 Ahmed Diab
Ahmed Diab Carla Valenzuela Ripoll
Carla Valenzuela Ripoll Zhen Guo
Zhen Guo Ali Javaheri
Ali Javaheri
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 08 March 2022
Sec. Lipids in Cardiovascular Disease
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.846990
This article is part of the Research Topic Insights in Lipids in Cardiovascular Disease: 2021 View all 11 articles
Although research on high-density lipoprotein (HDL) has historically focused on atherosclerotic coronary disease, there exists untapped potential of HDL biology for the treatment of heart failure. Anti-oxidant, anti-inflammatory, and endothelial protective properties of HDL could impact heart failure pathogenesis. HDL-associated proteins such as apolipoprotein A-I and M may have significant therapeutic effects on the myocardium, in part by modulating signal transduction pathways and sphingosine-1-phosphate biology. Furthermore, because heart failure is a complex syndrome characterized by multiple comorbidities, there are complex interactions between heart failure, its comorbidities, and lipoprotein homeostatic mechanisms. In this review, we will discuss the effects of heart failure and associated comorbidities on HDL, explore potential cardioprotective properties of HDL, and review novel HDL therapeutic targets in heart failure.
Cardiovascular disease (CVD) is a leading cause of mortality worldwide (1). Heart failure (HF) is a common result of cardiometabolic disease and a major contributor to CVD mortality (2). The prevalence of HF in the developed world is rising and is estimated to be at 2%, while the incidence approaches 5–10 per 1,000 persons per year (3). HF is a clinical syndrome, typically presenting with symptoms of dyspnea, fluid retention, and decreased exercise tolerance. It usually follows structural or functional disorders of the endocardium, myocardium, or pericardium and is divided into three categories: HF with reduced ejection fraction (HFrEF), HF with preserved ejection fraction (HFpEF), and HF with mid-range ejection fraction (4, 5).
Multiple rationale suggest a mechanistic link between lipoproteins and HF. Interestingly, in HF patients, plasma cholesterol concentrations are inversely associated with mortality (6, 7). This observation, termed the “cholesterol paradox,” could be related to malnutrition, cachexia (8, 9), and inflammation (10–14) observed in HF patients, as well as direct effects of lipoproteins on the myocardium. Moreover, recent Mendelian randomization studies support a causal effect of low-density lipoprotein cholesterol (LDL-C) and triglycerides on LV mass and myocardial remodeling (15). Analogously, a clinical trial showed that reconstituted high-density lipoprotein (HDL) infusion shortens cardiac repolarization, demonstrating the capability of HDL to alter cardiac electrophysiological properties (16). Both studies exemplify a direct role of lipoproteins on the myocardium. Furthermore, lipoproteins can function as a fuel source, an important consideration in HF patients, where the energy-starved myocardium primarily consumes ketone bodies and fatty acids (17).
Based on two large randomized trials, a case could even be made for statin use in HF patients, thus LDL-C lowering via statins is unlikely to exacerbate HF outcomes (18, 19). We hypothesized that decreased HDL or HDL-associated apolipoproteins could be a driver of adverse HF outcomes (18, 19). High-density lipoprotein cholesterol (HDL-C) is inversely associated with CVD risk, as large epidemiological studies, such as the Framingham Heart Study have shown (20). Nonetheless, multiple randomized trials have failed to show a decrease in CVD risk or major adverse cardiac events when increasing HDL-C levels as a therapeutic target (21, 22). One interpretation of these findings is that, rather than the steady-state cholesterol mass, HDL or its associated apolipoproteins could exert beneficial effects in the setting of HF (or even CVD or other cardiac inflammatory disorders). For instance, our group has shown that reduced pre-transplant HDL cholesterol efflux capacity is associated with the progression of cardiac allograft vasculopathy, a major cause of mortality for cardiac transplant recipients (23). This example served as a proof-of-paradigm that HDL functions may be relevant outside of traditional atherosclerosis. The cardioprotective role of HDL may be related to its anti-oxidant and anti-inflammatory properties, endothelial protection, as well as its reverse cholesterol transport capacity (24).
Many pre-clinical studies performed mainly in rodents focus on the effect of HDL in cardiac pathophysiology and have shown positive effects on the myocardium. For instance, HDL can reduce infarct size in the setting of cardiac ischemia/reperfusion injury, attenuate apoptosis, preserve mitochondrial function, and protect the myocardium against oxidative stress (25–31).
Although a broad range of anti-atherogenic properties have been attributed to HDL, many are independent of its cholesterol content and reverse cholesterol transport. The heterogeneous properties of HDL particles are relatively complex, due to the wide variety of proteomic and lipidomic cargo of the particles. These characteristics lead to specific cardioprotective functions, such as increased endothelial nitric oxide (NO) production, reduced inflammation in endothelial cells and macrophages, stimulation of insulin-independent glucose uptake in the myocardium, among others. For example, the anti-oxidative capacity of HDL is mainly attributed to its ability to protect LDL from oxidation by free radicals. Of note, antioxidant components of HDL, such as the HDL-associated enzyme Paraoxonase 1 (PON1), metabolize lipid hydroperoxides and prevent their accumulation in LDL particles, decreasing LDL endocytosis by macrophages and formation of foam cells, thus averting the formation of atherosclerotic plaque (32–37).
Recent advances in proteomic characterization have led to the identification of novel HDL subclasses that will, in all likelihood, eventually supersede the historical size and density-based characterization system (38, 39). For historical reference, larger HDL2 particles are inversely associated with CVD risk, while smaller, denser HDL3 subclass exerts anti-atherogenic, anti-oxidant, and anti-inflammatory functions (40, 41), and these subclasses are also associated with mortality in acute HF patients. Total and small HDL particles (diameter <8.8 nm, mostly HDL3), measured by nuclear magnetic resonance spectroscopy, were inversely associated with 3-month mortality in patients with acute HF, while both large HDL and HDL-C demonstrated no significant association (42). Similarly, in HFrEF and HFpEF patients, total and small HDL were inversely associated with adverse outcomes (43).
To the best of our knowledge, the rigorous analysis of HDL proteomics has yet to be performed in advanced HF cohorts. Nonetheless, multiple preclinical and human epidemiological studies support the concept of pleiotropic effects of HDL-associated apolipoproteins (44, 45), which may play a significant role in the pathogenesis of HF. These observations led us to hypothesize that specific apolipoproteins and enzymes associated with HDL particles may potentially explain the cholesterol paradox and the underlying cardioprotective effects of HDL, which could be relevant therapeutic targets in HF. In this review, we will discuss the effects of HF and associated comorbidities on HDL, explore potential cardioprotective properties of HDL, and review novel HDL therapeutic targets in HF.
Advanced HF is a multisystem syndrome often identified in patients with multiple cardiometabolic comorbidities; hence, both HF and its associated comorbidities can have complex effects on lipoprotein biology. Hepatic, renal, and gastrointestinal malperfusion secondary to reduced cardiac index and increased filling pressures all contribute to a vicious cycle of decreased nutritional intake, increased inflammation, metabolic stress, perturbations that can have important effects on lipoprotein homeostasis (Figure 1).
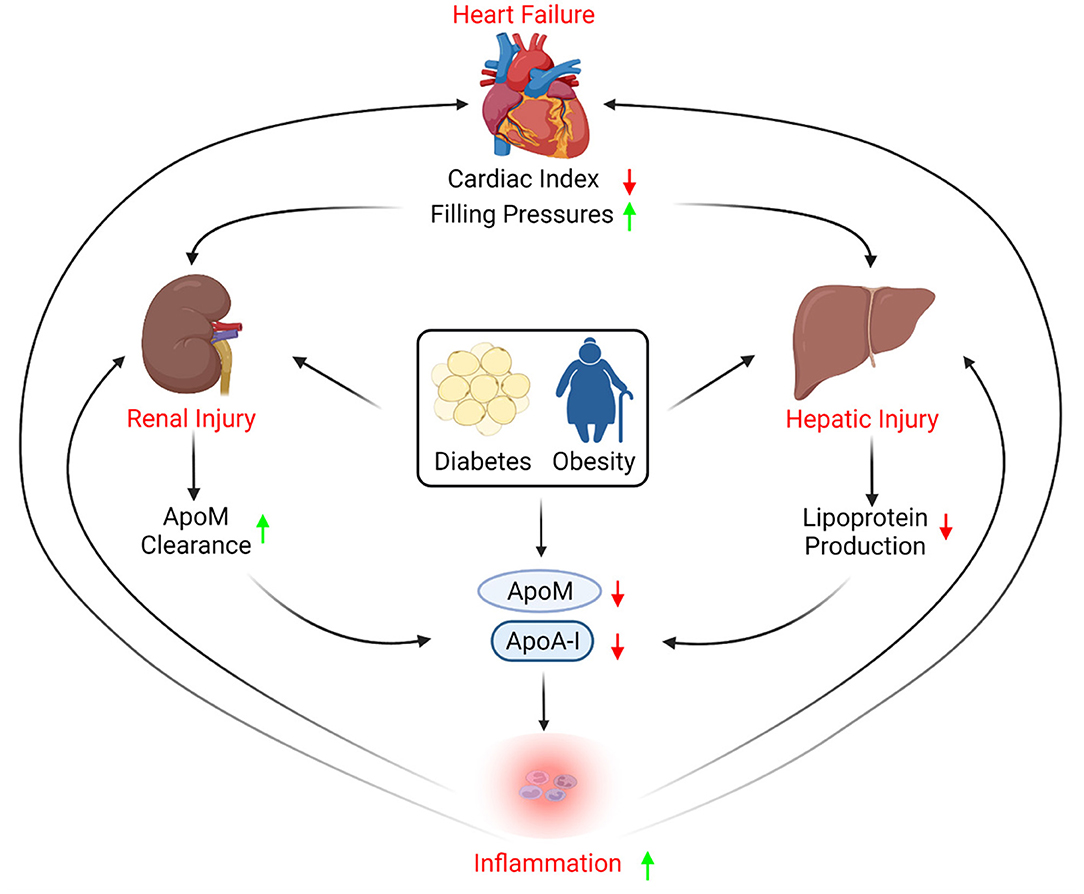
Figure 1. Apolipoproteins role in heart failure progression. Heart failure causes reduced cardiac index and increased filling pressures, which subsequently leads to hepatic injury that can affect apolipoprotein production. In addition, heart failure-induced kidney injury may increase renal excretion of apolipoprotein M (ApoM). Co-morbidities such as diabetes and obesity are also known to reduce circulating apolipoproteins, contributing to inflammation, thus exacerbating kidney and hepatic injury, and provoking further cardiac dysfunction.
HF is characterized by a chronic inflammatory state. While the increase in pro-inflammatory cytokines in HF has been well-documented, there is still debate regarding the extent to which increased cytokines are directly responsible for deleterious results or are simply a reflection of the ongoing pathophysiological processes (46–49). Nevertheless, chronic inflammatory states, such as that observed in HF, can affect plasma HDL levels, composition, and overall function. For instance, plasma HDL contains lower cholesterol ester levels, higher free cholesterol, triglycerides, and fatty acids under inflammatory states (50). Moreover, inflammation strips HDL from key proteins that are important for its normal function (e.g., lecithin-cholesterol acyltransferase (LCAT), cholesteryl ester transfer protein, and transferrin), as well as certain important apolipoproteins (e.g., apolipoproteins A-I and M) (51–55). Apolipoprotein A-I (ApoA-I) is the primary mediator of cholesterol efflux, the key rate-limiting step of reverse cholesterol transport, and the main protein component of HDL particles (56–59). In the same context, apolipoprotein M (ApoM), a cardioprotective apolipoprotein (45), is a negative acute response protein, levels of which decrease in response to inflammation and infection (52, 60, 61). The decrease in HDL levels and alteration of its structural composition in inflammatory states impair the reverse cholesterol transport process and HDL's anti-inflammatory and anti-oxidant properties. In the long run, this can lead to the development of atherosclerosis and increased risk of CVD and HF.
How inflammation affects HDL particle number and composition is not very well-understood. In mice, endotoxin directly impairs active cholesterol efflux by ATP-binding cassettes A1 and G1 (ABCA1 and ABCG1) transporters, as well as scavenger receptor class B type I (SR-B1) mediated passive diffusion (62–64). Meanwhile, inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukins 1 and 6 (IL-1β, IL-6), upregulate the expression of endothelial lipase (EL), which exhibits an inverse association with HDL levels (65, 66). Badellino et al. showed that experimental administration of low-dose endotoxin in humans decreases HDL phospholipid, corresponding with EL peak concentration (67). Tietge et al. (68) reported that mice that overexpress secretory phospholipase A2 have changes in HDL composition, and under inflammatory conditions, exhibit increased HDL catabolism. Interestingly, in a murine model of pressure overload-induced HF, EL knockout exacerbated cardiac dysfunction compared to wild-type controls, consistent with the hypothesis that EL provides an alternative pathway for free fatty acid uptake as a source of energy and protects the failing myocardium (69). Thus, it is plausible that chronic inflammation may be upregulating the expression of EL, which leads to HDL catabolism to release fatty acids to the energy-starved myocardium at the expense of other cardioprotective components of the HDL particle.
