 Tianyu Shen
Tianyu Shen Dayin Lyu
Dayin Lyu Mengping Zhang
Mengping Zhang Hui Shang
Hui Shang Qiulun Lu
Qiulun Lu- 1Department of Orthopaedic, Taihe Hospital, Hubei University of Medicine, Shiyan, China
- 2Key Laboratory of Cardiovascular and Cerebrovascular Medicine, Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, Nanjing Medical University, Nanjing, China
- 3Department of Hematology, Taihe Hospital, Hubei University of Medicine, Shiyan, China
Myocardial infarction is one of the most severe heart diseases, leading to sudden death. Currently, angiography and stenting are widely performed in clinics, yet more effective treatment is still needed. Herein, we presented that dioscin, a natural product, showed protective effect on infarcted hearts via mitochondrial maintenance. Upon dioscin treatment, cardiac dysfunction was alleviated, and remodeling is prevented. Mechanistically, disocin maintains mitochondria function through the maintenance of Kreb's cycle, and suppresion of ROS accumulation. In this way, by targeting mitochondrial dysfunction, dioscin is a potential drug for infarcted hearts.
Introduction
Nowadays, it is widely considered that cardiovascular diseases are one of the major causes for human mortality around the world (1). Particularly, myocardial infarction (MI) affects large population in both developing and developed countries (2). It characterized with dysfunction of energy supply and reactive oxygen species (ROS) generation relates to mitochondrial-based disorder, which leads to pathogenesis of MI and even heart failure (3). Therefore, novel drugs of protecting the heart against myocardial infarction are urgently needed.
Mitochondrial dysfunction is associated with the pathogenesis of myocardial infarction (4). Mitochondrial-derived ROS represent a major source of oxidative stress in cardiomyocytes (5). On the other hand, the primary source of energy generating processes is located at mitochondria among the cell, which is severe sensitive to free radical occurrence in cardiomyocytes (6). Taken together, mitochondria maintenance would moderate heart injury after infarction.
Recently, researches indicated that natural productions from some herbals have potential therapeutic effect for the prevention and treatment of cardiovascular diseases (7). The protective effects are associated with mitochondrial function, including alleviating mitochondrial oxidative stress, de-activation of respiratory chain enzymes, postponing mitochondrial Kreb's cycle, and maintenance of mitochondrial membrane potential (Δψ) and ATP content (8–10). Dioscin, a natural steroid saponin isolated from the root bark of wild dioscorea nipponica, is reported to treat MI by promoting angiogenesis (11). Additionally, it was reported that dioscin maintains mitochondrial dynamics, which rescues silicosis by reducing mitochondrial-dependent apoptosis (12). However, whether dioscin protects cardiac injury against MI by rescuing mitochondrial dysfunction remains unclear.
Here, our work revealed that dioscin attenuates cardiac dysfunction induced by myocardial infarction. Administration of dioscin benefits infarcted hearts could significantly enhance mitochondrial Kreb's cycle and activities of respiratory chain enzymes, reduce ROS by promoting the antioxidant enzyme activities, and suppress the cardiomyocytes apoptosis by rescuing mitochondrial member potential. Taken together, our results showed that dioscin could alleviate MI via attenuate mitochondrial malfunction.
Materials and Methods
Animal Model and Chemicals
Mice were male and at age of 8 weeks old. All male C57BL/6 were from Jiangsu Animal Experimental for Medical and Pharmaceutical Research Center. The in vivo experiments were approval of Institutional Animal Care and Use Committees at the University of Nanjing Medical University (Number: 1912034) and carried out in accordance with the guidelines. Mice were kept with 12 h light/12 h dark cycle at 23 ± 3°C and 30–70% humidity.
Infarction mouse model was generated through ligature of the left anterior descending (LAD) as described previously with male mice (11). Briefly, mice were anesthetized with 1.5–2% isoflurane, and then a left thoracotomy and pericardiotomy was performed. The left anterior descending was completely occluded by a knot with 7–0 non-absorbable surgical suture. After the ribs and skin were closed with 5–0 silk, the mouse remained under anesthesia until succinylcholine was completely metabolized. For the procedure of sham surgery, mice underwent the same procedure without the ligation of the artery.
Dioscin (80 mg/kg/day, Di'ao group, Chengdu, China) was administrated with mice for 2 weeks beginning on the day of operation. Briefly, the dioscin was first dissolved in dimethyl sulphoxide (DMSO) and then added 5% sodium carboxymethyl cellulose (CMC-Na) to make the volume ratio for 1:19. Consequently, the mice were given the dissolution by intragastric administration for continuous 14 days after myocardial infarction 1 day. The treatment groups were given dioscin solution and the control groups were given same volume of 5% CMC-Na vehicle solution. The preparation and application of the drug's dissolution should be completed in 1 day.
