypothesis that critical illnesses can drive CHIP must be taken seriously (280).
Both atherosclerosis and heart failure are mechanistically linked with clonality, Tet2 and Dnmt3a loss of function, consequent increased activation of the NLRP3 inflammasome and IL-1β and IL-6 production in monocytes (281–283). IL-1β antagonism, which mechanistically should reduce IL-6 and hsCRP, reduced cardiovascular event rates only in those with therapy sensitive decreases in hsCRP in the CANTOS trial (44). Considering that Tet2 is required for IL-6 driven inflammation repression whereas Dnmt3a represses mast cell inflammatory responses (284, 285), if treatment responders in CANTOS had a high incidence of CHIP while the treatment non-responders didn't, a mechanistic pathway with supportive data exits to explain this observation (286). This latter paradigm where CHIP drives post-acute illness adverse outcomes is supported by survival data from patients undergoing trans-catheter aortic valve implantation (287). The finding that CHIP is a cardiovascular risk factor, increases the propensity to certain types of infection, and that Tet2 dysfunction leads to sub-optimal emergency myelopoiesis (273), means investigating whether CHIP has a role in NLRP3 hyperactivation, peri-operative and ICU-acquired non-ischaemic myocardial injuries, the inability to resolve inflammation is a high priority particularly as it may be important targetable driver of hyperinflammation in both the acute and survivor phases of critical illnesses (288).
Studies in primary hyperaldosteronism show that mineralocorticoid receptor (MR) agonism without an acute pathological insult is deleterious to the cardiovascular and renal systems (289). Renal, cardiac and vascular T-cell infiltration, IFN-γ production and deleterious remodeling in the context of hypertension has been shown to be modifiable by action at T-cell mineralocorticoid receptors (MR), supporting the role of immune infiltrates in cardiovascular and renal chronic disease progression (290, 291). Similarly MRs are present in myeloid cells and are involved in polarization to a pro-inflammatory phenotype with receptor blockade by antagonists reducing cardiac hypertrophy, fibrosis, and vascular damage following pathological insults (292). Fibrotic remodeling in the context of MR agonism can occur without clinical hypertension, suggesting that vascular dysfunction and tissue remodeling is a feature of a specific immune response which often predates hypertensive change (293). This suggests that a disease process producing similar immune changes, such as critical illness, could also begin the cascade of tissue remodeling resulting in these disease phenotypes without overt signs of hypertension. It is also noteworthy that adverse tissue remodeling through MR activation depends on immune cell and adipose released neutrophil gelatinase-associated lipocalin (NGAL) (294, 295). NGAL is substantially upregulated during critical illness and has also been found at elevated levels in the bloodstream of patients with heart disease (independent of renal function) where it carries prognostic significance (294, 296–298). In addition to the genomic MR pathway, there are additional non-genomic MR pathways that are related to adverse tissue remodeling—namely the transactivation of epithelial growth factor receptors (EGFR), the IGF-1 receptor pathway, and the AT1 receptor/NOX pathway (299). NOX, AT1, and IGF-1 have already been described as carrying key roles in the proposed pathophysiology above and are heavily involved in immunometabolic change and epigenetic training.
The MR also has an important role in the regulation of the cardiovascular system at the level of the paraventricular nucleus (PVN) of the hypothalamus. Hypothalamic AT1-receptor activation and endotoxin-mediated TLR activation, as occurs in sepsis leads to increased hypothalamic NOX and NFκB activity, which in turn has been shown to deleteriously affect autonomic tone and cardiac remodeling (300). It also appears that circulating pro-inflammatory cytokines have the same effect on the PVN and this is mediated by perivascular macrophages (301). These changes in hypothalamic NOX, nuclear factor kappa B (NFκB), the associated sympathetic overactivity and dysautonomia are seen in aging, in sedentarism, in hypertensives, after myocardial infarction and in heart failure, and it is particularly notable that microglia (central nervous system macrophages), play a prominent role in this (302–305). Furthermore, continual centrally mediated sympathetic activation has been found in pre-hypertensives where it contributes to deleterious cardiovascular remodeling, and patients with dysautonomia have been shown to have cardiac hypertrophy which is mediated by MR mediated supine hypertension despite normal daytime blood pressure and aldosterone levels (306, 307). Activation of hypothalamic MRs enhances the AT1-mediated effect on NOX while antagonism of hypothalamic MRs is associated with improvements in dysautonomia, reduced hypothalamic NOX activation and reduced hypertension (308–310). This central mechanism of dysautonomia, partially driven by oxidative stress is likely an important one for the recovering critically ill patient who often has encountered a degree of autonomic dysfunction (311). Further evidence that this may be of significance is that significantly increased free radical mediated apoptotic change has been found in the microglial fraction of hypothalamuses of septic shock patients (312).
The acute response to severe illness involves activation of the sympathetic and renin-angiotensin-aldosterone systems. Different studies suggest aldosterone levels are either higher or lower than normal in severe disease (313, 314). However, pathological MR agonism is not solely an aldosterone related phenomenon. Cortisol and aldosterone both have the same affinity for and can signal via the MR. Furthermore, functional plasma levels of cortisol are 100-fold higher than those of aldosterone. Intracellular enzyme systems (11-β hydroxysteroid dehydrogenases I & II) and cellular redox status ensure aldosterone selectivity for the MR in health (315). During physiological stress increased circulating cortisol levels induce these enzymes so that intracellular cortisol levels increase further, whereas an alteration of the cellular redox status affects the mechanisms of transcriptional repression that normally inhibit cortisol-dependent pathways of MR activation (316, 317). Therefore under stress, peripheral and central MRs can become activated by glucocorticoids, as suggested by experimental studies (308, 318). The beneficial effects of MR antagonists in patients with chronic cardiovascular pathology where plasma aldosterone levels are normal is suspected to be mediated in this way (319). Notwithstanding that, the picture during critical illness is uncertain as dynamic changes in plasma cortisol and aldosterone during this time are well-recognized though poorly understood, and furthermore tissue resistance to steroid hormones in sepsis is strongly suspected to exist (320). Nevertheless, it is clear that the MR is a receptor with immunomodulatory actions and one which modifies both redox status (via NOX & AT1) and is involved in tissue remodeling (via EGFR/IGF-1 pathways). This and the known adverse cardiorenal outcomes following severe critical illnesses is the basis for the anticipated Dapa-SSCILL randomized controlled trial (12).
During periods of shock, large volumes of intravenous resuscitation fluids are used by clinicians to restore the patient's intravascular volume. Although some degree of fluid resuscitation is almost invariably necessary, the optimal level for any individual patient is not defined. Furthermore, the use of resuscitative fluid therapy leads to atrial stretch and natriuretic peptide induced endothelial dysfunction, which may contribute to further acute organ dysfunction, and persists to some degree during convalescence (321, 322). TLR4 agonists such as endotoxin and resuscitation fluid volume have also been shown to induce (or associate with) the transformation of mature and progenitor endothelial cells to a fibroblast-like form, in keeping with the process of endothelial-to-mesenchymal transition (EMT) (323, 324). This important process which also involves TGF-β signaling is suspected to be centrally involved in multiple cardiovascular pathologies including atherosclerosis, leading weight to the idea that acute severe disease transitions to chronic (325).
Following resuscitation there is often a prolonged period where intravenous fluids continue to be delivered for either maintenance or as part of drug delivery. This is an enormous sodium burden for the critically ill patient, which is not easily mobilized (326, 327). Although plasma levels of sodium vary in critically ill patients, it is unclear whether vascular interstitial sodium concentrations are in the same range as plasma, or like hypertensive patients are potentially much higher. This has not been widely studied in the critically ill population but if interstitial sodium is and remains elevated as suggested by one small study in patients with AKI, then this elevation could also trigger antigen presenting dendritic and effector Th17 cells to exacerbate deleterious immune recruitment to the vasculature as in hypertensive patients (213, 328, 329).
Taking all this evidence together suggests that over the course of critical illness, severe acute inflammation (ROS generation, altered self-antigen presentation, high circulating levels of IL-1β, IFN-γ, and other pro-inflammatory cytokines), Ang II, catecholamines, and potentially the sodium load of fluid resuscitation synergise and produce an adverse cardiovascular and renal immune infiltrate. This creates (or accelerates pre-existing) cardiovascular and renal disease by virtue of an immunometabolic driven, long-lived epigenetic reprogramming of the immune system which in turn drives chronic low grade inflammation beyond the phase of critical illness. This chronic inflammation continues to drive pathology just as it does in patients with chronic diseases who have never had a critical illness but who go on to develop T2DM, CKD and cardiovascular diseases. The aforementioned mechanisms would imply a dose response relating to the severity of inflammation and likelihood of consequent cardiovascular and renal disease—indeed this is seen. Furthermore, they provide explanations for chronic illness pathologies in critically ill patients despite the absence of traditional risk factors, and despite significant periods of time where blood pressure is not elevated (Figure 2).
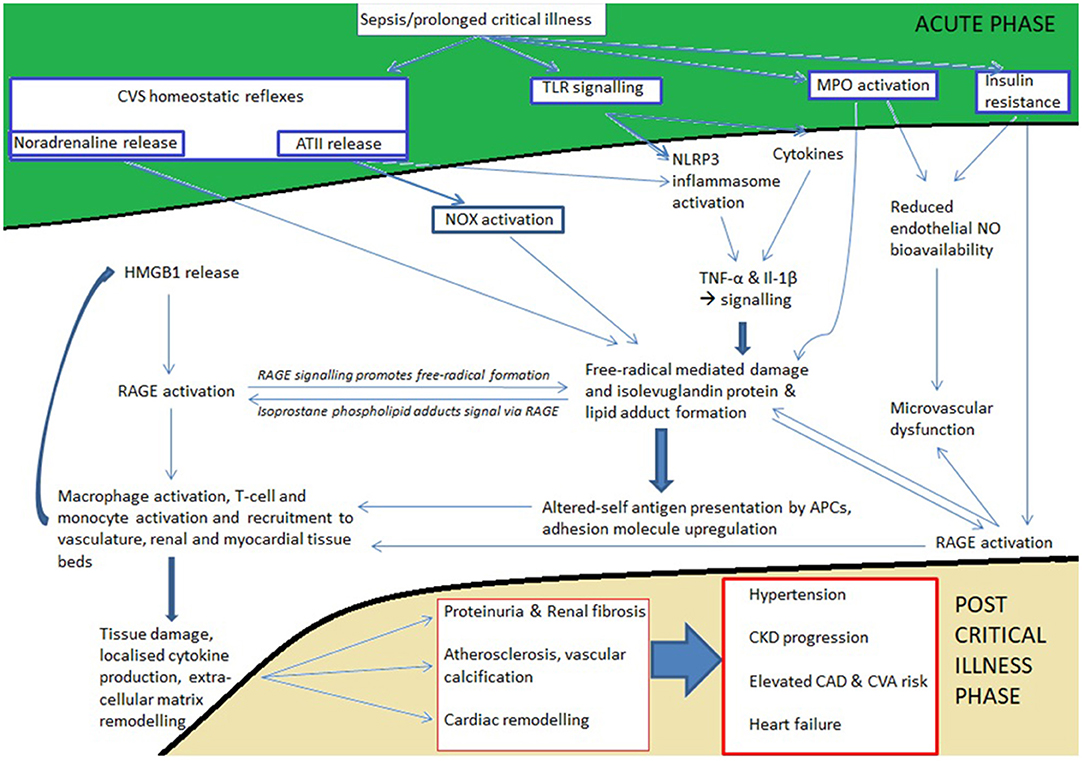
Figure 2. Outline of how acute inflammation leads to chronicity affecting the cardiovascular and renal systems. Areas in green are triggered during the onset of acute inflammation whereas areas in gold become apparent during survivorship. The white area between acute and chronic phases represents the cellular processes precipitated (de novo disease) or accelerated (disease already exists) by acute inflammation; these processes continue during survivorship because antigen presentation and deleterious immune infiltrates have become established. Note that none of the mechanisms in the white area are reliant on traditional risk factors of hypertension, dyslipidaemia, obesity, and diabetes for triggering the chronic diseases in gold. Nevertheless, the recently elucidated inflammatory and immune mechanisms underlying vascular dysfunction, hypertension, and atherosclerosis are central to triggering these chronic diseases. What is not specifically highlighted is the legacy effect of epigenetic reprogramming (trained immunity) due to the effects of altered cellular redox status and accelerated glycolysis on macrophages, monocytes, and T-cells.
