 Zifeng Yang1,2†
Zifeng Yang1,2† Ruifeng Tian
Ruifeng Tian Xiao-Jing Zhang
Xiao-Jing Zhang Jingjing Cai
Jingjing Cai Zhi-Gang She
Zhi-Gang She Hongliang Li
Hongliang Li- 1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Institute of Model Animal, Wuhan University, Wuhan, China
- 3School of Basic Medical Sciences, Wuhan University, Wuhan, China
- 4Department of Cardiology, The Third Xiangya Hospital, Central South University, Changsha, China
- 5Gannan Innovation and Translational Medicine Research Institute, Gannan Medical University, Ganzhou, China
In the past few decades, non-alcoholic fatty liver disease (NAFLD) and heart failure with preserved ejection fraction (HFpEF) have become the most common chronic liver disease and the main form of heart failure (HF), respectively. NAFLD is closely associated with HFpEF by sharing common risk factors and/or by boosting systemic inflammation, releasing other secretory factors, and having an expansion of epicardial adipose tissue (EAT). Therefore, the treatments of NAFLD may also affect the development and prognosis of HFpEF. However, no specific drugs for NAFLD have been approved by the Food and Drug Administration (FDA) and some non-specific treatments for NAFLD are applied in the clinic. Currently, the treatments of NAFLD can be divided into non-pharmacological and pharmacological treatments. Non-pharmacological treatments mainly include dietary intervention, weight loss by exercise, caloric restriction, and bariatric surgery. Pharmacological treatments mainly include administering statins, thiazolidinediones, glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter 2 inhibitors, and metformin. This review will mainly focus on analyzing how these treatments may affect the development and prognosis of HFpEF.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is one of the most prevalent chronic liver diseases without specific treatments and covers the disease spectrum from simple steatosis to non-alcoholic steatohepatitis (NASH), which can evolve into end-stage liver diseases (1–3). With a global incidence of 25.24%, NAFLD brings up a huge clinical and economic burden (2). The clinical and economic burden of NAFLD arises not only from liver-related morbidity and mortality but also from extrahepatic diseases, such as type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD) (4). Heart failure with preserved ejection fraction (HFpEF) is one of the major CVDs associated with NAFLD (5, 6). HFpEF is the most common type of heart failure (HF) and even has an incidence rate of 55% among the elderly (7, 8). A recent clinical study demonstrated that individuals with NAFLD are at increased risk of developing HF and that NAFLD is more likely to coexist with HFpEF (9). In addition, the coexistence of NAFLD and HFpEF often occurs in patients with HFpEF, and the prevalence of NAFLD in patients with HFpEF can reach 50% (10). All these facts indicate the tight association between NAFLD and HFpEF.
Theoretically, NAFLD is associated with HFpEF in two layers. On the one hand, NAFLD and HFpEF share common risk factors. Several documented risk factors of NAFLD, including age, male sex, obesity, and T2DM (11–13), are also known risk factors for HFpEF (14–16). On the other hand, accumulating compelling evidence indicates that the pathological consequence of NAFLD may promote the development of HFpEF. It is well known that NAFLD readily boosts systemic inflammation and has thicker epicardial adipose tissue (EAT) (17–19). In addition, a large number of other secretory factors are also released in NAFLD (20). These pathological processes play crucial roles in the progression of HFpEF (21–23). The close association between NAFLD and HFpEF indicates that treating NAFLD may simultaneously affect the progression of HFpEF.
However, there are no specific drugs approved for the treatment of NAFLD yet, management of NAFLD is currently achieved by employing non-specific non-pharmacological and/or pharmacological treatments. Non-pharmacological treatments mainly include dietary intervention, weight loss by exercise, caloric restriction, and bariatric surgery. Pharmacological treatments mainly include administering statins, thiazolidinediones, glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter 2 inhibitors, and metformin. These non-pharmacological and pharmacological treatments can effectively treat the liver in NAFLD/NASH patients (24–26). Interestingly, extensive literature shows that some treatments, which are commonly used in the treatment of NAFLD/NASH, coincidentally have a beneficial or detrimental impact on decreasing the effects of secretory factors, or reducing EAT accumulation, controlling common risk factors for NAFLD and HFpEF (27–29). Therefore, it can be speculated that some treatments for NAFLD may also affect the development of HFpEF.
This review mainly focuses on overviewing the possible effects of current interventions available for NAFLD on the development and prognosis of HFpEF.
2. NAFLD is closely associated with HFpEF
NAFLD and HFpEF are both systemic diseases (4, 30). Growing evidence shows that there is a close relationship between NAFLD and HFpEF (6, 9, 10). This association mainly exists in two layers. Firstly, NAFLD and HFpEF share common risk factors. Secondly, NAFLD may contribute to the development of HFpEF by enhancing systemic inflammation, releasing of other secretory factors, and having thicker EAT (Figure 1).
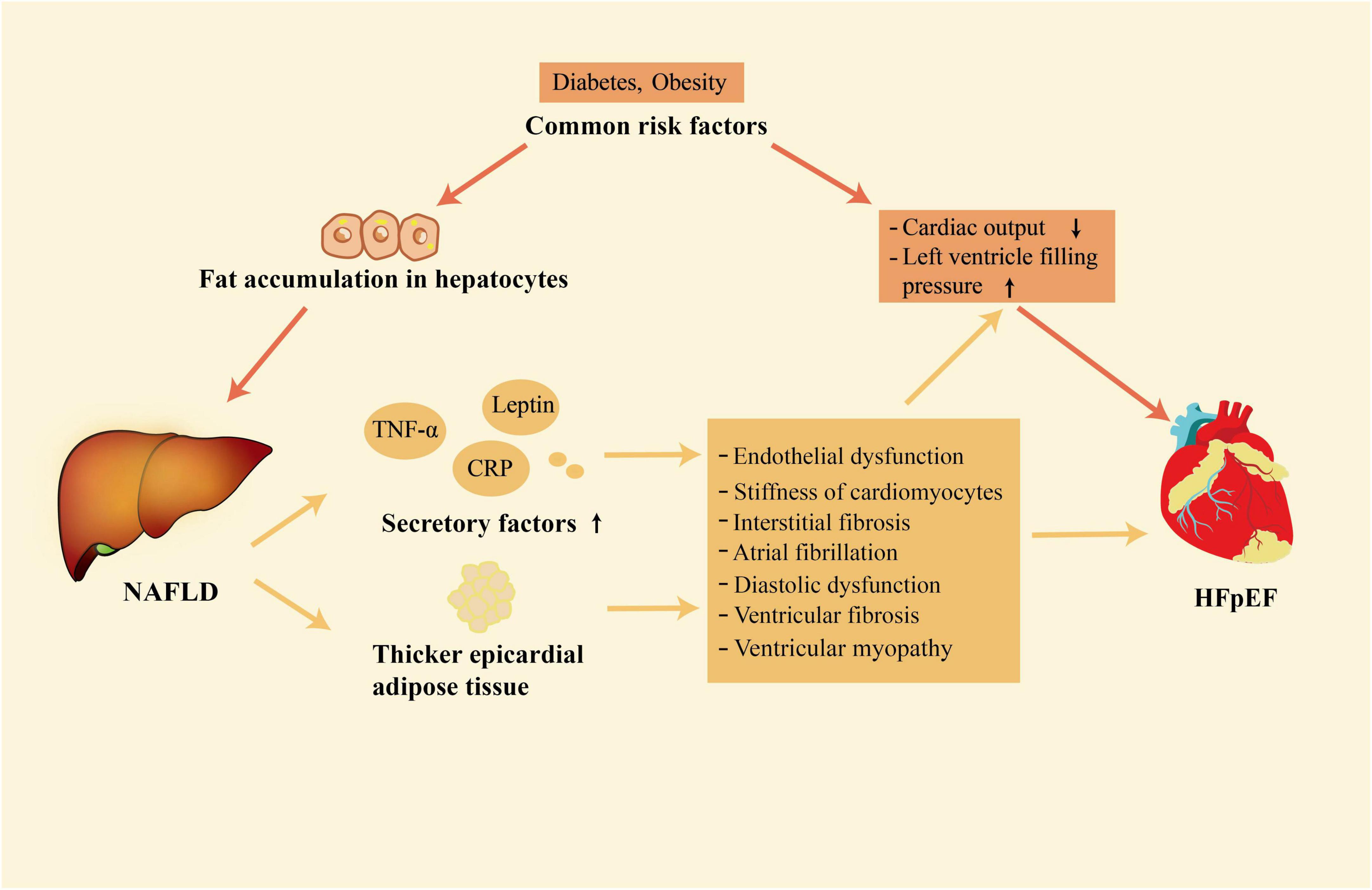
Figure 1. The close association between NAFLD and HFpEF. NAFLD and HFpEF have some common risk factors; in addition, NAFLD is associated with the development of HFpEF through its association with release of secretory factors and increased accumulation of epicardial adipose tissue. NAFLD, non-alcoholic fatty liver disease; HFpEF, heart failure with preserved ejection fraction.
2.1. NAFLD and HFpEF share common risk factors
NAFLD is characterized by excessive fat accumulation in hepatocytes without or with lobular inflammation and fibrosis and affects more than a billion people worldwide today (31, 32). NAFLD covers several liver diseases ranging from hepatic steatosis to NASH. Simple hepatic steatosis has been defined as NAFL, and NASH has been defined as a serious subtype of NAFLD with a more severe process of hepatocellular injury and inflammation (33). Moreover, NASH is often accompanied by liver fibrosis and even progresses to cirrhosis. Based on imaging, some clinical features such as elevated liver enzymes and the presence of metabolic comorbidities, or histology, NAFLD can be diagnosed in individuals (34). There are many risk factors for NAFLD, including obesity, and T2DM (35, 36). Together with NAFLD, these risk factors constitute major and causative risk factors of metabolic diseases and are mediators of the occurrence and development of CVDs, including HFpEF (37–40). Exposure to these risk factors, especially obesity and T2DM, contributes to an increased risk of NAFLD and HFpEF in the population (41, 42). More importantly, because they share risk factors, NAFLD and HFpEF are likely to coexist in the same individuals (10). These common risk factors should be one of the key mechanisms underlying the epidemiological link between NAFLD and HFpEF.
2.2. NAFLD is associated with the development of HFpEF
HFpEF has been defined as a multisystem disorder with multiple pathophysiological abnormalities. In the pathophysiology of HFpEF, left ventricular diastolic dysfunction plays a crucial and indispensable role (43). In addition to an abnormality in left ventricular diastolic function, HFpEF is a complex syndrome that has multiple injuries of cardiac reserve and rhythm, endothelial dysfunction, atrial dysfunction, chronotropic reserve deficits, ventricular and vascular stiffness, defects in vasodilation, pulmonary hypertension, peripheral microvascular dysfunction (43, 44). Coincidentally, NAFLD is associated with abnormalities in cardiac structure and function, which are common in HFpEF (45–47). NAFLD may be associated with the development of HFpEF through enhancing systemic inflammation, releasing of other secretory factors, and having thicker EAT (21–23, 48).
