 Manrose Singh1Akito T. Nicol1Jaclyn DelPozzo1Jia Wei2Mandeep Singh1
Manrose Singh1Akito T. Nicol1Jaclyn DelPozzo1Jia Wei2Mandeep Singh1 Tony Nguyen1
Tony Nguyen1 Satoru Kobayashi1
Satoru Kobayashi1 Qiangrong Liang1*
Qiangrong Liang1*- 1Department of Biomedical Sciences, College of Osteopathic Medicine, New York Institute of Technology, Old Westbury, NY, United States
- 2Department of Cardiology, The Second Affiliated Hospital of Xi'an Jiaotong University, Xian, China
Doxorubicin (DOX) is an extremely effective and wide-spectrum anticancer drug, but its long-term use can lead to heart failure, which presents a serious problem to millions of cancer survivors who have been treated with DOX. Thus, identifying agents that can reduce DOX cardiotoxicity and concurrently enhance its antitumor efficacy would be of great clinical value. In this respect, the classical antidiabetic drug metformin (MET) has stood out, appearing to have both antitumor and cardioprotective properties. MET is proposed to achieve these beneficial effects through the activation of AMP-activated protein kinase (AMPK), an essential regulator of mitochondrial homeostasis and energy metabolism. AMPK itself has been shown to protect the heart and modulate tumor growth under certain conditions. However, the role and mechanism of the hypothesized MET-AMPK axis in DOX cardiotoxicity and antitumor efficacy remain to be firmly established by in vivo studies using tumor-bearing animal models and large-scale prospective clinical trials. This review summarizes currently available literature for or against a role of AMPK in MET-mediated protection against DOX cardiotoxicity. It also highlights the emerging evidence suggesting distinct roles of the AMPK subunit isoforms in mediating the functions of unique AMPK holoenzymes composed of different combinations of isoforms. Moreover, the review provides a perspective regarding future studies that may help fully elucidate the relationship between MET, AMPK and DOX cardiotoxicity.
Introduction
The anthracycline doxorubicin (DOX) has been widely used for over 5 decades and is a highly effective chemotherapeutic agent for the treatment of a broad spectrum of cancers including various solid tumors and leukemia. Unfortunately, DOX chemotherapy can cause severe cardiotoxic effects (1–3). Acute toxicity occurs immediately after treatment and is generally transient. Chronic cardiotoxicity is more serious and culminates in irreversible congestive heart failure. Currently, only the iron chelator dexrazoxane has been approved for limited clinical use for reducing DOX cardiotoxicity in certain pediatric or breast cancer patients (4–7). Given the continuing widespread use of DOX in cancer chemotherapies, it is imperative to identify new strategies that can protect against DOX cardiotoxicity without compromising the anti-tumor activity of DOX. Metformin (MET), a drug used for the first-line treatment of type 2 diabetes, has been suggested as such a dual-function agent that can simultaneously decrease DOX cardiotoxicity (8–11) and increase its anticancer activity (12, 13). The differential effects of MET on cardiomyocytes and cancer cells may be related to the differences in cellular energy metabolism. Cardiomyocytes are highly dependent on mitochondria for energy supply, while cancer cells primarily use glycolysis-generated ATP. Therefore, drugs such as MET that modulate mitochondrial function may have substantially different effects on the heart as compared to tumors. AMP-activated protein kinase (AMPK), a cellular energy sensor, is activated by MET and implicated in both cardioprotection and tumor growth. Most cell-based studies have suggested AMPK as a downstream effector of MET that functions to reduce DOX cardiotoxicity (9, 11, 14–17). However, the role of AMPK in cancer has been controversial (18). It remains uncertain whether and how AMPK affects the ability of MET to modulate DOX cardiotoxicity or tumor growth in vivo. This mini-review will extract evidence from currently available literature for or against a role of AMPK in MET-mediated protection against DOX cardiotoxicity. For the effects of MET and AMPK in antitumor therapies, the readers are referred to other review articles published elsewhere (18–24).
Dox Cardiotoxicity is a Serious Clinical Problem
Dox is an extremely effective and wide-spectrum antineoplastic drug that can lead to dose-dependent cardiotoxicity, culminating in heart failure (1–3). This presents a serious problem to millions of cancer survivors who have been treated with DOX. Indeed, the cardiovascular mortality in cancer survivors exceeds that caused by cancer per se (25). DOX cardiotoxicity is even more significant in childhood cancer since about half of all pediatric patients are treated with anthracyclines and many childhood cancer survivors go on to develop cardiac dysfunction (26–28). Due to the dose-dependent risk, the lifetime cumulative dose of DOX has been recommended not to exceed 450 mg/m2 per patient (1). Thus, DOX cardiotoxicity is a significant life-long health concern for cancer survivors.
