
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 20 January 2022
Sec. General Cardiovascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.820416
This article is part of the Research Topic Insights in Heart Failure and Transplantation: 2021 View all 15 articles
 Mao Zhang1,2†
Mao Zhang1,2† Junxia Zhang1†Wenjia Zhang3
Junxia Zhang1†Wenjia Zhang3 Qingmei Hu1Li Jin1Peng Xie1Wen Zheng1Haibao Shang1
Qingmei Hu1Li Jin1Peng Xie1Wen Zheng1Haibao Shang1 Yan Zhang1,3,4*
Yan Zhang1,3,4*Heart failure is a syndrome in which the heart cannot pump enough blood to meet the body's needs, resulting from impaired ventricular filling or ejection of blood. Heart failure is still a global public health problem and remains a substantial unmet medical need. Therefore, it is crucial to identify new therapeutic targets for heart failure. Ca2+/calmodulin-dependent kinase II (CaMKII) is a serine/threonine protein kinase that modulates various cardiac diseases. CaMKII-δ9 is the most abundant CaMKII-δ splice variant in the human heart and acts as a central mediator of DNA damage and cell death in cardiomyocytes. Here, we proved that CaMKII-δ9 mediated cardiomyocyte death promotes cardiomyopathy and heart failure. However, CaMKII-δ9 did not directly regulate cardiac hypertrophy. Furthermore, we also showed that CaMKII-δ9 induced cell death in adult cardiomyocytes through impairing the UBE2T/DNA repair signaling. Finally, we demonstrated no gender difference in the expression of CaMKII-δ9 in the hearts, together with its related cardiac pathology. These findings deepen our understanding of the role of CaMKII-δ9 in cardiac pathology and provide new insights into the mechanisms and therapy of heart failure.
Heart failure is a complex and heterogeneous syndrome resulting from impairment of ventricular filling or ejection of blood associated with symptoms of dyspnea, fatigue, as well as peripheral and/or pulmonary edema. Heart failure is one of the most prominent causes of hospitalization globally, with 3.6 million newly diagnosed patients annually imposing an unprecedented cost burden on the health care system (1, 2). The pathophysiological mechanisms of heart failure consist of cardiac injuries at multiple levels, including the myocardium, vasculature, pericardium, heart valves, electrical system, or a combination of cardiac abnormalities, among which cardiomyocyte death and hypertrophy are two critical factors.
Cardiomyocyte death significantly contributes to the progression of heart failure (3, 4). Multiple myocardial injury insults lead to cardiomyocyte death. Adult mammalian cardiomyocytes are terminally differentiated cells and have a minimal capacity for self-replacement. The loss of mammalian cardiomyocytes cannot be replenished from living cells, resulting in compromised cardiac function and heart failure. On the other hand, in response to myocardial injury or chronically increased hemodynamic load, cardiac mass increases due to cardiomyocyte hypertrophy to help maintain ejection performance. However, continued hemodynamic overload leads to the dilation of the heart and the thinning of the cavity walls, resulting in the change of myocardial geometry, an increase of wall stress, and cardiac dysfunction (5, 6).
Cardiomyocyte death and hypertrophy interact with each other and synergistically promote the progression of heart failure. Sustained cardiac pathological stresses result in progressive myocardial hypertrophy that eventually exceeds the capacity of the coronary vasculature to adequately perfuse the myocardial mass, leading to multiple foci of myocardial ischemia, cardiomyocyte death, myocardial fibrosis, and the deterioration of cardiac dysfunction. On the other hand, multiple myocardial injury insults cause cardiomyocyte death, and the surviving myocytes compensate by becoming hypertrophic to maintain normal cardiac function. Despite extensive efforts of evidence-based pharmacologic and device therapies, an unacceptable number of patients suffer impaired functional capacity, poor quality of life, and early death due to heart failure. Furthermore, hospitalized heart failure patients continue to experience unacceptably high post-discharge mortality and readmission rates, which have not been improved in the last two decades (7, 8). Therefore, it is of great importance to identify new therapeutic targets of heart failure.
Ca2+/calmodulin-dependent kinase II (CaMKII) is a serine/threonine protein kinase that modulates various biological functions and pathological processes in the heart (9–11). Excessive CaMKII activation is critically involved in multiple cardiac pathological conditions, such as myocardial ischemic injury (12–15), arrhythmia (16, 17), cardiac hypertrophy and remodeling (18, 19), and cardiomyopathy and heart failure (15, 19), and inhibition of CaMKII over-activation profoundly alleviates these cardiac diseases in animal models (13–15, 19–23). In cardiomyocytes, CaMKII plays a central role in regulating cell survival (12, 24, 25) and hypertrophy (18, 26, 27). CaMKII is encoded by four genes, CaMKII-α, β, γ, and δ, and CaMKII-δ is predominantly expressed in the heart. CaMKII-δ is alternatively spliced to generate 11 different variants (28–30). Different isoforms and splice variants possess distinct or even opposite biological and pathological functions (25, 31–35). Our recent study shows that CaMKII-δ9 is the most abundant CaMKII-δ splice variant in the human heart and acts as a central mediator of DNA damage and cell death in cardiomyocytes (35). The cardiac-specific CaMKII-δ9 transgenic mice develop extensive cardiomyopathy and heart failure. But the role of CaMKII-δ9 in cardiac physiology and pathology, especially its function in myocardial hypertrophy and heart failure, remains far from clear.
Here, we proved that CaMKII-δ9 mediates cardiomyocyte death instead of hypertrophy to elicit cardiomyopathy and heart failure. Thus, this study not only deepens our understanding of the role of CaMKII-δ9 in cardiac pathology, but also provides new insights of the mechanisms of heart failure.
Animals were maintained in the Center for Experimental Animals (an Association for Assessment and Accreditation of Laboratory Animal Care-accredited experimental animal facility) at Peking University, Beijing, China. The animals were randomly allocated to experimental groups. Both males and females were used. No non-inclusion or exclusion parameters were used in our studies. Investigators were not blinded to treatments, but no subjective assessments were made. All procedures involving experimental animals (mice, rats, and rhesus monkeys) followed protocols approved by the Committee for Animal Research of Peking University and conformed to the Guide for the Care and Use of Laboratory Animals.
Adult C57BL/6 mice and Sprague-Dawley rats were from Vital River Laboratories, Beijing, China. Rhesus monkeys were from our in-house cohort as previously reported (36). The animals were euthanized by intravenous injection of an overdose of sodium pentobarbital, and the tissues were quickly frozen in liquid nitrogen for protein and total RNA extraction.
The cardiac-specific CaMKII-δ9 transgenic mice was generated as previously described (35).
Five-week-old wild-type and CaMKII-δ9 mice received ip. injection of either KN-93 (10 μmol/kg; Millipore, 422711) or a comparable volume of saline every other day. The survival rates were recorded for seven weeks, and cardiac function was then assessed by echocardiography.
