
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 10 March 2022
Sec. Cardiac Rhythmology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.840337
 Shaojie Chen1†
Shaojie Chen1† Yongping Lin1†
Yongping Lin1† Yue Zhu1†
Yue Zhu1† Le Geng2
Le Geng2 Chang Cui1
Chang Cui1 Zhaomin Li1
Zhaomin Li1 Hailei Liu1
Hailei Liu1 Hongwu Chen1
Hongwu Chen1 Weizhu Ju1*
Weizhu Ju1* Minglong Chen1*
Minglong Chen1*
PRKAG2 cardiomyopathy is a rare progressive disease characterized by increased ventricular wall thickness and preexcitation. Dysfunction of the protein 5′-AMP-activated protein kinase (AMPK) plays a decisive role in the progression of ventricular lesions. Although patients with the PRKAG2-R302Q mutation have a high incidence of atrial fibrillation (AF), the molecular mechanism contributing to the disease remains unclear. We carried out whole-genome sequencing with linkage analysis in three affected members of a family. Atrial samples were obtained from the proband via surgical intervention. Control atrium biopsies were obtained from patients with persistent AF. Pathological changes were analyzed using the hematoxylin and eosin (H&E), Masson, and periodic acid–Schiff (PAS) staining. The AMPK signaling pathway was investigated by western blot. A murine atrial cardiomyocyte cell line (HL-1) and human induced pluripotent stem derived atrial cardiomyocytes (hiPSC-ACMs) were transfected with an adenovirus carrying the same mutation. We used enzyme linked immunosorbent assay (ELISA) to determine the AMPK activity in HL-1 cells and hiPSC-ACMs overexpressing PRKAG2-R302Q. Pathological results showed a large quantity of glycogen accumulation and vacuolization in cardiomyocytes from the proband atrial tissue. Western blot analysis revealed that the AMPK activity was significantly downregulated compared with that of the controls. Furthermore, remarkable glycogen deposition and impairment of AMPK activity were reproduced in HL-1 cells overexpressing PRKAG2-R302Q. Taken together, PRKAG2-R302Q mutation directly impair atrial cardiomyocytes. PRKAG2-R302Q mutation lead to glycogen deposition and promote the growth of atrial lesions by disrupting the AMPK pathway.
The PRKAG2 gene was mapped to chromosome 7, encoding the γ2 subunit of AMP-activated protein kinase (AMPK) (1). AMPK occurs as a heterotrimeric kinase comprising 1 catalytic (α) and 2 regulatory (β and γ) subunits. The γ2 subunit, along with γ1 and γ3, has four individually conserved cystathionine β-synthase (CBS) domains, each of which can bind one molecule of AMP or ATP (2). Different ratios of AMP/ATP binding to the γ2 subunit result in different levels of AMPK activity. When activated, AMPK accelerates glycolysis and suppresses inflammatory and endoplasmic reticulum stress (3–5), exerting cardioprotective effects.
Cardiomyopathy caused by PRKAG2 gene mutation is called PRKAG2 cardiomyopathy. PRKAG2 cardiomyopathy is usually associated with ventricular hypertrophy, conduction disease, and progressive glycogen storage (6, 7). Familial ventricular preexcitation in PRKAG2 cardiomyopathy is thought to be associated with Mahaim fibers, which have atrioventricular node-like conduction properties (8). PRKAG2 mutations are autosomal dominant and tend to affect several family members of one pedigree. Increased activity of the insulin pathway, a widely accepted hypertrophic signal, was found to be responsible for cardiomyocyte hypertrophy caused by the PRKAG2 gene mutation (9).
Recently, a high incidence of atrial fibrillation was reported for patients with the PRKAG2-R302Q mutation (10). However, few of the molecular mechanisms involved in this process have been explored. The role of the AMPK pathway in PRKAG2 cardiomyopathy carrying the R302Q mutation remains controversial. Patient-specific induced pluripotent stem cell (iPSC)-derived cardiomyocytes have exhibited increased AMPK activities (11, 12). Acute adenoviral-mediated expression of the γ2 mutant (R302Q) in isolated cardiomyocytes resulted in a significant increase in AMPK activity (13). A transgenic mouse model demonstrated reduced AMPK activity (14). Although patient-specific iPSC-CMs harbor the same gene background as the host, these cells are immature compared to adult cardiomyocytes, including in terms of metabolic properties. Hence, pathological evidence from patient biopsies would be extremely useful for understanding the underlying mechanisms.
We clinically identified a Chinese pedigree with the PRKAG2 p. R302Q mutation. The mutation carriers showed histories of syncope, atrial tachycardia, embolic events and pacemaker implantation. We obtained atrial samples from the proband via surgical intervention and compared the histological and molecular characteristics to those of biopsies of AF patients undergoing cardiopulmonary bypass procedures. We verified the pathogenicity of the PRKAG2-R302Q mutation using two kinds of atrial muscle cells overexpressing PRKAG2-R302Q. Our results provide solid pathological evidence of atrial lesions caused by the PRKAG2-R302Q mutation, as well as evidence of an impaired AMPK protein activation mode in response to different stimuli, shedding light on potential therapeutic strategies for atrial cardiomyopathy.