Dox Induces Cardiotoxicity Via Multiple Mechanisms
Several mechanisms have been proposed to account for the ability of DOX to produce cardiotoxicity. DOX is concentrated in the mitochondria and its quinone moiety is reduced by the oxidoreductases to a semiquinone form which in turn donates its excess electron to O2, leading to the formation of reactive oxygen species (ROS) including superoxide anions (29, 30). Although the long-held ROS and oxidative stress theory of DOX cardiotoxicity is strongly supported by numerous animal studies (31–33), clinical trials have failed to demonstrate the efficacy of antioxidant supplements in reducing DOX-triggered cardiac injury (34, 35), suggesting that oxidative stress is not the only mechanism that mediates DOX cardiotoxicity. Interestingly, DOX has been shown to either bind with free iron (36) or cause mitochondrial iron accumulation in the heart (37), which may directly cause mitochondria-dependent ferroptosis or produce additional ROS intensifying the oxidative stress (38). The contribution of iron to DOX cardiotoxicity is demonstrated by the ability of the iron chelator dexrazoxane to attenuate DOX-induced cardiomyopathy (4, 5, 37). Another recognized culprit of DOX cardiotoxicity is mitochondrial dysfunction (39). Being the major site of DOX-induced ROS production, mitochondria themselves are vulnerable to oxidative injury. DOX interacts with the acidic lipoprotein cardiolipin in the inner mitochondrial membrane, resulting in its peroxidation and the opening of mitochondrial permeability transition pores which in turn triggers cytochrome c release and apoptosis (40, 41). The third mechanism proposed for DOX cardiotoxicity is through its effect on topoisomerase IIβ (TOPIIβ). While the antitumor effect of DOX is through DNA intercalation and TOPIIα inhibition (42–44), DOX also binds to TOPIIβ which is expressed mainly in quiescent cells such as cardiomyocytes. Mice null for TOPIIβ do not exhibit cardiotoxic effects with DOX treatment (45), suggesting that TOPIIβ is a major mediator of DOX cardiotoxicity. DOX is proposed to complex with TOPIIβ, leading to the activation of p53 mediated DNA damage pathways and the inhibition of genes implicated in mitochondrial biogenesis. Interestingly, dexrazoxane is shown to protect the heart by transiently depleting TOPIIβ levels in cardiomyocytes, suggesting that dexrazoxane may reduce DOX cardiotoxicity via both TOPIIβ depletion and iron chelation (46). The last potential mechanism of DOX cardiotoxicity relates to autophagy, a catabolic process for the cell to degrade long-lived proteins and organelles in the lysosome. The exact function of autophagy in DOX cardiotoxicity remains hotly debated, which is not surprising given the dynamic nature of the multi-step autophagic process and the numerous pathways implicated in its regulation. Indeed, DOX has been shown to either activate autophagy (17, 47–50) or inhibit autophagy (51–53), paradoxically, both of which contribute to cardiotoxicity. Adding to the confusion, DOX-triggered suppression of autophagy is seemingly cardioprotective (54). These conflicting results may be attributable to the differences in the experimental models used, the developmental stages of cardiomyopathy, and the dose and duration of DOX treatment, as well as the methods applied to manipulate different steps of the autophagic process and the techniques used to measure autophagic activities (49, 55). An early sign of DOX-induced mitochondrial damage is the loss of mitochondrial membrane potential (56–58). The latter is a major mechanism that triggers mitochondrial degradation by autophagy, a process known as mitophagy. However, as with autophagy, it remains controversial whether DOX activates or inhibits mitophagy and whether mitophagy contributes to or protects against DOX cardiotoxicity (59–62). Further investigation is needed to measure mitophagy flux and elucidate the role of mitophagy in DOX cardiotoxicity by using more reliable approaches and more clinically relevant animal models. In summary, it is likely that DOX induces cardiotoxicity via multiple mechanisms, including ROS generation, iron accumulation, cardiolipin peroxidation/mitochondrial injury, topoisomerase binding, and autophagy/mitophagy dysfunction (Figure 1).
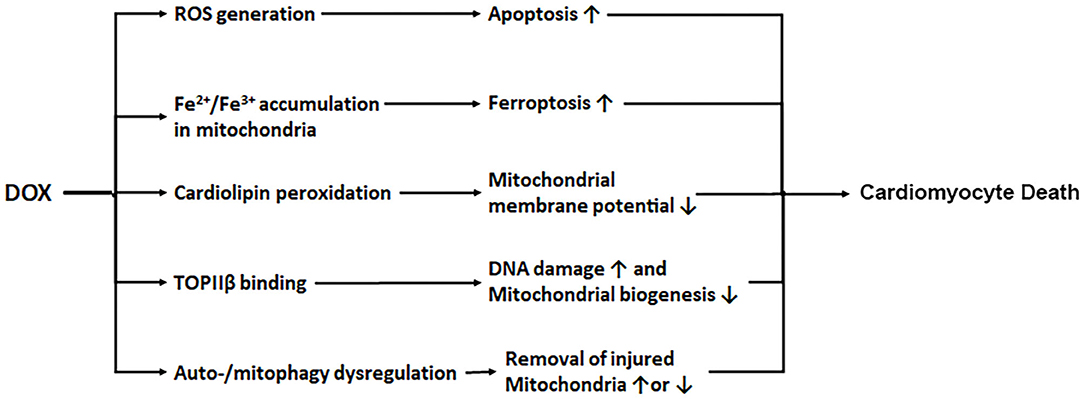
Figure 1. DOX induces cardiotoxicity via multiple mechanisms. DOX enters mitochondria triggering increased production of ROS, iron accumulation, cardiolipin peroxidation, and mitochondrial injury. DOX also binds to topoisomerase IIβ (TOPIIβ), resulting in DNA damage and reduced mitochondrial biogenesis. In addition, DOX causes autophagy/mitophagy dysfunction, leading to either reduced or excessive elimination of injured mitochondria, worsening cardiac injury.
New Strategies to Diminish Dox Cardiotoxicity in Cancer Patients are Desperately Needed
The current approach for reducing DOX cardiotoxicity is to limit the overall cumulative dose of the drug. However, this also narrows the therapeutic window for cancer treatment. Other strategies for limiting its cardiotoxicity have been pursued. Attempts to develop chemical analogs that retain anti-tumor properties but have reduced cardiotoxicity have had minimal success (63). Liposomal DOX has improved pharmacokinetics and reduced accumulation in the heart (64) but has failed to replace conventional DOX for treatment of most solid tumors (65). An additional approach is to combine DOX with a cardioprotective agent during treatment. Common neurohormonal antagonists, such as β-adrenergic receptor blockers and angiotensin-converting enzyme inhibitors, are routinely used for treating non-cancer-related heart failure, but they are not recommended for preventing and managing DOX cardiotoxicity due to the marginal benefits and related adverse events (66). Currently, only the iron chelator dexrazoxane has been approved for clinical use for reducing DOX cardiotoxicity (4, 5). Unfortunately, dexrazoxane is not a ubiquitous treatment for anthracycline cardiotoxicity, and its use has been limited to pediatric patients with high risk acute lymphoblastic leukemia and breast cancer patients on high doses of DOX, given the possibility of dexrazoxane to cause myelosuppression and secondary malignancies (6, 67, 68). Therefore, it is imperative to develop new strategies to protecting against DOX-induced heart damage without compromising the anti-tumor activity of DOX. In this regard, the antidiabetic drug metformin (MET) has appeared to be such a promising dual-function agent that can improve the clinical use of DOX.
Metformin Protects the Heart Against Various Pathological Conditions Including Dox Cardiotoxicity
Metformin (MET) is an oral biguanide agent that was first utilized to treat diabetes in France in 1957 (69) and approved by the US FDA in 1994 and has since been widely used as the first-line treatment for Type II diabetes due to its safety, efficacy and tolerability (70, 71). MET has been shown to protect the heart in people with or without diabetes mellitus (72). Indeed, MET is associated with decreased risk of heart failure (73) and reduced cardiovascular mortality independent of its glucose lowering effects (74). The cardioprotective effects of MET have been repeatedly confirmed by numerous pre-clinical studies under various cardiac conditions (75–79). Not surprisingly, MET can also reduce DOX cardiotoxicity in many animal studies (8–11). This may hold true in humans as well, given the ability of MET to attenuate radiation cardiotoxicity in breast cancer patients (80). Unfortunately, a phase II clinical trial “Use of Metformin to Reduce Cardiac Toxicity in Breast Cancer” was prematurely terminated due to its failure to meet target accrual (https://clinicaltrials.gov/ct2/show/NCT02472353). Apparently, further clinical trials are needed to confirm the cardioprotective effects of MET in cancer patients treated with DOX. MET has been suggested to antagonize DOX cardiotoxicity through several mechanisms (left panel in Figure 2), including attenuation of ROS generation and oxidative stress, inhibition of mitochondrial damage and maintenance of energy production (82), normalization of autophagy markers (8), increased expression of ferritin heavy chain in cardiomyocytes, and activation of AMP-activated protein kinase (AMPK) (11). The role of AMPK in MET-induced protection against DOX cardiotoxicity has been supported by numerous studies either in cultured cells or in animals (8–11).