Five-week-old wild-type and CaMKII-δ9 mice received ip. injection of either z-VAD (0.5 mg/kg; Sigma, V116) or a comparable volume of saline twice a week. The survival rates were recorded for seven weeks, and cardiac function was then assessed by echocardiography.
Normal human ventricular tissues were from the NIH NeuroBioBank at the University of Maryland, Baltimore, MD as previously described (35).
Transverse aortic constriction (TAC) was performed in 6-week-old male mice as described before (35). Mice were anesthetized under 3% isoflurane via intubation, the chest was opened, the aortic arch was visualized, and a 7-0 silk suture was passed under the arch between the innominate and left common carotid arteries. The suture was secured around both the aorta and a 28-gauge needle, the needle was removed, the chest was closed, and the mouse was extubated. Sham-surgery mice underwent an identical procedure except for the aortic ligation. Mice were provided buprenorphine via ip. injection during recovery.
According to the manufacturer's instructions, caspase 3/7 activity was measured with a kit from Promega (Cat#: G8091).
Echocardiographic analysis using a Vevo2100 digital imaging system (Visual Sonics, Toronto, ON, Canada) was performed under 1% isoflurane at 6 and 10 weeks of age, with mid-ventricular M and B mode measurements acquired in the parasternal short-axis view at the level of the papillary muscles. Once the mice were acclimated to the procedures, images were stored digitally on a magnetic, optical disk for review and analysis. Measurements of the LV internal end-diastolic diameter (LVIDd) were taken at the apparent maximal LV diastolic dimension. In contrast, the LV internal end-systolic diameter (LVIDs) measurements were taken at the time of the most anterior systolic excursion of the posterior wall. LV ejection fraction (EF) was calculated by the cubic method: LVEF (%) = {(LVIDd)3 − (LVIDs)3}/(LVIDd)3 × 100, and LV fractional shortening (FS) was calculated by FS (%) = (LVIDd - LVIDs)/LVIDd × 100. The data were averaged from five cardiac cycles.
Histological analysis of heart tissues was as previously described (12). The CardioTACSTM in situ apoptosis detection kit (Roche Applied Science, Cat#: 11684795910) was used for TUNEL staining as previously described (35). Immunohistochemistry was performed on heart tissues with anti-γH2AX antibody and the DNA damage levels were determined by the percentage of γH2AX positive cells.
The following primer pairs were used for quantitative real-time PCR:
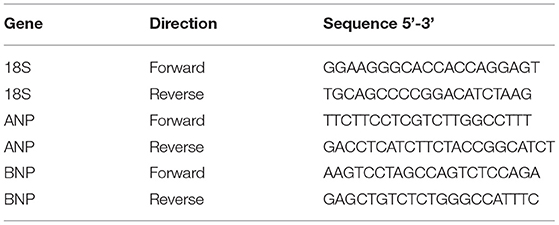
Amplification was performed as follows: 95°C for 3 s and 40 cycles at 95°C for 15 s and 60°C for 30 s. Data are the average of at least three independent experiments.
Neonatal rat ventricle myocytes (NRVMs) were isolated from 1-day-old Sprague-Dawley rats, and adenovirus-mediated gene transfer was implemented using methods described previously (35). NRVMs were exposed to KN-93 (5 μM) or isopropanol (ISO,10 μM) treatment.
Adult rat ventricle myocytes (ARVMs) were isolated from the hearts of 2–3-month-old Sprague-Dawley rats using a standard enzymatic technique, then cultured and infected with adenoviral vectors at a multiplicity of infection (MOI) indicated as described previously (35). Briefly, myocytes were plated at a density of 0.5 to 1 × 104/cm2 on coverslips or in dishes precoated with 10 μg/ml laminin. The culture medium was M199 (Sigma) plus 5 mmol/L creatine, 2 mmol/L l-carnitine, 5 mmol/L taurine, 0.1% insulin-transferrin-selenium-X, 1% penicillin and streptomycin, and 25 mmol/L HEPES, pH 7.4, at 37°C. Adenovirus-mediated gene transfer was implemented by adding adenoviral vectors (35) into the culture dish. The experiments were done with cells cultured 24 h after infection unless specified otherwise.
Western blot was performed as previously described (12).
For gene-silencing assays, siRNAs with 19 nucleotides in length, carrying a dTdT overhang at the 3' terminus, were designed using the Invitrogen website. Cardiomyocytes were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions (25).

Antibodies against the following proteins were used: UBE2T for mouse [Aviva Systems Biology, ARP-43145 (Lot#: QC13585-40506; 1:1000)], t-CaMKII-δ [GeneTex, GTX111401 (Lot#: 40058; 1:1000)], γH2AX (Millipore, 05-636, clone JBW301 (Lot#: 2884537; 1:1000 for western blots, 1:200 for immunohistochemistry), Cleaved Caspase-3 [Cell Signaling Technology, 9661 (1:1000)] and GAPDH [EASYBIO, BE0023, clone 2B8 (1:10000)]. Antibody against Exon 16 of CaMKII-δ9 was generated as described (35). ISO, z-VAD, and Doxorubicin were from Sigma-Aldrich. KN-93 was from Millipore (Cat# 422711).
Data are expressed as mean ± S.E.M. Statistical analysis was performed with GraphPad Prism version 8.4 (GraphPad Software, Inc.). Data sets were tested for normality of distribution with the Kolmogorov-Smirnov test. Data groups (two groups) with normal distributions were compared using the two-sided unpaired Student's t-test. The Mann-Whitney U-test was used for nonparametric data. One-way or two-way ANOVA assessed comparisons between multiple groups with Bonferroni post hoc analysis. *P < 0.05; **P < 0.01; NS, not significant. No statistical method was used to predetermine sample size.
First, we investigated whether CaMKII kinase activity was required for CaMKII-δ9-induced cardiomyocyte death, cardiomyopathy, and heart failure. KN-93 is the classic CaMKII inhibitor, inhibiting CaMKII kinase activity (37) and ameliorating multiple cardiac diseases in experimental models (38). We first set up cardiomyocyte injury models with cultured NRVMs with CaMKII-δ9 overexpression and found that pretreatment of KN-93 (5 μM) alleviates the CaMKII-δ9-induced cardiomyocyte death as indexed by caspase 3/7 activity (Figure 1A). We have previously shown that CaMKII-δ9 induced DNA damage in cardiomyocytes through the degradation of UBE2T (35). Here, we proved that in NRVMs, KN-93 suppressed CaMKII-δ9-mediated UBE2T degradation and subsequent DNA damage (Figures 1B,C).
