 Constanze Hoebinger
Constanze Hoebinger Dragana Rajcic
Dragana Rajcic Tim Hendrikx
Tim Hendrikx
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 10 January 2022
Sec. Lipids in Cardiovascular Disease
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.824481
This article is part of the Research Topic The Roles of Lipids in Immunometabolism: The crosstalk between lipid metabolisms and inflammation View all 17 articles
The prevalence of non-alcoholic fatty liver disease (NAFLD), ranging from simple steatosis to inflammatory steatohepatitis (NASH) and cirrhosis, continues to rise, making it one of the major chronic liver diseases and indications for liver transplantation worldwide. The pathological processes underlying NAFLD not only affect the liver but are also likely to have systemic effects. In fact, growing evidence indicates that patients with NAFLD are at increased risk for developing atherosclerosis. Indeed, cardiovascular complications are the leading cause of mortality in NAFLD patients. Here, we aim to address common pathophysiological molecular pathways involved in chronic fatty liver disease and atherosclerosis. In particular, we focus on the role of oxidized lipids and the formation of oxidation-specific epitopes, which are important targets of host immunity. Acting as metabolic danger signals, they drive pro-inflammatory processes and thus contribute to disease progression. Finally, we summarize encouraging studies indicating that oxidized lipids are promising immunological targets to improve intervention strategies for NAFLD and potentially limit the risk of developing atherosclerosis.
A sedentary lifestyle and excess caloric intake combined with reduced energy expenditure not only lead to overweight and obesity but also an increase in the prevalence of metabolic syndrome and various lipid-mediated diseases such as non-alcoholic fatty liver disease (NAFLD) (1, 2). As the liver is the most metabolic organ in the human body, increased circulatory lipid levels result in their accumulation in the liver, known as steatosis (fat accumulation in >5% of hepatocytes) (3, 4). While simple steatosis is still reversible by lifestyle modifications, the NAFLD spectrum also includes more progressive non-alcoholic steatohepatitis (NASH), characterized by inflammation, hepatocyte damage, and fibrosis (5). Importantly, the presence of NASH further increases the risk of developing an end-stage liver disease such as cirrhosis or hepatocellular carcinoma, ultimately requiring liver transplantation (4). By affecting ~25% of individuals, both adults and children, NAFLD has become the leading cause of chronic liver disease worldwide (6, 7). Considering that the prevalence is expected to rise further (7), NAFLD can be regarded as significant health and economic burden worldwide, resulting in a reduced quality of life (8).
While NAFLD primarily affects liver structure and function, leading to morbidity and mortality from liver failure, cardiovascular disease (CVD) is the most common cause of death in early NAFLD patients (9, 10). Moreover, there is increasing evidence that NAFLD is a risk factor for developing cardiovascular complications such as atherosclerosis (11). Atherosclerosis can be broadly described as a progressive chronic inflammatory disease of the large and medium-sized arteries that share metabolic patterns with NAFLD (12, 13). More precisely, atherosclerosis is characterized by the thickening and hardening of the arterial walls, mainly caused by developing complex lesions and accumulation of lipids and fibrous elements known as atheromatous plaques narrowing the arterial lumen (14, 15). Subsequently, plaque rupture and thrombosis can lead to acute clinical complications, such as heart attacks, strokes, unstable angina, arrhythmia, or sudden cardiac death, making CVD the primary cause of morbidity and mortality in Western countries (16).
Since atherosclerosis and NAFLD co-occur in patients with the metabolic syndrome, obesity, type 2 diabetes mellitus and insulin resistance, it is difficult to decipher the exact cause-effect relationship that leads to an increased risk of CVD in patients with NAFLD (17). Recent studies in children suffering from fatty liver disease support that NAFLD may cause CVD (18–20). Nevertheless, while growing evidence indicates that NAFLD can be considered a risk factor for atherosclerosis, the underlying disease mechanisms by which NAFLD contributes to CVD are not entirely understood. In this review, we will focus on the immunomodulatory effects of oxidized lipids and provide evidence for their involvement as common metabolic triggers for disease progression during NAFLD and atherosclerosis. In addition, we will discuss how targeting oxidized lipids via immunization strategies can be explored to improve interventions and potentially prevent the risk for CVD.
The defining hallmark of NAFLD is the accumulation of lipids containing triglycerides, cholesterol esters, and other lipid species in the liver. The increased hepatic triglyceride content, which determines the histological appearance of a steatotic liver, is a consequence of increased calorie intake, enhanced free fatty acids (FFA) influx from lipolysis of peripheral adipose tissue, elevated triglyceride synthesis by hepatic de novo lipogenesis, and reduced lipid export from the liver via very-low-density lipoprotein (VLDL) particles (21, 22). In parallel with deregulated hepatic lipid metabolism, NAFLD is associated with systemic dyslipidemia, as manifested by elevated triglyceride and cholesterol levels, lowered high-density lipoproteins (HDL), and increased low-density lipoprotein (LDL) particles in circulation (23–25). Similarly, elevated cholesterol, high LDL, and low HDL serum levels are described as risk factors for the onset of CVD (26), where the accumulation of LDL particles in arterial walls is a crucial process in the development of atherosclerosis (27). Thus, despite the strong correlation with dyslipidemia, it has become clear that disturbances in lipid metabolism and increased LDL levels cannot merely explain the local pro-inflammatory tissue environment, of which its presence seems to be a crucial factor enhancing disease progression.
Dyslipidemia that goes beyond the body's coping mechanisms can lead to lipotoxicity, an essential mechanism associated with NAFLD and atherosclerosis (28, 29). During NAFLD, lipotoxicity occurs when the massive influx of FFAs into hepatocytes peaks at a point where the liver can no longer use or store the FFAs or export them as triglycerides. Subsequently, a chain of intracellular responses is activated, leading to lipotoxic stress in mitochondria and the endoplasmic reticulum, ultimately resulting in hepatocyte cell death and the release of pro-inflammatory cytokines and extracellular vesicles (28, 30–33). In turn, this leads to the activation of resident Kupffer cells and the recruitment of infiltrating monocytes and neutrophils to the liver, which contribute to inflammation via the release of cytokines, chemokines, nitric oxide, and reactive oxygen species (ROS) (4). In a similar process, lipid retention in atherosclerotic plaques induces local inflammation characterized by the influx of circulating monocytes that differentiate into macrophages that release pro-inflammatory stimuli and ROS (4, 34). Although ROS are products of normal cell metabolism and serve as signal molecules as in redox signal pathways (35), continued oxidative stress, characterized by high ROS exposure in combination with reduced levels or scavenging capacity of antioxidants, will harm different vital macromolecules such as proteins, nucleic acids (DNA/ RNA), and lipids (36).
Especially phospholipids, as building blocks of cells, and lipoproteins, are popular targets of ROS as part of a process called lipid peroxidation, which occurs via both enzymatic and non-enzymatic mechanisms (37, 38). Whereas, the enzymatic process of lipid peroxidation covers the activation of myeloperoxidases, lipoxygenases, cyclooxygenases, and cytochrome p450 (38, 39), the non-enzymatic process requires free radicals. Therefore, it can only be activated indirectly via nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and nitric oxide synthases (40). Both processes result in lipid hydroperoxide molecules, which are then degraded. Notably, a large variety of secondary products are formed during the degradation process of lipid peroxidation, including malondialdehyde (MDA), malondialdehyde-acetaldehyde (MAA), 4-hydroxynonenal (4-HNE), and the remaining core aldehyde of oxidized phospholipids (OxPL) (41–43). These oxidized lipids and their degradation products can hamper the normal function of proteins and lipids and therefore modify them (44). Further, some of these lipid derivatives, such as highly reactive aldehydes, can alter their self-molecules and form so-called oxidation-specific epitopes (OSEs), which comprise protein adducts with degradation products of lipid peroxidation, such as MDA and phosphocholine-containing OxPL (PC-OxPL) (34, 45). If removal of these products, primarily carried by dying cells, extracellular vesicles, and damaged lipoproteins such as oxidized LDL (OxLDL), is insufficient, sterile inflammation is triggered, and oxidative damage is exacerbated (37, 46, 47).