Patient samples, including venous blood samples and left atrial appendage, were taken after obtaining informed consent. This study was approved by the Scientific Ethics committee of the First Affiliated Hospital of Nanjing Medical University (2014-SR-090). The detailed clinical assessments were included in the Supplementary Methods.
Peripheral blood samples were obtained from the peripheral venous blood of five family members (II-1, II-3, II-5, III-1, and III-2). DNA was extracted using a QIAamp DNA Blood Maxi Kit. A TruSeq™ Exome Enrichment Kit (Illumina, San Diego, CA, United States) was used to capture exomes. The DNA library was quantified using the Quant-iT™ PicoGreen® dsDNA Assay Kit (LIFE). Sanger sequencing was performed to validate the mutation.
The experimental details, including cell culture, transfection, staining, western blot, and ELISA are available in Supplementary Material.
The difference between the two groups was assessed using Student’s t-test. The statistical analysis was performed using SPSS Statistics 19.0 (IBM, Chicago, IL, United States). A p-value < 0.05 was considered statistically significant.
A 44-year-old patient from a family with suspected inherited cardiomyopathy was hospitalized for the presentation of palpitations and syncope. Atrial flutter and intermittent high-grade AV block (the longest RR was 11.275 s) were observed under 24-h Holter-ECG monitoring (Figure 1A). An electrocardiogram (ECG) of the proband simultaneously revealed typical atrial flutter (Supplementary Figure 1A). Echocardiography demonstrated that the proband’s atrium was enlarged [left atrium diameter (LAD): 39 mm (Figure 1B); right atrium diameter (RAD): 38 mm], whereas both the ejection fraction (EF) and left ventricular end-diastolic diameter (LVDd) were normal (Supplementary Table 1). The proband’s interventricular septum was hypertrophic (IVS: 18 mm, Figure 1C), but neither ST-segment change nor T-wave inversion was observed in the ECG. Considering that left atrial hemodynamics can be altered by atrial flutter, the left atrial appendage (LAA) was analyzed in detail using enhanced cardiac CT scans. The results indicated the presence of an LAA thrombus (Figure 1D). Considering the proband’s diseases and probable thrombus formation, radiofrequency ablation of atrial flutter was terminated. A permanent pacemaker was implanted instead. After implantation, a posteroanterior chest X-ray showed no evident abnormality in the size and shape of the ventricle (Figure 1E).
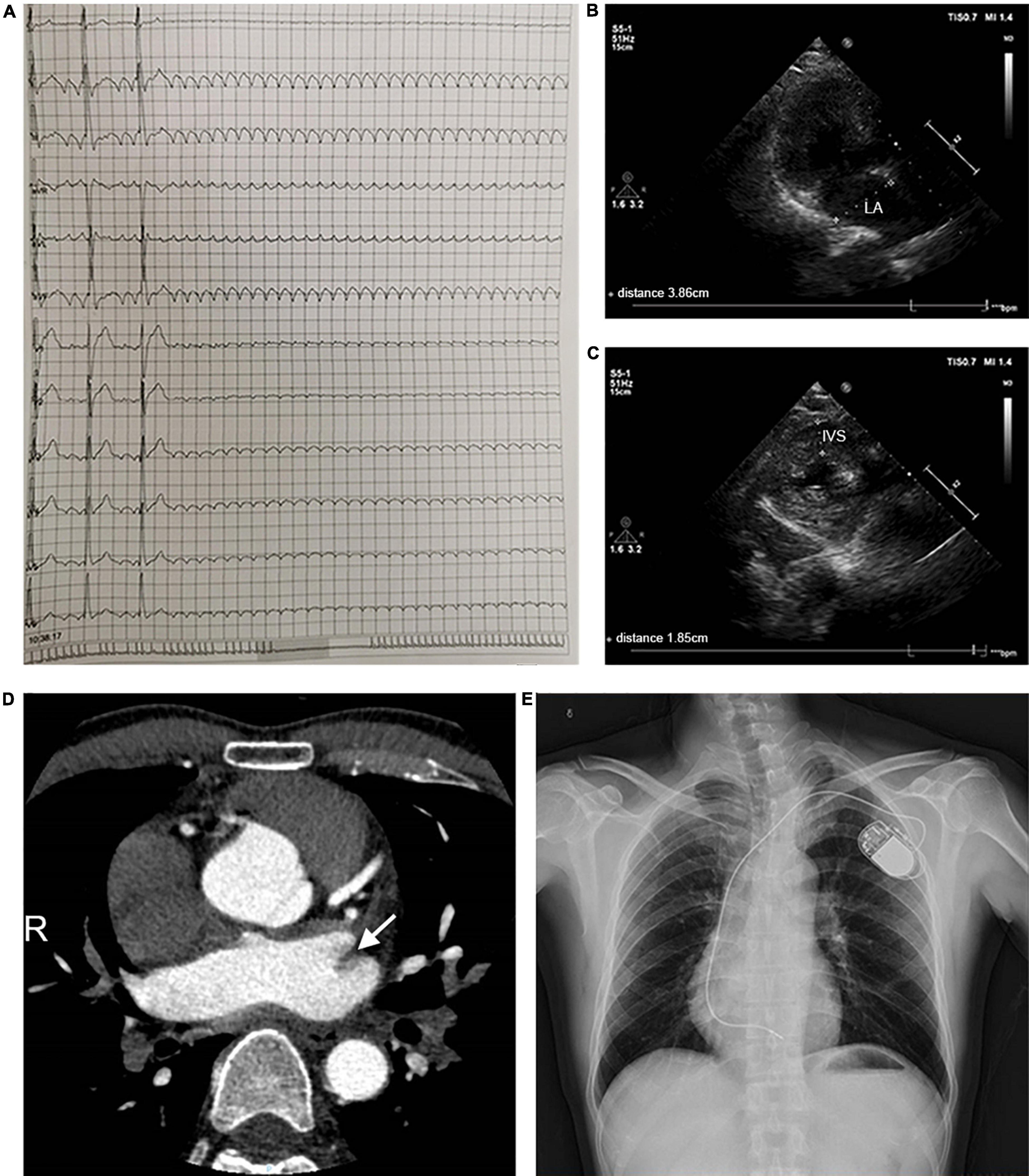
Figure 1. Clinical parameters for the pedigree. (A) 24-h Holter monitoring of the proband. (B,C) Echocardiographic parameters, including LAD and IVS. (D) The cardiac CT-scan indicated atrial thrombus (shown by the arrow). (E) A posteroanterior chest X-ray was taken immediately after pacemaker implantation. Abbreviations: LA, left atrium; IVS, interventricular septum.