Echocardiography
Transthoracic echocardiographic analysis was performed 2 weeks after the sham or MI surgery as previously described (13). Echocardiography on unconscious mice was used with a Vevo 2100 system (VisualSonics). The echocardiography was blinded to the mice in this study.
Histological Analysis
Hematoxylin and eosin and Masson trichrome staining were performed as described previously (14). The whole hearts were collected and fixed in 4% paraformaldehyde, embedded in paraffin, and then cut into 5-μm serial sections. Image analysis and quantification of fibrosis was performed by a blinded investigator using Image J software.
Mitochondrial Isolation
The purification of cardiac mitochondrial fraction from the heart tissues or cultured primary cardiomyocytes was followed as previously described (15), using Mitochondrial Extraction Kit (Solarbio Life Sciences, SM0020) according to the manufacturer's instructions.
Estimation of Reactive Oxygen Species
Dihydroethidium (DHE) staining (Beyotime, China) was performed as described previously (16). Briefly, heart samples were embedded in OCT compound and transverse sectioned at 5 μm thickness to perform staining and then conserved at −80°C. Next, heart sections were set in room temperature for 10 min and then washing by PBS three times for 5 min. Further, the sections were incubated with the DHE (1:1,000) for 30 min in wet box at 37°C. Finally, after washing by PBS three times for 5 min, co-stain with DAPI. The imagines are photographed by confocal, and we use the same intensity of laser.
Estimation of Mitochondrial Enzymatic and Non-enzymatic Antioxidant
The activity of glutathione peroxidase (GPx), superoxide dismutase (SOD), glutathione-S-transferase (GST), catalase (CAT), glutathione (GSH) was determined in cardiac tissues, using Micro Superoxide Dismutase (SOD) Assay Kit (Solarbio Life Sciences, BC0175, BC0205, BC1195, BC0355, and BC1175) according to the manufacturer's instructions.
TCA Cycle Enzyme Activities and Respiratory Chain Enzymes
Effect of dioscin on MI-induced mitochondrial damage was measured by detection of the activities of mitochondrial enzymes such as malate dehydrogenase (MDH), isocitrate dehydrogenase (ICDH), and succinate dehydrogenase (SDH) using Micro Isocitrate Dehydrogenase Mitochondrial (ICDHm) Assay Kit (Solarbio Life Sciences, BC2165), Micro Succinate Dehydrogenase (SDH) Assay Kit (Solarbio Life Sciences, BC0955) and Micro NADP-Malate Dehydrogenase (NAD-MDH) Assay Kit (Solarbio Life Sciences, BC1055) according to the manufacturer's instructions.
The nicotinamide adenine dinucleotide dehydrogenase (NADH) activity was determined in cardiac tissues as reported (17), using Micro Nicotinamide Adenine Dinucleotide Dehydrogenase (NADH) Assay Kit (Solarbio Life Sciences, BC0635) according to the manufacturer's instructions.
The cytochrome c-oxidase (NCR) activity was determined in cardiac tissues, using Micro NADPH-cytochrome C Reductase Assay Kit (Solarbio Life Sciences, BC2725) according to the manufacturer's instructions.
Cell Culture
H9C2 cells were cultured in DMEM (Servicebio) medium with 10% FBS (Gibco, Life Technologies) and 1% penicillin/streptomycin (Beyotime). Final concentration of 100 μM CoCl2 (Sigma-Aldrich) was added to simulate hypoxia condition. After 24 h of treatment, the extraction of mRNA was performed. And after 48 h, the corresponding staining was performed.
qRT-PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen). cDNA was generated using HiScript III 1st Strand cDNA Synthesis (Vazyme) and then qPCR was performed using on QuantStudio 5 applied biosystems (Thermo Fisher Scientific) with Hifair III One Step RT-qPCR SYBR Green Kit (Yeasen), all according to the manufacturer's instructions. Actin was regard as control and the data analysis was using 2CT. Obtaining form three independent experiments, and each sample was repeated duplicated in each experiment. Primer sequence are listed below.
For mouse qPCR:
Anp: TTCTTCCTCGTCTTGGCCTTT, GACCTCATCTTCTACCGGCATCT;
Bnp: CACCGCTGGGAGGTCACT, GTGAGGCCTTGGTCCTTCAAGGTCACT;
β-mhc: ATGTGCCGGACCTTGGAA, CCTCGGGTTAGCTGAGAGATCA;
Bax: CCCGAGAGGTCTTTTTCC, GCCTTGAGCACCAGTTTG;
Bcl2: CCTGGCTGTCTCTGAAGACC, CTCACTTGTGGCCCAGGTAT;
Actin: CATCGTCCACCGCAAATG, CACCTTCACCGTTCCAGTT.
