 Yuzhou Gui1,2
Yuzhou Gui1,2 Richard Y. Cao
Richard Y. Cao- 1Shanghai Xuhui Central Hospital, Zhongshan-Xuhui Hospital, Fudan University, Shanghai, China
- 2Shanghai Engineering Research Center of Phase I Clinical Research and Quality Consistency Evaluation for Drugs, Shanghai, China
- 3Department of Cardiovascular, Shanghai Xuhui Central Hospital, Zhongshan-Xuhui Hospital, Fudan University, Shanghai, China
Foam cells play a vital role in the initiation and development of atherosclerosis. This review aims to summarize the novel insights into the origins, consequences, and molecular mechanisms of foam cells in atherosclerotic plaques. Foam cells are originated from monocytes as well as from vascular smooth muscle cells (VSMC), stem/progenitor cells, and endothelium cells. Novel technologies including lineage tracing and single-cell RNA sequencing (scRNA-seq) have revolutionized our understanding of subtypes of monocyte- and VSMC-derived foam cells. By using scRNA-seq, three main clusters including resident-like, inflammatory, and triggering receptor expressed on myeloid cells-2 (Trem2hi) are identified as the major subtypes of monocyte-derived foam cells in atherosclerotic plaques. Foam cells undergo diverse pathways of programmed cell death including apoptosis, autophagy, necroptosis, and pyroptosis, contributing to the necrotic cores of atherosclerotic plaques. The formation of foam cells is affected by cholesterol uptake, efflux, and esterification. Novel mechanisms including nuclear receptors, non-coding RNAs, and gut microbiota have been discovered and investigated. Although the heterogeneity of monocytes and the complexity of non-coding RNAs make obstacles for targeting foam cells, further in-depth research and therapeutic exploration are needed for the better management of atherosclerosis.
Introduction
Atherosclerosis is the major cause of acute cardiovascular events including unstable angina, myocardial infarction, and ischemic stroke. It is a chronic disease of the arteries and remains asymptomatic for many years (1). Current therapies reduce the risk of cardiovascular disorders by surgical interventions and treatment of hyperlipidemia and hypertension. However, atherosclerotic cardiovascular disease (ASCVD) is still among the major cause of death worldwide. Additional therapeutic approaches are needed for reducing the risk of cardiovascular events (2).
The pathology of atherosclerosis can be divided into three stages: (a) lipid-streak stage, (b) fibrous plaque stage, and (c) advanced lesions and thrombosis (2). In the lipid-streak stage, various forms of lipids undergo retention and trapping by arterial walls. Macrophages infiltrate into the intima of the arterial wall and uptake excessive lipids, leading to the formation of foam cells (3). In the fibrous plaque stage, the vascular smooth muscle cells (VSMCs) migrate to the intima of the arterial wall and generate the fibrous cap at the atherosclerotic site. The thick fibrous cap contributes to a stable plaque. Excessive accumulation of foam cells leads to necrosis within atherosclerotic plaques (4). In the advanced lesions and thrombosis stage, the necrotic core grows and the fibrous cap diminishes. The components of the necrotic core interact with blood coagulation cells and factors and trigger thrombosis (5).
Foam cell formation is a key step in the initiation and progression of atherosclerosis (6). Lipid homeostasis is maintained by the proper function of lipid uptake, efflux, and esterification. But in the course of atherosclerosis, the excessive lipid accumulation in macrophages induces the expression of uptake receptors such as scavenger receptors (SRs) but downregulates the expression of efflux transporters like ATP-binding cassette A1 and G1 (ABCA1 and ABCG1). The dysfunction of macrophages leads to the formation of foam cells (7).
Although foam cells are the major sources of necrotic core in atherosclerotic plaques, therapies targeting foam cells are at early stages due to the limited knowledge of the foam cells-related biological mechanisms (8). In the past decades, tremendous efforts have been made on the better understanding of origins, consequences, and regulation of foam cells. Vascular smooth muscle cells (VSMC), stem/progenitor cells (SPCs), and endothelium also contribute to the formation of foam cells (9, 10). It is worth noting that monocytes can be further categorized into classical, intermediate, and non-classical subtypes in atherosclerotic plaques (11). More importantly, single-cell sequencing (sc-seq) technologies have revolutionized the knowledge of the phenotypic diversity of monocytes and VSMC-derived foam cells (12, 13). In advanced lesions, apoptosis, autophagy, necroptosis, and pyroptosis have been discovered in foam cells. Molecular mechanisms of foam cell regulation include cholesterol transporters, as well as non-coding RNAs (ncRNAs), and gut microbiota.
This review summarizes the novel insights into the origins, consequences, and regulation of foam cells in atherosclerosis, especially in the past decade. More importantly, this review identifies the obstacles to targeting foam cells. Collectively, these efforts will be helpful for further in-depth research and therapeutic exploration.
Origins of Foam Cells in Atherosclerosis
Monocyte-Derived Foam Cells and Their Subtypes
In atherosclerosis, monocytes are classified as classical and non-classical monocytes (14). Ly6Chigh monocytes in mice are categorized as classical monocytes, which are most comparable to the human CD14++/CD16– subtype of monocytes. The classical monocytes consist of the majority of monocytes in circulation (∼90%). Ly6Clow cluster of monocytes in mice is classified as non-classical monocytes, which are comparable to the human CD14+/CD16++ cluster (∼10%). The role of classical and non-classical monocytes in the progression of atherosclerosis is under debate. The classical or “proinflammatory” monocytes enter into areas of endothelial injury (15, 16). Ly6Chigh monocytes have migrated into the underneath of endothelium and differentiated into plaque macrophages and foam cells (17), while “non-classical” monocytes are correlated with plaque lesion size in mice. Only when the entry of Ly6high and Ly6low monocytes into sub-endothelial spaces are inhibited could atherosclerosis be ultimately halted (18). Recent studies show the existence of an intermediate subtype of monocytes in mice (Gr1high, CD43high) and humans (CD14++/CD16+) (19, 20). Higher CD14++/CD16+ subtype of monocytes in circulation is correlated with higher cardiovascular risks and events (21, 22). Compared with the classical and non-classical monocyte, the intermediate subtype of monocytes generates a great extent of inflammatory responses (23, 24). Collectively, classical, non-classical and intermediate subsets of monocytes exert effects on lipid accumulation and inflammatory response in atherosclerosis.
Macrophages, differentiated and matured from monocytes, are the central cells in atherosclerosis (25). Since the 1960s, macrophages are classified in M1 and M2 subtypes (26). It has been extensively accepted that M1 macrophages trigger and participate in the inflammation process while M2 macrophages are anti-inflammatory (27, 28). However, this classification oversimplifies the function of macrophages in atherosclerosis (29). Recent studies have challenged the established classification (27). The classification of M1 and M2 macrophages are extreme states of functions and under different stimuli, the M2 phenotype is not always athero-protective (30, 31).
In the past decade, macrophage heterogeneity has been discovered and demonstrated in the atherosclerotic plaques (11). Single-cell technologies allow accurate measurement of phenotypes of an individual cell. These advanced technologies have greatly improved the understanding of macrophage phenotype diversity. Single-cell RNA sequencing has been utilized to examine macrophage heterogeneity in multiple mouse models of atherosclerosis (32, 33) and human carotid atherosclerosis (12). More importantly, single-cell technologies including single-cell RNA sequencing, single-cell transcriptomics, make it possible for monitoring the dynamic changes of macrophage phenotypes in atherosclerotic plaques. Three main clusters of the macrophages, which are resident-like, inflammatory, and triggering receptor expressed on myeloid cells-2 (TREM2hi), have been identified (34, 35). The resident-like macrophages can infiltrate into the plaques and their phenotypes are similar to M2 macrophages. The inflammatory macrophages are non-foamy subtypes and located in the intima of the plaques. They are the major population expressing inflammatory cytokines (36). TREM2hi macrophages, the third subtype, are foamy lipid-laden macrophages, with impaired cholesterol efflux capacity, and cellular catabolic processes, which have an M2-like phenotype (32).
Vascular Smooth Muscle Cell-Derived Foam Cells
VSMCs are flexible, non-autonomous cells that are widely distributed in multiple tissues such as blood vessels, the trachea, and the digestive tract (37). VSMCs are not terminally differentiated cells. Phenotype switching occurs under a variety of physiological and pathological conditions (9). The markers of smooth muscle cells, such as actin alpha 2 (ACTA2), myosin heavy chain 11 (Myh11), are inhibited while the markers of terminally differentiated cells are elevated, which is accompanied by the transformation of cell function. In adipose differentiation medium, VSMC undergo phenotypic transformation into adipocytes, and the expression of markers such as CCAATenhancer-binding proteins (C/EBPα), peroxisome proliferator-activated receptor-gamma (PPARγ), and leptin on the cell surface will increase (38, 39). In the state of tissue damage, the expression of SMemb/non-muscle MHC isoform-B and CRBP-1 in VSMC increases (40). In osteoporosis, VSMC can be transformed into osteoblast-like cells, with the induction of markers such as runt-related transcription factor 2 (Runx2), SRY-Box 9 (Sox9), etc. (41, 42).
In atherosclerosis, VSMC is firstly identified in the fibrous cap by using immunofluorescence. The fibrous cap is mainly composed of VSMC and its secreted collagen, elastin, etc. (43). The smooth muscle cell markers ACTA2 and SM myosin heavy chain (MHC) are highly expressed. The foam cells in the necrotic core are mainly composed of macrophages with high expression of macrophage surface markers such as Mac2 and CD68 (44). Recent studies greatly expand the origin of foam cells in atherosclerotic plaque.
Lineage-tracing technology greatly expands the understanding of the origins of foam cells in the plaques. VSMCs undergo trans-differentiation and become macrophage-like cells during atherogenesis (37). A lineage-tracing system, induced by myh11 promoter, is developed in the apoE–/– mouse model of atherosclerosis. The VSMC derived foam cells in advanced atherosclerotic plaque are underestimated and a large number of VSMCs undergo phenotypic conversion with the loss of contractile markers in the plaques (45). This phenomenon of VSMC phenotype switching has also been verified in humans (46). Approximately 40% of the macrophages in the plaque are derived from VSMC, which is the product of VSMC phenotypic transformation (46). In the process of phenotypic transformation, the expression of VSMC markers is reduced or even lost. In mouse models, VSMCs, identified by traditional immunostaining, only account for about 20% of fluorescent protein-labeled VSMCs, indicating that traditional immunostaining may miss a large portion of VSMC-derived cells, thereby underestimating the VSMCs in plaques (47). The impact of transformation on disease has shown that VSMCs are an important source of plaque macrophages, and phenotypic transformation can transform VSMCs into macrophages to obtain the surface markers and functions of macrophages (48). Therefore, inhibiting the phenotypic transformation of VSMCs is beneficial to inhibit the number of macrophages in the plaque and maintain the number of VSMC in the fibrous cap, which is a potential new strategy to stabilize plaque (49). In vitro studies have confirmed that under the action of a variety of lipids and inflammatory stimuli, such as cholesterol (50), oxidized phospholipids (oxPL) (51), platelet-derived growth factor (PDGF) (52), VSMCs undergo the phenotypic transformation and transform into macrophage-like cells. The level of surface markers of macrophages, including CD68 and macrophage-2 (Mac2), is increased, while the expression of smooth muscle cell surface markers is decreased (53). Besides, krüppel-like factor 4 (KLF4) promotes the phenotypic switching of VSMC to macrophage-like proinflammatory cells and the formation of atherosclerotic plaques. A significant reduction in lesional size is observed in VSMC-specific knockout of KLF4 in apoE–/– mice (47).