The COVID-19 pandemic resulted in a global surge of often lengthy ICU admissions, predominantly for respiratory failure and ARDS. In common with ARDS, sepsis and other severe organ dysfunction syndrome patients, many critical-COVID-19 survivors have been left with chronic problems that continue to impede function at 2-year follow-up (330). Although critical-COVID-19 survivors frequently describe new problems, predominatly related to dyspnoea and limited functional capacity, some of these problems arise from SARS-CoV-2 viral infection with no, or only mild-moderate acute symptoms and therefore may not be related to critical illness per se (331). Nonetheless, studies clearly show that critical COVID-19 disease is associated with a significantly higher likelihood of a range of post-infection CVS diseases in addition to cognitive and functional impairments when compared to less severe disease, even in patients who have never experienced CVS disease before (26, 330). Although some of these functional problems may relate to the degree of slow-resolving pulmonary damage that was acutely sustained (332), this does suggest that critical illness and its associated biology induce maladaptive pathways of disease resolution in COVID-19 convalescence that go beyond those that are specific to long-COVID. Such common pathways might include those highlighted earlier which are involved in AKI to CKD transition (333, 334). Similarly, though SARS-CoV-2 infects cells by entry through the ACE receptor (335), direct pathogen-mediated CVS disease and inflammatory cell death with continued endotheliopathy (possibly in the face of a latent, active, viral reservoir) appears a convergent mode of post-ICU CVS dysfunction that may be especially pronounced after SARS-CoV-2 infection (336–338). Critical COVID-19 disease is also an important determinant of subjective long-term non-recovery, and residual inflammation (CRP) and coagulopathy are associated with worse subjective functional and cognitive outcomes too (332, 339). Importantly, the link between interferon-mediated (antiviral) inflammation, neurodegeneration and associated cognitive decline is increasingly appreciated but may also be a consequence of COVID-19 other inflammatory insults seen in ICU (e.g., trauma) that share similarity with nucleic acid activated stress pathways (340–342). These interferon-driven processes appear to be persistently active due to ongoing immune activation in those with long-COVID but it is unclear whether a similar persistence of interferon signaling occurs in convalescence after other critical illnesses (343). However, as in other critical illnesses, T-cell exhaustion is more likely found in severe acute COVID-19 disease (344), continues in convalescence, and is associated with mitochondrial and cellular metabolic dysfunction (345). It is therefore likely that further COVID-19 disease, long-COVID and post-critical COVID-19 recovery research will continue to highlight new and shared pathways of post-ICU CVS and other chronicities.
The high levels of inflammation and reactive oxygen species (ROS) seen in severe critical illness generate an immune-mediated onset of de novo, or acceleration of pre-existing, cardiovascular disease. The CVS and renal disease that is seen therefore does not necessarily relate to traditional risk factors which tend to be normal or depressed acutely (such as blood pressure, lipid profile, HbA1c) at the onset of critical illness survival. The presence of continued low grade inflammation and immune changes that are slow to resolve following critical illness create an environment where adverse tissue remodeling continues, endothelial dysfunction is sustained, metabolic syndrome can take hold, and the risk of CVS and renal disease is elevated. In order to arrest disease progression an anti-inflammatory therapy is mandated which is sensitive to the fragile and immunosuppressed state of the critically ill.
All authors contributed equally to writing, proof reading, and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Adhikari NKJ, Fowler RA, Bhagwanjee S, Rubenfeld GD. Critical care and the global burden of critical illness in adults. Lancet. (2010) 376:1339–46. doi: 10.1016/S0140-6736(10)60446-1
2. Needham DM, Feldman DR, Kho ME. The functional costs of ICU survivorship. Am J Respir Crit Care Med. (2011) 183:962–4. doi: 10.1164/rccm.201012-2042ED
3. Mikkelsen ME, Still M, Anderson BJ, Bienvenu OJ, Brodsky MB, Brummel N, et al. Society of critical care medicine's international consensus conference on prediction and identification of long-term impairments after critical illness. Crit Care Med. (2020) 48:1670–9. doi: 10.1097/CCM.0000000000004586
4. Arulkumaran N, Reay H, Brett SJ, on behalf of the JLA Intensive Care Research Priority Setting Partnership. Research priorities by professional background - a detailed analysis of the james lind alliance priority setting partnership. J Intensive Care Soc. (2016) 17:111–6. doi: 10.1177/1751143715609954
5. Wunsch H, Guerra C, Barnato AE, Angus DC, Li G, Linde-Zwirble WT. Three-year outcomes for medicare beneficiaries who survive intensive care. JAMA. (2010) 303:849–56. doi: 10.1001/jama.2010.216
6. Wang CY, Calfee CS, Paul DW, Janz DR, May AK, Zhuo H, et al. One-year mortality and predictors of death among hospital survivors of acute respiratory distress syndrome. Intensive Care Med. (2014) 40:388–96. doi: 10.1007/s00134-013-3186-3
7. Ranzani OT, Zampieri FG, Besen BA, Azevedo LC, Park M. One-year survival and resource use after critical illness: impact of organ failure and residual organ dysfunction in a cohort study in Brazil. Crit Care. (2015) 19:269. doi: 10.1186/s13054-015-0986-6
8. Yende S, D'Angelo G, Kellum JA, Weissfeld L, Fine J, Welch RD, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med. (2008) 177:1242–7. doi: 10.1164/rccm.200712-1777OC
9. Prescott HC, Sjoding MW, Langa KM, Iwashyna TJ, McAuley DF. Late mortality after acute hypoxic respiratory failure. Thorax. (2017) 73:618–25. doi: 10.1136/thoraxjnl-2017-210109
10. Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: propensity matched cohort study. BMJ. (2016) 353:i2375. doi: 10.1136/bmj.i2375
11. Shankar-Hari M, Harrison DA, Ferrando-Vivas P, Rubenfeld GD, Rowan K. Risk factors at index hospitalization associated with longer-term mortality in adult sepsis survivors. JAMA Netw Open. (2019) 2:e194900. doi: 10.1001/jamanetworkopen.2019.4900
12. Bangash MN, Owen A, Alderman JE, Chotalia M, Patel JM, Parekh D. COVID-19 recovery: potential treatments for post-intensive care syndrome. Lancet Respir Med. (2020) 8:1071–3. doi: 10.1016/S2213-2600(20)30457-4
13. Mayr VD, Dunser MW, Greil V, Jochberger S, Luckner G, Ulmer H, et al. Causes of death and determinants of outcome in critically ill patients. Crit Care. (2006) 10:R154. doi: 10.1186/cc5086
14. Fleischmann-Struzek C, Rose N, Freytag A, Spoden M, Prescott HC, Schettler A, et al. Epidemiology and costs of postsepsis morbidity, nursing care dependency, and mortality in Germany, 2013 to 2017. JAMA Netw Open. (2021) 4:e2134290–e2134290. doi: 10.1001/jamanetworkopen.2021.34290
15. Stewart IJ, Sosnov JA, Howard JT, Orman JA, Fang R, Morrow BD, et al. Retrospective analysis of long-term outcomes after combat injury: a hidden cost of war. Circulation. (2015) 132:2126–33. doi: 10.1161/CIRCULATIONAHA.115.016950
16. Duke JM, Randall SM, Fear MW, Boyd JH, Rea S, Wood FM. Understanding the long-term impacts of burn on the cardiovascular system. Burns. (2016) 42:366–74. doi: 10.1016/j.burns.2015.08.020
17. Duke JM, Randall SM, Fear MW, Boyd JH, O'Halloran E, Rea S, et al. Increased admissions for diabetes mellitus after burn. Burns. (2016) 42:1734–9. doi: 10.1016/j.burns.2016.06.005
18. Carlson N, Hommel K, Olesen JB, Soja AM, Vilsboll T, Kamper AL, et al. Trends in one-year outcomes of dialysis-requiring acute kidney injury in Denmark 2005-2012: a population-based nationwide study. PLoS ONE. (2016) 11:e0159944. doi: 10.1371/journal.pone.0159944
19. Gallagher M, Cass A, Bellomo R, Finfer S, Gattas D, Lee J, et al. Long-term survival and dialysis dependency following acute kidney injury in intensive care: extended follow-up of a randomized controlled trial. PLoS Med. (2014) 11:e1001601. doi: 10.1371/journal.pmed.1001601
20. Yende S, Linde-Zwirble W, Mayr F, Weissfeld LA, Reis S, Angus DC. Risk of cardiovascular events in survivors of severe sepsis. Am J Respir Crit Care Med. (2014) 189:1065–74. doi: 10.1164/rccm.201307-1321OC
21. Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang CC, et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA. (2015) 313:264–74. doi: 10.1001/jama.2014.18229
22. Kwong JC, Schwartz KL, Campitelli MA, Chung H, Crowcroft NS, Karnauchow T, et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N Engl J Med. (2018) 378:345–53. doi: 10.1056/NEJMoa1702090
23. Wang HE, Moore JX, Donnelly JP, Levitan EB, Safford MM. Risk of acute coronary heart disease after sepsis hospitalization in the reasons for geographic and racial differences in stroke (REGARDS) cohort. Clin Infect Dis. (2017) 65:29–36. doi: 10.1093/cid/cix248
24. Kosyakovsky LB, Angriman F, Katz E, Adhikari NK, Godoy LC, Marshall JC, et al. Association between sepsis survivorship and long-term cardiovascular outcomes in adults: a systematic review and meta-analysis. Intensive Care Med. (2021) 47:931–42. doi: 10.1007/s00134-021-06479-y
25. Bergh C, Fall K, Udumyan R, Sjoqvist H, Frobert O, Montgomery S. Severe infections and subsequent delayed cardiovascular disease. Eur J Prev Cardiol. (2017) 24:1958–66. doi: 10.1177/2047487317724009
26. Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. (2022) 28:583–90. doi: 10.1038/s41591-022-01689-3
27. Mammen C, Al Abbas A, Skippen P, Nadel H, Levine D, Collet JP, et al. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. Am J Kidney Dis. (2012) 59:523–30. doi: 10.1053/j.ajkd.2011.10.048
28. Calderon-Margalit R, Golan E, Twig G, Leiba A, Tzur D, Afek A, et al. History of childhood kidney disease and risk of adult end-stage renal disease. N Engl J Med. (2018) 378:428–38. doi: 10.1056/NEJMoa1700993
29. Hundeshagen G, Herndon DN, Clayton RP, Wurzer P, McQuitty A, Jennings K, et al. Long-term effect of critical illness after severe paediatric burn injury on cardiac function in adolescent survivors: an observational study. Lancet Child Adolesc Health. (2017) 1:293–301. doi: 10.1016/S2352-4642(17)30122-0
