
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 02 February 2023
Sec. Cardiovascular Imaging
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1075639
This article is part of the Research Topic Insights in Cardiovascular Imaging: 2022 View all 29 articles
 Muhammad Umer
Muhammad Umer Dinesh K. Kalra*
Dinesh K. Kalra*Fabry disease is a rare, progressive X-linked inherited disorder of glycosphingolipid metabolism due to a deficiency of α-galactosidase A enzyme. It leads to the accumulation of globotriaosylceramide within lysosomes of multiple organs, predominantly the vascular, renal, cardiac, and nervous systems. Fabry cardiomyopathy is characterized by increased left ventricular wall thickness/mass, functional abnormalities, valvular heart disease, arrhythmias, and heart failure. Early diagnosis and treatment are critical to avoid cardiac or renal complications that can significantly reduce life expectancy in untreated FD. This review will focus on the role of cardiovascular magnetic resonance imaging in the diagnosis, clinical decision-making, and monitoring of treatment efficacy.
Fabry disease (FD) is a rare, monogenic X-chromosome-linked lysosomal storage disorder caused by mutations in the GLA gene. It results in an absence or deficiency of the enzymatic activity of α-galactosidase A (α-GAL) (1). More than 1000 GLA gene variants have been identified–including pathogenic mutations, variants of unknown significance, and benign polymorphisms. The deficiency of α-GAL activity impairs the breakdown of the glycosphingolipid, globotriaosylceramide (GL3)–resulting in progressive accumulation throughout the body, including the blood vessels, heart, kidneys, skin, nervous system, gastrointestinal system, and eyes (2). The massive accumulation of GL3 in cardiomyocytes is detectable as early as childhood and adolescence (3). It activates secondary pathways, including cytokine production, coagulation activation, and oxidative stress (4). GL3-induced oxidative stress in cardiomyocytes causes tyrosine nitration and DNA damage—resulting in contractile dysfunction, myocardial stiffness, and cardiomyocyte apoptosis. GL3 accumulation in the microvasculature causes endothelial injury, intima-media thickening due to smooth muscle cell proliferation, and atheroma production. Cardiomyopathy results from progressive GL3 accumulation in myocytes, valvular fibroblasts, conductive tissue, the microvascular endothelium, and smooth muscle cells. Left ventricular hypertrophy (LVH) is present in 50% of males and 33% of females (1). LVH and diastolic dysfunction occur in the early stages of the disease and eventually progress to systolic dysfunction and heart failure over the next few decades of life.
The prevalence of FD is around 1 in 40,000 to 1 in 117,000 (1) in the general population. However, FD may be more prevalent than previously believed as it is the underlying diagnosis in about 0.5% of patients with non-obstructive hypertrophic cardiomyopathy (prevalence of 1 in 300 in the adult population) (5, 6). Classic FD is defined by absent or very low α-GAL activity (7), early-onset, and progressive multisystemic involvement. In comparison, atypical FD or cardiac variant has some residual or lower than normal α-GAL activity (6), variable onset, and predominantly involves the heart. Heterozygous females may have low-normal or variably deficient α-GAL activity, variable onset, and may develop significant multisystemic manifestations depending on the underlying GLA mutation and X-chromosome inactivation (8, 9). Furthermore, diastolic dysfunction and myocardial fibrosis can develop in females without LVH (10).
Fabry disease is a multisystem disease with frequent misdiagnoses and significant diagnostic delays in females (16.3 ± 14⋅7 years) and males (13.7 ± 12⋅9) (9, 11), that adversely affects patient outcomes. FD can significantly reduce life expectancy, by approximately 20 years in males and 15 years in females (12, 13). FD cardiomyopathy includes progressive left ventricular wall thickness, ventricular dysfunction, myocardial ischemia, arrhythmias, and valvular heart disease. Severe microvascular dysfunction is the primary underlying mechanism for myocardial ischemia in the absence of coronary artery disease.
The main diagnostic challenge from a cardiac perspective is distinguishing FD cardiomyopathy from other forms of unexplained LVH, given its infrequent clinical suspicion, especially in the absence of extracardiac manifestations in atypical FD patients and heterozygote females. Increased community awareness will be needed in order to recognize FD as a potential cause of seemingly idiopathic LVH. Confirmation of FD is made by enzyme activity assay and/or genetic testing; tissue biopsy is rarely required. However, genetic testing is the initial screening test in most US centers due to its wide availability. A comprehensive diagnostic approach is needed for early diagnosis and treatment of FD cardiomyopathy, including early recognition of clinical red flags, biomarkers, multimodality cardiac imaging, and assessment for the involvement of other organ systems such as the kidneys, nervous system, etc.
Cardiovascular magnetic resonance is an essential imaging modality for the quantitative and qualitative assessment of cardiomyopathies. In contrast to transthoracic echocardiography (TTE), CMR provides anatomical and structural evaluation, myocardial strain analysis, and quantitative tissue assessment using late gadolinium enhancement (LGE) and novel parametric mapping techniques like native T1 mapping and extracellular volume (ECV) measurement. It can detect the majority of genotype-positive patients with mild or subclinical cardiac phenotypes. CMR, with advanced mapping techniques, is a valuable diagnostic tool in asymptomatic carriers and preclinical deposition of GL3 in the myocardium, microvasculature, conduction system, and valves (14). Multiparametric CMR, along with biomarker testing, picks up the majority of cases of early organ involvement in mild FD (15). This significantly impacts decision-making in asymptomatic disease, as current guidelines recommend treatment when imaging features indicate myocardial involvement. However, CMR is less readily available than TTE and requires an experienced technician and interpreter, and medical device incompatibility or artifacts may limit accurate evaluation.
Cardiovascular magnetic resonance is the gold standard for the assessment of myocardial wall thickness and mass. In comparison, TTE is limited by acoustic windows, overestimating or underestimating wall thickness and mass, dropout artifacts in the basal inferolateral wall and RV myocardium, and lower reproducibility (17).
Left ventricular hypertrophy is the most common structural change reported in FD (18). Patients predominantly have concentric LVH at the beginning (1). Asymmetrical hypertrophy with a grossly thickened septum compared to the inferolateral wall develops in late stages–replacement fibrosis causing wall thinning of the latter. Kampmann et al. (19) noted that the severity of LVH progresses with age, occurring 10–15 years later in females than in males. Females are less likely to develop LVH than males (33% vs. 50%) (1). Left ventricular mass (LVM) is directly related to left atrial thickness and dimensions. CMR analysis is also valuable due to the higher contribution of papillary muscles and trabeculations to total LVM in FD patients (20, 21).