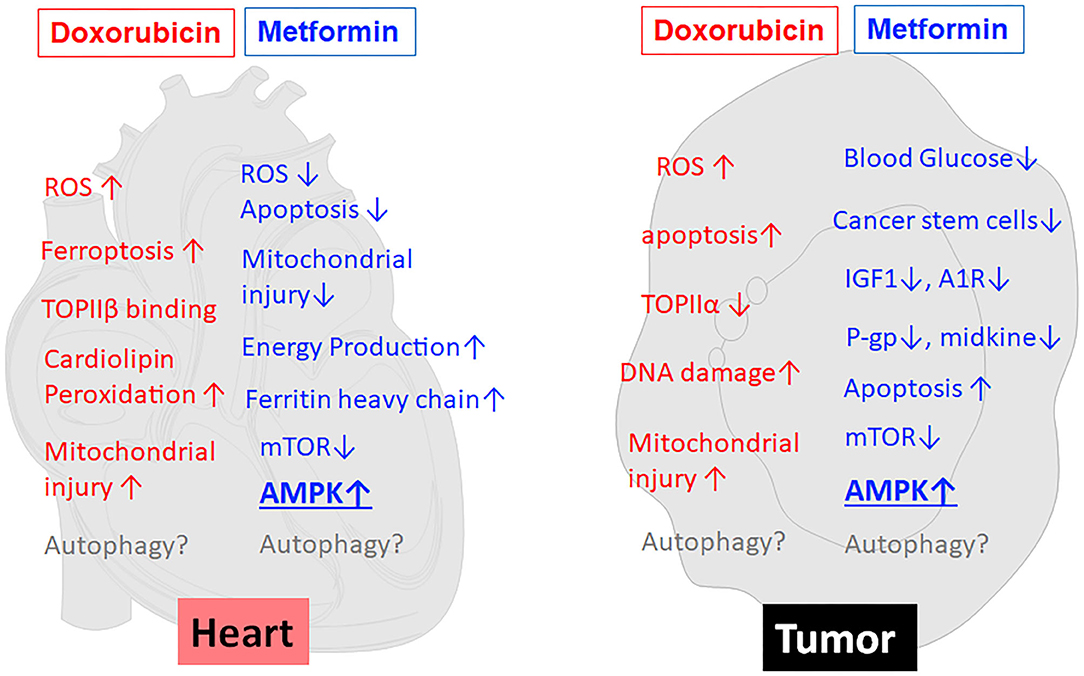
Figure 2. MET reduces the toxic effects of DOX on cardiomyocytes but concurrently enhances the anticancer effects of DOX on tumor cells. As shown in the left panel (heart), MET antagonizes DOX cardiotoxicity through several mechanisms, including attenuation of ROS generation and oxidative stress, inhibition of mitochondrial damage and maintenance of energy production, increased expression of ferritin heavy chain, and activation of AMPK. At the same time, MET enhances DOX antitumor effects (tumor, the right panel) through reduction of blood glucose, inhibition of cancer stem cells, reduction of IGF-1, modulation of adenosine A1 receptor (A1R), down-regulation of drug-resistant gene P-glycoprotein (P-gp), induction of apoptosis, inhibition of midkine, inhibition of mTOR, and activation of AMPK. Of note, AMPK activation has been suggested to be the major mechanism that mediates both the anti-tumor and cardioprotective effects of MET. On the other hand, the effects of MET on autophagy/mitophagy are not very clear. ↑, increase or upregulation; ↓, inhibition or downregulation; ROS, Reactive oxygen species; TOPII, Topoisomerase II; A1R, Adenosine A1 receptor; IGF1, Insulin-like growth factor 1; P-gp, P-glycoprotein.
Metformin has Antitumor Properties that may Synergize With the Antitumor Activity of Dox
Several epidemiological studies, meta-analyses and animal studies have revealed that MET has anti-neoplastic and chemopreventive activities (20, 81) despite mixed results observed in other studies (82, 83). Indeed, diabetic patients taking MET have significantly reduced risk of cancer and lower cancer-related mortality (84–89). Several small-scale clinical trials have shown the ability of MET to induce favorable cellular and molecular changes in cancer patients (90–93). For example, clinical trials in pre-surgical endometrial cancer patients exhibited a significant decrease in Ki67 with MET monotherapy (19). Another study showed the ability of MET to inhibit the increase of Insulin-like growth factor 1 (IGF-1) and maintain the levels of IGF binding protein-1 although the progression-free survival was not affected (91). In addition, numerous animal studies have shown that MET can enhance the anticancer activity of DOX (11–13, 94, 95). Thus, it is highly desirable that large scale randomized clinical trials be conducted to confirm the usefulness of MET in cancer chemotherapy. Nevertheless, given the demonstrated anti-tumor and cardioprotective properties of MET, it is reasonable to believe that MET can be used in DOX-containing chemotherapy to enhance the antitumor activity of DOX and at the same time to reduce its cardiotoxic effect (96). Metformin is believed to exert its antitumor effects via multiple mechanisms (right panel in Figure 2), including activation of AMPK and inhibition of mTOR (13, 97, 98), reduction of blood glucose (21), reduction of insulin and IGF-1(98), inhibition of cancer stem cells (99), modulation of adenosine A1 receptor (100), down-regulating drug-resistant gene P-glycoprotein (P-gp) (94), inhibition of midkine (101), and induction of apoptosis (102, 103). Among them, AMPK activation has been suggested to be the major mechanism that mediates both the anti-tumor and cardioprotective effects of metformin (11, 13, 21, 97). If this is true, modulation of AMPK per se should improve the application of DOX in antitumor therapy.