30. Duke JM, Randall SM, Fear MW, Boyd JH, Rea S, Wood FM. Long-term effects of pediatric burns on the circulatory system. Pediatrics. (2015) 136:e1323–30. doi: 10.1542/peds.2015-1945
31. Hippisley-Cox J, Coupland C, Brindle P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: prospective cohort study. BMJ. (2017) 357:j2099. doi: 10.1136/bmj.j2099
32. Sartipy U, Dahlstrom U, Edner M, Lund LH. Predicting survival in heart failure: validation of the MAGGIC heart failure risk score in 51,043 patients from the Swedish heart failure registry. Eur J Heart Fail. (2014) 16:173–9. doi: 10.1111/ejhf.32
33. Kivimaki M, Batty GD, Singh-Manoux A, Ferrie JE, Tabak AG, Jokela M, et al. Validating the framingham hypertension risk score: results from the whitehall II study. Hypertension. (2009) 54:496–501. doi: 10.1161/HYPERTENSIONAHA.109.132373
34. Hippisley-Cox J, Coupland C, Brindle P. Derivation and validation of QStroke score for predicting risk of ischaemic stroke in primary care and comparison with other risk scores: a prospective open cohort study. BMJ. (2013) 346:f2573. doi: 10.1136/bmj.f2573
35. McMahon GM, Preis SR, Hwang SJ, Fox CS. Mid-adulthood risk factor profiles for CKD. J Am Soc Nephrol. (2014) 25:2633–41. doi: 10.1681/ASN.2013070750
36. Moreton JR. Atherosclerosis and alimentary hyperlipemia. Science. (1947) 106:190–1. doi: 10.1126/science.106.2748.190
37. Kotchen TA. Historical trends and milestones in hypertension research: a model of the process of translational research. Hypertension. (2011) 58:522–38. doi: 10.1161/HYPERTENSIONAHA.111.177766
38. Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis–chicken or egg? N Engl J Med. (2008) 359:1953–5. doi: 10.1056/NEJMe0807235
39. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
40. Deyab G, Hokstad I, Whist JE, Smastuen MC, Agewall S, Lyberg T, et al. Methotrexate and anti-tumor necrosis factor treatment improves endothelial function in patients with inflammatory arthritis. Arthritis Res Ther. (2017) 19:232. doi: 10.1186/s13075-017-1439-1
41. Angel K, Provan SA, Gulseth HL, Mowinckel P, Kvien TK, Atar D. Tumor necrosis factor-alpha antagonists improve aortic stiffness in patients with inflammatory arthropathies: a controlled study. Hypertension. (2010) 55:333–8. doi: 10.1161/HYPERTENSIONAHA.109.143982
42. Kaynar AM, Yende S, Zhu L, Frederick DR, Chambers R, Burton CL, et al. Effects of intra-abdominal sepsis on atherosclerosis in mice. Crit Care. (2014) 18:469. doi: 10.1186/s13054-014-0469-1
43. Fuijkschot WW, Morrison MC, van der Linden R, Krijnen PAJ, Zethof IPA, Theyse LFH, et al. Orthopedic surgery increases atherosclerotic lesions and necrotic core area in ApoE-/- mice. Atherosclerosis. (2016) 255:164–70. doi: 10.1016/j.atherosclerosis.2016.07.909
44. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. (2018) 391:319–28. doi: 10.1016/S0140-6736(17)32814-3
45. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
46. Weber C, von Hundelshausen P. CANTOS trial validates the inflammatory pathogenesis of atherosclerosis: setting the stage for a new chapter in therapeutic targeting. Circ Res. (2017) 121:1119–21. doi: 10.1161/CIRCRESAHA.117.311984
47. Shankar-Hari M, Rubenfeld GD. Understanding long-term outcomes following sepsis: implications and challenges. Curr Infect Dis Rep. (2016) 18:37. doi: 10.1007/s11908-016-0544-7
48. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. (2013) 13:260–8. doi: 10.1016/S1473-3099(13)70001-X
49. Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. (2007) 27:1433–9. doi: 10.1161/ATVBAHA.106.138743
50. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care. (2011) 34:392–7. doi: 10.2337/dc10-1676
51. Asada M, Oishi E, Sakata S, Hata J, Yoshida D, Honda T, et al. Serum lipopolysaccharide-binding protein levels and the incidence of cardiovascular disease in a general japanese population: the hisayama study. J Am Heart Assoc. (2019) 8:e013628. doi: 10.1161/JAHA.119.013628
52. Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. (1996) 271:13018–22. doi: 10.1074/jbc.271.22.13018
53. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. Host response to injury large-scale collaborative research, a genomic storm in critically injured humans. J Exp Med. (2011) 208:2581–90. doi: 10.1084/jem.20111354
54. Crimi E, Sica V, Williams-Ignarro S, Zhang H, Slutsky AS, Ignarro LJ, et al. The role of oxidative stress in adult critical care. Free Radic Biol Med. (2006) 40:398–406. doi: 10.1016/j.freeradbiomed.2005.10.054
55. Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. (2002) 296:2391–4. doi: 10.1126/science.1106830
56. Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest. (2013) 123:3815–28. doi: 10.1172/JCI67478
57. Poliakov E, Brennan ML, Macpherson J, Zhang R, Sha W, Narine L, et al. Isolevuglandins, a novel class of isoprostenoid derivatives, function as integrated sensors of oxidant stress and are generated by myeloperoxidase in vivo. FASEB J. (2003) 17:2209–20. doi: 10.1096/fj.03-0086com
58. Guo L, Chen Z, Amarnath V, Yancey PG, Van Lenten BJ, Savage JR, et al. Isolevuglandin-type lipid aldehydes induce the inflammatory response of macrophages by modifying phosphatidylethanolamines and activating the receptor for advanced glycation endproducts. Antioxid Redox Signal. (2015) 22:1633–45. doi: 10.1089/ars.2014.6078
59. Salomon RG, Kaur K, Batyreva E. Isolevuglandin-protein adducts in oxidized low density lipoprotein and human plasma: a strong connection with cardiovascular disease. Trends Cardiovasc Med. (2000) 10:53–9. doi: 10.1016/S1050-1738(00)00040-2
60. Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. (2016) 126:50–67. doi: 10.1172/JCI80761
61. Salomon RG, Bi W. Isolevuglandin adducts in disease. Antioxid Redox Signal. (2015) 22:1703–18. doi: 10.1089/ars.2014.6154
62. May-Zhang LS, Yermalitsky V, Huang J, Pleasent T, Borja MS, Oda MN, et al. Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters HDL structure and function. J Biol Chem. (2018) 293:9176–87. doi: 10.1074/jbc.RA117.001099
63. Ware LB, Fessel JP, May AK, Roberts LJ II. Plasma biomarkers of oxidant stress and development of organ failure in severe sepsis. Shock. (2011) 36:12–7. doi: 10.1097/SHK.0b013e318217025a
64. Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. (2004) 95:764–72. doi: 10.1161/01.RES.0000146094.59640.13
65. Yao Z, Mates JM, Cheplowitz AM, Hammer LP, Maiseyeu A, Phillips GS, et al. Blood-Borne lipopolysaccharide is rapidly eliminated by liver sinusoidal endothelial cells via high-density lipoprotein. J Immunol. (2016) 197:2390–9. doi: 10.4049/jimmunol.1600702
66. Munford RS, Weiss JP, Lu M. Biochemical transformation of bacterial lipopolysaccharides by acyloxyacyl hydrolase reduces host injury and promotes recovery. J Biol Chem. (2020) 295:17842–51. doi: 10.1074/jbc.REV120.015254
67. Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Krankel N, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of toll-like receptor-2. Immunity. (2013) 38:754–68. doi: 10.1016/j.immuni.2013.02.009
68. Shroff R, Speer T, Colin S, Charakida M, Zewinger S, Staels B, et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J Am Soc Nephrol. (2014) 25:2658–68. doi: 10.1681/ASN.2013111212
69. Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation. (2010) 121:110–22. doi: 10.1161/CIRCULATIONAHA.108.836346
70. Winkler MS, Nierhaus A, Rosler G, Lezius S, Harlandt O, Schwedhelm E, et al. Symmetrical (SDMA) and asymmetrical dimethylarginine (ADMA) in sepsis: high plasma levels as combined risk markers for sepsis survival. Crit Care. (2018) 22:216. doi: 10.1186/s13054-018-2090-1
71. Zewinger S, Kleber ME, Rohrer L, Lehmann M, Triem S, Jennings RT, et al. Symmetric dimethylarginine, high-density lipoproteins and cardiovascular disease. Eur Heart J. (2017) 38:1597–607. doi: 10.1093/eurheartj/ehx118
72. Vellinga NA, Boerma EC, Koopmans M, Donati A, Dubin A, Shapiro NI, et al. International study on microcirculatory shock occurrence in acutely ill patients. Crit Care Med. (2015) 43:48–56. doi: 10.1097/CCM.0000000000000553
73. Yende S, Kellum JA, Talisa VB, Peck Palmer OM, Chang CH, Filbin MR, et al. Long-term host immune response trajectories among hospitalized patients with sepsis. JAMA Netw Open. (2019) 2:e198686. doi: 10.1001/jamanetworkopen.2019.8686
74. Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. (1997) 6:315–25. doi: 10.1016/S1074-7613(00)80334-9
75. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci USA. (2020) 117:22351–6. doi: 10.1073/pnas.2010229117
76. Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat-Baslanti T, Wang Z, et al. Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. J Trauma Acute Care Surg. (2018) 84:342–9. doi: 10.1097/TA.0000000000001758
77. Stehouwer CD, Smulders YM. Microalbuminuria and risk for cardiovascular disease: analysis of potential mechanisms. J Am Soc Nephrol. (2006) 17:2106–11. doi: 10.1681/ASN.2005121288
78. Mussbacher M, Salzmann M, Haigl B, Basilio J, Hochreiter B, Gleitsmann V, et al. Ikk2-mediated inflammatory activation of arterial endothelial cells promotes the development and progression of atherosclerosis. Atherosclerosis. (2020) 307:21–31. doi: 10.1016/j.atherosclerosis.2020.06.005
79. Sehnert B, Burkhardt H, Wessels JT, Schroder A, May MJ, Vestweber D, et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proc Natl Acad Sci USA. (2013) 110:16556–61. doi: 10.1073/pnas.1218219110
80. Chen J, Sivan U, Tan SL, Lippo L, De Angelis J, Labella R, et al. High-resolution 3D imaging uncovers organ-specific vascular control of tissue aging. Sci Adv. (2021) 7:eabd7819. doi: 10.1126/sciadv.abd7819
81. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction 2018. Eur Heart J. (2019) 40:237–69. doi: 10.1093/eurheartj/ehy856
82. Landesberg G, Vesselov Y, Einav S, Goodman S, Sprung CL, Weissman C. Myocardial ischemia, cardiac troponin, and long-term survival of high-cardiac risk critically ill intensive care unit patients. Crit Care Med. (2005) 33:1281–7. doi: 10.1097/01.CCM.0000166607.22550.87
83. Puelacher C, Lurati Buse G, Seeberger D, Sazgary L, Marbot S, Lampart A, et al. Perioperative myocardial injury after noncardiac surgery: incidence, mortality, and characterization. Circulation. (2018) 137:1221–32. doi: 10.1161/CIRCULATIONAHA.117.030114
84. Docherty AB, Sim M, Oliveira J, Adlam M, Ostermann M, Walsh TS, et al. Early troponin I in critical illness and its association with hospital mortality: a cohort study. Crit Care. (2017) 21:216. doi: 10.1186/s13054-017-1800-4
