 Li-na Miao1,2†
Li-na Miao1,2† Deng Pan1,2†
Deng Pan1,2† Junhe Shi1
Junhe Shi1 Jian-peng Du1,3
Jian-peng Du1,3 Peng-fei Chen1
Peng-fei Chen1 Jie Gao1,3Yanqiao Yu1,2Da-Zhuo Shi1,3*
Jie Gao1,3Yanqiao Yu1,2Da-Zhuo Shi1,3* Ming Guo1,3*
Ming Guo1,3*- 1Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Department of Graduate School, Beijing University of Chinese Medicine, Beijing, China
- 3China Heart Institute of Chinese Medicine, China Academy of Chinese Medical Sciences, Beijing, China
Protein kinase C (PKC) is a protein kinase with important cellular functions. PKC-δ, a member of the novel PKC subfamily, has been well-documented over the years. Activation of PKC-δ plays an important regulatory role in myocardial ischemia/reperfusion (IRI) injury and myocardial fibrosis, and its activity and expression levels can regulate pathological cardiovascular diseases such as atherosclerosis, hypertension, cardiac hypertrophy, and heart failure. This article aims to review the structure and function of PKC-δ, summarize the current research regarding its activation mechanism and its role in cardiovascular disease, and provide novel insight into further research on the role of PKC-δ in cardiovascular diseases.
Introduction
The term cardiovascular disease (CVD) includes many different diseases, such as coronary heart disease, hypertension, heart failure, and atherosclerosis (1). The global prevalence of CVD increased from 271 million in 1990 to 523 million in 2019, and the corresponding mortality rates were 12.1 million in 1990 and approximately 18.6 million in 2019 (2). Although the treatment of cardiovascular diseases has greatly improved in recent years, these diseases still exhibit high morbidity and mortality. There is still a lack of research exploring new targets for the prevention and treatment of CVD. In recent years, studies have found that protein kinase C (PKC) plays a multifaceted role in the pathophysiology of heart development and many cardiovascular diseases.
Protein kinase C is a member of the AGC protein kinase superfamily. To date, 10 PKC family members have been found in mammalian tissues. These family members can be divided into three categories according to their dependency on lipid second messengers and calcium and ion coactivators for activation: conventional PKCs (α, β1, β2, γ), novel PKCs (δ, ε, η, θ), and atypical PKCs (ζ, λ/ι) (3– 5). The activation of conventional PKCs requires diglyceride (DAG), phosphatidylserine (PS), and Ca2+. Novel PKCs are regulated by DAG and PS but do not require Ca2+ for activation, and the activity of atypical PKCs is only stimulated by PS (6–8) (Table 1). Protein kinase C activation is a complicated process that includes membrane association of the enzymes, priming by phosphorylation, conformational changes induced by binding of proteins or second messengers (e.g., Ca2+, PS), and the release of a pseudo substrate. In addition, DAT and DAT-based phospholipids can also activate PKC (9–11). The activated PKC isoform protein plays a vital role in many important human diseases, including diabetes (PKC-β) (12), cancer (PKC-ε) (13), autoimmune diseases (PKC-θ) (14), Parkinson (PKC-δ) (15, 16) etc. PKC-α, PKC-β, PKC-ε, and PKC-δ are among the most studied molecules in CVD, participate in various CVD, such as atherosclerosis, hypertension, atrial fibrillation, and cardiac hypertrophy (17–22). Protein kinase C mediates CVD by regulating apoptosis in smooth muscle cells and cardiomyocytes, regulating endothelial function, and participating in the regulation of cardiac ion channels. PKC-α is involved in platelet activation in atherosclerosis, and inhibition of PKC-α can reduce thrombogenesis at atherosclerotic plaques (23). Both PKC-δ and PKC-ε are activated in the ischemic heart of humans and myocardial infarction (MI) model of animals. However, opposite effects were observed (21, 24, 25). Selective activation of PKC-ε protects mitochondrial function prior to ischemic events or during reperfusion by activating mitochondrial aldehyde dehydrogenase 2 (ALDH2) and removing toxic aldehyde-lipid peroxidation products (26). In addition, PKC-ε inhibits the activation of L-type calcium channels in cardiomyocytes and prevents fatal ventricular arrhythmias associated with ischemia-reperfusion (I/R) injury (27). In contrast, PKC-δ activation mediates damage mainly by activating mitochondrial pyruvate dehydrogenase kinase, which inhibits pyruvate dehydrogenase, ATP regeneration, and thus induces necrosis (28). Rats and mice with vascular restenosis are benefited by inhibition of PKCβ and PKC-δ therapy. PKC-δ inhibition and PKC-ε activation can also alleviate vascular disease after transplantation or interventional therapy (29). PKC-δ is one of the most important and studied targets of the cardiac G protein-coupled receptor signaling pathway. PKC-δ can mediate apoptosis, inflammation, vasoconstriction, and vasodilation of blood vessels, which plays a key role in the regulation of cardiac contractility, ischemic preconditioning, and cardiac structure remodeling. Activation of PKC-δ plays an important regulatory role in myocardial ischemia/reperfusion (IRI) injury and myocardial fibrosis, and is involved in many cardiovascular diseases, such as atherosclerosis, hypertension, myocardial hypertrophy, and heart failure. The role of conventional PKC in the pathogenesis of pathological CVD had already been systematically summarized (11, 30). This paper reviews the research progress of PKC-δ in the pathogenesis of pathological CVD to provide ideas for further study on the role of PKC-δ in the pathogenesis of cardiovascular disease.
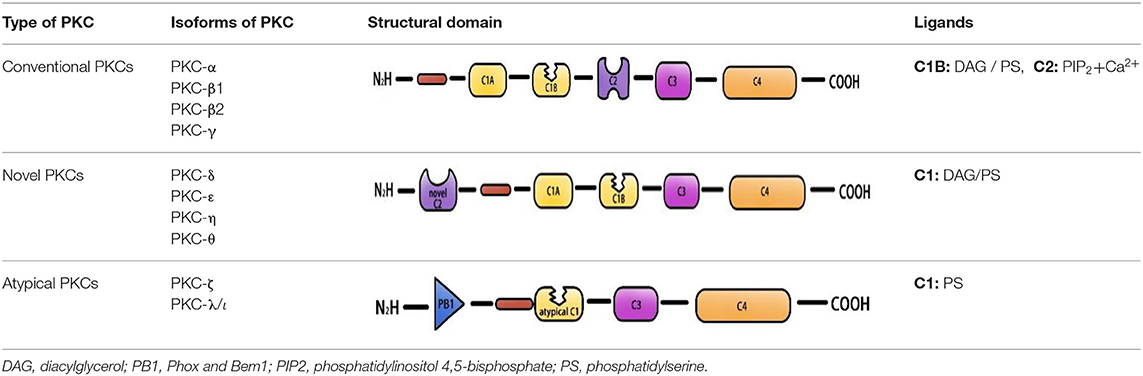
Table 1. PKC isoform sub-family members.
Structure and Function of PKC-δ
Structure of PKC-δ
Protein kinase C is a Ca2+ and lecithin-dependent protein kinase that is activated by Ca2+, acylglycerol (DAG), and phorbol ester (PS). PKC-δ is a member of the novel PKC family, which was cloned from a rat cDNA library in 1987 (31) and is widely present in various tissues, such as brain tissue and epithelial tissue cells, and its activation does not require Ca2+ involvement. Its molecular structure consists of a peptide chain, divided into four conserved regions (C1–C4) and five variable regions (V1–V5) (Figure 1). The C1 region of PKC-δ binds diacylglycerol and PS, which consists of a pseudosubstrate binding site and a zinc finger region, playing an essential role in its activation, and includes a tandem repeat of Cys-rich motifs named CIA and C1B (32). The C1A and C1B regions of PKC-δ are not equivalent, and the C1B region is the major PS-binding site of PKC-δ (33–35). PKC-δ lacks the authentic C2 region and instead has a C2-like region in the amino-terminal end of the molecule (36). The PKC-δ C2 domain differs from the C2 domain of traditional PKC, and it lacks key residues coordinating with Ca2+ (37). C3 is the ATP binding site. The C4 region contains a substrate-binding region that is required for the recognition of phosphorylated substrates. The C4 region harbors a phosphorylation motif site, Thr-505, in the activation loop, and the carboxyl-terminal end of the enzyme has two conserved phosphorylation sites, Ser-643, and Ser-662, which are hinge and hydrophobic motif sites, respectively (38).
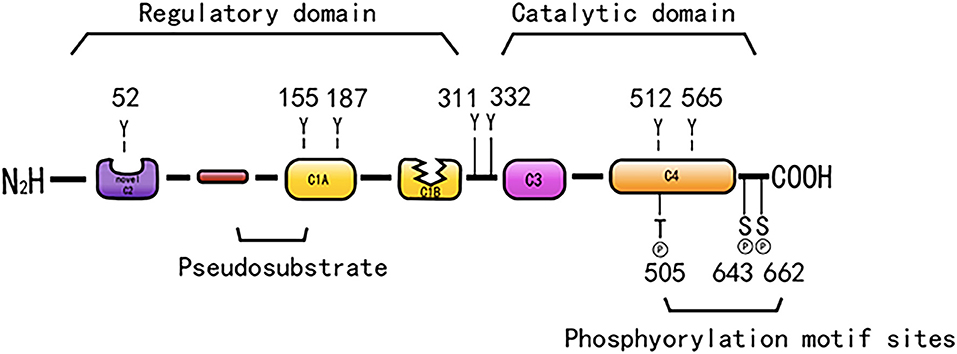
Figure 1. The structure of PKC-δ. Schematic showing pseudo substrate (red rectangle), C1 domain (yellow rectangle), C2 domain (purple rectangle), C3 domain (pink rectangle), C4 domain (orange rectangle). The cyan represents the connecting hinge segment, the kinase region, and the gray rectangle represents the C-terminal tail. In particular, w switch in C1B domain determines the affinity of DAG containing membrane.
PKC-δ consists of a regulatory region and a catalytic region that is separated by the V3 region. The regulatory region is composed of an NH2-terminal regulatory domain, which has a targeting effect that is related to the subcellular localization of the enzyme and regulates the activity of the enzyme. The catalytic region consists of a COOH-terminal catalytic domain, which can bind to a protein, a polypeptide substrate, and a phosphate donor ATP/GTP, and this domain can transfer a phosphate-based amino acid residue to the corresponding amino acid residue.
Both the catalytic and regulatory domains can be used to generate drugs that modulate PKC activity, leading to PKC inhibition or activation. The drug target regions of PKC-δ are mainly located at ATP-binding site in catalytic domain (39), DAG-binding site of C1 domain (40), and unique sequence of C2 domain (41). Generation of specific PKC subtype inhibitors is difficult due to high homolog of catalytic domain and conservation of ATP-binding site in PKC subtypes. In addition, it is also challenging to generate a drug that selectively binds to one of the isozymes C1 domains because of the high homolog of C1 domain and DAG-binding site. Drugs that target the C2 region and the inserted sequence (V region) between the domains show higher isozyme selectivity. Current drugs targeting PKC-δ include: Rottlerin (ATP-binding site), Bryostatin1 (C1 domain of PKC), and δV1-1 (Unique sequence of PKCδ -C2 domain) are the main drugs targeting PKC-δ.
In addition, PKC-δ has genetic polymorphisms, and conditional knockout mice can be generated through splicing of the PKC- δ gene, Prkcd. Conditional knockout mice are generated by regulating expression of exons (42). Conditional general PKC-δ knockout mice were generated by knockout of PKC-δ encoding gene (by deleting shared exons of PKC-δ I, II, IV, V, VI, and VII) through Cre-Lox recombination (42). In addition, conditional specific PKC-δ-suptype knockout mice were also reported. Leitges et al. generated PKC-δ II deletion mice by inserting a LacZ/neo cassette into the first transcribed exon of Prkcd (43). So far, nine subtypes of PKC-δ (I, II, III, IV, V, VI, VII, VIII, and IX) have been discovered (44–48). Among them, PKC-δ I, II, IV, V, VI, VII, and IX were found to be expressed in mice (44–46). PKC-δ I and III were found to be expressed in rats (47), whereas PKC-δ I and VIII were found to be expressed in human (48). And PKC-δ VIII is thought to be the human homolog of mouse PKC-δ II (48). In current studies, PKC-δ−/− mice are mostly the knockout of PKC-δ I and II subtypes. Recently, Niino et al. generated a conditional knockout mice lack of PKC-δ I or II, IV, V, VI, and VII (42). Their study found that a wide range of PKC-δ subtypes knockout can inhibit fetal development and is associated with adult mice heart elastic fiber hyperplasia and lung inflammation (42).