Normal renal function is crucial for proper HDL function (70). Renal dysfunction induces pathologic alterations in lipoprotein metabolism in general, and HDL in particular (71). HF can induce renal dysfunction, which is a strong independent predictor of poor cardiovascular outcomes (72, 73). Cardiorenal syndrome is a term that describes the mutual interaction between the heart and kidneys, considering that injury to one of the organs usually causes dysfunction of the other (74).
Renal dysfunction and the associated chronic inflammatory state present in cardiorenal syndrome correlate with increased oxidative stress across multiple systems (75). Oxidized HDL (ox-HDL) is a modified HDL observed during conditions of increased oxidative stress and reduced anti-oxidant capacity present in cardiorenal syndrome (76, 77). Various HDL and ApoA-1 post-translational modifications can result in ox-HDL formation (76, 78, 79), which has been linked to an increased risk of cardiovascular events (80). Myeloperoxidase (MPO) can modify ApoA-I leading to ox-HDL that is less avid in its ability to bind SR-BI receptors and dysfunctional for normal cholesterol efflux activity (81, 82). Conversely, hypochlorite-generated ox-HDL exhibits increased affinity toward SR-BI, albeit with less cholesterol efflux capacity than normal HDL (83). We propose that various post-translational modifications (for example, MPO adducts) might alter specific ox-HDL characteristics (for example, higher vs. lower affinity toward SR-BI); nonetheless, renal dysfunction can contribute toward “dysfunctional” HDL particles. Moreover, ox-HDL exhibits diminished endothelial nitric oxide synthase (eNOS) mediated endothelial protective function as well as anti-apoptotic activity, which leads to impaired endothelial repair and increased pro-inflammatory activation (84–86).
In a proteomic analysis of HDL in uremic patients, isolated HDL particles lost their anti-inflammatory properties and induced the production of inflammatory cytokines (87). HDL isolated from these patients contained high levels of serum amyloid A (SAA), a protein known to promote inflammatory cytokine production and impair the anti-inflammatory capacity of HDL (87). Furthermore, HDL from uremic patients may contribute to the systemic inflammatory state in chronic kidney disease patients by decreasing apoptosis of polymorphonuclear leukocytes (88).
Diabetes is a pathophysiological process that can significantly impact the biogenesis of HDL, and cause alterations in myocardial metabolism, impairing metabolic flexibility and leading to diabetic cardiomyopathy (89). Oxidative stress, intramyocardial inflammation, cardiac fibrosis, and cardiac apoptosis all contribute to diabetic cardiomyopathy (90), which can in theory be mitigated by the anti-inflammatory and anti-oxidative functions of HDL.
In diabetes, hyperglycemia-induced advanced glycation end products, oxidative stress, and inflammation can negatively affect normal HDL function and composition, potentially contributing to an increased risk of HF (91). Glycated HDL loses atheroprotective properties and cholesterol-accepting capacity, leading to the acceleration of atherosclerosis (92). HDL isolated from diabetic patients is also rendered ineffective concerning endothelial protective function (93). Many of the pleiotropic effects of HDL are attributed to ApoM-bound sphingosine-1-phosphate (S1P), which is diminished in diabetic patients mainly due to glycation of ApoM that results in the impaired binding capacity to S1P (94).
Obesity has been established as a major risk factor for hypertension, CVD, and left ventricular hypertrophy, all risk factors for the development of HF (95). Obesity is associated with reduced HDL-C. In a large cross-sectional study, HDL-C is inversely associated with body mass index (BMI) (96). Obesity can also affect HDL subclasses and metabolism likely reflecting an underlying change in key HDL proteins and lipids (97, 98). Plasma ApoA-I exhibits a linear inverse correlation with BMI (99), while ApoM is also reduced in obese individuals (100) and is inversely associated with non-alcoholic fatty liver disease (NAFLD) (101), another comorbidity associated with obesity and an emerging risk factor for HF, in particular HFpEF (102).
Proteomic studies of HDL in patients with obesity and other comorbidities have also been informative. In NAFLD patients, quantitative changes occur in the HDL proteome, relative to morbidly obese patients without steatosis (103). One challenging aspect of studying comorbidities related to obesity is selecting the best control or reference population. Comparing obese patients with comorbidities of obesity vs. more metabolically healthy obese patients will likely minimize differences between groups. Another challenge is that many unmeasured confounders might be associated with obesity. Further prospective studies are required to unravel the complex interactions between obesity and its comorbidities, including HF, and how these interactions might be mediated by lipoproteins. In particular, the need for prospective studies is highlighted by the focus of older literature on HDL subclasses, and new studies suggesting that meals of various fat compositions can acutely affect the HDL proteome (104). Altogether, obesity itself, or its comorbidities, may alter HDL proteomic and lipidomic contents, impairing potential cardioprotective functions. This area is both complex and rapidly evolving.
Metabolic disease, obesity, and HF can also result in arrhythmias. The most common arrhythmia observed in patients is atrial fibrillation (AF). Low baseline HDL-C is associated with an increased risk of AF (105–108). AF is associated with reduced HDL quality as AF was associated with reduced HDL cholesterol efflux capacity, HDL-particle number, ApoA-I levels, and reduced LCAT activity; interestingly, all these indices improved following the restoration of sinus rhythm (109). Further validation of these findings would be encouraging, especially because the mechanistic link between AF and HDL remains unclear, particularly in the acute setting. One possible theory is the role HDL plays in myocardial membrane stabilization (110). In the more chronic setting, other HDL attributes including anti-inflammatory, anti-oxidant, and anti-atherogenic properties could interact with AF development and severity.
Aging is a well-established risk factor for the development and progression of HF, resulting from the deterioration of both cardiac structure and function, as well as the high risk of co-morbidities. Elderly patients have increased HDL oxidation, which can impair the normal protective capacity against LDL oxidation, and lead to the acceleration of atherosclerosis and CVD, both risk factors for HF (111).
Holzer et al. (112) compared HDL isolated from healthy young and elderly patients and found that aging alters HDL composition and function. HDL from elderly subjects had higher SAA and sphingomyelin, while levels of total cholesterol were reduced (112). Furthermore, HDL isolated from older patients demonstrated reduced cholesterol efflux capacity, principally through the ABCA1 pathway (113). In the same context, aged murine models have exhibited reduced ApoM secretion from the liver, with consequent impairment of S1P signaling, which reduces resistance to injury-induced vascular leak and precipitates organ fibrosis (114).
Oxidative stress is one of the main pathophysiological processes associated with aging (115) and is involved in the development of HF (116). PON1 is one of the most prominent antioxidant components of HDL (112, 117, 118). In elderly patients, it has been shown that PON1 activity and ApoE levels, both having important antioxidant properties, are diminished (112, 119). Overall, these studies suggest that aging may alter HDL structure and properties, resulting in reduced antioxidant capacity and cholesterol efflux, which can contribute to higher susceptibility to CVD and advance processes associated with HF mortality.
Inflammation, renal dysfunction, diabetes, obesity, atrial fibrillation, and aging can either occur antecedent to HF or comorbid with it. These HF comorbidities, and others, can have a tremendous impact on lipid metabolism and HDL biology, which in turn may impact disease progression. In the next section, we discuss how changes in HDL may alter the development of HF or HF outcomes.
Atherosclerotic CVD can lead to ischemic cardiomyopathy, which is a major clinical cause of HF (120). LDL-C is a critical, causal factor in the pathogenesis of CVD (121). In animal models, HDL has been shown to have a protective role against the development of atherosclerosis and CVD (122). HDL exerts its protective effect on vascular endothelium mainly through stimulation of eNOS increasing NO production (123, 124). HDL is critical for the reverse cholesterol transport process, which removes excess cholesterol from the atherosclerotic plaques, reducing the risk and progression of CVD (125, 126). Additionally, HDL has anti-apoptotic, anti-inflammatory, and antithrombotic protective properties on the vascular endothelium (34, 124, 127).
Both in vitro and in vivo models have repeatedly demonstrated multiple cardioprotective properties of HDL particles on many levels. HDL has shown a direct protective effect on cardiomyocytes and endothelial cells, independent of its effect on the coronary vasculature or atherosclerosis (16, 26, 128–131). It has been proposed that HDL mediates direct action on cardiomyocytes through its different array of apolipoproteins (e.g., ApoA-I and ApoM), which interact with different receptors expressed on cardiomyocytes regulating various intracellular signaling pathways. Furthermore, HDL could also indirectly protect cardiomyocytes through its systemic and local anti-inflammatory and anti-oxidative effects (132, 133).
In murine models, HDL inhibited mechanical stress-induced myocardial cell hypertrophy and autophagy through downregulation of the angiotensin II type 1 receptor (129). Angiotensin II receptors are upregulated on cardiomyocytes exposed to mechanical stress, and blockade of the renin-angiotensin pathway is a sine qua non of HF therapy. Downregulation of these receptors by HDL would be an important mechanism by which this particle may improve HF outcomes. Further, multiple in vitro studies show that HDL also protected against doxorubicin-induced cell injury on cultured cardiomyocytes (26, 134, 135), mostly through reducing doxorubicin-induced apoptosis. These studies are clinically important given that anthracyclines, such as doxorubicin, remain an important cause of cardiotoxicity and clinical HF.
HDL has also been associated with the preservation of endothelial barrier integrity. HDL increases NO production in endothelial cells, which enhances endothelial vasodilation and preserves endothelial integrity mainly through an SR-BI-dependent mechanism (136–140). It also modulates the contractile state of subjacent myocytes via paracrine mechanisms (141). Additionally, HDL contributes to endothelial repair by increasing the number and function of endothelial progenitor cells at sites of endothelial injury (128). Moreover, HDL-carried glycosphingolipids have demonstrated an anti-apoptotic capacity against stress-induced endothelial death (142). Van Linthout et al. (130) reported that HDL protects against myocardial dysfunction and hyperglycemia-induced cardiomyocyte apoptosis in diabetic murine models mainly via the phosphoinositide 3-kinase / protein kinase B (PI3K/Akt) pathway. Altogether, these studies suggest multiple mechanisms by which HDL may directly protect cardiomyocytes in the failing myocardium.
It has previously been established that systemic inflammatory mediators (e.g., C reactive protein (CRP), TNF-α) can contribute to the development of HF, and inflammation can induce cardiomyocyte apoptosis and endothelial dysfunction (143). Multiple studies have demonstrated anti-inflammatory properties of HDL. For instance, HDL inhibits endothelial activation and decreases the expression of adhesion molecules (e.g., VCAM-1 and ICAM-1), which prevents the recruitment of leukocytes in response to myocardial cell injury, and can attenuate the insult due to reduced chemokine secretion and impede further recruitment of inflammatory cells. HDL also blocks T-cell binding and activation of monocytes, which results in diminished production of pro-inflammatory cytokines (144–146).
Moreover, recent studies suggest that the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome plays a role in the development of atherosclerosis, and it has also been tied to post-ischemic remodeling, and HF (147–149). HDL can suppress the activation of the NLRP3 inflammasome, likely by simultaneous downregulation of IL-1β, and reduced activation of caspase 1 (150). Other anti-inflammatory properties of HDL are promoting the expression of anti-inflammatory cytokines, such as transforming growth factor-β2 (TGF-β2) in endothelial cells (151, 152) and neutralizing pro-inflammatory activity of both, IL-6 and CRP (153). In summary, these and other anti-inflammatory properties of HDL merit further exploration and offer a variety of targets for developing pharmacologic therapies.
One of the hallmarks of HF pathophysiology is stress-induced myocardial cell death with subsequent proliferation, fibrosis, and remodeling (154, 155). HDL has demonstrated many antioxidant properties that may combat these processes. Treating cultured cardiomyocytes with HDL protects against stress-induced cell death (156). This effect has been suggested to be mediated by the anti-oxidative enzyme PON1 (157–160). Another antioxidant enzyme present on HDL is the platelet-activating factor acetylhydrolase that induces the hydrolysis of fatty acids and phospholipids peroxides (161, 162). Furthermore, HDL blocks eNOS uncoupling in myocardial cells, reducing the formation of reactive oxygen species (163–166). In addition, HDL-associated lipoproteins (ApoA-I, ApoA-II, ApoA-IV, ApoE, etc.) neutralize the remaining phospholipid hydroperoxides transferred to HDL (167). Finally, HDL can also indirectly reduce oxidative stress secondary to its anti-inflammatory properties discussed previously (168).