During the pathogenies of NAFLD, increased amounts of lipids accumulate in the liver and lead to oxidative stress, cell death of hepatocytes, recruitment of inflammatory cells, and increased secretion of inflammatory factors (49). The immune cell population of the liver such as Kupffer cells, dendritic cells, and macrophages switches from an immune tolerance phenotype to an immunogenic phenotype in the pathogenesis of NAFLD (50). The shift of proinflammatory phenotype and the activation of the immune response promote the production and release of cytokines, increasing the penetration of circulating immune cells into the liver and enhancing local inflammatory response (51). Then a large number of inflammatory mediators are carried by the blood from the hepatic sinus into the systemic circulation (52). The increased amount of inflammatory molecule secretion, including tumor necrosis factor-A (TNF-α), C-reactive protein (CRP), interleukin-6 (IL-6), and some other pro-inflammations (53), boosts systemic inflammation. Systemic inflammation not only contributes to the transition from simple steatosis into NASH but also poses a threat to the cardiovascular system (21). It has been reported that systemic inflammation would induce coronary microvascular endothelial dysfunction (22). Then, the bioavailability of nitric oxide and the activity of protein kinase G in adjacent cardiomyocytes are decreased by coronary microvascular endothelial inflammation (54). Low activity of protein kinase G contributes to elevated resting tension and cardiac hypertrophy, resulting in interstitial fibrosis and cardiomyocyte stiffness (22, 55, 56). Both interstitial fibrosis and stiff cardiomyocytes promote left ventricular stiffness and the development of HFpEF. A meta-analysis showed that high circulating levels of inflammatory markers, such as CRP and IL-6, also induce atrial fibrillation in the general population (57). Atrial fibrillation is not only closely associated with diastolic dysfunction, but also an important risk factor of HFpEF, which can predict HFpEF (58, 59). Meanwhile, elevated levels of inflammatory markers themselves, such as CRP, are independently associated with asymptomatic diastolic dysfunction (60).
Inflammatory factors play a key role in linking NAFLD and HFpEF. Meanwhile, NAFLD could also promote the development of HFpEF through the effects of other secretory factors such as angiotensinogen, leptin, and hepatokines. Angiotensinogen is mainly secreted by the liver and is the only precursor of the renin-angiotensin system (61). In the case of obesity, the adipose tissue can also produce a large amount of angiotensinogen and lead to elevation of the circulating angiotensinogen (62), which is pretty common in patients with NAFLD. Increased circulating angiotensinogen levels lead to excessive production of angiotensin I and angiotensin II (63). Eventually, this scenario leads to the activation of the renin-angiotensin system and increases the risk of CVD, including HFpEF. In patients with NAFLD, the sensitivity to leptin is usually reduced, leading to an increase in the levels of leptin (64). High leptin levels can lead to ventricular fibrosis and cardiomyocyte hypertrophy (65). At the same time, leptin is also associated with sodium retention and increased plasma volume, which would increase the risk of heart failure (66). The liver also secretes a class of proteins called hepatokines. Some cardiovascular risk-related hepatokines, such as fetuin-A and fibroblast growth factor-21, are increased in NAFLD (67, 68). Fetuin-A can increase the proliferation of smooth muscle cells and the deposition of collagen, thus accelerating vascular remodeling and resulting in the occurrence of HFpEF (69). Fibroblast growth factor-21, however, exerts an anti-inflammation function, and inflammation plays a key role in the development of HFpEF (70). Therefore, the net effects of hepatokines on the cardiovascular system in the case of NAFLD are still unclear and need to be further explored.
In addition to systemic inflammation and the effects of other secretory factors, EAT also plays a significant role in the association between NAFLD and HFpEF. In addition to energy storage, adipose tissue is also an endocrine organ that can secrete many proinflammatory adipokines in chronic systemic disorders (71). Compared with normal subjects, EAT is increased in individuals with NAFLD and the degree of expansion is related to the severity of liver steatosis and fibrosis (72). The expansion of EAT is often accompanied by the upregulation of proinflammatory cytokines and infiltration of immune cells such as macrophages, dendritic cells, and T lymphocytes. Consequently, EAT has become an important source of proinflammatory and profibrotic mediators such as IL-6, monocyte chemoattractant protein-1, TNF-α, and interleukin-1β (73). These mediators can cause ventricular fibrosis and coronary microvascular dysfunction, leading to the development of HFpEF (71, 74, 75). It was reported that adjoin of dysfunctional EAT with the left ventricle resulted in ventricular myopathy characterized by impaired cardiac dilatation, which would lead to HFpEF (76). Conversely, removing EAT can improve the structure of vascular tissue and restore diastolic function (77, 78).
NAFLD may also be associated with the development of HFpEF through other pathophysiological processes such as metabolic abnormalities and oxidative stress. The overproduction of glucose and triglycerides in NAFLD would cause hyperglycemia and hyperglyceridemia (79). Hyperglycemia and hyperglyceridemia are the key features of the pathophysiology of metabolic syndrome. Furthermore, excessive fat accumulation in hepatocytes induces oxidative stress and the overproduction of reactive oxygen species in the mitochondria (80). Both metabolic syndrome and oxidative stress are closely related to the development of HFpEF (81, 82).
In summary, accumulating studies have indicated that a close association is between NAFLD and HFpEF. Therefore, the treatments against NAFLD are also likely to exert beneficial or harmful effects on the development and prognosis of HFpEF.
2.3. NAFLD directly leads to the occurrence of HFpEF
The above evidence indicates the close association between NAFLD and HFpEF. Moreover, accumulating studies also show that the progression of NAFLD may result in the occurrence of HFpEF, probably by preventing blood flow back to the heart, inducing changes in hemodynamics, and releasing secretory factors.
About 25% of the total blood returns to the heart through the liver (83). When liver fibrosis progressively deteriorates, it would increase resistance in the hepatic sinus and lead to obstruction of blood flow through the liver (84, 85). This blockage prevents the return of blood from the veins to the heart. Some studies have shown that this phenomenon even existed in the early stage of NAFLD (84). During the development of NAFLD, the liver changes from simple hepatic steatosis to NASH, and eventually to cirrhosis (86). Portal hypertension is one of the typical characteristics of liver cirrhosis (87). Thus, spontaneous portosystemic shunt and arteriovenous shunt would be formed in the late stage of NAFLD, resulting in hemodynamic changes with increased cardiac output at rest (6, 88). These alterations limit the preload reserve of the heart and make it difficult to increase cardiac output during stress, eventually resulting in the occurrence of HFpEF. Finally, during the progression of NAFLD, many secretory factors would be increased in circulation, leading to the occurrence of HFpEF, as stated in section 2.2.
In summary, NAFLD can lead to the occurrence of HFpEF through the above-mentioned mechanism. In this aspect, more direct evidence is needed to confirm the causal relationship and to facilitate discovering the detailed molecular pathways. Employing the power of emerging frontier technologies, such as multi-omics technologies and single-cell technologies, may help us to achieve these goals.
3. Treatments for NAFLD and their impact on HFpEF
At present, there is no specific drug for the treatment of NAFLD. Instead, some non-specific non-pharmacological and pharmacological treatments are currently applied (Figure 2). Some of the treatments have been documented to play a vital role in controlling common risk factors, reducing the effects of secreted factors, or decreasing EAT accumulation. Therefore, these treatments may also have a therapeutic impact on HFpEF (Table 1).
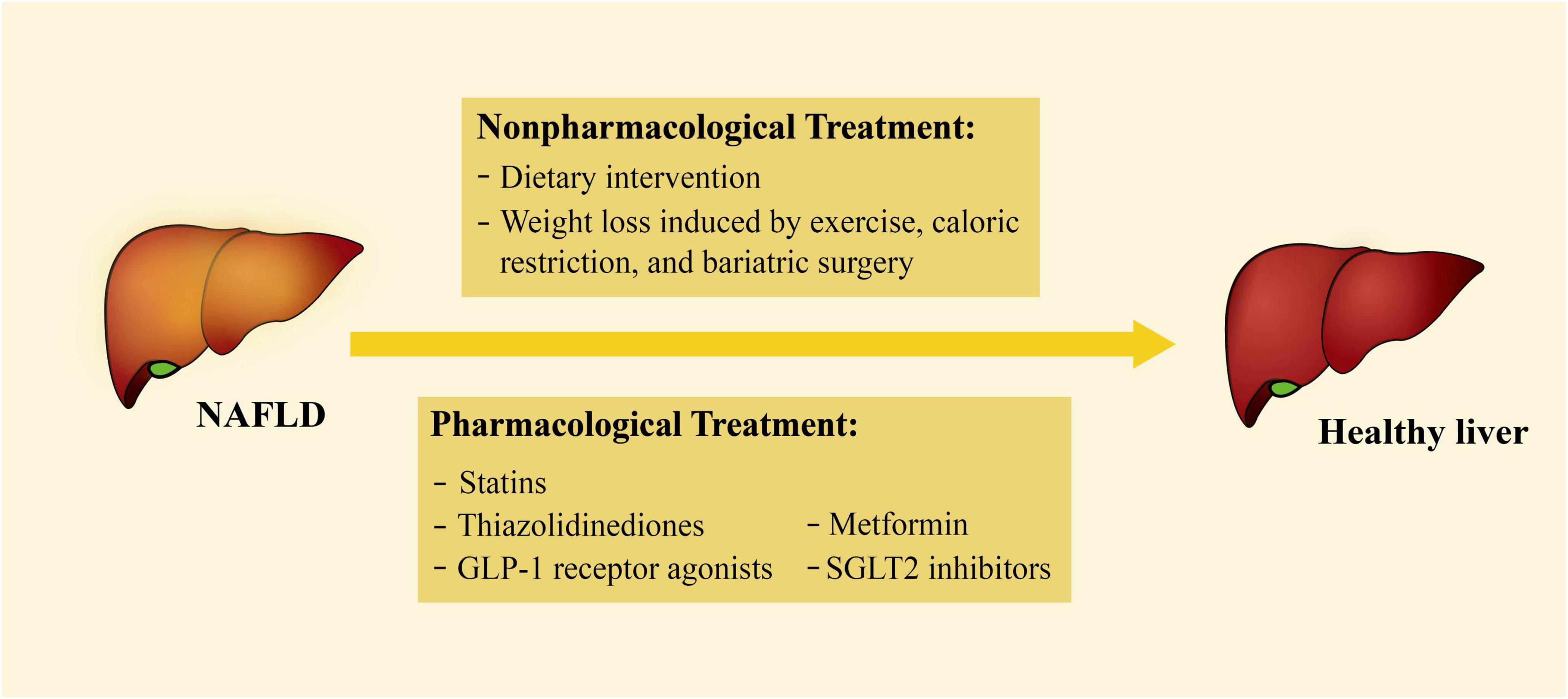
Figure 2. Current treatments for NAFLD. The treatments of NAFLD can be divided into non-pharmacological and pharmacological treatments. Non-pharmacological treatments include dietary intervention, weight loss by exercise, caloric restriction, and bariatric surgery; pharmacological treatments include statins, thiazolidinediones, GLP-1 receptor agonists, SGLT2 inhibitors, and metformin. NAFLD, non-alcoholic fatty liver disease; GLP-1, glucagon-like peptide-1; SGLT2, sodium-glucose cotransporter 2.
