
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 22 December 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1004024
 Pamela W. Sowa1,2,3*
Pamela W. Sowa1,2,3* Aleksander S. Kiełbik1,4
Aleksander S. Kiełbik1,4 Andrei G. Pakhomov1Emily Gudvangen1Uma Mangalanathan1
Andrei G. Pakhomov1Emily Gudvangen1Uma Mangalanathan1 Volker Adams2
Volker Adams2 Olga N. Pakhomova1
Olga N. Pakhomova1Electric shocks, the only effective therapy for ventricular fibrillation, also electroporate cardiac cells and contribute to the high-mortality post-cardiac arrest syndrome. Copolymers such as Poloxamer 188 (P188) are known to preserve the membrane integrity and viability of electroporated cells, but their utility against cardiac injury from cardiopulmonary resuscitation (CPR) remains to be established. We studied the time course of cell killing, mechanisms of cell death, and protection with P188 in AC16 human cardiomyocytes exposed to micro- or nanosecond pulsed electric field (μsPEF and nsPEF) shocks. A 3D printer was customized with an electrode holder to precisely position electrodes orthogonal to a cell monolayer in a nanofiber multiwell plate. Trains of nsPEF shocks (200, 300-ns pulses at 1.74 kV) or μsPEF shocks (20, 100-μs pulses at 300 V) produced a non-uniform electric field enabling efficient measurements of the lethal effect in a wide range of the electric field strength. Cell viability and caspase 3/7 expression were measured by fluorescent microscopy 2–24 h after the treatment. nsPEF shocks caused little or no caspase 3/7 activation; most of the lethally injured cells were permeable to propidium dye already at 2 h after the exposure. In contrast, μsPEF shocks caused strong activation of caspase 3/7 at 2 h and the number of dead cells grew up to 24 h, indicating the prevalence of the apoptotic death pathway. P188 at 0.2–1% reduced cell death, suggesting its potential utility in vivo to alleviate electric injury from defibrillation.
- AC16 human cardiomyocytes subjected to μsPEF shocks display strong activation of caspase 3/7, and the number of dead cells gradually increases up to 24 h, indicating the prevalence of apoptotic death.
- After exposure to nsPEF shocks most of the lethally injured cardiomyocytes die within 2 h, with little or no caspase 3/7 activation.
- Poloxamer 188 profoundly reduces cardiomyocytes death after ns- and μsPEF shocks, highlighting its potential use to enhance cardiomyocyte recovery after defibrillation.
Each year, approximately 350,000 adults in the United States suffer out-of-hospital cardiac arrests (OHCA). In 19%, the initial recorded cardiac rhythm is shockable by defibrillation (1). However, the survival statistics for OHCA remain disappointing. According to the American Heart Association, the survival to hospital discharge after an incidence with shockable rhythm reaches 25% (2). The high mortality from cardiac arrest can in part be contributed to the electrical injury from defibrillation, compromising myocardial function (3–8). Indeed, many adverse outcomes of defibrillation are associated with electroporation (9–12), including subepicardial tissue damage (11, 12) and proarrhythmic activity (13). At the same time, electroporation, transiently impairing the conduction, can be antiarrhythmic and support defibrillation (9, 14).
In comparison to conventional millisecond shocks used by defibrillation, much shorter nanosecond shocks require higher field strength but overall lower energy (15). “Nanoelectropores” in the cell membrane after nsPEF shocks are 1–1.5 nm in diameter (16, 17) and tend to be smaller than those formed by millisecond shocks (18–20), thus leakage through the membrane is less injurious than after conventional defibrillation. In mouse, pig, and rabbit cardiomyocytes, nanosecond shocks resulted in less uptake of membrane impermeable propidium dye compared to longer shocks thus indicating reduced cell injury (21). They are also expected to induce spatially uniform electroporation which implies more homogenous tissue penetration (20, 22, 23). nsPEF shocks were found to cause calcium entry into cardiac myocytes by routes other than calcium channels, followed by a slow sustained depolarization of unknown significance to cardiomyocytes (21, 24).
The efficiency and safety of ns defibrillation in the termination of fibrillation have already been demonstrated in isolated rabbit hearts (15). Interestingly, in vitro stimulation of single cardiomyocytes was impossible without electric damage (21, 24). The potential explanation is that stimulation of the whole heart results from electroporation of single cells, which further stimulate the adjacent ones. A single stimulation of the entire heart would therefore cause undetectable lesions (25). Whether the excitation by nsPEF shocks is possible without electroporation and which mechanism prevails is determined by the membrane charging time constant τ (26). Since this parameter is proportional to cell size, cardiomyocytes, which are among the largest cells, are likely to have a higher time constant. Moreover, in the heart tissue, due to the lower extracellular conductivity and gap junctions between neighboring cells, the charging time constant may be substantially increased. Increasing τ shifts the balance from electroporation towards excitation, The larger time constant in situ may be a feature that enabled defibrillation of the heart without damage (26).
Electroporated cells could still be capable of restoring their membrane continuity, thus possibly avoiding cell death (25, 27–31). During CPR, the delay needed for membrane restoration can be fatal. This prompted our study to search for an agent that could mitigate electroporation-induced membrane damage of cardiomyocytes. One of the promising substances for this task is Poloxamer P188 (P188), a member of the copolymer group. P188 was shown to stabilize cellular membranes (32) and protect against electrical injury, increasing the survival of electroporated isolated rat skeletal muscle cells (33) and restoring the structural integrity of muscle tissue (34). Regarding cardiac injury during CPR, the intervention with Poloxamer 188 has been evaluated only as part of a bundle therapy with standard advanced life support (35). Therefore, its role in preventing cardiac electric damage has not been established.
Here, we hypothesized that P188 would alleviate electric insult and reduce cellular death of human cardiomyocytes when infused straight after nanosecond or microsecond electric shocks. Using a human cardiomyocyte cell line (AC16), we observed a strong protective effect of P188 after ns- and μsPEF shocks. We established different dynamics and mechanisms of cell killing by ns- and μsPEF shocks. Our data suggest that exposure to nanosecond pulses induced necrosis and microsecond pulses likely activated apoptosis of human cardiomyocytes. Our research thus implies the protective effect of P188 against both types of cell death.
AC16, a human cardiomyocyte cell line derived from adult ventricular heart tissues (36) was purchased from Millipore (Burlington, MA, USA). Cells were maintained at 37°C, 5% CO2 in DMEM/F-12 medium (Gibco, New York, NY, USA) containing 2 mM L-Glutamine (Mediatech Cellgro, Herndon, VA, USA), 12.5% FBS (Atlanta Biologicals, Norcross, GA, USA) and 1X Penicillin-Streptomycin Solution (GIBCO, Gaithersburg, MD, USA), and expanded to the final confluency of 80–90%. Complete medium formulation is available at: https://www.thermofisher.com/de/de/home/technical-resources/media-formulation.55.html.
We used nanofiber 24-multiwell plates (Nanofiber Solutions 9602, Darmstadt, Germany) to create monolayers of AC16 human cardiomyocytes. The plates were precoated with 20-μm thick layers of nanofiber polymers of 700-nm diameter, which structurally mimic the architecture of the cardiac extracellular matrix (37). We also coated the plates with human fibronectin (Gibco, Carlsbad, CA, USA) dissolved in PBS (Gibco, Carlsbad, CA, USA) at 2 μg/cm2 to minimize cell detachment. One day before exposure cells were seeded on the plates at 0.15 × 106 cells per well to achieve a homogenous monolayer.
Anet A8 3D printer (Shenzhen Anet Technology Co, China) was customized with an electrode holder to precisely position stimulation electrodes orthogonal to cell monolayers (38). Contact electrodes produced the electric field gradually decaying with distance from them, allowing the comparison of cell killing in a range of electric field strengths in a single sample. A 24-well plate was precisely fixed in a frame attached to the printer bed. The printer was programmed to deliver the electrodes to the center of the first well, remain there for 40 s for PEF exposure, then move to the second well, and so on. This interval was set to 40 s to maintain a constant interval between PEF treatments, agent administration, and image acquisition. Since the maximal dose of P188 applied in vitro in earlier studies was about 1% (34, 39, 40), we tested three different concentrations of P188: 0.2% (0.2 mM), 0.5% (0.6 mM) and 1% (1.2 mM).
The compound was diluted in the medium in a double concentration and the volume of added solution was equal to the volume of medium already in a well. The agent was administrated manually using pipette 10 s after PEF exposure. The same volume of medium without P188 was added as a vehicle control.
In the experiments, we used two different electrode arrays, an asymmetric one for μsPEF shocks and a symmetrical one for nsPEF shocks. Previous studies with trains of long pulses (≥100 μs) at lethal intensities reported intense bubble formation at the cathode (21, 38). Bubbles covering the electrode surface could change the electric field distribution during a pulse train. To avoid this effect, we made the cathode electrode larger, thereby decreasing the current density at its surface (Figure 1). Lower current density reduces gas evolution at the electrode (41). Using a larger electrode, we did not observe bubble formation.