Mounting evidence indicates increased levels of oxidized lipids and elevated presence of various OSEs during the progression from simple steatosis to NASH (46, 48–50) as well as in atherosclerosis and CVD (47). Of the different types of OSEs that can be formed during lipid peroxidation, MDA and 4-HNE are prototypical markers of oxidative stress that can be measured, for example, by the commonly used 2-thiobarbituric acid reaction (TBAR) assay (37). While the presence of 4-HNE is associated with different stages of fatty liver disease (51) and mitochondrial 4-HNE adducts are increased in NASH (52), 4-HNE is also found in atherosclerotic lesions in humans and animal models of disease (34). Furthermore, MDA epitopes are increased in patients, mice, and rats suffering from NAFLD and NASH (53–58). In addition to elevated systemic MDA concentrations, we previously demonstrated that MDA adducts accumulate in the liver during human NASH and in hypercholesterolemic Ldlr−/− mice with steatohepatitis (46, 59). Similarly, atherosclerotic lesions were shown to contain MDA epitopes (46), and elevated serum MDA-LDL levels are associated with the progression of carotid atherosclerosis (60). Recently, OxPLs, which are found to be present in atherosclerotic lesions in humans and mice (61), have also been described to be elevated in circulation and livers of patients and mice with NASH (48). Since it has been demonstrated that only modified LDL and not native LDL has a major influence on the development of atherosclerotic plaques (27), the presence of oxidized lipids and various types of OSEs may represent an essential link between NAFLD and atherosclerosis (34, 62). While systemic dyslipidemia might be responsible for the generation of high levels of OSE and modified LDL, thereby leading to both fatty liver disease and CVD, another possibility is that hepatic lipid accumulation increases oxidative stress and OSEs in the liver, prior to their release into circulation and thus subsequently promoting atherosclerosis development. As lipid peroxidation and consequently the accumulation of altered self-molecules involves interference with structural and functional properties of the physiological state, immunological mechanisms are taking place to protect the body from potential detrimental consequences. In the following section, we provide an overview of existing immune recognition and the pattern recognition receptors (PRRs) responsible for the uptake and/or binding of oxidized lipoproteins and OSEs (63).
Since OSEs identify and label altered proteins and lipids that have been damaged by oxidative stress, cellular debris, and apoptotic cells, recognition mechanisms to provide effective clearance are required (34, 64, 65). Consequently, oxidized lipids and OSEs are recognized by various PRRs on different components of the immune system that mediate their removal to maintain homeostasis in situations of increased oxidative stress. As such, OSEs play an essential role in tissue repair and reconstruction (66). However, during pathological conditions in which OSEs accumulate, they can act as damage-associated molecular patterns (DAMPs), ultimately resulting in chronic inflammation (66). Previous characterization of various OSEs suggested that both cellular and soluble PRRs can recognize OSEs, which we will discuss in light of their involvement in NAFLD and atherosclerosis.
A variety of cell surface receptors present on innate immune cells recognize OSEs and act as sensors of oxidative stress (67). Here, we will focus on cellular PRRs expressed on macrophages, as they have been shown to play a pivotal role in initiating and sustaining the inflammatory process upon binding and subsequent phagocytosis of oxidatively altered molecules, including oxidized lipoproteins (68, 69).
Toll-like receptors (TLRs) represent a group of classic cellular PRRs of innate immunity capable of binding different pathogen-associated molecular patterns (PAMPs), including bacterial and viral components, as well as DAMPs such as OSEs. For example, oxidized cholesterol esters (OxCE) and OxPL on the surface of extracellular vesicles are ligands for TLR4 (68, 70), while OxPLs have also been reported to stimulate macrophages in a TLR2-dependent mechanism (71). Furthermore, there is evidence that hydro(pero)xylated phospholipids can be considered endogenous TLR4-activating danger signals, and thus TLR4 may act as a sensor for oxidative stress (70). In addition, certain TLRs have been found to respond to OxPL, OxLDL, and other OSEs as part of a multimeric complex with other PRRs (34). As such, it has been shown that the transmission of PC-OxPL-mediated inflammatory signaling requires the formation of a heterotrimer of TLR4-TLR6 and cluster of differentiation 36 (CD36), a scavenger receptor (72, 73). Similarly, ω-(2-Carboxyethyl) pyrrole (CEP) signaling necessitates the cooperation between TLR2 and CD36 (34). Importantly, once ligands bind to TLRs, they activate nuclear factor NF-κB, which stimulates cytokine production and macrophage proliferation (74). Thus, some OSEs represent endogenous ligands recognized by members of the TLR family that can trigger inflammatory responses either with or without cooperation via another class of PRRs expressed on macrophages, namely scavenger receptors.
Scavenger receptors comprise another prototypical class of different surface receptors that recognize and internalize OSEs (67). Similar to TLRs, scavenger receptors bind oxidized and non-native LDL particles and contribute to the activation of macrophages in the context of inflammation (67, 75). There are many different types of scavenger receptors, including CD36, scavenger receptor type A1 (SR-A1), SR-A2, SR-B1, CD68, and lectin-like oxidized LDL receptor 1 (LOX1) (67). Of these, SR-A1, SR-A2, and CD36 have shown to be primarily responsible for the uptake of OxLDL, as in vitro assays have shown that macrophages deficient in these receptors exhibit 75–90% decreased binding and degradation of OxLDL (76). In addition, PC-OxPs have been found to bind to CD36, whereas PC from non-oxidized phospholipids does not serve as a ligand (77). Moreover, CEP-modified proteins are recognized by CD36 (78) and MDA epitopes are shown to be recognized by SR-A1 and SR-A2 (79, 80).
Interestingly, receptor-mediated uptake of oxidized lipids by macrophages has been found to play a central role in the chronic inflammatory responses present during both NAFLD and atherosclerosis. In both conditions, excess uptake of oxidized lipoproteins causes a transformation of local macrophages into bloated, lipid-rich foam cells, resulting in the activation of a cascade of pro-inflammatory events (81–84) (Figure 1). Under normal conditions, once internalized, lipids are transported into the lysosomal compartment for degradation by lysosomal enzymes such as cathepsins (85, 86). In contrast to native or acetylated LDL, OxLDL is poorly degraded, leading to disabled intracellular trafficking and lysosomal accumulation of oxidized lipids. Consequently, under pathological conditions characterized by the increased presence of modified lipoproteins and OxLDL, lysosomes turn dysfunctional, and cholesterol crystals are formed, resulting in NLPR3 inflammasome activation, which contributes to inflammation via the maturation and release of IL-1β and IL-18 (76, 87, 88). Furthermore, foamy macrophages express more CD36 and SR-A1, the primary receptors mediating OxLDL uptake, and might, in turn, take up even more oxidized lipids, thereby further amplifying inflammation (76). Studies by others and us showed that NLPR3 inflammasome activation contributes to liver disease and atherosclerosis in various murine models, indicating the involvement of this process in both entities (88–91). Moreover, we showed that hematopoietic deficiency of CD36 and SR-A1 reduces foam cell formation and hepatic inflammatory responses during NASH in mice (81, 82). In line, macrophage CD36 and SR-A1/2 contribute significantly to atherosclerotic lesion formation (92, 93). Further, it was demonstrated that Ldlr−/− ApoB100/100 mice fed a Western diet showed a decrease in atherosclerosis when either SR-A or CD36 was silenced in bone marrow cells using lentivirus vectors encoding shRNA against them (94). Mechanistically, we previously demonstrated that MDA-induced cytokine secretion depends on CD36 and SR-A1. Bone marrow-derived macrophages from mice lacking either of these receptors secrete less CXCL1 upon MDA stimulation, suggesting the involvement of MDA-mediated pro-inflammatory signaling in macrophages in both disease pathologies (46).