Ampk Signaling may Protect Against Dox Cardiotoxicity
AMP-activated protein kinase (AMPK) is a heterotrimeric protein kinase composed of a catalytic α subunit and two regulatory subunits (β and γ). Each subunit has multiple isoforms encoded by distinct genes (α1, α2, β1, β2, γ1, γ2, and γ3), and they combine to form 12 different AMPK holoenzymes (104). All isoforms except for γ3 are expressed in mouse and human heart, which can form 8 AMPK holoenzymes (105). As an energy sensor, AMPK detects and reacts to fluctuations in intracellular ATP levels under normal and stress conditions. The activated AMPK affects multiple metabolic pathways to maintain an energy homeostasis conducive to stress resistance and cell survival (106). There has been continuous intense research targeting AMPK for the treatment of multiple prevalent diseases, such as obesity, diabetes, cancer and cardiovascular diseases (107–109). Using AMPK deficient mice and chemical activators of AMPK such as AICAR and MET, numerous studies have shown that AMPK exerts a cardioprotective effect against myocardial ischemic injury (110, 111), diabetic cardiomyopathy (112), pathological cardiac remodeling (113), and heart failure (109). However, the use of MK-8722, a pan-AMPK activator, induces cardiac hypertrophy despite its ability to improve glucose homeostasis in rodents and rhesus monkeys (114), casting some doubt on the notion that AMPK activation always benefits the heart. Indeed, the gain-of-function mutations of the AMPK γ2 subunit result in severe cardiomyopathy in humans (115, 116), suggesting that the activation of some AMPK isoforms or holoenzymes can be detrimental to the heart under certain conditions. Interestingly, AMPK holoenzymes containing the α2 rather than the α1 subunit are the primary mediators of the cardiac phenotype of γ2 mutations (117), suggesting that α1-AMPK may play a different role than α2-AMPK, which underscores the complexity of isoform-specific functions of AMPK. This isoform-specific phenomenon was also observed in skeletal muscle where α2 but not α1 AMPK is responsible for AICAR-induced glucose uptake (118). When it comes to DOX cardiotoxicity, most cell-based studies have suggested AMPK as cardioprotective (9, 11, 14–17) despite the fact that DOX has been reported to either increase or decrease cardiac AMPK activity depending on the dose and duration of DOX treatment as well as the experimental models used (59, 119, 120). Pharmacological agents including MET, statins and many others can simultaneously activate AMPK and protect against DOX cardiotoxicity, but this remains an association and the causality between these two effects has not been established (119, 120). For example, the proposed role of AMPK in MET-mediated protection against DOX cardiotoxicity remains to be determined by using genetic animal models lacking AMPK function. Also, it remains essentially unknown which of the 8 isoform-specific AMPK holoenzymes mediates the putative protective effects on DOX cardiotoxicity in vivo.
Ampk Plays Temporal and Isoform-Dependent Dichotomous Roles in Cancer
AMPK is considered to be both a tumor suppressor and an oncogene depending on the context (22). Studies have suggested AMPK as a tumor suppressor before disease arises, which is further enhanced by the biguanide phenformin. However, once cancer has occurred, AMPK becomes a tumor promoter to enhance cancer cell survival by protecting against metabolic, oxidative and genotoxic stresses (23). Indeed, the Liver Kinase B1 (LKB1)/AMPK pathway contributes to tumor cell survival by promoting cellular sensing of and adaptation to bioenergetic stress. Repression of LKB1 by miR-17~92 sensitizes MYC-dependent lymphoma to biguanide treatment (121). In addition, a loss of both AMPK α1 and α2 subunit isoforms in H-Ras-transformed mouse embryonic fibroblasts (MEFs) caused a complete failure of their growth in vivo in immunodeficient mice (122). However, a loss of AMPK α2 alone caused the tumors to grow more rapidly (123), suggesting isoform-dependent differential effects of AMPK on tumor growth. In summary, whether AMPK behaves as a tumor suppressor or a promoter depends on the developmental stage of the tumor and the specific isoform of the AMPK subunits.
Met Activates Ampk, But it is Unknown if Ampk is Responsible for Cardioprotection By Met
Met has been shown to activate the AMPK pathway, and this has been proposed as the major mechanism that mediates the cardioprotective (9, 11, 109, 119, 120) and antitumor (13, 96, 97, 124) effects of MET. Thus, pharmacologically activating the AMPK pathway seems to be a two-birds-with-one-stone strategy to simultaneously reduce DOX cardiotoxicity and enhance its antitumor activity. However, it remains to be determined whether AMPK is indeed responsible for the potential double benefits of MET in humans or in clinically relevant animal models. Indeed, MET is shown to reduce pathological cardiac remodeling in the absence of AMPKα2 (76), suggesting the possibility that MET may reduce DOX cardiotoxicity independently of AMPK. Given the dual role of AMPK in tumor growth, it is equally unclear if the antitumor effects of MET are mediated by AMPK or its subunit isoforms.
Summary and Future Perspectives
MET has been safely used to treat diabetes for several decades, making it a good candidate for repurposing (19). Indeed, many animal and preclinical studies suggest that MET has both cardioprotective and antitumor properties, which lends itself as a promising adjuvant drug for DOX anticancer therapies to reduce cardiotoxicity. MET is proposed to achieve these beneficial effects through the activation of AMPK that itself has been shown to protect the heart and modulate tumor growth under certain conditions. However, the role and mechanism of the hypothesized MET-AMPK axis in DOX cardiotoxicity and antitumor efficacy have not been firmly established. Convincing in vivo studies using tumor-bearing animal models and large-scale prospective clinical trials are needed to fully establish MET as an effective antitumor agent either alone or together with DOX. Also, the proposed role of AMPK in MET-mediated protection against DOX cardiotoxicity should be validated in genetic animal models lacking AMPK in the heart. Given the emerging evidence suggesting distinct functional roles of the AMPK isoforms, it is important to investigate how different AMPK holoenzymes containing unique combinations of isoforms will modulate the ability of DOX to affect either heart function or tumor growth. Future studies should also explore the cellular and molecular mechanisms that account for the differential responses of cardiomyocytes vs. cancer cells to DOX and MET, either individually or in combination. Without any doubt, answers to the above questions are expected to have a positive impact on the treatment of many types of cancers with DOX. For example, if it is firmly established that MET can reduce DOX cardiotoxicity and concurrently maintain its antitumor activity, the results could be rapidly translated into use for cancer patients because MET has been used in diabetic patients for decades. Specifically, including MET in a therapeutic protocol could reduce the amount of DOX needed to achieve the same antitumor effect. Alternatively, MET could make it possible to use larger doses of DOX to eradicate cancer more effectively without increasing cardiac damage. In short, MET could improve the therapeutic window for DOX, allowing greater flexibility in designing regimens for treating cancer. Finally, a comprehensive understanding of the relationship between DOX cardiotoxicity, antitumor efficacy, and individual isoforms of AMPK will guide novel mechanism-based therapeutic strategies that target AMPK.