Sc-seq is a powerful and effective way to accurately identify VSMC subsets in atherosclerotic plaques. A couple of research groups have combined sc-seq with lineage tracing technology. In human atherosclerotic plaques, VSMC can differentiate into fibroblast-like cells instead of macrophages as previously reported (54). VSMC-specific knockout of Transcription Factor 21 (TCF21) inhibits phenotypic modulation in mice. TCF21 expression is strongly associated with VSMC phenotypic modulation and cardiovascular risks in humans. A recent scRNA-seq study discovers an intermediate cell state of VSMC in atherosclerotic plaques termed “SEM” cells. It can be identified in human coronary and carotid arteries with plaques. SEM undergoes differentiation into macrophage and fibrochondrocyte-like cells (13, 55).
Other Origins of Foam Cells
SPCs and endothelial cells also lead to the formation of foam cells in atherosclerotic plaques (56). The reason why SPCs should be studied for atherosclerosis is that SPCs expressing stem cell antigen-1 (Sca1+) increase the areas of necrotic cores which consist of foam cells (57). A lineage-tracing study gives more evidence that VSMC derived Sca-1+ cells are capable of generating several cell types including macrophages (58). As maintaining VSMC phenotype is more atheroprotective, kruppel-like factor 4 (KLF-4) may be key in modulating this cell trans-differentiation (59). Endothelial cells have been reported to influence the entry of lipoproteins, but their contribution to foam cells is less discussed (60). The origin of foam cells in atherosclerotic plaques is depicted in Figure 1.
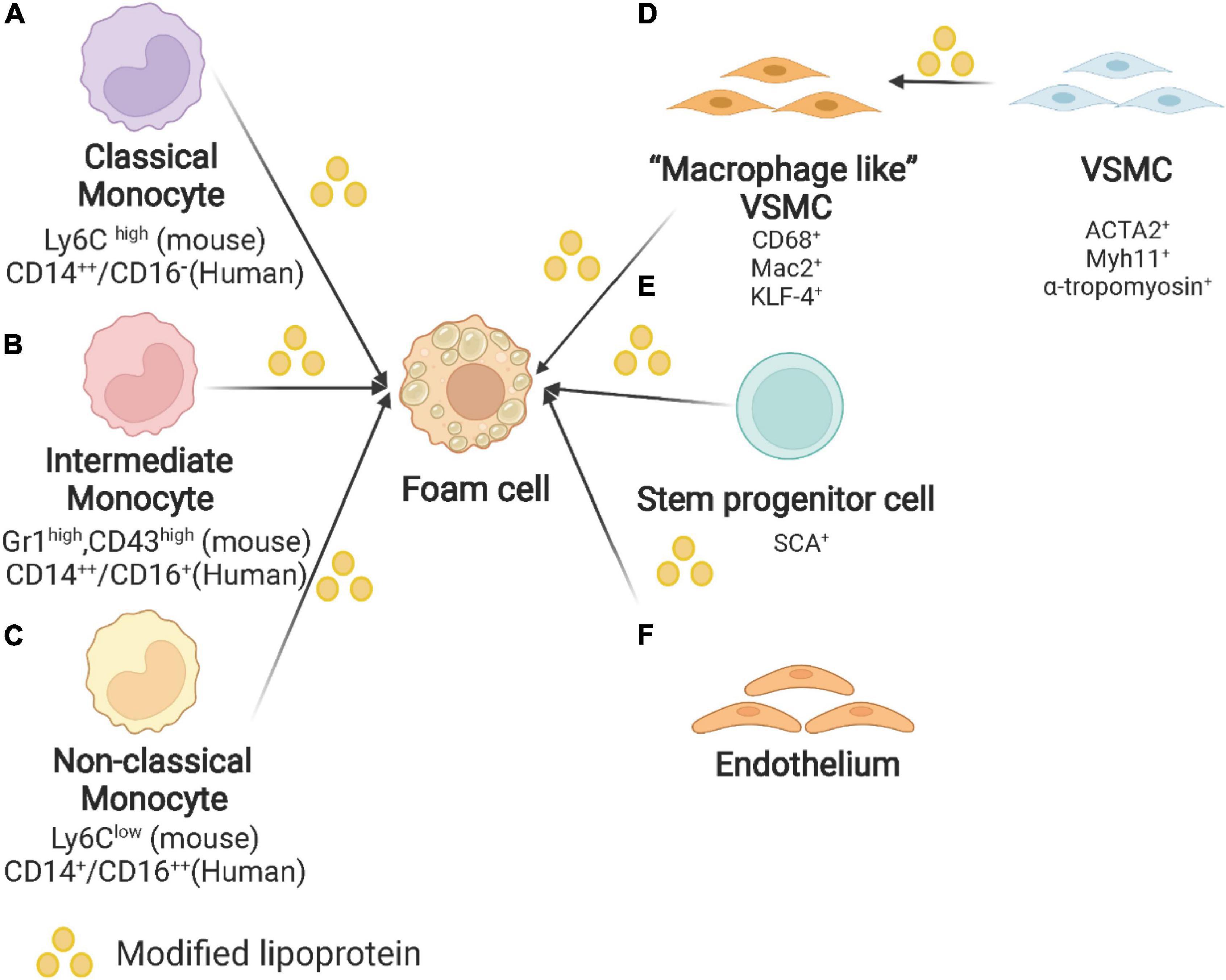
Figure 1. Origin of foam cells in atherosclerotic plaques. (A) Classical monocyte: Ly6Chigh monocytes in mice and CD14++/CD16– in humans. (B) Intermediate monocyte: Gr1high, CD43high in mouse and CD14++/CD16+ in human. (C) Non-classical monocyte: Ly6Clow monocytes in mice and CD14+/CD16++ in humans. (D) VSMC: VSMC undergoes phenotypic switching to macrophage-like VSMC and finally converts into foam cells. (E) Other origins of foam cells: stem/progenitor cells (SPCs) and endothelium. Created with BioRender.com.
Consequences of Foam Cells in Atherosclerosis
The foam cells, derived from macrophages or VSMCs, impairs efferocytosis and triggers multiple pathways of programmed cell death including apoptosis, autophagy, necroptosis, and pyroptosis. The increasing dead cells enlarge the necrotic cores in the atherosclerotic plaques and reduce the plaque stability.
Apoptosis
The lipid overload by macrophages and VSMCs induces foam cell formation and triggers apoptosis in atherosclerotic plaques (61). Numerous papers have shown that oxLDL can initiate and activate the apoptotic pathway through the caspase cascade (62–64). The cytochrome c, a mitochondrial intermembrane protein, is delivered into the cytosol and induces the expression of apoptotic protease activating factor 1 (APAF1) and apoptosome, which triggers apoptosis cascade by activating the serine protease caspase 9. Active caspase 9 stimulates caspase 3 and caspase 7, which results in DNA fragmentation (65).
However, previous attempts to reduce apoptosis of macrophages may not lead to beneficial effects in atherosclerotic plaques. Inhibition of B-cell lymphoma-2 (Bcl-2) associated X protein (Bax) in macrophages suppresses apoptosis in macrophages but accelerates atherosclerosis (62, 66). Interestingly, inhibition of Bcl-2 leads to excessive apoptosis which induces larger necrotic areas in atherosclerotic plaques of apoE–/– mice (67). Scientists have explored the reason for this diverse effect of inhibition and induction of apoptosis. In-depth studies reveal that apart from apoptosis, the ability to clearance of apoptotic cells is vital for its consequences.
Efferocytosis is an intracellular process that can resolve inflammation and prevent excessive apoptosis (68). Excessive apoptosis triggers large-scale and irreversible necrosis in plaques. Instead, with the proper function of efferocytosis, apoptotic cells are appropriately cleared (69). The accumulation of uncleared apoptotic cells triggers necrosis and releases pro-inflammatory mediators when efferocytosis capacity is impaired (70, 71). The impaired function of efferocytosis leads to a large necrotic core and unstable plaques due to insufficient clearance of dying macrophages. Efforts have been made to regulate apoptotic cells to promote efficient clearance to alleviate atherosclerosis (72). ATP citrate lyase deficiency stabilizes atherosclerotic plaque via enhanced apoptosis and improved efferocytosis (73). Circadian miR21 expression induces a diurnal rhythm of apoptosis with impaired function of efferocytosis and increases the size of the necrotic core (74). In early lesions, efferocytosis induces anti-inflammatory pathways and prevents cellular necrosis by activation of NF-κB signaling (75). In foam cells, Inflammatory pathways are induced by endoplasmic reticulum stress (ER stress). It promotes apoptosis by inhibiting NF-κB signaling and induction of the c-Jun N-terminal kinase (JNK) pathway (76).
Autophagy
Autophagy is a conserved pathway for protein lysosome degradation (77). Autophagy is initiated by the formation of autophagosomes, characterized by double-membrane vesicles (78). The membrane elongation in the process of autophagosome formation is mediated by the microtubule-associated protein 1A/1B light chain 3 (LC3) conjugation system. The autophagosome is then subjected to lysosomal degradation. Unlike apoptosis, autophagy is an intracellular process for the reuse of degraded proteins. Autophagy also removes abnormal organelles and proteins for cellular homeostasis. However, impaired clearance of autophagosome triggers autophagic cell death. Autophagy abnormality participates in various diseases including cardiovascular diseases, cancer, and aging-related diseases (79). The role of autophagy in atherosclerosis is under debate. Several studies demonstrate the athero-protective effect by maintaining basal autophagy in atherosclerosis (80). Impaired autophagy abolishes the clearance of dysfunctional organelles and results in the accumulation of cytotoxic aggregates (81). Rescue of impaired autophagy by rapamycin, a mechanistic target of rapamycin (mTOR) kinase inhibitor, shows the potential for maintaining the stability of atherosclerotic plaque (82). However, excessive autophagy may induce cell death. Multiple research works demonstrate that oxidized lipids and inflammation-induced autophagy hyper-activation lead to damage to the vascular and progression of atherosclerosis (83–85).
In foam cells, autophagy contributes to the reversal of normal lipid homeostasis. It participates in the transport of lipid droplets to lysosomes for cholesterol ester hydrolysis. Autophagy involves the delivery of lipid droplets to lysosomes for hydrolysis by lysosomal acid lipase (LPL). In foam cells, intracellular free cholesterol is then subject to ABCA1-dependent efflux (86). Autophagy-related 5 (Atg5) is a key mediator of membrane elongation. The deficiency of Atg5 promotes plaque necrosis. Atg5-knockout in macrophage induces expression of p62 and down-regulated the LC3, thus the autophagy function is abolished. Furthermore, Atg5 knockout in macrophages may result in increased plaques by activation of inflammasome (87). Another study reveals that phagocyte autophagy is evoked by Akt inhibitors, mTOR inhibitors, and mTOR-siRNA, which indicates that activating autophagy of phagocytes will stabilize vulnerable coronary-artery disease plaques (87).
Collectively, these results point toward the necessity of basal level of autophagy and its protective role in atherosclerosis. However, when autophagy participates in the destruction of the cell, it exacerbates the inflammation and accelerates cell death. Future studies are necessary to explore the way for maintaining basal autophagy while avoiding excessive autophagy in atherosclerosis.
Necroptosis
Necroptosis, which is also termed “programmed necrosis,” is an emerging pathway of programmed cell death (88, 89). It is triggered by various cell-surface receptors, including tumor necrosis factor –α (TNF-α), interferon (IFN), toll-like receptors (90). Although morphologically similar to necrosis, necroptosis is mediated by receptor-interacting serine/threonine-protein kinase 1 and 3 (RIPK1&3) (91). After the addition of caspase inhibitor zVAD, it could not only inhibit the apoptotic caspase cascade but also initiate the necroptosis pathway (92). TNF-α and TNF receptors interactions lead to three unique complexes (93). The RIP homotypic interaction motif (RHIM) induces the process of necroptosis by strengthening the interaction of RIPK3 and RIPK1 (94). RIPK1 and RIPK3 are activated by phosphorylation and sequentially phosphorylates the mixed lineage kinase domain-like protein (MLKL). This process leads to the assembly of the complex called necrosome, which is a critical step for the progression of necroptosis in humans (90). Necroptosis participates in the death of necrotic cells via RIPK1 and RIPK3, which is different from necrosis (91). Necroptosis has been discovered in many diseases, including cancer, cardiovascular diseases, central nerve system (CNS) syndromes, and other systematic inflammatory diseases (95).