Mitochondrial Membrane Potential
Mitochondrial membrane potential was measured by the method of fluorescent probe, using the MitoTracker® Green FM (Yeasen, 40742ES50) according to the manufacturer's instructions.
TUNEL Staining
TUNEL staining was performed as previously report (18). The suitable density H9C2 cells were induced to CoCl2 (100 uM, Sigma, 449776) under the condition of treatment with dioscin (5 mg/L) for 48 h, compared to control group. Further, the H9C2 cells were fixed in 4% paraformaldehyde and stained with One Step TUNEL Apoptosis Assay Kit (Beyotime, C1086). TUNEL positive were counted from different images per sample, and the averages were presented.
Estimation of Caspase-1 Activities
The cysteine-requiring aspartate protease 1 (Caspase-1) activity was determined in cardiac tissues, using Caspase-1 Reductase Assay Kit (Hypertime, China) according to the manufacturer's instructions.
Statistical Analysis
Data were presented as mean ± standard error of the mean (SEM) using GraphPad Prism 8. Two-tailed unpaired t-test were used to determine the differences between two groups, and two-ANOVA was employed for comparisons of the differences between multiple groups. Statistical significance is indicated by * p < 0.05.
Results
Dioscin Alleviates the Mitochondrial Impairment by CoCl2 in H9C2 Cells
Mitochondrial dysfunction is one of the primary causes for cardiac death in response to infarction. Firstly, we performed in vivo experiment to assess the mitochondrial injury after dioscin treatment in MI heart tissues. The protein level of Pink1 revealed that dioscin recovered the expression of Pink1 after MI, indicating dioscin has a protective role in mitochondria (Figure 1A). Then, the protective effect of dioscin was evaluated on mitochondrial membrane potential. The disturbance of the mitochondrial membrane potential (MMP) caused by CoCl2, and this injury was mitigated by dioscin (Figure 1B). Considering the impaired mitochondria could lead to cell death, the dead cells were detected by using TUNEL assay. This showed that dioscin recovered the numbers of H9C2 cells in response to hypoxic condition (Figure 1C). Additionally, the mRNA levels of Bax and Bcl2 as apoptotic markers were detected in hypoxic H9C2 cells after dioscin treatment. The results showed that dioscin exerts anti-apoptotic effects and prevents cells from death by downregulating Bax expression and increasing Bcl2 expression (Figure 1D). Furthermore, the activities of cysteine-requiring aspartate protease 1 (caspase-1) was repressed in hypoxic cells after dioscin incubation, suggesting that dioscin repressed apoptosis caused by hypoxia (Figure 1E). These data suggest that dioscin plays protective roles against hypoxia induced mitochondrial injury to mitigate cellular apoptosis.
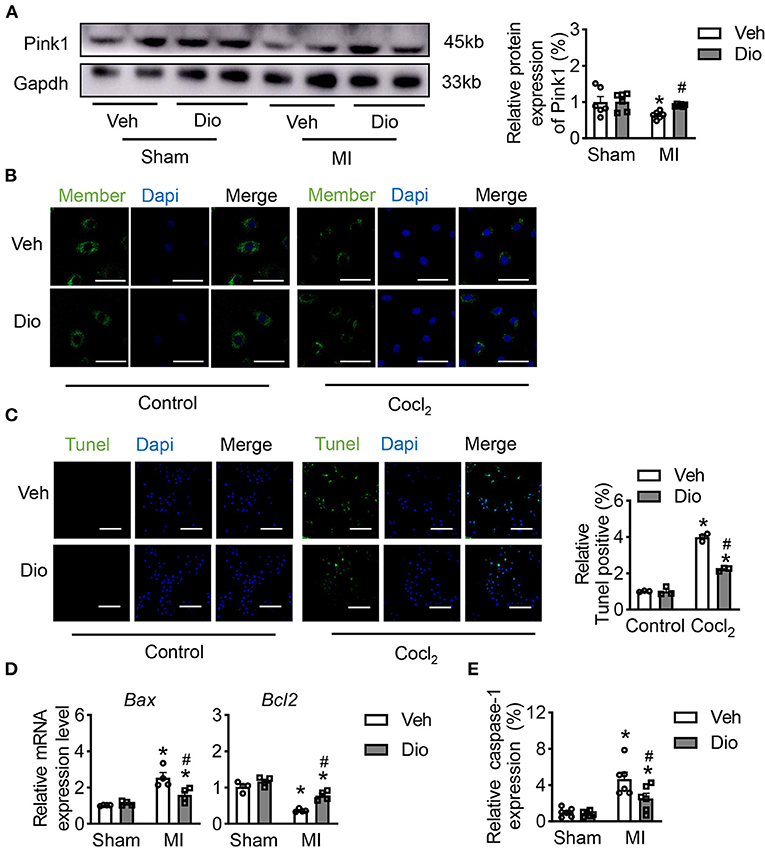
Figure 1. Dioscin alleviates the mitochondrial injury by CoCl2 in H9C2 cells. (A) The protein level of Pink1 in heart tissue (n = 6). (B) Representative images of the active mitochondrial, bar = 20 μm (n = 3). (C) Representative images of TUNEL staining to detect the anti-apoptosis role of dioscin in vitro, bar = 50 μm (n = 3). (D) The mRNA level of Bax and Bcl2 in vivo (n = 4). (E) The measurable of caspase-1 in vivo (n = 6). Data are mean ± SEM. *P < 0.05 vs. sham group, #P < 0.05 vs. MI group.
