 Cassandra L. Clift
Cassandra L. Clift Janet Saunders1
Janet Saunders1 Richard R. Drake
Richard R. Drake
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 10 November 2022
Sec. Heart Valve Disease
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1024049
This article is part of the Research Topic Frontiers in Cardiovascular Medicine: Rising Stars 2022 View all 75 articles
In heart valve biology, organization of the extracellular matrix structure is directly correlated to valve function. This is especially true in cases of pediatric congenital aortic valve stenosis (pCAVS), in which extracellular matrix (ECM) dysregulation is a hallmark of the disease, eventually leading to left ventricular hypertrophy and heart failure. Therapeutic strategies are limited, especially in pediatric cases in which mechanical and tissue engineered valve replacements may not be a suitable option. By identifying mechanisms of translational and post-translational dysregulation of ECM in CAVS, potential drug targets can be identified, and better bioengineered solutions can be developed. In this review, we summarize current knowledge regarding ECM proteins and their post translational modifications (PTMs) during aortic valve development and disease and contributing factors to ECM dysregulation in CAVS. Additionally, we aim to draw parallels between other fibrotic disease and contributions to ECM post-translational modifications. Finally, we explore the current treatment options in pediatrics and identify how the field of proteomics has advanced in recent years, highlighting novel characterization methods of ECM and PTMs that may be used to identify potential therapeutic strategies relevant to pCAVS.
Congenital aortic valve stenosis (CAVS) accounts for 10% of all congenital heart defects, which effect 1 in every 150 people (1). The dominant phenotype of CAVS is the fusion of two of the three leaflets, causing a narrowing of the aortic orifice, obstructing aortic outflow, leading to left ventricular hypertrophy and heart failure. Despite the clinical significance of this disease, no pharmacotherapeutics exist. The clinical paradigm for treatment is “watch and wait” until surgical valve replacement and repair is necessary to prevent heart failure. Most CAVS patients are stable until adulthood when they develop fibrocalcific AVS (FAVS), where the end-stage is valve calcification (2). However, ∼10% of all CAVS patients are pediatrics (pCAVS), where end-stage is defined as excessive ECM deposition. Bioengineered AV replacements are notably limited for pediatric patients due to their inability to grow with the patient, a lifelong requirement of anti-clotting agents, and repeated surgeries due to valve failure (3). There remains a critical need to identify pharmacotherapeutic targets in pCAVS that may halt disease progression. One knowledge gap is of collagen regulation in valvular disease. Collagens are the fundamental scaffolding of valvular structure that influences valvular, and thus cardiac function (4, 5). In the future sections, we aim to outline ECM regulation, with a focus on collagen subtypes, in valvular health and disease.
Just after the initial heart contractions occur (E16–22 days) in early embryonic valve development, the three endocardial aortic cushions form (E31–35 days) and begin to emerge as ECM is deposited. At this time, the aortic valves (AVs) are primarily composed of hydrophilic glycosaminoglycans (GAGs), hyaluronan (HA), lined by endothelial cells (6–10). Between E35 and 37, hyaluronan synthase-2 (Hyal2) activity (which catalyzes formation of the HA polymer) is elevated, forming a “jelly” in which cells of the developing AVs undergo endothelial to mesenchymal transition (EndMT) (6, 10–12). This results in two unique populations of cells – valvular endothelial cells (VECs) and valvular interstitial cells (VICs). As healthy valves are avascular, VECs are responsible for regulating nutrients and biochemical signals from the blood to the VICs (13). VICs organize and maintain valvular structure through regulation of ECM proteins (14–18). Studies show intercellular crosstalk between VECs and VICs is essential to valvular health (19–21). Additionally, cardiac neural crest cells (CNCCs) and second heart field (SHF) precursors migrate to the newly formed endocardial cushions, promoting cell apoptosis and ECM regulation of the developing valve (17, 22–24). Recent literature also shows a population of mesenchymal stem cells present within the human-derived aortic valve cultures (25). At E37–E42, cell populations concurrently proliferate and apoptose, additional ECM is deposited, and the endocardial cushion elongates (5, 26–31).
In late embryonic AV development (E20–39 weeks for humans) the valve leaflet stratifies into three distinctive layers – the collagen rich fibrosa, the GAG rich spongiosa, and the elastin rich ventricularis. Studies in mice have shown that ECM organization begins during embryonic development and continues after birth, when oxygen levels and hemodynamics (and corresponding biomechanics) adjust (5). Throughout development, the composition and distribution of GAG subtypes change. The once prominent HA is supplemented with chondroitin sulfate proteoglycans, such as aggrecan and versican, as well as small leucine-rich proteoglycans such as biglycan and decorin, amongst others (4, 32). Similarly, collagen type distributions change over the course of development. While fibril-type collagens Col1a1, Col1a2, and Col3a1 are dominant in early development, later embryonic and post-natal development involves the deposition of network and FACIT type collagens as well (Figure 1). This data, however, is interpreted from mouse studies (4). Similar studies have yet to be done extensively on post-natal human aortic valve tissues. In this review we focus on collagen-related changes in heart valve development and disease.
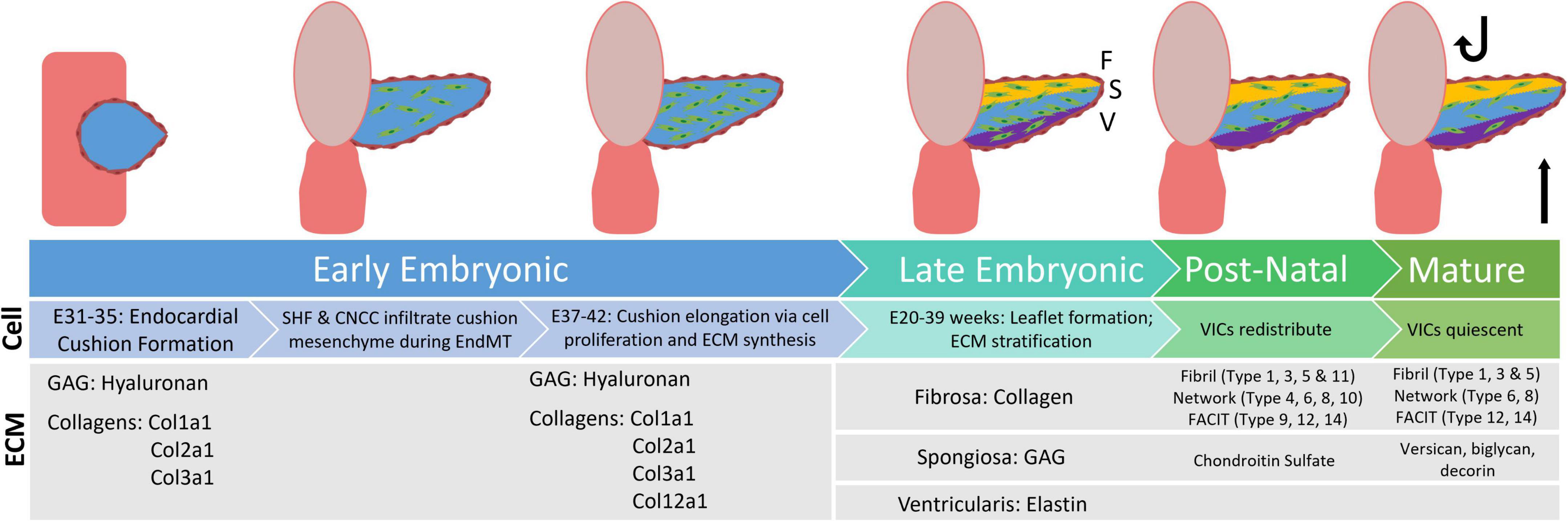
Figure 1. Flowchart displaying distribution of extracellular matrix proteins during the stages of valve development, from early embryonic through the mature aortic valve. GAG, glycosaminoglyca; SHF, second heart field; CNCC, cardiac neural crest cells; EndMT, endothelial to mesenchymal transition; VIC, valvular interstitial cells [development information edited from Combs et al. (6). ECM information edited from Peacock et al. (4)].
The aortic valve is the most mechanically stressed tissue in the body, undergoing approximately 35 million cyclic openings every year. In the mature valve during systole, total stresses reach up to 50 kPa; these stresses can range from 200 to 400 kPa during diastole (calculated based on human valve dynamics) (14, 33). During late embryonic development and post-natal development, peak shear stresses range from 30 to 1,500 dynes/cm2 (with higher values occurring during stenosis); these hemodynamics play a significant role in valve structure and stratification (14, 34). During systole, shear stresses are unidirectional and pulsatile, stretching the leaflet, which primarily affects the elastin rich ventricularis. Conversely, the outflow side of the aortic valve requires the stiffer collagen rich fibrosa to maintain structure and function, as oscillatory vortexes develop behind the leaflet during diastole. The internal middle layer of the valve is a GAG rich spongiosa that acts as shock absorbers during the entire cardiac cycle (35). Hemodynamics play a role in collagen alignment in the healthy valve, which has been shown to align along the circumferential axis, parallel to the endothelium (36, 37). In addition to hemodynamics, emerging subpopulations of VICs and differentially expressed genes can play a role in ECM stratification. In the ventricularis, Notch1 expression is dominant and may play a role in elastin deposition (6, 38). In the spongiosa layer, BMP2 signaling promotes Sox9 expression and deposition of proteoglycans, such as aggrecan, versican, chondroitin sulfate, and decorin (6, 39). In the fibrosa layer, Wnt signaling has been linked to promote the expression of fibroblast related ECM components such as fibrillar collagens type 1 and 3 and fibronectin (6, 40).
Extracellular matrix proteins make up the bulk of valvular tissues, and these proteins are abundantly post-translationally modified (Figure 2). Hydroxylation is a major post-translational modification (PTM) of collagens that maintains structural integrity of the valve. Hydroxylation of proline and hydroxylation of lysine are the two most abundant PTMs of collagen type proteins (41, 42). These PTMs are necessary for the organization of collagen’s triple helix structure as well as its thermal stability. In both collagen and elastin, hydroxyproline is a well-regulated modification, with between 20 and 24% of prolines being hydroxylated (41). Prolyl hydroxylases are responsible for hydroxylation of proline in elastin as well as collagen, with an X–P–G amino acid sequence required substrate in most cases (43). Elastin studies of the human aorta show that two specific prolines (P190, P615) have the highest degrees of hydroxylation, suggesting site-specific regulation, however, this has not been elucidated in the healthy or diseased AV (44). Similarly, only a single study has attempted to perform site specific hydroxyproline mapping in health or diseased AV (37). Other PTMs of structural ECM in the valve include O- and N-linked modifications, phosphorylation, allysine via lysyl oxidase, and collagen-lysyl intermolecular crosslinking (Figure 2). This other ECM PTMs have been attributed to site-specific functions related to protein folding, ECM secretion in stress conditions, collagen assembly, crosslinking and stabilization.