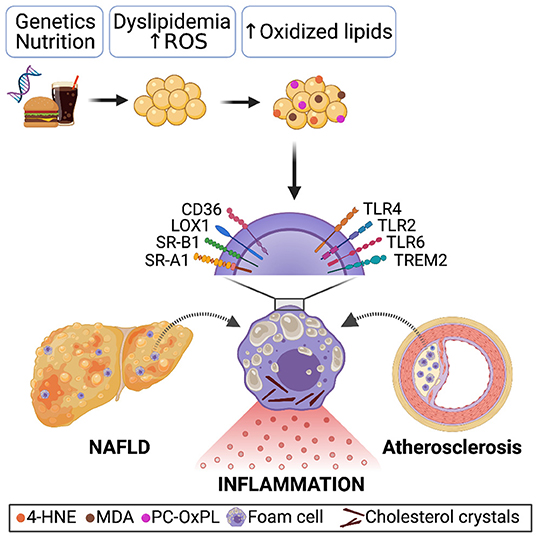
Figure 1. Genetics, sedentary lifestyle and/or unhealthy nutrition causes systemic dyslipidemia characterized by increased LDL levels. Due to oxidative stress and ROS production, oxidation-specific epitopes present on oxidized lipids, dying cells, and microvesicles accumulate. Receptor-mediated recognition and uptake of oxidized lipoproteins by macrophages in the steatotic liver and atherosclerotic plaque results in a foamy appearance, lysosomal dysfunction, and cholesterol crystals formation, causing pro-inflammatory signaling and thereby enhancing disease progression.
In addition to the common findings concerning the contribution of SR-mediated uptake of oxidized lipoproteins during NAFLD and atherosclerosis, multiple studies indicate that TLR-mediated immune recognition also contributes to disease progression. Indeed, NASH has been shown to improve in TLR4 knockout mice receiving a methionine- and choline-deficient (MCD) diet (95), and Ldlr−/− mice receiving an atherogenic diet were found protected from triglyceride accumulation in the liver in the absence of TLR4 (96). The contribution of TLR4 to the progression of atherosclerosis is supported by two studies showing that TLR4-deficient ApoE−/− mice have lower aortic lipid accumulation (70–80% reduction) and reduced levels of aortic atherosclerosis compared to the control mice (97, 98). In line, a case-control study of 183 patients showed that a single nucleotide polymorphism in Tlr4 leads to impaired signaling and is associated with reduced plaque formation and a decrease in acute coronary events (99). Moreover, Tlr4 expression is higher in macrophages in atherosclerotic plaques of ApoE−/− mice on an atherogenic diet and in humans (100), suggesting that TLR4 represents a pathophysiological link between oxidized lipids, inflammation, and atherosclerosis. Although most of the available studies focus on the role of TLR4 in NASH and atherosclerosis, it has also been shown that TLR2 deficiency in Ldlr−/− mice leads to a reduction in atherogenesis, while administration of TLR2 agonists results in increased atherosclerosis in mice (101). Concerning fatty liver disease, it was demonstrated that administration of an anti-TLR2 antibody ameliorates liver injury, inflammation, steatosis, and fibrosis in rats with obesity (102). Taken together, these studies indicate that TLR signaling contributes to the inflammatory responses observed in NAFLD and atherosclerosis.
More recently, using single-cell RNA sequencing technology, multiple studies in lipid-mediated disorders including NASH and atherosclerosis demonstrate the occurrence of a specialized type of macrophages characterized by high expression of the triggering receptor expressed on myeloid cells 2 (Trem2). Whereas, humans and mice with NASH or cirrhosis have increased hepatic expression of Trem2 that correlates positively with AST and ALT levels (103, 104), more Trem2-expressing macrophages are also found in atherosclerotic plaques (105). Interestingly, TREM2 has been shown to bind and recognize (apo-) lipoproteins, including ApoE, LDL, and MDA-LDL particles (106), suggesting TREM2 might also recognize OSEs, thereby potentially contributing to inflammatory responses and disease progression. Functional studies assessing the role of TREM2 in NAFLD and atherosclerosis are still required since this receptor might also be involved in a common lipid-induced mechanistic pathway.
Taken together, there are overarching mechanisms regulating the uptake of OSEs and OxLDL via PRRs on macrophages. As a result, macrophages and hepatic Kupffer cells turn into foam cells, become activated, and secrete pro-inflammatory factors in response to elevated oxidized lipids, thereby contributing to the development of NAFLD and atherosclerosis in a similar process, representing part of a shared etiology. In the next section, we will focus on soluble factors recognizing OSEs and their involvement in steatohepatitis and atherosclerosis.
Besides recognizing OSEs by cell surface receptors, soluble receptors capable of binding OSEs are described, such as C-reactive protein (CRP) and proteins of the complement system (62, 107). CRP, an acute-phase protein produced in the liver, was found to recognize PC-OxPL on OxLDL and apoptotic cells, indicating CRP's responsiveness to OSEs (108). Since CRP levels are a known marker of systemic inflammation, CRP in circulation might indirectly also be considered a reflection of tissue injury, oxidative stress, and/or ongoing lipid peroxidation and OSE levels. As a regulator of complement activity, an essential machinery for clearance of metabolic waste and dead cells, complement factor H (CFH) was identified to recognize and bind MDA epitopes. Moreover, genetic variants of one of the MDA-binding sites of CFH were shown to influence the capacity of CFH to bind MDA (109). Since MDA has pro-inflammatory effects during NASH, reduced CFH might contribute to the harmful effects of lipid peroxidation products in fatty liver disease progression. Nevertheless, the functional role of CFH-mediated OSE recognition and its consequences for NAFLD and CVD has not yet been fully described.
Insights from others and us give prominence to B cell-derived antibodies or immunoglobulins targeting OSEs in NAFLD and CVD (59, 107, 110–112). Antibodies exist in different isotypes (IgM, IgG, IgA, IgE, IgD) that implement key functions in defending the body against pathogens and are also responsible for maintaining homeostasis by eliminating metabolic waste (113). Owing to their broad specificity for pathogens and their ability to recognize highly conserved structures such as self-antigens, they provide another group of soluble factors able to detect OSEs residing on oxidized lipoproteins, apoptotic cells, and extracellular vesicles (64, 114). Studies in mice and humans have shown that various OSEs, including PC-OxPL, MDA, and 4-HNE adducts, are prominent targets of natural antibodies, which are pre-existing germline-encoded antibodies, predominantly of the IgM type, that are present without external antigens. In fact, several OSEs are bound by up to 30% of all natural IgM found in the plasma of both wild-type and gnotobiotic mice (64, 114), indicating their relevance for homeostasis and immune defense. In addition to innate natural IgM antibodies, produced and secreted by B1 cells, adaptive IgG isotypes secreted by B-2 cells are capable of recognizing and binding various OSEs such as MDA (115).
Interestingly, consistent observations concerning systemic levels of antibodies binding OxLDL and OSE are described between NAFLD and atherosclerosis. First of all, in line with the negative association between IgM levels in circulation and the severity of atherosclerosis and CVD in general (107), we have shown that NAFLD patients have lower serum IgM titers toward various OSEs including MDA and MAA compared to healthy controls (59). While liver disease data upon IgM deficiency is still lacking, a lack of secreted IgM antibodies resulted in increased atherosclerotic plaques in Ldlr−/− mice after Western-type diet (112). In addition, we previously demonstrated that an increase in B1-derived natural IgM with specificity for OxLDL in Ldlr−/− mice deficient for sialic acid-binding immunoglobulin-like lectin G (Siglec-G) protects against atherosclerosis and steatohepatitis after atherogenic diet, further supporting the protective role of IgM antibodies (110). By recognizing and neutralizing OSEs, IgM can limit OxLDL-induced foam cell formation, as well as pro-inflammatory macrophage responses, and generally contribute to reduced inflammation (115). More studies pointing out the protective properties of increased anti-OxLDL IgM levels are described in the section about immunotherapy.