Right-sided structural changes are common in FD, typically right ventricular hypertrophy (RVH), with preserved systolic function and normal chamber size. However, diastolic dysfunction often exists that may progress to advanced heart failure (22). Niemann et al. (23) noted that RVH was evident in 71% of the patients at baseline. A significant positive correlation existed between left and right ventricular wall thickness. ERT showed no beneficial effects on RV morphology and function in this study. However, in another study by Wuest et al. (24), ERT significantly reduced RV mass (baseline 31 ± 6 g/m2 vs. follow-up 27 ± 7 g/m2, p < 0.05).
Fabry disease is different from other interstitial cardiomyopathies–GL3 accumulation is intracellular, resulting in a true increase in LV myocyte mass and a reactive LVH. It impairs ventricular compliance, increases filling pressures, and restricts diastolic filling, causing heart failure (19). CMR is highly accurate and reproducible in measuring ejection fraction (EF) and ventricular volumes and does not rely on geometric assumptions as in TTE. It can determine small changes in ventricular function and volume on serial assessment and is especially helpful in quantifying the impact of therapy.
Cardiovascular magnetic resonance measurement of myocardial deformation and mechanics by strain and strain rate analysis is an emerging tool for the quantitative assessment of global and regional cardiac function in cardiomyopathies, often providing a preclinical diagnosis. Feature tracking-CMR (FT-CMR) is a very feasible and highly accurate technique for strain/strain rate analysis in cardiac diseases, especially the assessment of LV-GLS (global longitudinal strain) in LVH has excellent reproducibility (25). It is more accurate in assessing all myocardial segments and independent of intramyocardial features compared to TTE-speckle tracking. Mathur et al. (26) demonstrated the reproducibility of CMR strain abnormalities in FD. Base-to-apex circumferential strain (CS) gradient was lower in FD patients compared to healthy controls (2.1 ± 3.7% vs. 6.5 ± 2.2%, p = 0.002), and it was able to discriminate between FD patients without LVH or LGE from healthy controls, endorsing it as an early marker of cardiac involvement in FD. In a study by Roller et al. (16), GLS was significantly reduced in FD patients (p = 0.0009) and correlated with Lyso-GL3 elevation. GLS values increased with worsening LVH and LGE. Another study by Vijapurapu et al. (27) demonstrated that in LVH-negative FD patients, GLS impairment was correlated with a reduction in T1, suggesting that mechanical dysfunction occurs before GL3 accumulation. In conclusion, FT-CMR abnormalities are reproducible imaging biomarkers for early cardiac involvement in FD.
Late gadolinium enhancement reflects replacement fibrosis and helps differentiate FD cardiomyopathy from ischemic and other hypertrophic cardiomyopathies. LGE is present in almost half of FD patients and typically involves the basal and mid inferolateral myocardium in about 75% of these patients (28, 29) (Figure 1). About one-fourth of FD females can develop LGE without LVH (10). TTE can miss nearly half of the early-stage cardiomyopathy cases in females; however, the majority of these will be detected by CMR. Thus, the assessment of fibrosis by CMR is crucial in the screening and staging of FD, especially in female patients who may not meet conventional LVH criteria early on by TTE (10). Liu et al. (30) studied the association between diastolic dysfunction and myocardial fibrosis in FD. LGE was present in 38% of FD patients, mostly at the basal and mid-segments of the inferolateral wall. In 9% of patients, LGE was present without functional abnormalities. This indicates that LGE can be present in FD patients with normal diastolic and systolic function; thus, chronic inflammation likely contributes to the development of replacement fibrosis.
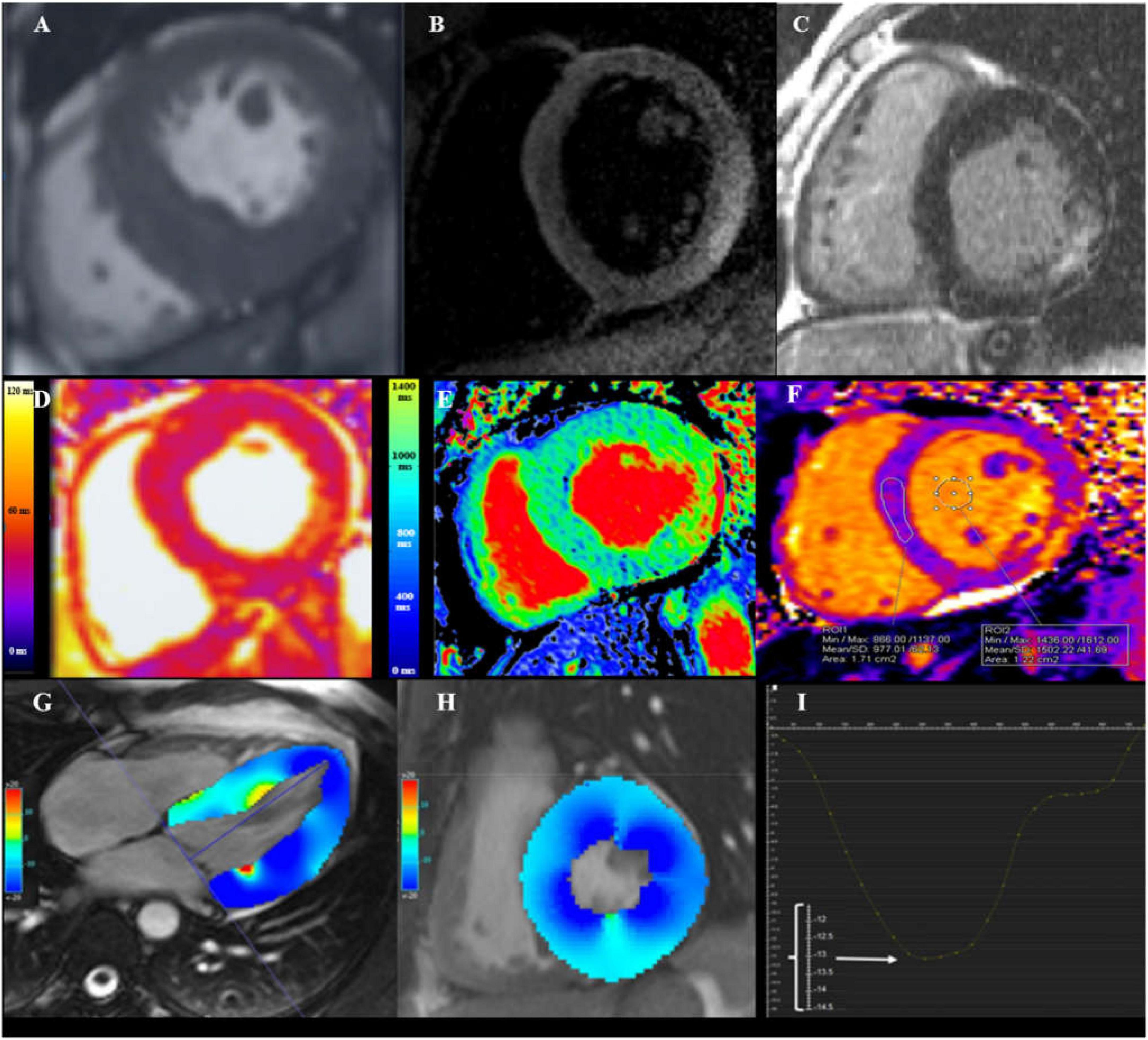
Figure 1. Cardiovascular magnetic resonance (CMR) assessment in Fabry disease (FD). (A) Steady state free precession (SSFP) CINE short-axis view showing increased wall thickness of mid-inferoseptum measuring 22 mm. (B) Dark-blood T2 short inversion-time, inversion-recovery (STIR) image showing myocardial edema (arrowheads) in the basal inferolateral wall (BIFL). (C) Late gadolinium enhancement (LGE) imaging showing mid-myocardium BIFL LGE in short-axis view. (D) Native T2 mapping showing high myocardial T2 value in BIFL (54 ms; normal reference value 45 ± 2 ms for this 1.5 T scanner). (E) Native T1 mapping showing low myocardial T1 value in the septum (812 ms; normal reference value 984 ± 18 ms for this 1.5 T scanner). (F) Native T1 mapping in advance disease showing pseudonormalization of T1 value in the septum and increased T1 in the BIFL. (G) Long-axis CINE SSFP image with color-coded myocardial longitudinal strain map. (H) Short-axis CINE SSFP image with color-coded myocardial circumferential strain map. (I) Decreased global longitudinal strain of −13.2%, as the enlarged scale on the Y-axis showed [adapted from Roller et al. (16)].
