 Xing Li†
Xing Li† Sha Lin
Sha Lin Tao Wang
Tao Wang Kaiyu Zhou
Kaiyu Zhou Yifei Li
Yifei Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 23 December 2021
Sec. Pediatric Cardiology
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.810291
This article is part of the Research Topic Case Reports in Pediatric Cardiology: 2021 View all 8 articles
Background: Hypertrophic cardiomyopathy (HCM) is the second most common cardiomyopathy in childhood with a life-threatening risk. Implantable cardioverter-defibrillator (ICD) placement is recommended for early prevention if there are two or more clinical risk factors. Pediatric patients with HCM are at a higher risk of sudden cardiac death (SCD), but there are limited reports on indications for ICD implantation in children. Herein we describe the case of Myh7 mutation-induced HCM and cardiac arrest in a patient and evaluated information originating from genetic background to guide ICD administration.
Case Presentation: The patient was a girl aged 7 years and 8 months who had been diagnosed with cardiomyopathy in utero 8 years prior. She had had recurrent cardiac arrests within the last 4 years. Electrocardiography indicated abnormalities in conduction, and ST segment changes. Echocardiography indicated significant left ventricular hypertrophy and hypertrophic systolic interventricular septum. Cardiac magnetic resonance imaging depicted general heart enlargement with hypertrophy, and delayed enhancement in myocardium with perfusion defect was also evident. Whole exon sequencing identified a de novo c.2723T>C (p.L908P) heterozygous mutation in the MYH7 gene. MYH7 p.L908P predicted unstable protein structure and impaired function. The patient was scheduled for ICD implantation. There were no complications after ICD implantation, and she was discharged from hospital on the 10th day. Regular oral beta-blockers, amiodarone, spironolactone, and enalapril were administered, and she was required to attend hospital regularly for follow-up. During follow-up there were no cardiac arrests. Literature review of clinical prognoses associated with genetic mutations of MYH7, MYBPC3, TNNI3, TNNT2, and TPM1 in pediatric HCM patients with and without ICD implantation indicated that they were totally differently. Previous reports also indicated that gene mutations predicted earlier onset of cardiac hypertrophy, and increase likelihood of SCD.
Conclusion: Variant burden and variant type contribute to the risk of adverse events in pediatric HCM. Early recognition and intervention are vital in children. Gene mutation could be considered an indication for early ICD placement during standard risk stratification of HCM patients. Whether this extends to the majority of pediatric patients requires further investigation.
Hypertrophic cardiomyopathy (HCM) is the second most common myocardial disease in childhood, with an annual incidence of ~0.24–0.47 per 100,000. Characterized by unexplained left ventricular hypertrophy (LVH), the typical histological manifestation is uneven distribution of myocardium. The causes of HCM are heterogeneous in children, and include congenital metabolic defects, chromosome-related syndromes, and neuromuscular diseases (1, 2). More than 1,400 rare variants in at least 8 sarcomere-related genes have been linked to HCM, with variations in the MYH7 and MYBPC3 genes the most common. Most cases of child-onset HCM are familial/hereditary however, and are caused by mutations in genes encoding the sarcomere, cytoskeleton, or desmosome protein (3).
Children with HCM are at greater risk of sudden cardiac death (SCD) and other adverse events (4). Both the European Society of Cardiology and the American Heart Association recommend implantable cardioverter defibrillators (ICDs) for early prevention if there are two or more clinical risk factors, including severe LVH, unexplained syncope, cardiac arrest, non-sustained ventricular tachycardia, and a family history of SCD (5, 6). However, there is no practical guideline for pediatric patients. Moreover, there is no genetic factor involved in analyzing the risks of pediatric HCM patients suffering SCD. Previous studies indicate that patients with gene mutations are more likely to experience poor outcomes (7). Herein we describe the case of a patient with de novo MYH7 mutation-induced HCM and recurrent cardiac arrest, then present related literature to assess the influence of genetic risk on early ICD intervention in pediatric HCM.
This report was approved by the Ethics Committee of the West China Second Hospital of Sichuan University (approval number 2014-034). Informed consent was obtained from the patient's parents prior to performing whole exon sequencing, and for the inclusion of the patient's clinical and imaging details in subsequent publications.