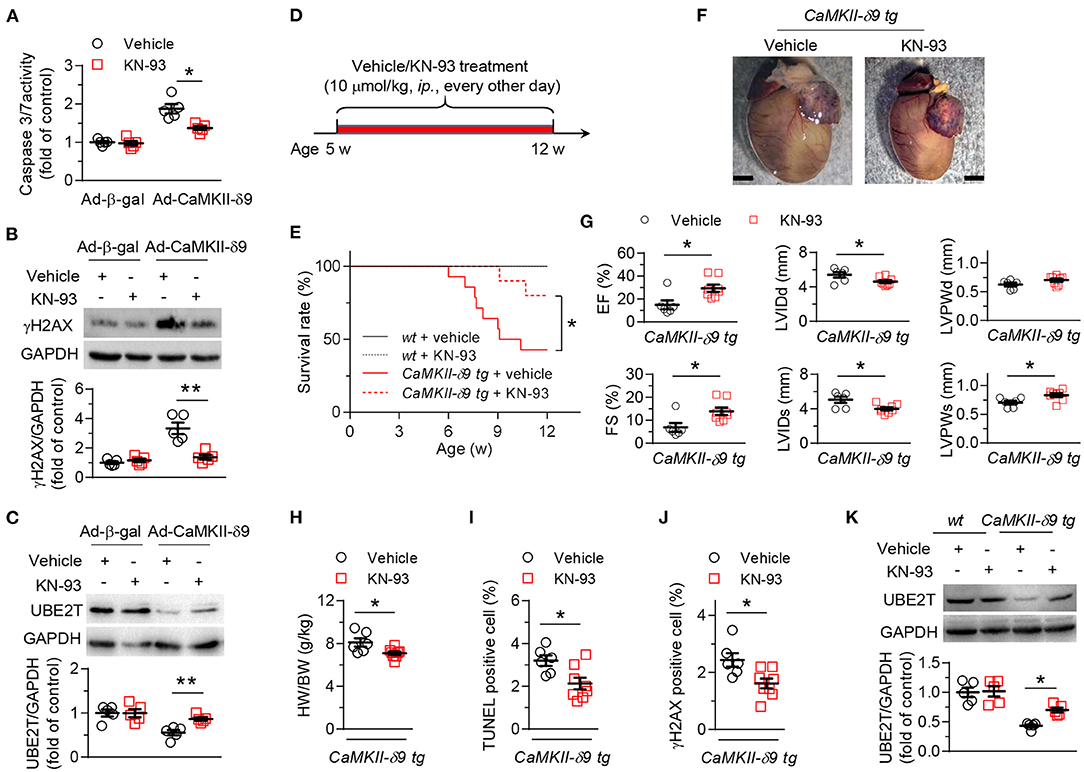
Figure 1. KN-93 treatment prevents CaMKII-δ9-induced cardiomyopathy and heart failure. (A–C), Cell viability assayed by caspase 3/7 activity (A) and representative western blots and statistical data showing the levels of γH2AX (B) and UBE2T (C) in NRVMs infected with Ad-β-gal or Ad-CaMKII-δ9 with or without KN-93 treatment (5 μM) (n = 5). (D) Experimental protocol for in vivo KN-93 treatment in CaMKII-d9 tg mice. The mice were treated with KN-93 (10 μmol/kg, i.p.) every other day from 5 to 12 weeks of age. (E) Kaplan-Meier survival curves of wt and CaMKII-δ9 tg mice with or without KN-93 treatment (n = 8 for wt vehicle, n = 7 for wt KN-93, n = 14 for CaMKII-δ9 tg vehicle, n = 10 for CaMKII-δ9 tg KN-93). (F–J) Gross morphology of the hearts (F), statistical data of the left ventricle echocardiography (G), ratio of heart weight to body weight (H), and statistical data of TUNEL (I) and γH2AX (J) staining of the hearts from CaMKII-δ9 tg mice with or without KN-93 treatment. EF, ejection fraction; FS, fractional shortening; LVIDd and LVIDs, diastolic and systolic left ventricular internal diameter; LVPWd and LVPWs, diastolic and systolic left ventricular posterior wall thickness. Scale bar, 2 mm (n = 6 for vehicle, n = 8 for KN-93). (K) Representative western blots and statistical data showing the levels of UBE2T from the hearts of wt and CaMKII-δ9 tg mice with or without KN-93 treatment (n = 5). Data are mean ± S.E.M. *P < 0.05, **P < 0.01; two-way ANOVA (A–C,K), log-rank (Mantel-Cox) test, or Student's t-test (E,G–J).
We have constructed the mice with cardiac-specific overexpression of CaMKII-δ9 (CaMKII-δ9 tg mice) (35). In CaMKII-δ9 tg mice, treatment with KN-93 (10 μmol/kg, ip., every other day) from the age of 5 weeks attenuates premature animal death, cardiac hypertrophy, myocardial dysfunction, and cardiomyocyte DNA damage and cell death (Figures 1D–K). Therefore, CaMKII kinase activity is required for CaMKII-δ9-induced cardiomyocyte death, cardiomyopathy, and heart failure.
Based on the phenotypes of the CaMKII-δ9 tg mice, there are three possible functions of CaMKII-δ9 in the heart: First, it directly induces cardiomyocyte hypertrophy, which indirectly causes myocyte death when hypertrophy progresses to decompensation, or it directly induces cardiomyocyte death, and the surviving myocytes compensate by becoming hypertrophic, or both.
To distinguish these possibilities, we first treated the CaMKII-δ9 tg mice with z-VAD (5 mg/kg, ip., twice a week), a caspase inhibitor, from the age of 5 weeks to inhibit cardiomyocyte death (Figure 2A). Our data showed that the premature animal death in CaMKII-δ9 tg mice was markedly attenuated by z-VAD treatment (Figure 2B). Furthermore, cardiomyocyte death, cardiac hypertrophy, and heart dysfunction in CaMKII-δ9 tg mice were also ameliorated by z-VAD (Figures 2C–E), indicating that the cytosolic execution of apoptosis contributes to CaMKII-δ9-induced cardiomyocyte death, and cardiomyocyte death is essential for CaMKII-δ9-induced cardiomyocyte death, cardiomyopathy, and heart failure.