Besides altered IgM antibody levels, ~40% of adults (49) and 60% of children have elevated IgG titers in circulation when diagnosed with NAFLD (116). Particularly, anti-OSE IgGs produced by B-2 lymphocytes have been shown to correlate with the onset of steatohepatitis and are considered as an independent predictor of fibrosis in NAFLD patients (49, 116, 117). These observations are in line with those in atherosclerosis, where higher IgG levels are associated with more severe atherosclerosis in humans and mice (118–120). Furthermore, a meta-analysis even suggests that systemic IgG antibody titers are a potential predictor of future atherosclerosis-related cardiovascular events since a positive correlation between IgG levels and events was found (121). However, the functional significance and contribution of IgG are still unclear as studies in humans and animal models indicate that anti-OSE IgG may also have atheroprotective properties (107, 122, 123). Given these diverse and partly inconsistent findings, the role of IgG in atherosclerosis has not been fully elucidated, which has been discussed in detail by Sage et al. (124). Nevertheless, in mice, anti-CD20 antibody-mediated depletion of B cells reduces the development of atherosclerosis (125), and ameliorates NASH progression (126). Importantly, anti-CD20 treatment preserves the production of anti-OxLDL IgM antibodies, while IgG targeting OxLDL are greatly diminished (125). In addition, TACI-Ig mice, characterized by a depletion of B2 cells, were found to have milder steatohepatitis and less progression of fibrosis (117), further supporting a more pro-inflammatory effect of anti-OSE IgG antibodies.
To summarize, altered anti-OSE IgM and IgG titers are associated with NAFLD and fibrotic NASH, consistent with findings in atherosclerosis (50, 59, 107, 127), suggesting another pathophysiological pathway in common between NAFLD and atherosclerotic CVD. While IgG-producing B-2 cells may promote pro-inflammatory mechanisms (50), natural IgM secreted by B1 cells seems to have protective properties (50). As such, antibodies recognizing OSEs provide an attractive target for B cell-mediated immunotherapeutic approaches to improve therapeutic strategies and/or prevent cardiovascular complications during NAFLD. Besides targeting B cells as done using anti-CD20 administration, immunization approaches to enhance immunity against OSEs have been investigated, which we will summarize in the next section.
As neither lifestyle modification nor currently existing pharmacotherapy is sufficient to reduce liver fibrosis and inflammation, NAFLD is becoming a global burden on the healthcare system and poses an urgent need for developing therapeutic interventions (128). Although several drugs and combination therapies are under investigation, no truly effective treatment has yet been identified (129, 130). However, regarding oxidative stress, nutrients and antioxidants such as vitamin E have beneficial effects on NAFLD (131) by lowering the NAFLD activity score (NAS) and reducing inflammation (132, 133). Consistent with these findings, a recent study has demonstrated that vitamin E is negatively correlated with serum MDA in women suffering from NAFLD (54). In addition, similar outcomes also indicate a protective role of vitamin E in atherosclerosis (134, 135). Nevertheless, since we currently lack complete understanding of the metabolic pathways affected and controlled by oxidative stress, more studies are needed to design an adequate therapeutic trial to assess antioxidants to combat atherogenesis (136, 137). Growing evidence shows that directly addressing OSEs yields a high potential for reducing the inflammatory response in NAFLD and atherosclerosis, thereby representing a promising avenue for treating both diseases (48, 63, 138). Here, we will provide an overview of current data on intervention approaches addressing OSEs through immunization strategies. Simplified, enhancing protective B cell antibody responses via immunization can be divided into two separate classes: passive immunization in which antibodies that directly target, bind, and inactivate an antigen are infused; and active immunization, wherein an antigen is used in a vaccination protocol to boost antibody titers and as such, provide long-term immunity (Figure 2).

Figure 2. In mice, passive immunization strategies with OSE-recognizing IgM antibodies LR04, IK17, and E06, or boosting IgM antibody titers toward oxidized lipids in active immunization approaches, such as with heat-inactivated S. pneumococci or specific peptides, can induce protection against NAFLD progression and reduce atherosclerotic plaque burden.
As described above, data from others and us support that anti-OSE IgM antibodies act protective against liver disease and atherosclerosis, and as such, are a viable tool to prevent disease (59, 83, 110, 127, 139). Besides our findings using Siglec-G−/− mice that have increased anti-OSE IgM levels, we showed that intravenous administration of LR04, a monoclonal IgM specifically targeting MDA, neutralizes endogenously produced MDA epitopes, leading to a decrease in liver inflammation in Ldlr−/− mice after a Western-type diet (46). Similarly, intravenous injection and intraperitoneal infusion of the human antibody IK17, directed to MDA-like epitopes, significantly reduced atherosclerosis progression by 30–50% (140, 141). Moreover, antibodies E06 and T15, which target PC epitopes on OxPL were found to protect against atherosclerosis by blocking lipid uptake by macrophages, preventing inflammation, and promoting the clearance of apoptotic cells (141). Expression of the single-chain variable fragment of E06 in Ldlr−/− mice was shown to be sufficient to suppress the development of atherosclerosis at multiple disease stages (139). These mice were also protected against various aspects of NASH, including steatosis, inflammation, fibrosis, hepatocyte cell death, and progression to hepatocellular carcinoma, further supporting the causal role of OxPL in the pathogenesis of NASH (49).
Besides evaluating the potential beneficial effect of IgM antibodies on disease outcome, certain studies focused on using IgG antibodies. For example, administration of the human IgG antibody 2D03 directed against MDA-modified ApoB exhibited atheroprotective immune responses against OxLDL (123). More recently, a study revealed the protective function against the development of atherosclerotic plaques using autoantibodies against the ApoB100 peptide p210 in ApoE−/− mice, which was accomplished by injecting IgG2b against p210. These results in ApoE−/− mice support previous human studies, which showed an inverse association between apoB100 native p210 IgG and plaques in coronary or carotid arteries (142).
Besides passive immunization approaches, several studies assessed the potential beneficial effect of enhanced immunity toward various epitopes of OxLDL using these as antigens. In one of the first studies applying active immunization, injection of homologous MDA-LDL into Ldlr−/− rabbits induced higher anti-MDA antibody titers and significantly reduced the extent of atherosclerotic lesions in the aortic tree of immunized animals (143). Consistent with this, Ldlr−/− mice injected with MDA-modified LDL showed smaller atherosclerotic lesion size after an atherogenic diet, although this was independent of changes in anti-OSE IgM levels (144). In relation to fatty liver disease, a study in which C57BL/6 mice were injected with MDA conjugated to bovine serum albumin (MDA-BSA) reported increased severity of NASH in immunized mice after MCD diet. The authors attributed this to the fact that MDA-BSA injection enhanced IgG responses and increased hepatic T cell infiltration, which may ultimately lead to increased inflammation (145). Importantly, immunization with MDA-BSA adducts did not influence IgM antibody levels toward MDA-derived antigens, potentially lacking protective capacities. On the other hand, studies that immunized Ldlr−/− mice with heat-inactivated S. pneumonia, which significantly induced PC-OxPL recognizing IgM titers due to molecular mimicry, showed that mice immunized were protected against diet-induced steatohepatitis and atherosclerosis after Western-type diet (83, 146), supporting the idea that raising IgM titers toward OxLDL has beneficial effects.
In addition to using various OSEs as antigens, several experimental studies assessed whether administration of stable peptides could be applied for immunization. One of these peptides is the p210-PADRE, which has already been mentioned in the section on passive immunization. Besides direct infusion of IgG antibodies, ApoE−/− mice were immunized with the p210-PADRE peptide, which induced a specific IgG1 response against p210, thereby preventing MDA-LDL accumulation in lesions and reducing atherosclerotic plaque formation in the aorta (142). Moreover, immunization with OxLDL- and MDA-modified ApoB100 peptides were described to have an atheroprotective effect associated with an increase in IgG and IgM antibodies specific for the antigen used (142, 147). Interestingly, two small immunogenic peptides, linear P1 and circular P2, were identified that immunologically mimic MDA-type epitopes (148). Since we have shown the critical role for endogenous MDA in NASH and P2-BSA immunization raised IgM antibody levels toward MDA, these mimotopes are a promising tool to induce immunity against this relevant antigen to reduce/prevent NAFLD progression (46). Although most of the above-mentioned immunization approaches have been reported to be successful for atherosclerosis, more studies focusing on liver disease are required to further confirm that the treatment principles of increased anti-OSE IgM to reduce atherosclerosis also apply to NASH.
Growing evidence indicates a significant association between the clinical patterns of NAFLD and atherosclerosis. Common molecular mechanistic pathways seem to play a central role in disease progression, and NAFLD might even be considered a risk factor for developing CVD.