PKC-δ Activation Mechanisms
PKC-δ typically exists in the cytoplasm in an inactive form. Mechanisms regarding how PKC-δ is activated by various stimuli include binding of diacylglycerol and PS, phosphorylation at motif sites, tyrosine phosphorylation, membrane translocation, and proteolysis. In addition, stimulation by ATP and H2O2 also activates PKC-δ (49) (Figure 2).
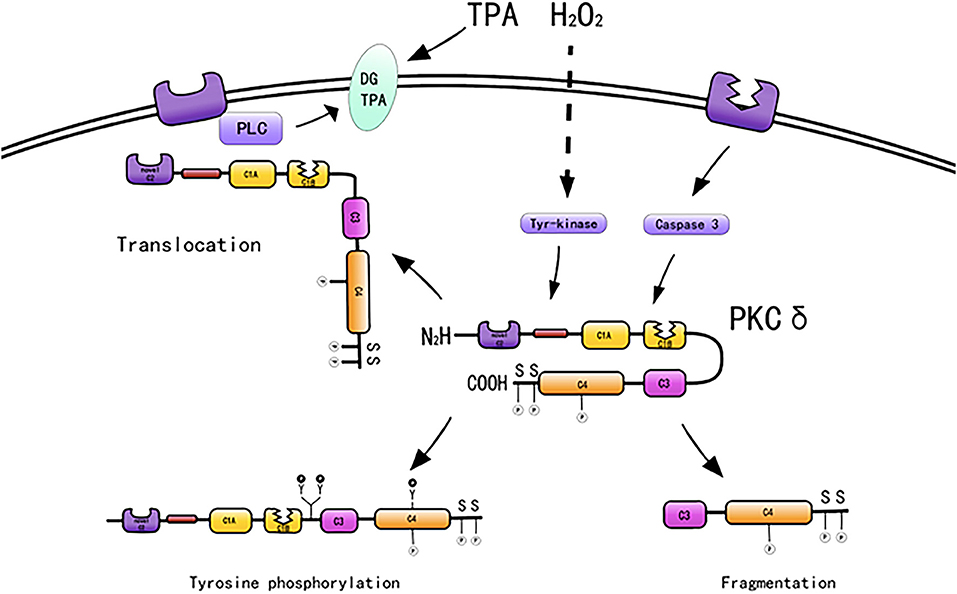
Figure 2. Activation mechanisms of PKC-δ [adapted from Ushio et al. (50)].
Diacylglycerol and Phorbol Ester
PKC-δ lacks a calcium-binding C2 domain and is maximally activated by DAG and PS in the absence of calcium (51). PKC-δ is activated by DAG, A variety of molecules, such as growth factors, PDGF, hormones, epidermal growth factors, and cytokines, when bound to their respective receptors, will activate members of the phospholipase C family, which will produce diacylglycerols, thereby activating PKC isoenzymes (52). PKC-δ serves as the receptor for tumor-promoting PSs, such as 12-O-tetradecanoylphorbol 13-acetate (TPA). 12-O-Tetradecanoylphorbol 13-acetate directly activates PKC-δ (53). Phorbol treatment induces PKC-δ overexpression and cell proliferation (54) and differentiation (55). For example, phorbol-treated NIH3T3 cells express PKC-δ and exhibit reduced cell growth (54), and TPA induces monocyte differentiation in 32D cells overexpressing PKC-δ (55).
Phosphorylation at Motif Sites
Protein kinase C consists of a series of conserved phosphorylation sites, which are located in the kinase activating ring, the trans motif and the hydrophobic motif. Phosphorylation causes the kinase to mature and prolong its cellular half-life (11). PKC-δ has phosphorylation motif sites of conserved serine or threonine residues (5, 38). PKC-δ is phosphorylated in the hinge and hydrophobic motif sites in the carboxyl-terminal end region. PKC-δ phosphorylates cTnI at Ser23/Ser24 when activated by lipid cofactors (56). PKC-δ harbors an activation loop, hinge, and hydrophobic motif sites at Thr-505, Ser-643, and Ser-662, respectively, and these sites are substantially phosphorylated in vivo (57). The phosphorylation of PKC-δ at Thr505 is induced by PMA or the α1-adrenergic receptor agonist noradrenaline (norepinephrine) in cardiomyocytes (58). PKC-δ is phosphorylated in vivo by c-Fyn (59, 60), c-Src (61, 62), and growth factor receptor (59, 60).
Tyrosine Phosphorylation
PKC-δ exhibits the most efficient tyrosine phosphorylation among the PKC family members, and different tyrosine residues are further phosphorylated upon cellular stimulation (57). Several tyrosine phosphorylation sites were found in the catalytic domain (tyr512 and tyr523), regulatory domain (tyr52, tyr155, and tyr187), and hinge region (tyr311 and tyr332) of PKC-δ. In contrast to phosphorylation at serine and threonine motif sites, the phosphorylated tyrosine residues identified to date, such as Tyr-311 and Tyr-332 of PKC-δ, are not conserved among members of the PKC family; however, Tyr-512 of PKC-δ represents an exception (57). PKC-δ is phosphorylated on tyrosine residues of cells stimulated by SRC or Ras transformation, H2O2, PMA, EGF, or PDGF. Frank et al. demonstrated the presence of distinct tyrosine kinase activation pathways of PKC-δ/PYK2/JAK2 utilized by H2O2 in vascular smooth muscle cells (VSMCs) (63). During oxidative stress, PKC-δ is activated by Src-mediated phosphorylation of an otherwise unutilized tyrosine residue in the enzyme (56, 64). A study indicated that tyrosine phosphorylation of PKC-δ may regulate its activity (59), and phosphorylation of tyrosine can both promote and inhibit the catalytic activity of PKC-δ. For example, the catalytic activity of PKC-δ was reduced by tyrosine phosphorylation in eRAS-transformed keratinocytes (60); however, PKC-δ phosphorylated at Tyr-311 in vitro shows enhanced catalytic activity (57). Tyrosine phosphorylation of PKC-δ modifies PKC-δ-dependent phosphorylation of cardiac troponin I (cTnI), a myofilament regulatory protein (56). In addition, tyrosine phosphorylation enhances the enzymatic activity in various cells stimulated by substances, such as PS, growth factors, and hormones (65–70). Many studies have relied on in vitro kinase analysis to address tyrosine phosphorylation-dependent changes in PKC-δ function. However, the catalytic activity of tyrosine-phosphorylated PKC-δ is variably described as reducing, increasing, or even changing in terms of substrate specificity and cofactor requirements, and information on PKC-δ tyrosine phosphate cannot be extracted from the literature (59, 60, 71, 72). Considering that PKC-δ is a tyrosine phosphorylated by TPA (59), it induces CHO cell division and arrest (40). Tyrosine phosphorylation of PKC-δ may be related to cell proliferation and differentiation.
In addition, ATP activates PKC-δ through G protein-coupled receptors (73). Proteolytic reactions and ultraviolet radiation also activate PKC-δ by different mechanisms. In general, PKC-δ is activated by diacylglycerol after serine and threonine phosphorylation at the motif site, which induces enzyme activation and produces a catalytic fragment (36). PKC-δ activated by different mechanisms has different functions; thus, the role of PKC-δ in each signaling pathway needs to be further studied.
Mechanism of PKC-Δ Involvement in Cardiovascular Disease
PKC-δ regulates myocardial contraction, plays a role in ischemic preconditioning, and participates in the pathogenesis of myocardial IRI and fibrosis.
Myocardial Ischemia/Reperfusion Injury
An important method for the treatment of acute MI is revascularization, i.e., myocardial I/R treatment. However, reperfusion therapy itself can cause myocardial damage. PKC-δ plays a key role in injury related to ischemic tissue reperfusion. At present, some controversies exist regarding the role of PKC-δ in the process of ischemia and reperfusion.
Some studies suggest that PKC-δ activity may promote reperfusion injury after ischemia (74), which is partly mediated by the translocation of PKC-δ to mitochondria, which results in reductions in ADP-linked mitochondrial respiration (75), tricarboxylic acid cycle activity (76), cell pH, and ATP production rates (77). Mitochondria generate increased reactive oxygen species (75) and induce apoptosis by releasing cytochrome c (78). Inhibition of PKC-δ translocation with specific peptide inhibitors during reperfusion can block these effects and protect the heart from injury (75–78).
Inagaki et al. applied δv1-1 (a peptide inhibitor selective for PKC-δ) in an in vivo porcine model of AMI and showed that a PKC-δ inhibitor may improve the outcome of reperfusion in patients with AMI (77), Specifically, δv1-1 reduces the infarct area and troponin T levels in blood. Importantly, δv1-1 treatment improved the 30-min ejection fraction after reperfusion in pigs. The I/R-induced PKC-δ shift was completely inhibited by δv1-1 treatment in these hearts (41, 77, 79), indicating that I/R-induced myocardial injury was mediated by PKC-δ in vivo. Treatment with δV1-1 during ex vivo reperfusion can significantly improve functional recovery and improve myocardial ATP, phosphocreatine, and intracellular pH recovery during reperfusion (77). Therefore, δV1-1 may improve cardiac function recovery by reducing the time required to restore myocardial energy and pH during reperfusion (77). Studies have indicated that the use of PKC-δ selective inhibitors during IRI can reduce the injury of isolated perfused rat hearts (41, 80). Another study also noted that PKC-δ inhibitors can prevent replication in vivo in pig AMI models (77). In PKC-δ knockout mice, reductions in myocardial injury and infarct area were noted after coronary artery ischemia (41, 77, 81). The activation of PKC-δ in mitochondria contributes to I/R injury by decreasing intracellular pH and ATP production; increasing the accumulation of mitochondrial ROS; inhibiting the AKT-BAD phosphorylation pathway; promoting the release of cytochrome c, the activation of caspase-3, and the induction of apoptosis; and promoting I/R injury (76, 77, 82, 83). These results indicate that PKC-δ is an important component of the apoptosis signaling pathway. During myocardial I/R, phosphorylated PKC-δ translocates to mitochondria, and the release of cytochrome C increases significantly. These studies indicate that reperfusion injury following cardiac ischemia is mediated at least in part by the activation of PKC-δ. The inhibition of PKC-δ may reduce injury-related pathology in the process of reperfusion.
Rottlerin is a natural polyphenol compound that can inhibit PKC-δ. Studies have indicated that Ca2+-activated large conductance K+ (BKCa) activation can reduce severe ischemia induced by MI, promote survival, and improve heart and blood vessel function (84–89). Rottlerin can protect the heart by directly activating BKCa channels (90–92). Clements et al. demonstrated that rottlerin increases myocardial contraction and coronary perfusion by activating BKCa++ channels after cold cardiac arrest in isolated rats (93). Zheng et al. demonstrated that rottlerin significantly reduced apoptosis induced by reperfusion (94). Kai-9803 is a PKC-δ inhibitor; mechanistically, kai-9803 destroys the binding of PKC-δ and its activated C kinase receptor to prevent PKC-δ from mitochondrial localization during myocardial I/R (78). KAI-9803 reduces I/R injury in animal models with acute MI (77). Research indicates that it is necessary to further test kai-9803 as an adjuvant therapy for ST segment elevation MI (74). The I/R-induced PKC-δ shift (50% greater than the base) was completely inhibited by δv1-1 treatment in these hearts (41, 77, 79), indicating that I/R-induced myocardial injury was mediated by PKC-δ in vivo. These results suggest that inhibition of PKC-δ translocation inhibits apoptosis induced by reperfusion injury.