The patient was a girl aged 7 years and 8 months who had been diagnosed with cardiomyopathy in utero 8 years prior. She had had recurrent cardiac arrest attacks within the last 4 years (from 2015 to 2021). On each of these occasions she had been saved via immediate chest compressions and other cardiopulmonary resuscitation methods. Seizures had been observed two or three times per year, which were ~2–3 min in duration. Her daily activity tolerance was limited, which indicated a NYHA II to III level of heart function. Her parents had no relevant clinical symptoms and were unaware of any family history of cardiovascular diseases or SCD. At rest the patient's ventricular rate was 100–110 beats/min, her breathing rate was 25–35 breaths/min, and her blood pressure was ~95/55 mmHg. Arrhythmia and dull heart sound were detected. Pulmonary, abdominal, musculoskeletal system, and neurological examination results were unremarkable.
Electrocardiography (ECG) revealed electrical axis deviation to the right, complete right bundle branch block (CRBBB), abnormal double atrium, ventricular hypertrophy, first-degree atrioventricular block, ST segment changes (lead I, aVL, and V3–V6 depression >0.05 mV) (Figure 1A). Notably however, ventricular tachycardia had been observed three times on a monitor and had failed to be recorded before electric cardioversion. Echocardiography have been performed by the same echocardiographist to make the data comparable, and the echocardiography pre-ICD implantation indicated significant LVH and hypertrophic interventricular septum (19 mm, Z-score = 5.64) at the end of diastole with normal left ventricular systolic function (62–71.6%) (Figure 1B). Cardiac magnetic resonance imaging demonstrated general heart enlargement with LVH. Delayed enhancement in the myocardium with perfusion defect were also detected, indicating microdiffusion disorder in myocardial tissue. Scattered flaky enhancement foci were evident in the myocardium, suggesting myocardial fibrosis (Figure 1C). Initial laboratory tests revealed dramatic elevation of troponin I (> 50 ug/L, n.v. < 0.02 μg/L) and B-type natriuretic peptide (10,330.6 pg/mL, n.v. < 146 pg/mL) immediately after her first cardiac arrest at age 6. During follow-up before ICD implantation, the troponin I range was 0.15–20.32 ug/L and the B-type natriuretic peptide range was 478.2–8746.1 pg/mL.
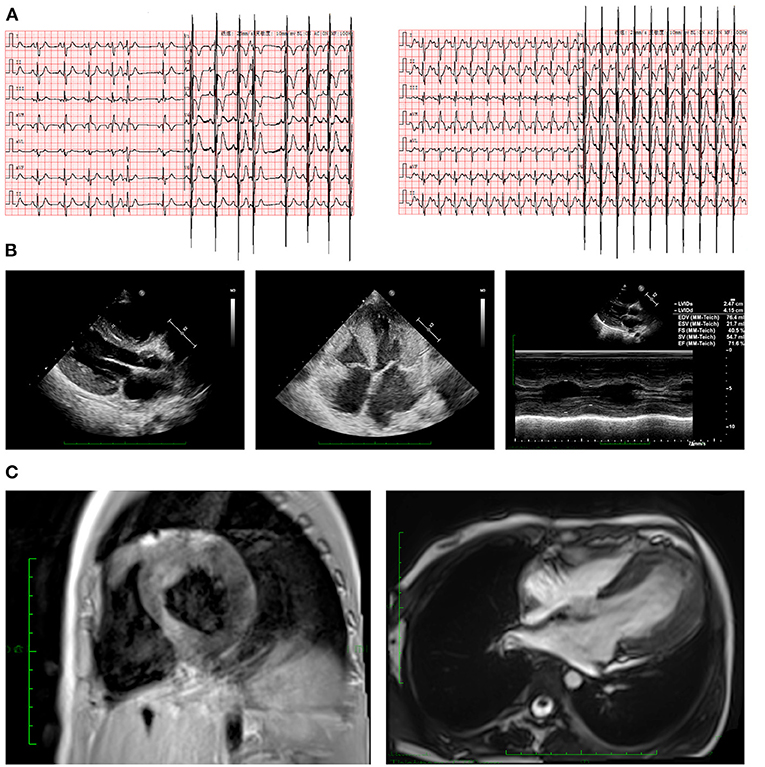
Figure 1. Clinical manifestations in ECG and imaging. (A) ECG revealed CRBBB, abnormal double atrium, ventricular hypertrophy, first-degree atrioventricular block, and ST segment changes (lead I, aVL, and V3–V6 depression >0.05 mV). (B) Echocardiography indicated severe LVH. (C) Cardiac magnetic resonance imaging depicted hypertrophic left ventricular wall in both short axis and long axis views. CRBBB, complete right bundle branch block; ECG, electrocardiogram; LVH, left ventricular hypertrophy.