HDL can protect against myocardial fibrosis through inhibition of the pro-fibrotic transforming growth factor-β1 (TGF-β1), which induces collagen production and deposition in the myocardium of murine models (169, 170). In a study on aortic endothelial cells in vitro, HDL reduced TGF-β1-induced endothelial-mesenchymal transition and attenuated fibrosis of the vascular wall in response to various insults (171). Alternatively, HDL may exhibit anti-fibrotic properties by binding and potentially sequestering S1P (through ApoM). Although this mechanism has not been directly demonstrated in the myocardium, this type of biology has been demonstrated in the retina, where ApoM can act as a negative regulator of S1P (172).
ApoA-I, the most abundant protein constituent of HDL, is involved in the systemic anti-inflammatory and anti-oxidative cardioprotective properties of HDL (173). ApoA-I is the main ligand of SR-BI and thus is very important for the cardioprotective functions of HDL previously described. Low ApoA-I levels are associated with left ventricular dysfunction and adverse outcomes in patients with non-ischemic HF (173, 174). Gombos et al. have shown that ApoA-I is inversely associated with NT-proBNP and mortality in HF (175). Similarly, Florvall et al. (176) have suggested that serum ApoA-I can predict CVD and mortality in elderly men.
ApoA-I's cardioprotective properties may be related to its anti-inflammatory and antioxidative properties. ApoA-I attenuates inflammation and is inversely correlated with CRP and fibrinogen levels (173). In addition, it blocks neutrophil activation and expression of the surface adhesion proteins that regulate leukocyte migration (177, 178). Bursill et al. (144) showed that when mice were injected with ApoA-I, the expression of chemokine receptors involved in leukocyte migration was significantly reduced. ApoA-I can also enhance the proliferation of endothelial progenitor cells and stimulate angiogenesis through the cell surface F1-ATP synthase, a high-affinity receptor of ApoA-I (179). Moreover, ApoA-I accelerates endothelial regeneration and prevents transplant vasculopathy in murine models (132, 180).
ApoA-I binds to SR-BI, which is mainly expressed in the liver. SR-BI mediates selective uptake of cholesterol, as well as HDL lipid hydroperoxides, and plays a major role in modulating HDL composition and therefore its function (Figure 2A). Muthuramu et al. described a cardioprotective role of SR-BI (181). They performed a study using SR-BI knockout mice that received either adeno-associated virus 8 (AAV8) expressing SR-BI (via a hepatocyte-specific promoter) or a control AAV8. Notably, when SR-BI knockout mice are exposed to pressure overload, they develop worse pathological ventricular hypertrophy, interstitial and perivascular fibrosis, and myocardial apoptosis than control mice. Interestingly, in mice that received AAV8-SR-BI, the plasma lipoprotein profile normalizes, attenuates cardiac dysfunction, and mortality is lower compared to mice that received Null injection. In addition, mice that underwent SR-BI gene transfer had lower oxidative stress than those that did not (181). Similarly, Durham et al. demonstrated that pretreatment with HDL protects against myocardial cell necrosis via the PI3K/Akt pathway (131). This finding was not observed in SR-BI knockout cells, suggesting that SR-BI is the upstream mediator of the PI3K/Akt signaling in cardiomyocytes and that HDL could be mediating this effect through interaction with SR-BI via ApoA-I (131). These studies suggest that ApoA-I, via SR-BI, may be an important mediator of the cardio-hepatic axis.
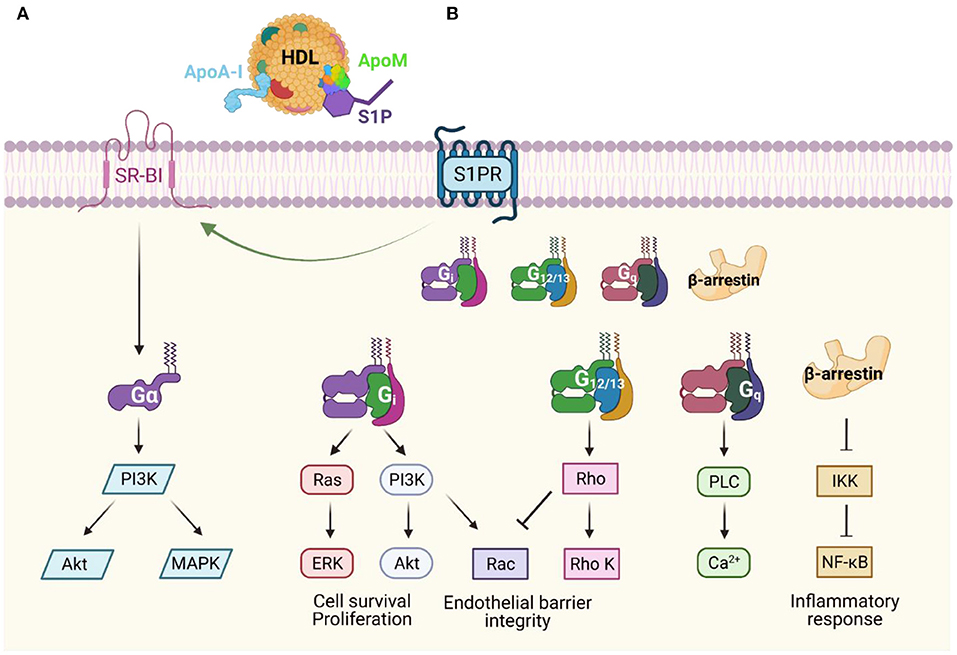
Figure 2. Apolipoprotein-dependent signal transduction pathways (A) Apolipoprotein A-I (ApoA-I) is the major component of high-density lipoproteins (HDL), and it binds scavenger receptor class B type I (SR-BI), which mediates selective uptake of cholesterol. SR-BI may also stabilize HDL particles allowing access to S1P receptors or activate other signaling cascades. (B) HDL and apolipoprotein M (ApoM)-dependent activation of S1P receptors (S1PR) leads to downstream G-protein coupled receptor signaling in both endothelial cells and cardiomyocytes. This signaling promotes diverse physiological responses including maintenance of endothelial barrier integrity, promotion of cell survival, and anti-inflammatory effects. Modified from (240–242).
ApoM is an apolipoprotein that binds S1P via its hydrophobic binding pocket, is secreted mostly by hepatocytes, and to a lesser extent by renal proximal tubular cells (182, 183). Although ApoM is only found in 5% of HDL particles, it exerts many of the beneficial effects of HDL through S1P signaling (Figure 2B). ApoM acts as a chaperone for S1P carrying about 70% of plasma S1P in the circulation, as well as increasing its efflux from erythrocytes to HDL (184). ApoM that is secreted from the proximal tubular cells in the kidney also prevents renal excretion of S1P (185).
Recent studies indicate a potential protective role of ApoM on atherosclerosis and CVD (186–188). ApoM has a protective effect on vascular endothelium as ApoM transgenic LDL receptor knock out mice developed smaller atherosclerotic lesions than control mice (189, 190). Atheroprotective functions of ApoM on vascular endothelium are likely mediated by S1P through S1P receptor 1 (S1PR1) signaling (191). ApoM also has a significant anti-inflammatory effect in vivo and in vitro mainly mediated by the S1P/S1PR axis (192).
Multiple mechanisms have been invoked for how ApoM may improve myocardial health or delay disease progression. Recent studies in murine models have demonstrated that ApoM/S1P enhances endothelial barrier function and improves cardiac outcomes through different signaling pathways. For example, in LPS-treated mice, ApoM attenuated LPS-induced organ injury as well as cardiomyocyte cell death via PI3K/Akt downstream of S1PR1/3 (193). Moreover, in vitro studies of human umbilical vein endothelial cells showed that ApoM/S1P markedly reduced pro-inflammatory cytokines, including TNF-α, inhibiting the inflammatory response, and reduced endothelial injury in a PI3K/Akt and S1PR2 dependent manner (194). Furthermore, ApoM knockout mice demonstrate impaired endothelial barrier integrity compared to wild-type mice (195). Reconstitution of plasma ApoM/S1P or treatment with an S1PR1 agonist rapidly reversed the vascular leak and restores endothelial integrity (195). S1P, acting through the receptors 1-3 (S1PR1-3), plays a crucial role in the regulation of the endothelial cell cytoskeleton, and is necessary for its proper function as well as new vessel formation. Multiple studies have demonstrated S1P to be a significant mediator of angiogenesis due to its potent chemoattractant properties for endothelial cells (196–198). S1P was found to have a higher capacity for stimulation of endothelial cell migration than known molecules such as vascular endothelial growth factor or basic fibroblast growth factor (199). Furthermore, S1P acting mainly via S1PR1 and S1PR3 have been repeatedly demonstrated to be crucial for endothelial migration (200, 201), endothelial integrity (202–204), and normal barrier function (205, 206). S1P effects on endothelial cells are mainly mediated by pathways involving Rho GTPases (207–209) as well as the PI3K/Akt pathway (210).
In addition to its protective role on the endothelium, S1P has shown multiple cardioprotective properties. Zhang et al. (211) showed that S1P signaling through S1PR1 in murine models activated the downstream PI3K/Akt pathway and attenuated myocardial cell injury in response to severe hypoxic stress. Similarly, Means et al. found that stimulation of S1PR2 and S1PR3 receptor activates PI3K/Akt pathway and protects against ischemia-reperfusion injury (212). Theilmeier et al. have also demonstrated that HDL and S1P, both acting through S1PR3, and NO-dependent mechanisms, protect against ischemia reperfusion-induced myocardial injury in ex vivo and in vivo mouse models (25). They also found that S1P reduced neutrophil recruitment to the site of injury and decreased cardiomyocyte apoptosis (25). Furthermore, S1PR2 showed some cardioprotective properties as well by activating signal transducer and activator of transcription 3 (STAT3) through ERK1/2 and Src-dependent mechanisms (26). STAT3 is important for myocardial adaptation to stress and has been shown to preserve cardiac function through its anti-apoptotic and anti-fibrotic effects (213–218). While these data support multiple mechanisms for S1P-mediated anti-apoptotic effects on cardiomyocytes through multiple S1P receptors, both in our experience and others, it is challenging to detect any significant S1PR2 mRNA expression in the myocardium in mice (219).
These studies support the concept that ApoM, via S1P, can reduce vascular leak, inflammation, and promote cell survival, all of which are likely critical targets for multiple organs in the syndrome of HF. Recently, our group measured circulating ApoM across 3 major HF cohorts comprising nearly 2,500 patients. In our study, reduced ApoM levels were significantly associated with the risk of all-cause mortality (45). These associations were independent of HDL-C and ApoA-I, natriuretic peptide levels, etiology of HF, and a commonly used HF risk score, and were observed in both HFrEF and HFpEF. Although we demonstrated a strong correlation between ApoM and S1P on HDL particles, mediation analysis suggested that ApoM could also have effects independent of S1P. Pathway analysis demonstrated that ApoM showed that the acute phase response was not only the most significant pathway associated with ApoM but also that ApoM was inversely associated with inflammation, as predicted by murine studies. In a follow-up study, we screened the plasma proteome to identify proteins that mediated the effect of diabetes on HFpEF outcomes. The only protein that fulfilled the criteria of this a priori analysis was ApoM, which was shown to mediate an astounding 70% of the effect of diabetes on HFpEF outcomes (220).
Multiple studies have shown a promising role for HDL-targeted therapies in HFrEF (221), HFpEF (221, 222), and diabetic cardiomyopathy murine models (223). In these animal models, HDL reversed pathologic features of myocardial hypertrophy, fibrosis, and stimulating reverse remodeling in pre-established HF. Multiple synthetic compounds have been designed to mimic the bioactive molecules of HDL and replicate their cardioprotective functions.
ApoA-I Milano is an ApoA-I mutant first described in Northern Italy in 1980 (224, 225). Heterozygous carriers of the mutation were thought to exhibit increased life expectancy and believed to develop atherosclerosis at lower rates compared to the normal population (226, 227). MDCO-216 is a recombinant HDL formulation of ApoA-I Milano in combination with phospholipids, which has been used to study ApoA-I Milano's potential therapeutic effects (183, 228–230).