Dioscin Protects Cardiac Dysfunction Against Myocardial Infarction
To determine the therapeutic role of dioscin in vivo, mice were subjected to MI surgery. Two weeks after dioscin administration, the echocardiography was performed to monitor cardiac function. Representative echocardiograph showed that the infarcted mice exhibited left chamber dilation, and dioscin administration alleviated the cardiac deformation (Figure 2A). Then, cardiac function analysis showed that the left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were recovered after administration of dioscin in infarcted mice (Figure 2B). Additionally, haematoxylin and eosin (HE) staining of heart sections from mice in each group showed that the normal arrangement of cells was destroyed in infarcted heart increased, which could be prevented by dioscin (Figure 2C). Representative images exhibited that less collagen deposition after dioscin treatment by the size of blue region compared to MI group, which was decreased about 50% (Figure 2D), Suggesting dioscin alleviated cardiac fibrosis in infarcted hearts. Additionally, the expression level of heart failure markers was detected in hearts, including natriuretic peptide A (Anp), natriuretic peptide B (Bnp) and beta-myosin heavy chain (β-Mhc). It showed that the increased levels of Anp, Bnp and β-Mhc mRNA levels in heart tissues from infarcted mice were suppressed by dioscin administration (Figure 2E). These results indicate that dioscin administration preserved the cardiac function by alleviating cardiac fibrosis after MI.
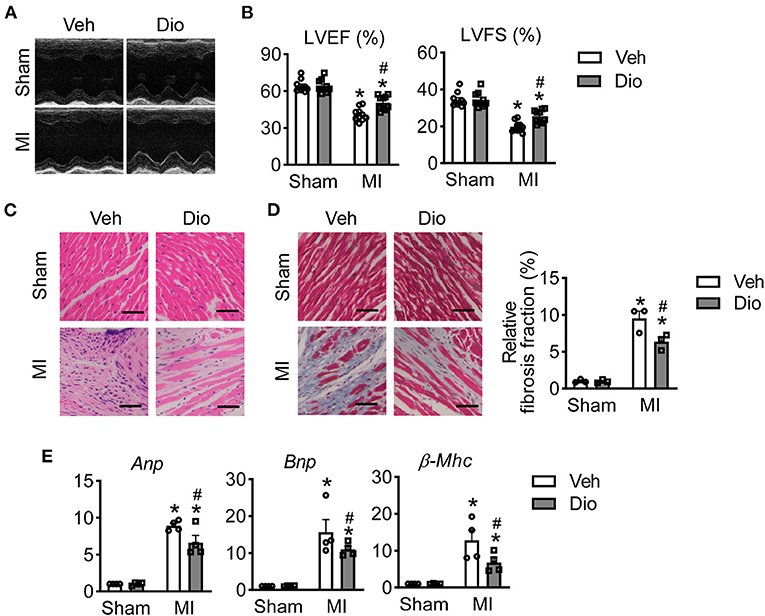
Figure 2. Dioscin protects mice against infarcted injury. (A) Representative images of echocardiographic measurement (n = 10). (B) Echocardiographic parameters for left ventricular ejection fraction (LVEF, %) and left ventricular fractional shortening (LVFS, %) (n = 10). (C) Representative images of HE staining, bar = 100 μm (n = 3). (D) Representative images of Masson trichrome stained and its quantification, bar = 100 μm (n = 3). (E) The mRNA level of heart failure markers for natriuretic peptide A (Anp), natriuretic peptide B (Bnp) and beta-myosin heavy chain (β-Mhc) (n = 4). Data are mean ± SEM. *P < 0.05 vs. sham group, #P < 0.05 vs. MI group.
Dioscin Prevents Cardiac Remodeling in MI Mouse Model
Considered the recovery of cardiac function occurs in infarcted hearts after disocin administration, cardiac remodeling was measured. There is no significant difference of heart rate among these four groups (Figure 3A). It was consistent with previous report that infarction increases left ventricular mass. Such increase was significantly repressed after administration of dioscin (Figure 3B). Other cardiac structure parameters, such as left ventricular posterior wall end-diastolic thickness (LVPW; d), left ventricular inner diameter end-diastolic thickness (LVID; d), left ventricular septum (IVS), and left ventricular end-systolic volume (LV Vol; d) were also sufficiently persevered after dioscin administration in infarcted hearts (Figures 3C–F). Additionally, mice subjected to MI showed a significant increase in heart weight/body weight (HW/BW) ratio and heart weight/tibia length (HW/TL), which could be ameliorated by dioscin e (Figures 3G,H). These results demonstrate that dioscin attenuated MI-induced heart remodeling.