A pedigree investigation demonstrated that family members I-1, II-3, and III-1, in addition to the proband (II-1), complained of a history of syncope (Figure 2A). II-3 was previously diagnosed with atrial flutter and hypertrophic cardiomyopathy (Supplementary Table 1) and was implanted with a pacemaker. The ECG data of III-1 showed sinus rhythm with slight, short PR intervals but did not meet the criteria for preexcitation. There was no evidence of structural heart defects in the echocardiogram of III-1 (Supplementary Table 1).
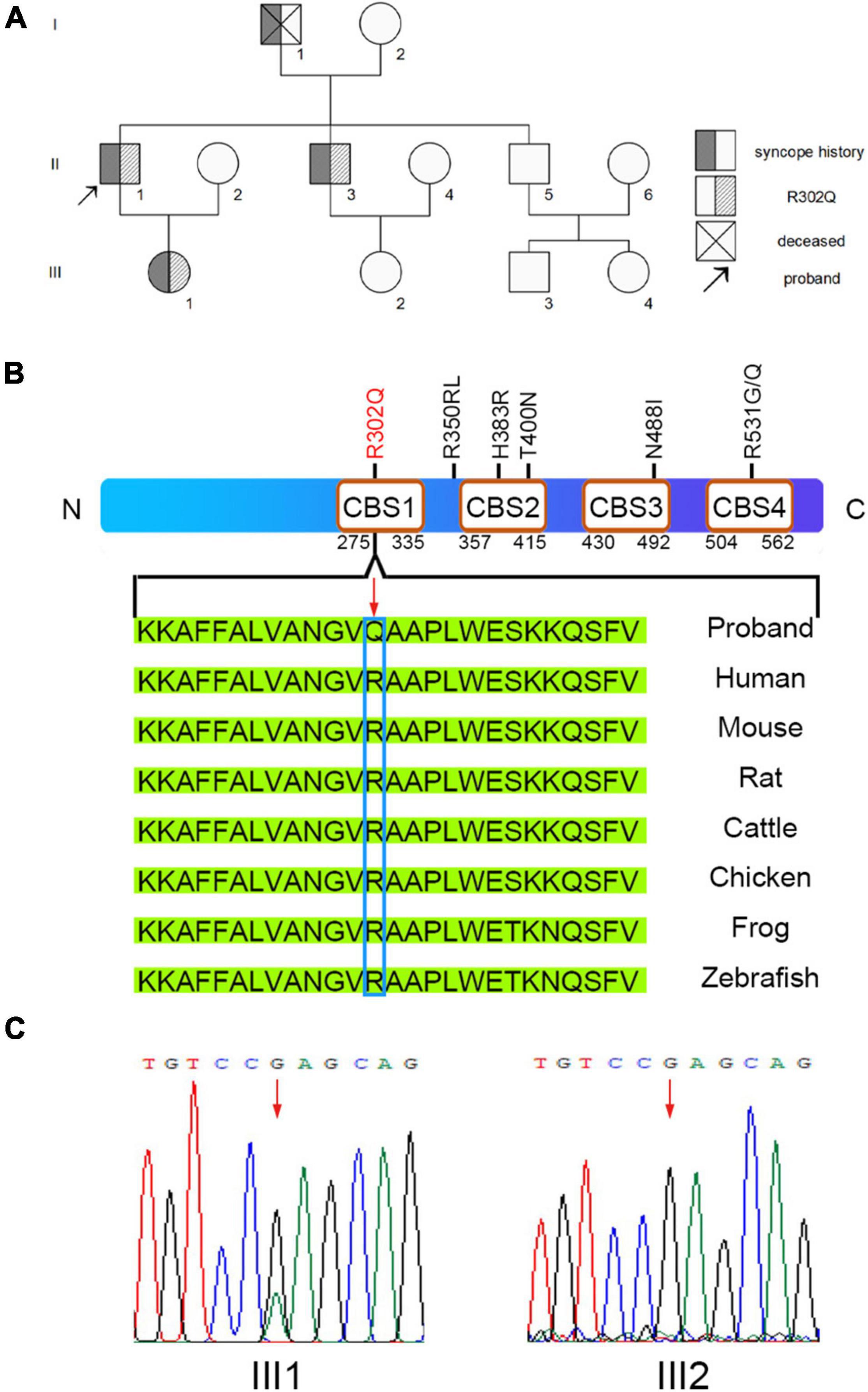
Figure 2. Sequence analysis for the pedigree. (A) Overview of the clinical symptoms for the pedigree. The proband is indicated by the black arrow. (B) The missense mutation PRKAG2-R302Q was found in the proband. (C) A heterozygous G > A transition at nucleotide 905 of PRKAG2 was confirmed by Sanger sequencing. A change in the amino acid sequence is highlighted by the red arrow.