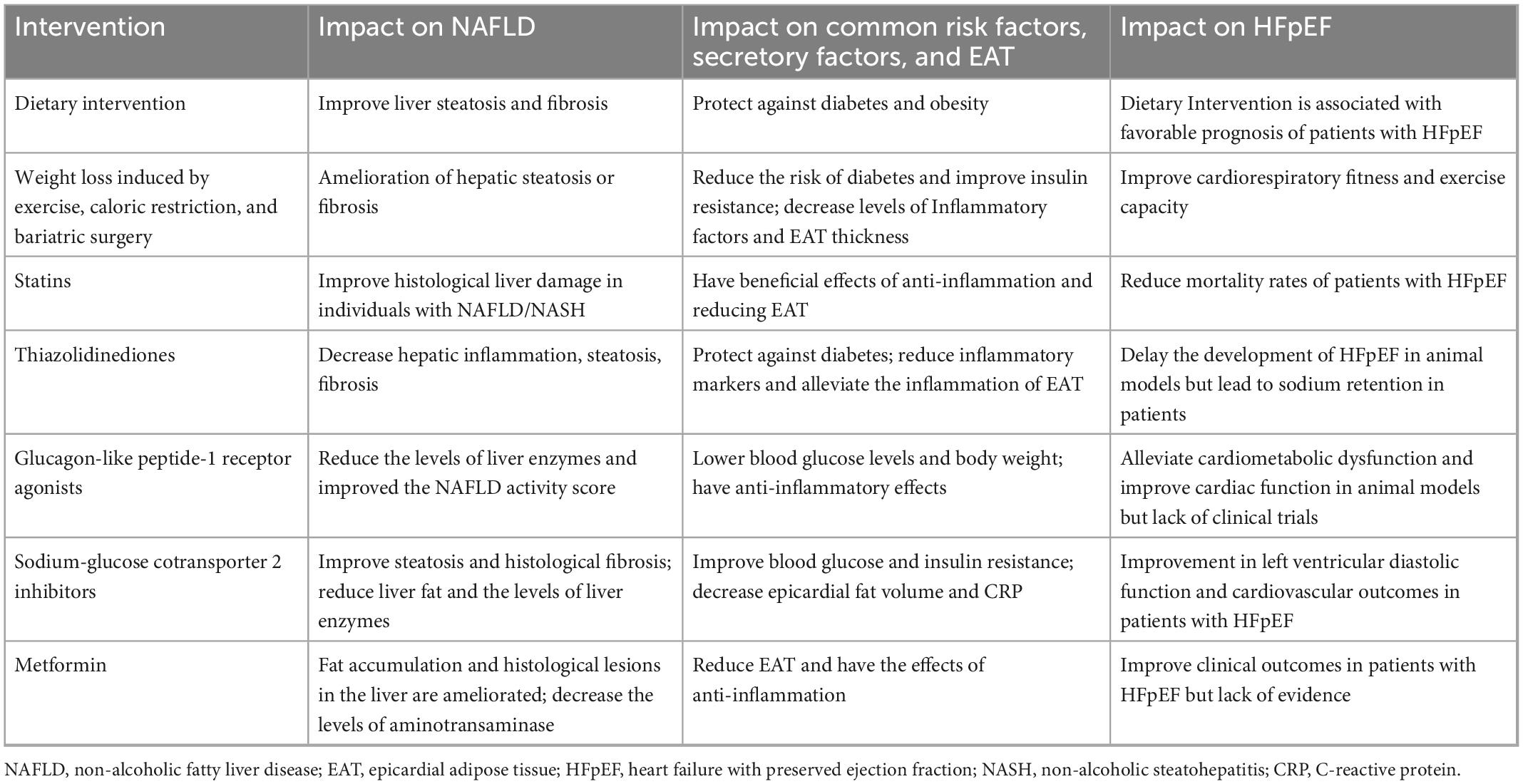
Table 1. Summary of treatments for NAFLD and their impact on HFpEF.
3.1. The influence of non-pharmacological treatment against NAFLD on the development of HFpEF
Non-pharmacological treatment is the cornerstone of NAFLD treatment, which mainly includes dietary intervention and weight loss induced by exercise, caloric restriction, and bariatric surgery. These non-pharmacological approaches are effective in the treatment of NAFLD and also showed promising results in delaying the development of HFpEF.
3.1.1. Dietary intervention
A large number of NAFLD patients overconsume fructose and saturated fatty acids and have reduced intake of dietary fiber and omega-3-rich food (89, 90). Obesity and metabolic syndrome can be caused by such dietary patterns, increasing the risk for NAFLD. Therefore, changes in diet with or without calorie restriction may be a viable and sustainable strategy for the treatment of NAFLD.
The Mediterranean diet, characterized by a large consumption of plant foods, consists of vegetables and fruits, fish, whole grains, legumes, etc. (91, 92). This diet is rich in monounsaturated fatty acids, mainly from low-fat dairy products. The potential association between the Mediterranean diet and the alleviation of NAFLD has been extensively studied. In a randomized, crossover 6-week dietary intervention study, the Mediterranean diet reduced liver steatosis and improved insulin sensitivity among subjects with biopsy-proven NAFLD, even without weight loss (93). At a ten-year follow-up visit, adherence to the Mediterranean diet has been inversely associated with liver steatosis and liver fibrosis (94). In addition to being favorable for NAFLD, the Mediterranean diet improves insulin sensitivity and protects against diabetes and CVD (93, 94). Moreover, it plays a very positive role in the prevention of hypertension and obesity (95, 96). Diabetes mellitus and obesity are the common risk factors for NAFLD and HFpEF. More importantly, in a prospective study, the Mediterranean diet positively affected the prognosis of patients with HFpEF, especially after a 10-year intervention (97). In this prospective study, the Mediterranean diet prevents the recurrence of coronary artery disease by directly ameliorating the effects of inflammation in patients with HFpEF (97). The previous cross-sectional study has also indicated that adherence to a Mediterranean diet can decrease the levels of heart failure biomarkers such as N-terminal pro-brain natriuretic peptide in individuals at high risk of CVD (98). It has also been reported that the Mediterranean diet is beneficial to diastolic and systolic function of the heart in individuals with chronic heart failure (99). Therefore, the Mediterranean diet may have a favorable impact on clinical outcomes in patients with NAFLD and/or HFpEF and should be recommended to these patients.
3.1.2. Weight loss induced by exercise, caloric restriction, and bariatric surgery
Excessive calorie intake and lack of exercise, which lead to obesity and related comorbidities, play a vital role in the development of NAFLD and NASH (100). Lifestyle changes, including weight loss by exercise and caloric restriction, remain the primary treatment for NAFLD management (101).
Weight loss caused by exercise and caloric restriction has a dose-dependent treatment effect on the liver histology of NAFLD patients. Patients with ≥ 5% of total body weight loss can have an improvement in hepatic steatosis, and patients with ≥ 7% of total body weight loss can have significant improvement in steatosis and NAFLD activity score (102). More importantly, among those with ≥ 10% of total body weight loss, liver fibrosis will even regress at least 1 stage (102). The therapeutic effect of weight loss is encouraging in NAFLD. Some common risk factors for NAFLD and HFpEF are also affected by weight loss, such as T2DM (103, 104). More importantly, weight loss caused by exercise and caloric restriction has an impact on EAT and inflammation. In a low-calorie diet weight loss clinical trial, a 6-month low-calorie diet decreased EAT thickness and improved left ventricular mass and diastolic function (105). Inflammatory markers, such as TNFα and CRP, were also significantly decreased by aerobic interval training and a low-calorie diet in a one-year randomized controlled trial (106). In addition, weight loss by exercise and caloric restriction is associated with improvements in cardiorespiratory fitness and exercise capacity among patients with HFpEF (107, 108). These may improve the physical condition of patients with HFpEF and their quality of life. Therefore, weight loss by exercise and a low-calorie diet is highly recommended for patients with NAFLD and may greatly decrease the risk of HFpEF.
The weight loss caused by exercise and caloric restriction has few side effects, but a majority of patients cannot achieve their weight loss goal through exercise and caloric restriction (109). At this time, bariatric surgery can help patients reach this goal. The effects of weight loss caused by bariatric surgery or lifestyle changes seem to be similar for NAFLD, and surgery is more drastic than lifestyle therapies (24). Weight loss caused by bariatric surgery can also alleviate systemic inflammation and control some shared risk factors for NAFLD and HFpEF (110, 111). However, there are conflicting reports in the literature regarding whether bariatric surgery can alleviate EAT. Gaborit et al. reported that epicardial fat decreased significantly after six months of bariatric surgery (112). Nevertheless, Foppa et al. reported that bariatric surgery can significantly decrease subcutaneous fat thickness instead of EAT (113). Moreover, only patients with a BMI > 35 kg/m2 can meet the indications for bariatric surgery (114). Therefore, bariatric surgery is not suitable for a majority of people, although it may be a promising treatment for patients with NAFLD and reduce the risk of HFpEF.
Weight loss induced by exercise, caloric restriction, and bariatric surgery has obvious benefits for patients. However, it is difficult to achieve the goal of weight loss through exercise and caloric restriction. In addition, due to strict indications for bariatric surgery, bariatric surgery cannot be applied to people with normal/lean weight. Therefore, some drugs may be needed during the treatment.
3.2. The influence of pharmacological treatment against NAFLD on the development of HFpEF
Although there are no drugs approved by the Food and Drug Administration for NAFLD, some non-specific pharmacological treatments currently have shown some benefits in NAFLD such as statins, thiazolidinediones, glucose-like peptide-1 receptor agonists, sodium-glucose cotransporter 2 inhibitors, and metformin. These drugs also have an impact on common risk factors for NAFLD and HFpEF, inflammation, and EAT. Therefore, these drugs may also be beneficial for delaying the development of HFpEF.
3.2.1. Statins
Because metabolic syndrome is related to NAFLD, abnormal lipid metabolism is common in individuals with NAFLD and can increase the risk of CVD (115–117). Therefore, lipid-lowering strategies are critical for individuals with NAFLD. Fortunately, statins can manage the metabolism of lipids such as lowering total cholesterol and low-density lipoprotein.
Statins, hydroxymethylglutaryl-CoA reductase inhibitors, are widely used in various chronic diseases including NAFLD (118, 119). In a multicenter cohort of 1,201 subjects, statins improved histological liver damage in individuals with NAFLD/NASH in a dose-dependent manner and protected against steatosis, steatohepatitis, and fibrosis progression (120). Moreover, statins can increase the therapeutic effect of other drugs on NAFLD by controlling the lipid profile (121). In addition to managing low-density lipoprotein cholesterol, statins have also shown other effects. A randomized controlled trial of atorvastatin and pravastatin therapy reported that statins can also lead to induced EAT regression in patients with hyperlipidemia (122). Furthermore, in other clinical trials, statins have also shown anti-inflammatory effects and alleviated systemic inflammation (28, 123), which will reduce the risk of HFpEF. More importantly, it is reported that statin use is related to improved survival rates in individuals with HFpEF (124, 125). Nochioka et al. also reported that statin use is associated with a better prognosis in HFpEF, independent of the presence of coronary artery disease (124). Statins can also prevent extracellular remodeling, delay the progression of cardiac hypertrophy, improve cardiac diastolic function, and reduce oxidative stress in animal models (126, 127). These findings show the promising results of statins in HFpEF.