Standard imaging evaluation of anatomical and functional abnormalities and the presence of replacement fibrosis lack sensitivity or specificity to diagnose FD. Quantifying longitudinal relaxation time (T1) generates a pixel-wise color-encoded map of the myocardium, allowing the detection of very subtle pathological changes at the microscopic level that may be indicative of the preclinical stage. Native myocardial T1 values, obtained without a paramagnetic contrast agent, are higher in fibrosis, edema, and amyloid but lower in iron overload and focal fat infiltration. Accumulation of sphingolipids inside lysosomes in FD significantly shortens the native T1 values. T1 mapping has overcome the limitations of gadolinium contrast agent used in patients with advanced renal disease. In addition, T1-mapping is superior to LGE, with higher diagnostic accuracy, when the myocardium has more uniform and diffuse involvement (31).
Pica et al. (32) found T1 mapping highly reproducible in FD patients. It had 48% sensitivity and 99% specificity in distinguishing LVH-negative FD subjects from healthy volunteers. Reduced native T1 was highly prevalent (89%) in LVH-positive FD patients. LVH-negative FD patients had a 48% prevalence of reduced native myocardial T1, which was associated with advanced echocardiographic parameters of cardiac dysfunction (GLS and E/e’ ratio). In FD, native T1 values are reduced in the early stages but begin to normalize with progressive GL3 accumulation. T1 values are increased in the advanced stages due to replacement fibrosis and ongoing inflammation.
In patients with LVH, T1 mapping could differentiate FD from other phenotypes. In a study by Sado et al. (33), FD patients had lower septal T1 values (FD vs. healthy volunteers vs. other patients; 882 ± 47, 968 ± 32, 1018 ± 74 ms, P < 0.0001), which were inversely related to LV wall thickness (r = -0.51; P = 0.0004). In 40% of the FD patients without LVH, T1 values were abnormal due to the early phase of GL3 accumulation in myocytes.
Thompson et al. (34) compared LV mass, wall thickness, mass/volume ratio, LVEF, myocardial T1 values, and ECV as potential disease-specific imaging biomarkers of FD. The study concluded that reduced native myocardial T1 values are the most sensitive and specific CMR parameter in FD patients, irrespective of sex, LV morphology, or function. Native myocardial T1 values were substantially lower in FD (1070 ± 50 ms) as compared to healthy controls (1177 ± 27 ms) and concentric remodeling or hypertrophy (1207 ± 33 ms). Pagano et al. (35) evaluated RV myocardium by T1 mapping in FD, pulmonary hypertension (PH), and healthy controls. FD patients with thickened RVs had similarly reduced native T1 values in the RV and LV. This was the first report of reduced native T1 values in the RV. PH patients with thickened RVs showed increased native T1 values in both ventricles suggesting fibrosis. However, T1 mapping of the RV remains challenging due to relative thinness and the possibility of contamination from the blood pool or epicardial fat.
T1 mapping parameters, as surrogates for GL3 accumulation, can be reduced in the absence of LVH and basal inferolateral wall (BIFL) LGE. Thus, it may play a potential role in detecting the most appropriate patients for treatment and early quantitative assessment of treatment efficacy.
T2 mapping sequences measure T2 relaxation times representing myocardial edema/inflammation (36). Nordin et al. (37) demonstrated elevated native T2 values in the early stage of myocardial involvement, later corresponding to areas of LGE in the BIFL. High-sensitivity troponin T was elevated in 40% of the patients (75th percentile: 32 ng/l; range 3–93 ng/l; normal reference 0–14 ng/l), and increased T2 value was the strongest predictor (B = 2.4; p < 0.001). All patients with elevated troponin had LGE representing inflammation instead of scar. Chronic T2 elevation in LGE areas and elevations of global T2 values are both associated with poor outcomes (38). These findings suggest FD as inflammatory and infiltrative cardiomyopathy.
Myocardial ECV is calculated from native and contrast enhanced T1 values of myocardium and blood as well as patient’s hematocrit. ECV, a measurement of the size of the extracellular space, is elevated in amyloidosis and other infiltrative diseases, but in their absence, it is a biomarker for myocardial fibrosis. ECV values are normal in FD as it is an intracellular storage disease (39). However, as cardiomyopathy progresses, ECV values may increase in the areas of myocardial fibrosis (37). Hypertrophic cardiomyopathy (HCM) has increased ECV values due to extracellular matrix expansion and myocardial disarray, whereas ECV is reduced in athlete’s heart due to an increase in healthy myocardium by cellular hypertrophy.