We collected the peripheral blood of the family members for whole-exome sequencing (WES). WES analysis identified a heterozygous, missense mutation c.905G > A (R302Q) in the PRKAG2 genes of II-1 and II-3 (Figure 2B). Further Sanger sequencing confirmed that III-1 was a PRKAG2-R302Q mutation heterozygous carrier, whereas III-2 was a negative non-carrier (Figure 2C). Given that PRKAG2 syndrome is usually associated with early-onset ventricular preexcitation, a long-term follow-up of III-1 was recommended.
After the proband had received 4 months of anticoagulation therapy, the CT scan data still indicated a possible thrombus in the LAA (Supplementary Figure 1B). Thus, left atrial appendectomy was performed for stroke prevention, together with surgical ablation of the tricuspid isthmus. We then compared the proband’s samples to LAA tissues from two age-matched individuals with AF undergoing surgical interventions (Supplementary Figure 2 and Supplementary Table 1).
Pathological changes, including glycogen storage and fibrosis, are one of the specific manifestations of myocardial lesions caused by the PRKAG2-R302Q mutation. In Periodic Acid-Schiff (PAS) stained sections of the LAA of the proband, large numbers of glycogen granules were observed in the mutant cardiomyocytes, whereas there was no evidence of gross glycogen deposition in the controls (Figure 3A). Unlike the controls, intense vacuolization of cardiomyocytes from the proband was observed by the H&E staining (Figure 3B). This could be generated by the presence of glycogen deposits, which were not visible in the H&E staining. Prominent blue-stained collagen fibers from Masson’s staining were observed in all three samples (Figure 3C).
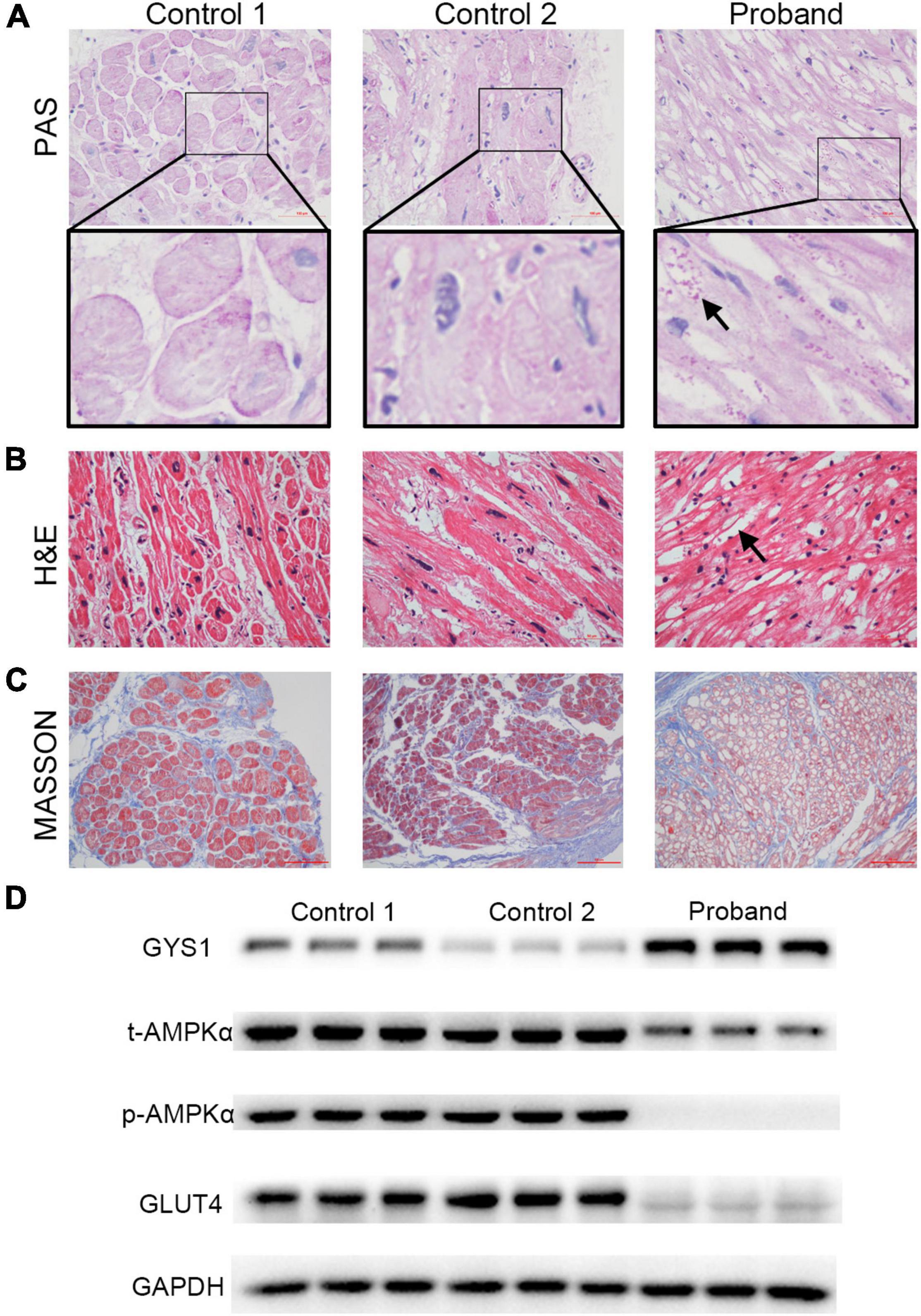
Figure 3. Alterations in the pathology and protein expression of the left atrial appendage tissues of the proband. (A) Representative photograph of PAS staining results. Many glycogen granules were observed (indicated by the black arrow, scale bar = 100 μm). (B) Representative images of H&E staining results. Distinct vacuolated myocytes (indicated by the black arrow) are visible (scale bar = 50 μm). (C) Observation of severe fibrosis in cardiac tissues by Masson staining (scale bar = 50 μm). (D) Western blotting of GYS1, t-AMPKα, p-AMPKα, GLUT4, and GAPDH. Abbreviations: GYS1, glycogen synthase 1; AMPK, 5′-AMP-activated protein kinase; GLUT4, glucose transporter 4.