Mishra et al. (222) reported that MDCO-216 attenuated cardiac hypertrophy, increased capillary density, and decreased interstitial fibrosis in murine models. In a subsequent study, Mishra et al. showed similar results of MDCO-216 in murine models of hypertension-associated cardiac hypertrophy (170). Aboumsallem et al. have demonstrated that MDCO-216 improves systolic and diastolic dysfunction, reduces myocardial fibrosis, and enhances myocardial vascularity in mice with HF (221). Further, Aboumsallem et al. showed that mice with diabetic cardiomyopathy that were treated with MDCO-216 presented regression of myocardial dysfunction and pathological cardiac remodeling (223). Altogether, these studies suggest MDCO-216 might be useful for HFrEF, HFpEF, or diabetic cardiomyopathy.
ApoA-I gene therapy strategies have also been employed in HF rodent models. Gordts et al. (231) evaluated if selective gene transfer may protect against the development of HF. In LDL receptor-deficient subjects to experimental MI, viral-mediated gene transfer of ApoA-I resulted in reduced infarct expansion and inhibition of left ventricular dilatation compared with controls. Similarly, Amin et al. studied the effect of selective AAV8-human ApoA-I (AAV8-ApoA-I) gene transfer on cardiac remodeling, induced by transverse aortic constriction in LDL deficient mice (232). They reported that AAV8-ApoA-I transduced mice had significantly attenuated septal wall thickness, cardiomyocyte cross-sectional area, and interstitial cardiac fibrosis compared to control mice, indicating reduced remodeling, and preserved systolic function reserve. Diastolic function was also significantly improved in mice transduced with the ApoA-I AAV8 (232).
ApoA-I mimetic peptides have also shown promise in preventing or attenuating myocardial dysfunction in murine models of MI and sepsis. Hamid et al. (233) have demonstrated that the ApoA-I mimetic peptide L-4F prevents prolonged and excessive inflammation after MI and improves post-MI LV remodeling. L-4F suppressed proliferation of myocardial pro-inflammatory monocytes and macrophages in murine models of reperfused MI (233). They suggested that L-4F could be used as a therapeutic adjunct in humans with MI to limit inflammation and alleviate the progression to HF (233). Another ApoA-I mimetic peptide D-4F has been also shown to improve vascular function, decrease myocardial inflammation, and restore angiogenic systemic sclerosis in murine models (234). Moreover, the ability of ApoA-I mimetic peptides to reduce sepsis-induced myocardial injury was studied by Moreira et al. (235). They demonstrated that the novel ApoA-I mimetic peptide D-4F reduced inflammation, attenuated vascular permeability, preserved myocardial function, and baroreceptor sensitivity in murine models of sepsis.
The AEGIS-I trial (Apo-AI Event Reducing in Ischemic Syndromes I), a multi-center, randomized, double-blind, placebo-controlled 2b trial assessed the safety of CSL112, an infusible plasma-derived ApoA-I, in patients with myocardial infarction (236). CSL112 was generally safe and well-tolerated (236). Currently, the AEGIS-II trial is underway to evaluate whether CSL112 can reduce the risk of major adverse cardiovascular events (237). To our knowledge, there are no active trials of CSL112 in human HF, although the hypothesis that CSL112 may benefit patients with acute HF should be pursued in randomized-controlled clinical trials (238).
Beyond therapeutics targeting only ApoA-I, Swendeman et al. (239) developed a recombinant ApoM fused to the constant domain of immunoglobins (ApoM-Fc) to prevent its rapid degradation. When this novel protein was tested in multiple systems, ApoM-Fc selectively activated S1PR1, leading to enhanced endothelial barrier integrity and downstream eNOS-dependent secretion of NO and subsequent vasodilation, which could be used therapeutically to control hypertension. In addition, they demonstrated improved outcomes in murine models of myocardial ischemia/reperfusion and stroke, by promoting endothelial function and reducing further tissue inflammation.
Altogether, apolipoprotein A-I and ApoM based therapeutics have demonstrated potential in preclinical models of cardiac dysfunction. Unfortunately, to our knowledge, the human translation of these therapeutics has not been tested in randomized controlled clinical trials in HF. In addition, understanding regarding the synergistic potential of these apolipoproteins (A-I and M), as well as other functions of HDL, remains poorly understood.
HDL apolipoproteins remain a promising therapeutic target in patients with HF. The advances in proteomic and lipidomic technologies have permitted the discovery of HDL components, and assessment of their impact on HF pathophysiology, predominantly in preclinical models. ApoA-I and ApoM are especially promising as they have shown multiple cardioprotective properties in murine models. Further studies are needed to elucidate the functional properties of HDL proteomic and lipidomic components and to explore possible therapeutic targets in patients with HF.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
AJ was supported by R01 HL155344 and K08HL138262 from the NHLBI and by the Diabetes Research Center at Washington University in St. Louis of the National Institutes of Health under award number P30DK020579, as well as the NIH grant P30DK056341 (Nutrition Obesity Research Center), and by the Children's Discovery Institute of Washington University (MC-FR-2020-919) and St. Louis Children's Hospital. ZG was supported by the American Heart Association Postdoctoral Fellowship (898679).
AJ has a patent for fusion protein nanodiscs and lipase inhibitors for the treatment of heart failure and has received grant support from AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Graphical figures were created with BioRender.com.
1. Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. (2017) 70:1–25. doi: 10.1016/J.JACC.2017.04.052
2. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. (2019) 139:e56–528. doi: 10.1161/CIR.0000000000000659
3. Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. (2020) 22:1342–56. doi: 10.1002/ejhf.1858
4. Hsu JJ, Ziaeian B, Fonarow GC. Heart Failure With Mid-Range (Borderline) Ejection Fraction: Clinical Implications and Future Directions. JACC: Heart Failure. (2017) 5:763–771. doi: 10.1016/j.jchf.2017.06.013
5. Borlaug BA, Redfield MM. Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation. (2011) 123:2006–14. doi: 10.1161/CIRCULATIONAHA.110.954388
6. Rauchhaus M, Clark AL, Doehner W, Davos C, Bolger A, Sharma R, et al. The relationship between cholesterol and survival in patients with chronic heart failure. J Am Coll Cardiol. (2003) 42:1933–40. doi: 10.1016/J.JACC.2003.07.016
7. Horwich TB, Hamilton MA, MacLellan WR, Fonarow GC. Low serum total cholesterol is associated with marked increase in mortality in advanced heart failure. J Cardiac Fail. (2002) 8:216–24. doi: 10.1054/jcaf.2002.0804216
8. Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. (1997) 349:1050–3. doi: 10.1016/S0140-6736(96)07015-8
9. Horwich TB, Kalantar-Zadeh K, MacLellan RW, Fonarow GC. Albumin levels predict survival in patients with systolic heart failure. Am Heart J. (2008) 155:883–9. doi: 10.1016/j.ahj.2007.11.043
10. May HT, Muhlestein JB, Carlquist JF, Horne BD, Bair TL, Campbell BA, et al. Relation of serum total cholesterol, C-reactive protein levels, and statin therapy to survival in heart failure. Am J Cardiol. (2006) 98:653–8. doi: 10.1016/j.amjcard.2006.03.046
11. Rauchhaus M, Koloczek V, Volk H-D, Kemp M, Niebauer J, et al. Inflammatory cytokines and the possible immunological role for lipoproteins in chronic heart failure. Int J Cardiol. (2000) 76:125–33. doi: 10.1016/S0167-5273(00)00224-2
12. Sato Y, Takatsu Y, Kataoka K, Yamada T, Taniguchi R, Sasayama S, et al. Serial circulating concentrations of c-reactive protein, interleukin (il)-4, and il-6 in patients with acute left heart decompensation. Clin Cardiol. (1999) 22:811–3. doi: 10.1002/clc.4960221211
13. Anand IS, Latini R, Florea VG, Kuskowski MA, Rector T, Masson S, et al. C-reactive protein in heart failure: prognostic value and the effect of valsartan. Circulation. (2005) 112:1428–34. doi: 10.1161/CIRCULATIONAHA.104.508465
14. Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. (2001) 103:2055–9. doi: 10.1161/01.CIR.103.16.2055
15. Aung N, Sanghvi MM, Piechnik SK, Neubauer S, Munroe PB, Petersen SE. The effect of blood lipids on the left ventricle: a mendelian randomization study. J Am Coll Cardiol. (2020) 76:2477–88. doi: 10.1016/J.JACC.2020.09.583
16. den Ruijter HM, Franssen R, Verkerk AO, van Wijk DF, Vaessen SF, Holleboom AG, et al. Reconstituted high-density lipoprotein shortens cardiac repolarization. J Am Coll Cardiol. (2011) 58:40–4. doi: 10.1016/J.JACC.2010.11.072
17. Bedi KC, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
18. GISSI-HF Investigators. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. (2008) 372:1231–9. doi: 10.1016/S0140-6736(08)61240-4
19. Kjekshus J, Apetrei E, Barrios V, Böhm M, Cleland JGF, Cornel JH, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. (2007) 357:2248–61. doi: 10.1056/NEJMoa0706201
20. Kannel WB, Dawber TR, Friedman GD, Glennon WE, McNamara PM. Risk factors in coronary heart disease: the framingham study. Ann Intern Med. (1964) 61:888–99. doi: 10.7326/0003-4819-61-5-888
21. AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. (2011) 365:2255–67. doi: 10.1056/NEJMoa1107579
22. Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. (2012) 367:2089–99. doi: 10.1056/NEJMoa1206797
23. Javaheri A, Molina M, Zamani P, Rodrigues A, Novak E, Chambers S, et al. Cholesterol efflux capacity of high-density lipoprotein correlates with survival and allograft vasculopathy in cardiac transplant recipients. J Heart Lung Transplant. (2016) 35:1295–302. doi: 10.1016/J.HEALUN.2016.06.022
24. Assmann G, Gotto AM. HDL cholesterol and protective factors in atherosclerosis. Circulation. (2004) 109:III8–14. doi: 10.1161/01.CIR.0000131512.50667.46
25. Theilmeier G, Schmidt C, Herrmann J, Keul P, Schäfers M, Herrgott I, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. (2006) 114:1403–9. doi: 10.1161/CIRCULATIONAHA.105.607135
26. Frias MA, James RW, Gerber-Wicht C, Lang U. Native and reconstituted HDL activate Stat3 in ventricular cardiomyocytes via ERK1/2: Role of sphingosine-1-phosphate. Cardiovasc Res. (2009) 82:313–23. doi: 10.1093/cvr/cvp024
27. Keul P, Sattler K, Levkau B. HDL and its sphingosine-1-phosphate content in cardioprotection. Heart Fail Rev. (2007) 12:301–6. doi: 10.1007/s10741-007-9038-x
28. Kennedy S, Kane KA, Pyne NJ, Pyne S. Targeting sphingosine-1-phosphate signalling for cardioprotection. Curr Opin Pharmacol. (2009) 9:194–201. doi: 10.1016/j.coph.2008.11.002
29. Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, et al. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. (2003) 23:1283–8. doi: 10.1161/01.ATV.0000079011.67194.5A
30. Brulhart-Meynet M-C, Braunersreuther V, Brinck J, Montecucco F, Prost J-C, Thomas A, et al. Improving reconstituted HDL composition for efficient post-ischemic reduction of ischemia reperfusion injury. PLoS ONE. (2015) 10:e0119664. doi: 10.1371/journal.pone.0119664
31. Calabresi L, Rossoni G, Gomaraschi M, Sisto F, Berti F, Franceschini G. High-density lipoproteins protect isolated rat hearts from ischemia-reperfusion injury by reducing cardiac tumor necrosis factor-α content and enhancing prostaglandin release. Circ Res. (2003) 92:330–7. doi: 10.1161/01.RES.0000054201.60308.1A
32. Brites F, Martin M, Guillas I, Kontush A. Antioxidative activity of high-density lipoprotein (HDL): mechanistic insights into potential clinical benefit. BBA Clin. (2017) 8:66–77. doi: 10.1016/j.bbacli.2017.07.002
33. Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. (2001) 7:853–7. doi: 10.1038/89986
34. Mineo C, Deguchi H, Griffin JH, Shaul PW. Endothelial and antithrombotic actions of HDL. Circ Res. (2006) 98:1352–64. doi: 10.1161/01.RES.0000225982.01988.93
35. Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. (2004) 95:764–72. doi: 10.1161/01.RES.0000146094.59640.13
36. Chiesa ST, Charakida M. High-density lipoprotein function and dysfunction in health and disease. Cardiovasc Drugs Ther. (2019) 33:207–19. doi: 10.1007/s10557-018-06846-w
37. Soran H, Schofield JD, Durrington PN. Antioxidant properties of HDL. Front Pharmacol. (2015) 6:222. doi: 10.3389/FPHAR.2015.00222
38. Furtado JD, Yamamoto R, Melchior JT, Andraski AB, Gamez-Guerrero M, Mulcahy P, et al. Distinct proteomic signatures in 16 HDL (high-density lipoprotein) subspecies. Arterioscler Thromb Vasc Biol. (2018) 38:2827–42. doi: 10.1161/ATVBAHA.118.311607