In both early and advanced atherosclerotic plaques, oxLDL and oxPL induce macrophages to undergo RIPK1/RIPK3/MLKL mediated necroptosis (96). A couple of studies discover that RIPK1 is abundant in early lesions of atherosclerosis in humans and mice (97–99). Antisense oligonucleotides (ASO) of RIPK1 significantly attenuates lesion areas in the aortic sinus and foam cell-rich lesions are also reduced (100). Besides, in macrophages, loss of RIPK1 reduces the production of interleukins and other proinflammatory cytokines (93). However, different studies generate diverse results. RIPK1 inhibition generates different results in the early and advanced stages of atherosclerosis. RIPK1 inhibitor attenuates atherosclerotic plaques after 2-weeks of treatment, with a lower level of inflammatory cytokines. Long-term administration (4-week) of RIPK1 inhibitor accelerates atherosclerosis with increased lesion area in the plaques and more lipid accumulation in macrophages (101). By suppressing necroptosis cell death, RIPK1 inhibitor promotes foam cell formation in isolated macrophages with incubation of oxLDL (102). An in-depth investigation reveals that MLKL knockdown attenuates necroptosis and inhibits the areas of necrotic cores in the plaques. However, the total lesion area is not altered (103). Besides, treatment with MLKL antisense oligonucleotide reduces circulating cholesterol levels compared with control antisense oligonucleotide but increases the accumulation of lipids within the plaque and in macrophage foam cells (103).
Pyroptosis
Pyroptosis is a highly inflammatory form of programmed necrosis which is initially identified in infections of bacterial and other pathogens (104). In the past decade, extensive studies have shown that pyroptosis is initiated by various kinds of inflammatory stimuli including lipopolysaccharides (LPS), oxLDL, reactive oxygen species (ROS). These stimuli activate the NF-κB pathway and the NOD-like receptor 3 (NLRP3) inflammasome. NLRP3 then causes pyroptosis through caspase-1- and caspase-11-mediated cytoplasmic protein gasdermin D (GSDMD). Caspase -1 and -11 cleave and activate GSDMD and GSDMD pores in the plasma membrane and consequently promotes pyroptotic cell death. Caspase-3 induces pyroptosis via cleavage of GSDME and granzyme A can cleave GSDMB to cause cell death (105).
Pyroptosis increases inflammation and induces foam cell formation in atherosclerosis (106). A large portion of cell death in atherosclerotic plaques is attributed to pyroptosis. It also promotes necrotic core formation in advanced atherosclerosis. In vitro study shows that low-concentration ox-LDL treatment induces pyroptosis in human monocyte-derived foam cells (107, 108). Absent in melanoma 2 (AIM2)-dependent macrophage pyroptosis exacerbates atherosclerosis as well. AIM2 inflammasome, which is induced by the Janus kinase 2 (Jak2) mutation Jak2V617F, induces the formation of necrotic cores (109). The consequences of foam cells in atherosclerosis including apoptosis, autophagy, necroptosis, and pyroptosis are depicted in Figure 2.
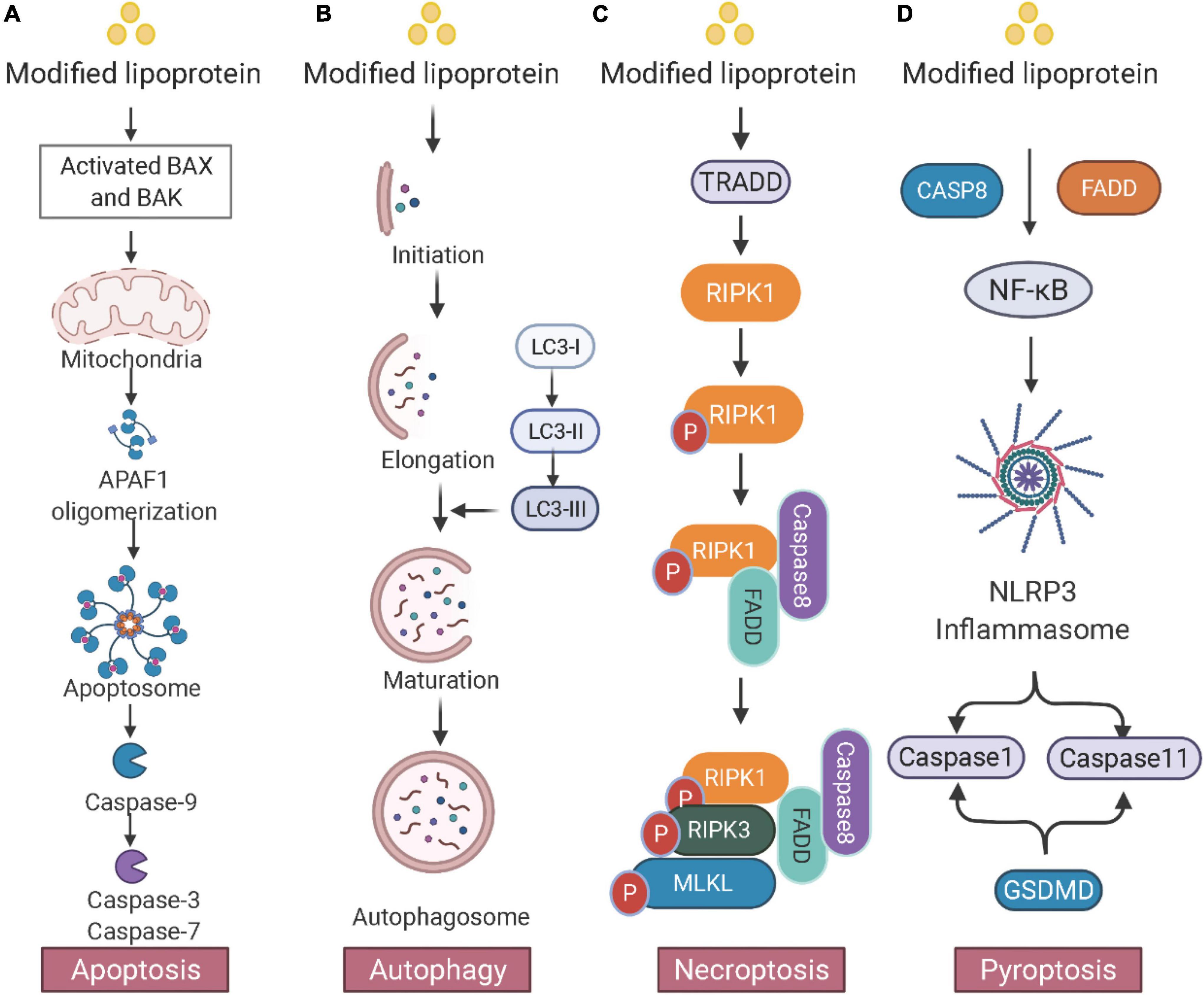
Figure 2. Consequences of foam cells in atherosclerosis. (A) Apoptosis: The mitochondrial intermembrane protein is released and triggers apoptotic protease activating factor 1 (APAF1) and apoptosome, leading to DNA fragmentation. (B) Autophagy: Double membrane autophagosomes are fused with lysosomes for degradation. The microtubule-associated protein 1A/1B light chain 3 (LC3) conjugation system participates in the membrane elongation and formation of the autophagosome. (C) Necroptosis: Activated TRADD (TNFRSF1A associated via death domain) triggers the phosphorylation of RIPK1 and RIPK3 and induces the downstream MLKL. (D) Pyroptosis: Inflammatory stimuli activate the NF-κB pathway and the NOD-like receptor 3 (NLRP3) inflammasome. NLRP3 then causes pyroptosis through caspase-1- and caspase-11-mediated cytoplasmic protein gasdermin D (GSDMD). Created with BioRender.com.
Regulation of Foam Cells in Atherosclerosis
Since the formation of foam cells is a hallmark in the initial stage of atherosclerosis, tremendous efforts have been made to unveil the regulatory pathways and mechanisms associated with the process. Foam cells are the incorporative effects of lipid uptake, lipid efflux, and cholesterol esterification. The abnormal accumulation of lipids and cholesterol esters in macrophages may demonstrate that cholesterol efflux is insufficient while lipid uptake and esterification are induced (110).
Cholesterol Uptake
Lipoproteins can be absorbed into macrophages and other cells through different transporters, which is called cholesterol uptake (111). In the 1970s, Brown and Goldstein laboratories demonstrated that modified LDLs were absorbed by macrophages via SRs (112). SRs are the major pathway of cholesterol uptake including CD36 (113) and scavenger receptors A (114). CD36 is a member of class B of the SR family. It is identified as a macrophage receptor of oxLDL and mediates its internalization and degradation (115). Apart from mediating lipid uptake, CD36 also modulates the proliferation, migration, and retention in arterial lesions (116). SRs also have different binding ligands of lipoproteins. By using SR-A and CD36 knockout (KO) mice, it has been demonstrated that CD36 and SR-A contribute to 75–90% of the uptake of modified lipoproteins (117). Macrophages also have new types of receptors. For example, lectin-like oxLDL receptor-1 (LOX-1) contributes to this process (118). LOX-1 overexpression significantly aggravates the development of atherosclerotic lesions (118), while LOX-1 knockout in low-density lipoprotein receptor knockout (LDLR–/–) mice has smaller atherosclerotic lesions, suggesting that LOX-1 accelerates the formation of foam cells and is proatherogenic (119). By comparing with LOX-1-deficient macrophages, LOX-1 in the normal macrophages accounts for 10% of oxLDL internalization under basal conditions, while this ratio increases by 40% in lysophosphatidylcholine (LPC)-treated macrophages. Efforts have been made to unveil the molecular mechanisms. Sorting nexin 10 (SNX10) plays a critical role in regulating macrophage function and foam cells. Knockout of SNX10 inhibits the function of CD36 in internalizing modified lipoproteins. SNX10 knockout significantly attenuates the progression of atherosclerosis in mice (120). CD146 facilitates the function of cholesterol uptake by CD36. Knockout of CD146 significantly attenuates the lesion areas in high-fat-diet apoE–/– mice (121). Casein kinase 2-interacting protein-1 (CKIP-1) suppresses the foam cell formations. By CKIP-1 deficiency, scavenger receptor LOX-1 is upregulated with faster progression of atherosclerosis (122). Although cholesterol uptake is mediated by CD36, SR-A, and LOX-1, multiple pathways exist, such as phagocytosis and pinocytosis. Current strategies targeting a single pathway of lipid uptake are difficult to inhibit foam cells. More efforts are needed to get a deeper understanding of cholesterol uptake and its function in foam cells.
Cholesterol Efflux
Multiple pathways mediates the cholesterol transport from the inside cell to the outside, including simple diffusion and transporter-dependent cholesterol efflux (123). In the normal condition, simple diffusion contributes to the majority of cholesterol efflux. In the lipid overload, ABCA1, ABCG1, and scavenger receptors BI (SR-BI) contribute 60–70% of cholesterol efflux (124). Cholesterol in the macrophage foam cells in the body can be transported into high-density lipoprotein (HDL) or apolipoprotein AI (Apo-AI) (125). This process is the initial step of HDL’s reverse cholesterol transport and one of the mechanisms by which it plays a protective role in the cardiovascular system (44). The three major transporters mediating this important process are ABCA1, ABCG1, and SR-BI (126).
ABCA1 transports a variety of substrates out of cellular membranes (127). In cholesterol efflux, ABCA1 facilitates free cholesterol to lipid-poor ApoA-1 and produces nascent HDL (128). Homozygous loss of function mutations in human ABCA1 result in the ultra-low level of HDL cholesterol (129). It has been widely acknowledged that the level of HDL-cholesterol is inversely correlated to cardiovascular risks. However, the effect of ABCA1 on cardiovascular risks and atherosclerosis development is controversial. The impact of losing ABCA1 on the patients of Tangier Disease is variable. Forty-four percent shows increased evidence of cardiovascular disease (CVD). However, four different heterozygous mutations in ABCA1 shows no association with CVD risks. Mutation of ABCA1 in apoE–/– mice has little effect on atherosclerosis (130). Therefore, further studies are needed for the effect of ABCA1 on CVD risks and the development of atherosclerosis (131, 132).