To gain insights into the molecular basis for the PRKAG2-R302Q mutation, AMPK activity was assessed by western blotting (Figure 3D). Total AMPKα (t-AMPKα) was weakly expressed in the mutant atrial myocardium. Subsequently, phosphorylation of AMPKα (p-AMPKα) expression was barely detectable. Consistent with the PAS staining results, expression of robust glycogen synthase 1 (GYS1), a rate-limiting enzyme of the glycogen synthesis pathway, was observed in the proband’s atrial tissue, unlike in the controls, indicating enhanced glycogen synthase activity. By contrast, glucose transporter 4 (GLUT4), a widely recognized downstream protein for AMPK, was highly expressed in two controls.
The HL-1 cell line was transfected with an adenovirus carrying the PRKAG2-R302Q mutation. The results demonstrated that overexpressing the PRKAG2-R302Q mutation in HL-1 murine atrial cardiomyocytes resulted in PAS-positive glycogen deposition compared with the vector control (Figures 4A,B).
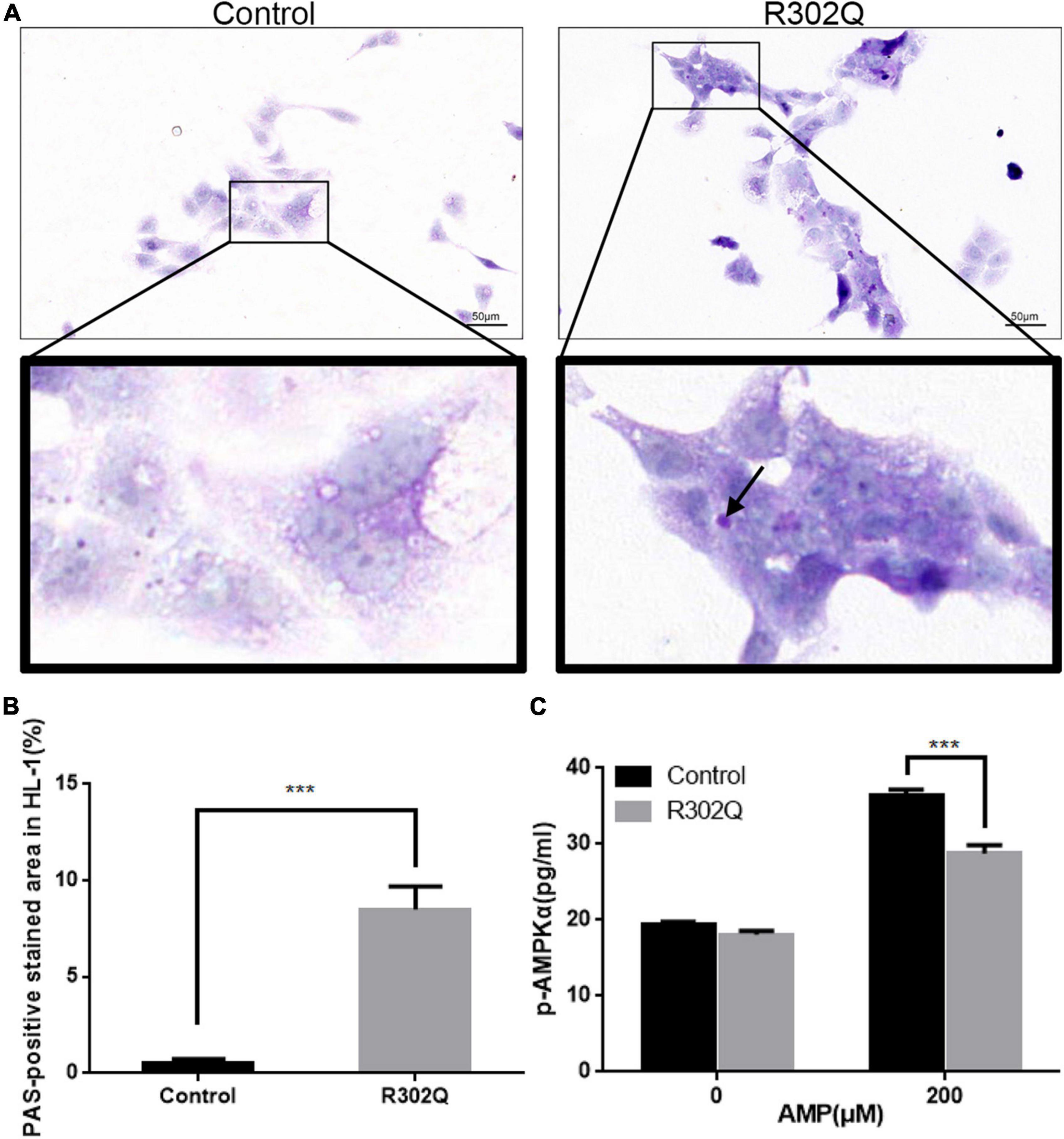
Figure 4. Molecular changes in HL-1 cells overexpressing PRKAG2-R302Q. (A) The glycogen content in HL-1 cells was analyzed by PAS staining (scale bar = 50 μm). (B) Quantitative analysis of the PAS-positive stained area in HL-1 cells. (C) The level of p-AMPKα expression was determined by ELISA (n = 3). All data were analyzed by the Student’s t–test (n = 3, ***p < 0.001).
A quantitative ELISA for the phosphorylation of AMPKα was used to evaluate AMPK activity under different conditions. At baseline, there were no differences in the p-AMPKα concentration of the mutant and control groups in HL-1 cells. Under stimulation with 200 μM AMP, both groups exhibited increased AMPK activity. However, the p-AMPKα concentration in the PRKAG2-R302Q group was considerably lower than that in the control group (Figure 4C). Moreover, hiPSC-ACMs overexpressing PRKAG2-R302Q replicated the similar outcomes (Supplementary Figure 5).
At the 3-year follow-up, the proband and II-3 underwent ECG and echocardiography examinations. The ECG showed that II-3 suffered from atrial flutter, whereas the proband maintained sinus rhythm (Supplementary Figure 3). However, atrial arrhythmia episodes were recorded in both patients using 24-h Holter-ECG monitoring (Supplementary Figure 4). Echocardiography suggested that both patients exhibited enlarged atriums and increased interventricular septum thicknesses (Supplementary Table 2).
Clinically, PRKAG2 mutations cause glycogen-storage cardiomyopathy, ventricular preexcitation, and conduction system degeneration (15, 16). Typical clinical symptoms can develop at approximately 40 years. In 70% of cases, ventricular preexcitation is the first symptom of PRKAG2 cardiomyopathy (17). To date, several mutation sites of the PRKAG2 gene have been confirmed to be pathogenic, among which R302Q is the most common mutation (18).