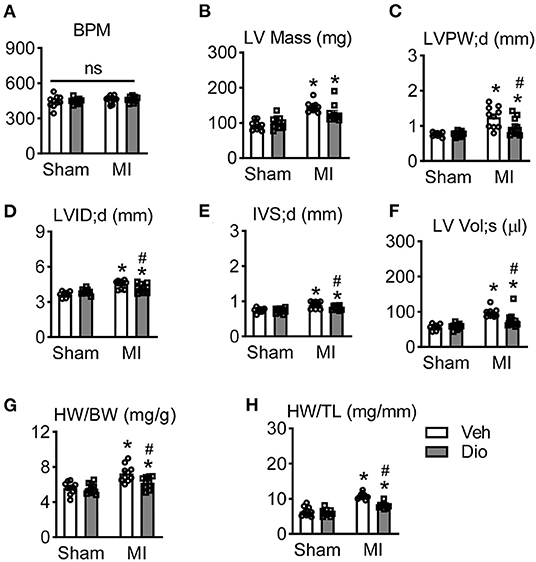
Figure 3. Dioscin prevents cardiac remodeling in response to myocardial infarction. (A) The analysis of beat per minute (BPM). Echocardiographic parameters of (B) left ventricular mass (LV Mass; mg), (C) end-diastolic left ventricular posterior wall (LVPW; d, mm), (D) end-diastolic left ventricular inner diameter (LVID; d, mm), (E) end-diastolic left ventricular septum (IVS; d, mm), and (F) left ventricular end-systolic volume (LV Vol; s, μl). (G) The ratio of heart weight to body weight (HW/BW; mg/g). (H) The ratio of heart weight to tibia length (HW/TL; mg/mm). n = 10 each group. Data are mean ± SEM. *P < 0.05 vs. sham group, #P < 0.05 vs. MI group.
Effect of Dioscin on the Activities of Mitochondrial Kreb's Cycle in Heart After MI
The impaired function of creatine kinase and decreased mitochondrial respiration are often observed in myocardium after pathological injury, including myocardial infarction and hypertrophy (19, 20). In attempt to explore the effect of dioscin in the process of energy metabolism, the activities of mitochondrial enzymes were measured in mice hearts. The activities of isocitrate dehydrogenase (ICDH), succinate dehydrogenase (SDH) and malate dehydrogenase (MDH) in mitochondria were significantly downregulated in the infarcted hearts compared to the sham group (Figures 4A–C). The activities of these enzymes were remarkable enhanced by treatment with dioscin in MI group (Figures 4A–C). These proved the beneficial role of dioscin on Kreb's cycle enzymes, which are the major source of ATP production in the cells. We further examined the activities of respiratory chain enzyme responsible for the synthesis of ATP. The activities of nicotinamide adenine dinucleotide dehydrogenase (NADH) (the Complex I) and cytochrome c-oxidase (the Complex III) were significantly reduced in the mitochondrial isolated from cardiac tissues obtained from the mice subjected to MI. Conversely, the decline was inhibited by treatment with dioscin (Figures 4D,E). Overall, these findings suggest that dioscin promotes the activities of mitochondrial Kreb's cycle in myocardial infarction.
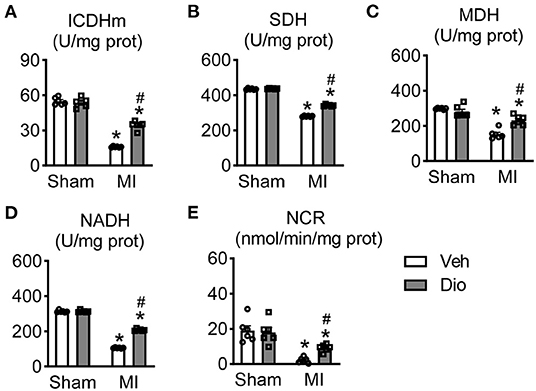
Figure 4. Effect of dioscin on the activities of mitochondrial KREB cycle and respiratory chain enzymes in hearts after MI. The key enzymes of tricarboxylic acid cycle (TCA) of (A) isocitrate dehydrogenase (ICDH; U/mg prot), (B) succinate dehydrogenase (SDH; U/mg prot), and (C) malate dehydrogenase (MDH; U/mg prot). The measureable of respiratory chain enzymes of (D) nitotinamide adenine dinucleotide dehydrogenase (the complex I, NADH; U/mg prot) and (E) cytochrome c-oxidase (the complex III, NCR; nmol/min/mg prot). n = 6. Data are mean ± SEM. *P < 0.05 vs. sham group, #P < 0.05 vs. MI group.