ABCG1 is a transporter of free cholesterol. It functions as a homodimer to transport free cholesterol to HDL particles (133). In humans and mice, the ABCG1 expresses at high levels in multiple organs including the lung, brain, spleen, adrenal glands, heart, and liver (134). Overexpression of ABCG1 leads to a high level of efflux of free cholesterol to HDL particles. (135). Besides, ABCG1 also mediates the efflux of other lipids including oxysterols, phospholipids, and sphingomyelin (136).
Cholesterol Esterification
Cholesterol esterification is the major process of cholesterol storage in cells. Cholesterol ester is synthesized by acetyl-CoA acetyltransferases (ACAT) at the endoplasmic reticulum (ER) (137). ACAT inhibitors, such as avasimibe and tomatidine inhibit the formation of foam cells and attenuate atherosclerosis in mice (138). However, they fail to attenuate carotid as well as coronary atherosclerosis in patients. Neutral CE hydrolases (NCEH) and hormone-sensitive lipase (HSL) are responsible for the hydrolysis of cholesterol ester in foam cells. NCEH knockout in vivo triggers larger plaque areas and more foam cell formation (138). Overexpression of NCEH1 significantly attenuates atherosclerotic lesion areas and induces cholesterol ester hydrolysis and efflux (137). But an NCEH inhibitor is not available and further studies are needed to unveil the mechanism of cholesterol esterification and its consequences on foam cells.
Molecular Mechanisms of Cholesterol Uptake, Efflux, and Esterification in Foam Cells
Cholesterol uptake, efflux, and esterification are regulated by multiple nuclear receptors, while non-coding RNAs (ncRNAs) and gut microbiota may also be involved. The process of cholesterol efflux, cholesterol influx and cholesterol esterification, and molecular mechanism in foam cell formation is depicted in Figure 3.
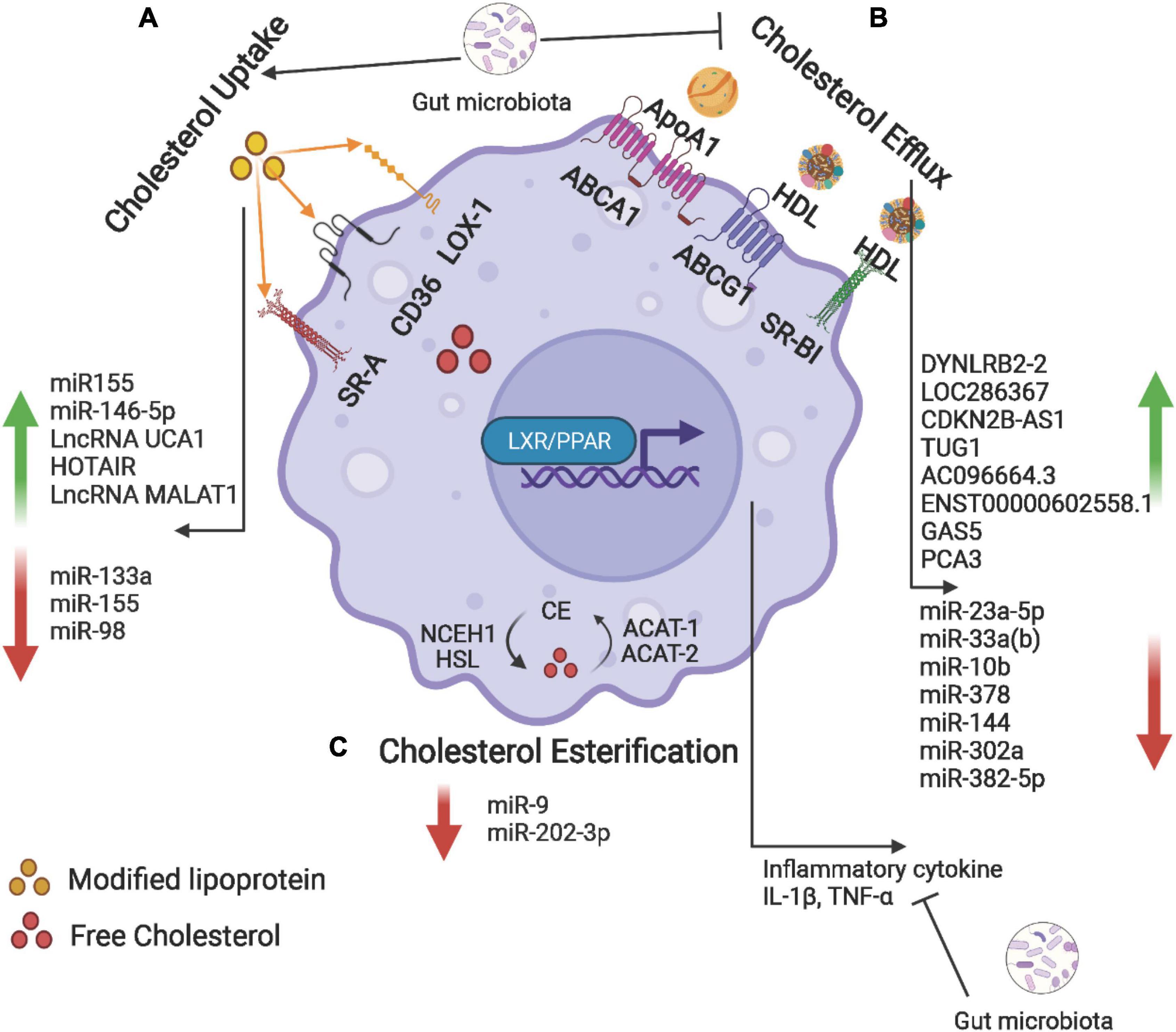
Figure 3. Cholesterol uptake, efflux, and esterification in foam cells. (A) Macrophage uptake modified lipoproteins (Ox-LDL) via scavenger receptors including SR-A, CD36, and LOX-1. (B) Intracellular free cholesterol is efflux to Apo-AI and HDL via ABCA1, ABCG1, and SR-BI. (C) The internalized free cholesterol is esterified by acetyl-coenzyme A acetyltransferase (ACAT-1 and -2) and stored in lipid droplets. The ester group is removed from cholesteryl by neutral cholesteryl ester hydrolase (NCEH) to release free cholesterol. Non-coding RNAs and gut microbiota may be involved in the regulation of these processes. Created with BioRender.com.
Nuclear Receptors
The receptors and transporters of cholesterol uptake, efflux, and esterification can be regulated in multiple pathways including transcriptional modulation and post-transcriptional modulation (139). Transcriptional modulation is mediated by a variety of nuclear receptors including liver X receptor (LXR), and PPARα, and PPARγ. LXR is an important nuclear receptor that maintains the hemostasis of intracellular lipids. Synthetic LXR agonists GW3965 and T0901317 can effectively inhibit atherosclerosis in apoE–/– mice and LDLR–/– mice (140). Subsequent studies discover that the mechanism is the induction of ABCA1 and lipid efflux (141). In-depth research on the promoter region of ABCA1 and ABCG1 reveals that the LXR binding site is located in the promoter region of ABCA1 and ABCG1 in humans and mice. However, LXR agonists increase sterol regulatory element-binding protein 1c (SREBP-1c) in the liver, and significantly induce lipid synthesis-related genes including fatty acid synthase (FAS) and acetyl-coenzyme A carboxylase (ACC), resulting in hypertriglyceridemia (142, 143). Selective LXR agonists are developed such as N, N-dimethyl-3-hydroxy-cholenamide (DMHCA), and WAY-252623. These compounds can promote cholesterol efflux from macrophages, and at the same time harm the liver (144). PPARα and PPARγ agonists can significantly induce the level of ABCA1 and cholesterol efflux (141). Mechanism studies reveals that this effect of PPARα and PPARγ agonists is achieved by promoting the expression of LXRα, and further by increasing the expression of LXRα to promote the expression of ABCA1 (145).
Non-coding RNAs
Different from transcriptional regulation, post-transcriptional regulation focuses on ncRNAs. The past decade has brought a greater understanding of ncRNAs in the regulation of foam cells in atherosclerosis. The function of ncRNAs on the progression of atherosclerosis has been explored (146, 147). NcRNAs are classified as small ncRNAs and long ncRNAs. Small ncRNAs (< 200 bp) include microRNA (miR, miRNA), siRNAs (small interfering RNAs), and piRNAs (PIWI-interacting RNA). ncRNAs, longer than > 200 bp, are called lncRNAs.
MicroRNA (miR, or miRNA) comprise a family of ncRNAs with 22 nucleotides. They can target a 3’ untranslated region of multiple RNAs to induce the degradation of target genes (148, 149). The target gene and the function of miRNAs in the regulation of foam cell formation are listed in Table 1. Single miRNA can be involved in different biological processes. MiRNA mimics or antagonism can also be targeted by different miRNAs, which is a limitation for its application (150).
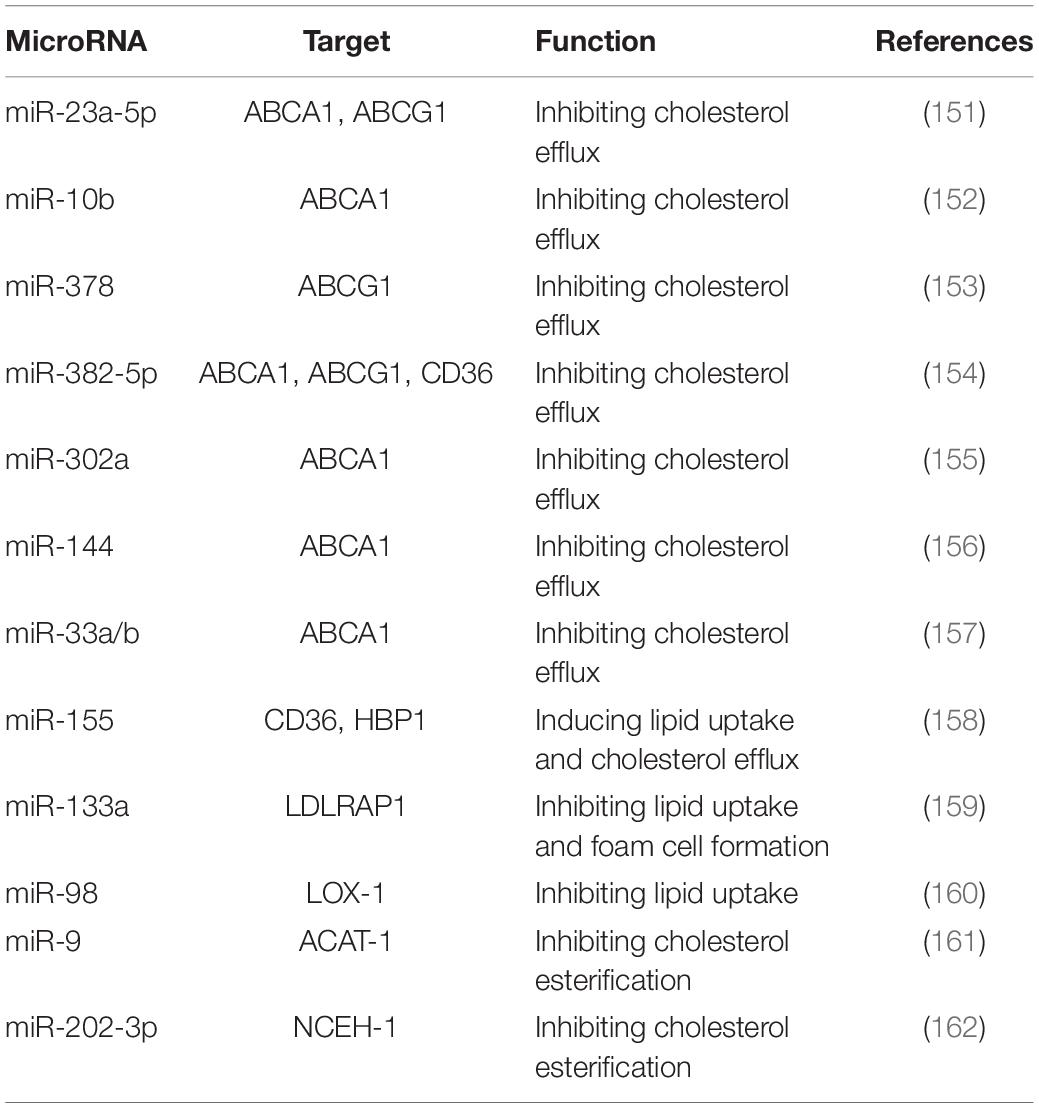
Table 1. MicroRNAs in the regulation of foam cells and lipid accumulation.