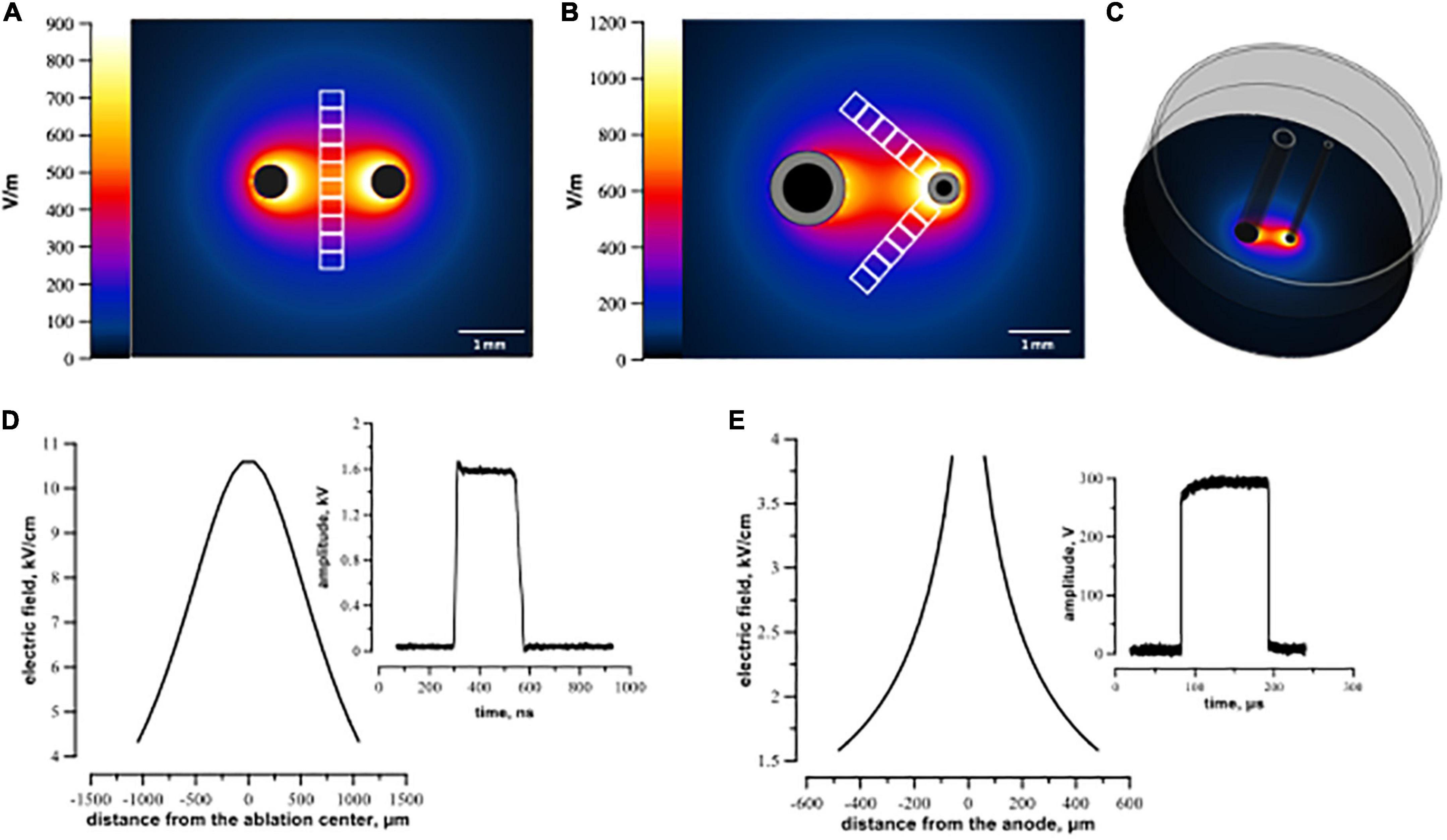
Figure 1. Electric field simulation for symmetric (A,D) and asymmetric (B,C,E) electrode arrays. Panels (A,B) visualize the calculated electric field distribution with 1 V applied between the electrodes. The direction in which the electric field thresholds were measured is indicated by white rectangles representing regions of interest (see text). Panel (C) illustrates a 3D configuration of the asymmetric electrode assembly orthogonal to the plastic well. Panels (D,E) show the electric field values for 300 V (100 μs pulse) and 1.74 kV (300 ns pulse) applied between electrodes. Distance from the ablation center (D,E) was measured along the regions of interest from the center of ablation (the middle of the line connecting centers of the electrodes) for symmetrical electrodes (D) and from the middle of the anode for the asymmetrical electrode (E).
The asymmetric electrodes used for μsPEF shocks were made of stainless steel hollow rods, 1.2 mm outer diameter for cathode and 0.5 mm for anode. They were placed 2.2 mm apart (center-to-center), parallel to each other and orthogonal to the cell monolayer.
Electroporation with nsPEF shocks was performed by applying electric pulses between two symmetrical cylindrical electrodes made of tungsten (500 μm diameter, 1.8 mm center-to-center distance). In preliminary experiments, electric field parameters for micro- and nanosecond pulses (number of pulses, frequency) were optimized to create comparable lesions. Trains of 200, 300-ns pulses at 1.74 kV were delivered at 10 Hz frequency from an EPULSUS-FPM4-7 pulse generator (Energy Pulse Systems, Lisbon, Portugal). Trains of 20, 100-μs pulses at 300 V were delivered at 1 Hz from a 6040 Universal Pulse Generator (BNC, San Rafael CA) triggered by a Model 577 Digital Delay/Pulse Generator (BNC). Pulse shape and amplitude were continuously monitored with a TDS 3052 oscilloscope (Tektronix, Beaverton, OR, USA).
For fluorescence imaging, 75 μM propidium iodide (PI) (Thermo Fisher Scientific, Waltham, MA, USA), an established membrane permeability marker dye, and 2.25 μM Hoechst-33342 (Thermo Fisher Scientific, Waltham, MA, USA), a membrane-permeant nucleic acid stain, were added 20 min prior to imaging, starting from 2 h after ns- and μsPEF shock treatments. PI is membrane impermeant and has weak fluorescence in the solution. However, once the cell membrane is permeabilized, Propidium enters the cell and its emission increases by binding to intracellular nucleic acids (21). Given that most membrane repairs are done within 10–15 min after exposure (31, 42–44), we considered that adding PI at 2 h after electroporation or later labeled only permanently permeabilized (i.e., dead) cells. All PI-positive cells were also positive for Hoechst, which made them look pink when both fluorescence channels were combined. PEF shocks were delivered in the culture medium. Each time point after PEF exposure was measured in a separate well to exclude possible cytotoxicity effect of Hoechst dye. We limited pulse treatments to 8 wells on each plate, so the plate remained outside the incubator no longer than 6 min.
For the detection of caspase activity at 1, 2, and 4h after exposure to PEF, the medium was replaced with 6 μM of Caspase 3/7 Green Detection Reagent CellEvent (Invitrogen) dissolved in 200 μl PBS with 5% FBS, mixed with PI and Hoechst and returned to the incubator for another 30 min before imaging. As a positive control, cells were incubated with 0.5 μM apoptotic inducer staurosporine (Sigma-Aldrich St. Louis, MO, USA) for 6 h.
We used wide-field fluorescence microscopy for measuring cell death caused by nano- and microsecond electric shocks (Figures 2A, B). Images were acquired using an IX83 microscope (Olympus America, Hamden, CT, USA) custom configured with an automated MS-2000 scanning stage (ASI, Eugene, OR, USA), X-Cite 110LED illuminator (Excelitas Technologies Corporation, Waltham, MA, USA), and an ORCA-Flash4 sCMOS camera (Hamamatsu, Shizuoka, Japan). A total of 9 images of adjacent regions (a 3 × 3 square) were taken using a 10x, 0.38 NA objective and merged into a single high-resolution image with cellSens software (Olympus America, Hamden, CT, USA). The samples were imaged using DAPI, Cy3 and FITC filter sets for Hoechst, PI, and caspase 3/7 signals, respectively.
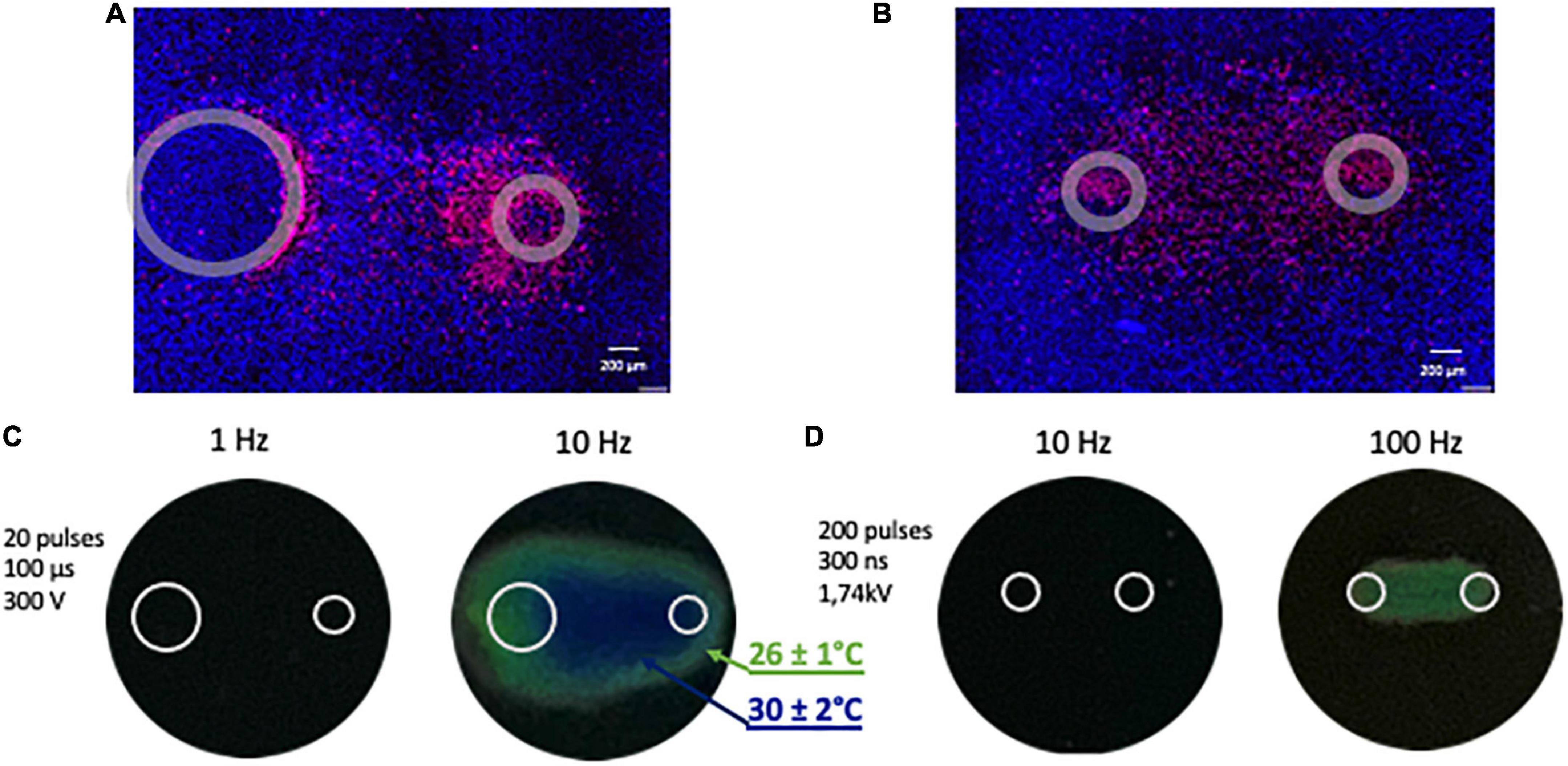
Figure 2. Representative images illustrating fluorescent staining of human cardiomyocyte monolayers and temperature monitoring. (A) Cells were subjected to 20 pulses (100 μs duration, 1 Hz, 300 V) and (B) 200 pulses (300 ns duration, 10 Hz, 1.74 kV). Panels (A,B) show merged signals from Hoechst-33342 (blue) and propidium iodide (red). Gray circles mark the footprints of the asymmetrical (A) and symmetrical (B) electrodes. (C,D) Monitoring the temperature rise following PEF (pulsed electric field) exposure with thermochromic liquid crystal sheets. Images were taken at the end of the exposure. Temperatures at the color transitions are labeled in the legend.