Previous reports have shown that apoptosis is a component of cardiomyocyte death during reperfusion (95). Myocardial I/R injury can activate endoplasmic reticulum (ER) stress. Continuous ER stress with a subsequent increase in intracellular Ca2+ concentration leads to apoptosis. In vivo, CaSR can activate PKC-δ to induce cardiomyocyte apoptosis through the ER stress-related apoptosis pathway (96). After IRI, the transformation of Caspase-3 into the active cleaved form increased significantly (97). However, the activity of Caspase-3 was greatly reduced by δ v1-1 treatment. These results suggest that inhibition of PKC-δ translocation can inhibit apoptosis induced by reperfusion injury. Murriel et al. demonstrated that PKC-δ activity and mitochondrial translocation at the beginning of reperfusion can promote the accumulation and dephosphorylation of apoptotic bad (Bcl-2-related death promoter), dephosphorylation of Akt, release of cytochrome c, cleavage of PARP (ADP ribose) polymerase, and DNA labeling (78). This finding suggests that PKC-δ activation plays a key role in promoting apoptosis in the cardiac response after ischemia and reperfusion. The application of PKC-δ inhibitors can inhibit this process and significantly reduce PKC-δ-induced apoptosis during reperfusion.
However, another study noted that PKC-δ knockout does not protect against but exaggerates I/R injury after pretreatment; in addition, the PKC-δ gene knockout-treated group had a larger infarct area than the untreated group (98). Mayr et al. provided evidence for loss of preconditioning-induced cardio protection in PKC-δ-deficient mice (99). In a related study, Fryer et al. used rottlerin, a PKC-δ selective antagonist, to observe its inhibitory effect on IPC-induced myocardial protection 30 min after ischemia and 2 h before reperfusion, suggesting that PKC-δ does not play an important role in IPC (100). A randomized controlled trial suggests that selective inhibition of PKC-δ does not reduce the biomarker of myocardial injury during PCI in patients with acute ST segment elevation MI (101).
Three recent studies suggest that PKC-δ may play a role in I/R through different signaling pathways. Remote ischemia per conditioning (RPerC) exhibits cardiac protective effects, and a study by Zhang et al. suggested that the underlying mechanism may occur through activation of opioid receptors and the NO-PKC-KATP channel signaling pathways (102). Diabetic myocardium shows higher vulnerability after IRI). It has been demonstrated that glucagon-like peptide-1 (GLP-1) has a protective effect on cardiomyocytes. High glucose-induced overexpression of PKC-δ in H9c2 cells destroys the GLP-1 receptor anti-apoptosis signaling pathway by inhibiting Akt phosphorylation (103). PKC-δ knockdown significantly restored the cardioprotective effects of GLP-1 in high glucose-cultured H9c2 cells (103). This finding suggests that high glucose-induced PKC-δ overexpression can reduce the protective effect of GLP-1 on the heart by downregulating GLP-1R and inhibiting the antiapoptotic signaling pathway of the GLP-1 receptor (103). Changes in mitochondrial metabolism are the primary cause of cardiovascular disease, and succinate is the mediator of mitochondrial dysfunction. The results showed that extracellular succinic acid promoted the transport of dynamic protein-related protein-1 (drp1) to mitochondria through the activation of PKC-δ. In addition, activation of extracellular signal regulated kinase-1/2 (ERK1/2) induced Gpr91-dependent phosphorylation of mitogen factor (MFF), leading to mitochondrial dysfunction and cardiomyocyte apoptosis. In the process of myocardial ischemia, succinic acid release activated Gpr91, which induced mitosis by regulating PKC-δ and ERK1/2 signaling branches. These findings indicate that inhibition of extracellular succinic acid mediated Gpr91 activation may represent a potential therapeutic strategy to protect myocardial cells from ischemia injury.
The role of PKC-δ in I/R may be affected by numerous factors. PKC-δ inhibitor content is related to the role of PKC-δ in I/R. Lower δV1 expression levels can inhibit ischemic-stimulated PKC-δ translocation, thereby protecting the heart from IRI. However, the severity of cardiomyopathy increases when δV1 is gradually expressed at higher levels in the heart (104). In the pretreated heart before ischemia, the ratio of proapoptotic PKC-δ and prosurvival PKC ε in mitochondria is regulated to determine the final fate of cells (83). In addition, different signal transduction pathways may also play an important role.
Fibrosis
Fibrosis can occur in a variety of organs. The main pathological changes are increased fibrous connective tissue and reduced parenchymal cells in organ tissues. Continuous progression of fibrosis can cause organ structure damage and functional decline and even failure, which seriously threatens human health and life. Fibrosis can lead to myocardial malfunction. Cardiac fibrosis occurs in a variety of cardiac pathological conditions. Cardiac fibrosis can reduce contractility and ultimately lead to heart failure. In addition, the period of CF will increase the production of extracellular matrix, destroy the myocardial circuits, induce arrhythmia, and even cause sudden cardiac death (105). Protein kinase C has been shown to modulate specific events leading to collagen deposition (106). The PKC-δ selective peptide inhibitor δV1-1 increased TGF-β1-induced proliferation (106). The data reported by Braun and Mochly-Rosen provide evidence that PKC-δ inhibits neonatal rat cardiac fibroblast proliferation (107).
Collagen is the main component of the extracellular matrix. The regulation of extracellular matrix remodeling is primarily mediated by cardiac fibroblasts (108–110). Under disease conditions, increased accumulation of interstitial collagens leads to fibrosis (111). Proliferation plays a key role in cardiac fibrosis. Ang II is an effective activator of CF, and activation of Ang II type 1 receptors can stimulate cell proliferation and de novo collagen synthesis (112, 113). Recent studies show that PKC-δ has a role in Ang II-induced cardiac fibrosis (114). Myofibroblasts (myoFb) represent the largest number of fibroblasts present in myocardial tissue. These cells can synthesize and maintain extracellular matrix, have autocrine and paracrine functions, and play an important role in maintaining normal heart function. Previous studies have demonstrated that Ang II induces myoFb proliferation and that myoFb is associated with cardiac fibrosis (115–119). When myocardial damage occurs for various reasons, fibroblasts proliferate, transform into myofibroblasts, increase extracellular matrix deposition, and cause cardiac fibrosis. PKC-δ-overexpressing myoFb showed a significant increase in PKC-δ activity and collagen expression. In addition, treatment with rottlerin, a PKC-δ inhibitor, significantly abrogated PKC-δ activity and Ang II-induced collagen expression in myoFb (114). A study by Olson et al. suggested that PKC-δ participates in transactivation of Ang II-induced cardiac fibroblasts ERK1/2 (a member of the tyrosine kinase pathway) (120). Chintalgattu and Katwa suggested that Ang II mediates the activity of PKC-δ and that inhibiting PKC-δ can abrogate Ang II-induced kinase activity. PKC-δ plays a key role in Ang II-induced collagen expression/fibrosis (114). Connective tissue growth factor (CTGF) is believed to play a variety of biological roles, including regulation of fibroblast proliferation, collagen synthesis, and apoptosis (121). Ang II increases CTGF expression in vivo (122). He et al. directly tested the regulation of CTGF expression by PKC-δ isoform activation after Ang II stimulation in vitro and in vivo, and the results indicated that PKC-δ inhibition may aggravate the expression of CTGF and potential fibrosis (123). Inhibition of PKC-δ leads to strong induction of CTGF expression after Ang II treatment (123).
PKC-δ in Cardiovascular Disease
PKC-δ is involved in many cardiovascular diseases, such as atherosclerosis, hypertension, myocardial hypertrophy, and heart failure (Table 2). Our current concepts regarding the cardiac actions of PKC-δ are derived in large part from studies that rely on peptide translocation inhibitors or studies that characterize the phenotype of nPKC knockout mice (140).
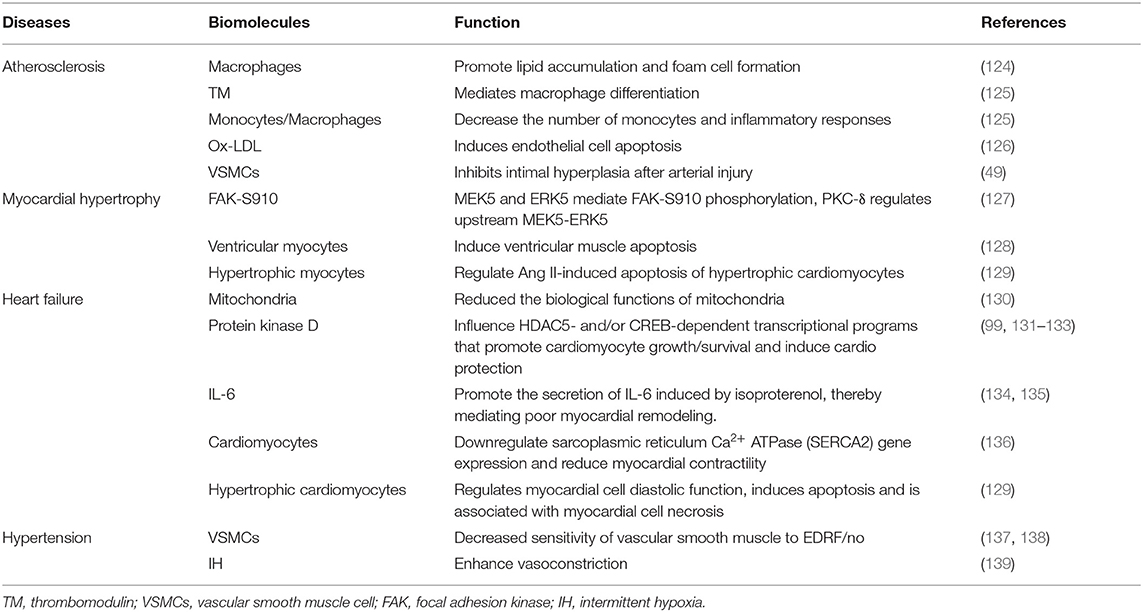
Table 2. The mechanism of PKC-δ in different cardiovascular diseases.
Atherosclerosis
Atherosclerosis is a chronic inflammatory reaction and the most common pathological cause of cardiovascular disease. The development of atherosclerosis involves many cells (including endothelial cells, VSMCs, macrophages, and others) (141). Macrophages participate in the intracellular accumulation of lipids and the formation of foam cells in early atherosclerotic lesions (124). Macrophage uptake of oxidized lipoprotein particles (ox-LDL) and foam cells is the first step in the development of atherosclerosis (142). Studies have shown that macrophage differentiation is an important process in the development of atherosclerosis, and thrombomodulin (TM), a transmembrane glycoprotein expressed on the cell surface, is a signaling molecule that mediates macrophage differentiation (125). Tsai et al. provided a new perspective on the role of TM in the differentiation of macrophages during the development of atherosclerosis and found that PKC-δ is highly expressed in human atherosclerotic arteries and infiltrates with TM in CD68-positive macrophages. TM-PKC-δ interactions in macrophages may contribute to the formation of atherosclerosis (125). Activation of monocytes/macrophages contributes to atherosclerosis, and the activation of the PKC-δ isoform in monocytes can decrease the number of monocytes and inflammatory responses in the arterial wall, thus slowing the development of atherosclerosis (143). Curcumin inhibits matrix invasion by inhibiting the PKC-δ/NADPH oxidase/ROS signaling pathway during monocyte macrophage differentiation to intervene in the process of atherosclerosis (144). PKC-δ may be closely related to the occurrence of atherosclerosis (144). Under pathological conditions, endothelial cell apoptosis is the initial step in atherosclerosis and contributes to the subsequent atherosclerosis process (145). Ox-LDL (an important atherosclerotic factor) induces endothelial cell apoptosis as an important process in the development of atherosclerosis. LOX-1 is overexpressed in atherosclerotic endothelial cells (146). Zhou and Chen's research provides evidence that lincRNA-p21 is the main mediator of human vascular endothelial cell apoptosis and that LOX-1 expression is induced by ox-LDL and works by activating PKC-δ (126). Smooth muscle cell (SMC) accumulation is a key event in the development of atherosclerosis. A study of a rat arterial injury model found that PKC-δ upregulation is accompanied by the apoptosis of vascular wall VSMCs and that PKC-δ inhibits intimal hyperplasia after arterial injury by stimulating apoptosis of VSMCs (49). Leitges et al. isografted vein segments from PKC-δ−/− mice and found more severe atherosclerosis compared with that noted in PKC-δ+/+ mouse vein grafts, which correlated with decreased SMC lesions (43). Above evidence indicated that PKC-δ activation enhances the apoptosis of SMCs and prevents the accumulation of SMCs, which is of great significance in the amelioration of atherosclerosis.