Fabry disease patients frequently experience angina, and microvascular dysfunction is the primary underlying mechanism correlating with the extent of replacement fibrosis (40). CMR perfusion mapping provides a rapid quantitative assessment of microvascular dysfunction. Knott et al. (41) demonstrated that FD patients had lower stress myocardial blood flow maps (MBF) than healthy controls, even in the absence of LVH. MBF decline, especially in the endocardium, correlates with disease severity and can be an early disease marker.
Society for cardiovascular magnetic resonance (SCMR) guidelines do not provide FD-specific recommendations for CMR evaluation in the pediatric population (42). In young athletes, CMR is the preferred imaging modality to differentiate LVH from physiological remodeling by assessing hypertrophy regression with deconditioning (43). CMR provides more accurate wall thickness measurements and LVM compared to TTE (44). FT-CMR and T1 mapping techniques can identify myocardial fibrosis without using contrast agents (45). CMR use is limited in children due to a higher risk of anesthesia, the lower signal-to-noise ratio in small children, reduced temporal resolution due to higher heart rates, and difficulty holding breath under anesthesia.
Multiparametric CMR has a vital role in differentiating FD cardiomyopathy from other etiologies of LVH and can strongly impact clinical decision-making and prognosis by initiating disease-specific therapies (46) (Table 1). Cardiac and renal disease may not manifest clinically until adolescence or adulthood. Furthermore, renal damage is typically subclinical in early stages and requires biopsy for identification. Children with FD mutations should be treated as soon as the symptoms develop. Although in asymptomatic boys with classic FD mutation, treatment should be considered as early as 8–10 years of age (47). To avoid potentially irreversible complications, CMR is essential in early recognition and clinical decision-making. The European Fabry Working Group consensus statement recommends initiation of therapy in both classic and non-classic FD patients of both sexes when there is an increased LV wall thickening >12 mm (Class 1 recommendation) (48). However, major cardiology guidelines do not provide FD-specific recommendations for CMR. Nonetheless, it is vital to further study the potential role of strain analysis and T1/T2 mapping in treatment initiation and as an early quantitative measure of its efficacy. LVM reduction varies among various studies (10–27%), likely depending on the timing of therapy, the intensity of therapy, stage of cardiomyopathy, and other confounding factors such as age, sex, hypertension, etc. (Table 1).
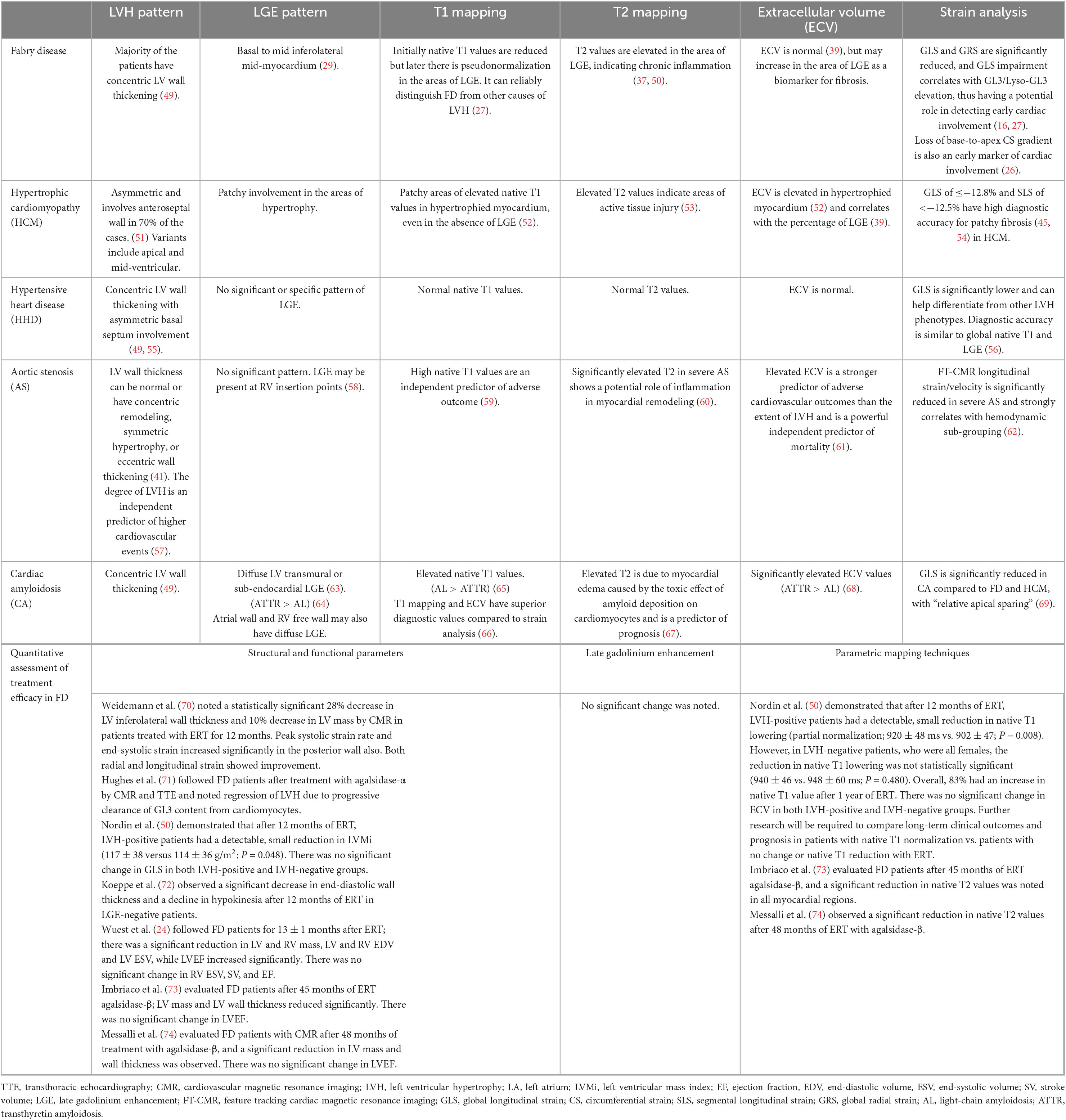
Table 1. Role of CMR parameters in characterizing the etiology of hypertrophic cardiomyopathy and the quantitative assessment of treatment efficacy in Fabry disease (FD).