Electric field (E) distribution maps were generated by a numerical simulation with Sim4Life v3.2 software (Zurich Med Tech, Swiss). The electrodes were placed in the same position as in experiments described in Section “2.2 Electroporation of cell monolayers.” Separate simulations were performed for symmetrical (Figures 1A, D) and asymmetrical (Figures1B, C, E) electrode assemblies. The electric field values in Figure 1 were calculated in a plane 5 μm above the bottom of the well for 1 V applied between the electrodes. For further data analyses, the simulated electric field values were multiplied by the applied voltage, i.e., by 300 V for μs- and 1.74 kV for nsPEF shocks.
For cell death analysis after nsPEF shocks, we drew 10 rectangular 600 μm × 180 μm regions of interest (ROI) in the middle of the line connecting the centers of two symmetrical electrodes footprints (Figure 1A). Since we used asymmetrical electrodes for μsPEF shocks, we set two lines arising from the anode at an angle of 45° to the line connecting the centers of electrodes and designated along them 12 rectangular 600 μm × 180 μm ROIs (Figure 1B). In each ROI, cells stained with PI and cells stained with Hoechst only were counted with cellSens software. The percentage of dead cells was calculated as a ratio of PI-stained cells to all cells. For data processing, we matched the values of cell death from each ROI with the corresponding electric field intensity obtained from simulations (see Section “2.6 Electric field simulations”). The cell death ratio leveled off to a plateau (about 5%) away from the electrodes, indicating that in this area the effect of electric field was negligible. Note that the cells located in this peripheral region were subjected to the same conditions. Consequently, ROI covering those areas were regarded as sham control.
For Caspase detection we applied the same principle, however, for asymmetrical electrodes ROIs (600 μm × 180 μm) were drawn in the middle between electrodes, since in the nearest area of electrodes the majority of cells were PI-permeable before caspase activity could be detected. In each ROI, using cellSens software, we calculated the integrated fluorescence intensity (the sum of each pixel intensities).
To rule out the thermal effect during exposure to electric shocks, maximum heating from PEF treatments was assessed (38, 45; Figures 2C, D). R25C5B calibrated thermochromic liquid crystal sheets (LCR Hallcrest, Glenview, IL, USA) were positioned at the bottom of the well in contact with electrodes, and color changes were registered with a digital camera. All measurements were taken at a room temperature of 22–23°C. No color changes were observed when a train of 200, 300 ns pulses at 10 Hz or 20, 100 us pulses at 1 Hz was applied, which are the repetition rates used in our experiments. For positive control, we increased the frequency of ns- and μsPEF shocks to 100 and 10 Hz, respectively, and the color changed from black, implying no detectable heating effect, to green (26 ± 1°C) and blue (30 ± 2°C).
In all experiments, different exposure conditions (i.e., P188 concentration) were randomized. Exposure of one well was considered as a single experiment. Each graph represents the results of 3 to more than 6 experiments. Data were analyzed using a two-tailed t-test with Holm-Sidak correction for multiple comparisons. Normal distribution of data was confirmed using D’Agostino and Pearson normality test. Grapher 11 software (Golden Software, Golden, CO, USA) was used to prepare the graphs.
Cell death caused by pulsed electric fields can occur over extended time intervals (46, 47). Our first experiments were designed to determine the time interval after PEF treatment when the cell death reaches maximum. A train of 200, 300-ns pulses at 10 Hz, or of 20, 100-μs pulses at 1 Hz was applied to cell monolayers.
After nsPEF shocks (Figure 3) the percentage of dead cells was gradually increasing up to 8 h. For example, at 9.5 kV/cm, the percentage of dead cells rose from 68% ± 3 at 2 h to 86% ± 3 at 8 h, p < 0.05. At longer intervals, up to 24 h this percentage did not vary. Most of cell death occurred already at 2–4 h, and the further (later) increase in cell death was minor.
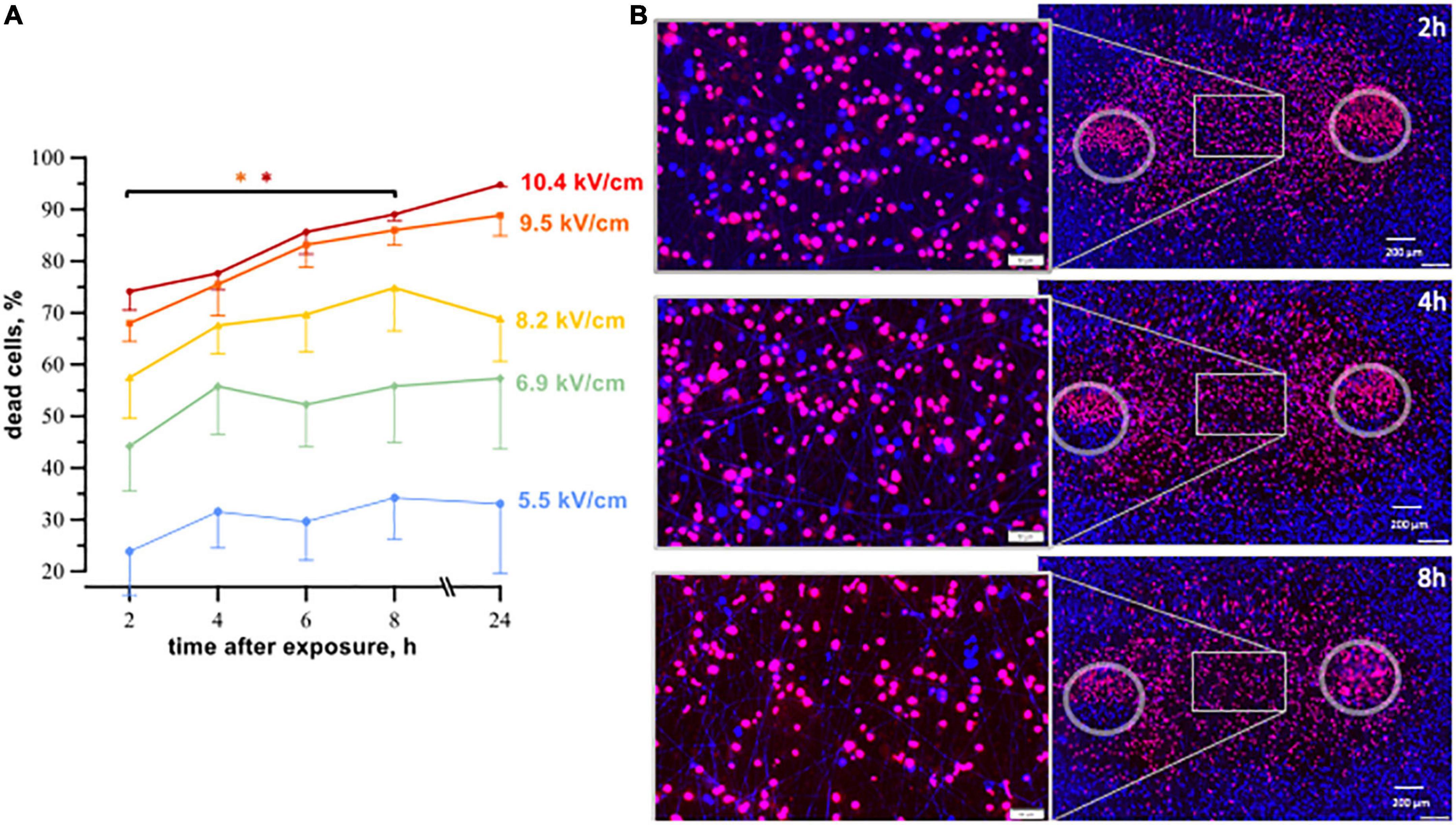
Figure 3. The time course of cell death in human cardiomyocytes subjected to nanosecond electric shocks (200 pulses at 10 Hz, 300 ns, 1.74 kV). (A) The percentage of dead cells in the regions with different electric field strength as a function of time after nsPEF shocks exposure, mean ± SEM, 5–6 independent experiments for each time interval, *p < 0.05 for the difference in the percent of dead cells between different time intervals. Cells stained with propidium (red dye) were considered dead, while Hoechst staining (blue) marked live cells. Panel (B) represents fluorescence images of human cardiomyocytes following nsPEF shocks applied to the cell monolayer. Gray circles mark the footprints of the symmetrical electrodes. Insets show magnifications of selected areas. The cell death reached its maximum at 8 h after exposure.