LncRNAs have a size over 200 bp and they can function in both cis and trans regulation of target genes (163, 164). The target gene and the function of lncRNAs in the regulation of foam cell formation are listed in Table 2.
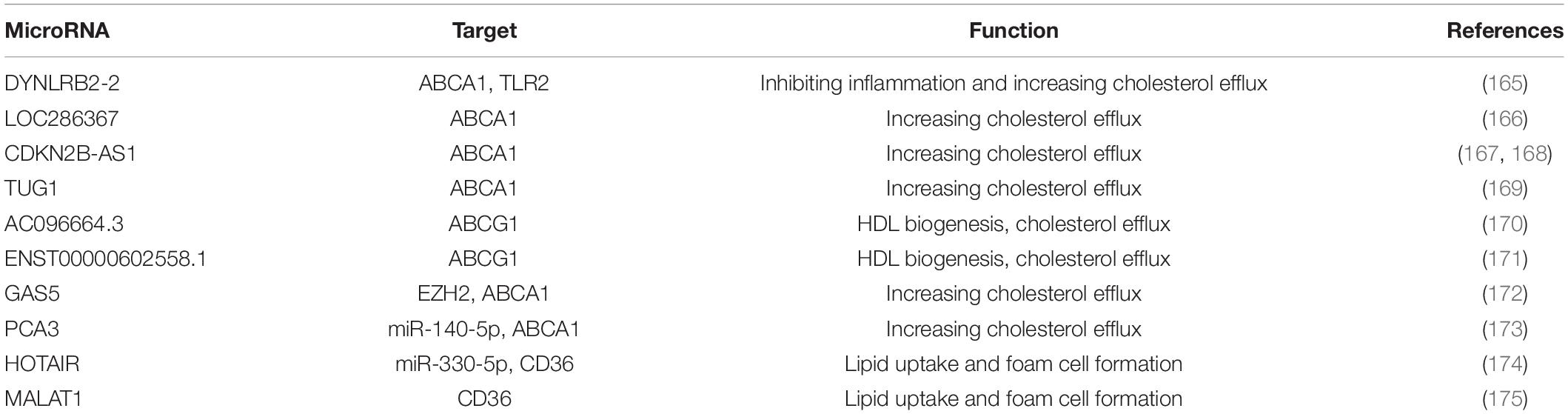
Table 2. LncRNAs in the regulation of foam cells and lipid accumulation.
NcRNAs have important roles in the regulation of foam cells. Targeting ncRNA is an attractive strategy of therapeutic approaches (8). Antisense oligonucleotides (ASO) target the mature miRNA and lead to the specific miRNA for degradation. Adeno-associated virus (AAV) has been explored to deliver miRNA to a specific organ. Multiple strategies including liposomes, nanoparticles have been explored and developed for the delivery of miRNAs. Furthermore, the stability of miRNA mimics and inhibitors are sufficient to maintain in plasma and can be delivered easily to the gene targets. However, it is challenging for viral and non-viral approaches to deliver miRNA mimics and inhibitors to a specific cell line (176, 177). Although preliminary studies show a positive effect on targeting cells and their gene expression (146, 148), more efforts on delivery systems are needed for clinical trials. Among all of the ncRNAs, miRNA-33 has been studied extensively. The decrease of efflux of cholesterol causes lipid accumulation in cells and accelerates the formation of foam cells. Studies have shown that miRNA-33 can inhibit the expression of ABCA1 and ABCG1, thereby inhibiting cholesterol efflux (178). Antisense oligonucleotides targeting miRNA-33 can effectively boost cholesterol efflux and alleviate the lipid accumulation in macrophage-foam cells via increasing the level of ABCA1 and ABCG1 and finally attenuating the atherosclerotic plaques in LDLR–/– mice. Administrations of miRNA-33 ASO for 12 weeks in non-human primates can effectively increase the expression of ABCA1, and further induce the concentration of HDL and reduce the concentration of VLDL triglycerides (179, 180). Clinical trials targeting miRNA have been explored on heart failure and hepatitis C infection (181–183) but miRNAs targeting foam cell formation require clinical evaluation in the future.
Gut Microbiota
Gut microbiota is a population of microorganisms that colonizes the gastrointestinal tracts. There is large and compelling evidence that the microbiota is involved in atherosclerosis. Several studies focus on the impact of the microbiome-derived metabolites on atherosclerosis including trimethylamine-N-Oxide (TMAO), indoxyl sulfate, and short-chain fatty acid (SCFA). Multiple studies have evaluated the effect of microbiome-derived metabolites on foam cell formation. TMAO induces the level of scavenger receptors and inhibits cholesterol efflux in macrophages leading to the accumulation of foam cells (184). Besides, TMAO is also involved in the migration of macrophages to the plaque areas and induces the secretion of inflammatory cytokines including IL-6 and TNF-α (185). Furthermore, TMAO can also accelerate atherosclerosis by inducing pyroptosis via succinate dehydrogenase complex subunit B (SDHB)/ROS signaling pathway (186). Indoxyl sulfate, a byproduct of dietary nutrients, can enhance oxidative stress and inflammation, besides, it also reduces cholesterol efflux and induces foam cell formation (187). Toxic microbiota metabolites, p-cresol has been demonstrated to correlate with the lipid accumulation in macrophages increasing CVD risk (188). Administration of butyrate, a four-carbon SCFA, may alter the species of gut microbiota and induce the cholesterol efflux function in macrophages by the mechanism of up-regulating ABCA1 in high fat diet-fed mice. These results demonstrate the potential of butyrate against atherosclerosis development (189).
Apart from influencing the cholesterol uptake and efflux in macrophages, gut microbiota modulates the foam cells via the inflammatory pathway. Since reducing inflammation inhibits the development of atherosclerosis, studies have explored the impact of gut microbiota on inflammation caused by foam cells. SCFAs, including acetate, butyrate, and propionate, are produced by the gut microbiota (190). In the past decade, SCFAs have been playing a key role in the modulation of inflammatory processes of foam cells. In apoE knockout mice, the addition of butyrate in the diet reduces atherosclerosis and lowers the secretion of inflammatory cytokines. Colonic infusions of SCFA in humans (high acetate containing SCFA mixture) significantly alleviate the level of pro-inflammatory cytokine IL-1β (191). Similarly, colonic acetate infusion generates a lower level of TNF-α in humans (192).
Conclusion and Future Perspectives
Foam cells are the hallmark of the initiation of atherosclerosis. The cell origins of foam cells are not only from monocytes but also from VSMCs, SPCs, and endothelium cells. Novel technologies such as lineage tracing and scRNA-seq have revolutionized our understanding of subtypes of monocytes and VSMCs-derived foam cells. Three main clusters of the macrophages include resident-like, inflammatory, and TREM2hi. The progression of foam cells leads to diverse processes of programmed cell death including apoptosis, autophagy, necroptosis, and pyroptosis. The formation of foam cells is affected by cholesterol uptake, efflux, and esterification. Novel molecular mechanisms including nuclear receptors, ncRNAs, and gut microbiota are involved in the regulation of foam cells.
Foam cells are an attractive target for treating atherosclerosis. But therapy targeting foam cells is missing for the treatment of atherosclerosis. The reason lies in the inadequate understanding of foam cells. In this review, the novel insights into the origins, consequences, and regulation have been summarized extensively. Since clinical trials of ASOs targeting miRNA have been explored on heart failure and hepatitis C infection (181–183), therapies targeting non-coding RNAs are worth developing and validating in clinical studies.
Nevertheless, the goal of controlling foam cells faces several obstacles. Since the heterogeneity of monocytes and VSMCs, it is difficult to target a specific subtype for therapeutics (11). Second, conclusive results are missing on whether the basal and excessive levels of programmed cell death is beneficial or harmful for controlling the progression of foam cell (43). Therefore, further in-depth research and therapeutic exploration targeting foam cells are needed for the better management of atherosclerosis.
Author Contributions
YG collected the literature and wrote the manuscript. HZ reviewed the manuscript. RC conceived the idea and reviewed the manuscript. All authors read and approved the final manuscript for publication.