Dioscin Modulates Mitochondrial Antioxidant Status in Heart
In the infarcted hearts, subcellular organelles undergo stress caused by the released of reactive oxygen species (ROS) from mitochondria. Hence, prevention of ROS accumulation in the subcellular compartments are essential for the restoration of the normal cardiomyocyte function. Representative images of dihydroethidium (DHE) staining showed that dioscin attenuated the increase of ROS in response to MI surgery (Figure 5A). We further detected the expression of antioxidant enzymes in mitochondria. Consistently, the activities of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) and glutathione-S-transferase (GST) were declined in the animals subjected to MI, but recovred after dioscin treatment (Figures 5B–E). The decrease in the endogenous antioxidant defense enzyme system was corroborated to a diminished glutathione (GSH) content (Figure 5F). Similarly, the ROS accumulation was measured in H9C2 cells. Consistent with in vivo experiments, dioscin suppressed the enhancement of ROS caused by CoCl2 (Figure 5G). These results confirmed that dioscin abrogated the oxidative stress, enhancing the endogenous antioxidant defense system. Taken together, our study demonstrates that dioscin protects heart dysfunction against myocardial infarction by suppressing the oxidative stress.
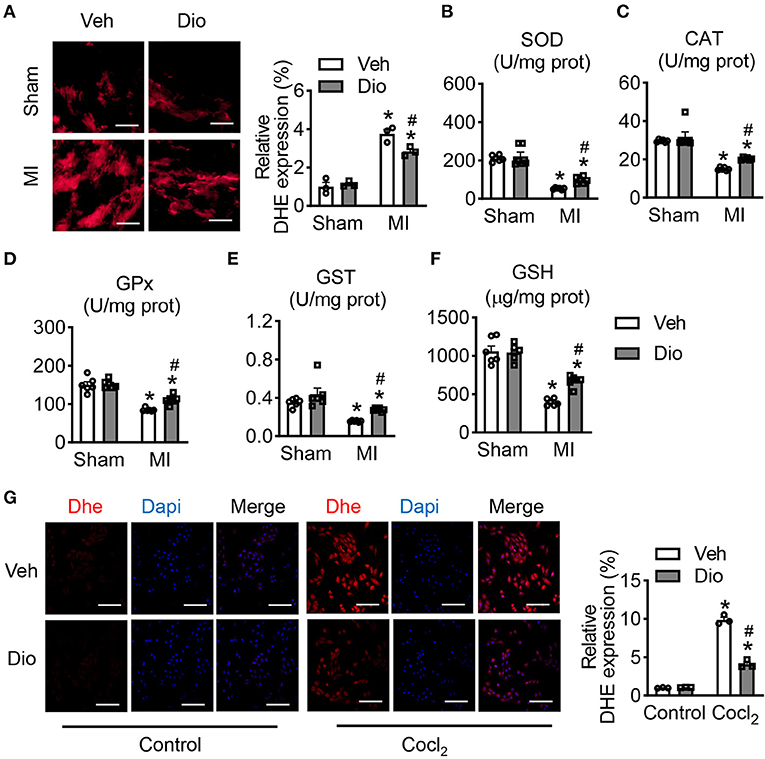
Figure 5. Dioscin modulates mitochondrial antioxidant status. (A) Representative images of DHE in heart section and its qualification, bar = 50 μm (n = 3). The antioxidant enzymes of (B) superoxide dismutase (SOD; U/mg prot), (C) catalase (CAT; U/mg prot), (D) peroxidase (GPx; U/mg prot), (E) glutathione-S-transferase (GST; U/mg prot), and (F) glutathione (GSH; U/mg prot) (n = 6). (G) Representative images of DHE in H9C2 cells and its quantification, bar = 50 μm (n = 3). Data are mean ± SEM. *P < 0.05 vs. sham group, #P < 0.05 vs. MI group.
Discussion
In this study, the activities of antioxidant enzymes were obviously enhanced by treatment of dioscin to further eliminate the production of ROS. Herein, dioscin alleviated cardiac dysfunction in mouse model of MI via mitochondrial protection. In detail, dioscin speeds up the Kreb's cycle by up-regulating the activities of enzymes in mitochondrial. Our study further confirmed the therapeutic effect of dioscin in mice infarcted/hypoxic modelshearts and provided the promising administration of dioscin for heart diseases in clinics.