Cell death after exposure to μsPEF shocks peaked after an even longer interval of time (Figure 4). For most electric field strengths, cardiomyocyte death was gradually increasing to its maximum at 16–24 h. The only exception was with the strongest tested electric field of 3.6 kV/cm, which killed nearly 100% of cells already at 2 h. The highest (5-fold) increase in cell death with time was observed at 2.2 kV/cm, where the percentage of dead cells rose from 18% ± 7 at 2 h to 91% ± 4 at 24 h (p < 0.05). The apparent decline in cell death from 24 to 30 h after μsPEF shocks can be explained by the repopulation of the ablated area by viable cells and/or by the detachment of dead cells.
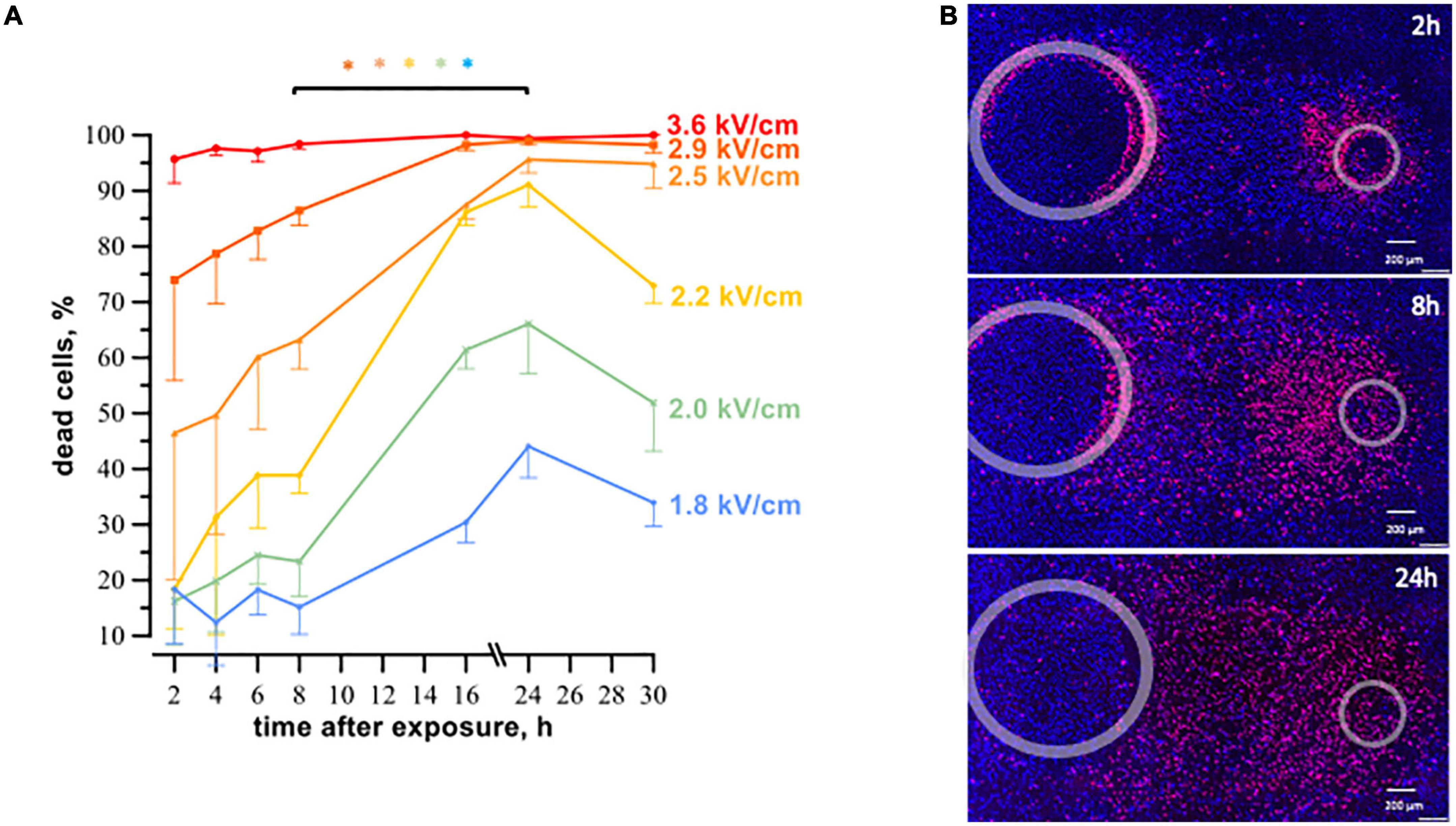
Figure 4. The time course of cell death in human cardiomyocyte subjected to microsecond electric shocks (20 pulses at 1 Hz, 100 μs, 300 V). (A) The plots display the percentage of dead cells in the regions with different electric field strength as a function of time after μsPEF shocks exposure, mean ± SEM, 3–5 independent experiments for each time interval, *p < 0.05 for the difference in the percent of dead cells between different time intervals. Cells were labeled with propidium and Hoechst at time intervals ranging from 2 to 30 h after μsPEF shocks see Figure 3 and text for more details. Panel (B) represents fluorescence images of human cardiomyocytes following μsPEF shocks. Gray circles mark the footprints of the asymmetrical electrodes. Note the gradual increase of cell death over time with a maximum at 24 h.
Considering that cardiomyocyte death in the experiments above did not occur immediately after PEF exposure, in the following experiments, cell death was measured 4- and 8 h after nsPEF shocks and 8- and 24 h after μsPEF shocks exposure.
P188 applied 10 s after nsPEF shocks decreased cell mortality in all tested concentrations, with more protective effect for stronger (more lethal) electric field exposures. Figure 5 presents cell death measurements at 4 and 8 h after nsPEF shocks exposures at different electric field strengths. The protective effect of P188 was both concentration- and electric field-dependent, becoming stronger and statistically significant for exposures to the electric field of 8.2 kV/cm or higher. At 8 h after exposure at 10.4 kV/cm, 1% P188 reduced cell death 1.7 times compared to the vehicle control (from 97% ± 2 to 58% ± 9, p < 0.05). Treatments with 0.5 and 0.2% P188 reduced cell killing 1.4 times (from 97% ± 2 to 68% ± 10) and 1.3 times (from 97% ± 2 to 75% ± 3), respectively (p < 0.05).
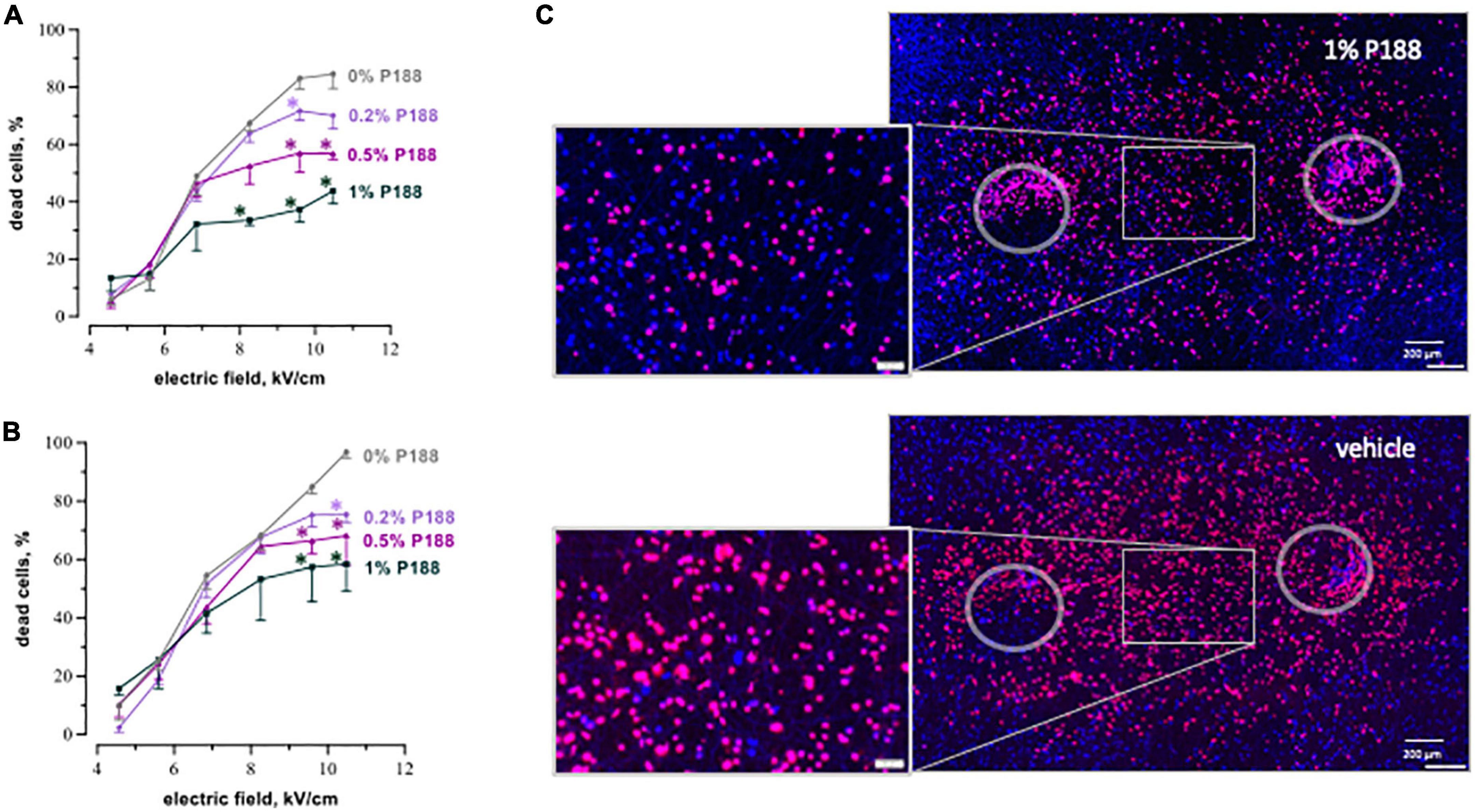
Figure 5. P188 reduces cardiac cell death after nsPEF shocks. The plots display the percent of dead cells at 4 h (A) and 8 h (B) after exposure to 200, 300-nsPEF shocks at 10 Hz. Poloxamer 188 (P188) was added at indicated concentrations (0% is vehicle control) 10 s after the end of nsPEF shocks exposure. Mean ± SEM, 3–5 independent experiments for each P188 concentration. *p < 0.05 for the difference between vehicle and P188 at the same electric field strength. Fluorescence images of cell monolayers (C) were taken at 8 h after exposure to nsPEF shocks followed in 10 s by either 1% P188 or vehicle. Selected areas are magnified in the insets. Note a profound reduction of cell death in samples treated with P188. See Figure 3 for more details.