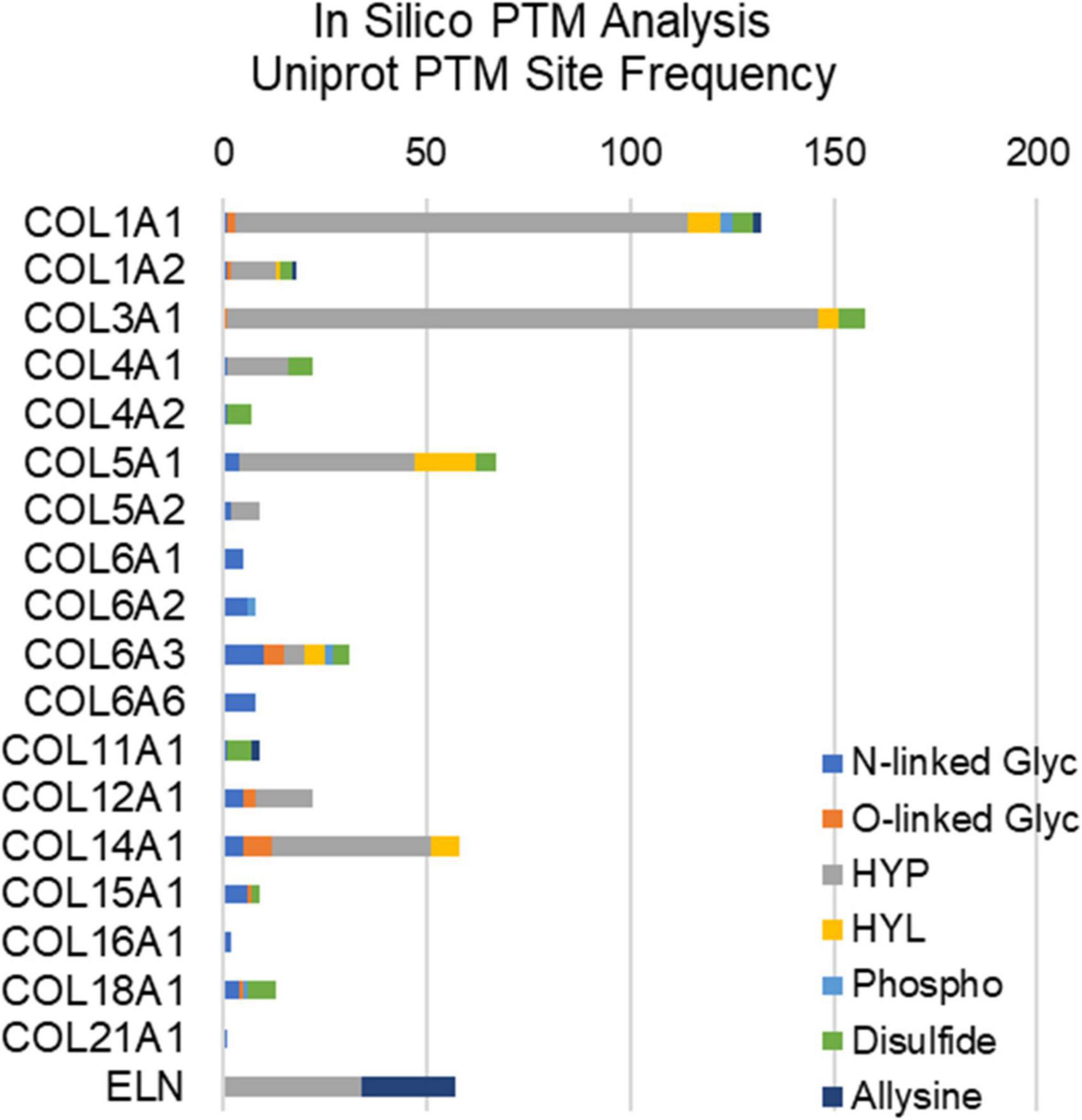
Figure 2. In silico post-translational modification (PTM) site frequency analysis of valvular structural proteins (collagens and elastin); uniport reporting. Glyc, glycosylation; HYP, hydroxyproline; HYL, hydroxylysine; Phospho, phosphorylation.
This review primarily focuses on major structural ECM proteins (collagen and elastin). For information regarding minor ECM proteins, glycoproteins, and proteoglycans (such as vitronectin, fibronection, periostin, osteonectin, osteopontin, and aggrecan) basement membrane proteins (laminin, perlecan, and nidogen) or ECM remodeling proteins, such as matrix metalloproteinases (MMPs) and their inhibitors (TIMPs), we point the reader to the following reviews and original research manuscripts (9, 36, 45–52).
Currently, congenital aortic valve stenosis (CAVS) accounts for 10% of all congenital heart defect cases, which affect 1 in 150 people (1). CAVS progresses as an obstructive narrowing of the aortic opening due to enlargement of the AV leaflets through deregulated ECM production and results in heart failure. Approximately 90% of all CAVS cases are due to bicuspid aortic valve (BAV) (53). Disease loci associated with BAV have been found on chromosomes 18q, 13q, and 5q and while the inheritability of BAV within certain families suggest a genetic component, only mutations in NOTCH1 signaling have been attributed directly to valvular stenosis (54, 55). However, other genetic mutations have been associated with BAV-aortopathy (56). Despite other genes being implicated in murine models of BAV (such as eNOS and GATA6 mutations), these have not been correlated to the valvular stenosis in humans (57, 58).
Most CAVS patients (90%) are stable until adulthood, where severe calcific lesions are the primary cause of valve replacement. In adult fibrocalcific AVS (FAVS), inflammation and adhesion are shown to increase along with mast cell infiltration (59, 60). Adult cases also tend to have comorbidities, such as high blood pressure or coronary artery disease, further confounding research (54). The remaining 10% of all CAVS patients are pediatrics, where end-stage is defined by excessive ECM deposition, and no calcification (pCAVS) (59, 61). A main feature of both pediatric and adult CAVS is bicuspid aortic valve (BAV). The dominant BAV morphology across all patients is the fusion of the right- and left-coronary leaflets (71%) (62). Potential changes in dominant cusp fusion morphology (RL, RN, and NL) have not been differentiated between pediatric and adult calcific CAVS progression; however, right and non-coronary fusion (R–N) are the most common predictor of pediatric valve dysfunction and intervention (35% of patients; >4-fold increase) (63). Literature acknowledges a distinct molecular profile between pediatric and adult calcific CAVS (59), but the molecular mechanisms behind the rapid progression of pCAVS remain to be elucidated. A summary of what is known regarding distinct molecular signatures between these two groups is outlined in Figure 3.
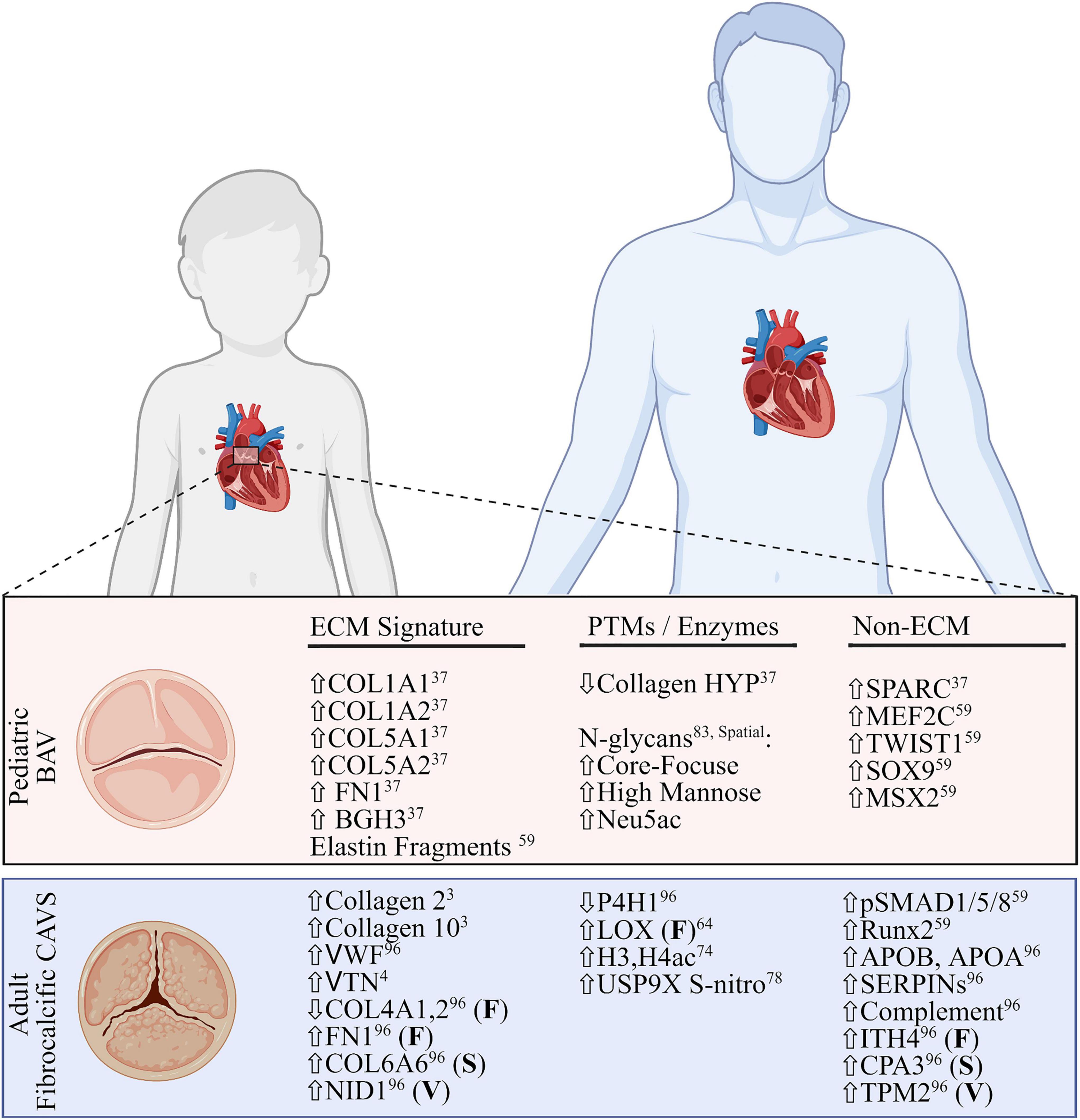
Figure 3. Comparative summary of transcriptomic and proteomic profiles of pediatric bicuspid aortic valve disease and adult fibrocalcific CAVS. Extracellular matrix (ECM) signatures (left), post-translational modifications or related enzymes (middle) and non-ECM/PTM signatures (right) are shown. All signatures are described as global changes unless otherwise specified (F, fibrosa; S, spongiosa; V, ventricularis). For the purposes of this review, the top layer-specific alteration is shown. FN1, fibronectin; BGH3, TGFB induced protein ig-h3; MEF2C, myocycte-specific enhancer factor 2C; TWIST1, twist-related protein 1; MSX2, homobox protein MSX2. VWF, Von Willebrand Factor; VTN, vitronectin; NID1, nidogen-1; P4H1, prolyl-4 hydroxylase-1; LOX, lipoxygenase; H3,H4, histone3,4; USP9X, ubiquitin carboxyl-terminal hydrolase FAF-X; Runx2, runt-related transcription factor-2; ITH4, inter-alpha trypsin inhibitor heavy chain 4; CPA3, carboxypeptidase A3; TPM2, tropomyosin 2. Figure created using BioRender.com (2022).