Author Contributions
ManrS, AN, JD, JW, MandS, and TN contributed to sections of the first draft. SK made the figures and edited the manuscript. QL wrote the outline, edited the draft, and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This line of research in the laboratory was supported by an in-house grant from New York Institute of Technology College of Osteopathic Medicine (NYITCOM) to QL and a research grant from the American Osteopathic Association (AOA, #2007816) to TN.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
DOX, doxorubicin; MET, metformin; ROS, reactive oxygen species; AMPK, AMP-activated protein kinase; TOPIIα/β, topoisomerase IIα/β; ACE, angiotensin-converting enzyme; IGF1, Insulin-like growth factor 1; P-gp, P-glycoprotein; LKB1, Liver Kinase B1; MEFs, mouse embryonic fibroblasts.
References
1. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. (2003) 97:2869–79. doi: 10.1002/cncr.11407
2. Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. (1998) 339:900–5. doi: 10.1056/NEJM199809243391307
3. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. (2004) 56:185–229. doi: 10.1124/pr.56.2.6
4. Injac R, Strukelj B. Recent advances in protection against doxorubicin-induced toxicity. Technol Cancer Res Treat. (2008) 7:497–516. doi: 10.1177/153303460800700611
5. Hasinoff BB, Herman EH. Dexrazoxane: how it works in cardiac and tumor cells. Is it a prodrug or is it a drug? Cardiovasc Toxicol. (2007) 7:140–4. doi: 10.1007/s12012-007-0023-3
6. Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. (2007) 25:493–500. doi: 10.1200/JCO.2005.02.3879
7. Swain SM, Whaley FS, Gerber MC, Ewer MS, Bianchine JR, Gams RA. Delayed administration of dexrazoxane provides cardioprotection for patients with advanced breast cancer treated with doxorubicin-containing therapy. J Clin Oncol. (1997) 15:1333–40. doi: 10.1200/JCO.1997.15.4.1333
8. Zilinyi R, Czompa A, Czegledi A, Gajtko A, Pituk D, Lekli I, et al. The cardioprotective effect of metformin in doxorubicin-induced cardiotoxicity: the role of autophagy. Molecules. (2018) 23:1184. doi: 10.3390/molecules23051184
9. Kobashigawa LC, Xu YC, Padbury JF, Tseng YT, Yano N. Metformin protects cardiomyocyte from doxorubicin induced cytotoxicity through an AMP-activated protein kinase dependent signaling pathway: an in vitro study. PLoS ONE. (2014) 9:e104888. doi: 10.1371/journal.pone.0104888
10. Asensio-Lopez MC, Lax A, Pascual-Figal DA, Valdes M, Sanchez-Mas J. Metformin protects against doxorubicin-induced cardiotoxicity: involvement of the adiponectin cardiac system. Free Radic Biol Med. (2011) 51:1861–871. doi: 10.1016/j.freeradbiomed.2011.08.015
11. Ajzashokouhi AH, Bostan HB, Jomezadeh V, Hayes AW, Karimi G. A review on the cardioprotective mechanisms of metformin against doxorubicin. Hum Exp Toxicol. (2019) 39:237–48. doi: 10.1177/0960327119888277
12. Li Y, Luo J, Lin MT, Zhi P, Guo WW, Han M, et al. Co-delivery of metformin enhances the antimultidrug resistant tumor effect of doxorubicin by improving hypoxic tumor microenvironment. Mol Pharm. (2019) 16:2966–79. doi: 10.1021/acs.molpharmaceut.9b00199
13. El-Ashmawy NE, Khedr NF, El-Bahrawy HA, Abo Mansour. HE. Metformin augments doxorubicin cytotoxicity in mammary carcinoma through activation of adenosine monophosphate protein kinase pathway. Tumour Biol. (2017) 39:1010428317692235. doi: 10.1177/1010428317692235
14. Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L, et al. AMPK/PGC1alpha activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. (2018) 129:59–72. doi: 10.1016/j.freeradbiomed.2018.08.032
15. Liu MH, Lin XL, Guo DM, Zhang Y, Yuan C, Tan TP, et al. Resveratrol protects cardiomyocytes from doxorubicin-induced apoptosis through the AMPK/P53 pathway. Mol Med Rep. (2016) 13:1281–86. doi: 10.3892/mmr.2015.4665
16. Liu D, Ma Z, Xu L, Zhang X, Qiao S, Yuan J. PGC1alpha activation by pterostilbene ameliorates acute doxorubicin cardiotoxicity by reducing oxidative stress via enhancing AMPK and SIRT1 cascades. Aging. (2019) 11:10061–73. doi: 10.18632/aging.102418
17. Chen K, Xu X, Kobayashi S, Timm D, Jepperson T, Liang Q. Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J Biol Chem. (2011) 286:21993–2006. doi: 10.1074/jbc.M111.225805
18. Russell FM, Hardie DG. AMP-activated protein kinase: do we need activators or inhibitors to treat or prevent cancer? Int J Mol Sci. (2020) 22:186. doi: 10.3390/ijms22010186
19. Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, et al. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget. (2016) 7:40767–80. doi: 10.18632/oncotarget.8194
20. Kheirandish M, Mahboobi H, Yazdanparast M, Kamal W, Kamal MA. Anti-cancer effects of metformin: recent evidences for its role in prevention and treatment of cancer. Curr Drug Metab. (2018) 19:793–7. doi: 10.2174/1389200219666180416161846
21. Aljofan M, Riethmacher D. Anticancer activity of metformin: a systematic review of the literature. Future Sci OA. (2019) 5:FSO410. doi: 10.2144/fsoa-2019-0053
22. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. (2013) 73:2929–35. doi: 10.1158/0008-5472.CAN-12-3876
23. Vara-Ciruelos D, Russell FM, Hardie DG. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? (dagger). Open Biol. (2019) 9:190099. doi: 10.1098/rsob.190099
24. Samuel SM, Varghese E, Kubatka P, Triggle CR, Busselberg D. Metformin: the answer to cancer in a flower? Current knowledge and future prospects of metformin as an anti-cancer agent in breast cancer. Biomolecules. (2019) 9:846. doi: 10.3390/biom9120846
25. Hoffman RK, Kim BJ, Shah PD, Carver J, Ky B, Ryeom S. Damage to cardiac vasculature may be associated with breast cancer treatment-induced cardiotoxicity. Cardiooncology. (2021) 7:15. doi: 10.1186/s40959-021-00100-3
26. van der Pal HJ, van Dalen EC, Hauptmann M, Kok WE, Caron HN, van den Bos C, et al. Cardiac function in 5-year survivors of childhood cancer: a long-term follow-up study. Arch Intern Med. (2010) 170:1247–255. doi: 10.1001/archinternmed.2010.233
27. Maeda M. Late effects of childhood cancer: life-threatening issues. J Nippon Med Sch. (2008) 75:320–4. doi: 10.1272/jnms.75.320
28. Mancilla TR, Iskra B, Aune GJ. Doxorubicin-induced cardiomyopathy in children. Comprehens Physiol. (2019) 9:905–31. doi: 10.1002/cphy.c180017
29. Link G, Tirosh R, Pinson A, Hershko C. Role of iron in the potentiation of anthracycline cardiotoxicity: identification of heart cell mitochondria as a major site of iron-anthracycline interaction. J Lab Clin Med. (1996) 127:272–8. doi: 10.1016/S0022-2143(96)90095-5
30. Songbo M, Lang H, Xinyong C, Bin X, Ping Z, Liang S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol Lett. (2019) 307:41–8. doi: 10.1016/j.toxlet.2019.02.013
31. Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair D. K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Investig. (1996) 98:1253–60. doi: 10.1172/JCI118909
32. Siveski-Iliskovic N, Hill M, Chow DA, Singal PK. Probucol protects against adriamycin cardiomyopathy without interfering with its antitumor effect. Circulation. (1995) 91:10–15. doi: 10.1161/01.CIR.91.1.10
33. Sun X, Zhou Z, Kang YJ. Attenuation of doxorubicin chronic toxicity in metallothionein-overexpressing transgenic mouse heart. Cancer Res. (2001) 61:3382–7.