A study has suggested that patients from a pedigree carrying the PRKAG2-R302Q mutation all showed left atrial enlargement, but some patients were diagnosed with no or mild ventricular hypertrophy (10). Thus, atrial impairments might occur in the early stage of PRKAG2 cardiomyopathy. In our present study, the 44-year-old proband and his younger brother were diagnosed with atrial flutter and implanted with pacemakers. Masson and H&E staining revealed fibrosis and vacuolization of the proband’s atrial tissue, suggesting severe atrial damage.
The PRKAG2 gene encodes the γ2 subunit of AMPK. AMPK is highly expressed in human cardiac muscle and plays a vital role in regulating cellular energy homeostasis and metabolism (19). In a hypertrophic cardiomyopathy (HCM) mouse model expressing mutant α-MyHC, activation of AMPK was limited. As a result, lipid uptake and the cardiomyocyte content were reduced, and ventricular systolic function was impaired (20). After AMPK was effectively activated, the phenotype of ventricular dysfunction could be restored. Patient-specific iPSCs carrying PRKAG2 mutation-derived cardiomyocytes exhibited increased AMPK activities (11, 12). Acute adenoviral-mediated expression of the γ2 mutant (R302Q) in isolated cardiomyocytes resulted in a significant increase in AMPK activity (13). In this study, western blot analysis was performed on the atrial tissue of a proband carrying the PRKAG2-R302Q mutation, and both the p-AMPK/t-AMPK ratio and t-AMPK content decreased significantly. Our result was consistent with studies on a transgenic mouse model (14, 21). Overall, the PRKAG2-R302Q mutation can directly impair the atrium by disrupting AMPK signaling.
Each γ subunit of AMPK combine with four molecules of AMP and ATP. Thus, the higher the quantity of AMPK bound to AMP is, the higher the activation of AMPK is. Free AMP had a low content in the myocardium but a higher affinity for AMPK than ATP (22). However, an essential feature of the PRKAG2-R302Q mutation is the significantly reduced affinity between AMP and the γ2 subunit of AMPK, which prevents effective activation of AMPK. In the present study, AMPK activation was limited in the mutant group compared with the control group in response to high concentration of AMP. These results confirmed that AMPK activity was suppressed in patients with the PRKAG2-R302Q mutation in response to endogenous or exogenous stimuli. Thus, cardiac function could not be effectively preserved.