Funding
This work was supported by the National Natural Science Foundation of China (81903601 to YG and 81672260 to RC), the Shanghai Sailing Program (19YF1444900 to YG), and the internal funding from Shanghai Xuhui Central Hospital to YG.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
2. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. (2011) 473:317–25. doi: 10.1038/nature10146
3. Wang T, Palucci D, Law K, Yanagawa B, Yam J, Butany J. Atherosclerosis: pathogenesis and pathology. Diagnostic Histopathol. (2012) 18:461–7. doi: 10.1016/j.mpdhp.2012.09.004
4. Clarke MCH, Bennett MR. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
5. Aldons J. Lusis atherosclerosis. Nature. (2000) 407:233–41. doi: 10.1038/35025203.Atherosclerosis
6. Poznyak AV, Nikiforov NG, Starodubova AV, Popkova TV, Orekhov AN. Macrophages and foam cells: brief overview of their role, linkage, and targeting potential in atherosclerosis. Biomedicines. (2021) 9:1221. doi: 10.3390/biomedicines9091221
7. Fayad ZA, Swirski FK, Calcagno C, Robbins CS, Mulder W, Kovacic JC. Monocyte and macrophage dynamics in the cardiovascular system: JACC macrophage in CVD series (part 3). J Am Coll Cardiol. (2018) 72:2198–212. doi: 10.1016/j.jacc.2018.08.2150
8. Javadifar A, Rastgoo S, Banach M, Jamialahmadi T, Johnston TP, Sahebkar A. Foam cells as therapeutic targets in atherosclerosis with a focus on the regulatory roles of non-coding rnas. Int J Mol Sci. (2021) 22:1–27. doi: 10.3390/ijms22052529
9. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. (2012) 95:156–64. doi: 10.1093/cvr/cvs115
10. Tang J, Wang H, Huang X, Li F, Zhu H, Li Y, et al. Arterial Sca1+ vascular stem cells generate de novo smooth muscle for artery repair and regeneration. Cell Stem Cell. (2020) 26:81–96.e4. doi: 10.1016/j.stem.2019.11.010
11. Nagenborg J, Goossens P, Biessen EAL, Donners MMPC. Heterogeneity of atherosclerotic plaque macrophage origin, phenotype and functions: implications for treatment. Eur J Pharmacol. (2017) 816:14–24. doi: 10.1016/j.ejphar.2017.10.005
12. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
13. Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. (2020) 142:2060–75. doi: 10.1161/CIRCULATIONAHA.120.048378
14. Trzebanski S, Jung S. Plasticity of monocyte development and monocyte fates. Immunol Lett. (2020) 227:66–78. doi: 10.1016/j.imlet.2020.07.007
15. Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. (2008) 117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091
16. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/JCI28549
17. Yu XH, Qian K, Jiang N, Zheng XL, Cayabyab FS, Tang CK. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clin Chim Acta. (2014) 428:82–8. doi: 10.1016/j.cca.2013.11.010
18. Guo J, Li Y, Ren YH, Sun Z, Dong J, Yan H, et al. Mutant LRP6 impairs endothelial cell functions associated with familial normolipidemic coronary artery disease. Int J Mol Sci. (2016) 17:1173. doi: 10.3390/ijms17071173
19. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. (2010) 116:5–7. doi: 10.1182/blood-2010-02-258558
20. Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. (2003) 197:1701–7. doi: 10.1084/jem.20022156
21. Heine GH, Ulrich C, Seibert E, Seiler S, Marell J, Reichart B, et al. CD14++CD16+ monocytes but not total monocyte numbers predict cardiovascular events in dialysis patients. Kidney Int. (2008) 73:622–9. doi: 10.1038/sj.ki.5002744
22. Rogacev KS, Seiler S, Zawada AM, Reichart B, Herath E, Roth D, et al. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur Heart J. (2011) 32:84–92. doi: 10.1093/eurheartj/ehq371
23. Sunderkötter C, Nikolic T, Dillon MJ, van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. (2004) 172:4410–7. doi: 10.4049/jimmunol.172.7.4410
24. Saha P, Modarai B, Humphries J, Mattock K, Waltham M, Burnand KG, et al. The monocyte/macrophage as a therapeutic target in atherosclerosis. Curr Opin Pharmacol. (2009) 9:109–18. doi: 10.1016/j.coph.2008.12.017
25. Moroni F, Ammirati E, Norata GD, Magnoni M, Camici PG. The role of monocytes and macrophages in human atherosclerosis, plaque neoangiogenesis, and atherothrombosis. Mediators Inflamm. (2019) 2019:7434376. doi: 10.1155/2019/7434376
27. Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell Mol Life Sci. (2020) 77:1919–32. doi: 10.1007/s00018-019-03371-3
28. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophages review 2014 macrophage phenotypes in atherosclerosis. Immunol Rev. (2014) 262:153–66. doi: 10.1111/imr.12218
29. Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell. (2016) 165:780–91. doi: 10.1016/j.cell.2016.04.019
30. Guo L, Akahori H, Harari E, Smith SL, Polavarapu R, Karmali V, et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J Clin Invest. (2018) 128:1106–24. doi: 10.1172/JCI93025
31. Pourcet B, Staels B. Alternative macrophages in atherosclerosis: not always protective! J Clin Invest. (2018) 128:910–2. doi: 10.1172/JCI120123
32. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127:402–26. doi: 10.1161/CIRCRESAHA.120.316903
33. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
34. Willemsen L, de Winther MPJ. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. (2020) 250:705–14. doi: 10.1002/path.5392
35. Fernandez DM, Giannarelli C. Immune cell profiling in atherosclerosis: role in research and precision medicine. Nat Rev Cardiol. (2022) 19:43–58. doi: 10.1038/s41569-021-00589-2
36. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. (2018) 123:1127–42. doi: 10.1161/CIRCRESAHA.118.312804
37. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
38. Davies JD, Carpenter KLH, Challis IR, Figg NL, McNair R, Proudfoot D, et al. Adipocytic differentiation and liver X receptor pathways regulate the accumulation of triacylglycerols in human vascular smooth muscle cells. J Biol Chem. (2005) 280:3911–9. doi: 10.1074/jbc.M410075200
39. Porse BT, Pedersen TÅ, Xu X, Lindberg B, Wewer UM, Friis-Hansen L, et al. E2F repression by C/EBPα is required for adipogenesis and granulopoiesis in vivo. Cell. (2001) 107:247–58. doi: 10.1016/S0092-8674(01)00516-5
40. Sekiguchi K, Kurabayashi M, Oyama Y, Aihara Y, Tanaka T, Sakamoto H, et al. Homeobox protein hex induces SMemb/nonmuscle myosin heavy chain-B gene expression through the caMP-responsive element. Circ Res. (2001) 88:52–8. doi: 10.1161/01.RES.88.1.52
41. Loebel C, Czekanska EM, Bruderer M, Salzmann G, Alini M, Stoddart MJ. In vitro osteogenic potential of human mesenchymal stem cells is predicted by Runx2/Sox9 ratio. Tissue Eng Part A. (2015) 21:115–23. doi: 10.1089/ten.tea.2014.0096
42. Komori T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. (2010) 339:189–95. doi: 10.1007/s00441-009-0832-8
43. Silvestre-Roig C, De Winther MP, Weber C, Daemen MJ, Lutgens E, Soehnlein O. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. (2014) 114:214–26. doi: 10.1161/CIRCRESAHA.114.302355
44. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Prim. (2019) 5:1–18. doi: 10.1038/s41572-019-0106-z
45. Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. (2013) 10:171–7. doi: 10.1038/nmeth.2332
46. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. (2014) 129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015
47. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. (2015) 21:628–37. doi: 10.1038/nm.3866
48. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
49. Farina FM, Hall IF, Serio S, Zani SM, Climent M, Salvarani N, et al. miR-128-3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ Res. (2020) 126:e120–35. doi: 10.1161/circresaha.120.316489
50. Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. (2015) 35:535–46. doi: 10.1161/ATVBAHA.114.304029
51. Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, et al. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. (2007) 101:792–801. doi: 10.1161/CIRCRESAHA.107.152736
52. Yoshida T, Gan Q, Shang Y, Owens GK. Platelet-derived growth factor-BB represses smooth muscle cell marker genes via changes in binding of MKL factors and histone deacetylases to their promoters. Am J Physiol Physiol. (2006) 292:C886–95. doi: 10.1152/ajpcell.00449.2006
53. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. (2014) 115:662–7. doi: 10.1161/CIRCRESAHA.115.304634
54. Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. (2019) 25:1280–9. doi: 10.1038/s41591-019-0512-5
55. Ma WF, Hodonsky CJ, Turner AW, Wong D, Song Y, Mosquera JV, et al. Enhanced single-cell RNA-seq workflow reveals coronary artery disease cellular cross-talk and candidate drug targets. Atherosclerosis. (2022) 340:12–22. doi: 10.1016/j.atherosclerosis.2021.11.025
56. Khetarpal SA, Qamar A, Bick AG, Fuster JJ, Kathiresan S, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential reshapes age-related CVD: JACC review topic of the week. J Am Coll Cardiol. (2019) 74:578–86. doi: 10.1016/j.jacc.2019.05.045
57. Psaltis PJ, Simari RD. Vascular wall progenitor cells in health and disease. Circ Res. (2015) 116:1392–412. doi: 10.1161/CIRCRESAHA.116.305368
58. Van Berlo JH, Molkentin JD. An emerging consensus on cardiac regeneration. Nat Med. (2014) 20:1386–93. doi: 10.1038/nm.3764
59. Murgai M, Ju W, Eason M, Kline J, Beury DW, Kaczanowska S, et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med. (2017) 23:1176–90. doi: 10.1038/nm.4400
60. Depuydt MAC, Prange KHM, Slenders L, Örd T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
61. Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell. (2019) 178:1344–61.e11. doi: 10.1016/j.cell.2019.08.004
62. Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, et al. A role for the apoptosis inhibitory factor AIM/Spα/Api6 in atherosclerosis development. Cell Metab. (2005) 1:201–13. doi: 10.1016/j.cmet.2005.02.002
63. Okura Y, Brink M, Itabe H, Scheidegger KJ, Kalangos A, Delafontaine P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation. (2000) 102:2680–6. doi: 10.1161/01.CIR.102.22.2680
64. Sanson M, Augé N, Vindis C, Muller C, Bando Y, Thiers JC, et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ Res. (2009) 104:328–36. doi: 10.1161/CIRCRESAHA.108.183749
65. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
66. Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. (2005) 25:174–9. doi: 10.1161/01.ATV.0000148548.47755.22
67. Thorp E, Li Y, Bao L, Yao PM, Kuriakose G, Rong J, et al. Brief report: Increased apoptosis in advanced atherosclerotic lesions of Apoe-/- mice lacking macrophage Bcl-2. Arterioscler Thromb Vasc Biol. (2009) 29:169–72. doi: 10.1161/ATVBAHA.108.176495
68. Yurdagul A, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31:518–33.e10. doi: 10.1016/j.cmet.2020.01.001
69. Kojima Y, Weissman IL, Leeper NJ. The role of efferocytosis in atherosclerosis. Circulation. (2017) 135:476–89. doi: 10.1161/CIRCULATIONAHA.116.025684
70. Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM, et al. Deletion of macrophage low-density lipoprotein receptor-related protein 1 (LRP1) accelerates atherosclerosis regression and increases C-C chemokine receptor type 7 (CCR7) expression in plaque macrophages. Circulation. (2018) 138:1850–63. doi: 10.1161/CIRCULATIONAHA.117.031702
71. Doran AC, Tall AR, Tabas I, Doran AC, Ozcan L, Cai B, et al. CAMKII g suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis find the latest version : CAMKII γ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Investig. (2017) 127:4075–89. doi: 10.1172/JCI94735
72. Tao W, Yurdagul A, Kong N, Li W, Wang X, Doran AC, et al. SiRNA nanoparticles targeting CaMKIIγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med. (2020) 12:eaay1063. doi: 10.1126/SCITRANSLMED.AAY1063