Myocardial Hypertrophy
PKC-δ is found in adult rat cardiac myocytes and cultured neonatal ventricular myocytes (34). PKC-δ is also detected in total protein extracts from isolated adult ventricular myocytes (147). PKC-δ has been implicated in the downregulation of SERCA2 [sarco(endo)plasmic reticulum Ca2+-ATPase] expression, a characteristic feature of the hypertrophic phenotype. A study of a pressure overload model of abdominal aortic coarctation showed that PKC-δ self-phosphorylation and activity increased in the process of ventricular hypertrophy, and PKC-δ expression also increased significantly with the gradual emergence and aggravation of left ventricular hypertrophy (148). The pressure overload model of ascending aortic stenosis also confirmed that PKC-δ expression increased during left ventricular hypertrophy (149). The Dahl salt-sensitive rat heart failure model also confirmed that PKC-δ expression continued to increase during the transformation from cardiac hypertrophy to heart failure. In the volume overload model, PKC-δ protein and mRNA levels also increased significantly (150). Yi et al. suggested that FAK may represent a candidate gene for nuclear hypertrophy gene transcription and RNA processing in chronic hypertensive cardiac hypertrophy (151). FAK-S910 phosphorylation is necessary for the formation of new costumeries in the process of myocardial cell remodeling. FAK-S910 phosphorylation is partially mediated by MEK5 and ERK5, and PKC-δ regulates upstream MEK5-ERK5 (127). Rottlerin treatment or PKC-δ-KD (kinase dead PKC-δ) expression inhibits PKC-δ, thereby inhibiting apoptosis induced by a variety of stimuli (152, 153). Adenovirus transfection studies have also shown that activation of PKC-δ in ventricular myocytes can directly induce ventricular muscle apoptosis (128). In summary, PKC-δ regulates myocardial cell diastolic function, induces apoptosis and is associated with myocardial cell necrosis. Apoptosis of hypertrophic cardiomyocytes is the key mechanism of heart failure. Cardiac hypertrophy is an adaptive process to increase hemodynamic overload. Rottlerin reduces the apoptotic rates induced by Ang II in mildly hypertrophic myocytes and in moderately hypertrophic myocytes, but rottlerin did not affect the apoptotic rate induced by Ang II in serum-free cultured myocytes. These results suggest that inhibition of PKC-δ reduces Ang II-induced apoptosis of hypertrophic cardiomyocytes and that PKC-δ is possibly involved in the apoptotic process of hypertrophic cardiomyocytes (129).
Heart Failure
As a stress protein, PKC-δ is activated in the early stage of pressure overload and heart failure (154). Yang et al. concluded that cardiac ROS-dependent activated PKC-δ colocalizes with mitochondria, and Drp1-dependent mitochondrial fission and fragmentation reduced the biological functions of mitochondria, resulting in cardiac failure (130). Myocardial ischemia and MI are the two most common reasons for heart failure (155). The degree of cardiac dysfunction after I/R injury reflects the level of myocyte injury and apoptosis (156). Although the role of PKC-δ in the pathogenesis of heart failure is not conclusive, many studies have confirmed that PKC-δ is associated with the occurrence of heart failure. Shende et al. reported that PKCβII deficiency and increased PKC-δ are important reasons for rector-deficient heart dysfunction after pressure overload (157). PKC-δ act as a lipid-independent enzyme in the soluble fraction of cardiomyocytes undergoing oxidative stress (57, 158). In addition, PKC-δ can contribute to receptor-dependent pathways that activate protein kinase D and thereby influence HDAC5- and/or CREB-dependent transcriptional programs that promote cardiomyocyte growth/survival and induce cardio protection (99, 131–133). IL-6 is a multifunctional cytokine that is an important biomarker that predicts the severity, prognosis, and mortality of heart failure (159). IL-6 is involved in mediating adverse myocardial remodeling (134). PKC-δ mediates IL-6 secretion induced by isoprenaline (135).
Compensatory cardiac hypertrophy caused by hypertrophic cardiomyopathy is the most important factor leading to heart failure. PKC-δ regulates myocardial cell diastolic function, induces apoptosis, and is associated with myocardial cell necrosis (129). Apoptosis of hypertrophic cardiomyocytes is the key mechanism of heart failure. Myocardial hypertrophy is a process that adapts to the increase in hemodynamic load. Rottlerin is a specific inhibitor of PKC-δ that can inhibit the apoptosis rate of mildly hypertrophic cardiomyocytes and moderately hypertrophic cardiomyocytes (129). These results suggest that inhibition of PKC-δ reduces Ang II-induced apoptosis of hypertrophic cardiomyocytes (129). Low-dose PKC-δ is beneficial to the adaptation of cardiomyocytes to exercise stimulation (160), This mechanism may protect the heart muscle by regulating the mitochondrial K+ channel (161). Long-term PKC-δ expression can damage cardiomyocytes, and PKC-δ can significantly downregulate sarcoplasmic reticulum Ca2+ ATPase (SERCA2) gene expression, and reduce myocardial contractility (136).
PKC-δ is related to cardiomyocyte hypertrophy, which can move from cytoplasm to mitochondria, and then trigger apoptosis signal pathway. Xie et al. shows that the mitochondrial pathway of PKC-δ determines the apoptosis signal of hypertrophic cardiomyocytes (162). PKC-δ may be involved in the transition from cardiac hypertrophy to apoptosis (129). In a dog model of pacing-induced heart failure, the apoptosis rate increased significantly (78, 163), and apoptosis was further increased in heart failure (164). In addition, Ozgen et al. found that the Galpha(q)- PKC-δ-PKD-CREB-Ser (126) phosphorylation pathway was involved in cardiac remodeling and could be used as a target for heart failure treatment (132).
Hypertension
A high level of PKC activity is a typical characteristic of different types of arterial hypertension (137, 165). The main reason for the reduced aortic endothelium-dependent diastolic function in spontaneously hypertensive rats is the decreased sensitivity of vascular smooth muscle to EDRF/no, which is caused by the increase in PKC activity (137, 138). Novokhatska et al. showed that increasing the activity of the PKC-δ isoform in the vascular wall can decrease the endothelium-dependent relaxation induced by acetylcholine, which is related to the tall outward potassium current of spontaneously hypertensive rats. In addition, an RNA interference technique can be used to silence the PKC-δ isoform gene to restore normal vascular contractility in hypertensive rats (166).
Phosphorylation of tyrosine residues in focal adhesion kinase (FAK) is necessary for cardiomyocytes to respond to growth factor and mechanical load M, and serine and tyrosine phosphorylation of FAK is significantly increased in rats with hypertension (151). Related reports showed that the phosphorylation level of serine residues in FAK increased significantly in hypertensive rats, and different phosphorylation sites can regulate the late subcellular localization of FAK (167). Gerasymenko et al. showed that transgenic lettuce plants producing shRNA targeting PKC-δ can be used as a raw material for the development of novel drugs for hypertensive treatment (168).
Obstructive sleep apnea (OSA) can cause intermittent hypoxia (IH), which is one of the risk factors for hypertension (169). Studies in rodent models have shown that prolonged sleep apnea can lead to vascular dysfunction (170). Kanagy et al. indicated that IH exposure increases plasma ET-1, which may contribute to hypertension (171). Given the increase in circulating ET-1 levels, rats exposed to IH showed an increased contractile response to ET-1 in the mesenteric artery (172). The enhancement of vasoconstriction induced by IH is mediated by PKC-δ, and ET-1 increases the level of self-phosphorylation of PKC-δ (139). In addition, rottlerin reduces ET-1-mediated contraction in the arteries of rats exposed to IH (169). These results indicate that PKC-δ is involved in the hypertension response to ET-1 after IH exposure.
Discussion
PKC-δ is a serine/threonine kinase that plays a key role in growth regulation and tissue remodeling. PKC-δ activation mediates the pathological process of various cardiovascular diseases. Traditional PKC-δ activation models focus on the anchoring of physiological second messengers (such as DAG diacylglycerol) or tumor-promoting PSs (such as PMA) in the active conformation of PKC membranes. However, this receptor-driven, lipid cofactor-dependent PKC activation mechanism involving membrane-associated anchoring proteins does not adequately explain PKC-dependent phosphorylation of no membranous proteins. Therefore, traditional PKC-δ activation models must be broadened to allow for stimulation-specific differences in PKC-δ signals during growth factor stimulation and oxidative stress. New activation methods, such as tyrosine phosphorylation, provide the possibility for further research on PKC-δ. Many studies have relied on in vitro kinase analysis to address tyrosine phosphorylation-dependent changes in PKC-δ function. However, the catalytic activity of tyrosine-phosphorylated PKC-δ is variably described as reducing, increasing, or even changing in terms of substrate specificity and cofactor requirements. At present, there is no uniform conclusion on PKC-δ tyrosine phosphorylation. Further research is needed. PKC-δ activation is achieved by coordinating phosphorylation and transport events. When evaluating PKC-δ signaling pathways, these two mechanisms must be considered. In addition, a drug inhibitor should be designed to eliminate the effects of PKC-δ.
PKC-δ can also be used as the target of SRC and RAS transformation or the phosphorylation of tyrosine residues stimulated by PMA, epidermal growth factor, platelet-derived growth factor, and hydrogen peroxide. However, the precise biological results of PKC-δ tyrosine phosphorylation have been difficult to decipher due to the presence of the regulatory domain (tyr52, tyr155, and tyr187), catalytic domain (tyr512 and tyr523), and hinge region (tyr311 and tyr332) in PKC-δ. However, exclusively focusing on the driving effect of PKC-δ phosphorylation may be myopic (or even misplaced). In view of recent evidence, PKC-δ phosphotyrosine can also be used as the docking site of other signal proteins. The role of PKC-δ as a signal-regulated scaffold can be the basis of some of its effector functions.
PKC-δ activation signaling regulates myocardial contraction and plays an important regulatory role in myocardial I/R injury and cardiac fibrosis. At present, research on the role of PKC-δ in I/R injury is mainly based on an in vivo AMI pig model, an in vitro rat heart model, and a PKC-δ gene knockout mouse model. At present, some controversies about the role of PKC-δ in the process of ischemia and reperfusion exist. Evidence in mouse and pig models suggest that PKC-δ promotes I/R injury, and inhibition of PKC-δ can block these effects. Inhibiting PKC-δ translocation with specific peptide inhibitors during reperfusion can block these effects and protect the heart from harm (75–78). The study of PKC-δ inhibitors in vivo and in vitro suggests that inhibition of PKC-δ plays a role in reducing the infarct area and troponin T level; inhibiting the I/R-induced PKC-δ shift; improving functional recovery during reperfusion; and improving myocardial ATP, creatine phosphate, and intracellular pH recovery. In addition, inhibition of PKC-δ reduces caspase-3 activity and has a positive role in inhibiting apoptosis.
These results suggest that inhibition of PKC-δ may be an important target for drug development to prevent irreversible cardiac injury during reperfusion. However, a random controlled trial (101) suggested that selective inhibition of PKC-δ does not reduce the biomarkers of myocardial injury in PCI of acute ST segment elevation MI, underscoring the differences in animal models and humans. Further clinical trials are necessary to verify the current conclusions and then determine future research directions. The role of PKC-δ in I/R may be related to the amount of PKC-δ inhibitor. A study (104) indicated that lower δV1 expression levels can inhibit ischemic-stimulated PKC-δ translocation, thereby protecting the heart from IRI. However, the severity of cardiomyopathy increases when δV1 is gradually expressed at higher levels in the heart. In addition, recent studies have found that PKC-δ may play a role in I/R through specific signaling pathways, which provides a new idea for the study of the mechanism of PKC-δ in I/R.