An interdisciplinary FD center should perform therapy planning and initiation. The main therapeutic goals are symptom reduction to improve quality of life and preventing or halting multiorgan involvement to improve life expectancy. Established treatment options to reduce GL3 accumulation include replacing deficient endogenous α-GAL with recombinant enzyme replacement therapy (ERT) or increasing α-GAL enzyme activity inside lysosomes by chaperone therapy. Current ERT options include intravenous agalsidase-α (71) or agalsidase-β (73, 74). Oral chaperone therapy with migalastat corrects the misfolding of α-GAL and increases its intra-lysosomal availability. Next-generation plant-derived forms of ERT include pegunigalsidase-α (75) and moss α-GAL (76) with increased plasma half-life and reduced immunogenicity. Other emerging therapies include substrate reduction and gene therapy. Substrate reduction therapy aims to decrease the substrate concentration and subsequently inhibit GL3 accumulation in the cells. Lucerastat (77) and venglustat (78, 79) inhibit glucosylceramide synthase (GCS) to reduce the biosynthesis of glucosylceramide (GL1) and downstream GL3. Gene therapies are being developed as a long-term treatment option to cause endogenous α-GAL expression within disease phenotype cells, including α-GAL cDNA insertion via lentivirus (80), adeno-associated virus (AAV) gene delivery (NCT04455230) and gene-editing technology such as CRISPR (clustered regularly interspaced palindromic repeats)/Cas (CRISPR-associated genes).
Cardiomyopathy is the leading cause of death in men (34%) and women (57%) with FD (81). Early diagnosis is vital to prevent cardiac involvement and stop disease progression to avoid life-threatening complications of arrhythmias, myocardial infarction, and heart failure. The efficacy of treatment decreases with advancing stages of cardiomyopathy (2), thus worsening the overall prognosis. Orsborne et al. (82) developed a prognostic model based on age, native myocardial T1 dispersion, and left ventricular mass index (LVMi) to provide an accurate estimate of the 5-year risk of adverse cardiac outcomes. CMR-derived myocardial T1 relaxation time with wider distribution may have a greater prognostic value as it can better reflect GL3 accumulation, fibrosis/inflammation, and thus disease severity. LVMi by CMR is independently associated with adverse cardiac events in FD (83). Other clinical indices of organ involvement such as renal function, proteinuria, and neurological dysfunction also portend long-term prognosis.
Cardiac involvement should be detected promptly in FD patients to prevent disease progression and life-threatening complications. Multiparametric CMR imaging can play a vital role in reaching the correct diagnosis of hypertrophic myocardium and differentiating it from other phenotypes. FT-CMR and parametric mapping are emerging techniques with the potential for preclinical detection of cardiac involvement and monitoring response to therapy. In particular, T1 mapping is a superior technique for detecting GL3 accumulation and diffuse fibrosis. It has the potential for quantitative assessment of treatment efficacy–current data is insufficient, and further research is required to establish this role.
Both authors contributed to review conception, design, research, writing, and editing and approved the submitted version.
The authors declare that the review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yousef Z, Elliott P, Cecchi F, Escoubet B, Linhart A, Monserrat L, et al. Left ventricular hypertrophy in Fabry disease: a practical approach to diagnosis. Eur Heart J. (2013) 34:802–8. doi: 10.1093/eurheartj/ehs166
2. Umer M, Motwani M, Jefferies J, Nagueh S, Kalra D. Cardiac involvement in Fabry disease and the role of multimodality imaging in diagnosis and disease monitoring. Curr Probl Cardiol. (2023) 48:101439.
3. Kampmann C, Wiethoff C, Whybra C, Baehner F, Mengel E, Beck M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. (2008) 97:463–9. doi: 10.1111/j.1651-2227.2008.00700.x
4. Thurberg B, Fallon J, Mitchell R, Aretz T, Gordon R, O’Callaghan M. Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation. (2009) 119:2561–7. doi: 10.1161/CIRCULATIONAHA.108.841494
5. Semsarian C, Ingles J, Maron M, Maron B. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. (2015) 65:1249–54. doi: 10.1016/j.jacc.2015.01.019
6. Gersh B, Maron B, Bonow R, Dearani J, Fifer M, Link M, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. J Am Coll Cardiol. (2011) 58:2703–38. doi: 10.1016/j.jacc.2011.10.825
7. Mehta A, Hughes D. Fabry disease. In: Adam M, Everman D, Mirzaa G, Pagon R, Wallace S, Bean L, et al. editors. GeneReviews®. Seattle, WA: University of Washington (1993).