However, statins have some side effects. In a randomized, double-blind trial, statins increased the prevalence of diabetes (128), which is supposed to increase the risk of HFpEF. Although statins have been proven to alleviate insulin resistance in animal models, it has been shown in clinical trials that statins can increase the risk of T2DM (129, 130). This may be because statins can impair insulin sensitivity and secretion by pancreatic β-cells and increase insulin resistance in peripheral tissues (131). Although this may cause concern about triggering T2DM, current literature indicates that statins have more advantages than disadvantages in treating NAFLD and reducing the risk of HFpEF. Of course, more studies on the association between statin therapy and the risk of T2DM are needed.
3.2.2. Thiazolidinediones
Peroxisome proliferator-activated receptor-γ (PPAR-γ), a member of the nuclear hormone receptor superfamily, is mainly expressed in adipose tissue. When PPAR-γ is activated, it can enhance lipogenesis, insulin sensitivity, and adipocyte differentiation (132). However, PPAR-γ inactivation in adipose tissue can lead to insulin resistance, metabolic disorders, and excessive fat accumulation in the liver (133). Therefore, PPAR-γ agonists are promising drugs for the treatment of NAFLD. Thiazolidinediones, PPAR-γ agonists, are primarily used to treat diabetes by increasing insulin sensitivity and have also achieved effective results in the treatment of NAFLD in several trials (134–136).
Pioglitazone, a thiazolidinedione, has been investigated in multiple trials. In a randomized, double-blind, placebo-controlled trial, 101 patients with NASH and prediabetes or T2DM received pioglitazone 45 mg/d or placebo for 18 months, followed by pioglitazone treatment for 18 months (134). Pioglitazone significantly improved liver histology including fibrosis in patients with NASH (134). Even low-dose pioglitazone (15 mg/day) can improve hepatic inflammation, steatosis, and insulin resistance in patients in the trial. In addition to the encouraging results in NAFLD treatment, pioglitazone also showed the effects of reducing biomarkers of systemic inflammation and reducing the inflammation of EAT. In a clinical trial of 73 patients with CVD and metabolic syndrome, EAT and plasma inflammatory markers were greatly reduced by pioglitazone, such as IL-6, TNF-α, resistin, and metalloproteinase-9 (137). Furthermore, the expression of proinflammatory and anti-inflammatory genes is reduced in EAT after pioglitazone treatment (138). Pioglitazone can also delay the development of left ventricular fibrosis and HFpEF in animal models (139). Therefore, from these aspects, pioglitazone may reduce the risk of HFpEF.
However, thiazolidinediones can lead to sodium retention and weight gain through anti-natriuretic effects on human proximal tubular cells. Thiazolidinediones can increase the risk of HF, which may be HFpEF or HFrEF (140). Nonetheless, in an Insulin Resistance Intervention After Stroke trial, the study indicated that thiazolidinediones did not increase the risk of developing HF (141). This may be because the action of alleviating systemic inflammation, reducing the secretion of inflammatory biomarkers of EAT, and reducing risk factors for HF by thiazolidinediones dwarfs the side effects of sodium retention, resulting in a neutral or favorable role of thiazolidinediones in HF. In summary, because the effect of thiazolidinediones on HFpEF is still unclear, whether thiazolidinediones can reduce the risk of HFpEF also requires further systemic studies.
3.2.3. Glucagon-like peptide-1 receptor agonists
Glucagon-like peptide-1 (GLP-1) is a hormone produced mainly by intestinal L cells and is an incretin. GLP-1, which is secreted in response to food digestion, stimulates insulin secretion by pancreatic β cells and inhibits glucagon secretion by α cells (142). GLP-1 analogs can lower blood sugar levels and body weight by regulating insulin secretion and suppressing appetite (143). Therefore, GLP-1 receptor agonists play a role in the treatment of NAFLD patients, especially those with T2DM and obesity.
Liraglutide, a long-acting GLP-1 receptor agonist, has shown beneficial effects on NAFLD/NASH in clinical trials. In a multicenter, randomized, double-blind, placebo-controlled trial, liraglutide decreased the levels of liver enzymes and improved the NAFLD activity score which included liver ballooning and lobular inflammation. (144). Furthermore, treatment with liraglutide, taking 1.2 mg daily for 6 months, significantly reduced liver fat content and body weight in patients (145). Similar results are also found in other GLP-1 receptor agonists, such as exenatide (146). The therapeutic effects of GLP-1 receptor agonists on NAFLD are very encouraging. More importantly, GLP-1 receptor agonists show anti-inflammatory effects in preclinical studies and clinical trials. GLP-1 receptor agonists have anti-inflammatory effects on the cardiovascular system, liver, pancreatic islets, and brain by reducing the production of inflammatory markers (147–150). Furthermore, GLP-1 receptor agonists reduce the levels of IL-6 and alleviate oxidative stress and endothelial dysfunction in patients with T2DM or type 1 diabetes mellitus in several clinical trials, which may reduce the risk of HFpEF (151–153). Although GLP-1 receptor agonists have not been invested in patients with HFpEF so far, GLP-1 receptor agonists alleviated cardiometabolic disorders and improved heart function in animal models (154). However, reports on the effects of GLP-1 receptor agonists on EAT are somewhat contradictory. In a clinical trial, 24 participants were randomized to receive liraglutide, and 26 were randomized to receive a placebo (155). Although liraglutide caused the historical resolution of NASH, liraglutide failed to reduce epicardial, hepatic, or myocardial fat. In addition, liraglutide even led to an increase in inflammatory factors in adipose tissue, such as TNF-α and macrophage chemoattractant protein-1 (MCP-1) (156). In contrast, Gianluca Iacobellis et al. reported a substantial and rapid EAT reduction caused by liraglutide (29). Therefore, the effect of GLP-1 receptor agonists on EAT needs further studies.
In summary, GLP-1 receptor agonists show encouraging results in NAFLD and may reduce the risk of HFpEF in patients by improving insulin sensitivity, anti-inflammation, and improving cardiac function. Therefore, GLP-1 receptor agonists deserve further investigation in patients with HFpEF.
3.2.4. Sodium-glucose cotransporter 2 inhibitors
Sodium-glucose cotransporter 2 (SGLT2) inhibitors inhibit the reabsorption of glucose in the proximal tubule of the kidney. SGLT2 inhibitors increase the excretion of glucose, thereby reducing blood glucose and improving insulin resistance (157). The improvement in hyperglycemia and insulin resistance may be related to the reduction of fatty acid synthesis and the blockage of de novo lipogenesis (158, 159). Therefore, SGLT2 inhibitors have promising therapeutic potential for the treatment of NAFLD.
Empagliflozin, an SGLT2 inhibitor, is an effective oral antidiabetic drug and has been explored for the treatment of NAFLD. In a clinical trial, NAFLD participants were randomly assigned to empagliflozin (10 mg/day) (N = 43) or placebo (N = 47) for 24 weeks (160). The result is that empagliflozin reduces liver fat and improves steatosis and histological fibrosis, regardless of the presence or absence of diabetes. In another clinical trial, after 20 weeks of empagliflozin treatment, liver fat and the levels of alanine transaminase were decreased in NAFLD patients (161). Moreover, SGLT2 inhibitors (dapagliflozin and luseogliflozin) can also reduce EAT accumulation and alleviate EAT inflammation, which would reduce the risk of HFpEF. During six months of dapagliflozin treatment in a clinical trial, dapagliflozin decreases the EAT volume in patients with diabetes mellitus (162). After 12 weeks of treatment, luseogliflozin can not only improve blood glucose and insulin resistance but also effectively reduce epicardial fat volume and the levels of CRP (163). SGLT2 inhibitors have also been reported to improve left ventricular diastolic function and cardiovascular outcomes such as lowering the risk of adverse events in patients with HFpEF (164–166). Most importantly, in 2022, SGLT2 inhibitors have been approved for the treatment of HF by the American Heart Association/American College of Cardiology (167). In summary, patients with NAFLD and/or HFpEF will benefit from SGLT2 inhibitors treatment.
3.2.5. Metformin
Metformin, a biguanide drug, is a first-line antidiabetic medicine that can improve insulin sensitivity, and regulate the utilization of glucose by the liver. Moreover, metformin has additional effects on metabolism, such as improvements in hepatic gluconeogenesis and fatty acid metabolism (168, 169). Growing evidence indicated that metformin has shown benefits against NAFLD.
In a clinical trial, NAFLD patients were treated with metformin (2000 mg/day) for 48 weeks (170). Liver biopsy, imaging studies, and metabolic profiling were performed at the beginning and end of treatment. The result is that among 30% of NASH patients, liver histology and the level of liver enzymes are improved by metformin. In another randomized trial, metformin reduced aminotransaminase and fat accumulation in the liver and improved histological lesions including fibrosis (171). Furthermore, metformin has the effects of reducing EAT and anti-inflammation, which may decrease the risk of HFpEF. In a double-blind placebo-controlled trial, metformin showed anti-inflammatory effects regardless of diabetes status (172). In clinical settings, metformin can also decrease EAT and reduce adipose tissue inflammation (173, 174). Most importantly, it is reported that the mortality of patients with HFpEF is reduced when treated with metformin (175). However, metformin has also been reported not to be associated with clinical outcomes in patients with HFpEF (176). Accordingly, although metformin shows potential therapeutic effects on NAFLD and HFpEF, the long-term clinical effect of metformin on HFpEF needs further study.
4. Conclusion and perspectives
Individuals with NAFLD account for approximately a quarter of the world’s population. Individuals with NAFLD have a higher risk of CVD such as HFpEF. Common risk factors, thicker EAT, and release of secretory factors play an important role in the close association between NAFLD and HFpEF. Hence, the treatments of NAFLD, being able to control common risk factors, decrease the effects of secretory factors, or reduce EAT accumulation, should also have a therapeutic effect on HFpEF.
This review summarizes the major non-pharmacologic and pharmacologic strategies currently applied in NAFLD and their impact on the development of HFpEF. In addition to showing encouraging results in NAFLD, non-pharmacologic therapies can also protect against obesity and diabetes, improve systemic inflammation, and decrease EAT accumulation. Non-pharmacologic therapies can even directly improve the condition of patients with HFpEF. Some drugs also show the potential effects of reducing the risk of HFpEF while showing a therapeutic effect on NAFLD, such as statins, GLP-1 receptor agonists, SGLT2 inhibitors, and metformin. However, some drugs have uncertain effects on the risk of HFpEF. Therefore, the impact of these drugs such as thiazolidinediones on the development of HFpEF needs to be rigorously assessed in subsequent clinical trials.