A loss of ECM stratification is seen in both pediatric and adult cases of CAVS (59). Hutson et al. set out to quantify changes in collagen architecture associated with adult calcific cases of CAVS (64). Their group showed that collagen content in the GAG-dominant spongiosa layer doubled in the disease state. Collagen fiber width and density also significantly increased, with lysyl oxidase expression also increasing in the diseased fibrosa layer (64); however, the corresponding PTM allysine was not studied. Studies in mice have shown that loss of crosslinking collagens Col5a1 and Col11a1 alter expression of fibrillar collagens type 1 and 3, resulting in AV stenosis (54). Similarly, studies of elastin in CAVS has shown increased fragmentation correlated to increased calcification (65). Fragmented elastin has also been seen in pediatric CAVS (59). Di Vito et al. recently published an extensive review of ECM alterations in adult calcific valve disease (66). Our group recently reported that total collagen deposition increases in pediatric CAVS, along with collagen fiber misalignment relative to the endothelium, and discrete regions of increased collagen fiber density (or “plaques”), as measured by histology (37). ECM targeted proteomics in this study showed differential collagen subtype regulation (in COL1A1, COL1A2, COL5A1, and COL5A2) between pCAVS, normal, and tricuspid aortic valve insufficiency (37). The role of site-specific collagen PTMs has yet to be studied in a localized manner relative to these collagen plaques, or layer specificity.
Congenital aortic valve stenosis-mediated alteration in hemodynamics influences all levels of molecular expression and feeds forward into deregulating the valvular structure (62). Velocity field analysis has shown that the total kinetic energy, Reynolds shear stress, and viscous shear stress are all significantly higher in cases of BAV as compared to the normal tricuspid configuration (35). Ex vivo analysis shows that abnormal hemodynamic shear stress may contribute to early adults CAVS pathogenesis (67). Additionally, pediatric and adult cases of BAV experience similar hemodynamic alterations (68). In environments with excessive shear stress such as the AV, enzyme-induced turnover of ECM is essential to maintain homeostasis. However, shear stresses experienced in BAV have been shown to exhibit myofibroblast induced-remodeling, attributed excessive ECM deposition (69). There remains a need to explore how similar shear stresses experienced in BAV effect ECM expression during different developmental stages, recapitulating pediatric vs. adult BAV fluid responses.
During in vitro bioreactor conditioning studies, increased pressure has been directly attributed to increased leaflet strain (70). Studies in VICs under variable strain (10, 14, and 20%) show significant increase in [3H]-proline incorporation, indicating increased collagen synthesis (70), but hydroxyproline site specificity in bioreactors conditions has yet to be explored. Transcriptional regulation of critical genes differs during strain as well. For example, Ku et al. showed Col3a1 is significantly upregulated as compared to a steady regulation of Col1a1 under strain (71). These studies offer insight into how the increased shear stresses seen in BAV contribute altered strain and to ECM deposition in CAVS.
The role of PTMs in ECM proteins is beginning to be explored in valve development and disease. While the PTM allysine has not been measured directly in CAVS, studies show an increase in the enzyme responsible for catalyzing the reactive aldehyde species, LOX (64). Similarly, PLOD1, the enzyme responsible for catalyze hydroxylysine residues in collagen, was found to be unchanged in adult CAVS compared to controls (64), however, hydroxylysine was not measured directly. While hydroxyproline content has been measured in healthy aortic valves, this is used as a measurement of insoluble collagen content, not in a site-specific analysis (72, 73). Our group recently reported the first study of collagen HYP quantification and site mapping with human pediatric CAVS tissue (37). Overall, a reduction of collagen HYP content was seen, particularly in regions within collagen sequence required for integrin and glycoprotein vi binding (37).
Potential modifications of intracellular proteins regulated in the heart can include both enzymatic (such as acetylation, phosphorylation, and ubiquitination) and non-enzymatic (such as nitrosylation) post-translational modifications. Acetylation of histones 3 and 4 has been reported in CAVS and non-calcified AVs from patients, although significantly higher in CAVS patients (74). Similarly, acetylation plays a role in modulating the heart’s response to I/R injury (75). Glycogen Synthase Kinase 3β (GSK3β) – which has been found in cardiac fibroblast and implicated in ventricular remodeling – is both phosphorylated and S-nitrosylated in a site-specific manner relevant to its function in cardiac fibrosis (76, 77). S-nitrosylation of USP9X has been shown to be enriched in NOTCH regulation of valvular calcification (78). Additionally, studies have shown increases in ubiquitin-mediated proteolysis in heart failure (79). While these studies are not specific to ECM proteins, data suggested that PTMs may be a potential therapeutic target in cardiac and valvular fibrosis.
Post-translational regulation via oxidation has been pinpointed as having a major role in other fibrotic diseases (80). While inflammatory-derived Reactive Oxygen Species (ROS) has been implicated in adult CAVS, ROS is unexplored in pediatric valve disease (66). However, ECM and PTM studies in pediatric valve disease suggest a potential role for ROS regulation. ROS can alter the expression of ECM both by activating transcription factors involved in ECM protein expression (such as TGFB1) as well as modifying ECM proteins post-translationally (80). Additionally, the hydroxylation of proline and lysine residues of collagen is highly redox dependent. Once lysine is hydroxylated, it serves as a synthesis starting point for proteins responsible for collagen and elastin crosslinking, such as desmosine (42, 80, 81). Hydroxylated lysine 2 has been shown to have glycosyl-transferase activity, serving as an anchor for O-glycosylation modification (41, 42). In silico analysis of valvular collagen type proteins demonstrates a potential abundance of N- and O-glycosylation sites, amongst other PTMs (Figure 2). At high concentrations, ROS can destroy these carbohydrate entities (80, 82). A recent publication by Angel et al. shows that N-glycosylation patterns are spatial regulated in pediatric aortic valve development and disease, with certain sialylated N-glycans amongst others being upregulated (83). At low concentrations, ROS can induce GAG-degrading enzymes, such as Hyal2, indirectly effecting ECM turnover (32). Recent studies have shown that GAGs can store growth factors and cytokines in the extracellular space; so, indirectly, depletion of GAGs via Hyal2 may modify receptor binding and activation (84). ROS can also induce collagen degrading enzymes, such as matrix-metalloproteinase (MMPs), further suggesting a role of oxidative stress in ECM remodeling (85).
Despite the clinical significance of CAVS, surgical valve replacement and repair is still the only available treatment. Mechanical-based engineered valve replacement devices are not suitable for young children as they lack any growth potential as well as requiring lifelong anti-coagulation therapy (86). However, in older pediatric cases, there has been success with a 55–90% freedom from reoperation at 15 years (3). Tissue engineered valve replacements are currently unavailable in sizes smaller than 19 mm, also making them unsuitable for young pediatric cases. Like mechanical prosthesis, there is a lack of growth potential with tissue engineered replacements, but anti-coagulation therapy is not required (87). Survival was reported to be 85% at 10 years, however the rate of reoperation is high – 65% at 10 years, with the median longevity being only 7.5 years (3). Homografts are able to retain normal hemodynamic profiles over time, with small sizes available for small children. Homografts also do not require anti-coagulation therapy and have the added advantage of being more resilient to infection that tissue engineered prosthesis, however, there is a limited donor pool (3, 88). Despite these advantages, homografts show only a 60% survival at 10 years, and 30% at 20 years and are not ideal for pediatric cases (89). Current therapeutic strategies are highlighted in Figure 4.