A peripheral blood sample was obtained from the patient in an ethylenediaminetetraacetic acid anticoagulant blood sample tube then stored at 4°C for < 6 h. DNA was extracted using the Blood Genome Column Medium Extraction Kit (Tiangen Biotech, Beijing, China) in accordance with the manufacturer's instructions. Protein-coding exome enrichment was performed using the xGen Exome Research Panel v.1.0, comprising 429,826 individually synthesized and quality-controlled probes targeting 49.11 Mb of protein-coding regions (> 23,000 genes) of the human genome. Whole exon sequencing was performed using the NovaSeq 6000 platform (Illumina, San Diego, CA, USA), and the raw data were processed using FastP to remove adapters and filter out low-quality reads. Paired-end reads were aligned to the Ensembl GRCh38/hg38 reference genome using the Burrows–Wheeler Aligner. Variant annotation was performed in accordance with database-sourced minor allele frequencies (MAFs) and practical guidelines on pathogenicity issued by the American College of Medical Genetics. The annotation of MAFs was performed based on the 1000 Genomes, dbSNP, ESP, ExAC, Provean, Sift, Polypen2_hdiv, Polypen2_hvar, and Chigene in-house MAF databases using R software (R Foundation for Statistical Computing, Vienna, Austria). The sequencing data have been deposited in the GSA database (http://ngdc.cncb.ac.cn/gsub/).
MutationTaster was used with R software to predict the pathogenicity of MYH7 c.2723T>C and assess the effects of this mutations on protein structure. Besides, according to the recent report from Lioncino et al., we also evaluated the related mutations of RASopathies, and the result was negative (8). There is no MYH7 protein crystal structure available. Modeling analysis was performed using SWISS-MODEL (https://swissmodel.expasy.org/) for the three domains with a 5tby.1.A template. The capability of the protein structure was estimated using Ramachandran plots (Figure 2A). Change in the free energy of the model was estimated using the mutation cut-off scanning matrix (mCSM) method (http://biosig.unimelb.edu.au/mcsm/). Site Directed Mutator (SDM, http://marid.bioc.cam.ac.uk/sdm2) was used to enable assessment of the effects of mutations on the stability of MYH7, and the DUET server was also used (http://biosig.unimelb.edu.au/duet/) to integrate mCSM and SDM to improve the overall prediction accuracy of the mutations under consideration. The signature vector that was ultimately generated was used to train the predictive classification and regression model for calculating the change in Gibbs folding free energy induced by the mutations. Missense3D (http://missense3d.bc.ic.ac.uk/) was used to visualize the structure of amino acid residue changes (wild-type and L908P variant). Clash scores were obtained via Missense3D to measure the stability of mutated proteins.

Figure 2. Effects of MYH7 c.2723T>C mutation on molecular protein structure. (A) Ramachandran plots of MYH7 with and without p.L908P mutation. (B) SWISS-MODEL to predict the variant's wild-type and mutated protein crystal structures using 5tby.1.A template, and structural changes identified in the helix. (C,D) The mCSM tool used to predict protein stability revealed a decline in Gibbs free energy, indicating an unstable protein structure associated with this mutation. (E) Missense 3D analysis indicated that MYH7 p.L908P caused a structure clash.
Based on laboratory analyses and the patient's clinical manifestations a genetic disorder was strongly suspected. Whole exon sequencing was performed using the Illumina NovaSeq 6000 platform, and a de novo c.2723T>C (p.L908P) heterozygous mutation was identified in the MYH7 gene. Neither the patient's mother nor her father had this mutation. According to the American College of Medical Genetics these variants have uncertain pathogenicity (PM1+PM2+PM3). MYH7 c.2327T>C has not been reported in any populations; this is the first report of this variant. Analysis performed with MutationTaster revealed that this mutation is considered pathogenic due to amino acid sequence changes, protein features affected, and loss of helix superstructure (probability = 1 for c.2327T>C). PolyPhen 2.0 predicted this mutation of p.L908P to be “probably damaging” (score = 1.0, sensitivity 0.00, specificity 1.00). The SWISS-MODEL tool was used to analyze stability after amino acid changes. Ramachandran plots indicated that amino acid positions were altered (Figure 2A). Rebuilding molecular structure based on a 5tby.1.A templet resulted in residue changes between Leu and Pro at 908 (Figure 2B). Using SWISS-MODEL protein stability prediction tools, ensemble changes among all the coded amino acids exhibited significant variance (Figure 2C). Three types of calculation methods all demonstrated significant destabilizing change (mCSM, −1.571 Kcal/mol; DUET, −2.044 Kcal/mol; SDM −3.140 Kcal/mol; Figure 2D). The Missense3D tool was also used to evaluate the molecular probity clash score. MYH7 p.L908P predicted obvious protein clash, with an elevated clash score > 18.00 (Figure 2E).