Although there is a close relationship between NAFLD and HFpEF, the potential mechanism between NAFLD and HFpEF is still unclear and needs to be extensively studied. HFpEF is a systemic disease and the potential mechanisms of HFpEF itself have not been fully clarified so far. Based on a clearer understanding of the mechanism underlying HFpEF, it can be easier to deduce which NAFLD drugs have therapeutic effects on HFpEF. More importantly, uncovering the mechanism of HFpEF should also boost the development of new drugs for HFpEF. In addition, some drugs, such as GLP-1 receptor agonists, have shown encouraging results in preclinical studies. Therefore, authoritative clinical trial studies are needed to further verify whether these drugs have positive effects on HFpEF. Last but not least, as the mechanisms underlying both NAFLD and HFpEF are complex and may vary between different persons, personalized therapies should be applied to different populations.
Author contributions
ZY and RT wrote the original manuscript. X-JZ, JC, HL, and Z-GS critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82270390, 81970364, 82170595, 82170436, and 81970070) and the Hubei Province Innovation Platform Construction Project (20204201117303072238).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. (2016) 15:249–74. doi: 10.1038/nrd.2015.3
2. Younossi Z, Koenig A, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
3. Zhu B, Chan S, Li J, Li K, Wu H, Cui K, et al. Non-alcoholic steatohepatitis pathogenesis, diagnosis, and treatment. Front Cardiovasc Med. (2021) 8:742382. doi: 10.3389/fcvm.2021.742382
4. Byrne C, Targher G. Nafld: a multisystem disease. J Hepatol. (2015) 62(1 Suppl.):S47–64. doi: 10.1016/j.jhep.2014.12.012
5. Packer M. Atrial fibrillation and heart failure with preserved ejection fraction in patients with nonalcoholic fatty liver disease. Am J Med. (2020) 133:170–7. doi: 10.1016/j.amjmed.2019.09.002
6. Salah H, Pandey A, Soloveva A, Abdelmalek M, Diehl A, Moylan C, et al. Relationship of nonalcoholic fatty liver disease and heart failure with preserved ejection fraction. JACC Basic Transl Sci. (2021) 6:918–32. doi: 10.1016/j.jacbts.2021.07.010
7. Cermakova P, Lund L, Fereshtehnejad S, Johnell K, Winblad B, Dahlstrom U, et al. Heart failure and dementia: survival in relation to types of heart failure and different dementia disorders. Eur J Heart Fail. (2015) 17:612–9. doi: 10.1002/ejhf.222
8. Owan T, Hodge D, Herges R, Jacobsen S, Roger V, Redfield M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. (2006) 355:251–9. doi: 10.1056/NEJMoa052256
9. Fudim M, Zhong L, Patel K, Khera R, Abdelmalek M, Diehl A, et al. Nonalcoholic fatty liver disease and risk of heart failure among medicare beneficiaries. J Am Heart Assoc. (2021) 10:e021654. doi: 10.1161/JAHA.121.021654
10. Miller A, McNamara J, Hummel S, Konerman M, Tincopa M. Prevalence and staging of non-alcoholic fatty liver disease among patients with heart failure with preserved ejection fraction. Sci Rep. (2020) 10:12440. doi: 10.1038/s41598-020-69013-y
11. Chalasani N, Younossi Z, Lavine J, Diehl A, Brunt E, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. (2012) 55:2005–23. doi: 10.1002/hep.25762
12. Rinella M. Nonalcoholic fatty liver disease: a systematic review. JAMA. (2015) 313:2263–73. doi: 10.1001/jama.2015.5370
13. Hassan K, Bhalla V, El Regal M, Hh A. Nonalcoholic fatty liver disease: a comprehensive review of a growing epidemic. World J Gastroenterol. (2014) 20:12082–101. doi: 10.3748/wjg.v20.i34.12082
14. Gladden J, Chaanine A, Redfield M. Heart failure with preserved ejection fraction. Annu Rev Med. (2018) 69:65–79. doi: 10.1146/annurev-med-041316-090654
15. Savji N, Meijers W, Bartz T, Bhambhani V, Cushman M, Nayor M, et al. The association of obesity and cardiometabolic traits with incident Hfpef and Hfref. JACC Heart Fail. (2018) 6:701–9. doi: 10.1016/j.jchf.2018.05.018
16. Polovina M, Lund L, Dikic D, Petrovic-Dordevic I, Krljanac G, Milinkovic I, et al. Type 2 diabetes increases the long-term risk of heart failure and mortality in patients with atrial fibrillation. Eur J Heart Fail. (2020) 22:113–25. doi: 10.1002/ejhf.1666
17. Cai J, Zhang X, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. (2019) 70:1026–37. doi: 10.1002/hep.30506
18. Emamat H, Tangestani H, Behrad Nasab M, Ghalandari H, Hekmatdoost A. The association between epicardial adipose tissue and non-alcoholic fatty liver disease: a systematic review of existing human studies. EXCLI J. (2021) 20:1096–105. doi: 10.17179/excli2021-3815
19. Muzurovic E, Mikhailidis D, Mantzoros C. Non-alcoholic fatty liver disease, insulin resistance, metabolic syndrome and their association with vascular risk. Metabolism. (2021) 119:154770. doi: 10.1016/j.metabol.2021.154770
20. Kim K, Kim K. Targeting of secretory proteins as a therapeutic strategy for treatment of nonalcoholic steatohepatitis (NASH). Int J Mol Sci. (2020) 21:2296. doi: 10.3390/ijms21072296
21. Gehrke N, Schattenberg J. Metabolic inflammation-a role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease? Gastroenterology. (2020) 158:1929–47.e6. doi: 10.1053/j.gastro.2020.02.020
22. Paulus W, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
23. Petta S, Argano C, Colomba D, Camma C, Di Marco V, Cabibi D, et al. Epicardial fat, cardiac geometry and cardiac function in patients with non-alcoholic fatty liver disease: association with the severity of liver disease. J Hepatol. (2015) 62:928–33. doi: 10.1016/j.jhep.2014.11.030
24. Romero-Gomez M, Zelber-Sagi S, Trenell M. Treatment of Nafld with diet, physical activity and exercise. J Hepatol. (2017) 67:829–46. doi: 10.1016/j.jhep.2017.05.016
25. Konerman M, Jones J, Harrison S. Pharmacotherapy for Nash: current and emerging. J Hepatol. (2018) 68:362–75. doi: 10.1016/j.jhep.2017.10.015
26. Ferguson D, Finck B. Emerging therapeutic approaches for the treatment of Nafld and type 2 diabetes mellitus. Nat Rev Endocrinol. (2021) 17:484–95. doi: 10.1038/s41574-021-00507-z
27. Borjesson M, Onerup A, Lundqvist S, Dahlof B. Physical activity and exercise lower blood pressure in individuals with hypertension: narrative review of 27 Rcts. Br J Sports Med. (2016) 50:356–61. doi: 10.1136/bjsports-2015-095786
28. Okura H, Asawa K, Kubo T, Taguchi H, Toda I, Yoshiyama M, et al. Impact of statin therapy on systemic inflammation, left ventricular systolic and diastolic function and prognosis in low risk ischemic heart disease patients without history of congestive heart failure. Intern Med. (2007) 46:1337–43. doi: 10.2169/internalmedicine.46.0021
29. Iacobellis G, Mohseni M, Bianco S, Banga P. Liraglutide causes large and rapid epicardial fat reduction. Obesity. (2017) 25:311–6. doi: 10.1002/oby.21718
30. Lin Y, Fu S, Yao Y, Li Y, Zhao Y, Luo L. Heart failure with preserved ejection fraction based on aging and comorbidities. J Transl Med. (2021) 19:291. doi: 10.1186/s12967-021-02935-x
31. Chalasani N, Younossi Z, Lavine J, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. (2018) 67:328–57. doi: 10.1002/hep.29367
32. Loomba R, Sanyal A. The global Nafld epidemic. Nat Rev Gastroenterol Hepatol. (2013) 10:686–90. doi: 10.1038/nrgastro.2013.171
33. Powell E, Wong V, Rinella M. Non-alcoholic fatty liver disease. Lancet. (2021) 397:2212–24. doi: 10.1016/S0140-673632511-3
34. Papatheodoridi M, Cholongitas E. Diagnosis of non-alcoholic fatty liver disease (Nafld): current concepts. Curr Pharm Des. (2018) 24:4574–86. doi: 10.2174/1381612825666190117102111
35. Younossi Z, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. (2011) 9:524–30.e1; quiz e60. doi: 10.1016/j.cgh.2011.03.020
36. Satapathy S, Sanyal A. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. (2015) 35:221–35. doi: 10.1055/s-0035-1562943
37. Alshehri A. Metabolic syndrome and cardiovascular risk. J Fam Community Med. (2010) 17:73–8. doi: 10.4103/1319-1683.71987
38. Lam C, Chandramouli C. Fat, female, fatigued: features of the obese Hfpef phenotype. JACC Heart Fail. (2018) 6:710–3. doi: 10.1016/j.jchf.2018.06.006
39. Boonman-de Winter L, Rutten F, Cramer M, Landman M, Liem A, Rutten G, et al. High prevalence of previously unknown heart failure and left ventricular dysfunction in patients with type 2 diabetes. Diabetologia. (2012) 55:2154–62. doi: 10.1007/s00125-012-2579-0
40. Kim N, Park J, Kim S, Kim Y, Kim D, Cho G, et al. Non-alcoholic fatty liver disease, metabolic syndrome and subclinical cardiovascular changes in the general population. Heart. (2014) 100:938–43. doi: 10.1136/heartjnl-2013-305099
41. Dunlay S, Roger V, Redfield M. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2017) 14:591–602. doi: 10.1038/nrcardio.2017.65
42. Lee Y, Kim K, Yoo M, Kim G, Yoon H, Jo K, et al. Association of non-alcoholic steatohepatitis with subclinical myocardial dysfunction in non-cirrhotic patients. J Hepatol. (2018) 68:764–72. doi: 10.1016/j.jhep.2017.11.023
43. Obokata M, Reddy Y, Borlaug B. Diastolic dysfunction and heart failure with preserved ejection fraction: understanding mechanisms by using noninvasive methods. JACC Cardiovasc Imaging. (2020) 13(1 Pt 2):245–57. doi: 10.1016/j.jcmg.2018.12.034
44. Borlaug B. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2014) 11:507–15. doi: 10.1038/nrcardio.2014.83
45. VanWagner L, Wilcox J, Colangelo L, Lloyd-Jones D, Carr J, Lima J, et al. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: a population-based study. Hepatology. (2015) 62:773–83. doi: 10.1002/hep.27869
46. Chiu L, Pedley A, Massaro J, Benjamin E, Mitchell G, McManus D, et al. The association of non-alcoholic fatty liver disease and cardiac structure and function-Framingham heart study. Liver Int. (2020) 40:2445–54. doi: 10.1111/liv.14600
47. Borlaug B, Paulus W. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. (2011) 32:670–9. doi: 10.1093/eurheartj/ehq426
48. Graner M, Nyman K, Siren R, Pentikainen M, Lundbom J, Hakkarainen A, et al. Ectopic fat depots and left ventricular function in nondiabetic men with nonalcoholic fatty liver disease. Circ Cardiovasc Imaging. (2015) 8:e001979. doi: 10.1161/CIRCIMAGING.114.001979