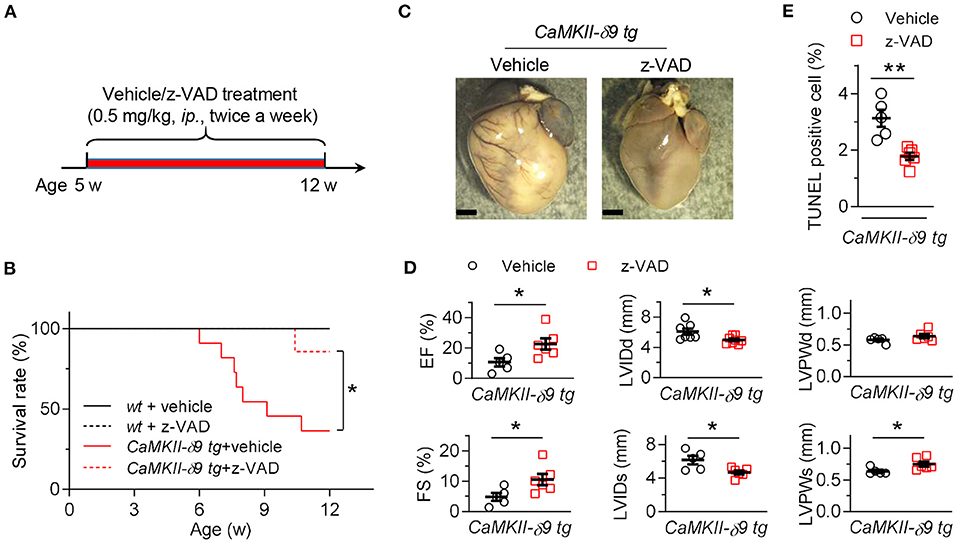
Figure 2. Inhibition of cardiomyocyte death alleviates CaMKII-δ9-induced cardiomyopathy and heart failure. (A) Experimental protocol for in vivo z-VAD treatment in CaMKII-δ9 tg mice. The mice were treated with z-VAD (0.5 mg/kg, i.p.) twice a week from 5 to 12 weeks of age. (B) Kaplan-Meier survival curves of wt and CaMKII-δ9 tg mice with or without z-VAD treatment (n = 6 for wt vehicle, n = 5 for wt z-VAD, n = 11 for CaMKII-δ9 tg vehicle, n = 7 for CaMKII-δ9 tg z-VAD). (C–E) Gross morphology of the hearts (C), statistical data of the left ventricle echocardiography (D), and statistical data of TUNEL staining of the hearts (E) from CaMKII-δ9 tg mice with or without z-VAD treatment. Scale bar, 2 mm (n = 5 for vehicle, n = 6 for z-VAD). Data are mean ± S.E.M. NS, not significant; *P < 0.05, **P < 0.01; log-rank (Mantel-Cox) test (B), or Student's t-test (D,E).
We next investigated the role of CaMKII-δ9 in cardiomyocyte hypertrophy. In the mouse hearts two weeks after sham or TAC surgery, CaMKII-δ protein abundance was markedly increased (Figure 3A). To avoid complex in vivo compensations, we used cultured NRVMs in conjunction with adenoviral gene transfer. We found that although total CaMKII-δ protein was upregulated in hypertrophic hearts, overexpression of CaMKII-δ9 did not alter the expression of cardiac hypertrophic genes, including atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). In contrast, as a positive control, isoproterenol treatment profoundly increased their expression (Figure 3B). Moreover, the knockdown of CaMKII-δ9 did not alter the hypertrophy phenotype in NRVM treated with isopropanol (Figures 3C,D). These data indicate that cardiac CaMKII-δ9 is not directly involved in cardiomyocyte hypertrophy. The myocardial hypertrophy in CaMKII-δ9 tg mice is the compensatory response of the surviving cardiomyocyte to maintain cardiac function.
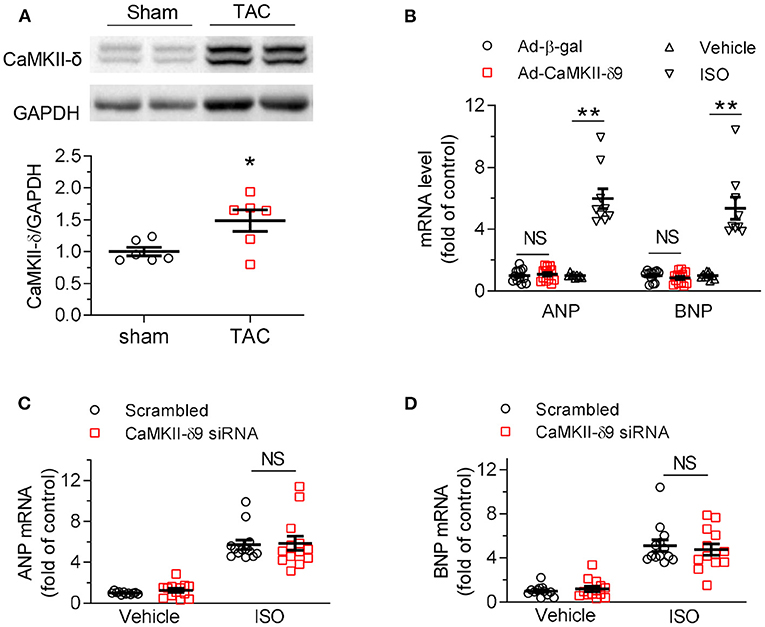
Figure 3. CaMKII-δ9 does not directly regulate cardiac hypertrophy. (A) Representative western blots and statistical data showing the levels of CaMKII-δ in the hearts of the mice 2 weeks after sham or transverse aortic constriction (TAC) surgery. n = 6. (B) Averaged data of mRNA levels of ANP and BNP assayed by real-time PCR in NRVMs infected with Ad-β-gal or Ad-CaMKII-δ9 (50 MOI, 48 h; Ad-β-gal, n = 12; Ad-CaMKII-δ9, n = 13). The NRVMs treated with isoproterenol (ISO, 10 mM, 24 h) were used as positive control (n = 9). (C,D) Averaged data of mRNA levels of ANP (C) and BNP (D) assayed by real-time PCR in NRVMs transfected with scrambled or CaMKII-δ9 siRNA with or without ISO treatment (10 μM, 24 h; Vehicle scrambled, n = 12; other groups, n = 13). Data are mean ± S.E.M. *P < 0.05, **P < 0.01; NS, not significant; Student's t-test (A,B), or two-way ANOVA (C,D).
Our previous study has shown that CaMKII-δ9 binds to ubiquitin-conjugating enzyme E2T (UBE2T) to promote its phosphorylation and degradation, disrupting UBE2T-dependent DNA repair and leading to the accumulation of DNA damage and genome instability, which results in cardiomyocyte death (35). However, the study was performed in the context of immature cardiomyocytes, including NRVMs and embryonic stem cell-derived cardiomyocytes. Given the differences between immature and mature cardiomyocytes in terms of morphology, gene expression, and proliferation capacity, here, we investigated the role of CaMKII-δ9 in adult cardiomyocytes, together with the underlying mechanisms.
First, we compared the protein abundance of CaMKII-δ9 in NRVMs with the adult hearts of multiple species, including human, monkey, rat, and mice (Figure 4A). Since these four species share the same amino-acid sequence encoded by exon 16 of CaMKII-δ (Figure 4B), the anti-exon 16 antibodies would be expected to work equally in these species. The protein abundance of CaMKII-δ9 in the heart of human, monkey, mice, and rat (adult) was 3.54 ± 0.19, 3.25 ± 0.44, 2.00 ± 0.20 and 1.53 ± 0.31-fold of that in NRVMs, respectively (Figure 4A), suggesting that the levels of CaMKII-δ9 are higher in adult hearts.