85. Grobben RB, van Waes JAR, Leiner T, Peelen LM, de Borst GJ, Vogely HC, et al. Unexpected cardiac computed tomography findings in patients with postoperative myocardial injury. Anesth Analg. (2018) 126:1462–8. doi: 10.1213/ANE.0000000000002580
86. Bertinchant JP, Polge A, Mohty D, Nguyen-Ngoc-Lam R, Estorc J, Cohendy R, et al. Evaluation of incidence, clinical significance, and prognostic value of circulating cardiac troponin I and T elevation in hemodynamically stable patients with suspected myocardial contusion after blunt chest trauma. J Trauma. (2000) 48:924–31. doi: 10.1097/00005373-200005000-00018
87. White M, Wiechmann RJ, Roden RL, Hagan MB, Wollmering MM, Port JD, et al. Cardiac beta-adrenergic neuroeffector systems in acute myocardial dysfunction related to brain injury. Evidence for catecholamine-mediated myocardial damage. Circulation. (1995) 92:2183–9. doi: 10.1161/01.CIR.92.8.2183
88. Masson S, Caironi P, Fanizza C, Carrer S, Caricato A, Fassini P, et al. Sequential N-terminal Pro-B-type natriuretic peptide and high-sensitivity cardiac troponin measurements during albumin replacement in patients with severe sepsis or septic shock. Crit Care Med. (2016) 44:707–16. doi: 10.1097/CCM.0000000000001473
89. Friden V, Starnberg K, Muslimovic A, Ricksten SE, Bjurman C, Forsgard N, et al. Clearance of cardiac troponin T with and without kidney function. Clin Biochem. (2017) 50:468–74. doi: 10.1016/j.clinbiochem.2017.02.007
90. Muslimovic A, Friden V, Tenstad O, Starnberg K, Nystrom S, Wesen E, et al. The liver and kidneys mediate clearance of cardiac troponin in the rat. Sci Rep. (2020) 10:6791. doi: 10.1038/s41598-020-63744-8
91. van der Linden N, Cornelis T, Kimenai DM, Klinkenberg LJJ, Hilderink JM, Luck S, et al. Origin of cardiac troponin T elevations in chronic kidney disease. Circulation. (2017) 136:1073–5. doi: 10.1161/CIRCULATIONAHA.117.029986
92. Landesberg G, Gilon D, Meroz Y, Georgieva M, Levin PD, Goodman S, et al. Diastolic dysfunction and mortality in severe sepsis and septic shock. Eur Heart J. (2012) 33:895–903. doi: 10.1093/eurheartj/ehr351
93. Metkus TS, Guallar E, Sokoll L, Morrow D, Tomaselli G, Brower R, et al. Prevalence and prognostic association of circulating troponin in the acute respiratory distress syndrome. Crit Care Med. (2017) 45:1709–17. doi: 10.1097/CCM.0000000000002641
94. Ostermann M, Ayis S, Tuddenham E, Lo J, Lei K, Smith J, et al. Cardiac troponin release is associated with biomarkers of inflammation and ventricular dilatation during critical illness. Shock. (2017) 47:702–8. doi: 10.1097/SHK.0000000000000811
95. Ackland GL, Abbott TEF, Cain D, Edwards MR, Sultan P, Karmali SN, et al. Preoperative systemic inflammation and perioperative myocardial injury: prospective observational multicentre cohort study of patients undergoing non-cardiac surgery. Br J Anaesth. (2019) 122:180–7. doi: 10.1016/j.bja.2018.09.002
96. Johansson PI, Stensballe J, Ostrowski SR. Shock induced endotheliopathy (SHINE) in acute critical illness - a unifying pathophysiologic mechanism. Crit Care. (2017) 21:25. doi: 10.1186/s13054-017-1605-5
97. Naganathar S, De'Ath HD, Wall J, Brohi K. Admission biomarkers of trauma-induced secondary cardiac injury predict adverse cardiac events and are associated with plasma catecholamine levels. J Trauma Acute Care Surg. (2015) 79:71–7. doi: 10.1097/TA.0000000000000694
98. May SM, Abbott TEF, Del Arroyo AG, Reyes A, Martir G, Stephens RCM, et al. MicroRNA signatures of perioperative myocardial injury after elective noncardiac surgery: a prospective observational mechanistic cohort study. Br J Anaesth. (2020) 125:661–71. doi: 10.1016/j.bja.2020.05.066
99. Chotalia M, Ali M, Hebballi R, Singh H, Parekh D, Bangash MN, et al. Hyperdynamic left ventricular ejection fraction in ICU patients with sepsis. Crit Care Med. (2021) 50:770–9. doi: 10.1097/CCM.0000000000005315
100. Helwani MA, Amin A, Lavigne P, Rao S, Oesterreich S, Samaha E, et al. Etiology of acute coronary syndrome after noncardiac surgery. Anesthesiology. (2018) 128:1084–91. doi: 10.1097/ALN.0000000000002107
101. Sheth T, Natarajan MK, Hsieh V, Valettas N, Rokoss M, Mehta S, et al. Incidence of thrombosis in perioperative and non-operative myocardial infarction. Br J Anaesth. (2018) 120:725–33. doi: 10.1016/j.bja.2017.11.063
102. Park JH, Kang SJ, Song JK, Kim HK, Lim CM, Kang DH, et al. Left ventricular apical ballooning due to severe physical stress in patients admitted to the medical ICU. Chest. (2005) 128:296–302. doi: 10.1378/chest.128.1.296
103. Lei Q, Yi T, Chen C. NF-kappaB-Gasdermin D (GSDMD) axis couples oxidative stress and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome-mediated cardiomyocyte pyroptosis following myocardial infarction. Med Sci Monit. (2018) 24:6044–52. doi: 10.12659/MSM.908529
104. Shen J, Wu JM, Hu GM, Li MZ, Cong WW, Feng YN, et al. Membrane nanotubes facilitate the propagation of inflammatory injury in the heart upon overactivation of the beta-adrenergic receptor. Cell Death Dis. (2020) 11:958. doi: 10.1038/s41419-020-03157-7
105. Amgalan D, Pekson R, Kitsis RN. Troponin release following brief myocardial ischemia: apoptosis versus necrosis. JACC Basic Transl Sci. (2017) 2:118–21. doi: 10.1016/j.jacbts.2017.03.008
106. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. (2017) 23:1481–7. doi: 10.1038/nm.4428
107. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
108. Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. (2020) 52:475–86.e5. doi: 10.1016/j.immuni.2020.02.002
109. Schmittinger CA, Dunser MW, Torgersen C, Luckner G, Lorenz I, Schmid S, et al. Histologic pathologies of the myocardium in septic shock: a prospective observational study. Shock. (2013) 39:329–35. doi: 10.1097/SHK.0b013e318289376b
110. Siddiqui Y, Crouser ED, Raman SV. Nonischemic myocardial changes detected by cardiac magnetic resonance in critical care patients with sepsis. Am J Respir Crit Care Med. (2013) 188:1037–9. doi: 10.1164/rccm.201304-0744LE
111. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169:510–22.e20. doi: 10.1016/j.cell.2017.03.050
112. Celes MR, Torres-Duenas D, Alves-Filho JC, Duarte DB, Cunha FQ, Rossi MA. Reduction of gap and adherens junction proteins and intercalated disc structural remodeling in the hearts of mice submitted to severe cecal ligation and puncture sepsis. Crit Care Med. (2007) 35:2176–85. doi: 10.1097/01.CCM.0000281454.97901.01
113. Braun CK, Kalbitz M, Halbgebauer R, Eisele P, Messerer DAC, Weckbach S, et al. Early structural changes of the heart after experimental polytrauma and hemorrhagic shock. PLoS ONE. (2017) 12:e0187327. doi: 10.1371/journal.pone.0187327
114. Lillo MA, Himelman E, Shirokova N, Xie LH, Fraidenraich D, Contreras JE. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight. (2019) 4:e130091. doi: 10.1172/jci.insight.130091
115. Moss TJ, Calland JF, Enfield KB, Gomez-Manjarres DC, Ruminski C, DiMarco JP, et al. New-Onset atrial fibrillation in the critically ill. Crit Care Med. (2017) 45:790–7. doi: 10.1097/CCM.0000000000002325
116. Arrigo M, Ishihara S, Feliot E, Rudiger A, Deye N, Cariou A, et al. New-onset atrial fibrillation in critically ill patients and its association with mortality: a report from the FROG-ICU study. Int J Cardiol. (2018) 266:95–9. doi: 10.1016/j.ijcard.2018.03.051
117. Devereaux PJ, Duceppe E, Guyatt G, Tandon V, Rodseth R, Biccard BM, et al. Dabigatran in patients with myocardial injury after non-cardiac surgery (MANAGE): an international, randomised, placebo-controlled trial. Lancet. (2018) 391:2325–34. doi: 10.1016/S0140-6736(18)30832-8
118. Walkey AJ, Hammill BG, Curtis LH, Benjamin EJ. Long-term outcomes following development of new-onset atrial fibrillation during sepsis. Chest. (2014) 146:1187–95. doi: 10.1378/chest.14-0003
119. Li WJ, Chen XQ, Xu LL, Li YQ, Luo BH. SGLT2 inhibitors and atrial fibrillation in type 2 diabetes: a systematic review with meta-analysis of 16 randomized controlled trials. Cardiovasc Diabetol. (2020) 19:130. doi: 10.1186/s12933-020-01105-5
120. Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, et al. Atrial Myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res. (2020) 127:1036–55. doi: 10.1161/CIRCRESAHA.120.316710
121. Ferreira VM, Marcelino M, Piechnik SK, Marini C, Karamitsos TD, Ntusi NAB, et al. Pheochromocytoma is characterized by catecholamine-mediated myocarditis, focal and diffuse myocardial fibrosis, myocardial dysfunction. J Am Coll Cardiol. (2016) 67:2364–74. doi: 10.1016/j.jacc.2016.03.543
122. Scally C, Abbas H, Ahearn T, Srinivasan J, Mezincescu A, Rudd A, et al. Myocardial and systemic inflammation in acute stress-induced (takotsubo) cardiomyopathy. Circulation. (2018) 139:1581–92. doi: 10.1161/CIRCULATIONAHA.118.037975
123. Jia L, Wang Y, Wang Y, Ma Y, Shen J, Fu Z, et al. Heme oxygenase-1 in macrophages drives septic cardiac dysfunction via suppressing lysosomal degradation of inducible nitric oxide synthase. Circ Res. (2018) 122:1532–44. doi: 10.1161/CIRCRESAHA.118.312910
124. Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. (2018) 215:423–40. doi: 10.1084/jem.20171274
125. Dal-Secco D, DalBo S, Lautherbach NES, Gava FN, Celes MRN, Benedet PO, et al. Cardiac hyporesponsiveness in severe sepsis is associated with nitric oxide-dependent activation of G protein receptor kinase. Am J Physiol Heart Circ Physiol. (2017) 313:H149–63. doi: 10.1152/ajpheart.00052.2016
126. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. (2019) 568:351–6. doi: 10.1038/s41586-019-1100-z
127. Scally C, Rudd A, Mezincescu A, Wilson H, Srivanasan J, Horgan G, et al. Persistent long-term structural, functional, and metabolic changes after stress-induced (takotsubo) cardiomyopathy. Circulation. (2018) 137:1039–48. doi: 10.1161/CIRCULATIONAHA.117.031841
128. Reddy YNV, Carter RE, Obokata M, Redfield MM, Borlaug BA. A simple, evidence-based approach to help guide diagnosis of heart failure with preserved ejection fraction. Circulation. (2018) 138:861–70. doi: 10.1161/CIRCULATIONAHA.118.034646
129. Prud'homme M, Coutrot M, Michel T, Boutin L, Genest M, Poirier F, et al. Acute kidney injury induces remote cardiac damage and dysfunction through the galectin-3 pathway. JACC Basic Transl Sci. (2019) 4:717–32. doi: 10.1016/j.jacbts.2019.06.005
130. Besler C, Lang D, Urban D, Rommel KP, von Roeder M, Fengler K, et al. Plasma and cardiac galectin-3 in patients with heart failure reflects both inflammation and fibrosis: implications for its use as a biomarker. Circ Heart Fail. (2017) 10:e003804. doi: 10.1161/CIRCHEARTFAILURE.116.003804