39. Sacks FM, Liang L, Furtado JD, Cai T, Sean Davidson W, He Z, et al. Protein-defined subspecies of HDLs (high-density lipoproteins) and differential risk of coronary heart disease in 4 prospective studies. Arterioscler Thromb Vasc Biol. (2020) 40:2714–27. doi: 10.1161/ATVBAHA.120.314609
40. Rosenson RS, Brewer HB, Chapman MJ, Fazio S, Hussain MM, Kontush A, et al. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin Chem. (2011) 57:392–410. doi: 10.1373/clinchem.2010.155333
41. Duprez DA, Otvos J, Tracy RP, Feingold KR, Greenland P, Gross MD, et al. High-density lipoprotein subclasses and noncardiovascular, noncancer chronic inflammatory-related events versus cardiovascular events: the multi-ethnic study of atherosclerosis. J Am Heart Assoc. (2015) 4:e002295. doi: 10.1161/JAHA.115.002295
42. Potočnjak I, Degoricija V, Trbušić M, Pregartner G, Berghold A, Marsche G, et al. Serum concentration of HDL particles predicts mortality in acute heart failure patients. Sci Rep. (2017) 7:46642. doi: 10.1038/srep46642
43. Hunter WG, McGarrah RW, Kelly JP, Khouri MG, Craig DM, Haynes C, et al. High-density lipoprotein particle subfractions in heart failure with preserved or reduced ejection fraction. J Am Coll Cardiol. (2019) 73:177–86. doi: 10.1016/J.JACC.2018.10.059
44. Frias MA, Lecour S, James RW, Pedretti S. High density lipoprotein/sphingosine-1-phosphate-induced cardioprotection. JAK STAT. (2012) 1:92–100. doi: 10.4161/jkst.19754
45. Chirinos JA, Zhao L, Jia Y, Frej C, Adamo L, Mann D, et al. Reduced apolipoprotein M and adverse outcomes across the spectrum of human heart failure. Circulation. (2020) 141:1463–76. doi: 10.1161/CIRCULATIONAHA.119.045323
46. Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, et al. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framingham Heart Study. Circulation. (2003) 107:1486–91. doi: 10.1161/01.CIR.0000057810.48709.F6
47. Mann DL. Innate immunity and the failing heart. Circ Res. (2015) 116:1254–68. doi: 10.1161/CIRCRESAHA.116.302317
48. Edelmann F, Holzendorf V, Wachter R, Nolte K, Schmidt AG, Kraigher-Krainer E, et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail. (2015) 17:214–23. doi: 10.1002/ejhf.203
49. Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. (1996) 93:704–11. doi: 10.1161/01.CIR.93.4.704
50. Khovidhunkit W, Kim M-S, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al. Thematic review series: the Pathogenesis of Atherosclerosis. Effects of infection and inflammation on lipid and lipoprotein metabolism mechanisms and consequences to the host. J Lipid Res. (2004) 45:1169–96. doi: 10.1194/jlr.R300019-JLR200
51. Beers A, Haas MJ, Wong NCW, Mooradian AD. Inhibition of apolipoprotein AI gene expression by tumor necrosis factor alpha: roles for MEK/ERK and JNK signaling. Biochemistry. (2006) 45:2408–13. doi: 10.1021/BI0518040
52. Feingold KR, Shigenaga JK, Chui LG, Moser A, Khovidhunkit W, Grunfeld C. Infection and inflammation decrease apolipoprotein M expression. Atherosclerosis. (2008) 199:19–26. doi: 10.1016/J.ATHEROSCLEROSIS.2007.10.007
53. Masucci-Magoulas L, Moulin P, Jiang XC, Richardson H, Walsh A, Breslow JL, et al. Decreased cholesteryl ester transfer protein (CETP) mRNA and protein and increased high density lipoprotein following lipopolysaccharide administration in human CETP transgenic mice. J Clin Invest. (1995) 95:1587–94. doi: 10.1172/JCI117832
54. Hardardóttir I, Moser AH, Fuller J, Fielding C, Feingold K, Grünfeld C. Endotoxin and cytokines decrease serum levels and extra hepatic protein and mRNA levels of cholesteryl ester transfer protein in Syrian hamsters. J Clin Invest. (1996) 97:2585–92. doi: 10.1172/JCI118707
55. Levels JHM, Pajkrt D, Schultz M, Hoek FJ, van Tol A, Meijers JCM, et al. Alterations in lipoprotein homeostasis during human experimental endotoxemia and clinical sepsis. Biochim Biophys Acta. (2007) 1771:1429–38. doi: 10.1016/J.BBALIP.2007.10.001
56. Sammalkorpi K, Valtonen V, Kerttula Y, Nikkilä E, Taskinen M-R. Changes in serum lipoprotein pattern induced by acute infections. Metabolism. (1988) 37:859–65. doi: 10.1016/0026-0495(88)90120-5
57. Grunfeld C, Pang M, Doerrler W, Shigenaga JK, Jensen P, Feingold KR. Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. (1992) 74:1045–52. doi: 10.1210/jcem.74.5.1373735
58. Feingold KR, Hardardottir I, Memon R, Krul EJT, Moser AH, Taylor JM, et al. Effect of endotoxin on cholesterol biosynthesis and distribution in serum lipoproteins in Syrian hamsters. J Lipid Res. (1993) 34:2147–58. doi: 10.1016/S0022-2275(20)35355-4
59. Cabana VG, Siegel JN, Sabesin SM. Effects of the acute phase response on the concentration and density distribution of plasma lipids and apolipoproteins. J Lipid Res. (1989) 30:39–49. doi: 10.1016/S0022-2275(20)38390-5
60. Kumaraswamy SB, Linder A, Akesson P, Dahlback B. Decreased plasma concentrations of apolipoprotein M in sepsis and systemic inflammatory response syndromes. Crit Care. (2012) 16:R60. doi: 10.1186/cc11305
61. Feingold KR, Grunfeld C. Effect of inflammation on HDL structure and function. Curr Opin Lipidol. (2016) 27:521–30. doi: 10.1097/MOL.0000000000000333
62. Malik P, Berisha SZ, Santore J, Agatisa-Boyle C, Brubaker G, Smith JD. Zymosan-mediated inflammation impairs in vivo reverse cholesterol transport. J Lipid Res. (2011) 52:951–7. doi: 10.1194/jlr.M011122
63. Reilly MP, McGillicuddy FC, de La Moya ML, Hinkle CC, Joshi MR, Chiquoine EH, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation. (2009) 119:1135–45. doi: 10.1161/CIRCULATIONAHA.108.810721
64. Baranova I, Vishnyakova T, Bocharov A, Chen Z, Remaley AT, Stonik J, et al. Lipopolysaccharide down regulates both scavenger receptor B1 and ATP binding cassette transporter A1 in RAW cells. Infect Immunity. (2002) 70:2995–3003. doi: 10.1128/IAI.70.6.2995-3003.2002
65. Jin W, Sun G-S, Marchadier D, Octtaviani E, Glick JM, Rader DJ. Endothelial cells secrete triglyceride lipase and phospholipase activities in response to cytokines as a result of endothelial lipase. Circ Res. (2003) 92:644–50. doi: 10.1161/01.RES.0000064502.47539.6D
66. Hirata K, Ishida T, Matsushita H, Tsao PS, Quertermous T. Regulated expression of endothelial cell-derived lipase. Biochem Biophys Res Commun. (2000) 272:90–3. doi: 10.1006/bbrc.2000.2747
67. Badellino KO, Wolfe ML, Reilly MP, Rader DJ. Endothelial lipase is increased in vivo by inflammation in humans. Circulation. (2008) 117:678–85. doi: 10.1161/CIRCULATIONAHA.107.707349
68. Tietge UJF, Maugeais C, Lund-Katz S, Grass D, deBeer FC, Rader DJ. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA-I in response to inflammation in human apoA-I transgenic mice. Arterioscler Thromb Vasc Biol. (2002) 22:1213–8. doi: 10.1161/01.atv.0000023228.90866.29
69. Nakajima H, Ishida T, Satomi-Kobayashi S, Mori K, Hara T, Sasaki N, et al. Endothelial lipase modulates pressure overload–induced heart failure through alternative pathway for fatty acid uptake. Hypertension. (2013) 61:1002–7. doi: 10.1161/HYPERTENSIONAHA.111.201608
70. Zhong J, Yang H, Kon V. Kidney as modulator and target of “good/bad” HDL. Pediatr Nephrol. (2019) 34:1683–95. doi: 10.1007/s00467-018-4104-2
71. Holzer M, Birner-Gruenberger R, Stojakovic T, El-Gamal D, Binder V, Wadsack C, et al. Uremia alters HDL composition and function. J Am Soc Nephrol. (2011) 22:1631–41. doi: 10.1681/ASN.2010111144
72. Anavekar NS, McMurray JJV, Velazquez EJ, Solomon SD, Kober L, Rouleau J-L, et al. Relation between renal dysfunction and cardiovascular outcomes after myocardial infarction. N Engl J Med. (2004) 351:1285–95. doi: 10.1056/NEJMoa041365
73. McAlister FA, Ezekowitz J, Tonelli M, Armstrong PW. Renal insufficiency and heart failure: prognostic and therapeutic implications from a prospective cohort study. Circulation. (2004) 109:1004–9. doi: 10.1161/01.CIR.0000116764.53225.A9
74. Chan EJ, Dellsperger KC. Cardiorenal syndrome: the clinical cardiologists' perspective. Cardiorenal Med. (2011) 1:13–22. doi: 10.1159/000322820
75. Kao MPC, Ang DSC, Pall A, Struthers AD. Oxidative stress in renal dysfunction: mechanisms, clinical sequelae and therapeutic options. J Hum Hypertens. (2010) 24:1–8. doi: 10.1038/jhh.2009.70
76. Nicholls SJ, Zheng L, Hazen SL. Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc Med. (2005) 15:212–9. doi: 10.1016/J.TCM.2005.06.004
77. Pan B, Yu B, Ren H, Willard B, Pan L, Zu L, et al. High-density lipoprotein nitration and chlorination catalyzed by myeloperoxidase impair its effect of promoting endothelial repair. Free Rad Biol Med. (2013) 60:272–81. doi: 10.1016/j.freeradbiomed.2013.02.004
78. Bergt C, Fu X, Huq NP, Kao J, Heinecke JW. Lysine residues direct the chlorination of tyrosines in YXXK motifs of apolipoprotein A-I when hypochlorous acid oxidizes high density lipoprotein. J Biol Chem. (2004) 279:7856–66. doi: 10.1074/JBC.M309046200
79. Javaheri A, Rader DJ. Apolipoprotein A-I and cholesterol efflux: the good, the bad, and the modified. Circ Res. (2014) 114:1681–3. doi: 10.1161/CIRCRESAHA.114.303974
80. Vazzana N, Ganci A, Cefalù AB, Lattanzio S, Noto D, Santoro N, et al. Enhanced lipid peroxidation and platelet activation as potential contributors to increased cardiovascular risk in the low-HDL phenotype. J Am Heart Assoc. (2013) 2:e000063. doi: 10.1161/JAHA.113.000063
81. Undurti A, Huang Y, Lupica JA, Smith JD, DiDonato JA, Hazen SL. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem. (2009) 284:30825–35. doi: 10.1074/jbc.M109.047605
82. Anderson JLC, Gautier T, Nijstad N, Tölle M, Schuchardt M, van der Giet M, et al. High density lipoprotein (HDL) particles from end-stage renal disease patients are defective in promoting reverse cholesterol transport. Sci Rep. (2017) 7: 41481. doi: 10.1038/srep41481
83. Marsche G, Hammer A, Oskolkova O, Kozarsky KF, Sattler W, Malle E. Hypochlorite-modified high density lipoprotein, a high affinity ligand to scavenger receptor class B, type I, impairs high density lipoprotein-dependent selective lipid uptake and reverse cholesterol transport. J Biol Chem. (2002) 277:32172–9. doi: 10.1074/JBC.M200503200
84. Peterson SJ, Choudhary A, Kalsi AK, Zhao S, Alex R, Abraham NG. OX-HDL: a starring role in cardiorenal syndrome and the effects of heme oxygenase-1 intervention. Diagnostics. (2020) 10:976. doi: 10.3390/diagnostics10110976
85. Speer T, Rohrer L, Blyszczuk P, Shroff R, Kuschnerus K, Kränkel N, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity. (2013) 38:754–68. doi: 10.1016/J.IMMUNI.2013.02.009
86. Shroff R, Speer T, Colin S, Charakida M, Zewinger S, Staels B, et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J Am Soc Nephrol. (2014) 25:2658–68. doi: 10.1681/ASN.2013111212
87. Weichhart T, Kopecky C, Kubicek M, Haidinger M, Döller D, Katholnig K, et al. Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol. (2012) 23:934–47. doi: 10.1681/ASN.2011070668
88. Raupachova J, Kopecky C, Cohen G. High-density lipoprotein from chronic kidney disease patients modulates polymorphonuclear leukocytes. Toxins. (2019) 11:73. doi: 10.3390/TOXINS11020073
89. Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. (2007) 115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597
90. Athithan L, Gulsin GS, McCann GP, Levelt E. Diabetic cardiomyopathy: pathophysiology, theories and evidence to date. World J Diabetes. (2019) 10:490–510. doi: 10.4239/WJD.V10.I10.490
91. Srivastava RAK. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol Cell Biochem. (2018) 440:167–87. doi: 10.1007/S11010-017-3165-Z
92. Duell PB, Oram JF, Bierman EL. Nonenzymatic glycosylation of HDL and impaired HDL-receptor-mediated cholesterol efflux. Diabetes. (1991) 40:377–384. doi: 10.2337/DIAB.40.3.377
93. Vaisar T, Couzens E, Hwang A, Russell M, Barlow CE, DeFina LF, et al. Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS ONE. (2018) 13:e0192616. doi: 10.1371/JOURNAL.PONE.0192616
94. Kobayashi T, Kurano M, Nanya M, Shimizu T, Ohkawa R, Tozuka M, et al. Glycation of HDL polymerizes apolipoprotein m and attenuates its capacity to bind to sphingosine 1-phosphate. J Atheroscler Thromb. (2021) 28:730–41. doi: 10.5551/JAT.55699
95. Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and cardiovascular disease: a scientific statement from the american heart association. Circulation. (2021) 143:E984–1010. doi: 10.1161/CIR.0000000000000973
96. Glueck CJ, Taylor HL, Jacobs D, Morrison JA, Beaglehole R, Williams OD. Plasma high-density lipoprotein cholesterol: association with measurements of body mass. The Lipid Research Clinics Program Prevalence Study. Circulation. (1980) 62::IV-62–9.