On the other hand, cardiac glucose uptake is primarily mediated by glucose transporter 4 (GLUT4), which is found to be regulated by AMPK. Previous studies confirmed that expression of GLUT4 was enhanced by upregulated AMPK activity (23, 24) and understandably limited by disrupted activation of AMPK. Moreover, GYS1 is identified as a key enzyme involved in glycogen synthesis. In a transgenic mice model harboring a knock-in mutation in GYS1 and mutation of the PRKAG2, the GYS1 activity was significantly reduced and glycogen accumulation was markedly eliminated (25). All these evidences suggested that glycogen deposits in PRKAG2 cardiomyopathy was due to enhanced expression of GYS1. In addition, a large accumulation of glycogen has been reported to cause preexcitation by damaging annulus fibrosis at the atrioventricular connection (25, 26). Using PAS staining, we also found a large accumulation of glycogen in the proband’s atrial tissue and HL-1 cells overexpressing PRKAG2-R302Q, which may also be a substrate of arrhythmia.
In the clinical view, the present study elucidated the mechanism of PRKAG2-R302Q mutation leading to atrial lesions. Clinicians were also reminded to pay attention to the possibility of early onset of atrial lesions in addition to ventricular lesions caused by PRKAG2 cardiomyopathy, and timely intervention treatment.
First, only one cell line was used in this study. However, HL-1 cell line, derived from mouse atrial myocytes, has been widely used to explore cardiac morphological, biochemical, and electrophysiological properties. The results are representative and stable. Moreover, hiPSC-ACM, though immature, overexpressing PRKAG2-R302Q mutation could well mimic the properties of atrial cardiomyocytes of the patients. Second, not all the family member underwent WES or Sanger sequencing. Their gene backgrounds need further identification. Third, under the condition of disrupted AMPK signaling due to PRKAG2-R302Q mutation, the mechanism of atrial myocytes being more vulnerable to stimuli needs to be further investigated.
In the present study, we obtained solid evidence that the PRKAG2-R302Q mutation directly caused atrial lesions. Impaired AMPK activation is the underlying pathogenic mechanism. These results are useful for the early diagnosis and intervention of patients with PRKAG2 cardiomyopathy.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SRR17257561; https://www.ncbi.nlm.nih.gov/, SRR17257560; and https://www.ncbi.nlm.nih.gov/, SRR17257559.
The studies involving human participants were reviewed and approved by the Bioethics Committee of the First Affiliated Hospital of Nanjing Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
SC, YL, and YZ performed the experiments, analyzed the data, and drafted the manuscript. LG contributed to the acquisition of human tissue samples. CC made contributions to the conception, design of the study, and revision of the manuscript. ZL, HL, and HC were involved in revising the manuscript. MC and WJ contributed to the guidance on the whole study. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (Grant Number 82070343 was awarded to MC, Grant Number 81870252 was awarded to WJ, and Grant Number 81900295 was awarded to CC) and the Natural Science Foundation of Jiangsu Province of China (Grant Number BK20181495 was awarded to WJ and Grant Number BK20191071 was awarded to CC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Qian Xuetian from Nanjing First Hospital for polishing the language.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.840337/full#supplementary-material
AMPK, AMP-activated protein kinase; CBS, cystathionine β -synthase; ECG, electrocardiogram; LAD, left atrium diameter; RAD, right atrium diameter; EF, ejection fraction; LVDd, left ventricular end-diastolic diameter; IVS, interventricular septum; LAA, left atrial appendage; WES, whole-exome sequencing; HCM, hypertrophic cardiomyopathy; iPSC, induced pluripotent stem cell; GYS1, glycogen synthase 1; GLUT4, glucose transporter 4.