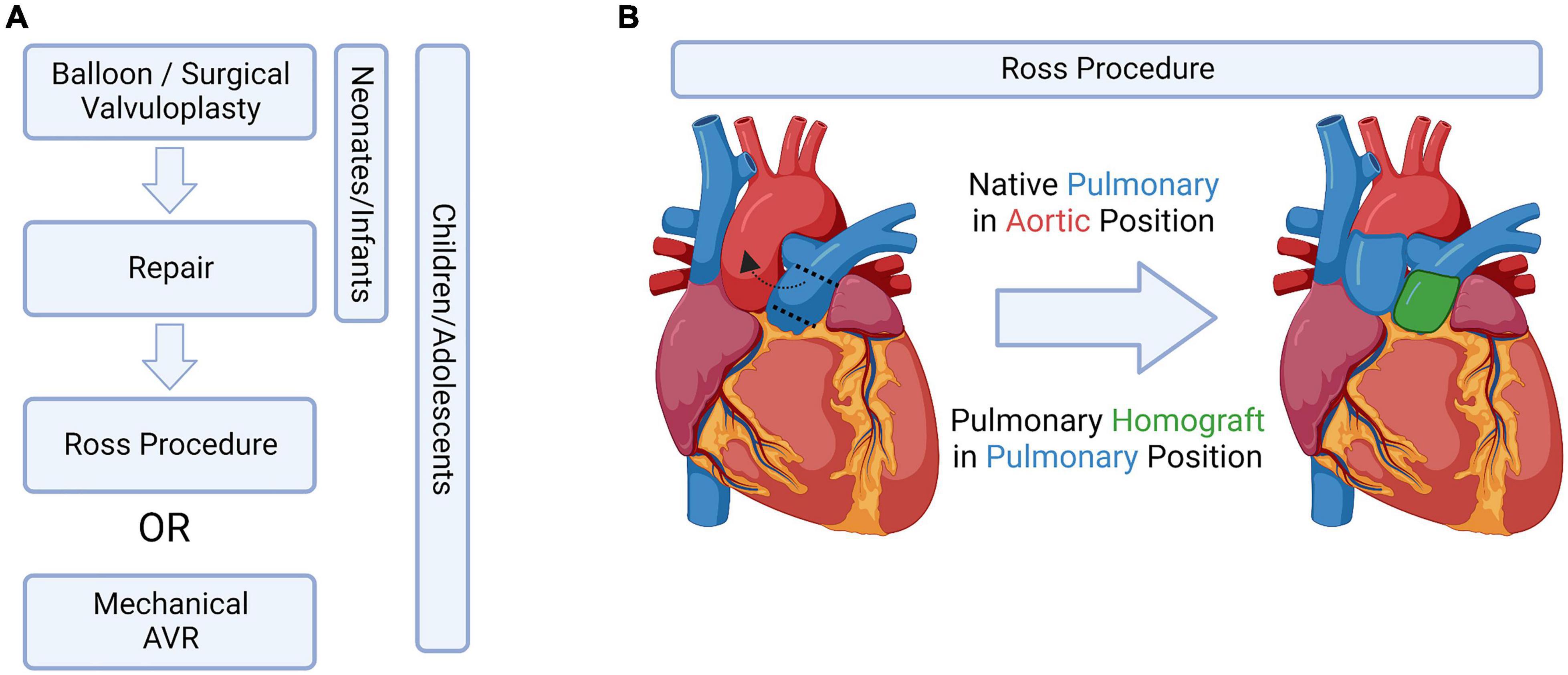
Figure 4. Current treatment options, bioengineered solutions, and the Ross Procedure. (A) Potential aortic valve intervention strategies in pediatric patients, adapted from Bouhout et al. (91). (B) Schematic drawing of the Ross procedure, which switches the native pulmonary valve into the aortic position. Figure created using BioRender.com (2022).
The limitations listed above are why the Ross procedure, which switches the native pulmonary valve into the aortic valve position, is currently the preferred treatment amongst young pediatric cases (90–92) (Figure 4B). However, this procedure does not come without complications: children having undergone this procedure have a 14.3% mortality rate in the first year – after surviving the first year, the likelihood of the child needing an aortic or pulmonary reintervention later in life is 15.3 and 27.5%, respectively (3).
Mechanisms of failure of pulmonary valves in the aortic position during Ross procedures can be inferred from studies regarding proteomic differences between the two valves. Ikhumetse et al. found that when pulmonary valve derived VICs were exposed to cyclic aortic pressures, sulfated GAGs as well as collagen synthesis were significantly increased (93). Angel et al. aimed to characterize the difference between AV and PV via chromatographic proteomic approaches. The study found that only 50% of the proteome of the AV is shared with the PV. The PV proteome was abundant in hypoxia associated proteins such as H2AX but only sparingly expressed proteins that may be critical to healthy AV function, such as MFGe8 and Emilin-1 (94). The role of PTMs in surgical valve failure has not been explored.
While transcriptional regulation of the valvular ECM has been well characterized, low cell density and high ECM content warrant translational level studies. Translational and post-translational regulation of these proteins in pediatric CAVS remains to be fully elucidated, as the majority of literature focuses on adult calcific cases of CAVS. Acquiring information on spatial organization of molecular patterns of ECM proteins and their PTMs is essential to defining both disease and development. In this section we aim to review advancements in the field of valvular and extracellular matrix focused proteomics, the relevance of spatial -omics in valve disease, and how multi-omics analysis may help identify novel therapeutic targets relevant to pediatric valve fibrosis (Figure 5).
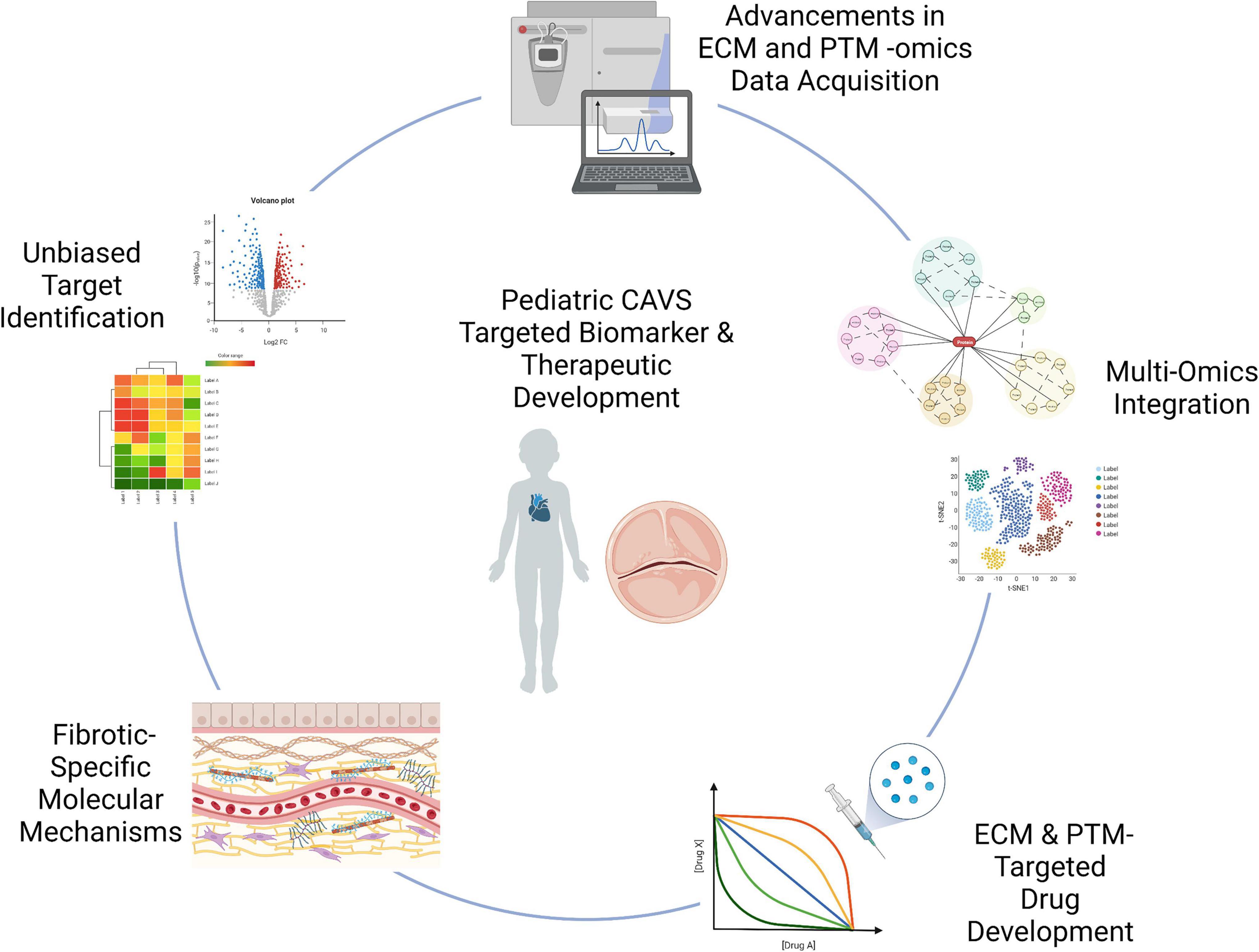
Figure 5. Graphical summary of technological advancements to be applied to pCAVS. Unbiased approaches, such as proteomics and transcriptomics, allow for novel discovery of disease markers. Advancements in acquisition techniques as described allow for a deeper mining of the extracellular matrisome, previously limited in accessibility. This increased accessibility allows for identification of post-translational modifications; while multi-omics integration probe upstream regulating enzymes, accelerating identification of therapeutic targets. Extracellular matrix targeted -omics techniques have the potential to elucidate fibrosis-specific biomarkers, highlighting the distinct molecular mechanisms between adult calcific and pediatric fibrotic CAVS. Graphics created using BioRender.com (2022).
The ECM proteome in pediatric valvular tissue has been difficult to characterize via conventional tryptic methods due to high levels of intra- and intermolecular crosslinking and PTMs. Brioschi et al. published a comparative analysis of gel-based and gel-free proteomic methods to characterize the human mitral valve proteome, showing large variability between the four methods and only 12% of all proteins identified were associated with ECM pathways (95). Schlotter et al. used label-free tryptic proteomics techniques with isolated VICs and micro-dissected valvular tissues. Amongst other findings, they showed that collagen biosynthesis, collagen modification, GAG metabolism, and ECM degradation pathways were enriched in the adult CAVS isolates (96). While these methods can implicate ECM pathways, ECM proteins were not specifically enriched during sample preparation for these studies. After years of advancement in valvular proteomic techniques (97, 98), the Barderas Lab enriched for ECM proteins in their 2D-LC MS/MS method on calcific AVS samples. The group found 13 differentially regulated ECM proteins in the adult calcific CAVS samples compared to controls. Of these, biglycan, periostin, prolargin, decorin, and lumican (implicated in collagen fiber assembly) formed a protein–protein interaction network cluster. One collagen protein, Col6a1, was identified to be upregulated in adult CAVS (99). Similarly, Bouchareb et al. used guanidine hydrochloride to extract the ECM for proteomic analysis, and identified two potential biomarkers of adult calcific CAVS – FNDC1 and MXRA5 (100). While not on native valvular tissue, Syedain et al. recently performed ECM enriched proteomic analysis on tri-tube valve conduits to analyze their regenerative capacity (101). The Hansen Lab recently did a comparative study of a dozen ECM enrichment techniques for proteomics, comparing decellularization, extraction and single-shot analysis methods (102). Alternative enrichment approaches used by our group use ECM-targeting enzymes, such as collagenase type III and MMP12, to enzymatically enrich for the extracellular matrix within valvular tissues for both spatial and LC-MS/MS proteomics. These studies have been applied to clinically available pediatric BAV formalin-fixed paraffin embedded (FFPE) tissues, with all proteins identified being of the extracellular matrix, and roughly half being collagen subtypes (37).