Just as receptor-mediated uptake of oxidized lipids leads to macrophages' foamy appearance and pro-inflammatory processes is one consistent observation in the liver during NAFLD and the vessel wall during atherosclerosis, other mechanistic patterns might be similarly occurring in both pathologies (81–84). Besides, in both NAFLD and atherosclerosis, B-1-derived IgM antibodies seem to have a protective role, while IgG produced by B2 cells seems to promote inflammation (115). Therefore, studies assessing both conditions might significantly enhance our understanding of their interrelationship and potentially lead to the identification of novel targets to improve treatment strategies. In particular, the Ldlr−/− mice on a high-fat, high-cholesterol diet is a suitable model to study the underlying pathways occurring in fatty liver disease and linked atherosclerosis (149). We have previously shown that these mice develop steatotic livers with increased inflammation and oxidative stress already after 2 weeks of diet, while atherosclerotic plaques develop after 6–8 weeks of dietary intervention (46). Moreover, Ldlr−/− mice present a human-like lipid profile, which is essential to facilitate translational studies regarding lipid-mediated diseases (150). In addition, since CVD is still the primary cause of death in patients with early NAFLD (9, 10), assessing liver disease in human cardiovascular study cohorts and vice versa could further enhance our understanding and potentially identify patients with advanced disease stage and/or increased risk for cardiovascular events. In this review, we focused on the potential immunotherapeutic approach by enhancing immunity toward OSEs and oxidized lipids. Since similar observations for systemic anti-OSE antibody titers are described, one might hypothesize that results from immunization studies, which mostly come from work in the field of atherosclerosis, can be translated and tested to prevent NAFLD progression. Nevertheless, further studies are needed to understand the role of OSEs and OSE-reactive immunity in maintaining homeostasis and controlling inflammatory responses to design translational studies and eventually offer novel treatment strategies for patients.
CH and TH wrote and edited the manuscript. TH composed the figures. DR helped with figure composition. All authors contributed to the article and approved the submitted version.
TH was funded by a Veni (NWO; 91619012), MLDS Right-On-Time grant (MLDS; 19-28), and Zukunftskollegs grant (FWF; ZK81B).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
4-HNE, 4-Hydroxynonenal; BSA, Bovine serum albumin; CD36, Cluster of differentiation 36; CEP, ω-(2-Carboxyethyl) pyrrole; CFH, Complement factor H; CRP, C-reactive protein; CVD, Cardiovascular disease; DAMP, Damage-associated molecular pattern; FFA, Free fatty acid; HDL, High-density lipoprotein; IgG, Immunoglobulin type G; IgM, Immunoglobulin type M; IL, Interleukin; LDL, Low-density lipoprotein; LDLR, Low-density lipoprotein receptor; MAA, Malondialdehyde-acetaldehyde; MDA, Malondialdehyde; NADPH, Nicotinamide adenine dinucleotide phosphate; NAFLD, Non-alcoholic fatty liver disease; NAS, NAFLD activity score; NASH, Non-alcoholic steatohepatitis; OSE, Oxidation-specific epitope; OxLDL, Oxidized LDL; OxPL, Oxidized phospholipids; PAMP, Pathogen-associated molecular pattern; PC-OxPL, Phosphocholine-containing OxPL; ROS, Reactive oxygen species; SR, Scavenger receptor; TBAR, 2-Thiobarbituric acid reaction; TLR, Toll-like receptor; TREM2, Triggering receptor expressed on myeloid cells 2; VLDL, Very low-density lipoprotein.
1. Mozaffarian D. Dietary and policy priorities for cardiovascular disease, diabetes, and obesity: a comprehensive review. Circulation. (2016) 133:187–225. doi: 10.1161/CIRCULATIONAHA.115.018585
2. Lemieux I, Després J-P. Metabolic syndrome: past, present and future. Nutrients. (2020) 12:3501. doi: 10.3390/nu12113501
3. Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin Liver Dis. (2016) 20:205–14. doi: 10.1016/j.cld.2015.10.001
4. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
5. Marengo A, Jouness RIK, Bugianesi E. Progression and natural history of nonalcoholic fatty liver disease in adults. Clin Liver Dis. (2016) 20:313–24. doi: 10.1016/j.cld.2015.10.010
6. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
7. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
8. Mantovani A, Scorletti E, Mosca A, Alisi A, Byrne CD, Targher G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism. (2020) 111:154170. doi: 10.1016/j.metabol.2020.154170
9. Tang K, Lin J, Ji X, Lin T, Sun D, Zheng X. Non-alcoholic fatty liver disease with reduced myocardial FDG uptake is associated with coronary atherosclerosis. J Nucl Cardiol. (2021) 28:610–20. doi: 10.1007/s12350-019-01736-6
10. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. (2020) 69:1691–705. doi: 10.1136/gutjnl-2020-320622
11. Arai T, Atsukawa M, Tsubota A, Kato K, Abe H, Ono H. Liver fibrosis is associated with carotid atherosclerosis in patients with liver biopsy-proven nonalcoholic fatty liver disease. Sci Rep. (2021) 11:15938. doi: 10.1038/s41598-021-95581-8
12. Kapuria D, Takyar VK, Etzion O, Surana P, O'Keefe JH, Koh C. Association of hepatic steatosis with subclinical atherosclerosis: systematic review and meta-analysis. Hepatol Commun. (2018) 2:873–83. doi: 10.1002/hep4.1199
13. Getz GS, Reardon CA. Natural killer T cells in atherosclerosis. Nat Rev Cardiol. Mai. (2017) 14:304–14. doi: 10.1038/nrcardio.2017.2
14. Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. (2001) 104:503–16. doi: 10.1016/S0092-8674(01)00238-0
15. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
16. Konkoth A, Saraswat R, Dubrou C, Sabatier F, Leroyer AS, Lacroix R. Multifaceted role of extracellular vesicles in atherosclerosis. Atherosclerosis. (2021) 319:121–31. doi: 10.1016/j.atherosclerosis.2020.11.006
17. Paik JM, Henry L, Avila LD, Younossi E, Racila A, Younossi ZM. Mortality related to nonalcoholic fatty liver disease is increasing in the United States. Hepatol Commun. (2019) 3:1459–71. doi: 10.1002/hep4.1419
18. Conjeevaram Selvakumar PK, Kabbany MN, Alkhouri N. Nonalcoholic fatty liver disease in children: not a small matter. Paediatr Drugs. (2018) 20:315–29. doi: 10.1007/s40272-018-0292-2
19. Castillo-Leon E, Connelly MA, Konomi JV, Caltharp S, Cleeton R, Vos MB. Increased atherogenic lipoprotein profile in children with non-alcoholic steatohepatitis. Pediatr Obes. (2020) 15:e12648. doi: 10.1111/ijpo.12648
20. Mann JP, Raponi M, Nobili V. Clinical implications of understanding the association between oxidative stress and pediatric NAFLD. Expert Rev Gastroenterol Hepatol. (2017) 11:371–82. doi: 10.1080/17474124.2017.1291340
21. Mendez-Sanchez N, Cruz-Ramon VC, Ramirez-Perez OL, Hwang JP, Barranco-Fragoso B, Cordova-Gallardo J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int J Mol Sci. (2018) 19:2034. doi: 10.3390/ijms19072034
22. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. (2014) 146:726–35. doi: 10.1053/j.gastro.2013.11.049
23. Zhang Q-Q, Lu L-G. Nonalcoholic fatty liver disease: dyslipidemia, risk for cardiovascular complications, and treatment strategy. J Clin Transl Hepatol. (2015) 3:78–84. doi: 10.14218/JCTH.2014.00037
24. Chatrath H, Vuppalanchi R, Chalasani N. Dyslipidemia in patients with nonalcoholic fatty liver disease. Semin Liver Dis. (2012) 32:22–9. doi: 10.1055/s-0032-1306423
25. Ho C-M, Ho S-L, Jeng Y-M, Lai Y-S, Chen Y-H, Lu S-C. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J Inflamm. (2019) 16:7. doi: 10.1186/s12950-019-0211-5