After several examinations and molecular tests the child was diagnosed with HCM, ventricular tachycardia, and Stokes-Adams syndrome. Before ICD implantation she received captopril, beta-blockers, and amiodarone to control heart rhythm. Clopidogrel, spironolactone, and vitamin C were provided to improve heart function. Electrical cardioversion has been used to terminate ventricular tachycardia. In accordance with 2020 American College of Cardiology/American Heart Association HCM guidelines (9) the patient was scheduled for ICD placement after the diagnosis of HCM with the novel MYH7 mutation c.2327C>T, suffering several cardiac arrests accompanied by sustained ventricular tachycardia, and echocardiography demonstrating an interventricular septum thickness > 15 mm.
After ICD implantation the patient suffered no complications and was discharged from hospital on the 10th day. Oral beta-blockers, amiodarone, and enalapril were then administered for regularity, and spironolactone had been provided at the 1st month post ICD implantation. in conjunction with The patient had been required to visit hospital strictly for follow-up. During the 1 year of follow-up period there was no recurrent cardiac arrest, and the ICD remained functional. The most recent ECG indicated sinus rhythm, CRBBB, LVH, and ST segment changes, and prolonged QT and QTc intervals. During the follow-up, Holters had been performed every 2 months, which were absence of ventricular tachycardia. Echocardiography indicated general hypertrophic changes in left and right ventricular hypertrophy at the end of diastole (interventricular septum 14 mm (Z-score = 4.22), left ventricular posterior wall 9 mm (Z score = 2.90), right ventricular posterior wall 6 mm) and normal left ventricular systolic function = 64%.
HCM is the most common type of cardiomyopathy in children. It is associated with SCD and other adverse outcomes that are usually life-threatening such as severe heart failure, which lead to early death. A recent statement from Monda et al. demonstrated the HCM was the major cause of SCD among all kinds of cardiomyopathies (10). Besides, this statement emphasized the importance of genetic variants evaluation in preventing future SCD. Notably however, predicting such cardiac events in HCM patients remains challenging. Furthermore, the European Society of Cardiology and the American Heart Association developed a risk score system to guide the clinical management of HCM patients. Unfortunately there are no specific recommendations for preventing SCD in pediatric HCM patients, and no specific recommendations pertaining to ICD implantation in such patients. In view of this limitation several case series and reports have discussed this issue and suggested reasonable indications for ICD placement. Because the technique for ICD placement in pediatric patients is challenging and high-risk, the procedure involves several external considerations that do not apply in adults.
Next generation sequencing tests are rapid, and are widely used in both developed and developing countries to investigate genetic associations. Mathew et al. (5) reported that the primary genes affected were MYH7 in 35% and MYBPC3 in 49% in a pediatric cohort. MYH7 mutation-positive patients exhibited earlier disease onset and were at a higher risk of major adverse cardiac events (HR, 2.7, 95% CI 1.3–5.7). The risk of major adverse cardiac events was also higher in patients with multiple variants (n = 16; HR, 2.5, 95% CI 1.1–5.9), and de novo variant subsets revealed a higher risk (HR, 5.7, 95% CI 2.6–12.7). Ferradini et al. (11) reported that MYH7 was the dominant genetic site overlapping HCM and arrhythmogenic cardiomyopathy. To predict the risk of SCD among HCM patients, several predictive models had been reported. Recently, two groups reported two individual validated model of SCD in children. The research from Norrish et al. demonstrated the maximal wall thickness and left atrium as the most important indicator to predict 5-years risk of SCD (12). However, this article did not take pathogenic variant into the formula of risk calculation, and they found the family history was not related to the risk of SCD (12). Another research from Miron et al. revealed a validated model for SCD risk. In this model, it confirmed the pathogenic variant elevated the likelihood of experiencing SCD events (HR, 1.32 [95% CI, 1.00–2.12]) (7). Among the reported pathogenic variants, MYH7 and MYBPC3 were the most common mutations had been elucidated (7). So that, more detail evaluation of genetic variants, including the molecular function of mutated genes (structural proteins, metabolic molecules or calcium handling channel, etc.) should be taken into different or hierarchical considerations to predict risk of SCD and guiding the application of ICD implantation. This research highlight the importance of genetic analyses and significance of mutations in guiding the implantation of ICD. However, the implantation of ICD among high risk (>6%) population to save patients from SCD was not very satisfied, as only 1 patient may potentially be saved from SCD at 5 years among every 10 ICDs implanted in patients with 6% or more of a 5-year SCD risk (12). So that, the clinical outcomes of ICD application is mainly relied on the patient risk stratification. Analysis of the role of sarcomere genes in the pathogenesis of arrhythmogenic cardiomyopathy is recommended in arrhythmogenic cardiomyopathy patients who test negative for desmosomal mutations. In other research utilizing the International Sarcomeric Human Cardiomyopathy Registry (13, 14) the top three genetic variants indicating HCM onset were MYH7, MYBPC3, and Thin Filament. The average age at initial diagnosis was 40.1 ± 15.2 years however, thus corresponding pediatric information remains lacking. That research demonstrated that new-onset atrial fibrillation developed in 19% of HCM patients with sarcomere mutations, and compared with other sarcomere genes, patients with pathogenic or likely pathogenic variation in MYH7 had a higher rate of incident atrial fibrillation independent of clinical and echocardiographic factors. Accordingly, MYH7 mutation is a major indicator of arrhythmia in HCM patients, which called positive recommendation for ICD application. Besides, the mutations of above genes, a newest research from Lioncino et al. demonstrated the RASopathies was critical to underline the clinical outcomes HCM (8). So that, any HCM patients, especially for the children who had right ventricular hypertrophy involved, should be paid more attention to exclude RASopathies.