At present, studies have shown that PKC-δ is related to the occurrence of heart failure, and its mechanism may involve hypertrophic hearts. We should explore the mechanism of PKC-δ and provide new ideas for the prevention and treatment of heart failure. PKC-δ was highly expressed in human atherosclerotic arteries infiltrated with TM and macrophages positive for CD68, suggesting that the interaction of macrophage tm-PKC-δ may participate in the formation of atherosclerosis (125). The activation of monocytes/macrophages contributes to the formation of atherosclerosis. Activation of the PKC-δ subtype of monocytes can reduce the number of PKC-δ subtypes and the inflammatory response of the arterial wall, thus slowing the development of atherosclerosis (143). Curcumin inhibits matrix invasion during monocyte phage differentiation by inhibiting the PKC-δ/NADPH oxidase/ROS signaling pathway, thus interfering with atherosclerosis (144). Zhou and Chen showed that lincrna-p21 was the main mediator of oxidized LDL-induced apoptosis and LOX-1 expression in human vascular endothelial cells by activating PKC-δ (126). PKC-δ upregulation was accompanied by VSMC apoptosis, and PKC-δ inhibited intimal hyperplasia by stimulating vascular smooth muscle cell apoptosis (49). At present, there is no unified theory regarding the mechanism of PKC-δ in atherosclerosis. Research on specific PKC-δ-targeting drugs should be performed in the future. The pressure overload model of abdominal aortic coarctation (148), the pressure overload model of ascending aortic stenosis (149), the Dahl salt-sensitive rat heart failure model and the volume overload model have been studied (150). Studies using these models confirmed that PKC-δ participates in cardiac hypertrophy. However, literature to support the specific mechanism is lacking. PKC-δ is involved in endothelium-dependent systolic and diastolic function in spontaneously hypertensive rats and in the response of hypertension to ET-1.
The role and mechanisms of PKC-δ in CVD are complex and involve different cardiovascular biological processes by mediating different biological factors. PKC-δ mediates lipid accumulation and foam cell formation, macrophage differentiation, monocyte number, inflammatory response, and endothelial cell apoptosis through the regulation of macrophage (124), TM (125), monocyte/macrophage (125), OX-LDL (126), VSMCs (49), and other biological factors. However, its role in heart failure is mainly to regulate cardiac remodeling and myocardial contractility by regulating IL-6 secretion (134), SERCA2 gene expression (136), and diastolic function of cardiomyocytes. PKC-δ regulates hypertension by mediating VSMCs (137, 138) and IH (139), reduces the sensitivity of vascular smooth muscle to EDRF/NO, and enhances vasoconstriction.
Protein kinase C inhibitors and activators are used for the diagnosis and treatment of various PKC-associated diseases, such as cancers, neurological diseases, cardiovascular diseases, and infections. However, the application of PKC in cardiovascular diseases still needs exploration. Flosequinan is a drug that can generally inhibit PKC isoenzymes. Studies have found that it produces continuous hemodynamic benefits in heart failure (173). PKC-ε activators such as volatile, anesthetics, and adenosine acadesine are used in the treatment of coronary artery bypass grafting and acute MI (174–177). Lincoff et al. concluded that selective inhibition of PKC-δ with intravenous infusion of delcasertib during PCI for acute STEMI in a population of patients treated according to the contemporary standard of care did not reduce biomarkers of myocardial injury (101). At present, there are relatively few clinical trials of cardiovascular diseases that target PKC, and the results are not as satisfactory as the results of animal experiments. The high homology of PKC isoenzymes makes the clinical transformation of PKC challenging, and drugs targeting a single isoenzyme are difficult to achieve. In addition, unpredictable adverse effects, insufficient therapeutic effects, insufficient preclinical studies, and lack of blood biomarkers are also important reasons leading to unsatisfactory clinical results.
This article reviews the role of PKC-δ in the development of cardiovascular disease. In recent years, an increasing number of studies have found that PKC-δ plays an important role in the occurrence, development, and prevention of cardiovascular diseases. The discussion of its activation mechanism has expanded from traditional activation methods, such as diacylglycerols and PSs, to tyrosine phosphorylation. Disease research has also been extended to hypertrophic cardiomyopathy, atherosclerosis, hypertension, and heart failure. In-depth drug research on PKC-δ is also increasing, and the role of PKC-δ in the pathogenesis of CVD is currently being explored through drug research. The role of PKC-δ as a potential molecular target for cardiovascular intervention requires further research. This review article provides an important theoretical basis to better understand current progress in research on PKC-δ in cardiovascular disease.
Perspective
Although PKC-δ is a very promising target for cardiovascular diseases, there are still many limitations to be solved urgently. First, despite the role of PKC-δ has been proved in previous experiments, the results are not satisfying in pre-clinical trials. This may partly be explained by the difference of animal models and corresponding human diseases. Second, there is a high degree of substantial homology between PKC isoenzymes. Many PKC isoenzymes exist in the same cell and are activated by the same stimulus. This feature of high homology makes it difficult to develop a single pharmacological tool that affects PKC-δ. Post-translational modifications of PKC-δ and upstream molecular regulation of the PKC-δ activation pathway may be promising targets for drug discovery. Third, there are few studies concerning the role of PKC-δ in specific heart diseases, such as alcoholic cardiomyopathy and obese cardiomyopathy currently, which suggests further research are necessary. Fourth, PKC-δ is widely expressed in various tissues and cells throughout the body and lacks specificity. In the future, the development of inhibitors or activators for PKC-δ subtypes may provide a way to tackle this difficulty. In addition, PKC-δ-related blood biomarkers are insufficient. To date, there are no PKC specific biomarkers to indicate dose adequacy or predict phenotypic outcomes, which can also be improved in future studies.
Conclusion
The current evidence proves that the activation and inhibition of PKC-δ has an important regulatory role in CVD. PKC-δ-mediated apoptosis, inflammatory, cardiac remodeling, and vascular contraction and dilatation lead to several biology effects, and thus bring hope for the treatment of CVD. However, there are still many limitations in current research, more in-depth research can be further conducted to explore new biomarkers and the specific functions of different PKC-δ subtypes.
Author Contributions
MG and D-ZS: conceptualization and supervision. L-nM and DP: literature search and summary. J-pD: project administration. L-nM and P-fC: writing original draft. MG, D-ZS, and JG: writing review and editing. D-ZS, JS, and Y-qY revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (Grant no. 81904025), Fundamental Research Funds for the Central Public Welfare Research Institutes (Grant no. ZZ13-YQ-016 and ZZ13-YQ-016-C1), and National Natural Science Foundation of China Youth Fund Project (Grant no. 81904046).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singh RM, Cummings E, Pantos C, Singh J. Protein kinase C and cardiac dysfunction: a review. Heart Fail Rev. (2017) 22:843–59. doi: 10.1007/s10741-017-9634-3
2. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
3. Nishizuka Y, Nakamura S. Lipid mediators and protein kinase C for intracellular signalling. Clin Exp Pharmacol Physiol Suppl. (1995) 22:S202–3. doi: 10.1111/j.1440-1681.1995.tb02883.x
4. Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. (1998) 332(Pt 2):281–92. doi: 10.1042/bj3320281
5. Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. (2001) 101:2353–64. doi: 10.1021/cr0002801
6. Freeley M, Kelleher D, Long A. Regulation of protein kinase C function by phosphorylation on conserved and non-conserved sites. Cell Signal. (2011) 23:753–62. doi: 10.1016/j.cellsig.2010.10.013
7. Reyland ME. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci. (2009) 14:3385. doi: 10.2741/3385
8. Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. (2008) 88:1341–78. doi: 10.1152/physrev.00034.2007
9. Mamidi N, Panda S, Borah R, Manna D. Synthesis and protein kinase C (PKC)-C1 domain binding properties of diacyltetrol based anionic lipids. Mol Biosyst. (2014) 10:3002–13. doi: 10.1039/C4MB00382A
10. Mamidi N, Gorai S, Ravi B, Manna D. Physicochemical characterization of diacyltetrol-based lipids consisting of both diacylglycerol and phospholipid headgroups. RSC Adv. (2014) 4:21971–8. doi: 10.1039/C4RA02495H
11. Marrocco V, Bogomolovas J, Ehler E, Dos Remedios CG, Yu J, Gao C, et al. PKC and PKN in heart disease. J Mol Cell Cardiol. (2019) 128:212–26. doi: 10.1016/j.yjmcc.2019.01.029
12. Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. (2010) 106:1319–31. doi: 10.1161/CIRCRESAHA.110.217117
13. Totoń E, Ignatowicz E, Skrzeczkowska K, Rybczyńska M. Protein kinase Cε as a cancer marker and target for anticancer therapy. Pharmacol Rep. (2011) 63:19–29. doi: 10.1016/S1734-1140(11)70395-4
14. Zanin-Zhorov A, Dustin ML, Blazar BR. PKC-θ function at the immunological synapse: prospects for therapeutic targeting. Trends Immunol. (2011) 32:358–63. doi: 10.1016/j.it.2011.04.007
15. Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson's disease. J Pharmacol Exp Ther. (2007) 322:913–22. doi: 10.1124/jpet.107.124669
16. Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. (2011) 472:319–24. doi: 10.1038/nature09788
17. Son M, Oh S, Jang JT, Son KH, Byun K. Pyrogallol-phloroglucinol-6 6-bieckol on attenuates high-fat diet-induced hypertension by modulating endothelial-to-mesenchymal transition in the aorta of mice. Oxid Med Cell Longev. (2021) 2021:8869085. doi: 10.1155/2021/8869085
18. McCauley MD, Hong L, Sridhar A, Menon A, Perike S, Zhang M, et al. Ion channel and structural remodeling in obesity-mediated atrial fibrillation. Circ Arrhythm Electrophysiol. (2020) 13:e008296. doi: 10.1161/CIRCEP.120.008296
19. Zhang Y, Da Q, Cao S, Yan K, Shi Z, Miao Q, et al. HINT1 (Histidine triad nucleotide-binding protein 1) attenuates cardiac hypertrophy via suppressing HOXA5 (homeobox A5) expression. Circulation. (2021) 144:638–54. doi: 10.1161/CIRCULATIONAHA.120.051094
20. Zhang L, Huang D, Shen D, Zhang C, Ma Y, Babcock SA, et al. Inhibition of protein kinase C βII isoform ameliorates methylglyoxal advanced glycation endproduct-induced cardiomyocyte contractile dysfunction. Life Sci. (2014) 94:83–91. doi: 10.1016/j.lfs.2013.11.011
21. Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. (2006) 70:222–30. doi: 10.1016/j.cardiores.2006.02.015
22. Ferreira JC, Brum PC, Mochly-Rosen D. βIIPKC and εPKC isozymes as potential pharmacological targets in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. (2011) 51:479–84. doi: 10.1016/j.yjmcc.2010.10.020
23. Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Haemost. (2010) 8:454–62. doi: 10.1111/j.1538-7836.2009.03722.x
24. Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. (2010) 24:225–34. doi: 10.1007/s10557-010-6236-x
25. Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol Res. (2007) 55:523–36. doi: 10.1016/j.phrs.2007.04.005
26. Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. (2008) 321:1493–5. doi: 10.1126/science.1158554
27. Ferreira JC, Mochly-Rosen D, Boutjdir M. Regulation of cardiac excitability by protein kinase C isozymes. Front Biosci. (2012) 4:283. doi: 10.2741/s283
28. Chen L, Shi D, Guo M. The roles of PKC-δ and PKC-ε in myocardial ischemia/reperfusion injury. Pharmacol Res. (2021) 170:105716. doi: 10.1016/j.phrs.2021.105716
29. Deuse T, Koyanagi T, Erben RG, Hua X, Velden J, Ikeno F, et al. Sustained inhibition of epsilon protein kinase C inhibits vascular restenosis after balloon injury and stenting. Circulation. (2010) 122:S170–8. doi: 10.1161/CIRCULATIONAHA.109.927640
30. Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. (2012) 11:937–57. doi: 10.1038/nrd3871
31. Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. Identification of three additional members of rat protein kinase C family: delta-, epsilon- and zeta-subspecies. FEBS Lett. (1987) 226:125–8. doi: 10.1016/0014-5793(87)80564-1
32. Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. (1997) 6:477–80. doi: 10.1002/pro.5560060228
33. Szallasi Z, Bogi K, Gohari S, Biro T, Acs P, Blumberg PM. Non-equivalent roles for the first and second zinc fingers of protein kinase Cdelta. Effect of their mutation on phorbol ester-induced translocation in NIH 3T3 cells. J Biol Chem. 271:18299–301. doi: 10.1074/jbc.271.31.18299
34. Hunn M, Quest AF. Cysteine-rich regions of protein kinase Cdelta are functionally non-equivalent. Differences between cysteine-rich regions of non-calcium-dependent protein kinase Cdelta and calcium-dependent protein kinase Cgamma. FEBS letters. (1997) 400:226–32. doi: 10.1016/S0014-5793(96)01395-6
35. Shindo M, Irie K, Nakahara A, Ohigashi H, Konishi H, Kikkawa U, et al. Toward the identification of selective modulators of protein kinase C (PKC) isozymes: establishment of a binding assay for PKC isozymes using synthetic C1 peptide receptors and identification of the critical residues involved in the phorbol ester binding. Bioorg Med Chem. (2001) 9:2073–81. doi: 10.1016/S0968-0896(01)00100-6
36. Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem. (2002) 132:831–9. doi: 10.1093/oxfordjournals.jbchem.a003294
37. Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. (2005) 121:271–80. doi: 10.1016/j.cell.2005.02.019
38. Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. EMBO J. (2000) 19:496–503. doi: 10.1093/emboj/19.4.496
39. Gschwendt M, Müller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. (1994) 199:93–8. doi: 10.1006/bbrc.1994.1199
40. Szallasi Z, Denning MF, Smith CB, Dlugosz AA, Yuspa SH, Pettit GR, et al. Bryostatin 1 protects protein kinase C-delta from down-regulation in mouse keratinocytes in parallel with its inhibition of phorbol ester-induced differentiation. Mol Pharmacol. (1994) 46:840–50.
41. Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci USA. (2001) 98:11114–9. doi: 10.1073/pnas.191369098
42. Niino YS, Kawashima I, Iguchi Y, Kanda H, Ogura K, Mita-Yoshida K, et al. PKCδ deficiency inhibits fetal development and is associated with heart elastic fiber hyperplasia and lung inflammation in adult PKCδ knockout mice. PLoS ONE. (2021) 16:e0253912. doi: 10.1371/journal.pone.0253912
43. Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, et al. Exacerbated vein graft arteriosclerosis in protein kinase Cdelta-null mice. J Clin Invest. (2001) 108:1505–12. doi: 10.1172/JCI200112902
44. Sakurai Y, Onishi Y, Tanimoto Y, Kizaki H. Novel protein kinase C delta isoform insensitive to caspase-3. Biol Pharm Bull. (2001) 24:973–7. doi: 10.1248/bpb.24.973
45. Kawaguchi T, Niino Y, Ohtaki H, Kikuyama S, Shioda S. New PKCdelta family members, PKCdeltaIV, deltaV, deltaVI, and deltaVII are specifically expressed in mouse testis. FEBS Lett. (2006) 580:2458–64. doi: 10.1016/j.febslet.2006.03.084
46. Kim JD, Seo KW, Lee EA, Quang NN, Cho HR, Kwon B. A novel mouse PKCδ splice variant, PKCδIX, inhibits etoposide-induced apoptosis. Biochem Biophys Res Commun. (2011) 410:177–82. doi: 10.1016/j.bbrc.2011.04.096
47. Ueyama T, Ren Y, Ohmori S, Sakai K, Tamaki N, Saito N. cDNA cloning of an alternative splicing variant of protein kinase C delta (PKC deltaIII), a new truncated form of PKCdelta, in rats. Biochem Biophys Res Commun. (2000) 269:557–63. doi: 10.1006/bbrc.2000.2331
48. Apostolatos H, Apostolatos A, Vickers T, Watson JE, Song S, Vale F, et al. Vitamin A metabolite, all-trans-retinoic acid, mediates alternative splicing of protein kinase C deltaVIII (PKCdeltaVIII) isoform via splicing factor SC35. J Biol Chem. (2010) 285:25987–95. doi: 10.1074/jbc.M110.100735
49. Yamanouchi D, Kato K, Ryer EJ, Zhang F, Liu B. Protein kinase C delta mediates arterial injury responses through regulation of vascular smooth muscle cell apoptosis. Cardiovasc Res. (2010) 85:434–43. doi: 10.1093/cvr/cvp328
50. Ushio K, Hidenori M, ToshiyoshiK Y. Protein Kinase Cδ (PKCδ): Activation Mechanisms and Functions. J. Biochem. (2002) 831–39.
51. Mizuno K, Kubo K, Saido TC, Akita Y, Osada S, Kuroki T, et al. Structure and properties of a ubiquitously expressed protein kinase C, nPKC delta. Eur J Biochem. (1991) 202:931–40. doi: 10.1111/j.1432-1033.1991.tb16453.x
52. Takai Y, Kishimoto A, Kikkawa U, Mori T, Nishizuka Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem Biophys Res Commun 1979. (2012) 425:571–7. doi: 10.1016/0006-291x(79)91197-5
53. Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. (1982) 257:7847–51. doi: 10.1016/S0021-9258(18)34459-4
54. Mischak H, Goodnight JA, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, et al. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. (1993) 268:6090–6. doi: 10.1016/S0021-9258(18)53223-3
55. Mischak H, Pierce JH, Goodnight J, Kazanietz MG, Blumberg PM, Mushinski JF. Phorbol ester-induced myeloid differentiation is mediated by protein kinase C-alpha and -delta and not by protein kinase C-beta II, -epsilon, -zeta, and -eta. J Biol Chem. (1993) 268:20110–5. doi: 10.1016/S0021-9258(20)80701-7
56. Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, et al. Tyrosine phosphorylation modifies protein kinase C delta-dependent phosphorylation of cardiac troponin I. J Biol Chem. (2008) 283:22680–9. doi: 10.1074/jbc.M802396200
57. Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, et al. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci USA. (2001) 98:6587–92. doi: 10.1073/pnas.111158798
58. Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. (2004) 384:449–59. doi: 10.1042/BJ20040704
59. Li W, Mischak H, Yu JC, Wang LM, Mushinski JF, Heidaran MA, et al. Tyrosine phosphorylation of protein kinase C-delta in response to its activation. J Biol Chem. (1994) 269:2349–52. doi: 10.1016/S0021-9258(17)41948-X
60. Denning MF, Dlugosz AA, Threadgill DW, Magnuson T, Yuspa SH. Activation of the epidermal growth factor receptor signal transduction pathway stimulates tyrosine phosphorylation of protein kinase C delta. J Biol Chem. (1996) 271:5325–31. doi: 10.1074/jbc.271.10.5325
61. Gschwendt M, Kielbassa K, Kittstein W, Marks F. Tyrosine phosphorylation and stimulation of protein kinase C delta from porcine spleen by SRC in vitro. Dependence on the activated state of protein kinase C delta. FEBS Lett. (1994) 347:85–9. doi: 10.1016/0014-5793(94)00514-1
62. Zang Q, Lu Z, Curto M, Barile N, Shalloway D, Foster DA. Association between v-Src and protein kinase C delta in v-Src-transformed fibroblasts. J Biol Chem. (1997) 272:13275–80. doi: 10.1074/jbc.272.20.13275
63. Frank GD, Saito S, Motley ED, Sasaki T, Ohba M, Kuroki T, et al. Requirement of Ca(2+) and PKCdelta for Janus kinase 2 activation by angiotensin II: involvement of PYK2. Mol Endocrinol. (2002) 16:367–77. doi: 10.1210/mend.16.2.0768
64. Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. Stimulus-specific differences in protein kinase C delta localization and activation mechanisms in cardiomyocytes. J Biol Chem. (2004) 279:19350–61. doi: 10.1074/jbc.M311096200
65. Li W, Yu JC, Michieli P, Beeler JF, Ellmore N, Heidaran MA, et al. Stimulation of the platelet-derived growth factor beta receptor signaling pathway activates protein kinase C-delta. Mol Cell Biol. (1994) 14:6727–35. doi: 10.1128/mcb.14.10.6727-6735.1994
66. Soltoff SP, Toker A. Carbachol, substance P, and phorbol ester promote the tyrosine phosphorylation of protein kinase C delta in salivary gland epithelial cells. J Biol Chem. (1995) 270:13490–5. doi: 10.1074/jbc.270.22.13490
67. Moussazadeh M, Haimovich B. Protein kinase C-delta activation and tyrosine phosphorylation in platelets. FEBS Lett. (1998) 438:225–30. doi: 10.1016/S0014-5793(98)01302-7
68. Li W, Jiang YX, Zhang J, Soon L, Flechner L, Kapoor V, et al. Protein kinase C-delta is an important signaling molecule in insulin-like growth factor I receptor-mediated cell transformation. Mol Cell Biol. (1998) 18:5888–98. doi: 10.1128/MCB.18.10.5888
69. Popoff IJ, Deans JP. Activation and tyrosine phosphorylation of protein kinase C delta in response to B cell antigen receptor stimulation. Mol Immunol. (1999) 36:1005–16. doi: 10.1016/S0161-5890(99)00128-5
70. Barbazuk SM, Gold MR. Protein kinase C-delta is a target of B-cell antigen receptor signaling. Immunol Lett. (1999) 69:259–67. doi: 10.1016/S0165-2478(99)00090-5
71. Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci USA. (1997) 94:11233–7. doi: 10.1073/pnas.94.21.11233
72. Benes C, Soltoff SP. Modulation of PKCdelta tyrosine phosphorylation and activity in salivary and PC-12 cells by Src kinases. Amer J Physiol Cell Physiol. (2001) 280:C1498–510. doi: 10.1152/ajpcell.2001.280.6.C1498
73. Ohmori S, Shirai Y, Sakai N, Fujii M, Konishi H, Kikkawa U, et al. Three distinct mechanisms for translocation and activation of the delta subspecies of protein kinase C. Mol Cell Biol. (1998) 18:5263–71. doi: 10.1128/MCB.18.9.5263
74. Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. (2008) 117:886–96. doi: 10.1161/CIRCULATIONAHA.107.759167
75. Churchill EN, Szweda LI. Translocation of deltaPKC to mitochondria during cardiac reperfusion enhances superoxide anion production and induces loss in mitochondrial function. Arch Biochem Biophys. (2005) 439:194–9. doi: 10.1016/j.abb.2005.05.007
76. Churchill EN, Murriel CL, Chen CH, Mochly-Rosen D, Szweda LI. Reperfusion-induced translocation of deltaPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ Res. (2005) 97:78–85. doi: 10.1161/01.RES.0000173896.32522.6e
77. Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, et al. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. (2003) 108:2304–7. doi: 10.1161/01.CIR.0000101682.24138.36
78. Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. (2004) 279:47985–91. doi: 10.1074/jbc.M405071200
79. Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem. (1997) 272:30945–51. doi: 10.1074/jbc.272.49.30945
80. Inagaki K, Hahn HS, Dorn GW II, Mochly-Rosen D. Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation. (2003) 108:869–75. doi: 10.1161/01.CIR.0000081943.93653.73
81. Ikeno F, Inagaki K, Rezaee M, Mochly-Rosen D. Impaired perfusion after myocardial infarction is due to reperfusion-induced deltaPKC-mediated myocardial damage. Cardiovasc Res. (2007) 73:699–709. doi: 10.1016/j.cardiores.2006.12.011
82. Murriel CL, Mochly-Rosen D. Opposing roles of delta and epsilonPKC in cardiac ischemia and reperfusion: targeting the apoptotic machinery. Arch Biochem Biophys. (2003) 420:246–54. doi: 10.1016/j.abb.2003.08.038
83. Churchill EN, Mochly-Rosen D. The roles of PKCdelta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem Soc Trans. (2007) 35:1040–2. doi: 10.1042/BST0351040
84. Aon MA, Cortassa S, Wei AC, Grunnet M, O'Rourke B. Energetic performance is improved by specific activation of K+ fluxes through K(Ca) channels in heart mitochondria. Biochim Biophys Acta. (2010) 1797:71–80. doi: 10.1016/j.bbabio.2009.08.002
85. Wojtovich AP, Nadtochiy SM, Urciuoli WR, Smith CO, Grunnet M, Nehrke K, et al. A non-cardiomyocyte autonomous mechanism of cardioprotection involving the SLO1 BK channel. PeerJ. (2013) 1:e48. doi: 10.7717/peerj.48
86. Sakamoto K, Ohya S, Muraki K, Imaizumi Y. A novel opener of large-conductance Ca2+-activated K+ (BK) channel reduces ischemic injury in rat cardiac myocytes by activating mitochondrial K(Ca) channel. J Pharmacol Sci. (2008) 108:135–9. doi: 10.1254/jphs.08150SC
87. Singh H, Lu R, Bopassa JC, Meredith AL, Stefani E, Toro L. MitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc Natl Acad Sci USA. (2013) 110:10836–41. doi: 10.1073/pnas.1302028110
88. Soltysinska E, Bentzen BH, Barthmes M, Hattel H, Thrush AB, Harper ME, et al. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS ONE. (2014) 9:e103402. doi: 10.1371/journal.pone.0103402