34. Gianni L, Herman EH, Lipshultz SE, Minotti G, Sarvazyan N, Sawyer DB. Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol. (2008) 26:3777–84. doi: 10.1200/JCO.2007.14.9401
35. Ladas EJ, Jacobson JS, Kennedy DD, Teel K, Fleischauer A, Kelly KM. Antioxidants and cancer therapy: a systematic review. J Clin Oncol. (2004) 22:517–28. doi: 10.1200/JCO.2004.03.086
36. Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree? Redox Biol. (2020) 29:101394. doi: 10.1016/j.redox.2019.101394
37. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Investig. (2014) 124:617–30. doi: 10.1172/JCI72931
38. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5:e132747. doi: 10.1172/jci.insight.132747
39. Wallace KB, Sardao VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. (2020) 126:926–41. doi: 10.1161/CIRCRESAHA.119.314681
40. Lebrecht D, Kokkori A, Ketelsen UP, Setzer B, Walker UA. Tissue-specific mtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J Pathol. (2005) 207:436–44. doi: 10.1002/path.1863
41. Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. (2015) 309:H1453–67. doi: 10.1152/ajpheart.00554.2015
42. Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. (2010) 17:421–33. doi: 10.1016/j.chembiol.2010.04.012
43. Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. (2011) 21:440–6. doi: 10.1097/FPC.0b013e32833ffb56
44. Marinello J, Delcuratolo M, Capranico G. Anthracyclines as topoisomerase ii poisons: from early studies to new perspectives. Int J Mol Sci. (2018) 19:3480. doi: 10.3390/ijms19113480
45. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
46. Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Godtel-Armbrust U, et al. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer. (2014) 14:842. doi: 10.1186/1471-2407-14-842
47. Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem. (2010) 285:793–804. doi: 10.1074/jbc.M109.070037
48. Xu X, Chen K, Kobayashi S, Timm D, Liang Q. Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 S6 kinase 1-mediated autophagy. J Pharmacol Exp Ther. (2012) 341:183–95. doi: 10.1124/jpet.111.189589
49. Ding Y, Sun X, Xu X. TOR-autophagy signaling in adult zebrafish models of cardiomyopathy. Autophagy. (2012) 8:142–3. doi: 10.4161/auto.8.1.18536
50. Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, et al. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. (2009) 119:99–106. doi: 10.1161/CIRCULATIONAHA.108.799700
51. Sishi BJ, Loos B, van Rooyen J, Engelbrecht AM. Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity. Biochem Pharmacol. (2013) 85:124–34. doi: 10.1016/j.bcp.2012.10.005
52. Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. (2012) 96:456–65. doi: 10.1093/cvr/cvs282
53. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. (2016) 133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443
54. Xiao B, Hong L, Cai X, Mei S, Zhang P, Shao L. The true colors of autophagy in doxorubicin-induced cardiotoxicity. Oncol Lett. (2019) 18:2165–72. doi: 10.3892/ol.2019.10576
55. Montalvo RN, Doerr V, Kwon OS, Talbert EE, Yoo JK, Hwang MH, et al. Protection against doxorubicin-induced cardiac dysfunction is not maintained following prolonged autophagy inhibition. Int J Mol Sci. (2020) 21:8105. doi: 10.3390/ijms21218105
56. Solem LE, Henry TR, Wallace KB. Disruption of mitochondrial calcium homeostasis following chronic doxorubicin administration. Toxicol Appl Pharmacol. (1994) 129:214–22. doi: 10.1006/taap.1994.1246
57. Hasinoff BB, Schnabl KL, Marusak RA, Patel D, Huebner E. Dexrazoxane (ICRF-187) protects cardiac myocytes against doxorubicin by preventing damage to mitochondria. Cardiovasc Toxicol. (2003) 3:89–99. doi: 10.1385/CT:3:2:89
58. Carvalho RA, Sousa RP, Cadete VJ, Lopaschuk GD, Palmeira CM, Bjork JA, et al. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology. (2010) 270:92–8. doi: 10.1016/j.tox.2010.01.019
59. Koleini N, Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget. (2017) 8:46663–80. doi: 10.18632/oncotarget.16944
60. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. (2013) 4:2308. doi: 10.1038/ncomms3308
61. Yin J, Guo J, Zhang Q, Cui L, Zhang L, Zhang T, et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol In Vitro. (2018) 51:1–10. doi: 10.1016/j.tiv.2018.05.001
62. Catanzaro MP, Weiner A, Kaminaris A, Li C, Cai F, Zhao F, et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. (2019) 33:11096–108. doi: 10.1096/fj.201802663R
63. Ludke AR, Al-Shudiefat AA, Dhingra S, Jassal DS, Singal PK. A concise description of cardioprotective strategies in doxorubicin-induced cardiotoxicity. Can J Physiol Pharmacol. (2009) 87:756–63. doi: 10.1139/Y09-059
64. Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, et al. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. (2010) 10:337. doi: 10.1186/1471-2407-10-337
65. Fukuda A, Tahara K, Hane Y, Matsui T, Sasaoka S, Hatahira H, et al. Comparison of the adverse event profiles of conventional and liposomal formulations of doxorubicin using the FDA adverse event reporting system. PLoS ONE. (2017) 12:e0185654. doi: 10.1371/journal.pone.0185654
66. Bansal N, Adams MJ, Ganatra S, Colan SD, Aggarwal S, Steiner R, et al. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology. (2019) 5:18. doi: 10.1186/s40959-019-0054-5
67. Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. (1997) 15:1318–32. doi: 10.1200/JCO.1997.15.4.1318
68. Seif AE, Walker DM, Li Y, Huang YS, Kavcic M, Torp K, et al. Dexrazoxane exposure and risk of secondary acute myeloid leukemia in pediatric oncology patients. Pediatr Blood Cancer. (2015) 62:704–9. doi: 10.1002/pbc.25043