In recent years ICD interventions have proven effective for the prevention of cardiac arrest by monitoring and terminating ventricular fibrillation or malignant ventricular tachycardia. ICD therapy in children is associated with higher rates of complications than it is in adults however, necessitating complex consideration before proceeding with ICD implantation (7, 15, 16). Herein we attempted to evaluate the potential benefits of ICD implantation in pediatric patients. Literature review indicated that clinical prognoses associated with genetic mutations (MYH7, MYBPC3, TNNI3, TNNT2, and TPM1) in pediatric HCM patients with and without ICD implantation were totally different (17–20). Multiple studies indicate that in adults some gene mutations predict earlier disease onset and more severe phenotypes in HCM (21–25). Although gene mutations and phenotypes are heterogeneously influenced by modifier genes, environmental factors, and other influences (16), current evidence suggests that HCM patients with multiple variants are more likely to suffer malignant outcomes and SCD, potentially justifying early ICD implantation (26–29).
Systematic review indicates that ICD implantation is usually recommended in HCM patients with severe cardiac events. Notably however, several large cohort studies and meta-analyses suggest that the most common age range for receiving ICD implantation is ~40–45 years. The current case of HCM was relatively rare in that the patient presented with severe arrhythmic attacks resulting in recurrent cardiac arrest. It was fortunate that death had not occurred prior to ICD placement. ICD placement was deemed urgent in this very young patient (aged 7.7 years). Given her very young age, we acquired all published reports detailing cases of ICD implantation in patients aged under 18 years that provided clear clinical and molecular results. This amounted to a total of seven reports describing 14 cases of patients suffering malignant cardiac events, and almost all of them involved a positive family history. Among them, two patients with compound MYBPC3 variants died soon after birth due to aggressive heart failure with HCM. Another 11 patients presented HCM phenotypes with MYH7, ALPK3, MYBPC3, TNNI3, TNNT2, and TPM1 variants. Age at ICD implantation ranged from 8.5 to 17.0 years. Unfortunately, SCD occurred in 5 of 5 children in whom the ICD placement procedure was not completed. Conversely, only 1 of 7 children (14.3%) died after successful ICD implantation (Table 1). So that, compared with the reports from Norrish et al., which revealed the ICD could only save 1 out of 10 HCM with high risk of SCD without genetic stratification, the patients with positive genetic variant might receive more benefits from ICD implantation according to literature review.