131. Doerschug KC, Delsing AS, Schmidt GA, Ashare A. Renin-angiotensin system activation correlates with microvascular dysfunction in a prospective cohort study of clinical sepsis. Crit Care. (2010) 14:R24. doi: 10.1186/cc8887
132. Annane D, Trabold F, Sharshar T, Jarrin I, Blanc AS, Raphael JC, et al. Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med. (1999) 160:458–65. doi: 10.1164/ajrccm.160.2.9810073
133. Calvier L, Martinez-Martinez E, Miana M, Cachofeiro V, Rousseau E, Sadaba JR, et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. (2015) 3:59–67. doi: 10.1016/j.jchf.2014.08.002
134. Calvier L, Miana M, Reboul P, Cachofeiro V, Martinez-Martinez E, de Boer RA, et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol. (2013) 33:67–75. doi: 10.1161/ATVBAHA.112.300569
135. Zhao WB, Lu Q, Nguyen MN, Su Y, Ziemann M, Wang LN, et al. Stimulation of beta-adrenoceptors up-regulates cardiac expression of galectin-3 and BIM through the Hippo signalling pathway. Br J Pharmacol. (2019) 176:2465–81. doi: 10.1111/bph.14674
136. Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, et al. Renal denervation prevents immune cell activation and renal inflammation in angiotensin ii-induced hypertension. Circ Res. (2015) 117:547–57. doi: 10.1161/CIRCRESAHA.115.306010
137. Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. (2014) 124:4642–56. doi: 10.1172/JCI74084
138. Hoste E, Bihorac A, Al-Khafaji A, Ortega LM, Ostermann M, Haase M, et al. Identification and validation of biomarkers of persistent acute kidney injury: the RUBY study. Intensive Care Med. (2020) 46:943–53. doi: 10.1007/s00134-019-05919-0
139. Chawla LS, Bellomo R, Bihorac A, Goldstein SL, Siew ED, Bagshaw SM, et al. Acute kidney disease and renal recovery: consensus report of the acute disease quality initiative (ADQI) 16 workgroup. Nat Rev Nephrol. (2017) 13:241–57. doi: 10.1038/nrneph.2017.2
140. Chung S, Overstreet JM, Li Y, Wang Y, Niu A, Wang S, et al. TGF-beta promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight. (2018) 3:e123563. doi: 10.1172/jci.insight.123563
141. Chen H, Fang Y, Wu J, Chen H, Zou Z, Zhang X, et al. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. (2018) 9:878. doi: 10.1038/s41419-018-0936-8
142. Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Paton J, et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. (2020) 589:281–6. doi: 10.1038/s41586-020-2941-1
143. Adler M, Mayo A, Zhou X, Franklin RA, Meizlish ML, Medzhitov R, et al. Principles of cell circuits for tissue repair and fibrosis. iScience. (2020) 23:100841. doi: 10.1016/j.isci.2020.100841
144. Rabinovich GA, Toscano MA. Turning 'sweet' on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. (2009) 9:338–52. doi: 10.1038/nri2536
145. Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med. (2018) 24:1721–31. doi: 10.1038/s41591-018-0194-4
146. Cippa PE, Sun B, Liu J, Chen L, Naesens M, McMahon AP. Transcriptional trajectories of human kidney injury progression. JCI Insight. (2018) 3:e123151. doi: 10.1172/jci.insight.123151
147. Hultstrom M, Becirovic-Agic M, Jonsson S. Comparison of acute kidney injury of different etiology reveals in-common mechanisms of tissue damage. Physiol Genomics. (2018) 50:127–41. doi: 10.1152/physiolgenomics.00037.2017
148. Gayat E, Hollinger A, Cariou A, Deye N, Vieillard-Baron A, Jaber S, et al. Impact of angiotensin-converting enzyme inhibitors or receptor blockers on post-ICU discharge outcome in patients with acute kidney injury. Intensive Care Med. (2018) 44:598–605. doi: 10.1007/s00134-018-5160-6
149. Lockett MF. The aetiology of essential hypertension. Postgrad Med J. (1945) 21:287–98. doi: 10.1136/pgmj.21.240.287
150. Parikh CR, Puthumana J, Shlipak MG, Koyner JL, Thiessen-Philbrook H, McArthur E, et al. Relationship of kidney injury biomarkers with long-term cardiovascular outcomes after cardiac surgery. J Am Soc Nephrol. (2017) 28:3699–707. doi: 10.1681/ASN.2017010055
151. Haines R, Crichton S, Wilson J, Treacher D, Ostermann M. Cardiac biomarkers are associated with maximum stage of acute kidney injury in critically ill patients: a prospective analysis. Crit Care. (2017) 21:88. doi: 10.1186/s13054-017-1674-5
152. Solagna F, Tezze C, Lindenmeyer MT, Lu S, Wu G, Liu S, et al. Pro-cachectic factors link experimental and human chronic kidney disease to skeletal muscle wasting programs. J Clin Invest. (2021) 131:e135821. doi: 10.1172/JCI135821
153. Tsai VWW, Husaini Y, Sainsbury A, Brown DA, Breit SN. The MIC-1/GDF15-GFRAL pathway in energy homeostasis: implications for obesity, cachexia, and other associated diseases. Cell Metab. (2018) 28:353–68. doi: 10.1016/j.cmet.2018.07.018
154. Jia L, Takahashi M, Morimoto H, Takahashi S, Izawa A, Ise H, et al. Changes in cardiac lipid metabolism during sepsis: the essential role of very low-density lipoprotein receptors. Cardiovasc Res. (2006) 69:545–55. doi: 10.1016/j.cardiores.2005.11.014
155. Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, et al. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell. (2019) 178:1231–44.e11. doi: 10.1016/j.cell.2019.07.033
156. Bennett MJ, Hauton D, Hole DG, Evans RD. Utilization of very low density lipoprotein by rat heart: the effect of endotoxin. Am J Physiol Endocrinol Metab. (2000) 278:E802–10. doi: 10.1152/ajpendo.2000.278.5.E802
157. Suriben R, Chen M, Higbee J, Oeffinger J, Ventura R, Li B, et al. Antibody-mediated inhibition of GDF15-GFRAL activity reverses cancer cachexia in mice. Nat Med. (2020) 26:1264–70. doi: 10.1038/s41591-020-0945-x
158. Bloch SA, Lee JY, Syburra T, Rosendahl U, Griffiths MJ, Kemp PR, et al. Increased expression of GDF-15 may mediate ICU-acquired weakness by down-regulating muscle microRNAs. Thorax. (2015) 70:219–28. doi: 10.1136/thoraxjnl-2014-206225
159. Xie Y, Liu S, Zheng H, Cao L, Liu K, Li X. Utility of plasma GDF-15 for diagnosis and prognosis assessment of ICU-acquired weakness in mechanically ventilated patients: prospective observational study. Biomed Res Int. (2020) 2020:3630568. doi: 10.1155/2020/3630568
160. Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. (2018) 19:731–45. doi: 10.1038/s41580-018-0068-0
161. Van Dyck L, Gunst J, Casaer MP, Peeters B, Derese I, Wouters PJ, et al. The clinical potential of GDF15 as a “ready-to-feed indicator” for critically ill adults. Crit Care. (2020) 24:557. doi: 10.1186/s13054-020-03254-1
162. Langouche L, Perre SV, Thiessen S, Gunst J, Hermans G, D'Hoore A, et al. Alterations in adipose tissue during critical illness: an adaptive and protective response? Am J Respir Crit Care Med. (2010) 182:507–16. doi: 10.1164/rccm.200909-1395OC
163. Ilias I, Vassiliadi DA, Theodorakopoulou M, Boutati E, Maratou E, Mitrou P, et al. Adipose tissue lipolysis and circulating lipids in acute and subacute critical illness: effects of shock and treatment. J Crit Care. (2014) 29:1130.e5–9. doi: 10.1016/j.jcrc.2014.06.003
164. Trinder M, Boyd JH, Brunham LR. Molecular regulation of plasma lipid levels during systemic inflammation and sepsis. Curr Opin Lipidol. (2019) 30:108–16. doi: 10.1097/MOL.0000000000000577
165. Han SJ, Glatman Zaretsky A, Andrade-Oliveira V, Collins N, Dzutsev A, Shaik J, et al. White Adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity. (2017) 47:1154–68.e6. doi: 10.1016/j.immuni.2017.11.009
166. Reid CL, Murgatroyd PR, Wright A, Menon DK. Quantification of lean and fat tissue repletion following critical illness: a case report. Crit Care. (2008) 12:R79. doi: 10.1186/cc6929
167. Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. (2006) 4:417–9. doi: 10.1016/j.cmet.2006.11.008
168. Plummer MP, Finnis ME, Phillips LK, Kar P, Bihari S, Biradar V, et al. Stress induced hyperglycemia and the subsequent risk of type 2 diabetes in survivors of critical illness. PLoS ONE. (2016) 11:e0165923. doi: 10.1371/journal.pone.0165923
169. van Beusekom I, Bakhshi-Raiez F, van der Schaaf M, Dongelmans DA, Busschers WB, de Keizer NF. The influence of clinical variables on the risk of developing chronic conditions in ICU survivors. J Crit Care. (2020) 55:134–9. doi: 10.1016/j.jcrc.2019.10.014
170. Boos CJ, Schofield S, Cullinan P, Dyball D, Fear NT, Bull AM, et al. Association between combat-related traumatic injury and cardiovascular risk. Heart. (2021) 108:367–74. doi: 10.1136/heartjnl-2021-320296
171. Artunc F, Schleicher E, Weigert C, Fritsche A, Stefan N, Haring HU. The impact of insulin resistance on the kidney and vasculature. Nat Rev Nephrol. (2016) 12:721–37. doi: 10.1038/nrneph.2016.145
172. Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. (2006) 55:480–6. doi: 10.2337/diabetes.55.02.06.db05-1103
173. Yki-Jarvinen H, Sammalkorpi K, Koivisto VA, Nikkila EA. Severity, duration, and mechanisms of insulin resistance during acute infections. J Clin Endocrinol Metab. (1989) 69:317–23. doi: 10.1210/jcem-69-2-317
174. Flynn MC, Kraakman MJ, Tikellis C, Lee MKS, Hanssen NMJ, Kammoun HL, et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ Res. (2020) 127:877–92. doi: 10.1161/CIRCRESAHA.120.316653
175. Watanabe K, Nagao M, Toh R, Irino Y, Shinohara M, Iino T, et al. Critical role of glutamine metabolism in cardiomyocytes under oxidative stress. Biochem Biophys Res Commun. (2021) 534:687–93. doi: 10.1016/j.bbrc.2020.11.018
176. Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. (2018) 27:1007–25.e5. doi: 10.1016/j.cmet.2018.03.002
177. McKenna HT, O'Brien KA, Fernandez BO, Minnion M, Tod A, McNally BD, et al. Divergent trajectories of cellular bioenergetics, intermediary metabolism and systemic redox status in survivors and non-survivors of critical illness. Redox Biol. (2021) 41:101907. doi: 10.1016/j.redox.2021.101907
178. Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med. (2013) 5:195ra95. doi: 10.1126/scitranslmed.3005893
179. Moreau R, Claria J, Aguilar F, Fenaille F, Lozano JJ, Junot C, et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J Hepatol. (2020) 72:688–701. doi: 10.1016/j.jhep.2019.11.009
180. Dhainaut JF, Huyghebaert MF, Monsallier JF, Lefevre G, Dall'Ava-Santucci J, Brunet F, et al. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation. (1987) 75:533–41. doi: 10.1161/01.CIR.75.3.533
181. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. (2017) 551:115–8. doi: 10.1038/nature24057
182. Bosch M, Sanchez-Alvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, et al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science. (2020) 370:eaay8085. doi: 10.1126/science.aay8085
183. Jarc E, Petan T. Lipid droplets and the management of cellular stress. Yale J Biol Med. (2019) 92:435–52.
184. McKenzie R, Fried MW, Sallie R, Conjeevaram H, Di Bisceglie AM, Park Y, et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med. (1995) 333:1099–105. doi: 10.1056/NEJM199510263331702
185. Koskinas J, Gomatos IP, Tiniakos DG, Memos N, Boutsikou M, Garatzioti A, et al. Liver histology in ICU patients dying from sepsis: a clinico-pathological study. World J Gastroenterol. (2008) 14:1389–93. doi: 10.3748/wjg.14.1389
186. Paumelle R, Haas JT, Hennuyer N, Bauge E, Deleye Y, Mesotten D, et al. Hepatic PPARalpha is critical in the metabolic adaptation to sepsis. J Hepatol. (2019) 70:963–73. doi: 10.1016/j.jhep.2018.12.037
187. Gumucio JP, Qasawa AH, Ferrara PJ, Malik AN, Funai K, McDonagh B, et al. Reduced mitochondrial lipid oxidation leads to fat accumulation in myosteatosis. FASEB J. (2019) 33:7863–81. doi: 10.1096/fj.201802457RR
188. Rossi MA, Celes MR, Prado CM, Saggioro FP. Myocardial structural changes in long-term human severe sepsis/septic shock may be responsible for cardiac dysfunction. Shock. (2007) 27:10–8. doi: 10.1097/01.shk.0000235141.05528.47
189. Standage SW, Caldwell CC, Zingarelli B, Wong HR. Reduced peroxisome proliferator-activated receptor alpha expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock. (2012) 37:164–9. doi: 10.1097/SHK.0b013e31823f1a00
190. Puthucheary ZA, Astin R, McPhail MJW, Saeed S, Pasha Y, Bear DE, et al. Metabolic phenotype of skeletal muscle in early critical illness. Thorax. (2018) 73:926–35. doi: 10.1136/thoraxjnl-2017-211073
191. Zager RA, Johnson AC, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, ischemic injury. Kidney Int. (2005) 67:111–21. doi: 10.1111/j.1523-1755.2005.00061.x
192. Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. (2014) 55:561–72. doi: 10.1194/jlr.P040501
193. Afshinnia F, Rajendiran TM, Soni T, Byun J, Wernisch S, Sas KM, et al. Impaired beta-oxidation and altered complex lipid fatty acid partitioning with advancing CKD. J Am Soc Nephrol. (2018) 29:295–306. doi: 10.1681/ASN.2017030350
194. Chen KW, Demarco B, Broz P. Beyond inflammasomes: emerging function of gasdermins during apoptosis and NETosis. EMBO J. (2020) 39:e103397. doi: 10.15252/embj.2019103397
195. Akdogan A, Calguneri M, Yavuz B, Arslan EB, Kalyoncu U, Sahiner L, et al. Are familial Mediterranean fever (FMF) patients at increased risk for atherosclerosis? Impaired endothelial function and increased intima media thickness are found in FMF. J Am Coll Cardiol. (2006) 48:2351–3. doi: 10.1016/j.jacc.2006.09.013
196. Park YH, Remmers EF, Lee W, Ombrello AK, Chung LK, Shilei Z, et al. Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol. (2020) 21:857–67. doi: 10.1038/s41590-020-0705-6
197. Sanders R, Craigova L, Schessler B, Casey C, White M, Parker M, et al. Postoperative troponin increases after noncardiac surgery are associated with raised neurofilament light: a prospective observational cohort study. Br J Anaesth. (2020) doi: 10.1016/j.bja.2020.10.012
198. Toldo S, Mauro AG, Cutter Z, Abbate A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2018) 315:H1553–68. doi: 10.1152/ajpheart.00158.2018
199. Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, necroptosis (PANoptosis). Front Cell Infect Microbiol. (2020) 10:237. doi: 10.3389/fcimb.2020.00237
200. Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. (2018) 138:2227–42. doi: 10.1161/CIRCULATIONAHA.118.035202
201. Griffith DM, Lewis S, Rossi AG, Rennie J, Salisbury L, Merriweather JL, et al. Systemic inflammation after critical illness: relationship with physical recovery and exploration of potential mechanisms. Thorax. (2016) 71:820–9. doi: 10.1136/thoraxjnl-2015-208114
202. Melchor SJ, Saunders CM, Sanders I, Hatter JA, Byrnes KA, Coutermarsh-Ott S, et al. IL-1R regulates disease tolerance and cachexia in toxoplasma gondii infection. J Immunol. (2020) 204:3329–38. doi: 10.4049/jimmunol.2000159
203. Wu Z, Oeck S, West AP, Mangalhara KC, Sainz AG, Newman LE, et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat Metab. (2019) 1:1209–18. doi: 10.1038/s42255-019-0150-8
204. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1beta induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell. (2019) 74:801–15.e6. doi: 10.1016/j.molcel.2019.02.038
205. Chevillard C, Nunes JPS, Frade AF, Almeida RR, Pandey RP, Nascimento MS, et al. Disease tolerance and pathogen resistance genes may underlie trypanosoma cruzi persistence and differential progression to chagas disease cardiomyopathy. Front Immunol. (2018) 9:2791. doi: 10.3389/fimmu.2018.02791
206. Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. (2020) 41:2153–163. doi: 10.1093/eurheartj/ehz542
207. Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. (2020) 141:124–31. doi: 10.1161/CIRCULATIONAHA.119.044362
208. Wen Y, Liu Y, Tang T, Lv L, Liu H, Ma K, et al. NLRP3 inflammasome activation is involved in ang II-induced kidney damage via mitochondrial dysfunction. Oncotarget. (2016) 7:54290–302. doi: 10.18632/oncotarget.11091
209. Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. (2017) 8:e3074. doi: 10.1038/cddis.2017.470
210. Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J. (2018) 39:60–69. doi: 10.1093/eurheartj/ehx261
211. Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight. (2017) 2:e94716. doi: 10.1172/jci.insight.94716
212. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
213. Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. (2018) 215:21–33. doi: 10.1084/jem.20171773
214. Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. (2013) 62:126–33. doi: 10.1161/HYPERTENSIONAHA.113.00689
215. Riche F, Chousterman BG, Valleur P, Mebazaa A, Launay JM, Gayat E. Protracted immune disorders at one year after ICU discharge in patients with septic shock. Crit Care. (2018) 22:42. doi: 10.1186/s13054-017-1934-4
216. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
217. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature. (2021) 594:100–5. doi: 10.1038/s41586-021-03547-7
218. Alpert A, Pickman Y, Leipold M, Rosenberg-Hasson Y, Ji X, Gaujoux R, et al. A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat Med. (2019) 25:487–95. doi: 10.1038/s41591-019-0381-y
219. Sayed N, Huang Y, Nguyen K, Krejciova-Rajaniemi Z, Grawe AP, Gao T, et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat Aging. (2021) 1:598–615. doi: 10.1038/s43587-021-00082-y
220. Limaye AP, Stapleton RD, Peng L, Gunn SR, Kimball LE, Hyzy R, et al. Effect of ganciclovir on IL-6 levels among cytomegalovirus-seropositive adults with critical illness: a randomized clinical trial. JAMA. (2017) 318:731–40. doi: 10.1001/jama.2017.10569
221. Ong DSY, Bonten MJM, Spitoni C, Verduyn Lunel FM, Frencken JF, Horn J, et al. Epidemiology of multiple herpes viremia in previously immunocompetent patients with septic shock. Clin Infect Dis. (2017) 64:1204–10. doi: 10.1093/cid/cix120
222. Wall NA, Chue CD, Edwards NC, Pankhurst T, Harper L, Steeds RP, et al. Cytomegalovirus seropositivity is associated with increased arterial stiffness in patients with chronic kidney disease. PLoS ONE. (2013) 8:e55686. doi: 10.1371/journal.pone.0055686
223. Firth C, Harrison R, Ritchie S, Wardlaw J, Ferro CJ, Starr JM, et al. Cytomegalovirus infection is associated with an increase in systolic blood pressure in older individuals. QJM. (2016) 109:595–600. doi: 10.1093/qjmed/hcw026
224. Haarala A, Kahonen M, Lehtimaki T, Aittoniemi J, Jylhava J, Hutri-Kahonen N, et al. Relation of high cytomegalovirus antibody titres to blood pressure and brachial artery flow-mediated dilation in young men: the cardiovascular risk in young finns study. Clin Exp Immunol. (2012) 167:309–16. doi: 10.1111/j.1365-2249.2011.04513.x
225. Brunner S, Herndler-Brandstetter D, Weinberger B, Grubeck-Loebenstein B. Persistent viral infections and immune aging. Ageing Res Rev. (2011) 10:362–9. doi: 10.1016/j.arr.2010.08.003
226. Cowley NJ, Owen A, Shiels SC, Millar J, Woolley R, Ives N, et al. Safety and efficacy of antiviral therapy for prevention of cytomegalovirus reactivation in immunocompetent critically ill patients: a randomized clinical trial. JAMA Intern Med. (2017) 177:774–83. doi: 10.1001/jamainternmed.2017.0895
227. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. (2018) 40:203–14. doi: 10.1007/s00281-017-0656-7
228. van Vught LA, Klein Klouwenberg PM, Spitoni C, Scicluna BP, Wiewel MA, Horn J, et al. Risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA. (2016) 315:1469–79. doi: 10.1001/jama.2016.2691
229. Huang J, Liu K, Zhu S, Xie M, Kang R, Cao L, et al. AMPK regulates immunometabolism in sepsis. Brain Behav Immun. (2017) doi: 10.1016/j.bbi.2017.11.003
230. Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. (2014) 5:4436. doi: 10.1038/ncomms5436
231. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. (2016) 7:13280. doi: 10.1038/ncomms13280
232. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. (2016) 213:337–54. doi: 10.1084/jem.20150900
233. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
234. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
235. Khatib-Massalha E, Bhattacharya S, Massalha H, Biram A, Golan K, Kollet O, et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat Commun. (2020) 11:3547. doi: 10.1038/s41467-020-17402-2
236. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46.e9. doi: 10.1016/j.cell.2017.11.025
237. Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med. (2018) 6:691–8. doi: 10.1016/S2213-2600(18)30177-2
238. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. (2016) 254:228–36. doi: 10.1016/j.atherosclerosis.2016.10.019
239. van der Heijden C, Keating ST, Groh L, Joosten LAB, Netea MG, Riksen NP. Aldosterone induces trained immunity: the role of fatty acid synthesis. Cardiovasc Res. (2020) 116:317–28. doi: 10.1093/cvr/cvz137