69. Bailey CJ. Metformin: historical overview. Diabetologia. (2017) 60:1566–76. doi: 10.1007/s00125-017-4318-z
70. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. (2017) 60:1577–85. doi: 10.1007/s00125-017-4342-z
71. Fujita Y, Inagaki N. Metformin: clinical topics and new mechanisms of action. Diabetol Int. (2017) 8:4–6. doi: 10.1007/s13340-016-0300-0
72. Varjabedian L, Bourji M, Pourafkari L, Nader ND. Cardioprotection by metformin: beneficial effects beyond glucose reduction. Am J Cardiovasc Drugs. (2018) 18:181–93. doi: 10.1007/s40256-018-0266-3
73. Eurich DT, McAlister FA, Blackburn DF, Majumdar SR, Tsuyuki RT, Varney J, et al. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: systematic review. BMJ. (2007) 335:497. doi: 10.1136/bmj.39314.620174.80
74. Johnson JA, Majumdar SR, Simpson SH, Toth EL. Decreased mortality associated with the use of metformin compared with sulfonylurea monotherapy in type 2 diabetes. Diabetes Care. (2002) 25:2244–8. doi: 10.2337/diacare.25.12.2244
75. Nesti L, Natali A. Metformin effects on the heart and the cardiovascular system: a review of experimental and clinical data. Nutr Metab Cardiovasc Dis. (2017) 27:657–69. doi: 10.1016/j.numecd.2017.04.009
76. Xu X, Lu Z, Fassett J, Zhang P, Hu X, Liu X, et al. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase alpha2. Hypertension. (2014) 63:723–8. doi: 10.1161/HYPERTENSIONAHA.113.02619
77. Tzanavari T, Varela A, Theocharis S, Ninou E, Kapelouzou A, Cokkinos DV, et al. Metformin protects against infection-induced myocardial dysfunction. Metabolism. (2016) 65:1447–58. doi: 10.1016/j.metabol.2016.06.012
78. Soraya H, Clanachan AS, Rameshrad M, Maleki-Dizaji N, Ghazi-Khansari M, Garjani A. Chronic treatment with metformin suppresses toll-like receptor 4 signaling and attenuates left ventricular dysfunction following myocardial infarction. Eur J Pharmacol. (2014) 737:77–84. doi: 10.1016/j.ejphar.2014.05.003
79. Loi H, Boal F, Tronchere H, Cinato M, Kramar S, Oleshchuk O, et al. Metformin protects the heart against hypertrophic and apoptotic remodeling after myocardial infarction. Front Pharmacol. (2019) 10:154. doi: 10.3389/fphar.2019.00154
80. Yu JM, Hsieh MC, Qin L, Zhang J, Wu SY. Metformin reduces radiation-induced cardiac toxicity risk in patients having breast cancer. Am J Cancer Res. (2019) 9:1017–026.
81. Zi F, Zi H, Li Y, He J, Shi Q, Cai Z. Metformin and cancer: an existing drug for cancer prevention and therapy. Oncol Lett. (2018) 15:683–90. doi: 10.3892/ol.2017.7412
82. Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract. (2018) 143:409–19. doi: 10.1016/j.diabres.2018.05.023
83. Pacal L, Kankova K. Metformin in oncology - how far is its repurposing as an anticancer drug? Klin Onkol. (2020) 33:107–13. doi: 10.14735/amko2020107
84. Berstein LM, Boyarkina MP, Teslenko SY. Familial diabetes is associated with reduced risk of cancer in diabetic patients: a possible role for metformin. Med Oncol. (2012) 29:1308–13. doi: 10.1007/s12032-011-9840-0
85. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. (2005) 330:1304–5. doi: 10.1136/bmj.38415.708634.F7
86. Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, Morgan CL. Mortality after incident cancer in people with and without type 2 diabetes: impact of metformin on survival. Diabetes Care. (2012) 35:299–304. doi: 10.2337/dc11-1313
87. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. (2009) 32:1620–5. doi: 10.2337/dc08-2175
88. Tseng CH. Metformin is associated with a lower incidence of benign brain tumors: a retrospective cohort study in patients with type 2 diabetes mellitus. Biomolecules. (2021) 11:1405. doi: 10.3390/biom11101405
89. Tseng CH. Metformin and risk of malignant brain tumors in patients with type 2 diabetes mellitus. Biomolecules. (2021) 11:578831. doi: 10.3389/fphar.2020.578831
90. Reni M, Dugnani E, Cereda S, Belli C, Balzano G, Nicoletti R, et al. (Ir)relevance of metformin treatment in patients with metastatic pancreatic cancer: an open-label, randomized phase ii trial. Clin Cancer Res. (2016) 22:1076–85. doi: 10.1158/1078-0432.CCR-15-1722
91. Zheng Y, Zhu J, Zhang H, Liu Y, Sun H. Metformin plus first-line chemotherapy versus chemotherapy alone in the treatment of epithelial ovarian cancer: a prospective open-label pilot trial. Cancer Chemother Pharmacol. (2019) 84:1349–57. doi: 10.1007/s00280-019-03963-7
92. Nanni O, Amadori D, De Censi A, Rocca A, Freschi A, Bologna A, et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res Treat. (2019) 174:433–42. doi: 10.1007/s10549-018-05070-2
93. Trucco M, Barredo JC, Goldberg J, Leclerc GM, Hale GA, Gill J, et al. A phase I window, dose escalating and safety trial of metformin in combination with induction chemotherapy in relapsed refractory acute lymphoblastic leukemia: metformin with induction chemotherapy of vincristine, dexamethasone, PEG-asparaginase, and doxorubicin. Pediatr Blood Cancer. (2018) 65:e27224. doi: 10.1002/pbc.27224
94. Li Y, Wang M, Zhi P, You J, Gao JQ. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget. (2018) 9:2158–174. doi: 10.18632/oncotarget.23187
95. Pateliya B, Burade V, Goswami S. Enhanced antitumor activity of doxorubicin by naringenin and metformin in breast carcinoma: an experimental study. Naunyn Schmiedebergs Arch Pharmacol. (2021) 394:1949–61. doi: 10.1007/s00210-021-02104-3
96. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. (2012) 122:253–70. doi: 10.1042/CS20110386