49. Targher G, Day C, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. (2010) 363:1341–50. doi: 10.1056/NEJMra0912063
50. Cai J, Zhang X, Ji Y, Zhang P, She Z, Li H. Nonalcoholic fatty liver disease pandemic fuels the upsurge in cardiovascular diseases. Circ Res. (2020) 126:679–704. doi: 10.1161/CIRCRESAHA.119.316337
51. Jager J, Aparicio-Vergara M, Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. J Intern Med. (2016) 280:209–20. doi: 10.1111/joim.12483
52. Cai J, Zhang X, Li H. Role of innate immune signaling in non-alcoholic fatty liver disease. Trends Endocrinol Metab. (2018) 29:712–22. doi: 10.1016/j.tem.2018.08.003
53. Fricker Z, Pedley A, Massaro J, Vasan R, Hoffmann U, Benjamin E, et al. Liver fat is associated with markers of inflammation and oxidative stress in analysis of data from the Framingham heart study. Clin Gastroenterol Hepatol. (2019) 17:1157–64.e4. doi: 10.1016/j.cgh.2018.11.037
54. Franssen C, Chen S, Unger A, Korkmaz H, De Keulenaer G, Tschope C, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. (2016) 4:312–24. doi: 10.1016/j.jchf.2015.10.007
55. van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira A, Begieneman M, Bronzwaer J, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. (2012) 126:830–9. doi: 10.1161/CIRCULATIONAHA.111.076075
56. Brutsaert D. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. (2003) 83:59–115. doi: 10.1152/physrev.00017.2002
57. Wu N, Xu B, Xiang Y, Wu L, Zhang Y, Ma X, et al. Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: a meta-analysis. Int J Cardiol. (2013) 169:62–72. doi: 10.1016/j.ijcard.2013.08.078
58. Vermond R, Geelhoed B, Verweij N, Tieleman R, Van der Harst P, Hillege H, et al. Incidence of atrial fibrillation and relationship with cardiovascular events, heart failure, and mortality: a community-based study from the Netherlands. J Am Coll Cardiol. (2015) 66:1000–7. doi: 10.1016/j.jacc.2015.06.1314
59. Ho J, Lyass A, Lee D, Vasan R, Kannel W, Larson M, et al. Predictors of new-onset heart failure: differences in preserved versus reduced ejection fraction. Circ Heart Fail. (2013) 6:279–86. doi: 10.1161/CIRCHEARTFAILURE.112.972828
60. Sciarretta S, Ferrucci A, Ciavarella G, De Paolis P, Venturelli V, Tocci G, et al. Markers of inflammation and fibrosis are related to cardiovascular damage in hypertensive patients with metabolic syndrome. Am J Hypertens. (2007) 20:784–91. doi: 10.1016/j.amjhyper.2007.01.023
61. Rong J, Tao X, Lin Y, Zheng H, Ning L, Lu H, et al. Loss of hepatic angiotensinogen attenuates sepsis-induced myocardial dysfunction. Circ Res. (2021) 129:547–64. doi: 10.1161/CIRCRESAHA.120.318075
62. Ailhaud G, Teboul M, Massiera F. Angiotensinogen, adipocyte differentiation and fat mass enlargement. Curr Opin Clin Nutr Metab Care. (2002) 5:385–9. doi: 10.1097/00075197-200207000-00006
63. Bhushan S, Xiao Z, Gao K, Mao L, Chen J, Ping W, et al. Role and interaction between Ace1, Ace2 and their related genes in cardiovascular disorders. Curr Probl Cardiol. (2022) 48. doi: 10.1016/j.cpcardiol.2022.101162
64. Cernea S, Roiban A, Both E, Hutanu A. Serum leptin and leptin resistance correlations with Nafld in patients with type 2 diabetes. Diabetes Metab Res Rev. (2018) 34:e3050. doi: 10.1002/dmrr.3050
65. Kain D, Simon A, Greenberg A, Ben Zvi D, Gilburd B, Schneiderman J. Cardiac leptin overexpression in the context of acute MI and reperfusion potentiates myocardial remodeling and left ventricular dysfunction. PLoS One. (2018) 13:e0203902. doi: 10.1371/journal.pone.0203902
66. Packer M. Leptin-aldosterone-neprilysin axis: identification of its distinctive role in the pathogenesis of the three phenotypes of heart failure in people with obesity. Circulation. (2018) 137:1614–31. doi: 10.1161/CIRCULATIONAHA.117.032474
67. Peter A, Kovarova M, Staiger H, Machann J, Schick F, Konigsrainer A, et al. The hepatokines fetuin-A and fetuin-B are upregulated in the state of hepatic steatosis and may differently impact on glucose homeostasis in humans. Am J Physiol Endocrinol Metab. (2018) 314:E266–73. doi: 10.1152/ajpendo.00262.2017
68. Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol. (2010) 53:934–40. doi: 10.1016/j.jhep.2010.05.018
69. Naito C, Hashimoto M, Watanabe K, Shirai R, Takahashi Y, Kojima M, et al. Facilitatory effects of fetuin-A on atherosclerosis. Atherosclerosis. (2016) 246:344–51. doi: 10.1016/j.atherosclerosis.2016.01.037
70. Yoo H, Choi K. Hepatokines as a link between obesity and cardiovascular diseases. Diabetes Metab J. (2015) 39:10–5. doi: 10.4093/dmj.2015.39.1.10
71. Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol. (2018) 71:2360–72. doi: 10.1016/j.jacc.2018.03.509
72. Liu B, Li Y, Li Y, Liu Y, Yan Y, Luo A, et al. Association of epicardial adipose tissue with non-alcoholic fatty liver disease: a meta-analysis. Hepatol Int. (2019) 13:757–65. doi: 10.1007/s12072-019-09972-1
73. Ansaldo A, Montecucco F, Sahebkar A, Dallegri F, Carbone F. Epicardial adipose tissue and cardiovascular diseases. Int J Cardiol. (2019) 278:254–60. doi: 10.1016/j.ijcard.2018.09.089
74. Duyuler S, Turker Duyuler P. Epicardial adipose tissue in coronary microvascular dysfunction. Int J Obes. (2022) 46:1564. doi: 10.1038/s41366-022-01125-z
75. Shah S, Lam C, Svedlund S, Saraste A, Hage C, Tan R, et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. (2018) 39:3439–50. doi: 10.1093/eurheartj/ehy531
76. Packer M. The epicardial adipose inflammatory triad: coronary atherosclerosis, atrial fibrillation, and heart failure with a preserved ejection fraction. Eur J Heart Fail. (2018) 20:1567–9. doi: 10.1002/ejhf.1294
77. McKenney-Drake M, Rodenbeck S, Bruning R, Kole A, Yancey K, Alloosh M, et al. Epicardial adipose tissue removal potentiates outward remodeling and arrests coronary atherogenesis. Ann Thorac Surg. (2017) 103:1622–30. doi: 10.1016/j.athoracsur.2016.11.034
78. Smail H, Baciu A, Dacher J, Litzler P. Surgical resection of circumferential epicardial adipose tissue hypertrophy: case report and systematic review of the literature. J Thorac Cardiovasc Surg. (2016) 151:e27–30. doi: 10.1016/j.jtcvs.2015.08.083
79. Yki-Jarvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. (2014) 2:901–10. doi: 10.1016/S2213-858770032-4
80. Yu Y, Cai J, She Z, Li H. Insights into the epidemiology, pathogenesis, and therapeutics of nonalcoholic fatty liver diseases. Adv Sci. (2019) 6:1801585. doi: 10.1002/advs.201801585
81. Budde H, Hassoun R, Mugge A, Kovacs A, Hamdani N. Current understanding of molecular pathophysiology of heart failure with preserved ejection fraction. Front Physiol. (2022) 13:928232. doi: 10.3389/fphys.2022.928232
82. Abudureyimu M, Luo X, Wang X, Sowers J, Wang W, Ge J, et al. Heart failure with preserved ejection fraction (Hfpef) in type 2 diabetes mellitus: from pathophysiology to therapeutics. J Mol Cell Biol. (2022) 14:mjac028. doi: 10.1093/jmcb/mjac028
83. Fudim M, Sobotka P, Dunlap M. Extracardiac abnormalities of preload reserve: mechanisms underlying exercise limitation in heart failure with preserved ejection fraction, autonomic dysfunction, and liver disease. Circ Heart Fail. (2021) 14:e007308. doi: 10.1161/CIRCHEARTFAILURE.120.007308
84. Hirooka M, Koizumi Y, Miyake T, Ochi H, Tokumoto Y, Tada F, et al. Nonalcoholic fatty liver disease: portal hypertension due to outflow block in patients without cirrhosis. Radiology. (2015) 274:597–604. doi: 10.1148/radiol.14132952
85. Garcia-Pagan J, Santos C, Barbera J, Luca A, Roca J, Rodriguez-Roisin R, et al. Physical exercise increases portal pressure in patients with cirrhosis and portal hypertension. Gastroenterology. (1996) 111:1300–6. doi: 10.1053/gast.1996.v111.pm8898644
86. Manne V, Handa P, Kowdley K. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis. (2018) 22:23–37. doi: 10.1016/j.cld.2017.08.007
87. Tsochatzis E, Bosch J, Burroughs A. Liver cirrhosis. Lancet. (2014) 383:1749–61. doi: 10.1016/S0140-673660121-5
88. Nardelli S, Riggio O, Gioia S, Puzzono M, Pelle G, Ridola L. Spontaneous porto-systemic shunts in liver cirrhosis: clinical and therapeutical aspects. World J Gastroenterol. (2020) 26:1726–32. doi: 10.3748/wjg.v26.i15.1726
89. Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. (2003) 37:909–16. doi: 10.1053/jhep.2003.50132
90. Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Blendis L, Halpern Z, et al. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (Nafld): a population based study. J Hepatol. (2007) 47:711–7. doi: 10.1016/j.jhep.2007.06.020
91. Martinez-Gonzalez M, Gea A, Ruiz-Canela M. The Mediterranean diet and cardiovascular health. Circ Res. (2019) 124:779–98. doi: 10.1161/CIRCRESAHA.118.313348
92. Naska A, Trichopoulou A. Back to the future: the Mediterranean diet paradigm. Nutr Metab Cardiovasc Dis. (2014) 24:216–9. doi: 10.1016/j.numecd.2013.11.007
93. Ryan M, Itsiopoulos C, Thodis T, Ward G, Trost N, Hofferberth S, et al. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J Hepatol. (2013) 59:138–43. doi: 10.1016/j.jhep.2013.02.012
94. Kouvari M, Boutari C, Chrysohoou C, Fragkopoulou E, Antonopoulou S, Tousoulis D, et al. Mediterranean diet is inversely associated with steatosis and fibrosis and decreases ten-year diabetes and cardiovascular risk in Nafld subjects: results from the Attica prospective cohort study. Clin Nutr. (2021) 40:3314–24. doi: 10.1016/j.clnu.2020.10.058
95. Papavagelis C, Avgeraki E, Augoulea A, Stamatelopoulos K, Lambrinoudaki I, Yannakoulia M. Dietary patterns, Mediterranean diet and obesity in postmenopausal women. Maturitas. (2018) 110:79–85. doi: 10.1016/j.maturitas.2018.02.001
96. Grosso G, Mistretta A, Frigiola A, Gruttadauria S, Biondi A, Basile F, et al. Mediterranean diet and cardiovascular risk factors: a systematic review. Crit Rev Food Sci Nutr. (2014) 54:593–610. doi: 10.1080/10408398.2011.596955
97. Kouvari M, Chrysohoou C, Aggelopoulos P, Tsiamis E, Tsioufis K, Pitsavos C, et al. Mediterranean diet and prognosis of first-diagnosed acute coronary syndrome patients according to heart failure phenotype: Hellenic heart failure study. Eur J Clin Nutr. (2017) 71:1321–8. doi: 10.1038/ejcn.2017.122