240. van der Heijden C, Groh L, Keating ST, Kaffa C, Noz MP, Kersten S, et al. Catecholamines induce trained immunity in monocytes in vitro and in vivo. Circ Res. (2020) 127:269–83. doi: 10.1161/CIRCRESAHA.119.315800
241. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61.e12. doi: 10.1016/j.cell.2017.11.034
242. Phan AT, Goldrath AW, Glass CK. Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity. (2017) 46:714–29. doi: 10.1016/j.immuni.2017.04.016
243. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
244. Lu YT, Li LZ, Yang YL, Yin X, Liu Q, Zhang L, et al. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. (2018) 9:672. doi: 10.1038/s41419-018-0708-5
245. Wang Y, Subramanian M, Yurdagul A Jr, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. (2017) 171:331–45.e22. doi: 10.1016/j.cell.2017.08.041
246. Diskin C, Palsson-McDermott EM. Metabolic modulation in macrophage effector function. Front Immunol. (2018) 9:270. doi: 10.3389/fimmu.2018.00270
247. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. (2007) 450:736–40. doi: 10.1038/nature06322
248. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
249. Klein Geltink RI, O'Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial priming by CD28. Cell. (2017) 171:385–97.e11. doi: 10.1016/j.cell.2017.08.018
250. Rambold AS, Pearce EL. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol. (2018) 39:6–18. doi: 10.1016/j.it.2017.08.006
251. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
252. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. (2017) 170:142–57.e19. doi: 10.1016/j.cell.2017.06.007
253. Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. (2016) 17:406–13. doi: 10.1038/ni.3398
254. Sun L, Yang X, Yuan Z, Wang H. Metabolic reprogramming in immune response and tissue inflammation. Arterioscler Thromb Vasc Biol. (2020) 40:1990–2001. doi: 10.1161/ATVBAHA.120.314037
255. Jensen IJ, Jensen SN, Sjaastad FV, Gibson-Corley KN, Dileepan T, Griffith TS, et al. Sepsis impedes EAE disease development and diminishes autoantigen-specific naive CD4 T cells. Elife. (2020) 9:e55800. doi: 10.7554/eLife.55800
256. Wang A, Luan HH, Medzhitov R. An evolutionary perspective on immunometabolism. Science. (2019) 363:eaar3932. doi: 10.1126/science.aar3932
257. Ganeshan K, Nikkanen J, Man K, Leong YA, Sogawa Y, Maschek JA, et al. Energetic trade-offs and hypometabolic states promote disease tolerance. Cell. (2019) 177:399–413.e12. doi: 10.1016/j.cell.2019.01.050
258. Timblin GA, Tharp KM, Ford B, Winchester JM, Wang J, Zhu S, et al. Mitohormesis reprogrammes macrophage metabolism to enforce tolerance. Nat Metab. (2021) 3:618–35. doi: 10.1038/s42255-021-00392-w
259. Colaco HG, Barros A, Neves-Costa A, Seixas E, Pedroso D, Velho T, et al. Tetracycline antibiotics induce host-dependent disease tolerance to infection. Immunity. (2021) 54:53–67.e7. doi: 10.1016/j.immuni.2020.09.011
260. Newson J, Motwani MP, Kendall AC, Nicolaou A, Muccioli GG, Alhouayek M, et al. Inflammatory resolution triggers a prolonged phase of immune suppression through COX-1/mPGES-1-derived prostaglandin E2. Cell Rep. (2017) 20:3162–75. doi: 10.1016/j.celrep.2017.08.098
261. Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. (2012) 287:25758–69. doi: 10.1074/jbc.M112.362343
262. Abdel-Hakeem MS, Manne S, Beltra JC, Stelekati E, Chen Z, Nzingha K, et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol. (2021) 22:1008–19. doi: 10.1038/s41590-021-00975-5
263. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. (2010) 464:121–5. doi: 10.1038/nature08778
264. Frodermann V, Rohde D, Courties G, Severe N, Schloss MJ, Amatullah H, et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat Med. (2019) 25:1761–71. doi: 10.1038/s41591-019-0633-x
265. Trudel G, Payne M, Madler B, Ramachandran N, Lecompte M, Wade C, et al. Bone marrow fat accumulation after 60 days of bed rest persisted 1 year after activities were resumed along with hemopoietic stimulation: the women international space simulation for exploration study. J Appl Physiol. (2009) 107:540–8. doi: 10.1152/japplphysiol.91530.2008
266. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. (2014) 20:754–8. doi: 10.1038/nm.3589
267. Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA. (2013) 110:16574–9. doi: 10.1073/pnas.1310655110
268. McAlpine CS, Kiss MG, Rattik S, He S, Vassalli A, Valet C, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. (2019) 566:383–87. doi: 10.1038/s41586-019-0948-2
269. Alexopoulou C, Bolaki M, Akoumianaki E, Erimaki S, Kondili E, Mitsias P, et al. Sleep quality in survivors of critical illness. Sleep Breath. (2018) 23:463–71. doi: 10.1007/s11325-018-1701-z
270. Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. (2018) 362:911–7. doi: 10.1126/science.aau3879
271. Bahar I, Elay G, Baskol G, Sungur M, Donmez-Altuntas H. Increased DNA damage and increased apoptosis and necrosis in patients with severe sepsis and septic shock. J Crit Care. (2018) 43:271–5. doi: 10.1016/j.jcrc.2017.09.035
272. Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp Hematol. (2018) 59:60–5. doi: 10.1016/j.exphem.2017.11.002
273. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. (2018) 554:123–7. doi: 10.1038/nature25434
274. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–49.e5. doi: 10.1016/j.stem.2018.10.013
275. Fuster JJ, Walsh K. Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age-related cardiovascular disease. Circ Res. (2018) 122:523–32. doi: 10.1161/CIRCRESAHA.117.312115
276. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
277. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature. (2018) 557:580–4. doi: 10.1038/s41586-018-0125-z
278. Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, et al. TET2-Loss-of-Function-driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep. (2020) 33:108326. doi: 10.1016/j.celrep.2020.108326
279. Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. (2021) 184:1348–61.e22. doi: 10.1016/j.cell.2021.01.049
280. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. (2021) 28:1428–42.e6. doi: 10.1016/j.stem.2021.03.002
281. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J Am Coll Cardiol. (2018) 71:875–86. doi: 10.1016/j.jacc.2017.12.037
282. Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res. (2018) 123:335–41. doi: 10.1161/CIRCRESAHA.118.313225
283. Abplanalp WT, Cremer S, John D, Hoffmann J, Schuhmacher B, Merten M, et al. Clonal hematopoiesis-driver DNMT3A mutations alter immune cells in heart failure. Circ Res. (2021) 128:216–28. doi: 10.1161/CIRCRESAHA.120.317104
284. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. (2015) 525:389–93. doi: 10.1038/nature15252
285. Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, et al. Dnmt3a restrains mast cell inflammatory responses. Proc Natl Acad Sci USA. (2017) 114:E1490–9. doi: 10.1073/pnas.1616420114
286. Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, et al. TET2-driven clonal hematopoiesis and response to canakinumab: an exploratory analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. (2022) 7:521–8. doi: 10.1001/jamacardio.2022.0386
287. Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. (2020) 41:933–9. doi: 10.1093/eurheartj/ehz591
288. Zekavat SM, Lin SH, Bick AG, Liu A, Paruchuri K, Wang C, et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat Med. (2021) 27:1012–24. doi: 10.1038/s41591-021-01371-0
289. Jaisser F, Farman N. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol Rev. (2016) 68:49–75. doi: 10.1124/pr.115.011106
290. Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, et al. T-Cell mineralocorticoid receptor controls blood pressure by regulating interferon-gamma. Circ Res. (2017) 120:1584–97. doi: 10.1161/CIRCRESAHA.116.310480
291. Li C, Sun XN, Zeng MR, Zheng XJ, Zhang YY, Wan Q, et al. Mineralocorticoid receptor deficiency in T cells attenuates pressure overload-induced cardiac hypertrophy and dysfunction through modulating T-cell activation. Hypertension. (2017) 70:137–47. doi: 10.1161/HYPERTENSIONAHA.117.09070
292. Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. (2010) 120:3350–64. doi: 10.1172/JCI41080
293. Pimenta E, Gordon RD, Stowasser M. Salt, aldosterone and hypertension. J Hum Hypertens. (2013) 27:1–6. doi: 10.1038/jhh.2012.27
294. Buonafine M, Martinez-Martinez E, Amador C, Gravez B, Ibarrola J, Fernandez-Celis A, et al. Neutrophil gelatinase-associated lipocalin from immune cells is mandatory for aldosterone-induced cardiac remodeling and inflammation. J Mol Cell Cardiol. (2018) 115:32–8. doi: 10.1016/j.yjmcc.2017.12.011
295. Sun WY, Bai B, Luo C, Yang K, Li D, Wu D, et al. Lipocalin-2 derived from adipose tissue mediates aldosterone-induced renal injury. JCI Insight. (2018) 3:e120196. doi: 10.1172/jci.insight.120196
296. Matsa R, Ashley E, Sharma V, Walden AP, Keating L. Plasma and urine neutrophil gelatinase-associated lipocalin in the diagnosis of new onset acute kidney injury in critically ill patients. Crit Care. (2014) 18:R137. doi: 10.1186/cc13958
297. Buonafine M, Martinez-Martinez E, Jaisser F. More than a simple biomarker: the role of NGAL in cardiovascular and renal diseases. Clin Sci. (2018) 132:909–23. doi: 10.1042/CS20171592
298. Martinez-Martinez E, Buonafine M, Boukhalfa I, Ibarrola J, Fernandez-Celis A, Kolkhof P, et al. Aldosterone target NGAL (neutrophil gelatinase-associated lipocalin) is involved in cardiac remodeling after myocardial infarction through NFkappaB pathway. Hypertension. (2017) 70:1148–56. doi: 10.1161/HYPERTENSIONAHA.117.09791
299. Ong GS, Young MJ. Mineralocorticoid regulation of cell function: the role of rapid signalling and gene transcription pathways. J Mol Endocrinol. (2017) 58:R33–57. doi: 10.1530/JME-15-0318
300. Masson GS, Nair AR, Dange RB, Silva-Soares PP, Michelini LC, Francis J. Toll-like receptor 4 promotes autonomic dysfunction, inflammation and microglia activation in the hypothalamic paraventricular nucleus: role of endoplasmic reticulum stress. PLoS ONE. (2015) 10:e0122850. doi: 10.1371/journal.pone.0122850