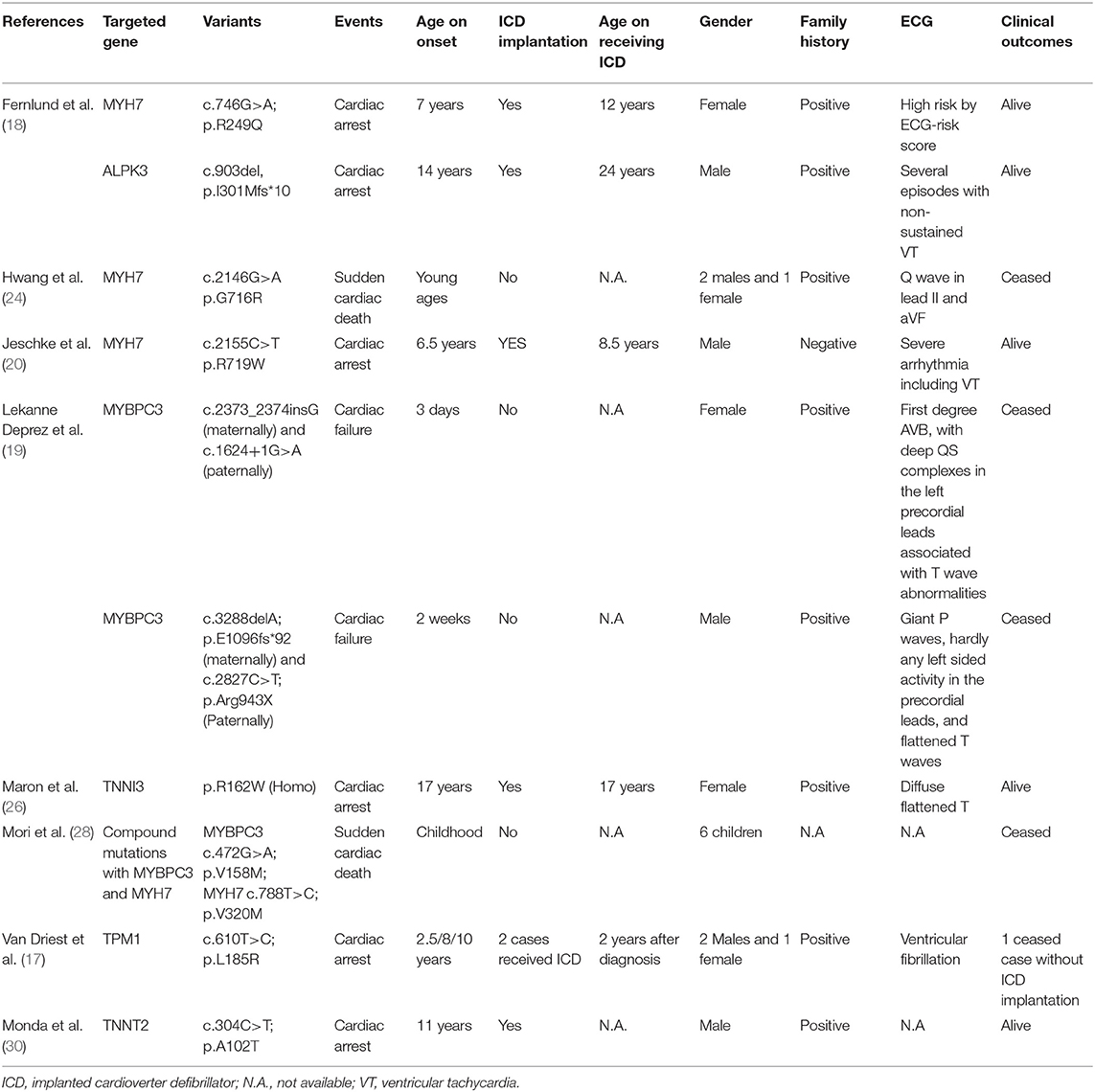
Table 1. Association between gene mutations and malignant events in children of hypertrophic cardiomyopathy.
The present case and the associated literature review facilitated the derivation of the genetic clues for a potential future perspective for ICD placement, as the patients with genetic mutations associated with HCM and arrhythmia, especially MYH7 and MYBPC3 mutations might be benefit from early ICD implantation. Besides the screening for positive genetic variant, several indicators required to be clarified to demonstrate the risk stratification for SCD. The echocardiographic parameters, especially for left atrium and right ventricle; the onset age with or without syncope; and whether there were notable abnormal ECG changes. However, two studies provided a controversial results on the application of ECG in predicting SCD (31, 32). So further prospective studies are required to provide more evidences on SCD application indication among HCM children.
Variant burden and variant type contribute to the risk of adverse events in pediatric HCM patients. Early recognition and intervention are vital in such patients. We suggest that gene mutation could be considered as an potential indication for early ICD placement in pediatric HCM patients during standard risk stratification, but this requires further investigation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: http://ngdc.cncb.ac.cn/gsub, GSA database.
The studies involving human participants were reviewed and approved by the Ethics Committee of West China Second Hospital of Sichuan University (2014-034). And informed consent from the patient's parents prior to conducting the WES had been obtained, including the patient's clinical and imaging details in the manuscript for the purpose of publication. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YH, KZ, and TW were the patient's physicians. XL and JT reviewed the literature and contributed to manuscript drafting. JL and SL performed the mutation analysis. YL conceptualized and designed the study, coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content. YH and YL were responsible for the revision of the manuscript for important intellectual content. All authors issued final approval for the version to be submitted.