26. Stols-Gonçalves D, Hovingh GK, Nieuwdorp M, Holleboom AG. NAFLD and atherosclerosis: two sides of the same dysmetabolic coin? Trends Endocrinol Metab. (2019) 30:891–902. doi: 10.1016/j.tem.2019.08.008
27. Zakiev ER, Sukhorukov VN, Melnichenko AA, Sobenin IA, Ivanova EA, Orekhov AN. Lipid composition of circulating multiple-modified low density lipoprotein. Lipids Health Dis. (2016) 15:134. doi: 10.1186/s12944-016-0308-2
28. Engin AB. What is lipotoxicity? Adv Exp Med Biol. (2017) 960:197–220. doi: 10.1007/978-3-319-48382-5_8
29. Symons JD, Abel ED. Lipotoxicity contributes to endothelial dysfunction : a focus on the contribution from ceramide. Rev Endocr Metab Disord. (2013) 14:59–68. doi: 10.1007/s11154-012-9235-3
30. Rada P, González-Rodríguez Á, García-Monzón C, Valverde ÁM. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis. (2020) 11:1–15. doi: 10.1038/s41419-020-03003-w
31. García-Monzón C, Lo Iacono O, Mayoral R, González-Rodríguez Á, Miquilena-Colina ME, Lozano-Rodríguez T, et al. Hepatic insulin resistance is associated with increased apoptosis fibrogenesis in nonalcoholic steatohepatitis chronic hepatitis C. J Hepatol. (2011) 54:142–52. doi: 10.1016/j.jhep.2010.06.021
32. Bellanti F, Villani R, Facciorusso A, Vendemiale G, Serviddio G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radical Biol Med. (2017) 111:173–85. doi: 10.1016/j.freeradbiomed.2017.01.023
33. Alkhouri N, Eng K, Lopez R, Nobili V. Non-high-density lipoprotein cholesterol (non-HDL-C) levels in children with nonalcoholic fatty liver disease (NAFLD). Springerplus. (2014) 3:407. doi: 10.1186/2193-1801-3-407
34. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
35. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radical Biol Med. (2020) 152:116–41. doi: 10.1016/j.freeradbiomed.2020.02.025
36. Ore A, Akinloye OA. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina. (2019) 55:26. doi: 10.3390/medicina55020026
37. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. (1991) 11:81–128. doi: 10.1016/0891-5849(91)90192-6
38. Bochkov VN, Oskolkova OV, Birukov KG, Levonen A-L, Binder CJ, Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. (2010) 12:1009–59. doi: 10.1089/ars.2009.2597
39. Niki E, Yoshida Y, Saito Y, Noguchi N. Lipid peroxidation: mechanisms, inhibition, and biological effects. Biochem Biophys Res Commun. (2005) 338:668–76. doi: 10.1016/j.bbrc.2005.08.072
40. O'Donnell VB, Murphy RC. New families of bioactive oxidized phospholipids generated by immune cells: identification and signaling actions. Blood. (2012) 120:1985–92. doi: 10.1182/blood-2012-04-402826
41. Salomon RG. Structural identification and cardiovascular activities of oxidized phospholipids. Circ Res. (2012) 111:930–46. doi: 10.1161/CIRCRESAHA.112.275388
42. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. (2011) 111:5944–72. doi: 10.1021/cr200084z
43. Pratt DA, Tallman KA, Porter NA. Free radical oxidation of polyunsaturated lipids: new mechanistic insights and the development of peroxyl radical clocks. Acc Chem Res. (2011) 44:458–67. doi: 10.1021/ar200024c
44. Bartolini Gritti B, Binder CJ. Oxidation-specific epitopes are major targets of innate immunity in atherothrombosis. Hamostaseologie. (2016) 36:89–96. doi: 10.5482/HAMO-14-11-0069
45. Chou M-Y, Hartvigsen K, Hansen LF, Fogelstrand L, Shaw PX, Boullier A. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med. (2008) 263:479–88. doi: 10.1111/j.1365-2796.2008.01968.x
46. Busch CJ-L, Hendrikx T, Weismann D, Jäckel S, Walenbergh SMA, Rendeiro AF. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology. (2017) 65:1181–95. doi: 10.1002/hep.28970
47. Leibundgut G, Witztum JL, Tsimikas S. Oxidation-specific epitopes and immunological responses: translational biotheranostic implications for atherosclerosis. Curr Opin Pharmacol. (2013) 13:168–79. doi: 10.1016/j.coph.2013.02.005
48. Sun X, Seidman JS, Zhao P, Troutman TD, Spann NJ, Que X. Neutralization of oxidized phospholipids ameliorates non-alcoholic steatohepatitis. Cell Metab. (2020) 31:189–206.e8. doi: 10.1016/j.cmet.2019.10.014
49. Albano E, Mottaran E, Vidali M, Reale E, Saksena S, Occhino G. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. (2005) 54:987–93. doi: 10.1136/gut.2004.057968
50. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol. (2020) 17:81–92. doi: 10.1038/s41575-019-0210-2
51. Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. (2002) 37:56–62. doi: 10.1016/S0168-8278(02)00073-9
52. Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia–reperfusion injury. Gut. (2008) 57:957–65. doi: 10.1136/gut.2007.147496
53. Köroglu E, Canbakan B, Atay K, Hatemi I, Tuncer M, Dobrucali A. Role of oxidative stress and insulin resistance in disease severity of non-alcoholic fatty liver disease. Turk J Gastroenterol. (2016) 27:361–6. doi: 10.5152/tjg.2016.16106
54. Zelber-Sagi S, Ivancovsky-Wajcman D, Fliss-Isakov N, Hahn M, Webb M, Shibolet O. Serum malondialdehyde is associated with non-alcoholic fatty liver and related liver damage differentially in men and women. Antioxidants. (2020) 9:578. doi: 10.3390/antiox9070578
55. Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am J Gastroenterol. (2005) 100:850–5. doi: 10.1111/j.1572-0241.2005.41500.x
56. Schröder T, Kucharczyk D, Bär F, Pagel R, Derer S, Jendrek ST. Mitochondrial gene polymorphisms alter hepatic cellular energy metabolism and aggravate diet-induced non-alcoholic steatohepatitis. Mol Metab. (2016) 5:283–95. doi: 10.1016/j.molmet.2016.01.010
57. Elshazly SM. Ameliorative effect of nicorandil on high fat diet induced non-alcoholic fatty liver disease in rats. Euro J Pharmacol. (2015) 748:123–32. doi: 10.1016/j.ejphar.2014.12.017
58. Song L, Qu D, Zhang Q, jiang J, Zhou H, Jiang R. Phytosterol esters attenuate hepatic steatosis in rats with non-alcoholic fatty liver disease rats fed a high-fat diet. Sci Rep. (2017) 7:41604. doi: 10.1038/srep41604
59. Hendrikx T, Watzenböck ML, Walenbergh SMA, Amir S, Gruber S, Kozma MO. Low levels of IgM antibodies recognizing oxidation-specific epitopes are associated with human non-alcoholic fatty liver disease. BMC Med. (2016) 14:107. doi: 10.1186/s12916-016-0652-0
60. Tanaga K, Bujo H, Inoue M, Mikami K, Kotani K, Takahashi K, et al. Increased circulating malondialdehyde-modified LDL levels in patients with coronary artery diseases their association with peak sizes of LDL particles. ATVB. (2002) 22:662–6. doi: 10.1161/01.ATV.0000012351.63938.84
61. Lee S, Birukov KG, Romanoski CE, Springstead JR, Lusis AJ, Berliner JA. Role of phospholipid oxidation products in atherosclerosis. Circul Res. (2012) 111:778–99. doi: 10.1161/CIRCRESAHA.111.256859
62. Hendrikx T, Binder CJ. Oxidation-specific epitopes in non-alcoholic fatty liver disease. Front Endocrinol. (2020) 11:607011. doi: 10.3389/fendo.2020.607011
63. Papac Milicevic N, Busch CJ-L, Binder CJ. Malondialdehyde epitopes as targets of immunity and the implications for atherosclerosis. Adv Immunol. (2016) 131:1–59. doi: 10.1016/bs.ai.2016.02.001
64. Chou M-Y, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. (2009) 119:1335–49. doi: 10.1172/JCI36800
65. Nicolai O, Pötschke C, Raafat D, van der Linde J, Quosdorf S, Laqua A. Oxidation-specific epitopes (OSEs) dominate the B cell response in murine polymicrobial sepsis. Front Immunol. (2020) 11:1570. doi: 10.3389/fimmu.2020.01570
66. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
67. Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res. (2009) 50 (Suppl):S282–6. doi: 10.1194/jlr.R800066-JLR200