73. Baardman J, Verberk SGS, Van Der Velden S, Gijbels MJJ, Van Roomen CPPA, Sluimer JC, et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat Commun. (2020) 11:6296. doi: 10.1038/s41467-020-20141-z
74. Schober A, Blay RM, Saboor Maleki S, Zahedi F, Winklmaier AE, Kakar MY, et al. MicroRNA-21 controls circadian regulation of apoptosis in atherosclerotic lesions. Circulation. (2021) 144:1059–73. doi: 10.1161/CIRCULATIONAHA.120.051614
75. Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr Biol. (2001) 11:795–805. doi: 10.1016/S0960-9822(01)00474-2
76. Xu H, Jiang J, Chen W, Li W, Chen Z, Medbury H. Vascular macrophages in atherosclerosis. J Immunol Res. (2019) 2019:4354786. doi: 10.1155/2019/4354786
77. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
78. Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta Mol Cell Res. (2009) 1793:664–73. doi: 10.1016/j.bbamcr.2008.07.014
79. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. (2018) 14:207–15. doi: 10.1080/15548627.2017.1378838
80. Kumar S, Nanduri R, Bhagyaraj E, Kalra R, Ahuja N, Chacko AP, et al. Vitamin D3-VDR-PTPN6 axis mediated autophagy contributes to the inhibition of macrophage foam cell formation. Autophagy. (2021) 17:2273–89. doi: 10.1080/15548627.2020.1822088
81. Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Hear Circ Physiol. (2013) 305:H459–76. doi: 10.1152/ajpheart.00936.2012
82. De Meyer GRY, Grootaert MOJ, Michiels CF, Kurdi A, Schrijvers DM, Martinet W. Autophagy in vascular disease. Circ Res. (2015) 116:468–79. doi: 10.1161/CIRCRESAHA.116.303804
83. Grootaert MOJ, Roth L, Schrijvers DM, De Meyer GRY, Martinet W. Defective autophagy in atherosclerosis: to die or to senesce? Oxid Med Cell Longev. (2018) 2018:7687083. doi: 10.1155/2018/7687083
84. Pi S, Mao L, Chen J, Shi H, Liu Y, Guo X, et al. The P2RY12 receptor promotes VSMC-derived foam cell formation by inhibiting autophagy in advanced atherosclerosis. Autophagy. (2020) 17:980–1000. doi: 10.1080/15548627.2020.1741202
85. Fang S, Wan X, Zou X, Sun S, Hao X, Liang C, et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. (2021) 12:1–18. doi: 10.1038/s41419-020-03357-1
86. Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. (2011) 13:655–67. doi: 10.1016/j.cmet.2011.03.023
87. Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. (2012) 15:545–53. doi: 10.1016/j.cmet.2012.01.022
88. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
89. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. (2013) 14:727–36. doi: 10.1038/nrm3683
90. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. (2016) 352:aaf2154. doi: 10.1126/science.aaf2154
91. Khan N, Lawlor KE, Murphy JM, Vince JE. More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr Opin Immunol. (2014) 26:76–89. doi: 10.1016/j.coi.2013.10.017
92. Martinet W, Schrijvers DM, Herman AG, De Meyer GRY. z-VAD-fmk-induced non-apoptotic cell death of macrophages: possibilities and limitations for atherosclerotic plaque stabilization. Autophagy. (2006) 2:312–4. doi: 10.4161/auto.2966
93. Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov. (2020) 19:553–71. doi: 10.1038/s41573-020-0071-y
94. Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci. (2020) 41:209–24. doi: 10.1016/j.tips.2020.01.002
95. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. doi: 10.1038/nrm.2016.149
96. Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. (2016) 2:e1600224. doi: 10.1126/sciadv.1600224
97. Kim SK, Seo G, Oh E, Jin SH, Chae GT, Lee SB. Palmitate induces RIP1-dependent necrosis in RAW 264.7 cells. Atherosclerosis. (2012) 225:315–21. doi: 10.1016/j.atherosclerosis.2012.09.021
98. Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat Rev Immunol. (2014) 14:601–18. doi: 10.1038/nri3720
99. Madrigal AG, Barth K, Papadopoulos G, Genco CA. Pathogen-mediated proteolysis of the cell death regulator RIPK1 and the host defense modulator RIPK2 in human aortic endothelial cells. PLoS Pathog. (2012) 8:e1002723. doi: 10.1371/journal.ppat.1002723
100. Coornaert I, Puylaert P, Marcasolli G, Grootaert MOJ, Vandenabeele P, De Meyer GRY, et al. Impact of myeloid RIPK1 gene deletion on atherogenesis in ApoE-deficient mice. Atherosclerosis. (2021) 322:51–60. doi: 10.1016/j.atherosclerosis.2021.02.021
101. Zhang Y, Li H, Huang Y, Chen H, Rao H, Yang G, et al. Stage-dependent impact of RIPK1 inhibition on atherogenesis: dual effects on inflammation and foam cell dynamics. Front Cardiovasc Med. (2021) 8:715337. doi: 10.3389/fcvm.2021.715337
102. Ren Y, Su Y, Sun L, He S, Meng L, Liao D, et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of receptor-interacting protein 1 (RIP1) for the treatment of systemic inflammatory response syndrome. J Med Chem. (2017) 60:972–86. doi: 10.1021/acs.jmedchem.6b01196
103. Rasheed A, Robichaud S, Nguyen MA, Geoffrion M, Wyatt H, Cottee ML, et al. Loss of MLKL (mixed lineage kinase domain-like protein) decreases necrotic core but increases macrophage lipid accumulation in atherosclerosis. Arterioscler Thromb Vasc Biol. (2020) 40:1155–67. doi: 10.1161/ATVBAHA.119.313640
104. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
105. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
106. Wang Q, Wu J, Zeng Y, Chen K, Wang C, Yang S, et al. Pyroptosis: a pro-inflammatory type of cell death in cardiovascular disease. Clin Chim Acta. (2020) 510:62–72. doi: 10.1016/j.cca.2020.06.044
107. Nogieć A, Bzowska M, Demczuk A, Varol C, Guzik K. Phenotype and response to PAMPs of human monocyte-derived foam cells obtained by long-term culture in the presence of oxLDLs. Front Immunol. (2020) 11:1592. doi: 10.3389/fimmu.2020.01592
108. Liu J, Wang C, Li J, Yu Y, Liu Y, Liu H, et al. Autophagy blockage promotes the pyroptosis of ox-LDL-treated macrophages by modulating the p62/Nrf2/ARE axis. J Physiol Biochem. (2021) 77:419–29. doi: 10.1007/s13105-021-00811-2
109. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
110. van Eijk M, Aerts JMFG. The unique phenotype of lipid-laden macrophages. Int J Mol Sci. (2021) 22:4039. doi: 10.3390/ijms22084039
111. Li J, Meng Q, Fu Y, Yu X, Ji T, Chao Y, et al. Novel insights: dynamic foam cells derived from the macrophage in atherosclerosis. J Cell Physiol. (2021) 236:6154–67. doi: 10.1002/jcp.30300
112. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
113. Ben J, Zhu X, Zhang H, Chen Q. Class A1 scavenger receptors in cardiovascular diseases. Br J Pharmacol. (2015) 172:5523–30. doi: 10.1111/bph.13105
114. Shen W, Azhar S, Kraemer FB. SR-B1 : a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol. (2018) 80:95–116. doi: 10.1146/annurev-physiol-021317-121550
115. Rao X, Zhong J, Maiseyeu A, Gopalakrishnan B, Villamena FA, Chen LC, et al. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ Res. (2014) 115:770–80. doi: 10.1161/CIRCRESAHA.115.304666
116. Zhao L, Varghese Z, Moorhead JF, Chen Y, Ruan XZ. CD36 and lipid metabolism in the evolution of atherosclerosis. Br Med Bull. (2018) 126:101–12. doi: 10.1093/bmb/ldy006
117. Shu H, Peng Y, Hang W, Nie J, Zhou N, Wang DW. The role of CD36 in cardiovascular disease. Cardiovasc Res. (2020) 118:115–29. doi: 10.1093/cvr/cvaa319
118. Pothineni NVK, Karathanasis SK, Ding Z, Arulandu A, Varughese KI, Mehta JL. LOX-1 in atherosclerosis and myocardial ischemia: biology, genetics, and modulation. J Am Coll Cardiol. (2017) 69:2759–68. doi: 10.1016/j.jacc.2017.04.010
119. Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. (2007) 100:1634–42. doi: 10.1161/CIRCRESAHA.107.149724
120. You Y, Bao WL, Zhang SL, Li HD, Li H, Dang WZ, et al. Sorting nexin 10 mediates metabolic reprogramming of macrophages in atherosclerosis through the lyn-dependent TFEB signaling pathway. Circ Res. (2020) 127:534–49. doi: 10.1161/CIRCRESAHA.119.315516
121. Luo Y, Duan H, Qian Y, Feng L, Wu Z, Wang F, et al. Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res. (2017) 27:352–72. doi: 10.1038/cr.2017.8
122. Fan J, Liu L, Liu Q, Cui Y, Yao B, Zhang M, et al. CKIP-1 limits foam cell formation and inhibits atherosclerosis by promoting degradation of Oct-1 by REGγ. Nat Commun. (2019) 10:1–14. doi: 10.1038/s41467-018-07895-3
123. Castaño D, Rattanasopa C, Monteiro-Cardoso VF, Corlianò M, Liu Y, Zhong S, et al. Lipid efflux mechanisms, relation to disease and potential therapeutic aspects. Adv Drug Deliv Rev. (2020) 159:54–93. doi: 10.1016/j.addr.2020.04.013
124. Jin X, Freeman SR, Vaisman B, Liu Y, Chang J, Varsano N, et al. ABCA1 contributes to macrophage deposition of extracellular cholesterol. J Lipid Res. (2015) 56:1720–6. doi: 10.1194/jlr.M060053
125. Josefs T, Basu D, Vaisar T, Arets B, Kanter JE, Huggins LA, et al. Atherosclerosis regression and cholesterol efflux in hypertriglyceridemic mice. Circ Res. (2021) 128:690–705. doi: 10.1161/CIRCRESAHA.120.317458
126. Pownall HJ, Rosales C, Gillard BK, Gotto AM. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat Rev Cardiol. (2021) 18:712–23. doi: 10.1038/s41569-021-00538-z
127. Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. (2007) 117:2216–24. doi: 10.1172/JCI32057
128. Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. (2007) 117:3900–8. doi: 10.1172/JCI33372
129. Lanthaler B, Steichen-Gersdorf E, Kollerits B, Zschocke J, Witsch-Baumgartner M. Maternal ABCA1 genotype is associated with severity of smith-lemli-opitz syndrome and with viability of patients homozygous for null mutations. Eur J Hum Genet. (2013) 21:286–93. doi: 10.1038/ejhg.2012.169
130. Aiello RJ, Brees D, Francone OL. ABCA1-deficient mice: insights into the role of monocyte lipid efflux in HDl formation and inflammation. Arterioscler Thromb Vasc Biol. (2003) 23:972–80. doi: 10.1161/01.ATV.0000054661.21499.FB
131. Akao H, Polisecki E, Schaefer EJ, Trompet S, Robertson M, Ford I, et al. ABCA1 gene variation and heart disease risk reduction in the elderly during pravastatin treatment. Atherosclerosis. (2014) 235:176–81. doi: 10.1016/j.atherosclerosis.2014.04.030
132. Fouladseresht H, Khazaee S, Javad Zibaeenezhad M, Hossein Nikoo M, Khosropanah S, Doroudchi M. Association of ABCA1 haplotypes with coronary artery disease. Lab Med. (2020) 51:157–68. doi: 10.1093/labmed/lmz031
133. Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport: basic mechanisms and their roles in vascular health and disease. Circ Res. (2019) 124:1505–18. doi: 10.1161/CIRCRESAHA.119.312617
134. Bloise E, Ortiga-Carvalho TM, Reis FM, Lye SJ, Gibb W, Matthews SG. ATP-binding cassette transporters in reproduction: a new frontier. Hum Reprod Update. (2016) 22:164–81. doi: 10.1093/humupd/dmv049
135. Frambach SJCM, de Haas R, Smeitink JAM, Rongen GA, Russel FGM, Schirris TJJ. Brothers in arms: ABCA1-and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatments. Pharmacol Rev. (2020) 72:152–90. doi: 10.1124/pr.119.017897
136. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. (2018) 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
137. Chang TY, Li BL, Chang CCY, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab. (2009) 297:1–9. doi: 10.1152/ajpendo.90926.2008
138. Rong JX, Blachford C, Feig JE, Bander I, Mayne J, Kusunoki J, et al. ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler Thromb Vasc Biol. (2013) 33:4–12. doi: 10.1161/ATVBAHA.112.252056
139. Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta. (2013) 424:245–52. doi: 10.1016/j.cca.2013.06.006