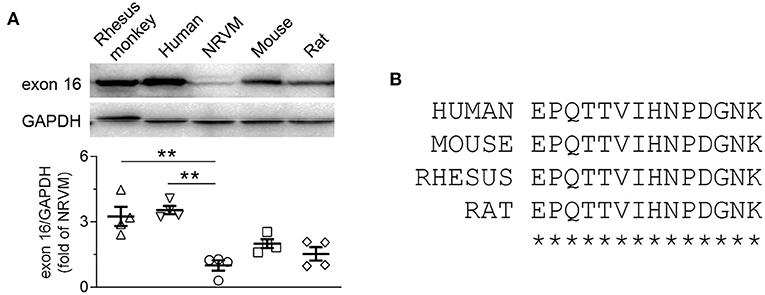
Figure 4. CaMKII-δ9 is abundant in the adult hearts of different species. (A) Representative western blots and statistical data showing the levels of CaMKII-δ9 in the hearts of rhesus monkey and human, neonatal rat ventricular myocytes (NRVMs), adult mouse together with rat (n = 4). Data are mean ± S.E.M. **P < 0.01; one-way ANOVA. (B) Amino acid sequences of the peptides encoded by the exon 16 of CaMKII-δ of human, mouse, rhesus monkey and rat.
Functionally, our data indicated that similar to the findings in NRVMs, at a comparable expression level, CaMKII-δ9 elicited much more severe cell death, DNA damage, and UBE2T degradation than CaMKII-δ2 in ARVMs (Figures 5A–C). Consistently, knockdown of CaMKII-δ9 with its specific siRNA prevented doxorubicin-induced cell death, DNA damage, and UBE2T degradation in ARVMs (Figures 5D–F). These data indicate that enhanced CaMKII-δ9 activation triggers the death of both adult and neonatal cardiomyocytes by suppressing UBE2T-dependent DNA repair signaling. Therefore, although CaMKII-δ9 differs in protein abundance between neonatal and adult cardiomyocytes, its function and downstream signaling are the same.
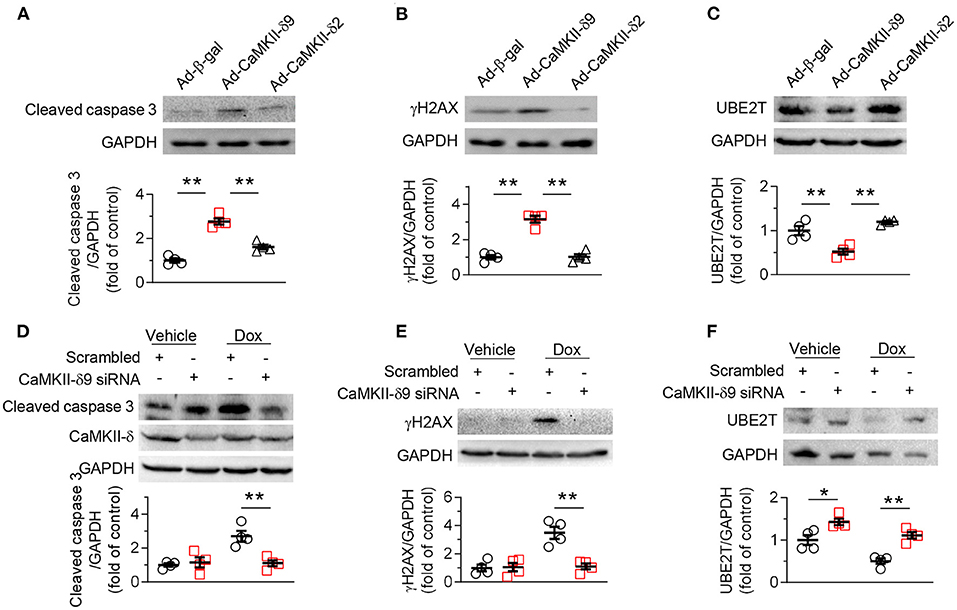
Figure 5. CaMKII-δ9 mediates DNA damage and cell death in adult cardiomyocytes. (A–C) Representative western blots and statistical data showing the levels of cleaved caspase 3 (A) γH2AX (B) and UBE2T (C) in adult rat ventricular myocytes (ARVMs) infected with Ad-β-gal, Ad-CaMKII-δ9, or Ad-CaMKII-δ2 (100 MOI, 24 h) (n = 4). (D–F) Representative western blots and statistical data showing the levels of cleaved caspase 3 (D), γH2AX (E) and UBE2T (F) in ARVMs infected with scrambled or CaMKII-δ9 siRNA with or without Dox treatment (1 mM, 16 h) (n = 4). Data are mean ± S.E.M. *P < 0.05, **P < 0.01; one-way ANOVA (A–C) or two-way ANOVA (D–F).
Previous studies have shown significant differences in the role of CaMKII-δ2 and -δ3 in males and females, and CaMKII activation is not necessarily deleterious in female cardiopathology (39). Thus, in order to fully understand the clinical significance of CaMKII-δ9 in the prevention and therapy of heart failure, we compared the possible gender differences of CaMKII-δ9 in terms of its expression and function in animals.
We found that there was no gender difference of myocardial CaMKII-δ9 abundance in wt and CaMKII-δ9 tg mice (Figures 6A,B). In addition, animal viability, cardiac morphology and function, as well as cardiomyocyte apoptosis and DNA damage did not differ, either (Figures 6C–F). In this way, there is no gender difference in terms of the expression and function of CaMKII-δ9 in the mice hearts.
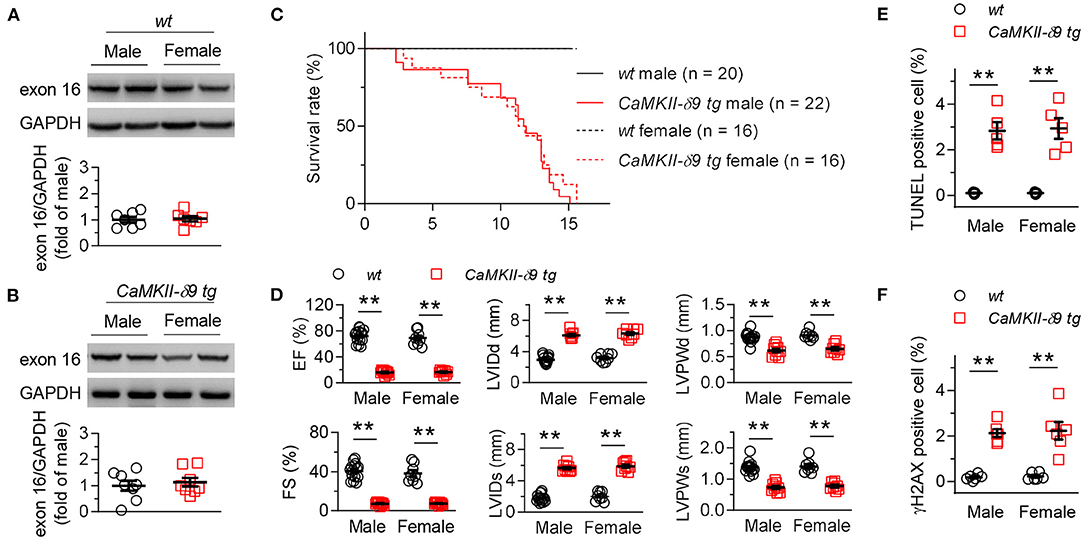
Figure 6. No gender difference in the CaMKII-δ9-induced cardiac pathology. (A,B) Representative western blots and statistical data showing the levels of CaMKII-δ9 from the hearts of wt (A, n = 7) and CaMKII-δ9 tg mice (B, n = 8). (C) Kaplan-Meier survival curves of male and female wt and CaMKII-δ9 tg mice (n = 20 for wt male, n = 22 for CaMKII-δ9 tg male, n = 16 for wt female and CaMKII-δ9 tg female). (D) Statistical data of the left ventricle echocardiography of male and female wt and CaMKII-δ9 tg mice at 10 weeks of age (n = 15 for wt male, n = 9 for CaMKII-δ9 tg male, n = 8 for wt female and CaMKII-δ9 tg female). EF, ejection fraction; FS, fractional shortening; LVIDd and LVIDs, diastolic and systolic left entricular internal diameter; LVPWd and LVPWs, diastolic and systolic left ventricular posterior wall thickness. (E,F) Statistical data of TUNEL (E, n = 5) and γH2AX (F, n = 6) staining of the hearts of male and female wt and CaMKII-δ9 tg mice at the age of 10 weeks (n = 5). Data are mean ± S.E.M. **P < 0.01; Student's t-test (A,B), log-rank (Mantel-Cox) test (C), or two-way ANOVA (D–F).