It has been reported that the mitochondrial ROS could be an index of oxidative stress in hearts. Notably, mitochondrial ROS (mtROS) is the main resource of oxidative stress. Previous studies showed dioscin protects against pulmonary injury by inhibition of the mtROS by activating autophagy (12). Dioscin also activates NRF2 and SIRT2 signal pathway to suppress ROS in doxorubicin-induced cardiotoxicity (21). Moreover, dioscin preventing the accumulation of ROS via activating FXR/LKB1 signal in ischemia/reperfusion injury (22). Although dioscin could improve mitochondrial function in pathological condition, whether it is involved in Kreb's cycle of mitochondria remained unclear.
The Kreb's cycle is a series of complex chemical reaction, which is occurred in organisms to generate energy (23). We detected the essential enzyme activities in mitochondrial Kreb's cycle and respiratory chain, which were enhanced after diosicin administration against the infarction/hypoxia injury. The selective accumulation of the citric acid cycle intermediate succinate is a typical metabolic issue after infarction in a range of tissues, responsible for the accumulation of mitochondrial ROS production after reperfusion (23). The in vivo reduction in the activities of Kreb's cycle in infarcted hearts may represent at an early maladaptive phase in the metabolic alterations after MI (24). It has been reported that high-throughput liquid chromatography-mass spectrometry (LC-MS)-based metabolomics demonstrate that dioscin could be associated with gluconeogenesis, Kreb's cycle, and butanoate metabolism (24). Consistently with the previous results, we further confirmed that dioscin is involved with Kreb's cycle. However, the insight underlying mechanism between disocin and Kreb's cycle requires further study.
Many studies showed that dioscin could, prevent infarction/hypoxia-induced apoptosis via down-regulating the Bax expression (25). Furthermore, dioscin is presented as a therapeutic nature product to maintain mitochondrial membrane potential (26). Our study suggested that the mitochondrial member potential was protected by dioscin administration in response to infarction/hypoxia, and further suppressed the apoptosis of cardiomyocytes.
In summary, the findings of current study indicate the cardioprotective role of dioscin against cardiac injury by suppressing the production of ROS in mitochondria, enhancing the activities of Kreb's cycle enzymes and respiratory chain complexes, and maintaining the structure and function of mitochondria. Our study could help to better understanding the mechanism underlying the dioscin treatment of infarcted heart diseases.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committees at the University of Nanjing Medical University (Ethical Approval Number: 1912034).
Author Contributions
QL and HS designed the study and revised the manuscript. DL, TS, and MZ carried out the experiments and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (81970414 to QL) and the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (19KJA35000 to QL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. Heart disease and stroke statistics-2016 update: a report from the American heart association. Circulation. (2016) 133:e38–360. doi: 10.1161/CIR.0000000000000350
2. Othman AI, Elkomy MM, El-Missiry MA, Dardor M. Epigallocatechin-3-gallate prevents cardiac apoptosis by modulating the intrinsic apoptotic pathway in isoproterenol-induced myocardial infarction. Eur J Pharmacol. (2017) 794:27–36. doi: 10.1016/j.ejphar.2016.11.014
3. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. (1981) 290:457–65. doi: 10.1038/290457a0
4. Yan K, An T, Zhai M, Huang Y, Wang Q, Wang Y, et al. Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. (2019) 10:500. doi: 10.1038/s41419-019-1734-7
5. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
6. Luczak ED, Wu Y, Granger JM, Joiner MA, Wilson NR, Gupta A, et al. Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy. Nat Commun. (2020) 11:4416. doi: 10.1038/s41467-020-18165-6
7. Wu L, Li F, Zhao C, Ming Y, Zheng C, Li Y, et al. Effects and mechanisms of traditional Chinese herbal medicine in the treatment of ischemic cardiomyopathy. Pharmacol Res. (2020) 151:104488. doi: 10.1016/j.phrs.2019.104488
8. Shanmugam K, Ravindran S, Kurian GA, Rajesh M. Fisetin confers cardioprotection against myocardial ischemia reperfusion injury by suppressing mitochondrial oxidative stress and mitochondrial dysfunction and inhibiting glycogen synthase kinase 3beta activity. Oxid Med Cell Longev. (2018) 2018:9173436. doi: 10.1155/2018/9173436