P188 at all concentrations reduced or eliminated the increase of cell killing as the electric field was made stronger, resulting in a plateau in cell death curves (Figures 5A, B). This result could indicate that the additional cell death at these stronger electric fields was due to a different mechanism than with weaker electric fields, and that the cell death mechanism triggered by stronger fields was selectively sensitive to P188. A more in-depth analysis of this phenomenon was beyond the scope of this study but is considered for future work.
P188 protected cardiomyocytes against μsPEF shocks injuries as well (Figure 6). The protective effect became significant only at 24 h (Figure 6B). P188 at 0.2 and 0.5% reduced cell death 1.2–1.6 times (p < 0.05) at the electric field strengths from 2.0 to 2.5 kV/cm. There was no protective effect at either lower or higher field strengths, probably because of just modest lethality and damage too severe to repair, respectively. Unlike the data for 8 h after nsPEF shocks exposure, 1% P188 did not reduce cell death at 24 h after μsPEF shocks. The protection could be counterbalanced by cytotoxicity of P188 at this highest concentration (39).
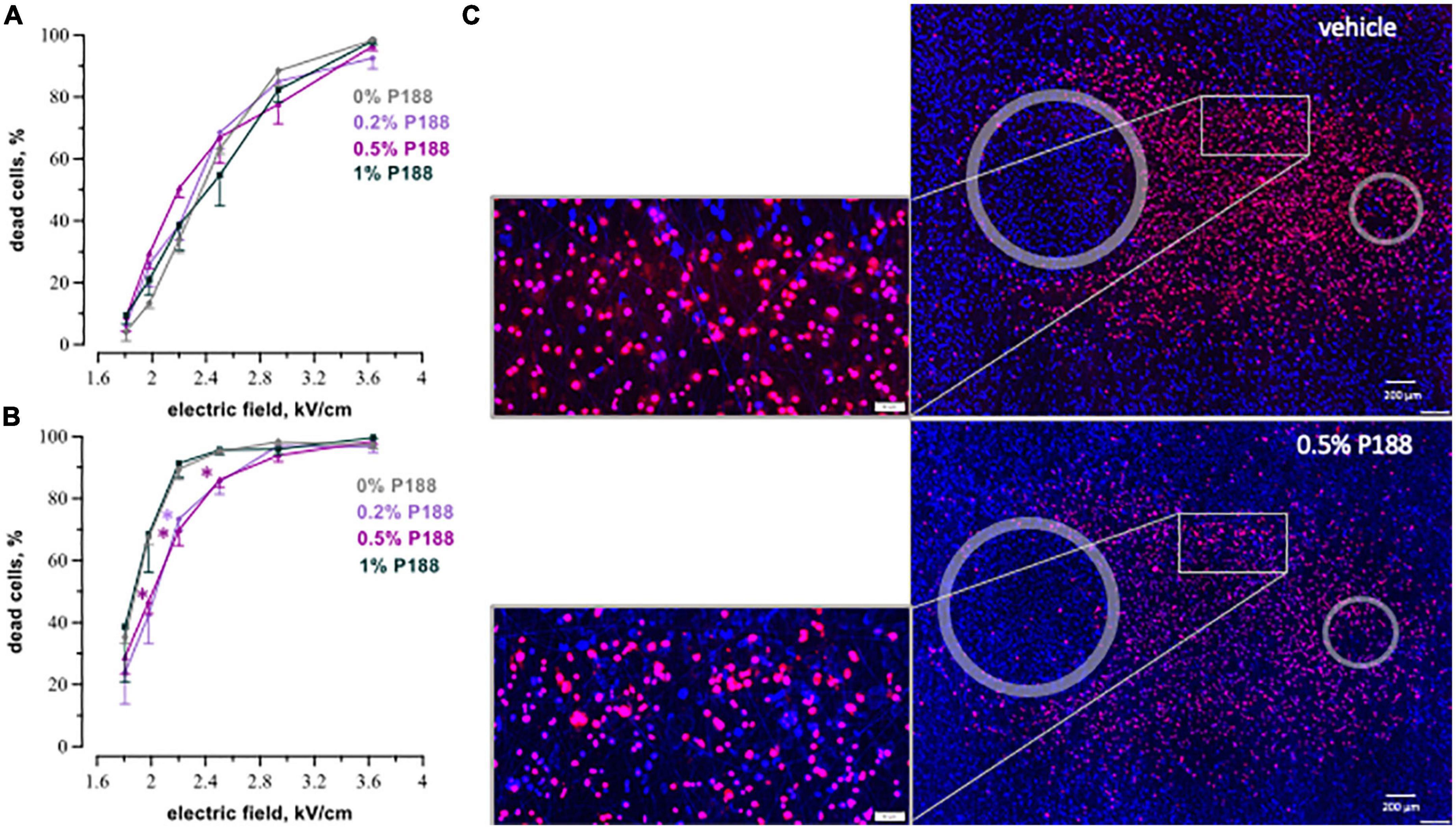
Figure 6. P188 protects cardiac cells against microsecond electric pulses. The plots display the percent of dead cells at 8 h (A) and 24 h (B) after exposure to 20, 100-μsPEF shocks at 1 Hz. P188 was added at indicated concentrations (0% is vehicle control) 10 s after the end of μsPEF exposure. Mean ± SEM, 3 experiments for each P188 concentration. *p < 0.05 for the difference between vehicle and P188 at the same electric field strength. The protective effect became significant at 24 h for P188 at 0.5% at the electric field strengths from 2.0 to 2.5 kV/cm and at 0.2% at 2.2 kV/cm. Panel (C) displays the example fluorescence images of cell monolayers at 24 h after exposure to μsPEF shocks.
Promising outcomes of P188 treatment together with the different timing of cell death after μs- and nsPEF shocks motivated further study to investigate the cell death mechanisms. A vast fraction of cardiomyocytes treated with nsPEF shocks died at intervals shorter than required to accomplish the apoptotic death pathway (48), whereas a 16–24 h delay to cell death after μsPEF shocks suggested apoptosis.
We measured caspase 3/7 activity for the detection of apoptosis. Results confirmed the apoptotic death pathway after μsPEF shocks (Figure 7). Cells exposed to 20, 100 μs pulses at 1 Hz displayed a significant increase in caspase 3/7 activity at 2 and 4 h compared to 1 h after pulse delivery (p < 0.05), with the highest fluorescence intensity at 2 h. This time interval of caspase detection corresponds to the early stage of apoptosis (49, 50). Caspase 3/7 activation following nsPEF shocks was marginal (Figures 2A, D). A large number of cells became PI-permeable before caspase activity could be detected.
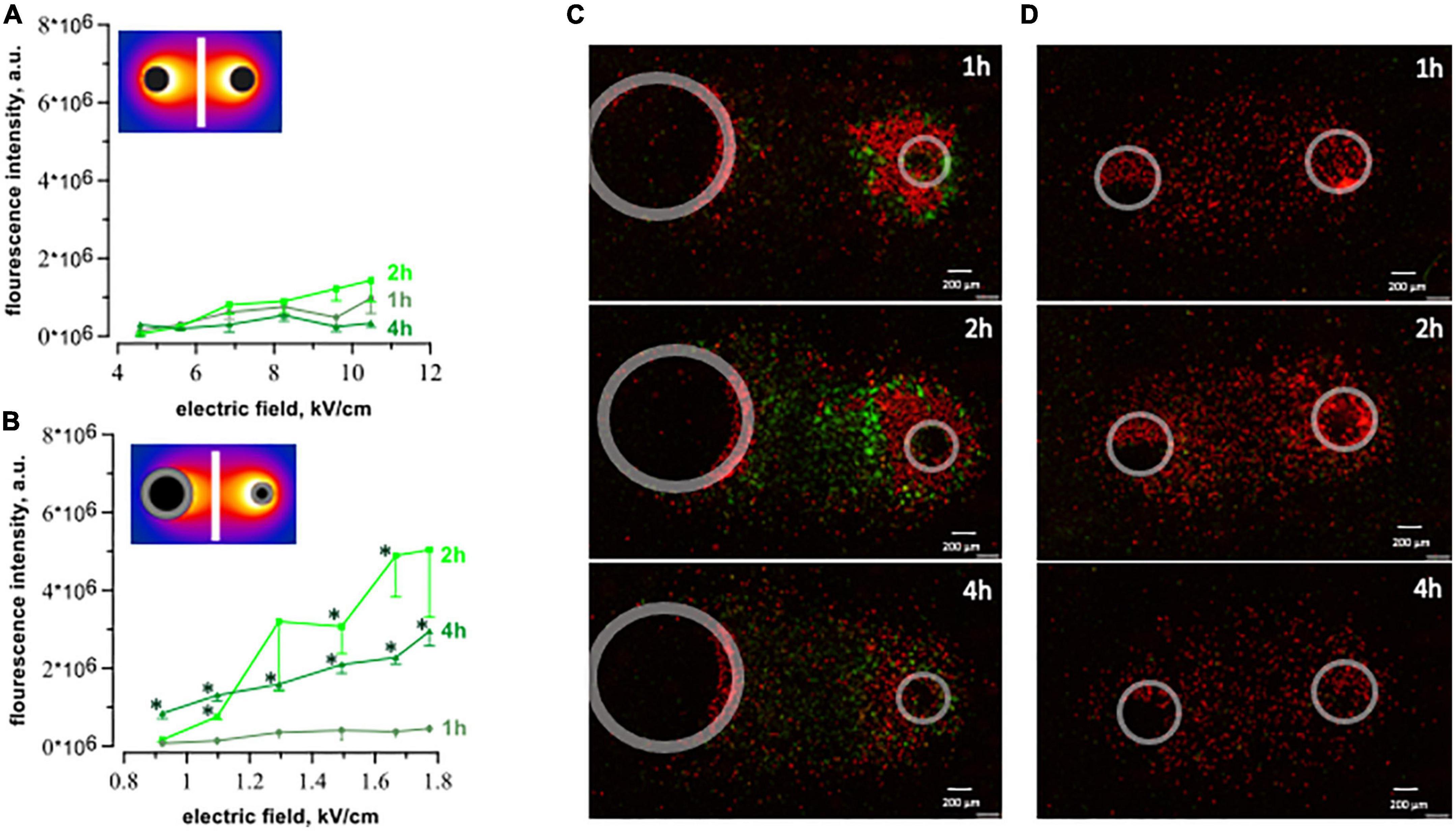
Figure 7. Caspase 3/7 activation after exposure to 20 pulses with 100 μs duration (1 Hz, 300 V) and 200 pulses with 300 ns duration (10 Hz, 1.74 kV). The plots display integrated fluorescence intensity of caspase 3/7 activity calculated for each region of interest (see text for details). Fluorescence was measured at 1-, 2-, and 4 h after exposure to ns- (A) and μsPEF shocks (B). Every single plot represents 3–5 independent experiments, *p < 0.05 for the difference between 1 h and 2- or 4 h. We observed an increase in caspase activity (green dye) following μsPEF shocks (C), but not after nsPEF shocks (D).