Serum proteomics is a potential source for biomarker discovery in tissue diseases unable to be biopsied, such as valvular disease. To date, no proteomic analysis has been done on serum correlating to pediatric CAVS tissue. However, serum profiling has been done for cardiac failure (103, 104) and other fibrotic diseases such as idiopathic pulmonary fibrosis (105) and liver cirrhosis (106). While there are many large-scale proteomic studies of serum profiles, ECM proteins are inherently difficult to enrich for (107). Multimodal -omics approaches, such as epigenomics, transcriptomics, proteomics, and metabolomics, offer promising advantages to traditional characterization techniques, and together moves the field away from strictly characterization toward biomarker and pharmacotherapeutic target discovery, as well as to inform bioengineered solutions (108, 109).
Despite advancements in multi-omics techniques listed above, bulk LC-MS/MS proteomics still exclude a key variable of disease pathology – spatial localization of dysregulated ECM within the tissue microarchitecture. Several mass spectrometry techniques exist that link the tissue proteome to its structure. One technique is Matrix-Assisted Laser Desorption/Ionization (MALDI) Imaging Mass Spectrometry (IMS). MALDI-IMS is a robust proteomic technique that spatially maps peptides from histological tissue sections. This is traditionally done by spraying the tissue with a thin, uniform layer of enzyme that, when analyzed in parallel to high resolution accurate mass proteomics, allows for the relative quantitation and localization of peptides and other analytes to specific regions of the tissue. In addition to tryptic peptides, novel MALDI-IMS and LC-MS/MS techniques have been developed to specifically map ECM proteins, that until recently were unable to be accessed via conventional proteomic workflows (37, 110–115).
Proteomic imaging via MALDI-IMS may allow for greater protein identification and improved PTM evaluation compared to antibody-based histology strategies. Angel et al. recently outlined the challenges of analyzing cardiac tissues with MALDI-IMS as well as the advancements that have been made in the field (116). Mourino-Alvarez et al. recently took advantage of MALDI-IMS technology to characterize fresh frozen calcific AVS tissue. The group was able to visualize m/z peaks correlating to calcified, collagenous, and elastin rich tissue areas. Two peptides of interest were from Col6a3 (responsible for calcium and hydroxyproline accumulation on human osteoblast-like cells) and NDRG2 (implicated in p53 mediated apoptosis and mineralization initiation) (117). Recently, several studies have emerged using MALDI-IMS and other imaging mass spectrometry methods to characterize molecules associated with healthy and diseased aortic valve, such as N-linked glycans (83, 112), lipids (118–120), glycosaminoglycans (112), and peptides (110, 112, 116). With diseased valve tissue samples becoming less abundant on the bench due to increases in transcatheter aortic valve replacements (121), these multi-modal MALDI-IMS techniques are a promising tool that can be used to characterize ECM deposition and post-translational modifications during CAVS progression, in conjunction to histopathological evaluation (111, 112).
Post translational modifications of ECM have been linked to various disease states, not limited to fibrotic diseases. While hydroxyproline and hydroxylysine modifications are essential to proper collagen fibril formation, over-hydroxylation of the protocollagen chains can delay collagen polymerization, resulting in excessive PTMs, such as glycosylation (41, 42, 122). This perturbs collagen’s triple-helical structure further which in turn contributes to various disease states, such as Ehlers-Danlos syndrome and osteogenesis imperfecta (41, 123). Of note, collagen-dysregulation associated connective tissue disorders have a high prevalence of aortic valve defects, including aortic valve stenosis (124). Similarly, under-hydroxylation can destabilize collagen’s triple helix, also contributing to disease states, as seen in vitamin C deficiencies leading to scurvy (125). Hydroxyproline modifications in collagen amongst other proteins has been implicated in oxygen-sensing pathways in cancer cells (126). Hydroxylysine aldehyde–derived collagen cross-links have been found to be upregulated in lung tumor stroma (127). Kamel et al. shows that increased hydroxylation of lysine in collagen increases the proliferation rate of uterine fibroids (128). Additionally, increased tissue stiffness due to over hydroxylation of the ECM may not only contribute to progression of tumors and fibrotic diseases but may also influence diffusion of drug molecules within the disease microenvironment (129, 130). Very few approaches are able to map which sites of hydroxylation contribute to disease status when modified. For these reasons, characterization of not only the ECM, but also site-specific PTMs, are critical for elucidating mechanisms of fibrotic diseases, such as pediatric end-stage CAVS.
Identified therapeutic strategies for adult CAVS, such as targeting of circulating lipid levels or osteogenic differentiation of VICs and mineral deposition directly, would not be appropriate for targeting the fibrosis seen pediatric end-stage (131). The late diagnosis of fibrotic diseases limits therapeutic intervention before rapid ECM remodeling occurs. The future of fibrotic treatments may rely on proteomic strategies to identify new ECM biomarkers, resulting in earlier detection. Walraven and Hinz propose in their 2018 review that inhibiting fibrosis will depend on two main strategies: (1) targeting the myofibroblasts directly through stimulatory or suppressing cytokines and stimulating apoptosis; and/or (2) targeting the fibrotic ECM directly using targeted proteolytic enzymes (132). Currently, the only FDA approved drugs on the market to treat any fibrotic disease (idiopathic pulmonary fibrosis) are Nintedanib and Pirfenidone. Previously thought to work through kinase-dependent fibrotic pathways (133), a recent study shows that these drugs inhibit collagen fibril assembly directly (134). A recent study suggests that clinically targeting the denatured collagen, which occurs in fibrotic disease during enzymatic remodeling, may be possible via collagen mimetic peptides or antibodies (135). Another proposed anti-fibrotic therapeutic strategy is the targeting of hydroxyprolines via collagen prolyl-4-hydroxylase (CP4H). However, CP4H inhibitors lack selectivity – while promising results are seen in vivo, off target affects to redox homeostasis and iron metabolism are also seen (136). Other strategies to target hydroxyproline involve scavenging the ROS superoxide, which inhibits CP4H (137, 138). Alternatively, targeting collagen-related partners whose binding is dependent on hydroxyprolination, such as integrins or glycoproteins, may modulate fibrotic signaling (138–140). While the role of hydroxylysine and allysine have yet to be elucidated in pCAVS or fibrocalcific CAVS, inhibition of lysyl oxidase inhibitors may be an additional potential therapeutic strategy, with promising results shown related to fibroblast activation in liver fibrosis (138). Realistically, the goal of anti-fibrotic therapies in pediatric CAVS may not be to cure stenosis but instead to delay the severity until patients reach an appropriate age and size for bioengineered options to be used without the need for repeated surgeries.
A major gap in knowledge hindering the development of therapies for pediatric CAVS is that the molecular mechanisms underlying the structure-function relationship remain poorly defined. One therapeutic avenue in need of further scientific exploration is the targeting of extracellular matrix proteins and their post translational modifications – specifically, collagens. Collagens are the fundamental scaffolding of valvular structure that influences valvular function, and thus cardiac function (4, 5). Multiple collagens are known for their cardiac remodeling potential and are involved in valvular development (7, 141, 142), yet translational regulation of these critical proteins due to pediatric CAVS remains unknown – thus preventing appropriate drug targets from being developed and administered upon initial diagnosis. Advancement of high-resolution accurate mass proteomics and spatial techniques like MALDI imaging mass spectrometry offer unique advantages in understanding the structure/function relationship of the ECM in cardiac tissue. These novel strategies may provide insight into AV development, the rapid progression of pediatric end-stage CAVS.
CC and PA: conceptualization and writing. CC, JS, RD, and PA: review and editing. RD and PA: supervision. CC, RD, and PA: funding acquisition. PA: project administration. All authors have read and agreed to the published version of the manuscript.