1. Ramratnam M, Sharma RK, D’Auria S, Lee SJ, Wang D, Huang XY, et al. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J Am Heart Assoc. (2014) 3:e000899. doi: 10.1161/jaha.114.000899
2. Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. (2004) 113:274–84. doi: 10.1172/JCI19874
3. Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Investig. (2004) 114:495–503. doi: 10.1172/jci19297
4. Ma ZG, Yuan YP, Xu SC, Wei WY, Xu CR, Zhang X, et al. CTRP3 attenuates cardiac dysfunction, inflammation, oxidative stress and cell death in diabetic cardiomyopathy in rats. Diabetologia. (2017) 60:1126–37. doi: 10.1007/s00125-017-4232-4
5. Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. (2010) 121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928
6. Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, et al. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. (2001) 104:3030–3. doi: 10.1161/hc5001.102111
7. Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, et al. Identification of a gene responsible for familial Wolff-Parkinson-white syndrome. N Engl J Med. (2001) 344:1823–31. doi: 10.1056/NEJM200106143442403
8. Tan HL, van der Wal AC, Campian ME, Kruyswijk HH, ten Hove Jansen B, van Doorn DJ, et al. Nodoventricular accessory pathways in PRKAG2-dependent familial preexcitation syndrome reveal a disorder in cardiac development. Circ Arrhythm Electrophysiol. (2008) 1:276–81. doi: 10.1161/CIRCEP.108.782862
9. Hinson JT, Chopra A, Lowe A, Sheng CC, Gupta RM, Kuppusamy R, et al. Integrative analysis of PRKAG2 cardiomyopathy iPS and microtissue models identifies AMPK as a regulator of metabolism, survival, and fibrosis. Cell Rep. (2016) 17:3292–304. doi: 10.1016/j.celrep.2016.11.066
10. Hu J, Tang B, Wang J, Huang K, Wang Y, Lu S, et al. Familial atrial enlargement, conduction disorder and symmetric cardiac hypertrophy are early signs of PRKAG2 R302Q. Curr Med Sci. (2020) 40:486–92. doi: 10.1007/s11596-020-2207-z
11. Zhan Y, Sun X, Li B, Cai H, Xu C, Liang Q, et al. Establishment of a PRKAG2 cardiac syndrome disease model and mechanism study using human induced pluripotent stem cells. J Mol Cell Cardiol. (2018) 117:49–61. doi: 10.1016/j.yjmcc.2018.02.007
12. Ben Jehuda R, Eisen B, Shemer Y, Mekies LN, Szantai A, Reiter I, et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm. (2018) 15:267–76. doi: 10.1016/j.hrthm.2017.09.024
13. Folmes KD, Chan AY, Koonen DP, Pulinilkunnil TC, Baczko I, Hunter BE, et al. Distinct early signaling events resulting from the expression of the PRKAG2 R302Q mutant of AMPK contribute to increased myocardial glycogen. Circ Cardiovasc Genet. (2009) 2:457–66. doi: 10.1161/CIRCGENETICS.108.834564
14. Sidhu JS, Rajawat YS, Rami TG, Gollob MH, Wang Z, Yuan R, et al. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation. (2005) 111:21–9. doi: 10.1161/01.CIR.0000151291.32974.D5
15. Ahamed H, Balegadde AV, Menon S, Menon R, Ramachandran A, Mathew N, et al. Phenotypic expression and clinical outcomes in a South Asian PRKAG2 cardiomyopathy cohort. Sci Rep. (2020) 10:20610. doi: 10.1038/s41598-020-77124-9
16. Lopez-Sainz A, Dominguez F, Lopes LR, Ochoa JP, Barriales-Villa R, Climent V, et al. Clinical features and natural history of PRKAG2 variant cardiac glycogenosis. J Am Coll Cardiol. (2020) 76:186–97. doi: 10.1016/j.jacc.2020.05.029
17. Thevenon J, Laurent G, Ader F, Laforet P, Klug D, Duva Pentiah A, et al. High prevalence of arrhythmic and myocardial complications in patients with cardiac glycogenosis due to PRKAG2 mutations. Europace. (2017) 19:651–9. doi: 10.1093/europace/euw067
18. Porto AG, Brun F, Severini GM, Losurdo P, Fabris E, Taylor MRG, et al. Clinical Spectrum of PRKAG2 Syndrome. Circ Arrhythm Electrophysiol. (2016) 9:e003121. doi: 10.1161/CIRCEP.115.003121
19. Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. (2000) 346(Pt 3):659–69. doi: 10.1042/0264-6021:3460659
20. Magida JA, Leinwand LA. Metabolic crosstalk between the heart and liver impacts familial hypertrophic cardiomyopathy. EMBO Mol Med. (2014) 6:482–95. doi: 10.1002/emmm.201302852
21. Thorn SL, Gollob MH, Harper ME, Beanlands RS, Dekemp RA, Dasilva JN. Chronic AMPK activity dysregulation produces myocardial insulin resistance in the human Arg302Gln-PRKAG2 glycogen storage disease mouse model. EJNMMI Res. (2013) 3:48. doi: 10.1186/2191-219X-3-48
22. Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. (2007) 449:496–500. doi: 10.1038/nature06161
23. Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. (1999) 87:1990–5. doi: 10.1152/jappl.1999.87.5.1990
24. Ojuka EO, Nolte LA, Holloszy JO. Increased expression of GLUT-4 and hexokinase in rat epitrochlearis muscles exposed to AICAR in vitro. J Appl Physiol. (2000) 88:1072–5. doi: 10.1152/jappl.2000.88.3.1072
25. Kim M, Hunter RW, Garcia-Menendez L, Gong G, Yang YY, Kolwicz SC Jr, et al. Mutation in the γ2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res. (2014) 114:966–75. doi: 10.1161/circresaha.114.302364
Keywords: PRKAG2, pedigree, atrial arrhythmia, histology, glycogen deposition
Citation: Chen S, Lin Y, Zhu Y, Geng L, Cui C, Li Z, Liu H, Chen H, Ju W and Chen M (2022) Atrial Lesions in a Pedigree With PRKAG2 Cardiomyopathy: Involvement of Disrupted AMP-Activated Protein Kinase Signaling. Front. Cardiovasc. Med. 9:840337. doi: 10.3389/fcvm.2022.840337
Received: 22 December 2021; Accepted: 18 February 2022;
Published: 10 March 2022.
Edited by:
Hung-Fat Tse, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Mihaela Gherghiceanu, Carol Davila University of Medicine and Pharmacy, RomaniaCopyright © 2022 Chen, Lin, Zhu, Geng, Cui, Li, Liu, Chen, Ju and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weizhu Ju, anV3ZWl6aHVAMTI2LmNvbQ==; Minglong Chen, Y2hlbm1pbmdsb25nQG5qbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.