301. Yu Y, Zhang ZH, Wei SG, Serrats J, Weiss RM, Felder RB. Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension. (2010) 55:652–9. doi: 10.1161/HYPERTENSIONAHA.109.142836
302. Su Q, Huo CJ, Li HB, Liu KL, Li X, Yang Q, et al. Renin-angiotensin system acting on reactive oxygen species in paraventricular nucleus induces sympathetic activation via AT1R/PKCgamma/Rac1 pathway in salt-induced hypertension. Sci Rep. (2017) 7:43107. doi: 10.1038/srep43107
303. Yu XJ, Suo YP, Qi J, Yang Q, Li HH, Zhang DM, et al. Interaction between AT1 receptor and NF-kappaB in hypothalamic paraventricular nucleus contributes to oxidative stress and sympathoexcitation by modulating neurotransmitters in heart failure. Cardiovasc Toxicol. (2013) 13:381–90. doi: 10.1007/s12012-013-9219-x
304. Zhang ZH, Francis J, Weiss RM, Felder RB. The renin-angiotensin-aldosterone system excites hypothalamic paraventricular nucleus neurons in heart failure. Am J Physiol Heart Circ Physiol. (2002) 283:H423–33. doi: 10.1152/ajpheart.00685.2001
305. Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. (2013) 497:211–6. doi: 10.1038/nature12143
306. Vagaonescu TD, Saadia D, Tuhrim S, Phillips RA, Kaufmann H. Hypertensive cardiovascular damage in patients with primary autonomic failure. Lancet. (2000) 355:725–6. doi: 10.1016/S0140-6736(99)05320-9
307. Arnold AC, Okamoto LE, Gamboa A, Black BK, Raj SR, Elijovich F, et al. Mineralocorticoid receptor activation contributes to the supine hypertension of autonomic failure. Hypertension. (2016) 67:424–9. doi: 10.1161/HYPERTENSIONAHA.115.06617
308. Zhang ZH, Kang YM, Yu Y, Wei SG, Schmidt TJ, Johnson AK, et al. 11beta-hydroxysteroid dehydrogenase type 2 activity in hypothalamic paraventricular nucleus modulates sympathetic excitation. Hypertension. (2006) 48:127–33. doi: 10.1161/01.HYP.0000224296.96235.dd
309. Zhang ZH, Yu Y, Kang YM, Wei SG, Felder RB. Aldosterone acts centrally to increase brain renin-angiotensin system activity and oxidative stress in normal rats. Am J Physiol Heart Circ Physiol. (2008) 294:H1067–74. doi: 10.1152/ajpheart.01131.2007
310. Xue B, Beltz TG, Johnson RF, Guo F, Hay M, Johnson AK. PVN adenovirus-siRNA injections silencing either NOX2 or NOX4 attenuate aldosterone/NaCl-induced hypertension in mice. Am J Physiol Heart Circ Physiol. (2012) 302:H733–41. doi: 10.1152/ajpheart.00873.2011
311. Schmidt H, Hoyer D, Wilhelm J, Soffker G, Heinroth K, Hottenrott K, et al. The alteration of autonomic function in multiple organ dysfunction syndrome. Crit Care Clin. (2008) 24:149–63, ix. doi: 10.1016/j.ccc.2007.10.003
312. Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, et al. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. (2003) 362:1799–805. doi: 10.1016/S0140-6736(03)14899-4
313. Lichtarowicz-Krynska EJ, Cole TJ, Camacho-Hubner C, Britto J, Levin M, Klein N, et al. Circulating aldosterone levels are unexpectedly low in children with acute meningococcal disease. J Clin Endocrinol Metab. (2004) 89:1410–4. doi: 10.1210/jc.2003-030505
314. Chung KS, Song JH, Jung WJ, Kim YS, Kim SK, Chang J, et al. Implications of plasma renin activity and plasma aldosterone concentration in critically ill patients with septic shock. Korean J Crit Care Med. (2017) 32:142–53. doi: 10.4266/kjccm.2017.00094
315. Funder JW. Aldosterone and mineralocorticoid receptors-physiology and pathophysiology. Int J Mol Sci. (2017) 18:1032. doi: 10.3390/ijms18051032
316. Funder JW. Mineralocorticoid receptors: distribution and activation. Heart Fail Rev. (2005) 10:15–22. doi: 10.1007/s10741-005-2344-2
317. Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci USA. (2003) 100:9202–7. doi: 10.1073/pnas.1633591100
318. Mihailidou AS, Loan Le TY, Mardini M, Funder JW. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension. (2009) 54:1306–12. doi: 10.1161/HYPERTENSIONAHA.109.136242
319. Funder JW. Mineralocorticoid receptor antagonists: emerging roles in cardiovascular medicine. Integr Blood Press Control. (2013) 6:129–38. doi: 10.2147/IBPC.S13783
320. Annane D, Pastores SM, Arlt W, Balk RA, Beishuizen A, Briegel J, et al. Critical illness-related corticosteroid insufficiency (CIRCI): a narrative review from a multispecialty task force of the society of critical care medicine (SCCM) and the European society of intensive care medicine (ESICM). Crit Care Med. (2017) 45:2089–98. doi: 10.1097/CCM.0000000000002724
321. Chappell D, Bruegger D, Potzel J, Jacob M, Brettner F, Vogeser M, et al. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit Care. (2014) 18:538. doi: 10.1186/s13054-014-0538-5
322. Byrne L, Obonyo NG, Diab SD, Dunster KR, Passmore MR, Boon AC, et al. Unintended consequences: fluid resuscitation worsens shock in an ovine model of endotoxemia. Am J Respir Crit Care Med. (2018) 198:1043–54. doi: 10.1164/rccm.201801-0064OC
323. Echeverria C, Montorfano I, Tapia P, Riedel C, Cabello-Verrugio C, Simon F. Endotoxin-induced endothelial fibrosis is dependent on expression of transforming growth factors beta1 and beta2. Infect Immun. (2014) 82:3678–86. doi: 10.1128/IAI.02158-14
324. Tapia P, Gatica S, Cortes-Rivera C, Otero C, Becerra A, Riedel CA, et al. Circulating endothelial cells from septic shock patients convert to fibroblasts are associated with the resuscitation fluid dose and are biomarkers for survival prediction. Crit Care Med. (2019) 47:942–50. doi: 10.1097/CCM.0000000000003778
325. Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, et al. Endothelial to mesenchymal transition in cardiovascular disease: jacc state-of-the-art review. J Am Coll Cardiol. (2019) 73:190–209. doi: 10.1016/j.jacc.2018.09.089
326. Gosling P. Salt of the earth or a drop in the ocean? A pathophysiological approach to fluid resuscitation. Emerg Med J. (2003) 20:306–15. doi: 10.1136/emj.20.4.306
327. Van Regenmortel N, Verbrugghe W, Roelant E, Van den Wyngaert T, Jorens PG. Maintenance fluid therapy and fluid creep impose more significant fluid, sodium, and chloride burdens than resuscitation fluids in critically ill patients: a retrospective study in a tertiary mixed ICU population. Intensive Care Med. (2018) 44:409–17. doi: 10.1007/s00134-018-5147-3
328. Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. (2017) 21:1009–20. doi: 10.1016/j.celrep.2017.10.002
329. Hammon M, Grossmann S, Linz P, Seuss H, Hammon R, Rosenhauer D, et al. 3 Tesla (23)Na magnetic resonance imaging during acute kidney injury. Acad Radiol. (2017) 24:1086–93. doi: 10.1016/j.acra.2017.03.012
330. Huang L, Li X, Gu X, Zhang H, Ren L, Guo L, et al. Health outcomes in people 2 years after surviving hospitalisation with COVID-19: a longitudinal cohort study. Lancet Respir Med. (2022). doi: 10.1016/S2213-2600(22)00126-6. [Epub ahead of print].
331. Groff D, Sun A, Ssentongo AE, Ba DM, Parsons N, Poudel GR, et al. Short-term and long-term rates of postacute sequelae of SARS-CoV-2 infection: a systematic review. JAMA Netw Open. (2021) 4:e2128568. doi: 10.1001/jamanetworkopen.2021.28568
332. Evans RA, McAuley H, Harrison EM, Shikotra A, Singapuri A, Sereno M, et al. Physical, cognitive, and mental health impacts of COVID-19 after hospitalisation (PHOSP-COVID): a UK multicentre, prospective cohort study. Lancet Respir Med. (2021) 9:1275–87. doi: 10.1016/S2213-2600(21)00383-0
333. Yende S, Parikh CR. Long COVID and kidney disease. Nat Rev Nephrol. (2021) 17:792–3. doi: 10.1038/s41581-021-00487-3
334. Gasparini M, Khan S, Patel JM, Parekh D, Bangash MN, Stupsilonmpfle R, et al. Renal impairment and its impact on clinical outcomes in patients who are critically ill with COVID-19: a multicentre observational study. Anaesthesia. (2021) 76:320–6. doi: 10.1111/anae.15293
335. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
336. Restrepo MI, Reyes LF. Pneumonia as a cardiovascular disease. Respirology. (2018) 23:250–9. doi: 10.1111/resp.13233
337. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. (2021) 7:43. doi: 10.1038/s41420-021-00428-w
338. Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-Coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. (2019) 5:101. doi: 10.1038/s41420-019-0181-7
339. Prasannan N, Heightman M, Hillman T, Wall E, Bell R, Kessler A, et al. Impaired exercise capacity in post-COVID syndrome: the role of VWF-ADAMTS13 axis. Blood Adv. (2022). doi: 10.1182/bloodadvances.2021006944. [Epub ahead of print].
340. Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. (2020) 130:1912–30. doi: 10.1172/JCI133737
341. Barrett JP, Knoblach SM, Bhattacharya S, Gordish-Dressman H, Stoica BA, Loane DJ. Traumatic brain injury induces cGAS activation and type I interferon signaling in aged mice. Front Immunol. (2021) 12:710608. doi: 10.3389/fimmu.2021.710608
342. Murira A, Lamarre A. Type-I interferon responses: from friend to foe in the battle against chronic viral infection. Front Immunol. (2016) 7:609. doi: 10.3389/fimmu.2016.00609
343. Phetsouphanh C, Darley DR, Wilson DB, Howe A, Munier CML, Patel SK, et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol. (2022) 23:210–6. doi: 10.1038/s41590-021-01113-x
344. Kusnadi A, Ramirez-Suastegui C, Fajardo V, Chee SJ, Meckiff BJ, Simon H, et al. Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8(+) T cells. Sci Immunol. (2021) 6:eabe4782. doi: 10.1126/sciimmunol.abe4782
Keywords: critical illness, chronicity, atherosclerosis, insulin resistance, inflammation, heart failure, immune aging, CKD
Citation: Owen A, Patel JM, Parekh D and Bangash MN (2022) Mechanisms of Post-critical Illness Cardiovascular Disease. Front. Cardiovasc. Med. 9:854421. doi: 10.3389/fcvm.2022.854421
Received: 13 January 2022; Accepted: 22 June 2022;
Published: 15 July 2022.
Edited by:
Hiroshi Iwata, Juntendo University, JapanReviewed by:
Naoki Ishimori, Hokkaido University, JapanCopyright © 2022 Owen, Patel, Parekh and Bangash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mansoor N. Bangash, bWFuc29vci5iYW5nYXNoMUBuaHMubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.