97. Bertière MC, Fumeron F, Rigaud D, Malon D, Apfelbaum M, Girard-Globa A. Low high density lipoprotein-2 concentrations in obese male subjects. Atherosclerosis. (1988) 73:57–61. doi: 10.1016/0021-9150(88)90163-3
98. Zipid J. Altered properties of high density lipoprotein subfractions in obese subjects. J Lipid Res. (1997) 38:600–11. doi: 10.1016/S0022-2275(20)37268-0
99. Lamon-Fava S, Wilson PWF, Schaefer EJ. Impact of body mass index on coronary heart disease risk factors in men and women. The Framingham Offspring Study. Arterioscler Thromb Vasc Biol. (1996) 16:1509–15. doi: 10.1161/01.ATV.16.12.1509
100. Sramkova V, Berend S, Siklova M, Caspar-Bauguil S, Carayol J, Bonnel S, et al. Apolipoprotein M: a novel adipokine decreasing with obesity and upregulated by calorie restriction. Am J Clin Nutr. (2019) 109:1499–510. doi: 10.1093/AJCN/NQY331
101. Niu L, Geyer PE, Wewer Albrechtsen NJ, Gluud LL, Santos A, Doll S, et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol Syst Biol. (2019) 15:e8793. doi: 10.15252/MSB.20188793
102. Miller A, McNamara J, Hummel SL, Konerman MC, Tincopa MA. Prevalence and staging of non-alcoholic fatty liver disease among patients with heart failure with preserved ejection fraction. Sci Rep. (2020) 10:12440. doi: 10.1038/s41598-020-69013-y
103. Rao PK, Merath K, Drigalenko E, Jadhav AYL, Komorowski RA, Goldblatt MI, et al. Proteomic characterization of high-density lipoprotein particles in patients with non-alcoholic fatty liver disease. Clin Proteomics. (2018) 15:10. doi: 10.1186/S12014-018-9186-0
104. Averill M, Rubinow KB, Cain K, Wimberger J, Babenko I, Becker JO, et al. Postprandial remodeling of high-density lipoprotein following high saturated fat and high carbohydrate meals. J Clin Lipidol. (2020) 14:66–76.e11. doi: 10.1016/J.JACL.2019.11.002
105. Alonso A, Yin X, Roetker NS, Magnani JW, Kronmal RA, Ellinor PT, et al. Blood lipids and the incidence of atrial fibrillation: the Multi-Ethnic Study of Atherosclerosis and the Framingham Heart Study. J Am Heart Assoc. (2014) 3:e001211. doi: 10.1161/JAHA.114.001211
106. Barkas F, Elisaf M, Korantzopoulos P, Tsiara S, Liberopoulos E. The CHADS 2 and CHA 2 DS 2-VASc scores predict atrial fibrillation in dyslipidemic individuals: role of incorporating low high-density lipoprotein cholesterol levels. Int J Cardiol. (2017) 241:194–9. doi: 10.1016/J.IJCARD.2017.04.062
107. Haywood LJ, Ford CE, Crow RS, Davis BR, Massie BM, Einhorn PT, et al. Atrial fibrillation at baseline and during follow-up in ALLHAT (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial). J Am Coll Cardiol. (2009) 54:2023–31. doi: 10.1016/J.JACC.2009.08.020
108. Annoura M, Ogawa M, Kumagai K, Zhang B, Saku K, Arakawa K. Cholesterol paradox in patients with paroxysmal atrial fibrillation. Cardiology. (1999) 92:21–7. doi: 10.1159/000006942
109. Trieb M, Kornej J, Knuplez E, Hindricks G, Thiele H, Sommer P, et al. Atrial fibrillation is associated with alterations in HDL function, metabolism, and particle number. Basic Res Cardiol. (2019) 114:27. doi: 10.1007/S00395-019-0735-0
110. Mora S, Akinkuolie AO, Sandhu RK, Conen D, Albert CM. Paradoxical association of lipoprotein measures with incident atrial fibrillation. Circ Arrhyth Electrophysiol. (2014) 7:612–9. doi: 10.1161/CIRCEP.113.001378
111. Khalil A, Jay-Gerin JP, Fülöp T. Age-related increased susceptibility of high-density lipoproteins (HDL) to in vitro oxidation induced by gamma-radiolysis of water. FEBS Lett. (1998) 435:153–8. doi: 10.1016/S0014-5793(98)01058-8
112. Holzer M, Trieb M, Konya V, Wadsack C, Heinemann A, Marsche G. Aging affects high-density lipoprotein composition and function. Biochim Biophys Acta. (2013) 1831:1442–8. doi: 10.1016/J.BBALIP.2013.06.004
113. Berrougui H, Isabelle M, Cloutier M, Grenier G, Khalil A. Age-related impairment of HDL-mediated cholesterol efflux. J Lipid Res. (2007) 48:328–36. doi: 10.1194/JLR.M600167-JLR200
114. Ding B, Yang D, Swendeman SL, Christoffersen C, Nielsen LB, Friedman SL, et al. Aging suppresses sphingosine-1-phosphate chaperone ApoM in circulation resulting in maladaptive organ repair. Dev Cell. (2020) 53:677–90.e4. doi: 10.1016/J.DEVCEL.2020.05.024
115. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. (2018) 13:757–72. doi: 10.2147/CIA.S158513
116. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:2181–90. doi: 10.1152/AJPHEART.00554.2011
117. Seres I, Paragh G, Deschene E, Fulop T, Khalil A. Study of factors influencing the decreased HDL associated PON1 activity with aging. Exp Gerontol. (2004) 39:59–66. doi: 10.1016/J.EXGER.2003.08.001
118. Jaouad L, de Guise C, Berrougui H, Cloutier M, Isabelle M, Fulop T, et al. Age-related decrease in high-density lipoproteins antioxidant activity is due to an alteration in the PON1's free sulfhydryl groups. Atherosclerosis. (2006) 185:191–200. doi: 10.1016/J.ATHEROSCLEROSIS.2005.06.012
119. Cherki M, Berrougui H, Isabelle M, Cloutier M, Koumbadinga GA, Khalil A. Effect of PON1 polymorphism on HDL antioxidant potential is blunted with aging. Exp Gerontol. (2007) 42:815–24. doi: 10.1016/J.EXGER.2007.04.006
121. Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2020) 41:2313–30. doi: 10.1093/eurheartj/ehz962
122. Vergeer M, Holleboom AG, Kastelein JJP, Kuivenhoven JA. The HDL hypothesis: does high-density lipoprotein protect from atherosclerosis? J Lipid Res. (2010) 51:2058–73. doi: 10.1194/JLR.R001610
123. Besler C, Lüscher TF, Landmesser U. Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med. (2012) 4:251–68. doi: 10.1002/emmm.201200224
124. Nofer J-R, Kehrel B, Fobker M, Levkau B, Assmann G, von Eckardstein A. HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis. (2002) 161:1–16. doi: 10.1016/S0021-9150(01)00651-7
125. Verdier C, Martinez LO, Ferrières J, Elbaz M, Genoux A, Perret B. Targeting high-density lipoproteins: update on a promising therapy. Arch Cardiovasc Dis. (2013) 106:601–11. doi: 10.1016/j.acvd.2013.06.052
126. Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. (2011) 364:127–35. doi: 10.1056/NEJMOA1001689
127. Rye K-A, Barter PJ. Antiinflammatory actions of HDL. Arterioscler Thromb Vasc Biol. (2008) 28:1890–1. doi: 10.1161/ATVBAHA.108.173575
128. Tso C, Martinic G, Fan W-H, Rogers C, Rye K-A, Barter PJ. High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler Thromb Vasc Biol. (2006) 26:1144–9. doi: 10.1161/01.ATV.0000216600.37436.cf
129. Lin L, Liu X, Xu J, Weng L, Ren J, Ge J, et al. High-density lipoprotein inhibits mechanical stress-induced cardiomyocyte autophagy and cardiac hypertrophy through angiotensin II type 1 receptor-mediated PI3K/Akt pathway. J Cell Mol Med. (2015) 19:1929–38. doi: 10.1111/JCMM.12567
130. Van Linthout S, Spillmann F, Riad A, Trimpert C, Lievens J, Meloni M, et al. Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation. (2008) 117:1563–73. doi: 10.1161/CIRCULATIONAHA.107.710830
131. Durham KK, Chathely KM, Trigatti BL. High-density lipoprotein protects cardiomyocytes against necrosis induced by oxygen and glucose deprivation through SR-B1, PI3K, and AKT1 and 2. Biochem J. (2018) 475:1253–65. doi: 10.1042/BCJ20170703
132. Feng Y, van Eck M, van Craeyveld E, Jacobs F, Carlier V, van Linthout S, et al. Critical role of scavenger receptor-BI-expressing bone marrow-derived endothelial progenitor cells in the attenuation of allograft vasculopathy after human apo A-I transfer. Blood. (2009) 113:755–64. doi: 10.1182/BLOOD-2008-06-161794
133. Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. (2006) 98:63–72. doi: 10.1161/01.RES.0000199272.59432.5B
134. Durham KK, Kluck G, Mak KC, Deng YD, Trigatti BL. Treatment with apolipoprotein A1 protects mice against doxorubicin-induced cardiotoxicity in a scavenger receptor class B, type I-dependent manner. Am J Physiol Heart Circ Physiol. (2019) 316:H1447–57. doi: 10.1152/AJPHEART.00432.2018
135. Taniyama Y, Walsh K. Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol. (2002) 34:1241–7. doi: 10.1006/JMCC.2002.2068
136. Massion PB, Feron O, Dessy C, Balligand J-L. Nitric oxide and cardiac function. Circ Res. (2003) 93:388–98. doi: 10.1161/01.RES.0000088351.58510.21
137. Tran-Dinh A, Diallo D, Delbosc S, Varela-Perez LM, Dang Q, Lapergue B, et al. HDL and endothelial protection. Br J Pharmacol. (2013) 169:493–511. doi: 10.1111/bph.12174
138. Saddar S, Carriere V, Lee W-R, Tanigaki K, Yuhanna IS, Parathath S, et al. Scavenger receptor class B type i is a plasma membrane cholesterol sensor. Circ Res. (2013) 112:140–51. doi: 10.1161/CIRCRESAHA.112.280081
139. Rämet ME, Rämet M, Lu Q, Nickerson M, Savolainen MJ, Malzone A, et al. High-density lipoprotein increases the abundance of eNOS protein in human vascular endothelial cells by increasing its half-life. J Am Coll Cardiol. (2003) 41:2288–97. doi: 10.1016/S0735-1097(03)00481-9
140. Zhu W, Saddar S, Seetharam D, Chambliss KL, Longoria C, Silver DL, et al. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res. (2008) 102:480–7. doi: 10.1161/CIRCRESAHA.107.159079
141. Brutsaert DL, Fransen P, Andries LJ, de Keulenaer GW, Sys SU. Cardiac endothelium and myocardial function. Cardiovasc Res. (1998) 38:281–90. doi: 10.1016/s0008-6363(98)00044-3
142. Nofer JR, Levkau B, Wolinska I, Junker R, Fobker M, von Eckardstein A, et al. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem. (2001) 276:34480–5. doi: 10.1074/JBC.M103782200
143. Kim JB, Hama S, Hough G, Navab M, Fogelman AM, Maclellan WR, et al. Heart failure is associated with impaired anti-inflammatory and antioxidant properties of high-density lipoproteins. Am J Cardiol. (2013) 112:1770–7. doi: 10.1016/J.AMJCARD.2013.07.045
144. Bursill CA, Castro ML, Beattie DT, Nakhla S, van der Vorst E, Heather AK, et al. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2010) 30:1773–8. doi: 10.1161/ATVBAHA.110.211342
145. Nicholls SJ, Cutri B, Worthley SG, Kee P, Rye K-A, Bao S, et al. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol. (2005) 25:2416–21. doi: 10.1161/01.ATV.0000184760.95957.d6
146. Carpintero R, Gruaz L, Brandt KJ, Scanu A, Faille D, Combes V, et al. HDL interfere with the binding of T cell microparticles to human monocytes to inhibit pro-inflammatory cytokine production. PLoS ONE. (2010) 5:e11869. doi: 10.1371/JOURNAL.PONE.0011869
147. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. (2018) 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