89. Clements RT, Terentyev D, Sellke FW. Ca(2+)-activated K(+) channels as therapeutic targets for myocardial and vascular protection. Circ J. (2015) 79:455–62. doi: 10.1253/circj.CJ-15-0015
90. Cordeiro B, Shinn C, Sellke FW, Clements RT. Rottlerin-induced BKCa channel activation impairs specific contractile responses and promotes vasodilation. Ann Thorac Surg. (2015) 99:626–34. doi: 10.1016/j.athoracsur.2014.07.091
91. Wu SN, Wang YJ, Lin MW. Potent stimulation of large-conductance Ca2+-activated K+ channels by rottlerin, an inhibitor of protein kinase C-delta, in pituitary tumor (GH3) cells and in cortical neuronal (HCN-1A) cells. J Cell Physiol. (2007) 210:655–66. doi: 10.1002/jcp.20866
92. Goldklang MP, Perez-Zoghbi JF, Trischler J, Nkyimbeng T, Zakharov SI, Shiomi T, et al. Treatment of experimental asthma using a single small molecule with anti-inflammatory and BK channel-activating properties. FASEB J. (2013) 27:4975–86. doi: 10.1096/fj.13-235176
93. Clements RT, Cordeiro B, Feng J, Bianchi C, Sellke FW. Rottlerin increases cardiac contractile performance and coronary perfusion through BKCa++ channel activation after cold cardioplegic arrest in isolated hearts. Circulation. (2011) 124:S55–61. doi: 10.1161/CIRCULATIONAHA.110.012112
94. Zheng H, Liu J, Liu C, Lu F, Zhao Y, Jin Z, et al. Calcium-sensing receptor activating phosphorylation of PKCdelta translocation on mitochondria to induce cardiomyocyte apoptosis during ischemia/reperfusion. Mol Cell Biochem. (2011) 358:335–43. doi: 10.1007/s11010-011-0984-1
95. Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. (1994) 94:1621–8. doi: 10.1172/JCI117504
96. Liu C, Li H, Zheng H, Zhai M, Lu F, Dong S, et al. CaSR activates PKCdelta to induce cardiomyocyte apoptosis via ER stressassociated apoptotic pathways during ischemia/reperfusion. Int J Mol Med. (2019) 44:1117–26. doi: 10.3892/ijmm.2019.4255
97. Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. (1995) 376:37–43. doi: 10.1038/376037a0
98. Zhao J, Renner O, Wightman L, Sugden PH, Stewart L, Miller AD, et al. The expression of constitutively active isotypes of protein kinase C to investigate preconditioning. J Biol Chem. (1998) 273:23072–9. doi: 10.1074/jbc.273.36.23072
99. Mayr M, Metzler B, Chung YL, McGregor E, Mayr U, Troy H, et al. Ischemic preconditioning exaggerates cardiac damage in PKC-delta null mice. Amer J Physiol Heart Circ Physiol. (2004) 287:H946–56. doi: 10.1152/ajpheart.00878.2003
100. Fryer RM, Hsu AK, Wang Y, Henry M, Eells J, Gross GJ. PKC-delta inhibition does not block preconditioning-induced preservation in mitochondrial ATP synthesis and infarct size reduction in rats. Basic Res Cardiol. (2002) 97:47–54. doi: 10.1007/s395-002-8387-x
101. Lincoff AM, Roe M, Aylward P, Galla J, Rynkiewicz A, Guetta V, et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: results of the PROTECTION AMI Randomized Controlled Trial. Eur Heart J. (2014) 35:2516–23. doi: 10.1093/eurheartj/ehu177
102. Zhang L, Guo H, Yuan F, Hong ZC, Tian YM, Zhang XJ, et al. Limb remote ischemia per-conditioning protects the heart against ischemia-reperfusion injury through the opioid system in rats. Can J Physiol Pharmacol. (2018) 96:68–75. doi: 10.1139/cjpp-2016-0585
103. Pan X, Chen J, Wang T, Zhang M, Wang H, Gao H. Essential role of high glucose-induced overexpression Of PKCbeta and PKCdelta in GLP-1 resistance in rodent cardiomyocytes. Diabetes Metab Syndr Obes. (2019) 12:2289–302. doi: 10.2147/OTT.S183625
104. Hahn HS, Yussman MG, Toyokawa T, Marreez Y, Barrett TJ, Hilty KC, et al. Ischemic protection and myofibrillar cardiomyopathy: dose-dependent effects of in vivo deltaPKC inhibition. Circ Res. (2002) 91:741–8. doi: 10.1161/01.RES.0000037091.64492.69
105. Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. (2002) 27:92–8. doi: 10.1007/s00059-002-2354-y
106. Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: a therapeutic target? Cardiovasc Res. (2009) 82:229–39. doi: 10.1093/cvr/cvp001
107. Braun MU, Mochly-Rosen D. Opposing effects of delta- and zeta-protein kinase C isozymes on cardiac fibroblast proliferation: use of isozyme-selective inhibitors. J Mol Cell Cardiol. (2003) 35:895–903. doi: 10.1016/S0022-2828(03)00142-1
108. Brilla CG, Zhou G, Rupp H, Maisch B, Weber KT. Role of angiotensin II and prostaglandin E2 in regulating cardiac fibroblast collagen turnover. Amer J Cardiol. (1995) 76:8D−13D. doi: 10.1016/S0002-9149(99)80485-8
109. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. (2005) 45:657–87. doi: 10.1146/annurev.pharmtox.45.120403.095802
110. Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. (2005) 65:40–51. doi: 10.1016/j.cardiores.2004.08.020
111. Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. (2007) 117:568–75. doi: 10.1172/JCI31044
112. Schorb W, Conrad KM, Singer HA, Dostal DE, Baker KM. Angiotensin II is a potent stimulator of MAP-kinase activity in neonatal rat cardiac fibroblasts. J Mol Cell Cardiol. (1995) 27:1151–60. doi: 10.1016/0022-2828(95)90051-9
113. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci USA. (2005) 102:437–42. doi: 10.1073/pnas.0408704102
114. Chintalgattu V, Katwa LC. Role of protein kinase C-delta in angiotensin II induced cardiac fibrosis. Biochem Biophys Res Commun. (2009) 386:612–6. doi: 10.1016/j.bbrc.2009.06.076
115. Irie N, Sakai N, Ueyama T, Kajimoto T, Shirai Y, Saito N. Subtype- and species-specific knockdown of PKC using short interfering RNA. Biochem Biophys Res Commun. (2002) 298:738–43. doi: 10.1016/S0006-291X(02)02531-7
116. Bouzeghrane F, Mercure C, Reudelhuber TL, Thibault G. Alpha8beta1 integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. J Mol Cell Cardiol. (2004) 36:343–53. doi: 10.1016/j.yjmcc.2003.11.007
117. Kim S, Ohta K, Hamaguchi A, Yukimura T, Miura K, Iwao H. Angiotensin II induces cardiac phenotypic modulation and remodeling in vivo in rats. Hypertension. (1995) 25:1252–9. doi: 10.1161/01.HYP.25.6.1252
118. Schunkert H, Jackson B, Tang SS, Schoen FJ, Smits JF, Apstein CS, et al. Distribution and functional significance of cardiac angiotensin converting enzyme in hypertrophied rat hearts. Circulation. (1993) 87:1328–39. doi: 10.1161/01.CIR.87.4.1328
119. Weber KT, Brilla CG, Campbell SE, Guarda E, Zhou G, Sriram K. Myocardial fibrosis: role of angiotensin II and aldosterone. Basic Res Cardiol. (1993) 88(Suppl 1):107–24. doi: 10.1007/978-3-642-72497-8_8
120. Olson ER, Shamhart PE, Naugle JE, Meszaros JG. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension. (2008) 51:704–11. doi: 10.1161/HYPERTENSIONAHA.107.098459
121. Brigstock DR. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev. (1999) 20:189–206. doi: 10.1210/er.20.2.189
122. Ruperez M, Lorenzo O, Blanco-Colio LM, Esteban V, Egido J, Ruiz-Ortega M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation. (2003) 108:1499–505. doi: 10.1161/01.CIR.0000089129.51288.BA
123. He Z, Way KJ, Arikawa E, Chou E, Opland DM, Clermont A, et al. Differential regulation of angiotensin II-induced expression of connective tissue growth factor by protein kinase C isoforms in the myocardium. J Biol Chem. (2005) 280:15719–26. doi: 10.1074/jbc.M413493200
124. Takahashi K, Takeya M, Sakashita N. Multifunctional roles of macrophages in the development and progression of atherosclerosis in humans and experimental animals. Med Electron Microsc. (2002) 35:179–203. doi: 10.1007/s007950200023
125. Tsai CS, Lin YW, Huang CY, Shih CM, Tsai YT, Tsao NW, et al. Thrombomodulin regulates monocye differentiation via PKCdelta and ERK1/2 pathway in vitro and in atherosclerotic artery. Sci Rep. (2016) 6:38421. doi: 10.1038/srep38421
126. Zhou T, Chen X. Long intergenic noncoding RNA p21 mediates oxidized LDLinduced apoptosis and expression of LOX1 in human coronary artery endothelial cells. Mol Med Rep. (2017) 16:8513–9. doi: 10.3892/mmr.2017.7623
127. Chu M, Iyengar R, Koshman YE, Kim T, Russell B, Martin JL, et al. Serine-910 phosphorylation of focal adhesion kinase is critical for sarcomere reorganization in cardiomyocyte hypertrophy. Cardiovasc Res. (2011) 92:409–19. doi: 10.1093/cvr/cvr247
128. Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential activation of mitogen-activated protein kinase cascades and apoptosis by protein kinase C epsilon and delta in neonatal rat ventricular myocytes. Circ Res. (2001) 89:882–90. doi: 10.1161/hh2201.099434
129. Guo WG, Yu ZB, Xie MJ. [Protein kinase Cdelta is possibly involved in the transition from hypertrophy to apoptosis of myocardiocytes]. Sheng Li Xue Bao [Acta Physiol Sin]. (2006) 58:269–74. doi: 10.1016/S0379-4172(06)60097-8
130. Yang YC, Tsai CY, Chen CL, Kuo CH, Hou CW, Cheng SY, et al. Pkcdelta activation is involved in ROS-mediated mitochondrial dysfunction and apoptosis in cardiomyocytes exposed to advanced glycation end products (ages). Aging Dis. (2018) 9:647–63. doi: 10.14336/AD.2017.0924
131. Guo J, Gertsberg Z, Ozgen N, Sabri A, Steinberg SF. Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J Biol Chem. (2011) 286:6500–9. doi: 10.1074/jbc.M110.208058
132. Ozgen N, Obreztchikova M, Guo J, Elouardighi H, Dorn GW II, Wilson BA, Steinberg SF. Protein kinase D links Gq-coupled receptors to cAMP response element-binding protein (CREB)-Ser133 phosphorylation in the heart. J Biol Chem. (2008) 283:17009–19. doi: 10.1074/jbc.M709851200
133. Harrison BC, Kim MS, van Rooij E, Plato CF, Papst PJ, Vega RB, et al. Regulation of cardiac stress signaling by protein kinase d1. Mol Cell Biol. (2006) 26:3875–88. doi: 10.1128/MCB.26.10.3875-3888.2006
134. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. (2010) 56:225–31. doi: 10.1161/HYPERTENSIONAHA.109.148635
135. Chen C, Du J, Feng W, Song Y, Lu Z, Xu M, et al. beta-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br J Pharmacol. (2012) 166:676–88. doi: 10.1111/j.1476-5381.2011.01785.x
136. D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, et al. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA. (1997) 94:8121–6. doi: 10.1073/pnas.94.15.8121