CC was supported by a T32 from NHLBI (HL007260) and an F31 from NHLBI (HL156524). PA appreciates support by UL1RR024975, AHA 16GRNT31380005, NIH/NCI R21 CA240148, and additional support to RD provided by the South Carolina Centers of Economic Excellence SmartState program. The MUSC Imaging Mass Spectrometry Research Resource was supported in part by the NIH/NIDDK Digestive Disease Research Core Center P30DK123704 and is a division of the Mass Spectrometry Facility, supported by the University and P20GM103542 (NIH/NIGMS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hoffman JIE, Kaplan S, Liberthson RR. Prevalence of congenital heart disease. Am Heart J. (2004) 147:425–39. doi: 10.1016/j.ahj.2003.05.003
2. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. (2006) 368:1005–11. doi: 10.1016/S0140-6736(06)69208-8
3. Sharabiani MTA, Dorobantu DM, Mahani AS, Turner M, Peter Tometzki AJ, Angelini GD, et al. Aortic valve replacement and the ross operation in children and young adults. J Am Coll Cardiol. (2016) 67:2858–70. doi: 10.1016/j.jacc.2016.04.021
4. Peacock JD, Lu Y, Koch M, Kadler KE, Lincoln J. Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Dev Dyn. (2008) 237:3051–8. doi: 10.1002/dvdy.21719
5. Hinton RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. (2006) 98:1431–8. doi: 10.1161/01.RES.0000224114.65109.4e
6. Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. (2009) 105:408–21. doi: 10.1161/CIRCRESAHA.109.201566
7. Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: Implications for postnatal adaptation, pathology, and tissue engineering. Circulation. (2006) 113:1344–52. doi: 10.1161/CIRCULATIONAHA.105.591768
8. Schoen FJ, Gotlieb AI. Heart valve health, disease, replacement, and repair: a 25-year cardiovascular pathology perspective. Cardiovasc Pathol. (2016) 25:341–52. doi: 10.1016/j.carpath.2016.05.002
9. Schoen FJ. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. (2008) 118:1864–80. doi: 10.1161/CIRCULATIONAHA.108.805911
10. Bischoff J. Endothelial-to-Mesenchymal Transition. Circ Res. (2019) 124:1163–5. doi: 10.1161/CIRCRESAHA.119.314813
11. Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. (1995) 77:1–6. doi: 10.1161/01.RES.77.1.1
12. Markwald RR, Fitzharris TPMF. Structural development of endocardial cushions. Am J Anat. (1977) 148:85–119. doi: 10.1002/aja.1001480108
13. Tao G, Kotick JD, Lincoln J, Gao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: The role of endothelial cells. Curr Top Dev Biol. (2012) 100:203–32. doi: 10.1016/B978-0-12-387786-4.00006-3
14. Butcher JT, Mahler GJ, Hockaday LA. Aortic valve disease and treatment: The need for naturally engineered solutions. Adv Drug Deliv Rev. (2011) 63:242–68. doi: 10.1016/j.addr.2011.01.008
15. Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. (2007) 171:1407–18. doi: 10.2353/ajpath.2007.070251
16. Wyss K, Yip CYY, Mirzaei Z, Jin X, Chen JH, Simmons CA. The elastic properties of valve interstitial cells undergoing pathological differentiation. J Biomech. (2012) 45:882–7. doi: 10.1016/j.jbiomech.2011.11.030
17. Hulin A, Hortells L, Gomez-Stallons MV, O’Donnell A, Chetal K, Adam M, et al. Maturation of heart valve cell populations during postnatal remodeling. Development. (2019) 146:dev173047. doi: 10.1242/dev.173047
18. Ma H, Killaars AR, DelRio FW, Yang C, Anseth KS. Myofibroblastic activation of valvular interstitial cells is modulated by spatial variations in matrix elasticity and its organization. Biomaterials. (2017) 131:131–44. doi: 10.1016/j.biomaterials.2017.03.040
19. Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. (2015) 242:251–60. doi: 10.1016/j.atherosclerosis.2015.07.008
20. Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. (2006) 12:905–15. doi: 10.1089/ten.2006.12.905
21. Shapero K, Wylie-Sears J, Levine RA, Mayer JE Jr., Bischoff J. Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J Mol Cell Cardiol. (2015) 80:175. doi: 10.1016/j.yjmcc.2015.01.006
22. Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, et al. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. (2011) 121:422–30. doi: 10.1172/JCI44244
23. Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. (1983) 220:1059–61. doi: 10.1126/science.6844926
24. Leatherbury L, Gauldin HE, Waldo K, Kirby ML, Cardiology P. Microcinephotography of the developing heart in neural crest-ablated chick embryos. Circulation. (1990) 81:1047–57. doi: 10.1161/01.cir.81.3.1047
25. Huang Y, Xu K, Zhou T, Zhu P, Dong N, Shi J. Comparison of rapidly proliferating, multipotent aortic valve-derived stromal cells and valve interstitial cells in the human aortic valve. Stem Cells Int. (2019) 2019:7671638. doi: 10.1155/2019/7671638
26. Fisher SA, Langille BL, Srivastava D. Apoptosis during cardiovascular development. Circ Res. (2000) 87:856–64. doi: 10.1161/01.RES.87.10.856
27. Abdelwahid E, Pelliniemi LJ, Jokinen E. Cell death and differentiation in the development of the endocardial cushion of the embryonic heart. Microsc Res Tech. (2002) 58:395–403. doi: 10.1002/jemt.10159
28. Tuleta I, Ghaddioui AKA, Bauriedel G, Wernert N, Preusse CJ, Welz A, et al. The imbalance between proliferation and apoptosis contributes to degeneration of aortic valves and bioprostheses. Cardiol J. (2013) 20:268–76. doi: 10.5603/CJ.2013.0072
29. Armstrong EJ, Bischoff J. Heart valve development: Endothelial cell signaling and differentiation. Circ Res. (2004) 95:459–70. doi: 10.1161/01.RES.0000141146.95728.da
30. Wang H, Anseth KS. Cardiac valve cells and their microenvironment - insights from invitro studies. Nat Rev Cardiol. (2014) 11:715–27. doi: 10.1038/nrcardio.2014.162
31. Wang Y, Fang Y, Lu P, Wu B, Zhou B. NOTCH Signaling in aortic valve development and calcific aortic Valve Disease. Front Cardiovasc Med. (2021) 8:682298. doi: 10.3389/fcvm.2021.682298
32. Amofa D, Hulin A, Nakada Y, Sadek HA, Yutzey KE. Hypoxia promotes primitive glycosaminoglycan-rich extracellular matrix composition in developing heart valves. Am J Physiol Heart Circ Physiol. (2017) 313:1143–54. doi: 10.1152/ajpheart.00209.2017
33. Thubrikar M, Piepgrass WC, Deck JD, Nolan SP. Stresses of natural versus prosthetic aortic valve leaflets in vivo. Ann Thorac Surg. (1980) 30:230–9. doi: 10.1016/s0003-4975(10)61250-7
34. Weston MW, LaBorde DV, Yoganathan AP. Estimation of the shear stress on the surface of an aortic valve leaflet. Ann Biomed Eng. (1999) 27:572–9. doi: 10.1114/1.199
35. Arjunon S, Rathan S, Jo H, Yoganathan AP. Aortic valve: mechanical environment and mechanobiology. Ann Biomed Eng. (2013) 41:1331–46. doi: 10.1007/s10439-013-0785-7
36. Kodigepalli KM, Thatcher K, West T, Howsmon DP, Schoen FJ, Sacks MS, et al. Biology and biomechanics of the heart valve extracellular matrix. J Cardiovasc Dev Dis. (2020) 7:1–22. doi: 10.3390/jcdd7040057
37. Clift CL, Su YR, Bichell D, Jensen Smith HC, Bethard JR, Norris-Caneda K, et al. Collagen fiber regulation in human pediatric aortic valve development and disease. Sci Rep. (2021) 11:9751. doi: 10.1038/s41598-021-89164-w
38. Mead TJ, Yutzey KE. Notch pathway regulation of neural crest cell development in vivo. Dev Dyn. (2012) 241:376–89. doi: 10.1002/dvdy.23717
39. Lincoln J, Alfieri CM, Yutzey KE. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev Biol. (2006) 292:290–302. doi: 10.1016/j.ydbio.2005.12.042
40. Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. (2010) 338:127–35.
41. Shoulders MD, Raines RT. Collagen Structure and Stability. Annu Rev Biochem. (2009) 78:929–58. doi: 10.1146/annurev.biochem.77.032207.120833
42. Yamauchi M, Sricholpech M. Lysine post-translational modifications of collagen. Essays Biochem. (2012) 52:113–33.
43. Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. (2010) 45:106–24. doi: 10.3109/10409231003627991
44. Schmelzer CEH, Nagel MBM, Dziomba S, Merkher Y, Sivan SS, Heinz A. Prolyl hydroxylation in elastin is not random. Biochim Biophys Acta. (2016) 1860:2169–77.
45. Wiltz D, Alexander C, Balaoing RL, Blancas ACM, Zhang X, et al. Extracellular Matrix Organization, Structure, and Function. Calcific Aortic Valve Disease. E Aikawa Editor. London: InTech (2013).
46. Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. (2011) 108:1510–24. doi: 10.1161/CIRCRESAHA.110.234237
47. Koch CD, Lee CM, Apte SS. Aggrecan in cardiovascular development and disease. J Histochem Cytochem. (2020) 68:777–95.
48. Granath C, Noren H, Björck H, Simon N, Olesen K, Rodin S, et al. Characterization of laminins in healthy human aortic valves and a modified decellularized rat scaffold. BioRes Open Access. (2020) 9:269–78. doi: 10.1089/biores.2020.0018
49. Norris RA, Moreno-Rodriguez R, Hoffman S, Markwald RR. The many facets of the matricelluar protein periostin during cardiac development, remodeling, and pathophysiology. J Cell Commun Signal. (2009) 3:275–86. doi: 10.1007/s12079-009-0063-5
50. Conway SJ, Doetschman T, Azhar M. The inter-relationship of periostin, TGFβ, and BMP in Heart Valve Development and Valvular Heart Diseases. Scientificworldjournal. (2011) 11:1509–24. doi: 10.1100/tsw.2011.132
51. Wilton E, Bland M, Thompson M, Jahangiri M. Work in progress report - Valves: Matrix metalloproteinase expression in the ascending aorta and aortic valve. Interact Cardiovasc Thorac Surg. (2008) 7:37–40.
52. Fondard O, Detaint D, Iung B, Choqueux C, Adle-Biassette H, Jarraya M, et al. Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur Heart J. (2005) 26:1333–41. doi: 10.1093/eurheartj/ehi248
53. Mathieu P, Bossé Y, Huggins GS, Della Corte A, Pibarot P, Michelena HI, et al. The pathology and pathobiology of the bicuspid aortic valve: state of the art and novel research perspectives. J Pathol Clin Res. (2015) 1:195–206. doi: 10.1002/cjp2.21
54. Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. (2011) 73:29.
55. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. (2005) 437:270–4.
56. Lo Presti F, Guzzardi DG, Bancone C, Fedak PWM, Della Corte A, Presti FL, et al. The science of BAV aortopathy. Prog Cardiovasc Dis. (2020) 63:465–74.
57. Butcher JT, Tressel S, Johnson T, Turner D, Sorescu G, Jo H, et al. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: Influence of shear stress. Arterioscler Thromb Vasc Biol. (2006) 26:69–77. doi: 10.1161/01.ATV.0000196624.70507.0d
58. Gharibeh L, Komati H, Bossé Y, Boodhwani M, Heydarpour M, Fortier M, et al. GATA6 regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation. (2018) 138:1025–38. doi: 10.1161/CIRCULATIONAHA.117.029506
59. Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. (2011) 50:561–9. doi: 10.1016/j.yjmcc.2010.12.005
60. Ho SY. Structure and anatomy of the aortic root. Eur J Echocardiogr. (2009) 10:i3–10. doi: 10.1093/ejechocard/jen243
61. Fedak PW, Verma S, David TE, Leask RL, Weisel RD, Butany J. Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation. (2002) 106:900–4.