148. Javaheri A, Bajpai G, Picataggi A, Mani S, Foroughi L, Evie H, et al. TFEB activation in macrophages attenuates postmyocardial infarction ventricular dysfunction independently of ATG5-mediated autophagy. JCI Insight. (2019) 4:e127312. doi: 10.1172/JCI.INSIGHT.127312
149. Abbate A, Toldo S, Marchetti C, Kron J, van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. (2020) 126:1260–80. doi: 10.1161/CIRCRESAHA.120.315937
150. Thacker SG, Zarzour A, Chen Y, Alcicek MS, Freeman LA, Sviridov DO, et al. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology. (2016) 149:306–19. doi: 10.1111/IMM.12638
151. Norata GD, Callegari E, Marchesi M, Chiesa G, Eriksson P, Catapano AL. High-density lipoproteins induce transforming growth factor-beta2 expression in endothelial cells. Circulation. (2005) 111:2805–11. doi: 10.1161/CIRCULATIONAHA.104.472886
152. Kimura T, Tomura H, Mogi C, Kuwabara A, Damirin A, Ishizuka T, et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J Biol Chem. (2006) 281:37457–67. doi: 10.1074/JBC.M605823200
153. Gomaraschi M, Basilico N, Sisto F, Taramelli D, Eligini S, Colli S, et al. High-density lipoproteins attenuate interleukin-6 production in endothelial cells exposed to pro-inflammatory stimuli. Biochim Biophys Acta. (2005) 1736:136–43. doi: 10.1016/J.BBALIP.2005.08.003
154. Moris D, Spartalis M, Spartalis E, Karachaliou G-S, Karaolanis GI, Tsourouflis G, et al. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann Transl Med. (2017) 5:326–6. doi: 10.21037/atm.2017.06.27
155. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
156. Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol. (2003) 23:1881–8. doi: 10.1161/01.ATV.0000091338.93223.E8
157. Mackness MI, Durrington PN, Mackness B. The role of paraoxonase 1 activity in cardiovascular disease: potential for therapeutic intervention. Am J Cardiovasc Drugs. (2004) 4:211–7. doi: 10.2165/00129784-200404040-00002
158. Mackness MI, Arrol S, Abbott C, Durrington PN. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis. (1993) 104:129–35. doi: 10.1016/0021-9150(93)90183-U
159. Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, la Du BN. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions: a possible peroxidative role for paraoxonase. J Clin Invest. (1998) 101:1581–90. doi: 10.1172/JCI1649
160. Shih DM, Gu L, Xia YR, Navab M, Li WF, Hama S, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature. (1998) 394:284–7. doi: 10.1038/28406
161. Marathe GK, Zimmerman GA, McIntyre TM. Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J Biol Chem. (2003) 278:3937–47. doi: 10.1074/jbc.M211126200
162. Connelly PW, Draganov D, Maguire GF. Paraoxonase-1 does not reduce or modify oxidation of phospholipids by peroxynitrite. Free Rad Biol Med. (2005) 38:164–74. doi: 10.1016/j.freeradbiomed.2004.10.010
163. van Linthout S, Frias M, Singh N, de Geest B. Therapeutic potential of HDL in cardioprotection and tissue repair. Handb Exp Pharmacol. (2015) 224:527–65. doi: 10.1007/978-3-319-09665-0_17
164. Bowry VW, Stanley KK, Stocker R. High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors. Proc Natl Acad Sci USA. (1992) 89:10316–20. doi: 10.1073/pnas.89.21.10316
165. Proudfoot JM, Barden AE, Loke WM, Croft KD, Puddey IB, Mori TA. HDL is the major lipoprotein carrier of plasma F2-isoprostanes. J Lipid Res. (2009) 50:716–22. doi: 10.1194/jlr.M800607-JLR200
166. Hayek T, Oiknine J, Dankner G, Brook JG, Aviram M. HDL apolipoprotein A-I attenuates oxidative modification of low density lipoprotein: studies in transgenic mice. Clin Chem Lab Med. (1995) 33:721–5. doi: 10.1515/cclm.1995.33.10.721
167. Zerrad-Saadi A, Therond P, Chantepie S, Couturier M, Rye K-A, Chapman MJ, et al. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity. Arterioscler Thromb Vasc Biol. (2009) 29:2169–75. doi: 10.1161/ATVBAHA.109.194555
168. Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high-density lipoprotein. Curr Opin Lipidol. (2007) 18:427–34. doi: 10.1097/MOL.0b013e3282364a17
169. Spillmann F, de Geest B, Muthuramu I, Amin R, Miteva K, Pieske B, et al. Apolipoprotein A-I gene transfer exerts immunomodulatory effects and reduces vascular inflammation and fibrosis in ob/ob mice. J Inflamm. (2016) 13:25. doi: 10.1186/s12950-016-0131-6
170. Mishra M, Muthuramu I, Kempen H, de Geest B. Administration of apo A-I (Milano) nanoparticles reverses pathological remodelling, cardiac dysfunction, and heart failure in a murine model of HFpEF associated with hypertension. Sci Rep. (2020) 10:8382. doi: 10.1038/s41598-020-65255-y
171. Spillmann F, Miteva K, Pieske B, Tschöpe C, van Linthout S. High-density lipoproteins reduce endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. (2015) 35:1774–7. doi: 10.1161/ATVBAHA.115.305887
172. Terao R, Honjo M, Aihara M. Apolipoprotein M Inhibits angiogenic and inflammatory response by sphingosine 1-phosphate on retinal pigment epithelium cells. Int J Mol Sci. (2018) 19:112. doi: 10.3390/IJMS19010112
173. Iwaoka M, Obata J-E, Abe M, Nakamura T, Kitta Y, Kodama Y, et al. Association of low serum levels of apolipoprotein A-I with adverse outcomes in patients with nonischemic heart failure. J Cardiac Fail. (2007) 13:247–53. doi: 10.1016/j.cardfail.2007.01.007
174. Wedel H, McMurray JJV, Lindberg M, Wikstrand J, Cleland JGF, Cornel JH, et al. Predictors of fatal and non-fatal outcomes in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA): incremental value of apolipoprotein A-1, high-sensitivity C-reactive peptide and N-terminal pro B-type natriuretic peptide. Eur J Heart Fail. (2009) 11:281–91. doi: 10.1093/EURJHF/HFN046
175. Gombos T, Förhécz Z, Pozsonyi Z, Jánoskuti L, Prohászka Z, Karádi I. Long-term survival and apolipoprotein A1 level in chronic heart failure: interaction with tumor necrosis factor α−308 G/A polymorphism. J Cardiac Fail. (2017) 23:113–20. doi: 10.1016/J.CARDFAIL.2016.06.004
176. Florvall G, Basu S, Larsson A. Apolipoprotein A1 is a stronger prognostic marker than are HDL and LDL cholesterol for cardiovascular disease and mortality in elderly men. J Gerontol Ser A Biol Sci Med Sci. (2006) 61:1262–6. doi: 10.1093/GERONA/61.12.1262
177. Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SPA, et al. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. (2008) 28:2071–7. doi: 10.1161/ATVBAHA.108.168690
178. Murphy AJ, Woollard KJ, Suhartoyo A, Stirzaker RA, Shaw J, Sviridov D, et al. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A-I in in vitro and in vivo models of inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:1333–41. doi: 10.1161/ATVBAHA.111.226258
179. González-Pecchi V, Valdés S, Pons V, Honorato P, Martinez LO, Lamperti L, et al. Apolipoprotein A-I enhances proliferation of human endothelial progenitor cells and promotes angiogenesis through the cell surface ATP synthase. Microvasc Res. (2015) 98:9–15. doi: 10.1016/J.MVR.2014.11.003
180. Feng Y, Jacobs F, van Craeyveld E, Brunaud C, Snoeys J, Tjwa M, et al. Human ApoA-I transfer attenuates transplant arteriosclerosis via enhanced incorporation of bone marrow-derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol. (2008) 28:278–83. doi: 10.1161/ATVBAHA.107.158741
181. Muthuramu I, Amin R, Aboumsallem JP, Mishra M, Robinson EL, de Geest B. Hepatocyte-specific SR-BI gene transfer corrects cardiac dysfunction in Scarb1-deficient mice and improves pressure overload-induced cardiomyopathy. Arterioscler Thromb Vasc Biol. (2018) 38:2028–40. doi: 10.1161/ATVBAHA.118.310946
182. Christoffersen C, Dahlbäck B, Nielsen LB. Apolipoprotein M: progress in understanding its regulation and metabolic functions. Scand J Clin Lab Invest. (2006) 66:631–8. doi: 10.1080/00365510600885500
183. Kempen HJ, Gomaraschi M, Simonelli S, Calabresi L, Moerland M, Otvos J, et al. Persistent changes in lipoprotein lipids after a single infusion of ascending doses of MDCO-216 (apoA-IMilano/POPC) in healthy volunteers and stable coronary artery disease patients. Atherosclerosis. (2016) 255:17–24. doi: 10.1016/J.ATHEROSCLEROSIS.2016.10.042
184. Christensen PM, Bosteen MH, Hajny S, Nielsen LB, Christoffersen C. Apolipoprotein M mediates sphingosine-1-phosphate efflux from erythrocytes. Sci Rep. (2017) 7:14983. doi: 10.1038/s41598-017-15043-y
185. Hajny S, Christoffersen C. A novel perspective on the ApoM-S1P axis, highlighting the metabolism of ApoM and its role in liver fibrosis and neuroinflammation. Int J Mol Sci. (2017) 18:1636. doi: 10.3390/IJMS18081636
186. Elsøe S, Christoffersen C, Luchoomun J, Turner S, Nielsen LB. Apolipoprotein M promotes mobilization of cellular cholesterol in vivo. Biochim Biophys Acta. (2013) 1831:1287–92. doi: 10.1016/J.BBALIP.2013.04.009
187. Su W, Jiao G, Yang C, Ye Y. Evaluation of apolipoprotein M as a biomarker of coronary artery disease. Clin Biochem. (2009) 42:365–70. doi: 10.1016/J.CLINBIOCHEM.2008.11.010
188. Zheng L, Luo G, Zhang J, Mu Q, Shi Y, Berggren-Söderlund M, et al. Decreased activities of apolipoprotein m promoter are associated with the susceptibility to coronary artery diseases. Int J Med Sci. (2014) 11:365–72. doi: 10.7150/IJMS.7696
189. Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for preβ-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med. (2005) 11:418–22. doi: 10.1038/nm1211
190. Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, et al. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem. (2008) 283:1839–47. doi: 10.1074/JBC.M704576200
191. Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnström J, Sevvana M, et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA. (2011) 108:9613–8. doi: 10.1073/PNAS.1103187108
192. Wang M, Luo GH, Liu H, Zhang YP, Wang B, Di DM, et al. Apolipoprotein M induces inhibition of inflammatory responses via the S1PR1 and DHCR24 pathways. Mol Med Rep. (2019) 19:1272–83. doi: 10.3892/MMR.2018.9747
193. Kurano M, Tsuneyama K, Morimoto Y, Shimizu T, Jona M, Kassai H, et al. Apolipoprotein M protects lipopolysaccharide-treated mice from death and organ injury. Thromb Haemost. (2018) 118:1021–35. doi: 10.1055/S-0038-1641750
194. Liu Y, Tie L. Apolipoprotein M and sphingosine-1-phosphate complex alleviates TNF-α-induced endothelial cell injury and inflammation through PI3K/AKT signaling pathway. BMC Cardiovasc. ers. (2019) 19:279. doi: 10.1186/S12872-019-1263-4/FIGURES/4
195. Christensen PM, Liu CH, Swendeman SL, Obinata H, Qvortrup K, Nielsen LB, et al. Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J. (2016) 30:2351–9. doi: 10.1096/FJ.201500064/-/DC1