137. Soloviev AI, Parshikov AV, Stefanov AV. Evidence for the involvement of protein kinase C in depression of endothelium-dependent vascular responses in spontaneously hypertensive rats. J Vasc Res. (1998) 35:325–31. doi: 10.1159/000025602
138. Soloviev AI, Tishkin SM, Zelensky SN, Ivanova IV, Kizub IV, Pavlova AA, et al. Ionizing radiation alters myofilament calcium sensitivity in vascular smooth muscle: potential role of protein kinase C. Amer J Physiol Regul Integr Comp Physiol. (2005) 289:R755–62. doi: 10.1152/ajpregu.00748.2004
139. Allahdadi KJ, Duling LC, Walker BR, Kanagy NL. Eucapnic intermittent hypoxia augments endothelin-1 vasoconstriction in rats: role of PKCdelta. Amer J Physiol Heart Circ Physiol. (2008) 294:H920–7. doi: 10.1152/ajpheart.01264.2007
140. Newton AC, Antal CE, Steinberg SF. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin Sci. (2016) 130:1499–510. doi: 10.1042/CS20160036
141. Yang S, Yuan HQ, Hao YM, Ren Z, Qu SL, Liu LS, et al. Macrophage polarization in atherosclerosis. Clin Chim Acta. (2019). 501:142–6. doi: 10.1016/j.cca.2019.10.034
142. Szilagyi K, Meijer AB, Neele AE, Verkuijlen P, Leitges M, Dabernat S, et al. PKCdelta is dispensible for oxLDL uptake and foam cell formation by human and murine macrophages. Cardiovasc Res. (2014) 104:467–76. doi: 10.1093/cvr/cvu213
143. Li Q, Park K, Xia Y, Matsumoto M, Qi W, Fu J, et al. Regulation of macrophage apoptosis and atherosclerosis by lipid-induced PKCdelta isoform activation. Circ Res. (2017) 121:1153–67. doi: 10.1161/CIRCRESAHA.117.311606
144. Huang SL, Chen PY, Wu MJ, Tai MH, Ho CT, Yen JH. Curcuminoids modulate the PKCdelta/NADPH oxidase/reactive oxygen species signaling pathway and suppress matrix invasion during monocyte-macrophage differentiation. J Agric Food Chem. (2015) 63:8838–48. doi: 10.1021/acs.jafc.5b04083
145. Wang Y, Zhang Y, Zhu Y, Zhang P. Lipolytic inhibitor G0/G1 switch gene 2 inhibits reactive oxygen species production and apoptosis in endothelial cells. Amer J Physiol Cell Physiol. (2015) 308:C496–504. doi: 10.1152/ajpcell.00317.2014
146. Pirillo A, Norata GD, Catapano AL. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. (2013) 2013:152786. doi: 10.1155/2013/152786
147. Steinberg SF, Goldberg M, Rybin VO. Protein kinase C isoform diversity in the heart. J Mol Cell Cardiol. (1995) 27:141–53. doi: 10.1016/S0022-2828(08)80014-4
148. Bayer AL, Heidkamp MC, Patel N, Porter M, Engman S, Samarel AM. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Mol Cell Biochem. (2003) 242:145–52. doi: 10.1007/978-1-4757-4712-6_19
149. Braun MU, LaRosee P, Schon S, Borst MM, Strasser RH. Differential regulation of cardiac protein kinase C isozyme expression after aortic banding in rat. Cardiovasc Res. (2002) 56:52–63. doi: 10.1016/S0008-6363(02)00511-4
150. Braun MU, LaRosee P, Simonis G, Borst MM, Strasser RH. Regulation of protein kinase C isozymes in volume overload cardiac hypertrophy. Mol Cell Biochem. (2004) 262:135–43. doi: 10.1023/B:MCBI.0000038229.23132.9f
151. Yi XP, Wang X, Gerdes AM, Li F. Subcellular redistribution of focal adhesion kinase and its related nonkinase in hypertrophic myocardium. Hypertension. (2003) 41:1317–23. doi: 10.1161/01.HYP.0000072772.74183.5F
152. Reyland ME, Anderson SM, Matassa AA, Barzen KA, Quissell DO. Protein kinase C delta is essential for etoposide-induced apoptosis in salivary gland acinar cells. J Biol Chem. (1999) 274:19115–23. doi: 10.1074/jbc.274.27.19115
153. Matassa AA, Carpenter L, Biden TJ, Humphries MJ, Reyland ME. PKCdelta is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J Biol Chem. (2001) 276:29719–28. doi: 10.1074/jbc.M100273200
154. Sheng JJ, Chen Y, Chang H, Wang YY, Jiao B, Yu ZB. Multisite phosphorylation of Bcl-2 via protein kinase Cdelta facilitates apoptosis of hypertrophic cardiomyocytes. Clin Exp Pharmacol Physiol. (2014) 41:891–901. doi: 10.1111/1440-1681.12295
155. Henning RJ, Bourgeois M, Harbison RD. Poly(ADP-ribose) polymerase (PARP) and PARP inhibitors: mechanisms of action and role in cardiovascular disorders. Cardiovasc Toxicol. (2018) 18:493–506. doi: 10.1007/s12012-018-9462-2
156. Chen-Scarabelli C, Knight R, Stephanou A, Scarabelli G, Onorati F, Tessari M, et al. Diabetic hearts have lower basal urocortin levels that fail to increase after cardioplegic arrest: association with increased apoptosis and postsurgical cardiac dysfunction. J Thorac Cardiovasc Surg. (2014) 148:2296–308. doi: 10.1016/j.jtcvs.2014.05.018
157. Shende P, Xu L, Morandi C, Pentassuglia L, Heim P, Lebboukh S, et al. Cardiac mTOR complex 2 preserves ventricular function in pressure-overload hypertrophy. Cardiovasc Res. (2016) 109:103–14. doi: 10.1093/cvr/cvv252
158. Goldberg M, Zhang HL, Steinberg SF. Hypoxia alters the subcellular distribution of protein kinase C isoforms in neonatal rat ventricular myocytes. J Clin Invest. (1997) 99:55–61. doi: 10.1172/JCI119133
159. Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, et al. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol. (1998) 31:391–8. doi: 10.1016/S0735-1097(97)00494-4
160. Carson LD, Korzick DH. Dose-dependent effects of acute exercise on PKC levels in rat heart: is PKC the heart's prophylactic? Acta Physiol Scand. (2003) 178:97–106. doi: 10.1046/j.1365-201X.2003.01131.x
161. Melling CW, Thorp DB, Milne KJ, Noble EG. Myocardial Hsp70 phosphorylation and PKC-mediated cardioprotection following exercise. Cell Stress Chaperones. (2009) 14:141–50. doi: 10.1007/s12192-008-0065-x
162. Xie MJ, Chang H, Wang YY, Zhang L, Song Z, Guo WG, et al. Evidence that apoptotic signalling in hypertrophic cardiomyocytes is determined by mitochondrial pathways involving protein kinase Cdelta. Clin Exp Pharmacol Physiol. (2010) 37:1120–8. doi: 10.1111/j.1440-1681.2010.05447.x
163. Heinke MY, Yao M, Chang D, Einstein R, dos Remedios CG. Apoptosis of ventricular and atrial myocytes from pacing-induced canine heart failure. Cardiovasc Res. (2001) 49:127–34. doi: 10.1016/S0008-6363(00)00242-X
164. Li Z, Bing OH, Long X, Robinson KG, Lakatta EG. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol. (1997) 272:H2313–9. doi: 10.1152/ajpheart.1997.272.5.H2313
165. Soloviev AI, Bershtein SA. The contractile apparatus in vascular smooth muscle cells of spontaneously hypertensive rats possess increased calcium sensitivity: the possible role of protein kinase C. J Hypertens. (1992) 10:131–6. doi: 10.1097/00004872-199202000-00004
166. Novokhatska T, Tishkin S, Dosenko V, Boldyriev A, Ivanova I, Strielkov I, et al. Correction of vascular hypercontractility in spontaneously hypertensive rats using shRNAs-induced delta protein kinase C gene silencing. Eur J Pharmacol. (2013) 718:401–7. doi: 10.1016/j.ejphar.2013.08.003
167. Yi XP, Zhou J, Huber L, Qu J, Wang X, Gerdes AM, et al. Nuclear compartmentalization of FAK and FRNK in cardiac myocytes. Amer J Physiol Heart Circ Physiol. (2006) 290:H2509–15. doi: 10.1152/ajpheart.00659.2005
168. Gerasymenko IM, Kleschevnikov VV, Kedlian VR, Sakhno LO, Arbuzova IA, Sheludko Y. V., et al. Establishment of transgenic lettuce plants producing potentially antihypertensive ShRNA. Cytol Genet. (2017) 51:1–7. doi: 10.3103/S0095452717010054
169. Webster BR, Osmond JM, Paredes DA, DeLeon XA, Jackson-Weaver O, Walker BR, et al. Phosphoinositide-dependent kinase-1 and protein kinase Cdelta contribute to endothelin-1 constriction and elevated blood pressure in intermittent hypoxia. J Pharmacol Exp Ther. (2013) 344:68–76. doi: 10.1124/jpet.112.195412
170. Wion D, Dematteis M, Nissou MF, Cretallaz C, Berger F, Issartel JP. [Oxygen tension and cancer-cell culture: half a century of artifacts?]. Med Sci. (2008) 24:1093–5. doi: 10.1051/medsci/200824121093
171. Kanagy NL, Walker BR, Nelin LD. Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension. (2001) 37:511–5. doi: 10.1161/01.HYP.37.2.511
172. Allahdadi KJ, Walker BR, Kanagy NL. Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension. (2005) 45:705–9. doi: 10.1161/01.HYP.0000153794.52852.04
173. Packer M, Narahara KA, Elkayam U, Sullivan JM, Pearle DL, Massie BM, et al. Double-blind, placebo-controlled study of the efficacy of flosequinan in patients with chronic heart failure. Principal investigators of the REFLECT study. J Am Coll Cardiol. (1993) 22:65–72. doi: 10.1016/0735-1097(93)90816-J
174. Guarracino F, Landoni G, Tritapepe L, Pompei F, Leoni A, Aletti G, et al. Myocardial damage prevented by volatile anesthetics: a multicenter randomized controlled study. J Cardiothorac Vasc Anesth. (2006) 20:477–83. doi: 10.1053/j.jvca.2006.05.012
175. Jin Z, Duan W, Chen M, Yu S, Zhang H, Feng G, et al. The myocardial protective effects of adenosine pretreatment in children undergoing cardiac surgery: a randomized controlled clinical trial. Eur J Cardiothorac Surg. (2011) 39:e90–6. doi: 10.1016/j.ejcts.2010.12.052
176. Mangano DT, Miao Y, Tudor IC, Dietzel C. Post-reperfusion myocardial infarction: long-term survival improvement using adenosine regulation with acadesine. J Am Coll Cardiol. (2006) 48:206–14.
177. Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol. (2005) 45:1775–80. doi: 10.1016/j.jacc.2005.02.061
Keywords: cardiovascular disease, PKC-δ, kinase, mechanism, PKC
Citation: Miao L-n, Pan D, Shi J, Du J-p, Chen P-f, Gao J, Yu Y, Shi D-Z and Guo M (2022) Role and Mechanism of PKC-δ for Cardiovascular Disease: Current Status and Perspective. Front. Cardiovasc. Med. 9:816369. doi: 10.3389/fcvm.2022.816369
Received: 18 November 2021; Accepted: 11 January 2022;
Published: 15 February 2022.
Edited by:
Hongliang Li, Wuhan University, ChinaCopyright © 2022 Miao, Pan, Shi, Du, Chen, Gao, Yu, Shi and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Da-Zhuo Shi, c2hpZGF6aHVvQDEyNi5jb20=; Ming Guo, NTQ3NDUzMjAyQHFxLmNvbQ==
†These authors have contributed equally to this work