68. Miller YI, Choi S-H, Wiesner P, Fang L, Harkewicz R, Hartvigsen K. Oxidation-specific epitopes are danger associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. (2011) 108:235–48. doi: 10.1161/CIRCRESAHA.110.223875
69. Weismann D, Binder CJ. The innate immune response to products of phospholipid peroxidation. Biochim Biophys Acta. (2012) 1818:2465–75. doi: 10.1016/j.bbamem.2012.01.018
70. Manček-Keber M, Frank-Bertoncelj M, Hafner-Bratkovič I, Smole A, Zorko M, Pirher N. Toll-like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles. Sci Signal. (2015) 8:ra60. doi: 10.1126/scisignal.2005860
71. Kadl A, Sharma PR, Chen W, Agrawal R, Meher AK, Rudraiah S. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic Biol Med. (2011) 51:1903–9. doi: 10.1016/j.freeradbiomed.2011.08.026
72. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
73. Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. (2010) 12:467–82. doi: 10.1016/j.cmet.2010.09.010
74. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
75. Kragh Petersen S, Bilkei-Gorzo O, Govaere O, Härtlova A. Macrophages and scavenger receptors in obesity-associated non-alcoholic liver fatty disease (NAFLD). Scand J Immunol. (2020) 92:e12971. doi: 10.1111/sji.12971
76. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. (2002) 277:49982–8. doi: 10.1074/jbc.M209649200
77. Boullier A, Friedman P, Harkewicz R, Hartvigsen K, Green SR, Almazan F. Phosphocholine as a pattern recognition ligand for CD36. J Lipid Res. (2005) 46:969–76. doi: 10.1194/jlr.M400496-JLR200
78. Kim Y-W, Yakubenko VP, West XZ, Gugiu GB, Renganathan K, Biswas S. Receptor-mediated mechanism controlling tissue levels of bioactive lipid oxidation products. Circ Res. (2015) 117:321–32. doi: 10.1161/CIRCRESAHA.117.305925
79. Duryee MJ, Freeman TL, Willis MS, Hunter CD, Hamilton BC, Suzuki H. Scavenger receptors on sinusoidal liver endothelial cells are involved in the uptake of aldehyde-modified proteins. Mol Pharmacol. (2005) 68:1423–30. doi: 10.1124/mol.105.016121
80. Shechter I, Fogelman AM, Haberland ME, Seager J, Hokom M, Edwards PA. The metabolism of native and malondialdehyde-altered low density lipoproteins by human monocyte-macrophages. J Lipid Res. (1981) 22:63–71. doi: 10.1016/S0022-2275(20)34741-6
81. Bieghs V, Verheyen F, van Gorp PJ, Hendrikx T, Wouters K, Lütjohann D. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in kupffer cells. PLoS ONE. (2012) 7:e34378. doi: 10.1371/journal.pone.0034378
82. Bieghs V, Wouters K, van Gorp PJ, Gijbels MJJ, de Winther MPJ, Binder CJ. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology. (2010) 138:2477–86, (2486) e1-3. doi: 10.1053/j.gastro.2010.02.051
83. Bieghs V, Gorp PJ van, Walenbergh SMA, Gijbels MJ, Verheyen F, Buurman WA. Specific immunization strategies against oxidized low-density lipoprotein: a novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology. (2012) 56:894–903. doi: 10.1002/hep.25660
84. Mushenkova NV, Bezsonov EE, Orekhova VA, Popkova TV, Starodubova AV, Orekhov AN. Recognition of oxidized lipids by macrophages and its role in atherosclerosis development. Biomedicines. (2021) 9:915. doi: 10.3390/biomedicines9080915
85. Yadati T, Houben T, Bitorina A, Oligschlaeger Y, Gijbels MJ, Mohren R. Inhibition of extracellular cathepsin D reduces hepatic lipid accumulation and leads to mild changes in inflammationin NASH mice. Front Immunol. (2021) 12:2846. doi: 10.3389/fimmu.2021.675535
86. Zhao CF, Herrington DM. The function of cathepsins B, D, and X in atherosclerosis. Am J Cardiovasc Dis. (2016) 6:163–70.
87. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE. (2010) 5:e11765. doi: 10.1371/journal.pone.0011765
88. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
89. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Rooyen DMV. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
90. Hendrikx T, Bieghs V, Walenbergh SMA, van Gorp PJ, Verheyen F, Jeurissen MLJ. Macrophage specific caspase-1/11 deficiency protects against cholesterol crystallization and hepatic inflammation in hyperlipidemic mice. PLoS ONE. (2013) 8:e78792. doi: 10.1371/journal.pone.0078792
91. Hendrikx T, Jeurissen MLJ, van Gorp PJ, Gijbels MJ, Walenbergh SMA, Houben T, et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. (2015) 282:2327–38. doi: 10.1111/febs.13279
92. Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. (2000) 105:1049–56. doi: 10.1172/JCI9259
93. Babaev VR, Gleaves LA, Carter KJ, Suzuki H, Kodama T, Fazio S, et al. Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arteriosclerosis Thromb Vasc Biol. (2000) 20:2593–9. doi: 10.1161/01.ATV.20.12.2593
94. Mäkinen PI, Lappalainen JP, Heinonen SE, Leppänen P, Lähteenvuo MT, Aarnio JV. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc Res. (2010) 88:530–8. doi: 10.1093/cvr/cvq235
95. Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G433–41. doi: 10.1152/ajpgi.00163.2009
96. Ferreira DF, Fiamoncini J, Prist IH, Ariga SK, de Souza HP, de Lima TM. Novel role of TLR4 in NAFLD development: modulation of metabolic enzymes expression. Biochim Biophys Acta Mol Cell Biol Lipids. (2015) 1851:1353–9. doi: 10.1016/j.bbalip.2015.07.002
97. Higashimori M, Tatro JB, Moore KJ, Mendelsohn ME, Galper JB, Beasley D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2011) 31:50–7. doi: 10.1161/ATVBAHA.110.210971
98. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. (2004) 101:10679–84. doi: 10.1073/pnas.0403249101
99. Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont M-C, Seknadji P. Association of the toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscl Thromb Vasc Biol. (2003) 23:e61–4. doi: 10.1161/01.ATV.0000101191.92392.1D
100. Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine human lipid-rich atherosclerotic plaques upregulated by oxidized LDL. Circulation. (2001) 104:3103–8. doi: 10.1161/hc5001.100631
101. Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. (2005) 115:3149–56. doi: 10.1172/JCI25482
102. Wu L, Sun J, Liu L, Du X, Liu Y, Yan X. Anti-toll-like receptor 2 antibody ameliorates hepatic injury, inflammation, fibrosis and steatosis in obesity-related metabolic disorder rats via regulating MAPK and NF-κB pathways. Int Immunopharmacol. (2020) 82:106368. doi: 10.1016/j.intimp.2020.106368
103. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell. (2019) 75:644–60.e5. doi: 10.1016/j.molcel.2019.07.028
104. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
105. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K. Single-Cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circul Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
106. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
107. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. (2014) 114:1743–56. doi: 10.1161/CIRCRESAHA.113.301145
108. Chang M-K, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA. (2002) 99:13043–8. doi: 10.1073/pnas.192399699
109. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HPN, Charbel Issa P. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. (2011) 478:76–81. doi: 10.1038/nature10449
110. Gruber S, Hendrikx T, Tsiantoulas D, Ozsvar-Kozma M, Göderle L, Mallat Z. Sialic acid-binding immunoglobulin-like lectin G promotes atherosclerosis and liver inflammation by suppressing the protective functions of B-1 cells. Cell Rep. (2016) 14:2348–61. doi: 10.1016/j.celrep.2016.02.027
111. Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. (2002) 109:745–53. doi: 10.1172/JCI7272
112. Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2009) 120:417–26. doi: 10.1161/CIRCULATIONAHA.109.868158
113. Tsiantoulas D, Perkmann T, Afonyushkin T, Mangold A, Prohaska TA, Papac-Milicevic N. Circulating microparticles carry oxidation-specific epitopes and are recognized by natural IgM antibodies 1. J Lipid Res. (2015) 56:440–8. doi: 10.1194/jlr.P054569
114. Chang MK, Bergmark C, Laurila A, Hörkkö S, Han KH, Friedman P. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci USA. (1999) 96:6353–8. doi: 10.1073/pnas.96.11.6353
115. Tsiantoulas D, Sage AP, Mallat Z, Binder CJ. Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arterioscler Thromb Vasc Biol. (2015) 35:296–302. doi: 10.1161/ATVBAHA.114.303569
116. Nobili V, Parola M, Alisi A, Marra F, Piemonte F, Mombello C. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int J Mol Med. (2010) 26:471–6. doi: 10.3892/ijmm_00000487
117. Bruzzì S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med. (2018) 124:249–59. doi: 10.1016/j.freeradbiomed.2018.06.015
118. Mayr M, Kiechl S, Tsimikas S, Miller E, Sheldon J, Willeit J. Oxidized low-density lipoprotein autoantibodies, chronic infections, and carotid atherosclerosis in a population-based study. J Am Coll Cardiol. (2006) 47:2436–43. doi: 10.1016/j.jacc.2006.03.024
119. Lappalainen J, Lindstedt KA, Oksjoki R, Kovanen PT. OxLDL-IgG immune complexes induce expression and secretion of proatherogenic cytokines by cultured human mast cells. Atherosclerosis. (2011) 214:357–63. doi: 10.1016/j.atherosclerosis.2010.11.024
120. Centa M, Jin H, Hofste L, Hellberg S, Busch A, Baumgartner R. Germinal center–derived antibodies promote atherosclerosis plaque size and stability. Circulation. (2019) 139:2466–82. doi: 10.1161/CIRCULATIONAHA.118.038534
121. Iseme RA, McEvoy M, Kelly B, Agnew L, Walker FR, Handley T. A role for autoantibodies in atherogenesis. Cardiovasc Res. (2017) 113:1102–12. doi: 10.1093/cvr/cvx112
122. Nilsson J. Can antibodies protect us against cardiovascular disease? EBioMedicine. (2016) 9:29–30. doi: 10.1016/j.ebiom.2016.06.039
123. Schiopu A, Bengtsson J, Söderberg I, Janciauskiene S, Lindgren S, Ares MPS. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. (2004) 110:2047–52. doi: 10.1161/01.CIR.0000143162.56057.B5
124. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:180–96. doi: 10.1038/s41569-018-0106-9
125. Ait-Oufella H, Herbin O, Bouaziz J-D, Binder CJ, Uyttenhove C, Laurans L. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. (2010) 207:1579–87. doi: 10.1084/jem.20100155
126. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P. Microbiota-driven activation of intrahepatic B cells aggravates NASH through innate and adaptive signaling. Hepatology. (2021) 74:704–22. doi: 10.1002/hep.31755
127. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circul Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
128. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. (2015) 149:367–78.e5; quiz e14–5. doi: 10.1053/j.gastro.2015.04.005
129. Francque S, Vonghia L. Pharmacological treatment for non-alcoholic fatty liver disease. Adv Ther. (2019) 36:1052–74. doi: 10.1007/s12325-019-00898-6
130. Neuschwander-Tetri BA. Therapeutic landscape for NAFLD in 2020. Gastroenterology. (2020) 158:1984–98.e3. doi: 10.1053/j.gastro.2020.01.051
131. Sanyal AJ, Friedman SL, McCullough AJ, Dimick-Santos L, American American Association for the Study of Liver Diseases United States Food Drug Administration. Challenges opportunities in drug biomarker development for nonalcoholic steatohepatitis: findings recommendations from an American Association for the Study of Liver Diseases-U.S. Food and Drug Administration joint workshop. Hepatology. (2015) 61:1392–405. doi: 10.1002/hep.27678
132. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. (2019) 394:2184–96. doi: 10.1016/S0140-6736(19)33041-7
133. Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. (2011) 305:1659–68. doi: 10.1001/jama.2011.520
135. Su M, Wang D, Chang W, Liu L, Cui M, Xu T. Preparation of vitamin E-containing high-density lipoprotein and its protective efficacy on macrophages. ASSAY Drug Dev Technol. (2018) 16:107–14. doi: 10.1089/adt.2017.831
136. Wang W, Kang PM. Oxidative stress and antioxidant treatments in cardiovascular diseases. Antioxidants. (2020) 9:1292. doi: 10.3390/antiox9121292
137. Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arteriosclerosis Thromb Vasc Biol. (2010) 30:2311–6. doi: 10.1161/ATVBAHA.108.179697
138. Hartley A, Haskard D, Khamis R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis – novel insights and future directions in diagnosis and therapy. Trends Cardiovasc Med. (2019) 29:22–6. doi: 10.1016/j.tcm.2018.05.010
139. Tsiantoulas D, Bot I, Ozsvar-Kozma M, Göderle L, Perkmann T, Hartvigsen K. Increased plasma IgE accelerate atherosclerosis in secreted IgM deficiency. Circul Res. (2017) 120:78–84. doi: 10.1161/CIRCRESAHA.116.309606
140. Shaw PX, Hörkkö S, Tsimikas S, Chang M-K, Palinski W, Silverman GJ. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arteriosclerosis Thromb Vasc Biol. (2001) 21:1333–9. doi: 10.1161/hq0801.093587
141. Tsimikas S, Miyanohara A, Hartvigsen K, Merki E, Shaw PX, Chou M-Y. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J Am Coll Cardiol. (2011) 58:1715–27. doi: 10.1016/j.jacc.2011.07.017
142. Dunér P, Mattisson IY, Fogelstrand P, Glise L, Ruiz S, Farina C. Antibodies against apoB100 peptide 210 inhibit atherosclerosis in apoE-/- mice. Sci Rep. (2021) 11:9022. doi: 10.1038/s41598-021-88430-1
143. Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci USA. (1995) 92:821–5. doi: 10.1073/pnas.92.3.821
144. Freigang S, Hörkkö S, Miller E, Witztum JL, Palinski W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol. (1998) 18:1972–82. doi: 10.1161/01.ATV.18.12.1972
145. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. (2014) 59:886–97. doi: 10.1002/hep.26749
146. Binder CJ, Hörkkö S, Dewan A, Chang M-K, Kieu EP, Goodyear CS, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae oxidized LDL. Nat Med. (2003) 9:736–43. doi: 10.1038/nm876
147. Tse K, Gonen A, Sidney J, Ouyang H, Witztum J, Sette A. Atheroprotective vaccination with MHC-II restricted peptides from ApoB-100. Front Immunol. (2013) 4:493. doi: 10.3389/fimmu.2013.00493
148. Amir S, Hartvigsen K, Gonen A, Leibundgut G, Que X, Jensen-Jarolim E. Peptide mimotopes of malondialdehyde epitopes for clinical applications in cardiovascular disease. J Lipid Res. (2012) 53:1316–26. doi: 10.1194/jlr.M025445
149. van den Hoek AM, Verschuren L, Worms N, van Nieuwkoop A, de Ruiter C, Attema J. A translational mouse model for NASH with advanced fibrosis and atherosclerosis expressing key pathways of human pathology. Cells. (2020) 9:2014. doi: 10.3390/cells9092014
Keywords: NAFLD (non-alcoholic fatty liver disease), oxidized lipids, foamy macrophages, immunoglobulins, atherosclerosis, NASH (non-alcoholic steatohepatitis)
Citation: Hoebinger C, Rajcic D and Hendrikx T (2022) Oxidized Lipids: Common Immunogenic Drivers of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. Front. Cardiovasc. Med. 8:824481. doi: 10.3389/fcvm.2021.824481
Received: 29 November 2021; Accepted: 15 December 2021;
Published: 10 January 2022.
Edited by:
Jue Zhang, Versiti Blood Research Institute, United StatesReviewed by:
Shunxing Rong, University of Texas Southwestern Medical Center, United StatesCopyright © 2022 Hoebinger, Rajcic and Hendrikx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim Hendrikx, dGltLmhlbmRyaWt4QG1lZHVuaXdpZW4uYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.