97. Cho SY, Lee HJ, Lee HJ, Jung DB, Kim H, Sohn EJ, et al. Activation of AMP-activated protein kinase alpha and extracelluar signal-regulated kinase mediates CB-PIC-induced apoptosis in hypoxic SW620 colorectal cancer cells. Evid Based Complement Altern Med. (2013) 2013:974313. doi: 10.1155/2013/974313
98. Sosnicki S, Kapral M, Weglarz L. Molecular targets of metformin antitumor action. Pharmacol Rep. (2016) 68:918–25. doi: 10.1016/j.pharep.2016.04.021
99. Malinowski B, Musiala N, Wicinski M. Metformin's modulatory effects on miRNAs function in cancer stem cells-a systematic review. Cells. (2020) 9:1401. doi: 10.3390/cells9061401
100. Lan B, Zhang J, Zhang P, Zhang W, Yang S, Lu D, et al. Metformin suppresses CRC growth by inducing apoptosis via ADORA1. Front Biosci. (2017) 22:248–57. doi: 10.2741/4484
101. Karadeniz Z, Aynacioglu AS, Bilir A, Tuna MY. Inhibition of midkine by metformin can contribute to its anticancer effects in malignancies: a proposal mechanism of action of metformin in context of endometrial cancer prevention and therapy. Med Hypotheses. (2020) 134:109420. doi: 10.1016/j.mehy.2019.109420
102. Sharma P, Kumar S. Metformin inhibits human breast cancer cell growth by promoting apoptosis via a ROS-independent pathway involving mitochondrial dysfunction: pivotal role of superoxide dismutase (SOD). Cell Oncol. (2018) 41:637–650. doi: 10.1007/s13402-018-0398-0
103. Tsai CC, Chuang TW, Chen LJ, Niu HS, Chung KM, Cheng JT, et al. Increase in apoptosis by combination of metformin with silibinin in human colorectal cancer cells. World J Gastroenterol. (2015) 21:4169–77. doi: 10.3748/wjg.v21.i14.4169
104. Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J. (2016) 283:2987–3001. doi: 10.1111/febs.13698
105. Kim M, Shen M, Ngoy S, Karamanlidis G, Liao R, Tian R. AMPK isoform expression in the normal and failing hearts. J Mol Cell Cardiol. (2012) 52:1066–73. doi: 10.1016/j.yjmcc.2012.01.016
106. Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, et al. AMPK: lessons from transgenic and knockout animals. Front Biosci. (2009) 14:19–44. doi: 10.2741/3229
107. Day EA, Ford RJ, Steinberg GR. AMPK as a therapeutic target for treating metabolic diseases. Trends Endocrinol Metab. (2017) 28:545–60. doi: 10.1016/j.tem.2017.05.004
108. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. (2016) 48:e245. doi: 10.1038/emm.2016.81
109. Li X, Liu J, Lu Q, Ren D, Sun X, Rousselle T, et al. AMPK: a therapeutic target of heart failure-not only metabolism regulation. Biosci Rep. (2019) 39:BSR20181767. doi: 10.1042/BSR20181767
110. Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. (2009) 104:403–411. doi: 10.1161/CIRCRESAHA.108.190918
111. Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, et al. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. (2006) 291:H2875–83. doi: 10.1152/ajpheart.01032.2005
112. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. (2011) 60:1770–8. doi: 10.2337/db10-0351
113. Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, et al. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. (2008) 52:918–24. doi: 10.1161/HYPERTENSIONAHA.108.114702
114. Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. (2017) 357:507–11. doi: 10.1126/science.aah5582
115. Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Investig. (2002) 109:357–62. doi: 10.1172/JCI0214571
116. Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. (2001) 10:1215–20. doi: 10.1093/hmg/10.11.1215
117. Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, et al. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. (2005) 112:3140–8. doi: 10.1161/CIRCULATIONAHA.105.550806
118. Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, et al. Knockout of the alpha2 but not alpha1 5'-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. (2004) 279:1070–9. doi: 10.1074/jbc.M306205200
119. Timm KN, Tyler DJ. The role of AMPK activation for cardioprotection in doxorubicin-induced cardiotoxicity. Cardiovasc Drugs Ther. (2020) 34:255–69. doi: 10.1007/s10557-020-06941-x
120. Chen J, Zhang S, Pan G, Lin L, Liu D, Liu Z, et al. Modulatory effect of metformin on cardiotoxicity induced by doxorubicin via the MAPK and AMPK pathways. Life Sci. (2020) 249:117498. doi: 10.1016/j.lfs.2020.117498
121. Izreig S, Gariepy A, Kaymak I, Bridges HR, Donayo AO, Bridon G, et al. Repression of LKB1 by miR-17 approximately 92 sensitizes MYC-dependent lymphoma to biguanide treatment. Cell Rep Med. (2020) 1:100014. doi: 10.1016/j.xcrm.2020.100014
122. Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5'-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. (2006) 26:5336–47. doi: 10.1128/MCB.00166-06
123. Phoenix KN, Devarakonda CV, Fox MM, Stevens LE, Claffey KP. AMPKalpha2 suppresses murine embryonic fibroblast transformation and tumorigenesis. Genes Cancer. (2012) 3:51–62. doi: 10.1177/1947601912452883
Keywords: doxorubicin, metformin, AMPK, doxorubicin cardiotoxicity, cardio-oncology
Citation: Singh M, Nicol AT, DelPozzo J, Wei J, Singh M, Nguyen T, Kobayashi S and Liang Q (2022) Demystifying the Relationship Between Metformin, AMPK, and Doxorubicin Cardiotoxicity. Front. Cardiovasc. Med. 9:839644. doi: 10.3389/fcvm.2022.839644
Received: 20 December 2021; Accepted: 04 January 2022;
Published: 24 January 2022.
Edited by:
Dong Han, People's Liberation Army General Hospital, ChinaReviewed by:
Xing Fu, Louisiana State University Agricultural Center, United StatesCopyright © 2022 Singh, Nicol, DelPozzo, Wei, Singh, Nguyen, Kobayashi and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiangrong Liang, cWxpYW5nMDNAbnlpdC5lZHU=