In the current study, we demonstrate the pathophysiological function of CaMKII-δ9 in the development of cardiomyopathy and heart failure, especially distinguished its role in hypertrophy and cardiomyocyte death. Specifically, CaMKII-δ9 mediates cardiomyocyte death, instead of hypertrophy, to promote the progression of cardiomyopathy and heart failure. In addition, we show that the function and the corresponding signaling pathway of CaMKII-δ9 are similar in immature and mature cardiomyocytes without gender difference.
Heart failure is a global public health problem and heavy finical burden to the patients. Despite significant efforts in the basic and clinical research to pursue the strategy of its prevention and therapy, it remains a huge unmet medical need. Cardiomyocyte death plays an essential role in the progression of cardiomyopathy and heart failure, and the inhibition of cardiomyocyte death alleviated heart failure (40–42). CaMKII is a key player in mediating cardiomyocyte death, and inhibition of CaMKII profoundly protects the cardiomyocyte against cell death induced by multiple pathological insults.
Further studies showed that different CaMKII-δ splice variants exert opposite functions in regulating cardiac cell viability. The cytosolic variant, CaMKII-δ2 (also named CaMKII-δC) facilities cardiomyocyte death, whereas the nuclear variant, CaMKII-δ3 (also named CaMKII-δB), is protective (25, 31–33). We recently provided evidence that CaMKII-δ9, instead of CaMKII-δ2, is the most critical cytosolic CaMKII variant in the human heart (35). Functionally, CaMKII-δ9 is more potent in inducing cardiomyocyte death than CaMKII-δ2 and plays an essential role in developing cardiomyopathy and heart failure (35). Here, we further demonstrate that CaMKII-δ9 directly elicits cardiomyocyte death, but not cardiac hypertrophy, to mediate the progression of heart failure. Thus, we showed a clear picture of the pathophysiological action of CaMKII-δ9, the major variant in human hearts, in the mediation of cardiomyopathy and heart failure, suggesting a new therapeutic strategy to target CaMKII-δ9 against human heart failure. Importantly, our data proved that CaMKII-δ9 downregulated UBE2T, impaired DNA repair machinery, and consequently elicited cardiomyocyte death and heart failure in the adult hearts of both male and female animals, which further enhances the clinical perspective of CaMKII-δ9 in the therapy of cardiac diseases.
During heart failure, CaMKII (in the human heart, mainly CaMKII-δ9) was activated by multiple cardiac pathological insults, including neurohumoral agonist signaling (43), oxidant stress (44–47), hyperglycemia (48, 49), ischemic injury (12, 13, 50–52), cardiac toxic drugs (12, 53), and other adverse stimuli associated with increased intracellular calcium levels (54, 55). The activated CaMKII mediates the phosphorylation of Ca2+ homeostatic proteins to enhance their activity and improve the performance of physiological events such as excitation-contraction coupling and fight/flight mechanical responses, which helps maintain normal cardiac function. However, excessive CaMKII activation caused by continuous myocardial stress promotes cardiac myocyte death and the deterioration of cardiomyopathy. We recently identified DNA damage as the specific downstream effector of CaMKII-δ9 in the induction of cardiac injury, and CaMKII-δ9 is much more potent in the induction of cardiomyocyte death than CaMKII-δ2 (35). In addition, some other mechanisms including inflammation (46, 50, 52, 56), mitochondrial stress (57), endoplasmic reticulum stress (47, 58–60), and p53 activation (61) have been shown to act as the downstream signaling of CaMKII-induced cardiomyocyte death. But compared with other splice variants, especially CaMKII-δ2, whether CaMKII-δ9 exerts similar functions in regulating these mechanisms still merits further investigation.
CaMKII has also been established to be a central mediator of cardiomyocyte hypertrophy. CaMKII is activated during cardiac hypertrophy, and inhibition of CaMKII profoundly alleviates myocardial hypertrophy, cardiomyopathy, and heart failure (22, 62, 63). Mechanically, CaMKII phosphorylates multiple substrates, including myocyte enhancer factor 2 (MEF2), histone deacetylases (HDACs), and histone H3, to induce a hypertrophic transcriptional response in cardiomyocytes (64–66). But all the previous studies are based on CaMKII-δ2 and CaMKII-δ3. Here in our study, we showed that different from the other variants, CaMKII-δ9 did not directly increase the expression levels of the hypertrophic genes in cardiomyocytes, and inhibition of CaMKII-δ9 failed to block isoproterenol-induced cardiomyocyte hypertrophy, implicating that CaMKII-δ9 is not involved in the regulation of cardiac hypertrophic response. In human cardiomyocytes, CaMKII-δ3 and CaMKII-δ9 are the major CaMKII splice variants, localized in the nuclei and cytosol, respectively. Our current data combined with the previous studies suggest that there is functional specialization of these variants in addition to the distinct subcellular localization. CaMKII-δ9 is responsible for regulating calcium handling and contraction and induces cardiomyocyte death; on the other hand, CaMKII-δ3 mediates the hypertrophic gene expression and protects against cardiac cell death. Therefore, the specific intervention of CaMKII-δ splice variant to target specific cardiac physiological and pathological functions is a promising strategy to improve the therapy of heart failure and other cardiac diseases.