8. Wang R, Lelis A, Mirocha J, Wilcox W. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. (2007) 9:34–45. doi: 10.1097/gim.0b013e31802d8321
9. Eng C, Fletcher J, Wilcox W, Waldek S, Scott C, Sillence D, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry registry. J Inherit Metab Dis. (2007) 30:184–92. doi: 10.1007/s10545-007-0521-2
10. Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, et al. Differences in Fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging. (2011) 4:592–601. doi: 10.1016/j.jcmg.2011.01.020
11. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest. (2004) 34:236–42. doi: 10.1111/j.1365-2362.2004.01309.x
12. MacDermot K, Holmes A, Miners A. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. (2001) 38:769–75. doi: 10.1136/jmg.38.11.769
13. MacDermot K, Holmes A, Miners A. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. (2001) 38:750–60. doi: 10.1136/jmg.38.11.750
14. Kozor R, Grieve S, Tchan M, Callaghan F, Hamilton-Craig C, Denaro C, et al. Cardiac involvement in genotype-positive Fabry disease patients assessed by cardiovascular MR. Heart. (2016) 102:298–302. doi: 10.1136/heartjnl-2015-308494
15. Weidemann F, Beer M, Kralewski M, Siwy J, Kampmann C. Early detection of organ involvement in Fabry disease by biomarker assessment in conjunction with LGE cardiac MRI: results from the SOPHIA study. Mol Genet Metab. (2019) 126:169–82. doi: 10.1016/j.ymgme.2018.11.005
16. Roller F, Brose A, Richter M, Schüssler A, Harth S, Tanislav C, et al. Value of left ventricular feature tracking strain analysis for detection of early cardiac involvement in Fabry disease (FD). J Clin Med. (2021) 10:3734. doi: 10.3390/jcm10163734
17. O’Brien C, Britton I, Karur G, Iwanochko R, Morel C, Nguyen E, et al. Left ventricular mass and wall thickness measurements using echocardiography and cardiac MRI in patients with Fabry disease: clinical significance of discrepant findings. Radiol Cardiothorac Imaging. (2020) 2:e190149. doi: 10.1148/ryct.2020190149
18. Kampmann C, Linhart A, Baehner F, Palecek T, Wiethoff C, Miebach E, et al. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol. (2008) 130:367–73. doi: 10.1016/j.ijcard.2008.03.007
19. Kampmann C, Baehner F, Whybra C, Martin C, Wiethoff C, Ries M, et al. Cardiac manifestations of Anderson-Fabry disease in heterozygous females. J Am Coll Cardiol. (2002) 40:1668–74. doi: 10.1016/s0735-1097(02)02380-x
20. Kozor R, Callaghan F, Tchan M, Hamilton-Craig C, Figtree G, Grieve SM. A disproportionate contribution of papillary muscles and trabeculations to total left ventricular mass makes choice of cardiovascular magnetic resonance analysis technique critical in Fabry disease. J Cardiovasc Magn Reson. (2015) 17:22. doi: 10.1186/s12968-015-0114-4
21. Niemann M, Liu D, Hu K, Herrmann S, Breunig F, Strotmann J, et al. Prominent papillary muscles in Fabry disease: a diagnostic marker? Ultrasound Med Biol. (2011) 37:37–43. doi: 10.1016/j.ultrasmedbio.2010.10.017
22. Kampmann C, Baehner F, Whybra C, Bajbouj M, Baron K, Knuf M, et al. The right ventricle in Fabry disease. Acta Paediatr Suppl. (2005) 94:15–8. doi: 10.1111/j.1651-2227.2005.tb02104.x
23. Niemann M, Breunig F, Beer M, Herrmann S, Strotmann J, Hu K, et al. The right ventricle in Fabry disease: natural history and impact of enzyme replacement therapy. Heart. (2010) 96:1915–9. doi: 10.1136/hrt.2010.204586
24. Wuest W, Machann W, Breunig F, Weidemann F, Koestler H, Hahn D, et al. Right ventricular involvement in patients with Fabry’s disease and the effect of enzyme replacement therapy. Rofo. (2011) 183:1037–42. doi: 10.1055/s-0031-1281744
25. Maceira A, Tuset-Sanchis L, López-Garrido M, San Andres M, López-Lereu M, Monmeneu J, et al. Feasibility and reproducibility of feature-tracking-based strain and strain rate measures of the left ventricle in different diseases and genders. J Magn Reson Imaging. (2018) 47:1415–25. doi: 10.1002/jmri.25894
26. Mathur S, Dreisbach J, Karur G, Iwanochko R, Morel C, Wasim S, et al. Loss of base-to-apex circumferential strain gradient assessed by cardiovascular magnetic resonance in Fabry disease: relationship to T1 mapping, late gadolinium enhancement and hypertrophy. J Cardiovasc Magn Reson. (2019) 21:45. doi: 10.1186/s12968-019-0557-0
27. Vijapurapu R, Nordin S, Baig S, Liu B, Rosmini S, Augusto J, et al. Global longitudinal strain, myocardial storage and hypertrophy in Fabry disease. Heart. (2019) 105:470–6. doi: 10.1136/heartjnl-2018-313699
28. Moon J, Sachdev B, Elkington A, McKenna W, Mehta A, Pennell D, et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. (2003) 24:2151–5. doi: 10.1016/j.ehj.2003.09.017
29. Deva D, Hanneman K, Li Q, Ng M, Wasim S, Morel C, et al. Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in Anderson-Fabry disease. J Cardiovasc Magn Reson. (2016) 18:14. doi: 10.1186/s12968-016-0233-6
30. Liu D, Oder D, Salinger T, Hu K, Müntze J, Weidemann F, et al. Association and diagnostic utility of diastolic dysfunction and myocardial fibrosis in patients with Fabry disease. Open Heart. (2018) 5:e000803. doi: 10.1136/openhrt-2018-000803
31. Perea R, Ortiz-Perez J, Sole M, Cibeira M, de Caralt T, Prat-Gonzalez S, et al. T1 mapping: characterisation of myocardial interstitial space. Insights Imaging. (2015) 6:189–202. doi: 10.1007/s13244-014-0366-9
32. Pica S, Sado D, Maestrini V, Fontana M, White S, Treibel T, et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. (2014) 16:99. doi: 10.1186/s12968-014-0099-4
33. Sado D, White S, Piechnik S, Banypersad S, Treibel T, Captur G, et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging. (2013) 6:392–8. doi: 10.1161/CIRCIMAGING.112.000070
34. Thompson R, Chow K, Khan A, Chan A, Shanks M, Paterson I, et al. 1 mapping with cardiovascular MRI is highly sensitive for Fabry disease independent of hypertrophy and sex. Circ Cardiovasc Imaging. (2013) 6:637–45. doi: 10.1161/CIRCIMAGING.113.000482