62. Atkins SK, Sucosky P. Etiology of bicuspid aortic valve disease: Focus on hemodynamics. World J Cardiol. (2014) 6:1227–33. doi: 10.4330/wjc.v6.i12.1227
63. Fernandes SM, Khairy P, Sanders SP, Colan SD. Bicuspid aortic valve morphology and interventions in the young. J Am Coll Cardiol. (2007) 49:2211–4. doi: 10.1016/j.jacc.2007.01.090
64. Hutson HN, Marohl T, Anderson M, Eliceiri K, Campagnola P, Masters KS. Calcific aortic valve disease is associated with layer-specific alterations in collagen architecture. PLoS One. (2016) 11:e0163858. doi: 10.1371/journal.pone.0163858
65. Perrotta I, Russo E, Camastra C, Filice G, Di Mizio G, Colosimo F, et al. New evidence for a critical role of elastin in calcification of native heart valves: Immunohistochemical and ultrastructural study with literature review. Histopathology. (2011) 59:504–13. doi: 10.1111/j.1365-2559.2011.03977.x
66. Vito AD, Donato A, Presta I, Mancuso T, Brunetti FS, Mastroroberto P, et al. Extracellular matrix in calcific aortic valve disease: architecture, dynamic and perspectives. Int J Mol Sci. (2021) 22:913. doi: 10.3390/ijms22020913
67. Sun L, Chandra S, Sucosky P. Ex Vivo evidence for the contribution of hemodynamic shear stress abnormalities to the early pathogenesis of calcific bicuspid aortic valve disease. PLoS One. (2012) 7:e48843. doi: 10.1371/journal.pone.0048843
69. Bogdanova M, Kostina A, Zihlavnikova Enayati K, Zabirnyk A, Malashicheva A, Stensløkken KO, et al. Inflammation and mechanical stress stimulate osteogenic differentiation of human aortic valve interstitial cells. Front Physiol. (2018) 9:1635. doi: 10.3389/fphys.2018.01635
70. Butcher JT, Simmons CA, Warnock JN. Review: mechanobiology fo the aortic heart valve. J Heart Valve Dis. (2008) 17:62–73.
71. Ku CHH, Johnson PH, Batten P, Sarathchandra P, Chambers RC, Taylor PM, et al. Collagen synthesis by mesenchymal stem cells and aortic valve interstitial cells in response to mechanical stretch. Cardiovasc Res. (2006) 71:548–56.
72. Korossis S. Structure-Function relationship of heart valves in health and disease. Structural Insufficiency Anomalies in Cardiac Valves. K Kirali editor. London: InTech (2018).
73. Filova E, Burdikova Z, Stankova L, Hadraba D, Svindrych Z, Schornik D, et al. Collagen structures in pericardium and aortic heart valves and their significance for tissue engineering. In proceedings of the 2013 E-Health and Bioengineering Conference EHB. Iasi: IEEE (2013). 2013 p.
74. Gu J, Lu Y, Deng M, Qiu M, Tian Y, Ji Y, et al. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp Mol Med. (2019) 51:1–14. doi: 10.1038/s12276-019-0272-9
75. Herr DJ, Singh T, Dhammu T, Menick DR. Regulation of metabolism by mitochondrial enzyme acetylation in cardiac ischemia-reperfusion injury. Biochim Biophys Acta - Mol Basis Dis. (2020) 1866:165728. doi: 10.1016/j.bbadis.2020.165728
76. Wang SB, Venkatraman V, Crowgey EL, Liu T, Fu Z, Holewinski R, et al. Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3β Function Independent of Its Phosphorylation State. Circ Res. (2018) 122:1517–31. doi: 10.1161/CIRCRESAHA.118.312789
77. Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, et al. Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. (2014) 130:419–30. doi: 10.1161/CIRCULATIONAHA.113.008364
78. Majumdar U, Manivannan S, Basu M, Ueyama Y, Blaser MC, Cameron E, et al. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci Adv. (2021) 7:eabe3706. doi: 10.1126/sciadv.abe3706
79. Li J, Johnson JA, Su H. Ubiquitin and ubiquitin-like proteins in cardiac disease and protection. Curr Drug Targets. (2018) 19:989–1002.
80. Grosche J, Meißner J, Eble JA. More than a syllable in fib-ROS-is: The role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol Aspects Med. (2018) 63:30–46. doi: 10.1016/j.mam.2018.03.005
81. Risteli M, Ruotsalainen H, Salo AM, Sormunen R, Sipilä L, Baker NL, et al. Reduction of lysyl hydroxylase 3 causes deleterious changes in the deposition and organization of extracellular matrix. J Biol Chem. (2009) 284:28204–11. doi: 10.1074/jbc.M109.038190
82. Iozzo RV, Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. (2015) 42:11–55. doi: 10.1016/j.matbio.2015.02.003
83. Angel PM, Drake RR, Park Y, Clift CL, West C, Berkhiser S, et al. Spatial N-glycomics of the human aortic valve in development and pediatric endstage congenital aortic valve stenosis. J Mol Cell Cardiol. (2021) 154:6–20. doi: 10.1016/j.yjmcc.2021.01.001
84. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. (2006) 126:855–67. doi: 10.1016/j.cell.2006.08.019
85. Petushkova AI, Zamyatnin AA. Redox-mediated post-translational modifications of proteolytic enzymes and their role in protease functioning. Biomolecules. (2020) 10:650.
86. Zhang BL, Bianco RW, Schoen FJ. Preclinical assessment of cardiac valve substitutes: Current status and considerations for engineered tissue heart valves. Front Cardiovasc Med. (2019) 6:72. doi: 10.3389/fcvm.2019.00072
87. Schoen FJ. Heart valve tissue engineering: quo vadis? Curr Opin Biotechnol. (2011) 22:698–705. doi: 10.1016/j.copbio.2011.01.004
88. Siebelink MJ, Albers MJIJIJ, Roodbol PF, Van De Wiel HBMM. Key factors in paediatric organ and tissue donation: An overview of literature in a chronological working model. Transplant Int. (2012) 25:265–71. doi: 10.1111/j.1432-2277.2011.01407.x
89. Grocott-Mason R, Lund O, Elwidaa H, Mazhar R, Chandrasakeran V, Mitchell AG, et al. Long-term results after aortic valve replacement in patients with congestive heart failure. Homografts vs prosthetic valves. Eur Heart J. (2000) 21:1698–707.
90. Saiki H, Senzaki H. Congenital Aortic Stenosis in Children. Calcific Aortic Valve Disease. E Aikawa editor. London: InTech (2013).
91. Bouhout I, Ba PS, El-Hamamsy I, Poirier N. Aortic valve interventions in pediatric patients. Semin Thorac Cardiovasc Surg. (2018) 31:277–87.
92. Takkenberg JJM, Klieverik LMA, Schoof PH, Van Suylen RJ, Van Herwerden LA, Zondervan PE, et al. The Ross procedure: A systematic review and meta-analysis. Circulation. (2009) 119:222–8.
93. Ikhumetse JD, Konduri S, Warnock JN, Xing Y, Yoganathand AP. Cyclic aortic pressure affects the biological properties of porcine pulmonary valve leaflets. J Heart Valve Dis. (2006) 15:295–302.
94. Angel PM, Nusinow D, Brown CB, Violette K, Barnett JV, Zhang B, et al. Networked-based characterization of extracellular matrix proteins from adult mouse pulmonary and aortic valves. J Proteome Res. (2011) 10:812–23. doi: 10.1021/pr1009806
95. Brioschi M, Baetta R, Ghilardi S, Gianazza E, Guarino A, Parolari A, et al. Normal human mitral valve proteome: A preliminary investigation by gel-based and gel-free proteomic approaches. Electrophoresis. (2016) 37:2633–43. doi: 10.1002/elps.201600081
96. Schlotter F, Halu A, Goto S, Blaser MC, Body SC, Lee LH, et al. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation. (2018) 138:377–93. doi: 10.1161/CIRCULATIONAHA.117.032291
97. Gil-Dones F, Martin-Rojas T, Lopez-Almodovar LF, de la Cuesta F, Darde VM, Alvarez-Llamas G, et al. Valvular aortic Stenosis: A proteomic insight. Clin Med Insights Cardiol. (2010) 4:1–7.
98. Martín-Rojas T, Gil-Dones F, Lopez-Almodovar LF, Padial LR, Vivanco F, Barderas MG. Proteomic profile of human aortic stenosis: Insights into the degenerative process. J Proteome Res. (2012) 11:1537–50. doi: 10.1021/pr2005692
99. Martin-Rojas T, Mourino-Alvarez L, Alonso-Orgaz S, Rosello-Lleti E, Calvo E, Lopez-Almodovar LF, et al. iTRAQ proteomic analysis of extracellular matrix remodeling in aortic valve disease. Sci Rep. (2015) 5:17290.
100. Bouchareb R, Guauque-Olarte S, Snider J, Zaminski D, Anyanwu A, Stelzer P, et al. Proteomic architecture of valvular extracellular matrix: FNDC1 and MXRA5 Are new biomarkers of aortic stenosis. JACC Basic Transl Sci. (2021) 6:25–39. doi: 10.1016/j.jacbts.2020.11.008
101. Syedain ZH, Haynie B, Johnson SL, Lahti M, Berry J, Carney JP, et al. Pediatric tri-tube valved conduits made from fibroblast-produced extracellular matrix evaluated over 52 weeks in growing lambs. Sci Transl Med. (2021) 13:7225. doi: 10.1126/scitranslmed.abb7225
102. McCabe MC, Schmitt LR, Hill RC, Dzieciatkowska M, Maslanka M, Daamen WF, et al. Evaluation and refinement of sample preparation methods for extracellular matrix proteome coverage. Mol Cell Proteomics. (2021) 20:100079. doi: 10.1016/j.mcpro.2021.100079
103. Egerstedt A, Berntsson J, Smith ML, Gidlöf O, Nilsson R, Benson M, et al. Profiling of the plasma proteome across different stages of human heart failure. Nat Commun. (2019) 10:5830.