In addition to ischemic heart diseases, heart failure can be caused by many other diseases and pathological conditions, including hypertension, diabetes, and anti-cancer drugs. Since CaMKII has been shown to play a central role in cardiomyocyte death induced by multiple insults (12, 13, 24, 25), we postulate that CaMKII-δ9-mediated cardiomyocyte death may be involved in heart failure elicited by various pathological insults.
In conclusion, we provided the evidence that CaMKII-δ9 mediates cardiomyocyte death, but not cardiac hypertrophy, to elicit cardiomyopathy and heart failure. Furthermore, CaMKII-δ9 promotes cardiomyocyte death and heart failure in both male and female animals. Our findings not only deepen our understanding of the role of CaMKII-δ9 in cardiac pathology but also provide new insights into the mechanisms and therapy of heart failure.
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Peking University.
MZ, JZ, and YZ proposed the hypothesis, generated the initial idea, conducted key experiments and data analysis, and wrote the manuscript. WZha, QH, LJ, PX, WZhe, and HS researched data and contributed to discussion. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (2018YFA0800501), and the National Natural Science Foundation of China (92168114, 31671177, 81630008, 81790621, 81872752, and 31521062).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank D. Ma, X. Sun, and J. H. Liu for their excellent technical support.
1. Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, et al. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. (2013) 6:606–19. doi: 10.1161/HHF.0b013e318291329a
2. Savarese G, Lund LH. Global Public Health Burden of Heart Failure. Card Fail Rev. (2017) 3:7–11. doi: 10.15420/cfr.2016:25:2
3. Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. (2003) 111:1497–504. doi: 10.1172/JCI17664
4. Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. (2003) 92:715–24. doi: 10.1161/01.RES.0000067471.95890.5C
5. Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. (1975) 56:56–64. doi: 10.1172/JCI108079
6. Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. (2006) 367:356–67. doi: 10.1016/S0140-6736(06)68074-4
7. Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, et al. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. (2014) 63:1123–33. doi: 10.1016/j.jacc.2013.11.053
8. Blecker S, Paul M, Taksler G, Ogedegbe G, Katz S. Heart failure-associated hospitalizations in the United States. J Am Coll Cardiol. (2013) 61:1259–67. doi: 10.1016/j.jacc.2012.12.038
9. Erickson J, He B, Grumbach I, Anderson M. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev. (2011) 91:889–915. doi: 10.1152/physrev.00018.2010
10. Feng N, Anderson M. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J Mol Cell Cardiol. (2017) 103:102–9. doi: 10.1016/j.yjmcc.2016.12.007
11. Dewenter M. von der Lieth A, Katus H, Backs J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circulat Res. (2017) 121:1000–20. doi: 10.1161/CIRCRESAHA.117.310355
12. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. (2016) 22:175–82. doi: 10.1038/nm.4017
13. Joiner M, Koval O, Li J, He B, Allamargot C, Gao Z, et al. CaMKII determines mitochondrial stress responses in heart. Nature. (2012) 491:269–73. doi: 10.1038/nature11444
14. He B, Joiner M, Singh M, Luczak E, Swaminathan P, Koval O, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med. (2011) 17:1610–8. doi: 10.1038/nm.2506
15. Zhang R, Khoo M, Wu Y, Yang Y, Grueter C, Ni G, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. (2005) 11:409–17. doi: 10.1038/nm1215
16. van Oort R, McCauley M, Dixit S, Pereira L, Yang Y, Respress J, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. (2010) 122:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298
17. Mesubi O, Anderson M. Atrial remodelling in atrial fibrillation: CaMKII as a nodal proarrhythmic signal. Cardiovasc Res. (2016) 109:542–57. doi: 10.1093/cvr/cvw002
18. Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr, Bers DM, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. (2003) 92:912–9. doi: 10.1161/01.RES.0000069686.31472.C5
19. Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. (2009) 119:1230–40. doi: 10.1172/JCI38022
20. Neef S, Steffens A, Pellicena P, Mustroph J, Lebek S, Ort KR, et al. Improvement of cardiomyocyte function by a novel pyrimidine-based CaMKII-inhibitor. J Mol Cell Cardiol. (2018) 115:73–81. doi: 10.1016/j.yjmcc.2017.12.015
21. Lebek S, Plößl A, Baier M, Mustroph J, Tarnowski D, Lücht C, et al. The novel CaMKII inhibitor GS-680 reduces diastolic SR Ca leak and prevents CaMKII-dependent pro-arrhythmic activity. J Mol Cell Cardiol. (2018) 118:159–68. doi: 10.1016/j.yjmcc.2018.03.020
22. Beauverger P, Ozoux M, Bégis G, Glénat V, Briand V, Philippo M, et al. Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin-dependent protein kinase II inhibitor, RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc Res. (2020) 116:329–38. doi: 10.1093/cvr/cvz097
23. Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. (2010) 107:1150–61. doi: 10.1161/CIRCRESAHA.110.220418
24. Wu Y, Wang Q, Feng N, Granger JM, Anderson ME. Myocardial death and dysfunction after ischemia-reperfusion injury require CaMKIIdelta oxidation. Sci Rep. (2019) 9:9291. doi: 10.1038/s41598-019-45743-6
25. Peng W, Zhang Y, Zheng M, Cheng H, Zhu W, Cao CM, et al. Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ Res. (2010) 106:102–10. doi: 10.1161/CIRCRESAHA.109.210914
26. Baier MJ, Klatt S, Hammer KP, Maier LS, Rokita AG. Ca(2+)/calmodulin-dependent protein kinase II is essential in hyperacute pressure overload. J Mol Cell Cardiol. (2020) 138:212–21. doi: 10.1016/j.yjmcc.2019.12.002
27. Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. (2006) 116:1853–64. doi: 10.1172/JCI27438
28. Mayer P, Möhlig M, Idlibe D, Pfeiffer A. Novel and uncommon isoforms of the calcium sensing enzyme calcium/calmodulin dependent protein kinase II in heart tissue. Basic Res Cardiol. (1995) 90:372–9. doi: 10.1007/BF00788498
29. Mayer P, Möhlig M, Schatz H, Pfeiffer A. Additional isoforms of multifunctional calcium/calmodulin-dependent protein kinase II in rat heart tissue. Biochem J. (1994) 298:757-8. Epub 1994/03/15. doi: 10.1042/bj2980757
30. Hoch B, Meyer R, Hetzer R, Krause E, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. (1999) 84:713–21. doi: 10.1161/01.RES.84.6.713
31. Little G, Saw A, Bai Y, Dow J, Marjoram P, Simkhovich B, et al. Critical role of nuclear calcium/calmodulin-dependent protein kinase IIdeltaB in cardiomyocyte survival in cardiomyopathy. J Biol Chem. (2009) 284:24857–68. doi: 10.1074/jbc.M109.003186