The novel methodology of our experiments enabled labor-effective, concurrent measurements of cardiac cell death for a range of the electric field strength in a single experiment. A customized 3D printer with mounted electrodes delivered electric pulses directly to the cell monolayer bypassing the process of cells trypsinization and repetitive pipetting, which might affect cell physiology (51, 52). This approach excluded the influence of enzymatic proteolysis and shear stress, providing more reliable results.
P188 has been shown to promote membrane repair in various cells and tissues (32, 34, 39, 53–57). The work of Lee et al. (34) was of particular interest to us, showing that P188 effectively prevents skeletal muscle membrane disruption caused by electroporation when used in concentrations > 0.05%. P188 was also shown to restore structural integrity of electropermeabilized muscle tissue in vivo, yielding a peak blood level of 0.8% (34). Conversely, studies by Quinlan et al. (58) found no effect of P188 in preventing exercise-induced cell membrane breakdown in dystrophin-deficient mdx skeletal muscle fibers. However, the authors suggested that the lack of effect might be attributed to the low concentration of P188 used in the study. Serum levels measured immediately before and during the experiment averaged 0.15% P188.
Our study produced several major findings. We demonstrated a different time course of ns- and μsPEF shocks -induced cardiomyocyte death. The majority of cells exposed to nanosecond shocks was dead within the first 2 h even at the lowest field strength. Whereas after microsecond shocks only a small fraction of cells subjected to the highest electric field died within 2 h after exposure. When exposed to nsPEF shocks, cell death reached its maximum at 8 h. After μsPEF shocks cell death was gradually increasing to its peak at 16–24 h. It is commonly known that electric pulses can impose a different mechanism of cell death depending on the cell type (59, 60). Here, we presented that AC16 human cardiomyocytes exposed to pulses of different duration can undergo either apoptotic or necrotic cell death. Cell death mechanism after electroporation of different cardiomyocytes cell lines remains an area for future investigation.
Several studies support our finding that 100 μs pulses are capable of triggering the apoptotic death pathway (61–64). Nevertheless, the majority of reports associate nsPEF shocks with apoptosis or other programmed forms of cell death (22, 46, 47, 59, 60, 65–68) showing caspase activation (66), cytochrome C release into the cytoplasm (65), externalization of phosphatidylserine (PS) (66), poly ADP-ribose polymerase (PARP) cleavage (46), and inter-nucleosomal DNA fragmentation (66). However, further studies found that the nsPEF shocks -induced externalization of PS was associated with electroporative membrane damages rather than activation of the programmed death pathway (17, 69). At the same time, nsPEF shocks -treated cells have been observed to swell right after exposure (18, 47, 70, 71), which is a morphological hallmark of necrosis. The prevailing cell death mechanism in cells exposed to PEF may be influenced by ionic balance, ATP supply, and the level of intracellular damage (46). In addition, the increase in the number of pulses or their amplitude and frequency can determine the cell death mechanism (60, 72).
The interplay between the induction of apoptosis and necrosis is not simple and depends not only on pulse parameters but, for example, on the presence of Ca2+ in the medium and on the colloid-osmotic composition of the medium (47, 73). That said, numerous studies reported that it is the distinguishing feature of nsPEF shocks that they induce apoptosis, as opposed to necrotic cell death from longer pulses (65, 66, 68). Our study is the first report that for a comparable membrane cell killing efficiency (tested in the wide range of the electric field strengths) the pathway to cell death is the opposite, namely necrosis with nsPEF shocks and apoptosis with μsPEF shocks. Comparing our data with other studies, we suspect that it is unlikely just the train duration, pulse frequency or peak electric field (which do not differ much from other studies) but the unique physiology of the cell line we studied and which makes it react differently. This unique observation may bring the important keys to understanding what are the physiological features responsible for one or another cell death pathway, and they will certainly be our focus for future work.
Furthermore, we demonstrated that the application of 0.2-, 0.5-, and 1% P188 significantly reduced the death of cardiomyocytes exposed to nsPEF shocks. Similar protection against microsecond PEF shocks was demonstrated with 0.5 and 0.2% P188.
According to several earlier reports, P188 can prevent necrosis (53, 56, 74) as well as apoptosis (75, 76). In our study P188 efficiently suppressed cardiomyocyte death imposed by nsPEF shocks. P188 was also efficient against μsPEF shocks, capable to activate caspase 3/7 and thus suggesting protection against apoptosis, although it showed no effect in higher 1% concentration. Presumably, this higher concentration after a long 24-h incubation became toxic in apoptotic cells. Based on our study, it appears appropriate to conduct subsequent in vivo measurements to test the efficacy of this agent for longer observation time.
P188 is the only copolymer molecule approved by the U.S. Food and Drug Administration as safe for oral or intravenous administration (77, 78) when dosed for up to 30 mg/kg/h for 72 h to reduce blood viscosity before transfusion (79). The expanding list of patents for P188 suggests that it may also be useful in preventing cell membrane damage in a variety of medical conditions. The authors of the patent Methods and Compositions of a Polymer (Poloxamer) for Repair of Electrical Injury suggested its possible application for victims of electrical trauma (80). In cardiology, it is expected to prevent cardiomyopathy (81) and chronic progressive heart failure (82). Our study highlighted the potential use of P188 to enhance cardiomyocyte recovery after defibrillation.
The beneficial effect of P188 in preventing cardiomyocyte electric injury can be attributed to its structure. Hydrophilic ends of P188 adsorb to membrane bilayers and interact with hydrocarbon tails of lipids. This is thought to reduce the size and number of electropores (83). P188 raises the electroporation threshold (84) and facilitates resealing of the cell membrane (34). The action of poloxamers may be selective to damaged portions of electroporated membranes. Namely, P188 would only interact with compromised bilayers, increasing locally reduced lipid packing density (85). Once the membrane is reconstituted, higher surface pressure will likely displace Poloxamer from the membrane (85).
Cardiac damage after defibrillation raises concerns about its contribution to the poor outcome of CPR. Sarcolemma injuries have been found after ms and ns electric shocks (12, 24). Since both types of pulses damage the membrane, reinforcement of membrane restoration might be necessary to prevent cell death. The clear benefits of P188 application after nanosecond pulses warrant further evaluation of ns-defibrillation with the perspective of improvement in post-cardiac arrest mortality.
An alternative solution would be to reduce the harm of electrical pulses in general. One of the promising modalities of nanosecond shocks for defibrillation without cardiac damage is MHz compression of nsPEF shocks. Applying high-rate bursts enabled cardiomyocytes excitation at lower voltage than with a single ns shock, giving a large safety gap (86).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
PS, OP, and AP conceived the study and analyzed and interpreted the data. AK and PS performed experiments. EG created software. PS performed electric field simulations and created figures. OP supervised the study. PS wrote the manuscript, with contributions and editing by all authors. All authors contributed to the article and approved the submitted version.
This study was supported by the Kosciuszko Foundation (to AK).
We thank Prof. Stephen J. Beebe for his expert opinion on the mechanism of cell death, Prof. Thomas Vernier for a fruitful discussion on the Poloxamer interactions, and Dr. Vitalii Kim for the instruction on performing electric field simulations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.1004024/full#supplementary-material
P188, Poloxamer 188; CPR, cardiopulmonary resuscitation; μ sPEF, microsecond pulsed electric field; nsPEF, nanosecond pulsed electric field; OHCA, out-of-hospital cardiac arrests; PS, phosphatidylserine.
1. Virani S, Alonso A, Aparicio H, Benjamin E, Bittencourt M, Callaway C, et al. Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation. (2021) 143:e254–743. doi: 10.1161/CIR.0000000000000950
2. Tsao C, Aday A, Almarzooq Z, Alonso A, Beaton A, Bittencourt M, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. (2022) 145:e153–639.
3. Stoddard M, Labovitz A, Stevens L, Buckingham T, Redd R, Kennedy H. Effects of electrophysiologic studies resulting in electrical countershock or burst pacing on left ventricular systolic and diastolic function. Am Heart J. (1988) 116:364–70.