This work was supported by grants from the National Natural Science Foundation of China (No. 81700360), Technology Project of Sichuan Province of China (2020YFS0101 and 2020YFS0102).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cardoso B, Gomes I, Loureiro P, Trigo C, Ferreira Pinto F. Clinical and genetic diagnosis of familial hypertrophic cardiomyopathy: Results in pediatric cardiology. Rev Portuguesa Cardiol. (2017) 36:155–65. doi: 10.1016/j.repce.2016.09.006
2. Moak JP, Kaski JP. Hypertrophic cardiomyopathy in children. Heart. (2012) 98:1044–54. doi: 10.1136/heartjnl-2011-300531
3. Ware SM, Wilkinson JD, Tariq M, Schubert JA, Sridhar A, Colan SD, et al. Genetic causes of cardiomyopathy in children: first results from the pediatric cardiomyopathy genes study. J Am Heart Assoc. (2021) 10:e017731. doi: 10.1161/JAHA.120.017731
4. Maron BJ, Rowin EJ, Casey SA, Lesser JR, Garberich RF, McGriff DM, et al. Hypertrophic cardiomyopathy in children, adolescents, and young adults associated with low cardiovascular mortality with contemporary management strategies. Circulation. (2016) 133:62–73. doi: 10.1161/CIRCULATIONAHA.115.017633
5. Mathew J, Zahavich L, Lafreniere-Roula M, Wilson J, George K, Benson L, et al. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin Genet. (2018) 93:310–9. doi: 10.1111/cge.13157
6. Norrish G, Ding T, Field E, McLeod K, Ilina M, Stuart G, et al. A validation study of the European Society of Cardiology guidelines for risk stratification of sudden cardiac death in childhood hypertrophic cardiomyopathy. Europace. (2019) 21:1559–65. doi: 10.1093/europace/euz118
7. Miron A, Lafreniere-Roula M, Steve Fan CP, Armstrong KR, Dragulescu A, Papaz T, et al. A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation. (2020) 142:217–29. doi: 10.1161/CIRCULATIONAHA.120.047235
8. Lioncino M, Monda E, Verrillo F, Moscarella E, Calcagni G, Drago F, et al. Hypertrophic cardiomyopathy in RASopathies: diagnosis, clinical characteristics, prognostic implications, and management. Heart Fail Clin. (2022) 18:19–29. doi: 10.1016/j.hfc.2021.07.004
9. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Thorac Cardiovasc Surg. (2021) 162:e23–106. doi: 10.1016/j.jtcvs.2021.04.001
10. Monda E, Lioncino M, Rubino M, Caiazza M, Cirillo A, Fusco A, et al. The risk of sudden unexpected cardiac death in children: epidemiology, clinical causes, and prevention. Heart Fail Clin. (2022) 18:115–23. doi: 10.1016/j.hfc.2021.07.002
11. Ferradini V, Parca L, Martino A, Lanzillo C, Silvetti E, Calò L, et al. Variants in MHY7 gene cause arrhythmogenic cardiomyopathy. Genes. (2021) 12:60793. doi: 10.3390/genes12060793
12. Norrish G, Ding T, Field E, Ziólkowska L, Olivotto I, Limongelli G, et al. Development of a novel risk prediction model for sudden cardiac death in childhood hypertrophic cardiomyopathy (HCM Risk-Kids). JAMA Cardiol. (2019) 4:918–27. doi: 10.1001/jamacardio.2019.2861
13. Canepa M, Fumagalli C, Tini G, Vincent-Tompkins J, Day SM, Ashley EA, et al. Temporal trend of age at diagnosis in hypertrophic cardiomyopathy: an analysis of the international sarcomeric human cardiomyopathy registry. Circ Heart Fail. (2020) 13:e007230. doi: 10.1161/CIRCHEARTFAILURE.120.007230
14. Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. (2018) 138:1387–98. doi: 10.1161/CIRCULATIONAHA.117.033200
15. Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, Estes NA, et al. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2013) 61:1527–35. doi: 10.1016/j.jacc.2013.01.037
16. Nakano SJ, Menon SC. Risk stratification in pediatric hypertrophic cardiomyopathy: Insights for bridging the evidence gap? Prog Pediatr Cardiol. (2018) 49:31–7. doi: 10.1016/j.ppedcard.2018.03.001
17. Van Driest SL, Will ML, Atkins DL, Ackerman MJ. A novel TPM1 mutation in a family with hypertrophic cardiomyopathy and sudden cardiac death in childhood. Am J Cardiol. (2002) 90:1123–7. doi: 10.1016/S0002-9149(02)02780-7