104. McDonald K, Glezeva N, Collier P, O’Reilly J, O’Connell E, Tea I, et al. Tetranectin, a potential novel diagnostic biomarker of heart failure, is expressed within the myocardium and associates with cardiac fibrosis. Sci Rep. (2020) 10:7507. doi: 10.1038/s41598-020-64558-4
105. Landi C, Bergantini L, Cameli P, d’Alessandro M, Carleo A, Shaba E, et al. Idiopathic pulmonary fibrosis serum proteomic analysis before and after nintedanib therapy. Sci Rep. (2020) 10:9378. doi: 10.1038/s41598-020-66296-z
106. Cheung KJ, Tilleman K, Deforce D, Colle I, Van Vlierberghe H. The HCV serum proteome: a search for fibrosis protein markers. J Viral Hepat. (2009) 16:418–29.
107. Lindsey ML, Jung M, Hall ME, DeLeon-Pennell KY. Proteomic analysis of the cardiac extracellular matrix: clinical research applications. Exp Rev Proteomics. (2018) 15:105–12. doi: 10.1080/14789450.2018.1421947
108. Blaser MC, Kraler S, Lüscher TF, Aikawa E. Multi-Omics approaches to define calcific aortic valve disease pathogenesis. Circ Res. (2021) 128:1371–97. doi: 10.1161/CIRCRESAHA.120.317979
109. Chester AH, Grande-Allen KJ. Which biological properties of heart valves are relevant to tissue engineering? Front Cardiovasc Med. (2020) 7:63.
110. Angel PM, Comte-Walters S, Ball LE, Talbot K, Mehta AS, Brockbank KGMM, et al. Mapping extracellular matrix proteins in formalin-fixed, paraffin-embedded tissues by MALDI imaging mass spectrometry. J Proteome Res. (2018) 17:635–46.
111. Clift CL, Mehta A, Drake RR, Angel PM. Multiplexed imaging mass spectrometry of histological staining, n-glycan and extracellular matrix from one tissue section: A tool for fibrosis research. Methods Mol Biol. (2019) 2350:313–29. doi: 10.1007/978-1-0716-1593-5_20
112. Clift CL, Drake RR, Mehta A, Angel PM. Multiplexed imaging mass spectrometry of the extracellular matrix using serial enzyme digests from formalin-fixed paraffin-embedded tissue sections. Anal Bioanal Chem. (2020) 413:2709–19. doi: 10.1007/s00216-020-03047-z
113. Angel PM, Bruner E, Bethard J, Clift CL, Ball LE, Drake RR, et al. Extracellular matrix alterations in low-grade lung adenocarcinoma compared with normal lung tissue by imaging mass spectrometry. J Mass Spectrom. (2020) 55:e4450. doi: 10.1002/jms.4450
114. Angel PM, Spruill L, Jefferson M, Bethard JR, Ball LE, Hughes-Halbert C, et al. Zonal regulation of collagen-type proteins and posttranslational modifications in prostatic benign and cancer tissues by imaging mass spectrometry. Prostate. (2020) 80:1071–86. doi: 10.1002/pros.24031
115. Angel PM, Schwamborn K, Comte-Walters S, Clift CL, Ball LE, Mehta AS, et al. Extracellular matrix imaging of breast tissue pathologies by MALDI imaging mass spectrometry. Proteom Clin Appl. (2018) 13:e1700152. doi: 10.1002/prca.201700152
116. Angel PM, Baldwin HS, Gottlieb Sen D, Su YR, Mayer JE, Bichell D, et al. Advances in MALDI imaging mass spectrometry of proteins in cardiac tissue, including the heart valve. Biochim Biophys Acta Proteins Proteom. (2017) 1865:927–35. doi: 10.1016/j.bbapap.2017.03.009
117. Mourino-Alvarez L, Iloro I, De La Cuesta F, Azkargorta M, Sastre-Oliva T, Escobes I, et al. MALDI-Imaging Mass Spectrometry: a step forward in the anatomopathological characterization of stenotic aortic valve tissue. Sci Rep. (2016) 6:27106. doi: 10.1038/srep27106
118. Lim J, Aguilan JT, Sellers RS, Nagajyothi F, Weiss LM, Angeletti RH, et al. Lipid mass spectrometry imaging and proteomic analysis of severe aortic stenosis. J Mol Histol. (2020) 51:559–71. doi: 10.1007/s10735-020-09905-5
119. Angel PM, Bayoumi AS, Hinton RB, Su YR, Bichell D, Mayer JE, et al. MALDI-IMS as a lipidomic approach to heart valve research. J Heart Valve Dis. (2016) 25:240–52.
120. Chaurand P, Cornett DS, Angel PM, Caprioli RM. From whole-body sections down to cellular level, multiscale imaging of phospholipids by MALDI mass spectrometry. Mol Cell Proteom. (2011) 10:O110.004259 doi: 10.1074/mcp.O110.004259
121. Atkins SK, Singh SA, Aikawa E. Calcific aortic valve disease “omics” is timely, but are we looking too late? JACC Basic Transl Sci. (2020) 5:1178–80. doi: 10.1016/j.jacbts.2020.11.001
122. Raghunath M. Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J Mol Biol. (1994) 236:940–9.
123. Gjaltema RAF, Bank RA. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit Rev Biochem Mol Biol. (2016) 52:74–95. doi: 10.1080/10409238.2016.1269716
124. Grygiel-Górniak B, Oduah MT, Olagunju A, Klokner M. Disorders of the aorta and aortic valve in connective tissue diseases. Curr Cardiol Rep. (2020) 22:70.
125. Peterkofsky B. Ascorbate requirement for hydroxylation and secretion of procollagen: relationship to inhibition of collagen synthesis in scurvy. Am J Clin Nutr. (1991) 54:1135S–40S. doi: 10.1093/ajcn/54.6.1135s
126. Zhou T, Erber L, Liu B, Gao Y, Ruan HB, Chen Y. Proteomic analysis reveals diverse proline hydroxylation-mediated oxygen-sensing cellular pathways in cancer cells. Oncotarget. (2016) 7:79154–69. doi: 10.18632/oncotarget.12632
127. Chen Y, Guo H, Terajima M, Banerjee P, Liu X, Yu J, et al. Lysyl hydroxylase 2 is secreted by tumor cells and can modify collagen in the extracellular space. J Biol Chem. (2016) 291:25799–808. doi: 10.1074/jbc.M116.759803
128. Kamel M, Wagih M, Kilic GS, Diaz-Arrastia CR, Baraka MA, Salama SA, et al. Overhydroxylation of Lysine of collagen increases uterine fibroids proliferation: Roles of Lysyl Hydroxylases, Lysyl Oxidases, and Matrix Metalloproteinases. BioMed Res Int. (2017) 2017:1–13.
130. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther. (2014) 14:523–32. doi: 10.1158/1535-7163.MCT-14-0580
131. Myasoedova VA, Ravani AL, Frigerio B, Valerio V, Moschetta D, Songia P, et al. Novel pharmacological targets for calcific aortic valve disease: Prevention and treatments. Pharmacol Res. (2018) 136:74–82.
132. Walraven M, Hinz B. Therapeutic approaches to control tissue repair and fibrosis: Extracellular matrix as a game changer. Matrix Biol. (2018) 71–72:205–24. doi: 10.1016/j.matbio.2018.02.020
133. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet. (2011) 377:1760–9.
134. Knüppel L, Ishikawa Y, Aichler M, Heinzelmann K, Hatz R, Behr J, et al. A Novel antifibrotic mechanism of nintedanib and pirfenidone. inhibition of collagen fibril assembly. Am J Respir Cell Mol Biol. (2017) 57:77–90. doi: 10.1165/rcmb.2016-0217OC
135. Wahyudi H, Reynolds AA, Li Y, Owen SC, Yu SM. Targeting collagen for diagnostic imaging and therapeutic delivery. J Control Release. (2016) 240:323.
136. Vasta JD, Raines RT, James Vasta MD, Raines RT. Collagen Prolyl 4-Hydroxylase as a Therapeutic Target. J Med Chem. (2018) 61:10403–11. doi: 10.1021/acs.jmedchem.8b00822
137. Shi R, Gao S, Smith AH, Li H, Shao M, Shangguan J, et al. Superoxide-induced Type I collagen secretion depends on prolyl 4-hydroxylases. Biochem Biophys Res Commun. (2020) 529:1011–7. doi: 10.1016/j.bbrc.2020.07.002
138. Gallorini M, Carradori S. Expert opinion on drug discovery: Understanding collagen interactions and their targeted regulation by novel drugs. Exp Opin Drug Discov. (2021) 16:1239–60. doi: 10.1080/17460441.2021.1933426
139. Miller MW, Basra S, Kulp DW, Billings PC, Choi S, Beavers MP, et al. Small-molecule inhibitors of integrin 2 1 that prevent pathological thrombus formation via an allosteric mechanism. Proc Natl Acad Sci USA. (2009) 106:719–24. doi: 10.1073/pnas.0811622106
140. Nissinen L, Koivunen J, Käpylä J, Salmela M, Nieminen J, Jokinen J, et al. Novel 21 integrin inhibitors reveal that integrin binding to collagen under shear stress conditions does not require receptor preactivation * ? S. J Biol Chem. (2012) 287:44694–702. doi: 10.1074/jbc.M111.309450
141. Hanson KP, Jung JP, Tran QA, Hsu SPP, Iida R, Ajeti V, et al. Spatial and temporal analysis of extracellular matrix proteins in the developing murine heart: a blueprint for regeneration. Tissue Eng Part A. (2013) 19:1132–43. doi: 10.1089/ten.TEA.2012.0316
Keywords: extracellular matrix, post-translational modifications, proteomics, aortic valve, stenosis, pediatric, congenital
Citation: Clift CL, Saunders J, Drake RR and Angel PM (2022) Perspectives on pediatric congenital aortic valve stenosis: Extracellular matrix proteins, post translational modifications, and proteomic strategies. Front. Cardiovasc. Med. 9:1024049. doi: 10.3389/fcvm.2022.1024049
Received: 20 August 2022; Accepted: 24 October 2022;
Published: 10 November 2022.
Edited by:
Anna Malashicheva, Institute of Cytology, RussiaReviewed by:
Anton G. Kutikhin, Russian Academy of Medical Sciences, RussiaCopyright © 2022 Clift, Saunders, Drake and Angel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peggi M. Angel, YW5nZWxwQG11c2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.