9. Saranya S, Baskaran R, Poornima P, Vijaya Padma V. Berbamine ameliorates isoproterenol-induced myocardial infarction by inhibiting mitochondrial dysfunction and apoptosis in rats. J Cell Biochem. (2019) 120:3101–13. doi: 10.1002/jcb.27522
10. Xue W, Wang X, Tang H, Sun F, Zhu H, Huang D, et al. Vitexin attenuates myocardial ischemia/reperfusion injury in rats by regulating mitochondrial dysfunction induced by mitochondrial dynamics imbalance. Biomed Pharmacother. (2020) 124:109849. doi: 10.1016/j.biopha.2020.109849
11. Kong C, Lyu D, He C, Li R, Lu Q. Dioscin elevates lncRNA MANTIS in therapeutic angiogenesis for heart diseases. Aging Cell. (2021) 20:e13392. doi: 10.1111/acel.13392
12. Du S, Li C, Lu Y, Lei X, Zhang Y, Li S, et al. Dioscin alleviates crystalline silica-induced pulmonary inflammation and fibrosis through promoting alveolar macrophage autophagy. Theranostics. (2019) 9:1878–92. doi: 10.7150/thno.29682
13. Du L, Chen J, Wu Y, Xia G, Chen M, Zhao P, et al. Long non-coding RNA N1LR protects against myocardial ischemic/reperfusion injury through regulating the TGF-beta signaling pathway. Front Cardiovasc Med. (2021) 8:654969. doi: 10.3389/fcvm.2021.654969
14. Yao Y, Lu Q, Hu Z, Yu Y, Chen Q, Wang QK, et al. non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat Commun. (2017) 8:133. doi: 10.1038/s41467-017-00171-w
15. Xue X, Yang JY, He Y, Wang LR, Liu P, Yu LS, et al. Aggregated single-walled carbon nanotubes attenuate the behavioural and neurochemical effects of methamphetamine in mice. Nat Nanotechnol. (2016) 11:613–20. doi: 10.1038/nnano.2016.23
16. Tawarahara K, Kurata C, Okayama K, Watanabe H, Kobayashi A, Yamazaki N. Reversal of perfusion defect on resting thallium-201 emission computed tomography in stunned myocardium and coronary spasm. Am Heart J. (1993) 125:1425–7. doi: 10.1016/0002-8703(93)91016-8
17. Yin X, Zhang X, Lv C, Li C, Yu Y, Wang X, et al. Protocatechuic acid ameliorates neurocognitive functions impairment induced by chronic intermittent hypoxia. Sci Rep. (2015) 5:14507. doi: 10.1038/srep14507
18. Ding L, Lu S, Zhou Y, Lyu D, Ouyang C, Ma Z, et al. The 3' untranslated region protects the heart from angiotensin II-induced cardiac dysfunction via AGGF1 expression. Mol Ther. (2020) 28:1119–32. doi: 10.1016/j.ymthe.2020.02.002
19. Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W, et al. The creatine kinase system in normal and diseased human myocardium. N Engl J Med. (1985) 313:1050–4. doi: 10.1056/NEJM198510243131704
20. Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. (2008) 80:30–9. doi: 10.1093/cvr/cvn184
21. Zhao L, Tao X, Qi Y, Xu L, Yin L, Peng J. Protective effect of dioscin against doxorubicin-induced cardiotoxicity via adjusting microRNA-140-5p-mediated myocardial oxidative stress. Redox Biol. (2018) 16:189–98. doi: 10.1016/j.redox.2018.02.026
22. Dong L, Yin L, Li R, Xu L, Xu Y, Han X, et al. Dioscin alleviates lung ischemia/reperfusion injury by regulating FXR-mediated oxidative stress, apoptosis, and inflammation. Eur J Pharmacol. (2021) 908:174321. doi: 10.1016/j.ejphar.2021.174321
23. Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
24. Wang XX, Yu PC, Li J. High-Throughput metabolomics for identification of metabolic pathways and deciphering the effect mechanism of dioscin on rectal cancer from cell metabolic profiles coupled with chemometrics analysis. Front Pharmacol. (2020) 11:68. doi: 10.3389/fphar.2020.00068
25. Si L, Zheng L, Xu L, Yin L, Han X, Qi Y, et al. Dioscin suppresses human laryngeal cancer cells growth via induction of cell-cycle arrest and MAPK-mediated mitochondrial-derived apoptosis and inhibition of tumor invasion. Eur J Pharmacol. (2016) 774:105–17. doi: 10.1016/j.ejphar.2016.02.009
Keywords: myocardial infarction, dioscin, mitochondrial dysfunction, ROS, KREB'S cycle
Citation: Shen T, Lyu D, Zhang M, Shang H and Lu Q (2022) Dioscin Alleviates Cardiac Dysfunction in Acute Myocardial Infarction via Rescuing Mitochondrial Malfunction. Front. Cardiovasc. Med. 9:783426. doi: 10.3389/fcvm.2022.783426
Received: 26 September 2021; Accepted: 01 February 2022;
Published: 04 March 2022.
Edited by:
Haiyang Tang, University of Arizona, United StatesReviewed by:
Weiming Wang, Central South University, ChinaJun Zou, Georgia State University, United States
Stephen C. Kolwicz Jr., Ursinus College, United States
Copyright © 2022 Shen, Lyu, Zhang, Shang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Shang, c2hhbmdodWk2MzQwQDE2My5jb20=; Qiulun Lu, cWl1bHVubHVAbmptdS5lZHUuY24=
†These authors have contributed equally to this work