35. Pagano J, Chow K, Khan A, Michelakis E, Paterson I, Oudit G, et al. Reduced right ventricular native myocardial T1 in Anderson-Fabry disease: comparison to pulmonary hypertension and healthy controls. PLoS One. (2016) 11:e0157565. doi: 10.1371/journal.pone.0157565
36. Kim P, Hong Y, Im D, Suh Y, Park C, Kim J, et al. Myocardial T1 and T2 mapping: techniques and clinical applications. Korean J Radiol. (2017) 18:113–31. doi: 10.3348/kjr.2017.18.1.113
37. Nordin S, Kozor R, Bulluck H, Castelletti S, Rosmini S, Abdel-Gadir A, et al. Cardiac Fabry disease with late gadolinium enhancement is a chronic inflammatory cardiomyopathy. J Am Coll Cardiol. (2016) 68:1707–8. doi: 10.1016/j.jacc.2016.07.741
38. Augusto J, Nordin S, Vijapurapu R, Baig S, Bulluck H, Castelletti S, et al. Myocardial edema, myocyte injury, and disease severity in Fabry disease. Circ Cardiovasc Imaging. (2020) 13:e010171. doi: 10.1161/CIRCIMAGING.119.010171
39. Sado D, Flett A, Banypersad S, White S, Maestrini V, Quarta G, et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart. (2012) 98:1436–41. doi: 10.1136/heartjnl-2012-302346
40. Chimenti C, Morgante E, Tanzilli G, Mangieri E, Critelli G, Gaudio C, et al. Angina in Fabry disease reflects coronary small vessel disease. Circ Heart Fail. (2008) 1:161–9. doi: 10.1161/CIRCHEARTFAILURE.108.769729
41. Knott K, Augusto J, Nordin S, Kozor R, Camaioni C, Xue H, et al. Quantitative myocardial perfusion in Fabry disease. Circ Cardiovasc Imaging. (2019) 12:e008872. doi: 10.1161/CIRCIMAGING.119.008872
42. Dorfman A, Geva T, Samyn M, Greil G, Krishnamurthy R, Messroghli D, et al. SCMR expert consensus statement for cardiovascular magnetic resonance of acquired and non-structural pediatric heart disease. J Cardiovasc Magn Reson. (2022) 24:44. doi: 10.1186/s12968-022-00873-1
43. Pelliccia A, Maron M, Maron B. Assessment of left ventricular hypertrophy in a trained athlete: differential diagnosis of physiologic athlete’s heart from pathologic hypertrophy. Prog Cardiovasc Dis. (2012) 54:387–96. doi: 10.1016/j.pcad.2012.01.003
44. Windram J, Dragelescu A, Benson L, Forsey J, Shariat M, Yoo S, et al. Myocardial dimensions in children with hypertrophic cardiomyopathy: a comparison between echocardiography and cardiac magnetic resonance imaging. Can J Cardiol. (2016) 32:1507–12. doi: 10.1016/j.cjca.2016.06.014
45. Bogarapu S, Puchalski M, Everitt M, Williams R, Weng H, Menon S. Novel cardiac magnetic resonance feature tracking (CMR-FT) analysis for detection of myocardial fibrosis in pediatric hypertrophic cardiomyopathy. Pediatr Cardiol. (2016) 37:663–73. doi: 10.1007/s00246-015-1329-8
46. Umer M, Motwani M, Jefferies J, Kalra D. Cardiac involvement in Fabry disease and the role of multimodality imaging in diagnosis and disease monitoring. Curr Probl Cardiol. (2022) 48:101439.
47. Hopkin R, Jefferies J, Laney D, Lawson V, Mauer M, Taylor M, et al. The management and treatment of children with Fabry disease: a United States-based perspective. Mol Genet Metab. (2016) 117:104–13. doi: 10.1016/j.ymgme.2015.10.007
48. Biegstraaten M, Arngrímsson R, Barbey F, Boks L, Cecchi F, Deegan P, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry working group consensus document. Orphanet J Rare Dis. (2015) 10:36. doi: 10.1186/s13023-015-0253-6
49. Weidemann F, Niemann M, Ertl G, Störk S. The different faces of echocardiographic left ventricular hypertrophy: clues to the etiology. J Am Soc Echocardiogr. (2010) 23:793–801. doi: 10.1016/j.echo.2010.05.020
50. Nordin S, Kozor R, Vijapurapu R, Augusto J, Knott K, Captur G, et al. Myocardial Storage, inflammation, and cardiac phenotype in Fabry disease after one year of enzyme replacement therapy. Circ Cardiovasc Imaging. (2019) 12:e009430. doi: 10.1161/CIRCIMAGING.119.009430
51. Maron M, Maron B, Harrigan C, Buros J, Gibson C, Olivotto I, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. (2009) 54:220–8. doi: 10.1016/j.jacc.2009.05.006
52. Xu J, Zhuang B, Sirajuddin A, Li S, Huang J, Yin G, et al. T1 mapping in hypertrophic cardiomyopathy: evaluation in patients without late gadolinium enhancement and hemodynamic obstruction. Radiology. (2020) 294:275–86. doi: 10.1148/radiol.2019190651
53. Gommans D, Cramer G, Bakker J, Michels M, Dieker H, Timmermans J, et al. High T2-weighted signal intensity is associated with elevated troponin T in hypertrophic cardiomyopathy. Heart. (2017) 103:293–9. doi: 10.1136/heartjnl-2016-309900
54. Wabich E, Dorniak K, Zienciuk-Krajka A, Nowak R, Raczak G, Daniłowicz-Szymanowicz L. Segmental longitudinal strain as the most accurate predictor of the patchy pattern late gadolinium enhancement in hypertrophic cardiomyopathy. J Cardiol. (2021) 77:475–81. doi: 10.1016/j.jjcc.2020.11.004
55. Gaudron P, Liu D, Scholz F, Hu K, Florescu C, Herrmann S, et al. The septal bulge–an early echocardiographic sign in hypertensive heart disease. J Am Soc Hypertens. (2016) 10:70–80. doi: 10.1016/j.jash.2015.11.006
56. Neisius U, Myerson L, Fahmy A, Nakamori S, El-Rewaidy H, Joshi G, et al. Cardiovascular magnetic resonance feature tracking strain analysis for discrimination between hypertensive heart disease and hypertrophic cardiomyopathy. PLoS One. (2019) 14:e0221061. doi: 10.1371/journal.pone.0221061
57. Cioffi G, Faggiano P, Vizzardi E, Tarantini L, Cramariuc D, Gerdts E, et al. Prognostic effect of inappropriately high left ventricular mass in asymptomatic severe aortic stenosis. Heart. (2011) 97:301–7. doi: 10.1136/hrt.2010.192997
58. Grajewski K, Stojanovska J, Ibrahim E, Sayyouh M, Attili A. Left ventricular hypertrophy: evaluation with cardiac MRI. Curr Probl Diagn Radiol. (2020) 49:460–75. doi: 10.1067/j.cpradiol.2019.09.005
59. Lee H, Park J, Yoon Y, Park E, Kim H, Lee W, et al. Noncontrast myocardial T1 mapping by cardiac magnetic resonance predicts outcome in patients with aortic stenosis. JACC Cardiovasc Imaging. (2018) 11:974–83. doi: 10.1016/j.jcmg.2017.09.005