4. Niebauer M, Babbs C, Geddes A, Bourland J. Efficacy and safety of defibrillation with rectangular waves of 2- to 20-milliseconds duration. Crit Care Med. (1983) 11:95–8. doi: 10.1097/00003246-198302000-00008
5. Dosdall DJ, Fast VG, Ideker RE. Mechanisms of defibrillation. Annu Rev Biomed Eng. (2010) 12:233.
6. Prado L, Goulart J, Zoccoler M, Oliveira P. Ventricular myocyte injury by high-intensity electric field: effect of pulse duration. Gen Physiol Biophys. (2016) 35:121–30. doi: 10.4149/gpb_2015047
7. Berg M, Banville I, Chapman F, Walker R, Gaballa M, Hilwig R, et al. Attenuating the defibrillation dosage decreases postresuscitation myocardial dysfunction in a swine model of pediatric ventricular fibrillation Marc. Pediatr Crit Care Med. (2008) 9:429–34. doi: 10.1097/PCC.0b013e318172e9f8
10. Wang Y, Efimov I, Cheng Y. Electroporation induced by internal defibrillation shock with and without recovery in intact rabbit hearts. Am J Physiol Heart Circ Physiol. (2012) 303:439–49. doi: 10.1152/ajpheart.01121.2011
11. Dosdall D, Fast V, Ideker R. Mechanisms of defibrillation. Annu Rev Biomed Eng. (2010) 12:233–58.
12. Nikolski V, Sambelashvili A, Krinsky V, Efimov I. Effects of electroporation on optically recorded transmembrane potential responses to high-intensity electrical shocks. Am J Physiol Heart Circ Physiol. (2004) 286:412–8. doi: 10.1152/ajpheart.00689.2003
13. Kodama I, Shibata N, Sakuma I, Mitsui K, Iida M, Suzuki R, et al. Aftereffects of high-intensity DC stimulation on the electromechanical performance of ventricular muscle. Am J Physiol. (1994) 267:H248–58. doi: 10.1152/ajpheart.1994.267.1.H248
14. Al-Khadra A, Nikolski V, Efimov I. The role of electroporation in defibrillation. Circ Res. (2000) 87:797–804.
15. Varghese F, Neuber J, Xie F, Philpott J, Pakhomov A, Zemlin C. Low-energy defibrillation with nanosecond electric shocks. Cardiovasc Res. (2017) 113:1789–97. doi: 10.1093/cvr/cvx172
16. Pakhomov A, Gianulis E, Vernier P, Semenov I, Xiao S, Pakhomova O. Multiple nanosecond electric pulses increase the number but not the size of long-lived nanopores in the cell membrane. Biochim Biophys Acta. (2015) 1848:958–66.
17. Pakhomov A, Bowman A, Ibey B, Andre F, Pakhomova O, Schoenbach K. Lipid nanopores can form a stable, ion channel-like conduction pathway in cell membrane. Biochem Biophys Res Commun. (2009) 385:181–6. doi: 10.1016/j.bbrc.2009.05.035
18. Nesin O, Pakhomova O, Xiao S, Pakhomov A. Manipulation of cell volume and membrane pore comparison following single cell permeabilization with 60- and 600-ns electric pulses. Biochim Biophys Acta. (2011) 1808:792–801. doi: 10.1016/j.bbamem.2010.12.012
19. Semenov I, Zemlin C, Pakhomova O, Xiao S, Pakhomov A. Diffuse, non-polar electropermeabilization and reduced propidium uptake distinguish the effect of nanosecond electric pulses. Biochim Biophys Acta. (2015) 1848:2118–25. doi: 10.1016/j.bbamem.2015.06.018
20. Gowrishankar T, Weaver J. Electrical behavior and pore accumulation in a multicellular model for conventional and supra-electroporation. Biochem Biophys Res Commun. (2006) 349:643–53. doi: 10.1016/j.bbrc.2006.08.097
21. Semenov I, Grigoryev S, Neuber J, Zemlin C, Pakhomova O, Casciola M, et al. Excitation and injury of adult ventricular cardiomyocytes by nano- to millisecond electric shocks. Sci Rep. (2018) 8:1–12. doi: 10.1038/s41598-018-26521-2
22. Schoenbach K, Hargrave B, Joshi R, Kolb J, Nuccitelli R, Osgood C, et al. Bioelectric effects of intense nanosecond pulses. IEEE Trans Dielect Electr Insul. (2007) 14:1088–109.
23. Son R, Smith K, Gowrishankar T, Vernier P, Weaver J. Basic features of a cell electroporation model: illustrative behavior for two very different pulses. J Membr Biol. (2014) 247:1209–28. doi: 10.1007/s00232-014-9699-z
24. Azarov J, Semenov I, Casciola M, Pakhomov A. Excitation of murine cardiac myocytes by nanosecond pulsed electric field. J Cardiovasc Electrophysiol. (2019) 3:392–401.
25. Neuber J, Varghese F, Pakhomov A, Zemlin C. Using nanosecond shocks for cardiac defibrillation. Bioelectricity. (2019) 1:240–6.
26. Pakhomov A, Pakhomova O. The interplay of excitation and electroporation in nanosecond pulse stimulation. Bioelectrochemistry. (2020) 136:1–8. doi: 10.1016/j.bioelechem.2020.107598
27. Saulis G. Pore disappearance in a cell after electroporation: theoretical simulation and comparison with experiments. Biophys J. (1997) 73:1299–309. doi: 10.1016/S0006-3495(97)78163-3
28. Demiryurek Y, Nickaeen M, Zheng M, Yu M, Zahn J, Shreiber D, et al. Transport, resealing, and re-poration dynamics of two-pulse electroporation-mediated molecular delivery. Biochim Biophys Acta. (2015) 1848:1706–14. doi: 10.1016/j.bbamem.2015.04.007
29. Kotnik T, Kramar P, Pucihar G, Miklavčič D, Tarek M. Cell membrane electroporation–Part 1: the phenomenon. IEEE Electr Insul Mag. (2012) 28:14–23.
30. Batista Napotnik T, Polajžer T, Miklavčič D. Cell death due to electroporation–A review. Bioelectrochemistry. (2021) 141:107871. doi: 10.1016/j.bioelechem.2021.107871
31. Bhattacharya S, Silkunas M, Gudvangen E, Mangalanathan U, Pakhomova O, Pakhomov A. Ca2+ dependence and kinetics of cell membrane repair after electropermeabilization. Biochim Biophys Acta. (2022) 1864:183823. doi: 10.1016/j.bbamem.2021.183823
32. Kwiatkowski T, Rose A, Jung R, Capati A, Hallak D, Yan R, et al. Multiple poloxamers increase plasma membrane repair capacity in muscle and nonmuscle cells. Am J Physiol Cell Physiol. (2019) 318:C253–62. doi: 10.1152/ajpcell.00321.2019
33. Collins J, Despa F, Lee R. Structural and functional recovery of electropermeabilized skeletal muscle in-vivo after treatment with surfactant poloxamer 188. Biochim Biophys Acta. (2007) 1768:1238–46. doi: 10.1016/j.bbamem.2007.01.012
34. Lee R, River L, Pan F, Ji L, Wollmann R. Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc Natl Acad Sci USA. (1992) 89:4524–8.
35. Bartos J, Matsuura T, Sarraf M, Youngquist S, McKnite S, Rees J, et al. Bundled postconditioning therapies improve hemodynamics and neurologic recovery after 17 min of untreated cardiac arrest. Resuscitation. (2015) 87:7–13. doi: 10.1016/j.resuscitation.2014.10.019
36. Davidson M, Nesti C, Palenzuela L, Walker W, Hernandez E, Protas L, et al. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. (2005) 39:133–47.
37. Ding M, Andersson H, Martinsson S, Sabirsh A, Jonebring A, Wang Q, et al. Aligned nanofiber scaffolds improve functionality of cardiomyocytes differentiated from human induced pluripotent stem cell-derived cardiac progenitor cells. Sci Rep. (2020) 10:1–14. doi: 10.1038/s41598-020-70547-4
38. Gudvangen E, Kim V, Novickij V, Battista F, Pakhomov A. Electroporation and cell killing by milli–to nanosecond pulses and avoiding neuromuscular stimulation in cancer ablation. Sci Rep. (2022) 12:1763. doi: 10.1038/s41598-022-04868-x
39. Gu J, Ge J, Li M, Xu H, Wu F, Qin Z. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS One. (2013) 8:e61641. doi: 10.1371/journal.pone.0061641
40. Toth K, Wenby R, Meiselman H. Inhibition of polymer-induced red blood cell aggregation by poloxamer 188. Biorheology. (2000) 37:301–12.
41. Vogt H. The actual current density of gas-evolving electrodes–notes on the bubble coverage. Electrochim Acta. (2012) 78:183–7.
42. Pakhomov A, Kolb J, White J, Joshi R, Xiao S, Schoenbach K. Long-lasting plasma membrane permeabilization in mammalian cells by nanosecond pulsed electric field (nsPEF). Bioelectromagnetics. (2007) 28:655–63.
43. Bier M, Hammer S, Canaday D, Lee R. Kinetics of sealing for transient electropores in isolated mammalian skeletal muscle cells. Bioelectromagnetics. (1999) 20:194–201.
44. Saulis G, Venslauskas M, Naktinis J. Kinetics of pore resealing in cell membranes after electroporation. J Electroanal Chem. (1991) 321:1–13.