196. English D, Kovala AT, Welch Z, Harvey KA, Siddiqui RA, Brindley DN, et al. Induction of endothelial cell chemotaxis by sphingosine 1-phosphate and stabilization of endothelial monolayer barrier function by lysophosphatidic acid, potential mediators of hematopoietic angiogenesis. J Hematother Stem Cell Res. (1999) 8:627–34. doi: 10.1089/152581699319795
197. Lee OH, Kim YM, Lee YM, Moon EJ, Lee DJ, Kim JH, et al. Sphingosine 1-phosphate induces angiogenesis: its angiogenic action and signaling mechanism in human umbilical vein endothelial cells. Biochem Biophys Res Communic. (1999) 264:743–50. doi: 10.1006/BBRC.1999.1586
198. Wang F, van Brooklyn JR, Hobson JP, Movafagh S, Zukowska-Grojec Z, Milstien S, et al. Sphingosine 1-phosphate stimulates cell migration through a G(i)-coupled cell surface receptor. Potential involvement in angiogenesis. J Biol Chem. (1999) 274:35343–50. doi: 10.1074/JBC.274.50.35343
199. English D, Welch Z, Kovala AT, Harvey K, Volpert OV, et al. Sphingosine 1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemostasis and angiogenesis. FASEB J. (2000) 14:2255–65. doi: 10.1096/FJ.00-0134COM
200. Paik JH, Chae SS, Lee MJ, Thangada S, Hla T. Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of alpha vbeta3- and beta1-containing integrins. J Biol Chem. (2001) 276:11830–37. doi: 10.1074/JBC.M009422200
201. Garcia JGN, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. (2001) 108:689–701. doi: 10.1172/JCI12450
202. Donati C, Bruni P. Sphingosine 1-phosphate regulates cytoskeleton dynamics: implications in its biological response. Biochim Biophys Acta. (2006) 1758:2037–48. doi: 10.1016/j.bbamem.2006.06.015
203. Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. (2004) 169:1245–51. doi: 10.1164/RCCM.200309-1258OC
204. Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, et al. Pulmonary endothelial cell barrier enhancement by sphingosine 1-Phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. (2004) 279:24692–700. doi: 10.1074/JBC.M313969200
205. Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, et al. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. (2004) 18:2392–403. doi: 10.1101/GAD.1227804
206. Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. (2005) 280:17320–8. doi: 10.1074/JBC.M411674200
207. Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, et al. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science. (2001) 291:1800–3. doi: 10.1126/SCIENCE.1057559
208. Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. (2000) 106:951–61. doi: 10.1172/JCI10905
209. Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. (1999) 99:301–12. doi: 10.1016/S0092-8674(00)81661-X
210. Lee MJ, Thangada S, Paik JH, Sapkota GP, Ancellin N, Chae SS, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. (2001) 8:693–704. doi: 10.1016/S1097-2765(01)00324-0
211. Zhang J, Honbo N, Goetzl EJ, Chatterjee K, Karliner JS, Gray MO. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am J Physiol Heart Circ Physiol. (2007) 293:H3150–8. doi: 10.1152/AJPHEART.00587.2006
212. Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, et al. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2007) 292:H2944–51. doi: 10.1152/AJPHEART.01331.2006
213. Smith RM, Suleman N, Lacerda L, Opie LH, Akira S, Chien KR, et al. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res. (2004) 63:611–6. doi: 10.1016/J.CARDIORES.2004.06.019
214. Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, et al. Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase). Circulation. (2005) 112:3911–8. doi: 10.1161/CIRCULATIONAHA.105.581058
215. Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA. (2000) 97:315–9. doi: 10.1073/PNAS.97.1.315
216. Jacoby JJ, Kalinowski A, Liu MG, Zhang SSM, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA. (2003) 100:12929–34. doi: 10.1073/PNAS.2134694100
217. Negoro S, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T, Yamauchi-Takihara K. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res. (2000) 47:797–805. doi: 10.1016/S0008-6363(00)00138-3
218. Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. (2005) 107:131–7. doi: 10.1016/J.PHARMTHERA.2005.02.003
219. Ahmed N, Linardi D, Decimo I, Mehboob R, Gebrie MA, Innamorati G, et al. Characterization and expression of sphingosine 1-phosphate receptors in human and rat heart. Front Pharmacol. (2017) 8:312. doi: 10.3389/FPHAR.2017.00312/BIBTEX
220. Hanff TC, Cohen JB, Zhao L, Javaheri A, Zamani P, Prenner SB, et al. Quantitative proteomic analysis of diabetes mellitus in heart failure with preserved ejection fraction. JACC Basic Transl Sci. (2021) 6:89–99. doi: 10.1016/J.JACBTS.2020.11.011
221. Aboumsallem JP, Mishra M, Amin R, Muthuramu I, Kempen H, de Geest B. Successful treatment of established heart failure in mice with recombinant HDL (Milano). Br J Pharmacol. (2018) 175:4167–82. doi: 10.1111/BPH.14463
222. Mishra M, Muthuramu I, Aboumsallem J, Kempen H, de Geest B. Reconstituted HDL (Milano) treatment efficaciously reverses heart failure with preserved ejection fraction in mice. Int J Mol Sci. (2018) 19:3399. doi: 10.3390/ijms19113399
223. Aboumsallem JP, Muthuramu I, Mishra M, Kempen H, de Geest B. effective treatment of diabetic cardiomyopathy and heart failure with reconstituted HDL (Milano) in mice. Int J Mol Sci. (2019) 20:1273. doi: 10.3390/IJMS20061273
224. Franceschini G, Sirtori CR, Capurso A, Weisgraber KH, Mahley RW. A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. (1980) 66:892–900. doi: 10.1172/JCI109956
225. Weisgraber KH, Bersot TP, Mahley RW, Franceschini G, Sirtori CR. A-Imilano apoprotein. Isolation and characterization of a cysteine-containing variant of the A-I apoprotein from human high density lipoproteins. J Clin Invest. (1980) 66:901–7. doi: 10.1172/JCI109957
226. Franceschini G, Sirtori M, Gianfranceschi G, Sirtori CR. Relation between the HDL apoproteins and AI isoproteins in subjects with the AIMilano abnormality. Metabolism. (1981) 30:502–9. doi: 10.1016/0026-0495(81)90188-8
227. Sirtori CR, Calabresi L, Franceschini G, Baldassarre D, Amato M, Johansson J, et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: the Limone sul Garda study. Circulation. (2001) 103:1949–54. doi: 10.1161/01.CIR.103.15.1949
228. Kallend DG, Reijers JAA, Bellibas SE, Bobillier A, Kempen H, Burggraaf J, et al. A single infusion of MDCO-216 (ApoA-1 Milano/POPC) increases ABCA1-mediated cholesterol efflux and pre-beta 1 HDL in healthy volunteers and patients with stable coronary artery disease. Eur Heart J Cardiovasc Pharmacother. (2016) 2:23–9. doi: 10.1093/EHJCVP/PVV041
229. Kempen HJ, Asztalos BF, Moerland M, Jeyarajah E, Otvos J, Kallend DG, et al. High-density lipoprotein subfractions and cholesterol efflux capacities after infusion of MDCO-216 (Apolipoprotein A-IMilano/Palmitoyl-Oleoyl-Phosphatidylcholine) in healthy volunteers and stable coronary artery disease patients. Arterioscler Thromb Vasc Biol. (2016) 36:736–42. doi: 10.1161/ATVBAHA.115.307052
230. Reijers JAA, Kallend DG, Malone KE, Jukema JW, Wijngaard PLJ, Burggraaf J, et al. MDCO-216 does not induce adverse immunostimulation, in contrast to its predecessor ETC-216. Cardiovasc Drugs Ther. (2017) 31:381–9. doi: 10.1007/S10557-017-6746-X
231. Gordts SC, Muthuramu I, Nefyodova E, Jacobs F, van Craeyveld E, de Geest B. Beneficial effects of selective HDL-raising gene transfer on survival, cardiac remodelling and cardiac function after myocardial infarction in mice. Gene Ther. (2013) 20:1053–61. doi: 10.1038/GT.2013.30
232. Amin R, Muthuramu I, Aboumsallem JP, Mishra M, Jacobs F, de Geest B. Selective HDL-raising human Apo A-I gene therapy counteracts cardiac hypertrophy, reduces myocardial fibrosis, and improves cardiac function in mice with chronic pressure overload. Int J Mol Sci. (2017) 18:2012. doi: 10.3390/IJMS18092012
233. Hamid T, Ismahil MA, Bansal SS, Patel B, Goel M, White CR, et al. The apolipoprotein A-I mimetic L-4F attenuates monocyte activation and adverse cardiac remodeling after myocardial infarction. Int J Mol Sci. (2020) 21:3519. doi: 10.3390/ijms21103519
234. Weihrauch D, Xu H, Shi Y, Wang J, Brien J, Jones DW, et al. Effects of D-4F on vasodilation, oxidative stress, angiostatin, myocardial inflammation, and angiogenic potential in tight-skin mice. Am J Physiol Heart Circ Physiol. (2007) 293:1432–41. doi: 10.1152/AJPHEART.00038.2007
235. Moreira RS, Irigoyen M, Sanches TR, Volpini RA, Camara NOS, Malheiros DM, et al. Apolipoprotein A-I mimetic peptide 4F attenuates kidney injury, heart injury, and endothelial dysfunction in sepsis. Am J Physiol Regul Integrat Compar Physiol. (2014) 307:R514–24. doi: 10.1152/ajpregu.00445.2013
236. Gibson CM, Korjian S, Tricoci P, Daaboul Y, Yee M, Jain P, et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I, after acute myocardial infarction: the AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation. (2016) 134:1918–30. doi: 10.1161/CIRCULATIONAHA.116.025687
237. Study, to Investigate CSL112 in Subjects With Acute Coronary Syndrome - Full Text View - ClinicalTrials,.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT03473223 (accessed January 16, 2022).
238. Javaheri A, Kolansky DM, Cuchel M. Reconstituted high-density lipoprotein therapies: a cause for optimism. Arterioscler Thromb Vasc Biol. (2014) 34:1800–2. doi: 10.1161/ATVBAHA.114.304156
239. Swendeman SL, Xiong Y, Cantalupo A, Yuan H, Burg N, Hisano Y, et al. An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci Signal. (2017) 10:eaal2722. doi: 10.1126/SCISIGNAL.AAL2722
240. Obinata H, Hla T. Sphingosine 1-phosphate and inflammation. Int Immunol. (2019) 31:617–25. doi: 10.1093/intimm/dxz037
241. Shaul PW, Mineo C. HDL action on the vascular wall: is the answer NO? J Clin Invest. (2004) 113:509–13. doi: 10.1172/jci21072
Keywords: high-density lipoprotein (HDL), apolipoprotein A-I, apolipoprotein M, sphingosine-1-phosphate, heart failure, cardiomyopathy
Citation: Diab A, Valenzuela Ripoll C, Guo Z and Javaheri A (2022) HDL Composition, Heart Failure, and Its Comorbidities. Front. Cardiovasc. Med. 9:846990. doi: 10.3389/fcvm.2022.846990
Received: 31 December 2021; Accepted: 09 February 2022;
Published: 08 March 2022.
Edited by:
Tomas Vaisar, University of Washington, United StatesReviewed by:
Gunther Marsche, Medical University of Graz, AustriaCopyright © 2022 Diab, Valenzuela Ripoll, Guo and Javaheri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Javaheri, YWxpLmphdmFoZXJpQHd1c3RsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.