140. Chen X, Zhao Y, Guo Z, Zhou L, Okoro EU, Yang H. Transcriptional regulation of ATP-binding cassette transporter A1 expression by a novel signaling pathway. J Biol Chem. (2011) 286:8917–23. doi: 10.1074/jbc.M110.214429
141. Chawla A, Boisvert WA, Lee C-H, Laffitte BA, Barak Y, Joseph SB, et al. A PPARγ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. (2001) 7:161–71. doi: 10.1016/s1097-2765(01)00164-2
142. Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem. (2000) 275:28240–5. doi: 10.1074/jbc.M003337200
143. Zeng L, Liao H, Liu Y, Lee TS, Zhu M, Wang X, et al. Sterol-responsive Element-binding protein (SREBP) 2 down-regulates ATP-binding cassette transporter A1 in vascular endothelial cells: a novel role of SREBP in regulating cholesterol metabolism. J Biol Chem. (2004) 279:48801–7. doi: 10.1074/jbc.M407817200
144. Buñay J, Fouache A, Trousson A, de Joussineau C, Bouchareb E, Zhu Z, et al. Screening for liver X receptor modulators: where are we and for what use? Br J Pharmacol. (2021) 178:3277–93. doi: 10.1111/bph.15286
145. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. (2001) 7:53–8. doi: 10.1038/83348
146. Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding rnas in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. (2016) 96:1297–325. doi: 10.1152/physrev.00041.2015
147. Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigó R, Johnson R. Towards a complete map of the human long non-coding RNA transcriptome. Physiol Behav. (2017) 176:100–6. doi: 10.1038/s41576-018-0017-y.Towards
148. Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. (2006) 25:6163–9. doi: 10.1038/sj.onc.1209909
149. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. (2013) 152:1298–307. doi: 10.1016/j.cell.2013.02.012
150. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. (2004) 23:4051–60. doi: 10.1038/sj.emboj.7600385
151. Yang S, Ye ZM, Chen S, Luo XY, Chen SL, Mao L, et al. MicroRNA-23a-5p promotes atherosclerotic plaque progression and vulnerability by repressing ATP-binding cassette transporter A1/G1 in macrophages. J Mol Cell Cardiol. (2018) 123:139–49. doi: 10.1016/j.yjmcc.2018.09.004
152. Wang D, Wang W, Lin W, Yang W, Zhang P, Chen M, et al. Apoptotic cell induction of miR-10b in macrophages contributes to advanced atherosclerosis progression in ApoE-/- mice. Cardiovasc Res. (2018) 114:1794–805. doi: 10.1093/cvr/cvy132/5025268
153. Wang D, Yan X, Xia M, Yang Y, Li D, Li X, et al. Coenzyme Q10 promotes macrophage cholesterol efflux by regulation of the activator protein-1/miR-378/ATP-binding cassette transporter G1-signaling pathway. Arterioscler Thromb Vasc Biol. (2014) 34:1860–70. doi: 10.1161/ATVBAHA.113.302879
154. Hu YW, Zhao JY, Li SF, Huang JL, Qiu YR, Ma X, et al. RP5-833A20.1/miR-382-5p/NFIA-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis and inflammatory reaction. Arterioscler Thromb Vasc Biol. (2015) 35:87–101. doi: 10.1161/ATVBAHA.114.304296
155. Meiler S, Baumer Y, Toulmin E, Seng K, Boisvert WA. MicroRNA 302a is a novel modulator of cholesterol homeostasis and atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:323–31. doi: 10.1161/ATVBAHA.114.304878
156. Ramírez CM, Rotllan N, Vlassov AV, Dávalos A, Li M, Goedeke L, et al. Control of cholesterol metabolism and plasma high-density lipoprotein levels by microRNA-144. Circ Res. (2013) 112:1592–601. doi: 10.1161/CIRCRESAHA.112.300626
157. Price NL, Rotllan N, Zhang X, Canfrán-Duque A, Nottoli T, Suarez Y, et al. Specific disruption of Abca1 targeting largely mimics the effects of miR-33 knockout on macrophage cholesterol efflux and atherosclerotic plaque development. Circ Res. (2019) 124:874–80. doi: 10.1161/CIRCRESAHA.118.314415
158. Tian FJ, An LN, Wang GK, Zhu JQ, Li Q, Zhang YY, et al. Elevated microRNA-155 promotes foam cell formation by targeting HBP1 in atherogenesis. Cardiovasc Res. (2014) 103:100–10. doi: 10.1093/cvr/cvu070
159. Gabunia K, Herman AB, Ray M, Kelemen SE, England RN, DeLa Cadena R, et al. Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J Mol Cell Cardiol. (2017) 105:38–48. doi: 10.1016/j.yjmcc.2017.02.005
160. Dai Y, Wu X, Dai D, Li J, Mehta JL. MicroRNA-98 regulates foam cell formation and lipid accumulation through repression of LOX-1. Redox Biol. (2018) 16:255–62. doi: 10.1016/j.redox.2018.03.003
161. Xu J, Hu G, Lu M, Xiong Y, Li Q, Chang CCY, et al. MiR-9 reduces human acyl-coenzyme A: cholesterol acyltransferase-1 to decrease THP-1 macrophage-derived foam cell formation. Acta Biochim Biophys Sin (Shanghai). (2013) 45:953–62. doi: 10.1093/abbs/gmt096
162. Li L, Wu F, Xie Y, Xu W, Xiong G, Xu Y, et al. MiR-202-3p inhibits foam cell formation and is associated with coronary heart disease risk in a Chinese population. Int Heart J. (2020) 61:153–9. doi: 10.1536/ihj.19-033
163. Cech TR, Steitz JA. The noncoding RNA revolution – trashing old rules to forge new ones. Cell. (2014) 157:77–94. doi: 10.1016/j.cell.2014.03.008
164. Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. (2014) 15:7–21. doi: 10.1038/nrg3606
165. Li Y, Sun T, Shen S, Wang L, Yan J. LncRNA DYNLRB2-2 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. Biol Chem. (2019) 400:1047–57. doi: 10.1515/hsz-2018-0461
166. Ma X, Wang T, Zhao ZL, Jiang Y, Ye S. Propofol suppresses proinflammatory cytokine production by increasing ABCA1 expression via mediation by the long noncoding RNA LOC286367. Mediators Inflamm. (2018) 2018:8907143. doi: 10.1155/2018/8907143
167. Li H, Han S, Sun Q, Yao Y, Li S, Yuan C, et al. Long non-coding RNA CDKN2B-AS1 reduces inflammatory response and promotes cholesterol efflux in atherosclerosis by inhibiting ADAM10 expression. Aging (Albany NY). (2019) 11:1695–715. doi: 10.18632/aging.101863
168. Ou M, Li X, Zhao S, Cui S, Tu J. Long non-coding RNA CDKN2B-AS1 contributes to atherosclerotic plaque formation by forming RNA-DNA triplex in the CDKN2B promoter. EBioMedicine. (2020) 55:102694. doi: 10.1016/j.ebiom.2020.102694
169. Yang L, Li T. LncRNA TUG1 regulates ApoM to promote atherosclerosis progression through miR-92a/FXR1 axis. J Cell Mol Med. (2020) 24:8836–48. doi: 10.1111/jcmm.15521
170. Xu BM, Xiao L, Kang CM, Ding L, Guo FX, Li P, et al. LncRNA AC096664.3/PPAR−γ/ABCG1−dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis. J Cell Biochem. (2018) 120:13775—-13782. doi: 10.1002/jcb.28650
171. Cai C, Zhu H, Ning X, Li L, Yang B, Chen S, et al. LncRNA ENST00000602558.1 regulates ABCG1 expression and cholesterol efflux from vascular smooth muscle cells through a p65-dependent pathway. Atherosclerosis. (2019) 285:31–9. doi: 10.1016/j.atherosclerosis.2019.04.204
172. Meng XD, Yao HH, Wang LM, Yu M, Shi S, Yuan ZX, et al. Knockdown of GAS5 inhibits atherosclerosis progression via reducing EZH2-mediated ABCA1 transcription in ApoE-/- mice. Mol Ther Nucleic Acids. (2020) 19:84–96. doi: 10.1016/j.omtn.2019.10.034
173. Zhao ZW, Zhang M, Liao LX, Zou J, Wang G, Wan XJ, et al. Long non-coding RNA PCA3 inhibits lipid accumulation and atherosclerosis through the miR-140-5p/RFX7/ABCA1 axis. Biochim Biophys Acta Mol Cell Biol Lipids. (2021) 1866:158904. doi: 10.1016/j.bbalip.2021.158904
174. Liu J, Huang GQ, Ke ZP. Silence of long intergenic noncoding RNA HOTAIR ameliorates oxidative stress and inflammation response in ox-LDL-treated human macrophages by upregulating miR-330-5p. J Cell Physiol. (2019) 234:5134–42. doi: 10.1002/jcp.27317
175. Huangfu N, Xu Z, Zheng W, Wang Y, Cheng J, Chen X. LncRNA MALAT1 regulates oxLDL-induced CD36 expression via activating β-catenin. Biochem Biophys Res Commun. (2018) 495:2111–7. doi: 10.1016/j.bbrc.2017.12.086
176. Xu S, Kamato D, Little PJ, Nakagawa S, Pelisek J, Jin ZG. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther. (2019) 196:15–43. doi: 10.1016/j.pharmthera.2018.11.003
177. Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigó R, Johnson R. Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet. (2018) 19:535–48. doi: 10.1038/s41576-018-0017-y
178. Mao M, Lei H, Liu Q, Chen Y, Zhao L, Li Q, et al. Effects of miR-33a-5P on ABCA1/G1-mediated cholesterol efflux under inflammatory stress in THP-1 macrophages. PLoS One. (2014) 9:e109722. doi: 10.1371/journal.pone.0109722
179. Afonso MS, Sharma M, Schlegel M, Van Solingen C, Koelwyn GJ, Shanley LC, et al. miR-33 silencing reprograms the immune cell landscape in atherosclerotic plaques. Circ Res. (2021) 128:1122–38. doi: 10.1161/CIRCRESAHA.120.317914
180. Rotllan N, Ramírez CM, Aryal B, Esau CC, Fernández-Hernando C. Therapeutic silencing of MicroRNA-33 inhibits the progression of atherosclerosis in Ldlr-/- mice – brief report. Arterioscler Thromb Vasc Biol. (2013) 33:1973–7. doi: 10.1161/ATVBAHA.113.301732
181. Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting MicroRNA. N Engl J Med. (2013) 368:1685–94. doi: 10.1056/nejmoa1209026
182. Täubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. (2021) 42:178–88. doi: 10.1093/eurheartj/ehaa898
183. Abplanalp WT, Fischer A, John D, Zeiher AM, Gosgnach W, Darville H, et al. Efficiency and target derepression of anti-miR-92a: results of a first in human study. Nucleic Acid Ther. (2020) 30:335–45. doi: 10.1089/nat.2020.0871
184. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med. (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
185. Geng J, Yang C, Wang B, Zhang X, Hu T, Gu Y, et al. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed Pharmacother. (2018) 97:941–7. doi: 10.1016/j.biopha.2017.11.016
186. Wu P, Chen JN, Chen JJ, Tao J, Wu SY, Xu GS, et al. Trimethylamine N-oxide promotes apoE-/- mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J Cell Physiol. (2020) 235:6582–91. doi: 10.1002/jcp.29518
187. Yamamoto S. Molecular mechanisms underlying uremic toxin-related systemic disorders in chronic kidney disease: focused on β 2 -microglobulin-related amyloidosis and indoxyl sulfate-induced atherosclerosis—Oshima award address 2016. Clin Exp Nephrol. (2019) 23:151–7. doi: 10.1007/s10157-018-1588-9
188. Chaves LD, Abyad S, Honan AM, Bryniarski MA, Mcskimming DI, Stahura CM, et al. Unconjugated p -cresol activates macrophage macropinocytosis leading to increased LDL uptake. J Clin Invest. (2021) 6:e144410. doi: 10.1172/jci.insight.144410
189. Du Y, Li X, Wang L, Su C, Hong B, Xi M, et al. Butyrate protects against high-fat diet-induced atherosclerosis via up-regulating ABCA1 expression in apolipoprotein E-deficiency mice. Br J Pharmacol. (2020) 177:1754–72. doi: 10.1111/bph.14933
190. Lee-rueckert M, Blanco-vaca F, Kovanen PT, Carles J. The role of the gut in reverse cholesterol transport – focus on the enterocyte. Prog Lipid Res. (2013) 52:317–28. doi: 10.1016/j.plipres.2013.04.003
191. Canfora EE, Van Der Beek CM, Jocken JWE, Goossens GH, Holst JJ, Olde Damink SWM, et al. Colonic infusions of short-chain fatty acid mixtures promote energy metabolism in overweight/obese men: a randomized crossover trial. Sci Rep. (2017) 7:1–12. doi: 10.1038/s41598-017-02546-x
Keywords: atherosclerosis, foam cell, programmed cell death, non-coding RNAs, gut microbiota
Citation: Gui Y, Zheng H and Cao RY (2022) Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 9:845942. doi: 10.3389/fcvm.2022.845942
Received: 30 December 2021; Accepted: 17 March 2022;
Published: 13 April 2022.
Edited by:
Pier Leopoldo Capecchi, University of Siena, ItalyReviewed by:
Owais Bhat, Virginia Commonwealth University, United StatesTimothy P. Fitzgibbons, University of Massachusetts Medical School, United States
Copyright © 2022 Gui, Zheng and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongchao Zheng, aGN6aGVuZ0BzY3JjLmFjLmNu; Richard Y. Cao, cnljYW9Ac2NyYy5hYy5jbg==