18. Fernlund E, Kissopoulou A, Green H, Karlsson JE, Ellegard R, Arstrand HK, et al. Hereditary hypertrophic cardiomyopathy in children and young adults-the value of reevaluating and expanding gene panel analyses. Genes. (2020) 11:1472. doi: 10.3390/genes11121472
19. Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. (2006) 43:829–32. doi: 10.1136/jmg.2005.040329
20. Jeschke B, Uhl K, Weist B, Schröder D, Meitinger T, Döhlemann C, et al. A high risk phenotype of hypertrophic cardiomyopathy associated with a compound genotype of two mutated beta-myosin heavy chain genes. Human Gene. (1998) 102:299–304. doi: 10.1007/s004390050695
21. Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, Lai A, Amr A, Haas J, et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clin Res Cardiol. (2018) 107:30–41. doi: 10.1007/s00392-017-1155-5
22. Velicki L, Jakovljevic DG, Preveden A, Golubovic M, Bjelobrk M, Ilic A, et al. Genetic determinants of clinical phenotype in hypertrophic cardiomyopathy. BMC Cardiovasc Disord. (2020) 20:516. doi: 10.1186/s12872-020-01807-4
23. Wang L, Seidman JG, Seidman CE. Narrative review: harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann Internal Med. (2010) 152:513–20. doi: 10.7326/0003-4819-152-8-201004200-00008
24. Hwang TH, Lee WH, Kimura A, Satoh M, Nakamura T, Kim MK, et al. Early expression of a malignant phenotype of familial hypertrophic cardiomyopathy associated with a Gly716Arg myosin heavy chain mutation in a Korean family. Am J Cardiol. (1998) 82:1509–13. doi: 10.1016/S0002-9149(98)00695-X
25. Garcia-Giustiniani D, Arad M, Ortiz-Genga M, Barriales-Villa R, Fernandez X, Rodriguez-Garcia I, et al. Phenotype and prognostic correlations of the converter region mutations affecting the beta myosin heavy chain. Heart. (2015) 101:1047–53. doi: 10.1136/heartjnl-2014-307205
26. Maron BJ, Maron MS, Semsarian C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. (2012) 9:57–63. doi: 10.1016/j.hrthm.2011.08.009
27. Wang J, Wang Y, Zou Y, Sun K, Wang Z, Ding H, et al. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur J Heart Fail. (2014) 16:950–7. doi: 10.1002/ejhf.144
28. Mori AA, Castro LR, Bortolin RH, Bastos GM, Oliveira VF, Ferreira GM, et al. Association of variants in MYH7, MYBPC3 and TNNT2 with sudden cardiac death-related risk factors in Brazilian patients with hypertrophic cardiomyopathy. Forensic Sci Int Genet. (2021) 52:102478. doi: 10.1016/j.fsigen.2021.102478
29. Stafford F, Thomson K, Butters A, Ingles J. Hypertrophic cardiomyopathy: genetic testing and risk stratification. Curr Cardiol Rep. (2021) 23:9. doi: 10.1007/s11886-020-01437-4
30. Monda E, Sarubbi B, Russo MG, Caiazza M, Mazzaccara C, Magrelli J, et al. Unexplained sudden cardiac arrest in children: clinical and genetic characteristics of survivors. Eur J Prevent Cardiol. (2020) 28:1134–7. doi: 10.1177/2047487320940863
31. Norrish G, Topriceanu C, Qu C, Field E, Walsh H, Ziółkowska L, et al. The role of the electrocardiographic phenotype in risk stratification for sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur J Prev Cardiol. (2021) 107:1962–73. doi: 10.1136/heartjnl-2021-BCS.1
Keywords: hypertrophic cardiomyopathy, genetic disorder, MYH7 mutation, ICD implantation, literature review
Citation: Li X, Tang J, Li J, Lin S, Wang T, Zhou K, Li Y and Hua Y (2021) Genetic Clues on Implantable Cardioverter-Defibrillator Placement in Young-Age Hypertrophic Cardiomyopathy: A Case Report of Novel MYH7 Mutation and Literature Review. Front. Cardiovasc. Med. 8:810291. doi: 10.3389/fcvm.2021.810291
Received: 06 November 2021; Accepted: 06 December 2021;
Published: 23 December 2021.
Edited by:
Giuseppe Limongelli, Second University of Naples, ItalyReviewed by:
Michele Lioncino, University of Campania Luigi Vanvitelli, ItalyCopyright © 2021 Li, Tang, Li, Lin, Wang, Zhou, Li and Hua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifei Li, bGl5Zndjc2hAc2N1LmVkdS5jbg==; Yimin Hua, bmF0aGFuX2h1YUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.