45. Pakhomov A, Gudvangen E, Xiao S, Semenov I. Interference targeting of bipolar nanosecond electric pulses for spatially focused electroporation, electrostimulation, and tissue ablation. Bioelectrochemistry. (2021) 141:107876. doi: 10.1016/j.bioelechem.2021.107876
46. Ibey B, Pakhomov A, Gregory B, Khorokhorina V, Roth C, Rassokhin M, et al. Selective cytotoxicity of intense nanosecond-duration electric pulses in mammalian cells. Biochim Biophys Acta. (2010) 1800:1210–9. doi: 10.1016/j.bbagen.2010.07.008
47. Pakhomova O, Gregory B, Semenov I, Pakhomov A. Two modes of cell death caused by exposure to nanosecond pulsed electric field. PLoS One. (2013) 8:e70278. doi: 10.1371/journal.pone.0070278
49. Tyas L, Brophy V, Pope A, Rivett A, Tavaré J. Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Rep. (2000) 1:266–70. doi: 10.1093/embo-reports/kvd050
50. Hu X, Li Z, Lin R, Shan J, Yu Q, Wang R, et al. Guidelines for regulated cell death assays: a systematic summary, a categorical comparison, a prospective. Front Cell Dev Biol. (2021) 9:634690. doi: 10.3389/fcell.2021.634690
51. Huang H, Hsing H, Lai T, Chen Y, Lee T, Chan H, et al. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci. (2010) 17:1–10. doi: 10.1186/1423-0127-17-36
52. Wiegmann L, de Zélicourt D, Speer O, Muller A, Goede J, Seifert B, et al. Influence of standard laboratory procedures on measures of erythrocyte damage. Front Physiol. (2017) 8:731. doi: 10.3389/fphys.2017.00731
53. Greenebaum B, Blossfield K, Hannig J, Carrillo C, Beckett M, Weichselbaum R, et al. Poloxamer 188 prevents acute necrosis of adult skeletal muscle cells following high-dose irradiation. Burns. (2004) 30:539–47.
54. Yasuda S, Townsend D, Michele D, Favre E, Day S, Metzger J. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. (2005) 436:1025–9.
55. Murphy A, McCormack M, Bichara D, Nguyen J, Randolph M, Watkins M, et al. Poloxamer 188 protects against ischemia-reperfusion injury in a murine hind-limb model. Plast Reconstr Surg. (2010) 125:1651–60. doi: 10.1097/PRS.0b013e3181ccdbef
56. Plataki M, Lee Y, Rasmussen D, Hubmayr R. Poloxamer 188 facilitates the repair of alveolus resident cells in ventilator-injured lungs. Am J Respir Crit Care Med. (2011) 184:939–47. doi: 10.1164/rccm.201104-0647OC
57. Spurney C, Guerron A, Yu Q, Sali A, van der Meulen J, Hoffman E, et al. Membrane sealant poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord. (2011) 11:20. doi: 10.1186/1471-2261-11-20
58. Quinlan J, Wong B, Niemeier R, McCullough A, Levin L, Emanuele M. Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul Disord. (2006) 16:855–64. doi: 10.1016/j.nmd.2006.09.016
59. Hall E, Schoenbach K, Beebe S. Nanosecond pulsed electric fields (nsPEF) induce direct electric field effects and biological effects on human colon carcinoma cells. DNA Cell Biol. (2005) 24:283–91. doi: 10.1089/dna.2005.24.283
60. Thomas Vernier P, Li A, Marcu L, Craft C, Gundersen M. Ultrashort pulsed electric fields induce membrane phospholipid translocation and caspase activation: differential sensitivities of Jurkat t lymphoblasts and rat glioma C6 cells. IEEE Trans Dielectr Electr Insul. (2003) 10:795–809.
61. Chai W, Zhang W, Wei Z, Xu Y, Shi J, Luo X, et al. Irreversible electroporation of the uterine cervix in a rabbit model. Biomed Microdevices. (2017) 19:1–8. doi: 10.1007/s10544-017-0248-2
62. Kim H, Sung C, Baik K, Moon K, Kim H, Yi J, et al. Changes of apoptosis in tumor tissues with time after irreversible electroporation. Biochem Biophys Res Commun. (2013) 435:651–6.
63. Zhang W, Chai W, Zeng J, Chen J, Bi L, Niu L. Irreversible electroporation for the treatment of rabbit VX2 breast cancer. Biomed Microdevices. (2017) 19:1–12.
64. Kiełbik A, Szlasa W, Michel O, Szewczyk A, Tarek M, Saczko J, et al. In vitro study of calcium microsecond electroporation of prostate adenocarcinoma cells. Molecules. (2020) 25:5406. doi: 10.3390/molecules25225406
65. Beebe S, Fox P, Rec L, Willis E, Schoenbach K. Nanosecond, high-intensity pulsed electric fields induce apoptosis in human cells. FASEB J. (2003) 17:1493–5.
66. Beebe S, Fox P, Rec L, Somers K, Stark R, Schoenbach K. Nanosecond pulsed electric field (nsPEF) effects on cells and tissues: apoptosis induction and tumor growth inhibition. Pulsed Power Plasma Sci. (2002) 30:286–92.
67. Morotomi-Yano K, Akiyama H, Yano K. Different involvement of extracellular calcium in two modes of cell death induced by nanosecond pulsed electric fields. Arch Biochem Biophys. (2014) 555–556:47–54. doi: 10.1016/j.abb.2014.05.020
68. Beebe S, Sain N, Ren W. Induction of cell death mechanisms and apoptosis by nanosecond pulsed electric fields (nsPEFs). Cells. (2013) 2:136–62.
69. Vernier P, Sun Y, Marcu L, Craft C, Gundersen M. Nanoelectropulse-induced phosphatidylserine translocation. Biophys J. (2004) 86:4040–8.
70. Pakhomov A, Shevin R, White J, Kolb J, Pakhomova O, Joshi R, et al. Membrane permeabilization and cell damage by ultrashort electric field shocks. Arch Biochem Biophys. (2007) 465:109–18. doi: 10.1016/j.abb.2007.05.003
71. Deng J, Schoenbach K, Buescher E, Hair P, Fox P, Beebe S. The effects of intense submicrosecond electrical pulses on cells. Biophys J. (2003) 84:2709–14.
72. Kiełbik A, Szlasa W, Novickij V, Szewczyk A, Maciejewska M, Saczko J, et al. Effects of high-frequency nanosecond pulses on prostate cancer cells. Sci Rep. (2021) 11:1–10. doi: 10.1038/s41598-021-95180-7
73. Pakhomova O, Gregory B, Semenov I, Pakhomov A. Calcium-mediated pore expansion and cell death following nanoelectroporation. Biochim Biophys Acta. (2014) 1838:2547–54. doi: 10.1016/j.bbamem.2014.06.015
74. Phillips D, Haut R. The use of a non-ionic surfactant (P188) to save chondrocytes from necrosis following impact loading of chondral explants. J Orthop Res. (2004) 22:1135–42. doi: 10.1016/j.orthres.2004.02.002
75. Tang S, Liao W, Pao H, Hsu C, Wu S, Huang K, et al. Poloxamer 188 attenuates ischemia-reperfusion-induced lung injury by maintaining cell membrane integrity and inhibiting multiple signaling pathways. Front Pharmacol. (2021) 12:650573. doi: 10.3389/fphar.2021.650573
76. Serbest G, Horwitz J, Jost M, Barbee K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. (2005) 20:308–10.
77. Jewell R, Khor S, Kisor D, Lacroix K, Wargin W. Pharmacokinetics of RheothRx injection in healthy male volunteers. J Pharm Sci. (1997) 86:808–12. doi: 10.1021/js960491e
78. Grindel J, Jaworski T, Piraner O, Emanuele R, Balasubramanian M. Distribution, metabolism, and excretion of a novel surface-active agent, purified poloxamer 188, in rats, dogs, and humans. J Pharm Sci. (2002) 90:1936–47. doi: 10.1002/jps.10190
79. Adams-Graves P, Kedar A, Koshy M, Steinberg M, Veith R, Ward D, et al. RheothRx (Poloxamer 188) injection for the acute painful episode of sickle cell disease: a pilot study. Blood. (1997) 90:2041–6.
80. Lee R. Methods and Composition of a Polymer (Poloxamer) for Repair of Electrical Injury. Washington, DC: U.S. Patent and Trademark Office (1997).
81. Metzger J, Townsend D, Yasuda S, Michele D. Compositions and Methods for Treating and Preventing Cardiomyopathy and Heart Disease. Washington, DC: U.S. Patent and Trademark Office (2007). p. 221–5.
82. Markham BE. Treatment of Chronic Progressive Heart Failure. Novi, MI: IFI CLAIMS Patent Services (2009).
83. Tung L, Troiano G, Sharma V, Raphael R, Stebe K. Changes in electroporation thresholds of lipid membranes by surfactants and peptides. Ann N Y Acad Sci. (1999) 888:249–65. doi: 10.1111/j.1749-6632.1999.tb07960.x
84. Sharma V, Stebe K, Murphy J, Tung L. Poloxamer 188 decreases susceptibility of artificial lipid membranes to electroporation. Biophys J. (1996) 71:3229–41. doi: 10.1016/S0006-3495(96)79516-4
85. Maskarinec S, Hannig J, Lee R, Lee K. Direct observation of poloxamer 188 insertion into lipid monolayers. Biophys J. (2002) 82:1453–9. doi: 10.1016/S0006-3495(02)75499-4
Keywords: defibrillation, Poloxamer 188, cardiomyocytes, membrane repair, apoptosis, microsecond pulsed electric field (μsPEF), nanosecond pulsed electric field (nsPEF)
Citation: Sowa PW, Kiełbik AS, Pakhomov AG, Gudvangen E, Mangalanathan U, Adams V and Pakhomova ON (2022) How to alleviate cardiac injury from electric shocks at the cellular level. Front. Cardiovasc. Med. 9:1004024. doi: 10.3389/fcvm.2022.1004024
Received: 26 July 2022; Accepted: 08 December 2022;
Published: 22 December 2022.
Edited by:
Hari S. Sharma, Uppsala University, SwedenReviewed by:
Rosana A. Bassani, State University of Campinas, BrazilCopyright © 2022 Sowa, Kiełbik, Pakhomov, Gudvangen, Mangalanathan, Adams and Pakhomova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela W. Sowa, ✉ cGFtZWxhLnNvd2EuMzEucHNAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.