98. Fito M, Estruch R, Salas-Salvado J, Martinez-Gonzalez M, Aros F, Vila J, et al. Effect of the Mediterranean diet on heart failure biomarkers: a randomized sample from the predimed trial. Eur J Heart Fail. (2014) 16:543–50. doi: 10.1002/ejhf.61
99. Chrysohoou C, Pitsavos C, Metallinos G, Antoniou C, Oikonomou E, Kotroyiannis I, et al. Cross-sectional relationship of a Mediterranean type diet to diastolic heart function in chronic heart failure patients. Heart Vessels. (2012) 27:576–84. doi: 10.1007/s00380-011-0190-9
100. Eslamparast T, Tandon P, Raman M. Dietary composition independent of weight loss in the management of non-alcoholic fatty liver disease. Nutrients. (2017) 9:800. doi: 10.3390/nu9080800
101. European Association for the Study of the Liver, European Association for the Study of Diabetes, European Association for the Study of Obesity. EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. (2016) 64:1388–402. doi: 10.1016/j.jhep.2015.11.004
102. Younossi Z, Corey K, Lim J. Aga clinical practice update on lifestyle modification using diet and exercise to achieve weight loss in the management of nonalcoholic fatty liver disease: expert review. Gastroenterology. (2021) 160:912–8. doi: 10.1053/j.gastro.2020.11.051
103. Magkos F, Hjorth M, Astrup A. Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. (2020) 16:545–55. doi: 10.1038/s41574-020-0381-5
104. Gkouskou K, Grammatikopoulou M, Lazou E, Sanoudou D, Goulis D, Eliopoulos A. Genetically-guided medical nutrition therapy in type 2 diabetes mellitus and pre-diabetes: a series of N-of-1 superiority trials. Front Nutr. (2022) 9:772243. doi: 10.3389/fnut.2022.772243
105. Iacobellis G, Singh N, Wharton S, Sharma A. Substantial changes in epicardial fat thickness after weight loss in severely obese subjects. Obesity. (2008) 16:1693–7. doi: 10.1038/oby.2008.251
106. Pedersen L, Olsen R, Anholm C, Astrup A, Eugen-Olsen J, Fenger M, et al. Effects of 1 year of exercise training versus combined exercise training and weight loss on body composition, low-grade inflammation and lipids in overweight patients with coronary artery disease: a randomized trial. Cardiovasc Diabetol. (2019) 18:127. doi: 10.1186/s12933-019-0934-x
107. El Hajj E, El Hajj M, Sykes B, Lamicq M, Zile M, Malcolm R, et al. Pragmatic weight management program for patients with obesity and heart failure with preserved ejection fraction. J Am Heart Assoc. (2021) 10:e022930. doi: 10.1161/JAHA.121.022930
108. Kitzman D, Brubaker P, Morgan T, Haykowsky M, Hundley G, Kraus W, et al. Effect of caloric restriction or aerobic exercise training on peak oxygen consumption and quality of life in obese older patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2016) 315:36–46. doi: 10.1001/jama.2015.17346
109. Wing R, Phelan S. Long-term weight loss maintenance. Am J Clin Nutr. (2005) 82(1 Suppl.):222S–5S. doi: 10.1093/ajcn/82.1.222S
110. Freitas W Jr, Oliveira L, Perez E, Ilias E, Lottenberg C, Silva A, et al. Systemic inflammation in severe obese patients undergoing surgery for obesity and weight-related diseases. Obes Surg. (2018) 28:1931–42. doi: 10.1007/s11695-017-3104-9
111. Courcoulas A, Gallagher J, Neiberg R, Eagleton E, DeLany J, Lang W, et al. Bariatric surgery vs lifestyle intervention for diabetes treatment: 5-year outcomes from a randomized trial. J Clin Endocrinol Metab. (2020) 105:866–76. doi: 10.1210/clinem/dgaa006
112. Gaborit B, Jacquier A, Kober F, Abdesselam I, Cuisset T, Boullu-Ciocca S, et al. Effects of bariatric surgery on cardiac ectopic fat: lesser decrease in epicardial fat compared to visceral fat loss and no change in myocardial triglyceride content. J Am Coll Cardiol. (2012) 60:1381–9. doi: 10.1016/j.jacc.2012.06.016
113. Foppa M, Pond K, Jones D, Schneider B, Kissinger K, Manning W. Subcutaneous fat thickness, but not epicardial fat thickness, parallels weight reduction three months after bariatric surgery: a cardiac magnetic resonance study. Int J Cardiol. (2013) 168:4532–3. doi: 10.1016/j.ijcard.2013.06.099
114. Runkel N, Colombo-Benkmann M, Huttl T, Tigges H, Mann O, Sauerland S. Bariatric surgery. Dtsch Arztebl Int. (2011) 108:341–6. doi: 10.3238/arztebl.2011.0341
115. Wang Z, Ye M, Zhang X, Zhang P, Cai J, Li H, et al. Impact of Nafld and its pharmacotherapy on lipid profile and Cvd. Atherosclerosis. (2022) 355:30–44. doi: 10.1016/j.atherosclerosis.2022.07.010
116. Li W, Liu J, Cai J, Zhang X, Zhang P, She Z, et al. Nafld as a continuous driver in the whole spectrum of vascular disease. J Mol Cell Cardiol. (2022) 163:118–32. doi: 10.1016/j.yjmcc.2021.10.007
117. Ganjooei N, Jamialahmadi T, Nematy M, Jangjoo A, Goshayeshi L, Khadem-Rezaiyan M, et al. The role of lipid profile as an independent predictor of non-alcoholic steatosis and steatohepatitis in morbidly obese patients. Front Cardiovasc Med. (2021) 8:682352. doi: 10.3389/fcvm.2021.682352
118. Bril F, Portillo Sanchez P, Lomonaco R, Orsak B, Hecht J, Tio F, et al. Liver safety of statins in prediabetes or T2dm and nonalcoholic steatohepatitis: post hoc analysis of a randomized trial. J Clin Endocrinol Metab. (2017) 102:2950–61. doi: 10.1210/jc.2017-00867
119. Torres-Pena J, Martin-Piedra L, Fuentes-Jimenez F. Statins in non-alcoholic steatohepatitis. Front Cardiovasc Med. (2021) 8:777131. doi: 10.3389/fcvm.2021.777131
120. Dongiovanni P, Petta S, Mannisto V, Mancina R, Pipitone R, Karja V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol. (2015) 63:705–12. doi: 10.1016/j.jhep.2015.05.006
121. Rinella M, Trotter J, Abdelmalek M, Paredes A, Connelly M, Jaros M, et al. Rosuvastatin improves the Fgf19 analogue Ngm282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol. (2019) 70:735–44. doi: 10.1016/j.jhep.2018.11.032
122. Alexopoulos N, Melek B, Arepalli C, Hartlage G, Chen Z, Kim S, et al. Effect of intensive versus moderate lipid-lowering therapy on epicardial adipose tissue in hyperlipidemic post-menopausal women: a substudy of the belles trial (beyond endorsed lipid lowering with EBT scanning). J Am Coll Cardiol. (2013) 61:1956–61. doi: 10.1016/j.jacc.2012.12.051
123. Parisi V, Petraglia L, D’Esposito V, Cabaro S, Rengo G, Caruso A, et al. Statin therapy modulates thickness and inflammatory profile of human epicardial adipose tissue. Int J Cardiol. (2019) 274:326–30. doi: 10.1016/j.ijcard.2018.06.106
124. Nochioka K, Sakata Y, Miyata S, Miura M, Takada T, Tadaki S, et al. Prognostic impact of statin use in patients with heart failure and preserved ejection fraction. Circ J. (2015) 79:574–82. doi: 10.1253/circj.CJ-14-0865
125. Liu G, Zheng X, Xu Y, Ru J, Hui R, Huang X. Meta-analysis of the effect of statins on mortality in patients with preserved ejection fraction. Am J Cardiol. (2014) 113:1198–204. doi: 10.1016/j.amjcard.2013.12.023
126. Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. (2001) 108:1429–37. doi: 10.1172/JCI13350
127. Ichihara S, Noda A, Nagata K, Obata K, Xu J, Ichihara G, et al. Pravastatin increases survival and suppresses an increase in myocardial matrix metalloproteinase activity in a rat model of heart failure. Cardiovasc Res. (2006) 69:726–35. doi: 10.1016/j.cardiores.2005.08.001
128. Ridker P, Pradhan A, MacFadyen J, Libby P, Glynn R. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet. (2012) 380:565–71. doi: 10.1016/S0140-673661190-8
129. Guo H, Lv H, Tang W, Chi J, Liu L, Xu F, et al. Rosuvastatin may modulate insulin signaling and inhibit atherogenesis beyond its plasma cholesterol-lowering effect in insulin-resistant mice. Cardiovasc Drugs Ther. (2012) 26:375–82. doi: 10.1007/s10557-012-6406-0
130. Carter A, Gomes T, Camacho X, Juurlink D, Shah B, Mamdani M. Risk of incident diabetes among patients treated with statins: population based study. BMJ. (2013) 346:f2610. doi: 10.1136/bmj.f2610
131. Galicia-Garcia U, Jebari S, Larrea-Sebal A, Uribe K, Siddiqi H, Ostolaza H, et al. Statin treatment-induced development of type 2 diabetes: from clinical evidence to mechanistic insights. Int J Mol Sci. (2020) 21:4725. doi: 10.3390/ijms21134725
132. Gross B, Pawlak M, Lefebvre P, Staels B. Ppars in obesity-induced T2dm, dyslipidaemia and Nafld. Nat Rev Endocrinol. (2017) 13:36–49. doi: 10.1038/nrendo.2016.135
133. Wang F, Mullican S, DiSpirito J, Peed L, Lazar M. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of ppargamma. Proc Natl Acad Sci U.S.A. (2013) 110:18656–61. doi: 10.1073/pnas.1314863110
134. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. (2016) 165:305–15. doi: 10.7326/M15-1774
135. Torres D, Jones F, Shaw J, Williams C, Ward J, Harrison S. Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: a 12-month randomized, prospective, open- label trial. Hepatology. (2011) 54:1631–9. doi: 10.1002/hep.24558
136. Della Pepa G, Russo M, Vitale M, Carli F, Vetrani C, Masulli M, et al. Pioglitazone even at low dosage improves Nafld in type 2 diabetes: clinical and pathophysiological insights from a subgroup of the TOSCA.IT randomised trial. Diabetes Res Clin Pract. (2021) 178:108984. doi: 10.1016/j.diabres.2021.108984
137. Grosso A, de Oliveira S, Higuchi Mde L, Favarato D, Dallan L, da Luz P. Synergistic anti-inflammatory effect: simvastatin and pioglitazone reduce inflammatory markers of plasma and epicardial adipose tissue of coronary patients with metabolic syndrome. Diabetol Metab Syndr. (2014) 6:47. doi: 10.1186/1758-5996-6-47
138. Sacks H, Fain J, Cheema P, Bahouth S, Garrett E, Wolf R, et al. Inflammatory genes in epicardial fat contiguous with coronary atherosclerosis in the metabolic syndrome and type 2 diabetes: changes associated with pioglitazone. Diabetes Care. (2011) 34:730–3. doi: 10.2337/dc10-2083
139. Kamimura D, Uchino K, Ishigami T, Hall M, Umemura S. Activation of peroxisome proliferator-activated receptor gamma prevents development of heart failure with preserved ejection fraction; inhibition of WNT-beta-catenin signaling as a possible mechanism. J Cardiovasc Pharmacol. (2016) 68:155–61. doi: 10.1097/FJC.0000000000000397
140. Benbow A, Stewart M, Yeoman G. Thiazolidinediones for type 2 diabetes. All glitazones may exacerbate heart failure. BMJ. (2001) 322:236.