60. Fehrmann A, Treutlein M, Rudolph T, Rudolph V, Weiss K, Giese D, et al. Myocardial T1 and T2 mapping in severe aortic stenosis: potential novel insights into the pathophysiology of myocardial remodelling. Eur J Radiol. (2018) 107:76–83. doi: 10.1016/j.ejrad.2018.08.016
61. Everett R, Treibel T, Fukui M, Lee H, Rigolli M, Singh A, et al. Extracellular myocardial volume in patients with aortic stenosis. J Am Coll Cardiol. (2020) 75:304–16. doi: 10.1016/j.jacc.2019.11.032
62. Buckert D, Cieslik M, Tibi R, Radermacher M, Rasche V, Bernhardt P, et al. Longitudinal strain assessed by cardiac magnetic resonance correlates to hemodynamic findings in patients with severe aortic stenosis and predicts positive remodeling after transcatheter aortic valve replacement. Clin Res Cardiol. (2018) 107:20–9. doi: 10.1007/s00392-017-1153-7
63. Syed I, Glockner J, Feng D, Araoz P, Martinez M, Edwards W, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. (2010) 3:155–64. doi: 10.1016/j.jcmg.2009.09.023
64. Dungu J, Valencia O, Pinney J, Gibbs S, Rowczenio D, Gilbertson J, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. (2014) 7:133–42. doi: 10.1016/j.jcmg.2013.08.015
65. Fontana M, Banypersad S, Treibel T, Maestrini V, Sado D, White S, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. (2014) 7:157–65. doi: 10.1016/j.jcmg.2013.10.008
66. Korthals D, Chatzantonis G, Bietenbeck M, Meier C, Stalling P, Yilmaz A. CMR-based T1-mapping offers superior diagnostic value compared to longitudinal strain-based assessment of relative apical sparing in cardiac amyloidosis. Sci Rep. (2021) 11:15521. doi: 10.1038/s41598-021-94650-2
67. Kotecha T, Martinez-Naharro A, Treibel T, Francis R, Nordin S, Abdel-Gadir A, et al. Myocardial edema and prognosis in amyloidosis. J Am Coll Cardiol. (2018) 71:2919–31. doi: 10.1016/j.jacc.2018.03.536
68. Fontana M, Banypersad S, Treibel T, Abdel-Gadir A, Maestrini V, Lane T, et al. Differential myocyte responses in patients with cardiac transthyretin amyloidosis and light-chain amyloidosis: a cardiac MR imaging study. Radiology. (2015) 277:388–97. doi: 10.1148/radiol.2015141744
69. Williams L, Forero J, Popovic Z, Phelan D, Delgado D, Rakowski H, et al. Patterns of CMR measured longitudinal strain and its association with late gadolinium enhancement in patients with cardiac amyloidosis and its mimics. J Cardiovasc Magn Reson. (2017) 19:61. doi: 10.1186/s12968-017-0376-0
70. Weidemann F, Breunig F, Beer M, Sandstede J, Turschner O, Voelker W, et al. Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation. (2003) 108:1299–301. doi: 10.1161/01.CIR.0000091253.71282.04
71. Hughes D, Elliott P, Shah J, Zuckerman J, Coghlan G, Brookes J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. (2008) 94:153–8. doi: 10.1136/hrt.2006.104026
72. Koeppe S, Neubauer H, Breunig F, Weidemann F, Wanner C, Sandstede J, et al. MR-based analysis of regional cardiac function in relation to cellular integrity in Fabry disease. Int J Cardiol. (2012) 160:53–8. doi: 10.1016/j.ijcard.2011.03.023
73. Imbriaco M, Pisani A, Spinelli L, Cuocolo A, Messalli G, Capuano E, et al. Effects of enzyme-replacement therapy in patients with Anderson-Fabry disease: a prospective long-term cardiac magnetic resonance imaging study. Heart. (2009) 95:1103–7. doi: 10.1136/hrt.2008.162800
74. Messalli G, Imbriaco M, Avitabile G, Russo R, Iodice D, Spinelli L, et al. Role of cardiac MRI in evaluating patients with Anderson-Fabry disease: assessing cardiac effects of long-term enzyme replacement therapy. Radiol Med. (2012) 117:19–28. doi: 10.1007/s11547-011-0710-9
75. Schiffmann R, Goker-Alpan O, Holida M, Giraldo P, Barisoni L, Colvin R, et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: a 1-year Phase 1/2 clinical trial. J Inherit Metab Dis. (2019) 42:534–44. doi: 10.1002/jimd.12080
76. Hennermann J, Arash-Kaps L, Fekete G, Schaaf A, Busch A, Frischmuth T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J Inherit Metab Dis. (2019) 42:527–33. doi: 10.1002/jimd.12052
77. Guérard N, Oder D, Nordbeck P, Zwingelstein C, Morand O, Welford R, et al. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin Pharmacol Ther. (2018) 103:703–11. doi: 10.1002/cpt.790
78. Peterschmitt M, Crawford N, Gaemers S, Ji A, Sharma J, Pham T. Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral venglustat in healthy volunteers. Clin Pharmacol Drug Dev. (2021) 10:86–98. doi: 10.1002/cpdd.865
79. Deegan P, Goker-Alpan O, Geberhiwot T, Hopkin R, Lukina E, Tylki-Szymanska A, et al. Venglustat, an orally administered glucosylceramide synthase inhibitor: assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study. Mol Genet Metab. (2022). doi: 10.1016/j.ymgme.2022.11.002 [Epub ahead of print].
80. Khan A, Barber D, Huang J, Rupar C, Rip J, Auray-Blais C, et al. Lentivirus-mediated gene therapy for Fabry disease. Nat Commun. (2021) 12:1178. doi: 10.1038/s41467-021-21371-5
81. Mehta A, Clarke J, Giugliani R, Elliott P, Linhart A, Beck M, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS–Fabry outcome survey. J Med Genet. (2009) 46:548–52. doi: 10.1136/jmg.2008.065904
82. Orsborne C, Bradley J, Bonnett L, Pleva L, Naish J, Clark D, et al. Validated model for prediction of adverse cardiac outcome in patients with Fabry disease. J Am Coll Cardiol. (2022) 80:982–94. doi: 10.1016/j.jacc.2022.06.022
Keywords: Fabry disease, cardiomyopathy, left ventricular hypertrophy, magnetic resonance imaging, myocardial mapping, feature tracking
Citation: Umer M and Kalra DK (2023) Cardiac MRI in Fabry disease. Front. Cardiovasc. Med. 9:1075639. doi: 10.3389/fcvm.2022.1075639
Received: 20 October 2022; Accepted: 30 December 2022;
Published: 02 February 2023.
Edited by:
Christos Bourantas, University College London, United KingdomReviewed by:
Patrick Doeblin, German Heart Center Berlin, GermanyCopyright © 2023 Umer and Kalra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dinesh K. Kalra,  ZGluZXNoLmthbHJhQGxvdWlzdmlsbGUuZWR1
ZGluZXNoLmthbHJhQGxvdWlzdmlsbGUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.