32. Gray C, Suetomi T, Xiang S, Mishra S, Blackwood E, Glembotski C, et al. CaMKIIδ subtypes differentially regulate infarct formation following ex vivo myocardial ischemia/reperfusion through NF-κB and TNF-α. J Mol Cell Cardiol. (2017) 103:48–55. doi: 10.1016/j.yjmcc.2017.01.002
33. Zhu W, Wang S, Chakir K, Yang D, Zhang T, Brown J, et al. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. (2003) 111:617–25. doi: 10.1172/JCI200316326
34. Zalcman G, Federman N, Romano A. CaMKII isoforms in learning and memory: localization and function. Front Mol Neurosci. (2018) 11:445. doi: 10.3389/fnmol.2018.00445
35. Zhang M, Gao H, Liu D, Zhong X, Shi X, Yu P, et al. CaMKII-delta9 promotes cardiomyopathy through disrupting UBE2T-dependent DNA repair. Nat Cell Biol. (2019) 21:1152–63. doi: 10.1038/s41556-019-0380-8
36. Zhang X, Zhang R, Raab S, Zheng W, Wang J, Liu N, et al. Rhesus macaques develop metabolic syndrome with reversible vascular dysfunction responsive to pioglitazone. Circulation. (2011) 124:77–86. doi: 10.1161/CIRCULATIONAHA.110.990333
37. Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, et al. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. (1991) 181:968–75. doi: 10.1016/0006-291X(91)92031-E
38. Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. (2014) 5:21. doi: 10.3389/fphar.2014.00021
39. Bell JR, Raaijmakers AJ, Curl CL, Reichelt ME, Harding TW, Bei A, et al. Cardiac CaMKIIdelta splice variants exhibit target signaling specificity and confer sex-selective arrhythmogenic actions in the ischemic-reperfused heart. Int J Cardiol. (2015) 181:288–96. doi: 10.1016/j.ijcard.2014.11.159
40. Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, et al. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. (2011) 2:e244. doi: 10.1038/cddis.2011.130
41. Huang F, Yang R, Xiao Z, Xie Y, Lin X, Zhu P, et al. Targeting ferroptosis to treat cardiovascular diseases: a new continent to be explored. Front Cell Dev Biol. (2021) 9:737971. doi: 10.3389/fcell.2021.737971
42. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
43. Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, et al. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. (2006) 291:H3065–75. doi: 10.1152/ajpheart.00353.2006
44. Luczak ED, Anderson ME. CaMKII oxidative activation and the pathogenesis of cardiac disease. J Mol Cell Cardiol. (2014) 73:112–6. doi: 10.1016/j.yjmcc.2014.02.004
45. Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. (2008) 133:462–74. doi: 10.1016/j.cell.2008.02.048
46. Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, et al. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest. (2009) 119:986–96. doi: 10.1172/JCI35814
47. Timmins JM, Ozcan L, Seimon TA Li G, Malagelada C, Backs J, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. (2009) 119:2925–41. doi: 10.1172/JCI38857
48. Zhu W, Tsang S, Browe DM, Woo AY, Huang Y, Xu C, et al. Interaction of beta1-adrenoceptor with RAGE mediates cardiomyopathy via CaMKII signaling. JCI Insight. (2016) 1:e84969. doi: 10.1172/jci.insight.84969
49. Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. (2013) 123:1262–74. doi: 10.1172/JCI65268
50. Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, et al. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. (2013) 112:935–44. doi: 10.1161/CIRCRESAHA.112.276915
51. Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, et al. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. (2007) 73:689–98. doi: 10.1016/j.cardiores.2006.12.003
52. Weinreuter M, Kreusser MM, Beckendorf J, Schreiter FC, Leuschner F, Lehmann LH, et al. CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med. (2014) 6:1231–45. doi: 10.15252/emmm.201403848
53. Sag CM, Kohler AC, Anderson ME, Backs J, Maier LS. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J Mol Cell Cardiol. (2011) 51:749–59. doi: 10.1016/j.yjmcc.2011.07.016
54. Altschuld RA, Starling RC, Hamlin RL, Billman GE, Hensley J, Castillo L, et al. Response of failing canine and human heart-cells to beta (2)-adrenergic stimulation. Circulation. (1995) 92:1612–8. PubMed PMID: WOS:A1995RU67200037. doi: 10.1161/01.CIR.92.6.1612
55. Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. (2007) 282:10833–9. doi: 10.1074/jbc.M611507200
56. Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. (2012) 52:1135–44. doi: 10.1016/j.yjmcc.2012.01.021
57. Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, et al. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res. (2005) 97:1009–17. doi: 10.1161/01.RES.0000189270.72915.D1
58. Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. (2012) 32:1552–62. doi: 10.1161/ATVBAHA.111.224915
59. Roe ND, Ren J. Oxidative activation of Ca(2+)/calmodulin-activated kinase II mediates ER stress-induced cardiac dysfunction and apoptosis. Am J Physiol Heart Circ Physiol. (2013) 304:H828–39. doi: 10.1152/ajpheart.00752.2012
60. Bracken C, Beauverger P, Duclos O, Russo RJ, Rogers KA, Husson H, et al. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. Am J Physiol Renal Physiol. (2016) 310:F1414–22. doi: 10.1152/ajprenal.00426.2015
61. Toko H, Takahashi H, Kayama Y, Oka T, Minamino T, Okada S, et al. Ca2+/calmodulin-dependent kinase IIdelta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation. (2010) 122:891–9. doi: 10.1161/CIRCULATIONAHA.109.935296
62. Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. (2011) 51:468–73. doi: 10.1016/j.yjmcc.2011.01.012
63. Cheng J, Xu L, Lai D, Guilbert A, Lim HJ, Keskanokwong T, et al. CaMKII inhibition in heart failure, beneficial, harmful, or both. Am J Physiol Heart Circ Physiol. (2012) 302:H1454–65. doi: 10.1152/ajpheart.00812.2011
64. Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. (2000) 105:1395–406. doi: 10.1172/JCI8551
65. Nakagawa Y, Kuwahara K, Harada M, Takahashi N, Yasuno S, Adachi Y, et al. Class II HDACs mediate CaMK-dependent signaling to NRSF in ventricular myocytes. J Mol Cell Cardiol. (2006) 41:1010–22. doi: 10.1016/j.yjmcc.2006.08.010
Keywords: CaMKII, CaMKII-δ9, cardiomyocyte death, cardiomyopathy, heart failure, hypertrophy
Citation: Zhang M, Zhang J, Zhang W, Hu Q, Jin L, Xie P, Zheng W, Shang H and Zhang Y (2022) CaMKII-δ9 Induces Cardiomyocyte Death to Promote Cardiomyopathy and Heart Failure. Front. Cardiovasc. Med. 8:820416. doi: 10.3389/fcvm.2021.820416
Received: 23 November 2021; Accepted: 21 December 2021;
Published: 20 January 2022.
Edited by:
Hui Gong, Fudan University, ChinaReviewed by:
Heng Ma, Fourth Military Medical University, ChinaCopyright © 2022 Zhang, Zhang, Zhang, Hu, Jin, Xie, Zheng, Shang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhang, emhhbmd5YW45ODc2QHBrdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.