141. Young L, Viscoli C, Schwartz G, Inzucchi S, Curtis J, Gorman M, et al. Heart failure after ischemic stroke or transient ischemic attack in insulin-resistant patients without diabetes mellitus treated with pioglitazone. Circulation. (2018) 138:1210–20. doi: 10.1161/CIRCULATIONAHA.118.034763
142. Nauck M, Quast D, Wefers J, Meier J. Glp-1 receptor agonists in the treatment of type 2 diabetes – State-of-the-art. Mol Metab. (2021) 46:101102. doi: 10.1016/j.molmet.2020.101102
143. Brown E, Wilding J, Barber T, Alam U, Cuthbertson D. Weight loss variability with Sglt2 inhibitors and Glp-1 receptor agonists in type 2 diabetes mellitus and obesity: mechanistic possibilities. Obes Rev. (2019) 20:816–28. doi: 10.1111/obr.12841
144. Armstrong M, Gaunt P, Aithal G, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. (2016) 387:679–90. doi: 10.1016/S0140-673600803-X
145. Petit J, Cercueil J, Loffroy R, Denimal D, Bouillet B, Fourmont C, et al. Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes: the Lira-Nafld study. J Clin Endocrinol Metab. (2017) 102:407–15. doi: 10.1210/jc.2016-2775
146. Liu L, Yan H, Xia M, Zhao L, Lv M, Zhao N, et al. Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab Res Rev. (2020) 36:e3292. doi: 10.1002/dmrr.3292
147. Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, et al. The glucagon-like peptide 1 analog liraglutide reduces Tnf-alpha-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis. (2012) 221:375–82. doi: 10.1016/j.atherosclerosis.2011.12.039
148. Wang X, Gusdon A, Liu H, Qu S. Effects of glucagon-like peptide-1 receptor agonists on non-alcoholic fatty liver disease and inflammation. World J Gastroenterol. (2014) 20:14821–30. doi: 10.3748/wjg.v20.i40.14821
149. Cechin S, Perez-Alvarez I, Fenjves E, Molano R, Pileggi A, Berggren P, et al. Anti-inflammatory properties of exenatide in human pancreatic islets. Cell Transplant. (2012) 21:633–48. doi: 10.3727/096368911X576027
150. Parthsarathy V, Holscher C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. Eur J Pharmacol. (2013) 700:42–50. doi: 10.1016/j.ejphar.2012.12.012
151. Ceriello A, Novials A, Canivell S, La Sala L, Pujadas G, Esposito K, et al. Simultaneous Glp-1 and insulin administration acutely enhances their vasodilatory, antiinflammatory, and antioxidant action in type 2 diabetes. Diabetes Care. (2014) 37:1938–43. doi: 10.2337/dc13-2618
152. Brock C, Hansen C, Karmisholt J, Moller H, Juhl A, Farmer A, et al. Liraglutide treatment reduced interleukin-6 in adults with type 1 diabetes but did not improve established autonomic or polyneuropathy. Br J Clin Pharmacol. (2019) 85:2512–23. doi: 10.1111/bcp.14063
153. Ceriello A, Novials A, Ortega E, Canivell S, La Sala L, Pujadas G, et al. Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care. (2013) 36:2346–50. doi: 10.2337/dc12-2469
154. Withaar C, Meems L, Markousis-Mavrogenis G, Boogerd C, Sillje H, Schouten E, et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117:2108–24. doi: 10.1093/cvr/cvaa256
155. Bizino M, Jazet I, de Heer P, van Eyk H, Dekkers I, Rensen P, et al. Placebo-controlled randomised trial with liraglutide on magnetic resonance endpoints in individuals with type 2 diabetes: a pre-specified secondary study on ectopic fat accumulation. Diabetologia. (2020) 63:65–74. doi: 10.1007/s00125-019-05021-6
156. Pastel E, McCulloch L, Ward R, Joshi S, Gooding K, Shore A, et al. Glp-1 analogue-induced weight loss does not improve obesity-induced at dysfunction. Clin Sci. (2017) 131:343–53. doi: 10.1042/CS20160803
157. Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, et al. Effects of Sglt2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol. (2013) 715:246–55. doi: 10.1016/j.ejphar.2013.05.014
158. Yamashita H, Takenoshita M, Sakurai M, Bruick R, Henzel W, Shillinglaw W, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U.S.A. (2001) 98:9116–21. doi: 10.1073/pnas.161284298
159. Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor Srebp-1c. Diabetes Obes Metab. (2010) 12(Suppl. 2):83–92. doi: 10.1111/j.1463-1326.2010.01275.x
160. Taheri H, Malek M, Ismail-Beigi F, Zamani F, Sohrabi M, Reza Babaei M, et al. Effect of empagliflozin on liver steatosis and fibrosis in patients with non-alcoholic fatty liver disease without diabetes: a randomized, double-blind, placebo-controlled trial. Adv Ther. (2020) 37:4697–708. doi: 10.1007/s12325-020-01498-5
161. Kuchay M, Krishan S, Mishra S, Farooqui K, Singh M, Wasir J, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-lift trial). Diabetes Care. (2018) 41:1801–8. doi: 10.2337/dc18-0165
162. Sato T, Aizawa Y, Yuasa S, Kishi S, Fuse K, Fujita S, et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc Diabetol. (2018) 17:6. doi: 10.1186/s12933-017-0658-8
163. Bouchi R, Terashima M, Sasahara Y, Asakawa M, Fukuda T, Takeuchi T, et al. Luseogliflozin reduces epicardial fat accumulation in patients with type 2 diabetes: a pilot study. Cardiovasc Diabetol. (2017) 16:32. doi: 10.1186/s12933-017-0516-8
164. Sezai A, Sekino H, Unosawa S, Taoka M, Osaka S, Tanaka M. Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc Diabetol. (2019) 18:76. doi: 10.1186/s12933-019-0877-2
165. Zhou H, Peng W, Li F, Wang Y, Wang B, Ding Y, et al. Effect of sodium-glucose cotransporter 2 inhibitors for heart failure with preserved ejection fraction: a systematic review and meta-analysis of randomized clinical trials. Front Cardiovasc Med. (2022) 9:875327. doi: 10.3389/fcvm.2022.875327
166. Yang D, Zhang Y, Yan J, Liu M, An F. Sglt-2 inhibitors on prognosis and health-related quality of life in patients with heart failure and preserved ejection fraction: a systematic review and meta-analysis. Front Cardiovasc Med. (2022) 9:942125. doi: 10.3389/fcvm.2022.942125
167. Heidenreich P, Bozkurt B, Aguilar D, Allen L, Byun J, Colvin M, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association joint committee on clinical practice guidelines. Circulation. (2022) 145:e895–1032. doi: 10.1161/CIR.0000000000001063
168. Marshall S. 60 years of metformin use: a glance at the past and a look to the future. Diabetologia. (2017) 60:1561–5. doi: 10.1007/s00125-017-4343-y
169. Barbero-Becerra V, Santiago-Hernandez J, Villegas-Lopez F, Mendez-Sanchez N, Uribe M, Chavez-Tapia N. Mechanisms involved in the protective effects of metformin against nonalcoholic fatty liver disease. Curr Med Chem. (2012) 19:2918–23. doi: 10.2174/092986712800672094
170. Loomba R, Lutchman G, Kleiner D, Ricks M, Feld J, Borg B, et al. Clinical trial: pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2009) 29:172–82. doi: 10.1111/j.1365-2036.2008.03869.x
171. Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol. (2005) 100:1082–90. doi: 10.1111/j.1572-0241.2005.41583.x
172. Cameron A, Morrison V, Levin D, Mohan M, Forteath C, Beall C, et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. (2016) 119:652–65. doi: 10.1161/CIRCRESAHA.116.308445
173. Ziyrek M, Kahraman S, Ozdemir E, Dogan A. Metformin monotherapy significantly decreases epicardial adipose tissue thickness in newly diagnosed type 2 diabetes patients. Rev Port Cardiol. (2019) 38:419–23. doi: 10.1016/j.repc.2018.08.010
174. Chen W, Greulich S, van der Meer R, Rijzewijk L, Lamb H, de Roos A, et al. Activin a is associated with impaired myocardial glucose metabolism and left ventricular remodeling in patients with uncomplicated type 2 diabetes. Cardiovasc Diabetol. (2013) 12:150. doi: 10.1186/1475-2840-12-150
175. Halabi A, Sen J, Huynh Q, Marwick T. Metformin treatment in heart failure with preserved ejection fraction: a systematic review and meta-regression analysis. Cardiovasc Diabetol. (2020) 19:124. doi: 10.1186/s12933-020-01100-w
Keywords: nonalcoholic fatty liver disease, heart failure with preserved ejection fraction, risk factor, systemic inflammation, secretory factors, epicardial adipose tissue, treatment
Citation: Yang Z, Tian R, Zhang X-J, Cai J, She Z-G and Li H (2023) Effects of treatment of non-alcoholic fatty liver disease on heart failure with preserved ejection fraction. Front. Cardiovasc. Med. 9:1120085. doi: 10.3389/fcvm.2022.1120085
Received: 09 December 2022; Accepted: 30 December 2022;
Published: 12 January 2023.
Edited by:
Peng Gao, Army Medical University, ChinaReviewed by:
Jin Chai, Third Military Medical University, ChinaYuxuan Luo, Southeast University, China
Copyright © 2023 Yang, Tian, Zhang, Cai, She and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Gang She,  emdzaGVAd2h1LmVkdS5jbg==; Hongliang Li, bGlobEB3aHUuZWR1LmNu
emdzaGVAd2h1LmVkdS5jbg==; Hongliang Li, bGlobEB3aHUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship