 Zhongxiao Lin
Zhongxiao Lin Qian Ding1
Qian Ding1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 04 January 2022
Sec. Cardiovascular Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.806988
This article is part of the Research Topic Novel Epigenetic Medicine and Cardiovascular Diseases View all 4 articles
Environment, diseases, lack of exercise, and aged tendency of population have becoming crucial factors that induce vascular aging. Vascular aging is unmodifiable risk factor for diseases like diabetes, hypertension, atherosclerosis, and hyperlipidemia. Effective interventions to combat this vascular function decline is becoming increasingly urgent as the rising hospitalization rate caused by vascular aging-related diseases. Fortunately, recent transformative omics approaches have enabled us to examine vascular aging mechanisms at unprecedented levels and precision, which make our understanding of slowing down or reversing vascular aging become possible. Epigenetic viz. DNA methylation, histone modifications, and non-coding RNA-based mechanisms, is a hallmark of vascular aging, its deregulation leads to aberrant transcription changes in tissues. Epigenetics mechanisms by mediating covalent modifications to DNA and histone proteins, consequently, influence the sensitivity and activities of signaling pathways in cells and tissues. A growing body of evidence supports correlations between epigenetic changes and vascular aging. In this article, we will provide a comprehensive overview of epigenetic changes associated with vascular aging based on the recent findings with a focus on molecular mechanisms of action, strategies to reverse epigenetic changes, and future perspectives.
Aging processes are accompanied with the accumulation of degenerative processes and changes in both physiological and functional parameters in mammals. In humans, aging associated vascular diseases constitutes a significant risk to health and the quality of life for individuals. Indeed, approximately 4 million people die from cardiovascular diseases (CVDs) each year in China (1). With the dramatic growth in aged populations around the world, CVDs are one of the biggest global challenges to health care systems.
Undoubtedly, a substantial amount of aging research has focused on finding ways to remove or counteract the loss of biological function in cells and tissues, with the hope of maintaining health or to even extend lifespan. Advances in our understanding of genetics including, “the central dogma,” “heritable traits” and the molecular mechanisms of gene regulation has underpinned breakthroughs in this field and has spawned a growing branch of research known as epigenetics. Epigenetics refers to a phenotype or changes in gene expression caused by mechanisms other than the alteration in a genetic or DNA sequence. These changes result from random events, the impact of environmental factors, diet, and stress, each of which can have significant influence on health and diseases processes in humans (2). Our understanding of the biological processes associated with the aging has advanced in recent years. Indeed, the causes and consequences of aging have been categorized by some researchers into specific “aging phenotypes” grouped by association like for example, oxidative stress and energetics, mitochondrial dysfunction, homeostatic mechanisms, shortening of telomeres, cell senescence, DNA damage, defects in proteostasis, and exhaustion of progenitor cells (3, 4). One common thread linking each of these catagories is that the biological aging process takes many years before it finally translates into structural and functional deterioration. Therefore, to delay aging or, to prevent it from worsening, we first need to understand the fundamental processes that govern these changes.
During vascular aging dysfunctions in blood vessels, including cellular senescence, vascular remodeling, vascular homeostatic imbalance, inflammation, VSMCs invasion, fibrosis, calcification, the decline in oxygen and nutrient delivery that drives disease severity (5). In each of these scenarios, epigenetic changes have been reported to play important roles in these processes (6). To date, various studies have reported on the mechanisms by which vascular damage occurs during aging, particularly in vascular-related tissues viz. ECs, VSMCs, epidermal cells, and extracellular matrix (ECM). Vascular aging provides instructions for selective gene expression, and it is closely related to a variety of vessel-related diseases, such as hypertension, diabetes, atherosclerosis, and hyperlipidemia. Dysregulation in these systems affects DNA modifications, chromatin structure, and gene expression, and is partly responsible for the occurrence and development of vascular aging and age-related diseases in the cardiovascular system (7). Therefore, an understanding of the intrinsic mechanisms of epigenetic regulation in vascular tissues is paramount for the future development of strategies to reduce disease burden in the general population. Currently, epigenetic-mediated aging mechanisms have gained interest from the academic community fueled by advances in omics technologies. These epigenetic mechanisms including DNA methylation (DNAm), RNA methylation (RNAm), histone modifications, and non-coding RNAs (ncRNAs) regulation. In the current review, the pathophysiological changes and roles of epigenetic-mediated vascular aging will be discussed. In addition, advances in our understanding of the underlying molecular mechanisms, and potential therapeutic strategies to manage these changes will be covered. It is hoped this information will assist in the development of novel approaches to diagnosis, treat, and manage vascular-related diseases in humans as summarized in Figure 1.
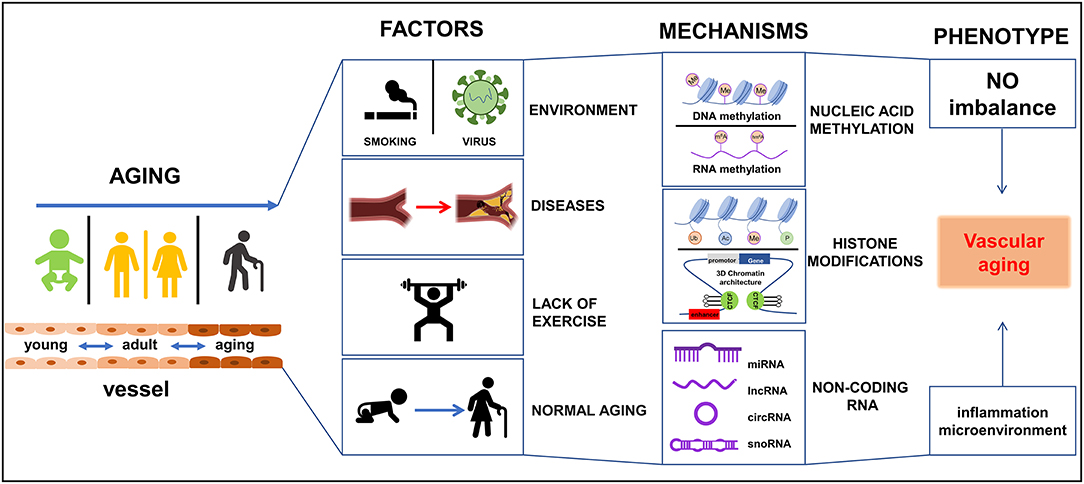
Figure 1. Schematic diagram of epigenetics-mediated vascular aging in cardiovascular system. Multiple factors contribute to vascular aging phenotypes, including environment factors like smoking and virus, various diseases like diabetes and metabolic syndrome, lack of exercise, and normal aging. Epigeneitc modifications of nucleic acid (DNA and RNA) like methylation and demethylation, histone modifications [like methylation, acetylation, ubiquitination, phosphorylation, 3-dimensional (3D) chromatin architecture], and non-coding RNAs, drive vascular aging processes. Ac, acetylation; Me, methylation; P, phosphorylation; Ub, ubiquitination.
The process of DNA methylation (DNAm) has been known since 1950 (8), and a further 17 types of chemical modifications have since been discovered that impact on DNA (9). DNAm acts via the attachment of methyl groups, hydroxymethyl, or other moieties to the carbon-5 position in the dinucleotide CpG. CpG islands (regions of DNA with a high G+C content and a high frequency of CpG dinucleotides) usually reside in or adjacent to the transcription start site (TSS). The DNAm status of DNA can therefore influence gene expression (either suppresses or activates) by influencing the interactions of transcription factors (TFs) with target sequences. The addition of methyl groups to DNA is regulated by DNA methyltransferases (DNMTs) including DNMT1, DNMT2, DNMT3A, DNMT3B, DNMT3L. Interestingly, both DNMT2 and DNMT3L are non-canonical family members that lack DNMT activity (10). DNAm can be divided into two main processes viz. hypermethylation and hypomethylation. DNA hypermethylation (promotor region) usually leads to the gene transcriptional repression, whereas hypomethylation causes gene transcriptional activation (11). Interestingly, Agha and colleagues recently showed that hypermethylation levels at 52 CpG regions in blood leukocytes can be used as a predictive risk marker for myocardial infarction and coronary heart disease (12). Similarly, in atherosclerotic patients, global DNA hypomethylation was found in peripheral white blood cells, vascular smooth muscle cells (VSMCs), and plaques (13), which means that deleterious genes could be overexpressed, of note, it depends on the balance between hazardous genes and the good ones.
DNA hydroxymethylation is also a crucial component of the epigenetic system, and it is governed by the activities of serine hydroxymethyltransferases (14). Ten-methylcytosine dioxygenase family members 2 (TET2) is one of the serine hydroxymethyltransferase involved in the oxidation of 5-methylcytosine (5-mC) to generate 5-hydroxymethylcytosine (5-hmC). Overexpression of TET2 attenuates intimal hyperplasia (15). DNA modification rarely affects vascular aging through a single enzymatic pathway, but rather alters multiple genes networks. DNMT1, DNMT3A, DNMT3B, TET2, and sensitive components of these networks are crucial as vascular aging targets. Decreased DNMT1 expression occurs in replicative senescent aortic SMCs and correlates with rates of hypomethylation of COL15A1 (over-expressed in the atherosclerotic lesion and localized to the atherosclerotic cap), this linking epigenetic regulation of DNMT1 with SMC phenotypes and prevalence rates of atherosclerosis (16). Epigenetic network including DNMT3A and TET2 are thought to act as tumor suppressor genes via their propensity to aid recruitment of histone deacetylases to gene promoters. Interestingly, in the cardiovascular system, reduced activity of DNMT3A and TET2 promotes artery smooth muscle cell proliferation and endothelial cell dysfunction. It is likely that these two genes also play corresponding roles in aging (17). Indeed, DNMT3B together with DNMT3A form a protein complex that interacts with histone deacetylases HDAC1, HDAC2, Sin3A, and the transcriptional suppressor proteins, Rb, TAZ-1, and heterochromatin proteins HP1, SUV39H1. As a result of these interactions, normal levels of DNA methylation and gene silencing are maintained. For example, complex in CpG sites in the promoter of p16(INK4A, an aging marker) leads to downregulation of p16(INK4A) expression (18). Methylation processes also affected by the supply of methyl groups, methylation patterns, folic acid metabolism, and hormone levels. There are still many unknown phenomena in the current research, and many studies need to be further conducted, such as the relationship between random DNA methylation drift in vascular aging. We believe that the methylation regulation is still in the experimental/initial stage, but has attracted the attention of many scholars.
Histone proteins are basic proteins abundant in lysine and arginine residues and include H1, H2A, H2B, H3, and H4. These proteins are responsible for maintaining chromatin structure, mediating dynamic and long-term gene regulation, post-translational mutations, and functional variation. These proteins are subjected to a myriad of modifications including acetylation, methylation, phosphorylation, ubiquitination, butyrylation, formylation, and succinylation. Histones are tightly associated with DNA and altered histone modifications impacts on DNA replication and repair, and the rates of transcription thus impacting on gene expression. Indeed, aberrant histone modifications, for example the dysregulation of histone deacetylase (HDACs), site- specific loss, and gains of heterochromatin are proposed as hallmarks of epigenetic change that can impact on 3D chromatin structure and control gene expression (19). However, many functional changes brought by histone modifications remained largely uncharacterized but research points to a complex regulatory network affected by the spatial structural features and external stress (20). For example, omics approaches reveals that the alteration in the three-dimensional (3D) chromatin architecture caused by histone modification is a crucial epigenetic element important in development, cancer, and aging (21).
Another important class of enzymes are the histone methyltransferases (HMTs) that catalyze histone methylation, typically at lysine and arginine residues present in histone proteins. The mono-methylation of H3K27, H3K9, H4K20, H3K79, and H2BK5 is usually related to gene activation, while that of tri-methylation of H3K27, H3K9, and H3K79 confers inhibition (22).
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are the most widely studied histone-modifying enzymes in vascular aging. HATs transfer acetyl group onto histone proteins, which in turn neutralizes the charge of histone, and weakens its interaction with DNA. Acetylation of H3K122 and H3K64 breaks the interaction between histone tails on adjacent nucleosomes and loosens the connection between nucleosomes (23). Histone acetylation can be identified by the bromodomain of TATA-binding protein-associated factor1 (TAF1), which attracts various TFs, and promotes transcription. HDACs have opposite effects to that of HATs and a dynamic balance between the activities of HATs and HDACs governs gene expression. In addition, histone phosphorylation plays a role in altering chromatin architecture like for example, the phosphorylation of H3T118 that enhances DNA accessibility, nucleosome mobility, and nucleosome disassembly (24). Other modifications including histone H2A ubiquitination reinforce nucleosome stability (25), and the hyper-ubiquitination of this target is considered an aging associated biomarker (26). In addition, chromatin architecture is also a crucial regulator. Chromatin is composed of different structural units, from the compartment, topologically associated domains to loops, and all these units have their characteristics and regulate chromatin function and gene expression (27). Indeed, the CCCTC-binding factor (CTCF) architectural protein can be up-regulated in pluripotent stem cells (iPSCs) helping in the formation of compact chromatin loops. However, reductions in CTCF disrupts the loop structure and promotes the overexpression of the p16 (INK4a), an aging-induced gene (28). Histone modification in the cardiovascular system is an active field of research with growing evidence implicating a role of HDACs in the regulation of vessel homeostasis. Indeed, HDAC4 regulates vascular inflammation via activation of autophagy in endothelial cells (29) and the deletion of HDAC9 promotes inflammation resolution and reverse cholesterol transport in atherosclerosis and coronary heart diseases (30, 31). In addition, elevation of acetyltransferase p300 accelerates aging (32). Histone H3 lysine 4 (H3K4) methyltransferase Smyd3 (SET- and MYND- domain-containing proteins) directly binds to the promoter region of Cdkn1a (coding for p21). This interaction causes an increase in H3K4me3 and leads to the activation of p21 giving rise to SASP. These SASPs can be reversed by the Smyd3-specific inhibitor EPZ031686 (33), but enhanced when the Hsp90α binds to Smyd3 (34). Collectively, the available research clearly shows that modification of histone proteins, either by methylation, acetylation, or ubiquitination, has downstream impacts on genes associated with vascular aging. Other systems include the Smyd family proteins that methylate H3K9, H3K27, H3K36, H4K20, and H3K79 (35). Interestingly, tri-methylation of H3K27 and H3K36 is associated with accelerated epigenetic aging in humans (36). These impacts are linked to changes in 3' untranslated region (UTR) length and the methyltransferase (MET-1) activities (37, 38). H3K27 is also a target for histone demethylase Jumonji domain-containing protein 3 (JMJD3) regulates vascular neointimal hyperplasia by mediating H3K27 tri-methylation (39). These modifications enlighten us that modifications at variant sites impact gene function in different styles.
In addition, NAD+ -dependent processes are critical for maintaining tissue and metabolic homeostasis relative to vascular aging. Diminished tissue concentrations of NAD+ leads to downregulation of SIRT family expression. The SIRT family proteins are positively associated with longevity and roles for SIRT1 in reducing vascular senescence, inflammation, DNA damage, and atherosclerosis are widely reported (40). Here we focused on SIRT6 and SIRT3 in vascular function. SIRT3 enhances the expression of the blood pressure regulator GATA5 (GATA-binding protein 5). The endothelial specific loss of GATA5 causes vascular endothelial dysfunction via the inhibition of transcriptional repressor Nkx3 mediated by deacetylation of histone H3K9. The SIRT6/GATA5 signaling pathways could be a way to reduce endothelial senescence and apoptosis (41). Another anti-atherosclerotic mechanism linked to SIRT6 is in the maintenance of endothelial function via its propensity to deacetylate H3K9 in the promoter region of the pro-atherogenic target TNFSF4 (42). Other components associated with vascular function include the p66shc, an epigenetic factor associated with diabetes-induced vascular senescence via its ability to inactivation miR-34a (43) and SIRT3 (44). SIRT3 is mainly expressed in mitochondria, while SIRT6 in the nucleus this fact highlighting the challenging nature of developing drugs targeting these proteins.
NcRNAs have been shown to be crucial for the maintenance of vascular function by regulating nuclear transcription and gene translation in cytoplasm. NcRNAs are divided into small or short ncRNAs [(smaller than 200 nucleotides (nt)], long ncRNAs (lncRNAs, longer than 200 nt), microRNAs (miRNAs, 21–25 nt) belonging to the small class of RNAs, and circular RNAs (circ RNAs, 300–500 nt) pertain to lncRNAs. To date, various classes of ncRNAs have been shown to influence inflammation, senescence, cellular function, and differentiation. For example, miR-92a blocks endothelial proliferation while miR-24 promotes vascular stress (45). miRNA bind to mRNA promoting degradation or the inhibition of translation. CircRNAs interact with miRNAs to form a circRNA-miRNA-mRNA loop regulatory unit, and crosstalk between components of this system plays a crucial role in the development of vascular diseases by altering pathways like cell adhesion, immune response, and regulation of cell adhesion (46). LncRNAs are often associated with homologous DNA and RNA sequences exerting their regulatory role in cells and tissues (47). Small nucleolar RNA (Sno-ncRNAs, 60–300 nt) are a class of nuclear-enriched intron-derived ncRNAs lacking 5' caps and 3' poly(A) tails. These molecules are widely expressed in tissues and are composed of two types, box C/D and box H/ACA snoRNAs; these types respond to 2'-O-ribose methylation and pseudouridylation, respectively (48). SnoRNAs are involved in oxidative stress responses and may therefore be important in regulate aging processes (49). MiR-22 and miR-128 possess therapeutic and prognostic potential as a novel target to treat post-infarct and aged-VSMC remodeling (50, 51). This ncRNA controls VSMC phenotypes and injury-induced arterial remodeling by modulating multiple genes including methyl-CpG binding protein 2 (MeCP2), SIRT1, HDAC4, and EVI1. Other epigenetic modifications including the hypomethylation of the lncRNA H19 (H19) promoter region can lead to the silence of NOTCH1. In this cascade, silencing of NOTCH1 prevents the recruitment of p53 to the NOTCH1 promoter. In turn, this leads to calcific aortic valve disease, endothelial injury, matrix remodeling, angiogenesis, and calcification (52). CircRNAs and snoRNAs are also regarded as critical components of ncRNAs. For example, circ ANRIL levels correlate with increased atherosclerosis risk (53). Moreover, the target SnoRNA U17 regulates cellular cholesterol trafficking (54). However, the molecular mechanism of action for each of these regulates is still unclear and warrants further investigation.
The epigenetic network encompasses a wide range of biological reactions, and it is likely that even with current technologies, researchers only capture a fraction of the changes that occurs in cells and tissues. Here we hold the opinion that some factors linked to epigenetic systems associated with vascular aging is of importance, as summarized in Table 1.
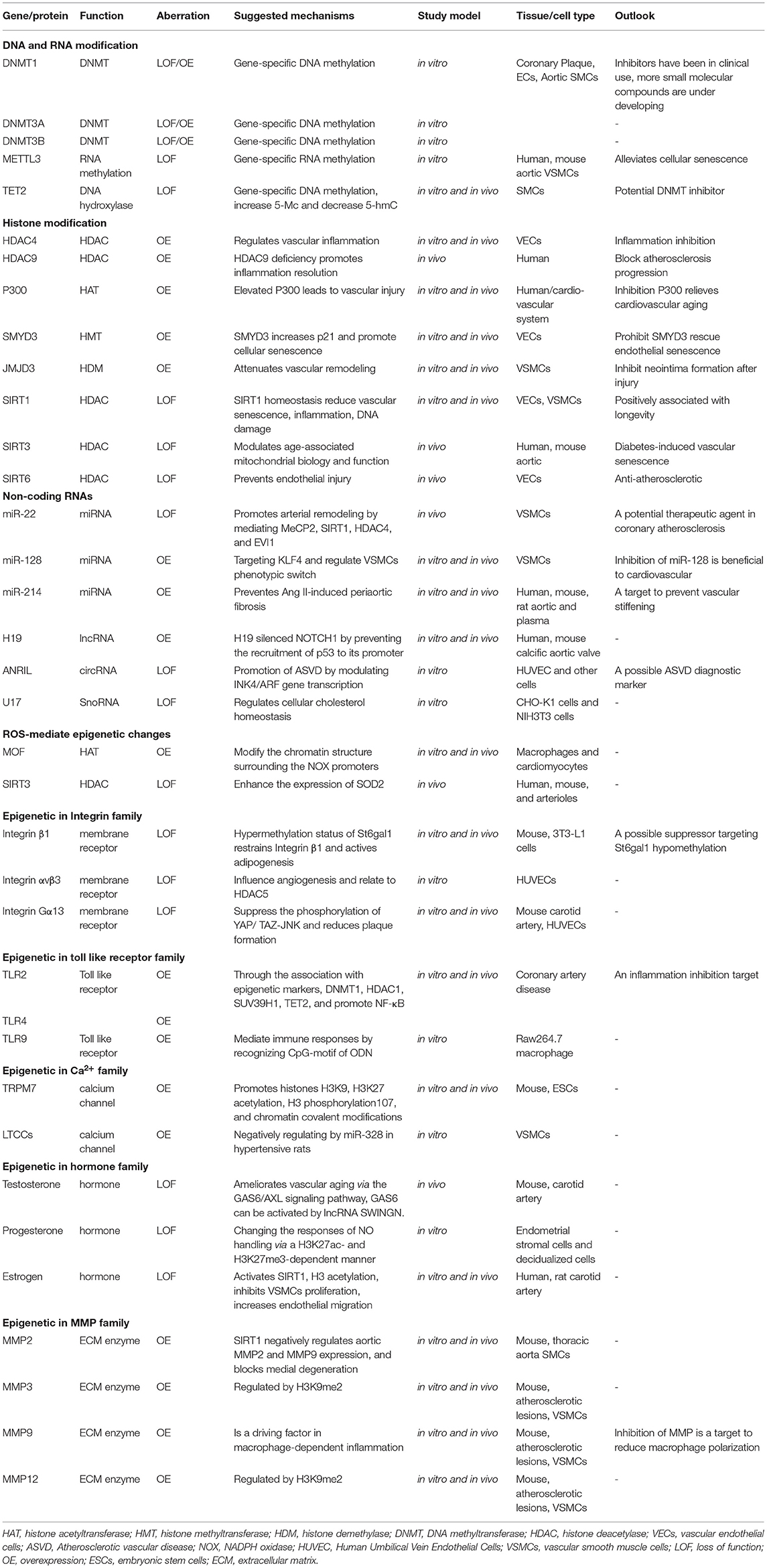
Table 1. Vascular aging-associated epigenetic alterations and its rationale.
ROS (reactive oxygen species) regulates vascular aging by impairing the function of endothelial nitric oxide synthesis (eNOS). Two regulatory mechanisms are involved. The first involves epigenetic mechanisms impacting on ROS accumulation like for example, NADPH oxidase (NOX). NOX is a source of ROS that is significantly increased in vascular aging. It is regulated by the transcription factor megakaryocytic leukemia 1 (MKL1). MKL1 recruits the histone acetyltransferase (MOF) to modify the chromatin structure surrounding NOX promoters, and this leads to the enhancement of NOX catalytic activity. Increased NOX activity drives rates of ROS accumulation in tissues (55). The second mechanism involved in ROS production involves the epigenetic processes that abolish ROS production or damage. In this regard, ROS scavenging enzymes like superoxide dismutase 2 (SOD2) are critical. Excessive ROS promotes increased levels of methylation at the SOD2 promoter region causing transcriptional silencing. Moreover, expression levels of SOD2 can be enhanced by SIRT3 (56). We speculate that SOD2 activator molecules would be useful therapeutic tools to manipulate SOD levels on tissues and deserves investigation. Indeed, SOD can reduce excess ROS and contributes to facilitating femoral artery endothelial function (57).
ECs synthesized NO at discrete concentrations whereby it suppresses VSMC relaxation and maintains low rates of cellular proliferation. However, under physiological conditions of excessive ROS production rates of NO production become dysregulated, promoting arterial stiffness, collagen synthesis, intimal hyperplasia, and apoptosis. Interestingly, treating with the NOX1/4 inhibitor, GKT137831, dampens ROS generation and is protective in this setting (58). Furthermore, endogenous ROS contribute to DNA repair and impairment, and this regulates cell development and differentiation in a PI3K/Akt-dependent manner (59). Aberrant activation of AKT disturbs rates of cellular proliferation, cell survival, and metabolic homeostasis by altering DNA methylation and histone modifications; processes impacted by the activities of Foxo1/3, p53/21-dependent pathways that drives rates of cellular senescence (60, 61).
Integrins are a family of membrane receptors that are expressed widely on the surface of cell membranes. These proteins mediate cellular adhesion to the extracellular matrix and function by allowing cells to sense changes to their localized environment. Integrins are crucial components with roles in thrombosis, leukocyte infiltration, VSMC aggregation, cell migration, ECM deposition, and in the vascular phenotype switch (62). Interestingly, integrin β1, has recently been found to mediate capillary aging and angiogenesis. Indeed, loss of endothelial integrin β1 leads to endothelial cell differentiation defects, cell adhesion suppression, reduced capacity to aid cellular migration, and survival inhibition during angiogenesis. In contrast, upregulation of integrin β1 results in SASP (63, 64). In terms of epigenetic mechanism, integrin β1 expression is regulated by β-galactoside α2,6-sialyltransferase-1 (St6gal1) DNAm (65). Other integrin proteins like integrin αvβ3 promote coronary arteriolar dilation and angiogenesis in a HDAC5-dependent manner (66). Furthermore, integrin Gα13 is atheroprotective (67).
TLRs can be activated during physiological and pathological aging, and induction results in a robust inflammatory response. For example, endothelial TLR4 drives lesion formation and causes stroke and seizure (68). TLR4 expression negatively correlates with regulatory factor X1 (RFX1). RFX1 reportedly increases rates of methylation of histone H3K9, and decreased levels of H3 and H4 acetylation in the TLR4 promoter via the involvement of DNMT1, HDAC1, and histone-lysine N-methyltransferase SUV39H1 (SUV39H1), respectively. In addition, TET2 impacts on TLR4 via the NF-κB p65 pathway (69). Dimerization of TLR2 and 4 induces the activation of the NLRP3-inflammasome resulting in diminished endothelial regeneration (70). TLR9 recognized CpG-motif of oligodeoxynucleotides (ODN) and elicited immune responses that lead to the induction of apoptosis (71). Clearly, given the intimate role that TLRs play in vascular damage and aging, these proteins could be useful drug targets.
Ion channels are involved in signaling networks and homeostatic regulation. Genetic variants coding for ion channels and epigenetics targets can impact on structural disorganization and functional defects. Ca2+ is a secondary messenger needed for proper cell function. The Ca2+ channel superfamily includes transient receptor potential (TRP) family of proteins namely, TRPC, TRPV, TRPM, TRPA, TRPML, TRPP, the ORAI family, and Ca2+-activated K+ (KCa) channel family (72). Combined these channels are critically important in the regulation of calcium signaling systems in cells and tissues. Some Ca2+ channels play a direct role in vascular aging. For example, TRPCs are non-selective cation channels permeable to calcium ions. Among them, TRPM7 affects histones H3K9, H3K27 acetylation, H3 phosphorylation, and chromatin covalent modifications (73). Increased acetylation status of H3K27 is associated with vascular aging. Tentative links between the Orai family members and vascular aging have also been proposed in recent times (74), however, the epigenetic mechanisms involved in their regulation are still unknown. Other calcium channels including the Ca2+-activated K+ (KCa) channels that act as transducers, that respond to elevated intracellular calcium ([Ca2+]i) signals causing hyperpolarization of Vm and decreased vascular resistance thereby enhancing blood flow. KCa channels facilitate Ca2+ influx into cells via non-selective cation channels that can stimulate increased synthesis of nitric oxide (NO) (75). Dysregulation of Ca2+ handling and the activities of KCa channels precipitate reductions in NO levels, leading to increased vascular stiffness, and eventually vascular aging. Lastly, dysfunction of L-type voltage-gated Ca2+ channels (LTCCs) exert an epigenetic influence by negatively regulating levels of miR-328 in artery-derived VSMCs (76).
After entering adolescence, males and females produce germ cells and secrete sex hormones (the sex steroid estrogens, androgens, and progesterone). Sex hormones promote the development of mature reproductive organs, reproductive function, and the secretion of sex hormones and regulatory systems. Mounting evidences have shown that the incidence of vascular diseases in elder male and postmenopausal female were age- and gender- specific. Both estradiol and testosterone, which modulate female and male endothelial function, typically fall in post-menopause females and elderly males. The low level of these two hormones correlates with vascular aging in women and vasomotor instability in male (77). The sex hormone cytosolic/nuclear receptors for estrogen, progesterone, and testosterone have been identified, and shown to function in vascular endothelium and smooth muscle cells signaling networks (78). Indeed, it is likely that these hormones are involved in vascular cell proliferation, development, and in cellular senescence. For example, testosterone ameliorates vascular aging via the GAS6/AXL signaling pathway (79). GAS6 signaling is reported to increase NO bioavailability via the androgen receptor-mediated activation of endothelial NO synthase (eNOS) (80). Testosterone may also function via lncRNA-SWI/SNF complex crosstalk (81). Interestingly, while therapeutic doses of testosterone can benefit elderly individuals, excessive circulatory testosterone has a detrimental effect. This negative impact of testosterone appears to be due to impaired endothelial progenitor cells function partly linked to the hypermethylation on the estrogen receptor β's promoter. Similarly, the administration of progesterone in postmenopausal women have been widely used therapeutically for the existence of epigenetic phenomena that progesterone influences the responses of cells to NO handling via a H3K27ac- and H3K27me3 dependent process (82, 83). Similarly, estrogen acts on vascular aging by activating SIRT1, H3 acetylation, and promotes miR-203 expression. These changes inhibiting VSMCs proliferation and promoting miR-126-3p expression that results in endothelial migration (84, 85). It is feasible that the activation of SIRT1 by estrogen partly explains why female have a higher life expectancy than that of males.
Metalloproteinases are a group of proteolytic enzymes that are involved in the metabolism of the extracellular matrix. These enzymes play essential roles in angiogenesis, wound healing, and fibrosis. Interestingly, overexpression of SIRT1 resulted in lower MMP2 expression in VSMCs and this correlates with the deacetylating of histone H3 lysine 9 (H3K9) sites within the MMP2 promoter (86). In addition, metalloproteinase 9 (MMP9) is involved in the decomposition of extracellular matrix and is essential for driving macrophage-dependent inflammation in the vascular system (87). MMP9 levels are regulated by TET2, miR-212, miR-132, and the long non-coding RNA, TETILA (88, 89). H3 lysine 9 di-methylation (H3K9me2), is a repressive epigenetic marker and the levels of which are often reduced in atherosclerotic lesions. H3K9me2 is down-regulation in the promotor regions of MMP3, MMP9, and MMP12 in VSMCs of arteries. Clearly, these three proteins play important roles in controlling VSMCs response to vascular inflammation.
In the 1970s, scientists discovered that RNA methylation can also occur in cells and tissues and that these modifications are now known to be crucial epigenetic regulators. At present, more than 160 RNA modifications have been identified, including N6-methyladenosine (m6A), 5-methylcytosine (m5C), N7-methylguanosine (m7G), and so on (90). Among these modifications, m6A is the most abundant mRNA modification in eukaryotes, which is mediated by methyltransferase like proteins namely METTL3, METTL14, WTAP (91), and this modification can be reversed by demethylase like FTO, ALKBH5. Besides, m6A binding proteins like YTHDF1/2/3, YTHDC1/2 also take part in the dynamic reversible m6A modification. METTL3 alleviates human mesenchymal stem cell senescence through m6A modification-dependent stabilization of the MIS12 transcript (92), the overexpression of METTL3 coupled with METTL14 promoted SASP-related cytokines (such as CXCL1, CXCL3, CXCL5, CXCL6, IL1α, IL1β, and IL6) releasing (93). The loss of FTO was also reported to antagonize the vascular dysfunction by improving the insulin sensitivity in obesity mice (94). These experimental results showed that RNA methylation is vital in regulating vascular aging processes.
Life expectancy is increasing due to advances in the clinical management of diseases, and due to our understanding of lifestyle factors that negatively impact on health. Numerous environmental factors including toxins, heavy metal ions, pollutants, gases, infections, smoking, and alcohol consumption are known to impact on vascular function and influence gene expression. In addition, some epigenetic alterations are heritable like for example, a parental high-fat diet that renders offspring more susceptible to developing obesity and diabetes (95). Of the known environmental drivers of cardiovascular diseases, smoking and microorganism infection are the most widely studied:
The adverse effects of smoking on human health have been known for thousands of years and this habit remains one of the leading drivers of premature death and disability worldwide. Tobacco use and its chemical constituents are associated with causing cellular damage including rates of apoptosis, cell cycle arrest, DNA damage, ER stress, and oxidative stress in vascular endothelial cells (96). Moreover, as shown in Figure 2, smoking hastens vascular aging by affecting multiple biochemical pathways. Indeed, the reactive metabolite benzo[a]pyrene diolepoxide (BPDE) causes the accumulation of DNA adducts, that can block the formation of the DNA replication forks in mammalian cells. Persistent blockage of the replication fork by bulky lesions causes DNA double-strand breaks. In turn, these strand breaks elicit histone H2AX phosphorylation (γH2AX, a marker of DNA double-strand breaks) and ataxia telangiectasia-mutated kinase (ATM)/CHK2-mediated events, and this is often seen in aging and in some cancers (97). Furthermore, reductions in DNMT1 and DNMT3B activities protect aging cells from BPDE-induced DNA damage via the inhibition of H2AX by ataxia telangiectasia-mutated kinase phosphorylation (98). BPDE also suppresses retinoic acid receptor-β2 (RAR-β2) expression by promoting DNMT 3A interactions with the promotor region of RAR-β2. This interaction causes the increased expression of c-Jun, extracellular signal-regulated protein kinases 1/2 (ERK1/2), and cyclooxygenase-2 (COX-2) (99). Of these targets, c-Jun is reported to associate with Fos-related antigen 1 (Fra1), allowing for its binding to the promoter region of Cdkn1a (coding for p21) and Cdkn2a (coding for p16), and this triggers the senescence-related phenotypes, often seen in many cardiovascular disorders and in vascular senescence (100).
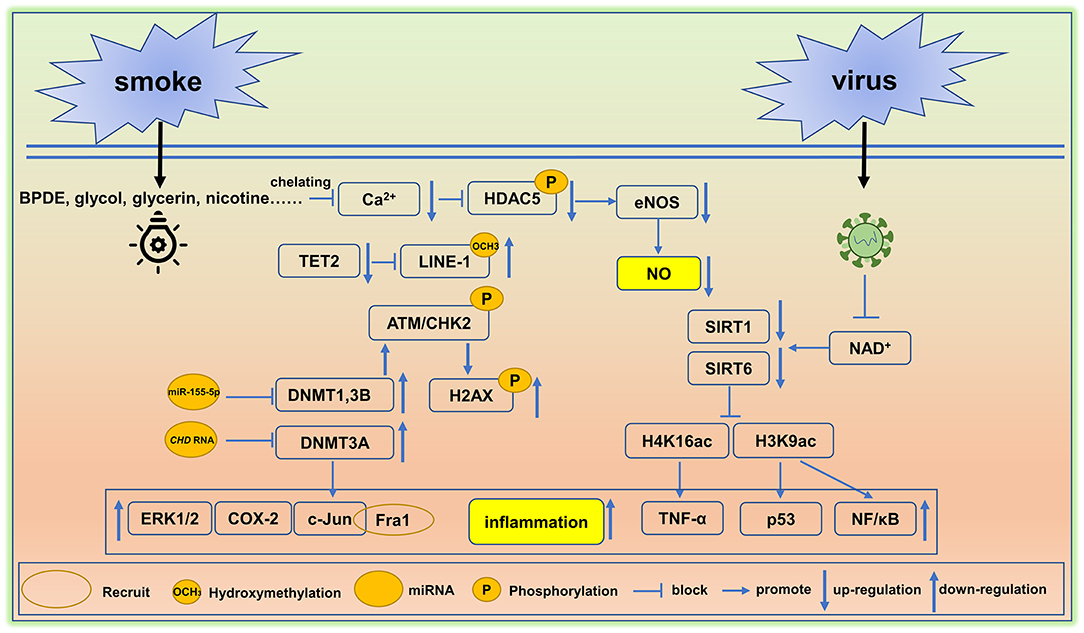
Figure 2. Environment-mediated vascular aging. Smoking and virus are two factors that cause epigenetic alterations in cells and tissues and induce vascular aging. BPDE, Benzo[a]pyrene diol epoxide, DNMT, DNA methyltransferase, eNOS, endothelial NO synthase, TET2, ten-methylcytosine- dioxygenase 2, SIRT, Sirtuin. A detail of these epigenetic pathway is elaborated in section environmental factors.
Decreased rates of hydroxymethylation in DNA correlates with smoking status. Cigarette smoke ingredients like propylene glycol, glycerin, additives, and nicotine trigger the hypomethylation of LINE-1 repeat elements by reducing global levels of DNA hydroxymethylation. Hypomethylation is typically caused by reductions in TET2 (101), leading to the overexpression of the LINE-1 gene (an aging biomarker) (102). Moreover, TET2 down-regulation is linked with arterial hypertension and immune activation (103, 104).
A study of 20 young healthy smokers including 10 males and 10 females was employed to study the alterations in microRNA signatures. The results showed that miR-29b (associated with aortic aneurysm and fibrosis) was significantly up-regulated and miR-223 is downregulated (105), the loss of miR-223 could lead to severe coronary artery pathology through a miR-223/PDGFRβ vascular smooth muscle cell axis (106). In endothelial cells, the miR-155 inhibition rescued the deleterious effects of cigarette smoke condensate on endothelial-mediated vascular relaxation and oxidative stress (107). Moreover, ncRNAs are critical to the regulation of transcription and in conjunction with DNA methylation rates can influence RNA–DNMT interactions. Indeed, it has been reported that NUP153, EF1a, and CHD RNA are bound to DNMT3A, yet only CHD RNA is capable of inhibiting the catalytic function of DNMT3A (108). These findings are similar to the inhibition of DNMT1 by miR-155-5p (109), suggesting that CHD RNA and miR-155-5p are potential therapeutic targets that could be used to restore DNMT3A, DNMT1 (107).
In other studies, the histone deacetylase SIRTUIN1 (SIRT1), which acts as a longevity gene and plays an instrumental role in cell cycle progression, energy metabolism, and aging (110). SIRT1 is reduced by smoking, resulting in an imbalance of downstream histone modifications, like H1K26, H3K9, H3K14, H3K56, and H4K16 (111). Under normal circumstances, SIRT1 binds to the promoter of p53 (a proapoptotic gene) decreasing H3K9 acetylation, and thereby inhibiting p53 gene transcription (112). A similar mechanism has been reported in the suppression of the NF-kappaB gene (113). Smoking clearly disturbs patterns of histone modification and this correlates with a vascular aging phenotype, inflammation, and apoptosis. In addition, smoking causes increased blood pressure leading to elevated fluid shear stress. This in turn, inhibits Ca2+ channel signaling, reduces eNOS, and blocks HDAC5 phosphorylation (114).
Microorganism infections contribute to vascular aging. For example, the hypercoagulable and hyperinflammatory states caused by the viral infections make vascular endothelial cells more susceptible to infection, resulting in endothelialitis (115). Persistent inflammation promotes the accumulation of toxic metabolites and reactive oxygen species, and further leads to genetic and epigenetic alterations. In this setting, DNA hypermethylation usually activates the senescence- associated secretory phenotype (SASP) (116). Some virus possesses an ADP-ribosylhydrolase that can deplete NAD+, and causes a decline in the activities of NAD+-dependent lysine deacetylases like the SIRT family proteins, SIRT1 and SIRT6 (117, 118). Loss of SIRT1 attenuates its deacetylation function on the target proteins H4K16 located in the TNF-α promotor region; this leads to increased TNF-α levels, a widely known proinflammatory mediator. Moreover, dramatic reduction in claudin-1 and vascular endothelial-cadherin inactivate DNA repair mechanisms and increase the rates of vascular inflammation and senescence (119). Importantly, TNF-α and IFN-γ triggered inflammation, causing cell death, tissue damage, and mortality (120). In a similar manner to SIRT1, SIRT6 attenuates inflammation via reducing NF-κB signaling following the deacetylation of the target H3K9 (113). Based on the available evidence, moderate activation of SIRT1 and SIRT6 could be used to dampen rates of vascular inflammation and could have potential in reducing rates of vascular aging in humans. A summary of environment factors linked to vascular aging are shown in Figure 2.
A wide range of epigenetic alterations has been discovered that are associated with the diseases-induced vascular aging phenotype (121). T-cell–derived miR-214 facilitates perivascular fibrosis in hypertension (122), nuclear miR-320 promotes lipid accumulation (hyperlipidemia) in the heart (123), and genome-wide DNA methylation profiling has discovered significant differences in promoter CpG islands in genes like HIF3A, CPT1A, CD38, PHGDH, ABCG1, SREBF1, CPT1A, PDX1 (124). In addition, the HDAC inhibitor trichostatin A, is reported to reduce atherosclerosis in ApoE-deficient mice, suggesting an important role of epigenetic modifications in the cardiovascular system (125). Other researchers have shown that the histone methyltransferases, SET7 and SET 9, and various histone deacetylases (HDAC4, HDAC7, HDAC10, and KMT2D), along with ncRNA, miRNA-199a-3p, miRNA 34a, circ HIPK3, and circ ZNF609 are linked with vascular dysfunction (126, 127).
Exercise is an exogenous stimulus that influences cellular metabolism by altering the expression of enzymes and proteins involved in numerous metabolic pathways involved in energy production. A lack of physical activity or a sedentary lifestyle can increase the risk of vascular aging disease, such as arterial stiffening and hypertension (128). However, moderate rates of exercise are seen as beneficial since this makes the myocardial systole more powerful, increases stroke output, enlarges the coronary artery diameter, improves the heart's blood supply, enhances the elasticity of systemic blood vessels, and delays arteriosclerosis. The epigenetic mechanism of exercise in reducing vascular aging will be illustrated in section Health lifestyle.
In addition to accelerating aging (above three mediators), the prevalence of vascular diseases is also on the increase as the number of elderly people is increasing worldwide. Even in the absence of overt injury, structural and functional changes occur in vessels as they age. Physiological functions, such as antioxidant enzyme activity, muscle composition, inflammatory factors expression, thickening of the intima, and changes in metabolic enzyme activities rise during normal aging. These age-related vascular changes often accompany or even precede CVD development. Over time, normal aging increases transcriptional noise and stochastic effects in tissues and drives dysregulation of genetic and epigenetic control in the ECM (129). Phenotypically, the aging vessels begin to show moderate increases in the level of peripheral resistance, increased lumen diameter, and changes to the media thickness in small-sized muscular arteries. No accompanying changes in the media-to-lumen ratio occur with this indicating that it is an outward hypertrophic remodeling. Currently, the epigenetic mechanisms that slow down normal vascular aging are usually associated with regulating the daily environment, exercise, and reducing the incidence of disease, which mediate alterations on DNA methylation, histone modification, and ncRNA changes.
Evidence points to the possibility that structural and functional vascular aging can be reversed. Age-related declines in vascular function can be halted by lifestyle changes, such as exercise, diet, sleep, and genetic/epigenetic pharmacological intervention. These activities slow down age-related changes and the loss of functional blood vessels. A summary of epigenetic intervention mechanisms is shown in Figure 3.
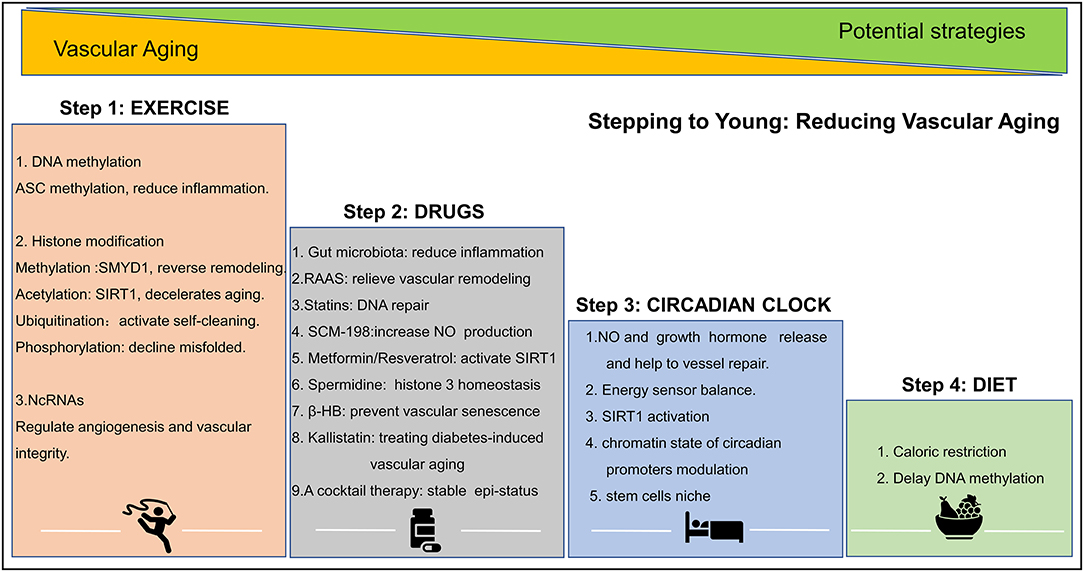
Figure 3. Epigenetic strategies to reduce vascular aging. Exercise, drug, circadian clock, and diet play important role in altering epigenetic modifications and in reducing vascular aging. ASC, factor–apoptosis-associated speck-like protein containing CARD, SMYD1, SET- and MYND- domain containing protein 1; SIRT, sirtuin; SCM-198, leonurine; β-HB, β-hydroxybutyrate. A detail of this figure is elaborated in section the epigenetic intervention strategies of vascular aging.
Regular physical activity improves vascular health and brings about discrete epigenetic modifications in tissues. For example, exercise increases the methylation levels of the pro-inflammatory factor apoptosis-associated speck-like protein containing CARD (ASC) gene (encoding IL-1β and IL-18), and thus suppress ASC-induced inflammation (130). Other evidence, shows that the histone methyltransferase, Smyd1, is protective roles in the pathological remodeling process (131), and that regular exercises increase SIRT1 expression and stimulates of NAD (+) biosynthesis (132). In addition, ubiquitination and phosphorylation have also been considered crucial regulatory mechanisms because they reduce protein misfolding, the accumulation of toxic proteins, and redundant “junk” proteins that occasionally accumulate in cells and tissues. Regular exercise stimulates ubiquitination and phosphorylation while promoting the elimination of defective proteins (133). MicroRNAs like miR-20a, miR-126, miR-210, and miR-221/222 reportedly regulating angiogenesis and maintaining vascular integrity and again the levels of these molecules are changed following exercise (134). Finally, exercise alters CTCF-based 3D chromatin architecture via an unknown mechanism (135).
The circadian clock significantly affects the expression of genes associated with obesity and type 2 diabetes. Poor circadian rhythm promotes hypomethylation in deleterious genes and hypermethylation in beneficial genes. This triggers an increase in transcriptional and genomic instability, chronic damage, and impacts on vascular homeostasis viz. the release of NO and growth hormone, MMP and ECM remodeling, and vascular stem cells niche. Circadian patterns also influence chromatin and promote self-repairing mechanisms like the activation of histone ubiquitination, and SIRT1 activation (136, 137).
Keeping a balanced and healthy diet delays vascular aging-related DNA methylation by regulating the methionine and S-adenosylmethionine cycle (95, 138). Caloric restriction is also thought to be a major factor in slowing down the aging processes of blood vessels (139).
RAAS inhibits vascular remodeling and increases the expression of pro-survival genes like nicotinamide phosphoribosyltransferase, and SIRT3. This occurs via reducing mitochondrial oxidative stress, and highlights that the suppression of the RAAS system could be a possible way of managing vascular aging (140).
Statins have an ability to decrease DNA damage by inducing DNA damage repair systems and by suppressing oxidative stress by increasing antioxidant defenses (141). However, use of statins is often associated with adverse reactions like rhabdomyolysis that are of clinical significance. Here, we speculate leonurine (SCM-198), which is still in the clinical trial but shows fewer adverse reactions, will be a more promising drug in the future based on the present findings that it regulates H3K27 demethylase JMJD3 (142, 143). Moreover, the phase I clinical trial of 36 subjects uncover that SCM-198 affected homocysteine-methionine metabolism (144), which means that it can reduce ROS-induced endothelial cell damage and lipid peroxidation, thereby reversing vascular damage and destruction. The basic experimental studies also confirm its therapeutical effect (145, 146).
A 78,241 people observational study has shown that the use of metformin increases life span (147). It is a known inducer of SIRT1, which increased SIRT1 promoter chromatin accessibility (148).
Spermidine prevents histone H3 hyperacetylation, can activate autophagy, and prolong life span (149). This anti-aging effect may be associated with its ability for restoring cellular metabolic dysfunction. A study of 85 elder adults reveals that spermidine significantly improved cognitive performance (150, 151).
Long-term studies have found that gut microbiota disorders are related to malnutrition, obesity, diabetes, and diseases. ProBiotic-4, composed of mixture of Bifidobacterium lactis, Lactobacillus casei, Bifidobacterium bifidum, and Lactobacillus acidophilus, has been shown to attenuate aging-induced gut microbiota dysbiosis by reducing toll-like receptor 4 (TLR4) expression and influence nuclear factor-κB (NF-κB) nuclear translocation (152).
Resveratrol is the only SIRT1 agonist that has been shown to delay vascular aging (110). Other molecules like, β-hydroxybutyrate (β-HB) have been found to prevent vascular senescence and can activate Oct4 expression level via stimulating DNA hydroxymethylation (153). Kallistatin attenuates TNF-α-induced endothelial progenitor cells (EPCs) senescence and STZ-induced aortic senescence by abolishing miR-34, and SIR-2.1 (154), Kallistatin is a candidate molecule that could be further developed as an epigenetic drug in the treatment of diabetes-induced vascular aging.
In summary, the following review has addressed the roles of epigenetic-mediated vascular aging, current discoveries, and possible roles of epigenetic processes in vascular aging. An understanding of these regulatory mechanisms critical in vascular aging will be paramount in developing novel treatment strategies. Epigenetic aging mechanisms that focus of much attention by researchers and growing evidence shows DNAm, post-translational histone modifications, ncRNAs, chromatin structure alteration are important in the cardiovascular system. In addition, several epigenetic-related drugs in clinical trials as highlighted by the WHO International Clinical Trial Registration Platform (WHO ICTRP). However, only two DNMTs inhibitors (azacytidine and decitabine), six HDACs inhibitors (vorinostat, romidepsin, belinostat, chidamide, panobinostat, and tazemetostat), and one ncRNA (siPCSK9) have been so far approved for clinical use. In the future, we are optimistic that other epigenetic based drugs will be developed. These developments arising from methodological improvements that allow for the analysis of epigenetic changes occurring at the single-cell and 3D level. These advances will make research easier and allow for studies addressing endothelial metabolism, angiogenesis, and pathological progress of arteriosclerosis during aging and collectively this will provide a dynamic pattern of change in cells and tissues. Most studies to date have stepped toward finding the role of 5mC reader (BAZ2A, MBD1/2, MBD4, MeCP2, SETDB1/2, UHRF1/2, and ZBTBR4/33/28), 5hmC and 5fmC readers (MHS6, PRP8, RPL26, UHRF2, EHMT1, FOXI3, FOXK1/2, FOXP1/4, L3MBTL2, MPG, and TDG), histone modification writer (EZH2, MMSET, DOT1L, SETD7, MLL1, PRMT1/3/5, and SMYD2/3), histone modification reader (CREBBP, EP200, MOZ, MOF, BRD2/3/4/7/9, and SMARCA2/4), histone modification eraser (HDAC4, JMJD3, and LSD1), and some ncRNAs in vascular aging. Many in vitro and in vivo studies have demonstrated some of these targets show the capacity in reducing vascular aging processes, but their pathologies and pharmacology are mostly unknown. Effects of these epigenetic targets that underpins the aging therapeutic should be considered in future studies.
YZ and ZL designed and revised the manuscript. QD, XL, YF, HH, and CH helped to improve the contexts and grammar. All authors contributed to the article and approved the submitted version.
This work was supported by Macau Science and Technology Development fund [FDCT (0067/2018/A2, 033/2017/AMJ, 0007/2019/AKP, 0052/2020/A, 0011/2020/A1, and 0030/2018/A1)]. The National Natural Science Foundation of China (No. 81973320).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li X, Wu C, Lu J, Chen B, Li Y, Yang Y, et al. Cardiovascular risk factors in China: a nationwide population-based cohort study. Lancet Public Health. (2020) 5:e672–81. doi: 10.1016/S2468-2667(20)30191-2
2. Harvey ZH, Chen YW, Jarosz DF. Protein-based inheritance: epigenetics beyond the chromosome. Mol Cell. (2018) 69:195–202. doi: 10.1016/j.molcel.2017.10.030
3. Margolick JB, Ferrucci L. Accelerating aging research: how can we measure the rate of biologic aging? Exp Gerontol. (2015) 64:78–80. doi: 10.1016/j.exger.2015.02.009
4. Ferrucci L, Levine ME, Kuo PL, Simonsick EM. Time and the metrics of aging. Circ Res. (2018) 123:740–4. doi: 10.1161/CIRCRESAHA.118.312816
5. Ding YN, Tang X, Chen HZ, Liu DP. Epigenetic regulation of vascular aging and age-related vascular diseases. Adv Exp Med Biol. (2018) 1086:55–75. doi: 10.1007/978-981-13-1117-8_4
6. Stubbs TM, Bonder MJ, Stark AK, Krueger F, Team BIAC, von Meyenn F, et al. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. (2017) 18:68. doi: 10.1186/s13059-017-1203-5
7. Fadini GP, Ceolotto G, Pagnin E, de Kreutzenberg S, Avogaro A. At the crossroads of longevity and metabolism: the metabolic syndrome and lifespan determinant pathways. Aging Cell. (2011) 10:10–7. doi: 10.1111/j.1474-9726.2010.00642.x
8. Wyatt GR. Occurrence of 5-methylcytosine in nucleic acids. Nature. (1950) 166:237–8. doi: 10.1038/166237b0
9. Zhao LY, Song J, Liu Y, Song CX, Yi C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell. (2020) 11:792–808. doi: 10.1007/s13238-020-00733-7
10. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. (2018) 19:81–92. doi: 10.1038/nrg.2017.80
11. Nanduri J, Semenza GL, Prabhakar NR. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L1096–100. doi: 10.1152/ajplung.00325.2017
12. Agha G, Mendelson MM, Ward-Caviness CK, Joehanes R, Huan T, Gondalia R, et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation. (2019) 140:645–57. doi: 10.1161/CIRCULATIONAHA.118.039357
13. Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci. (2009) 66:2249–61. doi: 10.1007/s00018-009-0015-5
14. Guiducci G, Paone A, Tramonti A, Giardina G, Rinaldo S, Bouzidi A, et al. The moonlighting RNA-binding activity of cytosolic serine hydroxymethyltransferase contributes to control compartmentalization of serine metabolism. Nucleic Acids Res. (2019) 47:4240–54. doi: 10.1093/nar/gkz129
15. Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. (2013) 128:2047–57. doi: 10.1161/CIRCULATIONAHA.113.002887
16. Connelly JJ, Cherepanova OA, Doss JF, Karaoli T, Lillard TS, Markunas CA, et al. Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum Mol Genet. (2013) 22:5107–20. doi: 10.1093/hmg/ddt365
17. Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res. (2018) 123:335–41. doi: 10.1161/CIRCRESAHA.118.313225
18. Suzuki M, Yamada T, Kihara-Negishi F, Sakurai T, Hara E, Tenen DG, et al. Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene. (2006) 25:2477–88. doi: 10.1038/sj.onc.1209272
19. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. (2014) 24:464–71. doi: 10.1016/j.tcb.2014.04.002
20. Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. (2016) 32:42–56. doi: 10.1016/j.tig.2015.10.007
21. Feng Y, Liu X, Pauklin S. 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell. (2021) 12:440–54. doi: 10.1007/s13238-020-00819-2
22. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. (2007) 129:823–37. doi: 10.1016/j.cell.2007.05.009
23. Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GC, Schneider R, et al. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet. (2016) 48:681–6. doi: 10.1038/ng.3550
24. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. (2014) 15:703–8. doi: 10.1038/nrm3890
25. Xiao X, Liu C, Pei Y, Wang YZ, Kong J, Lu K, et al. Histone H2A ubiquitination reinforces mechanical stability and asymmetry at the single-nucleosome level. J Am Chem Soc. (2020) 142:3340–5. doi: 10.1021/jacs.9b12448
26. Yang L, Ma Z, Wang H, Niu K, Cao Y, Sun L, et al. Ubiquitylome study identifies increased histone 2A ubiquitylation as an evolutionarily conserved aging biomarker. Nat Commun. (2019) 10:2191. doi: 10.1038/s41467-019-10136-w
27. Feng Y, Pauklin S. Revisiting 3D chromatin architecture in cancer development and progression. Nucleic Acids Res. (2020) 48:10632–47. doi: 10.1093/nar/gkaa747
28. Hirosue A, Ishihara K, Tokunaga K, Watanabe T, Saitoh N, Nakamoto M, et al. Quantitative assessment of higher-order chromatin structure of the INK4/ARF locus in human senescent cells. Aging Cell. (2012) 11:553–6. doi: 10.1111/j.1474-9726.2012.00809.x
29. Yang D, Xiao C, Long F, Su Z, Jia W, Qin M, et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc Res. (2018) 114:1016–28. doi: 10.1093/cvr/cvy051
30. Schiano C, Benincasa G, Franzese M, Della Mura N, Pane K, Salvatore M, et al. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol Ther. (2020) 210:107514. doi: 10.1016/j.pharmthera.2020.107514
31. Oksala NKJ, Seppala I, Rahikainen R, Makela KM, Raitoharju E, Illig T, et al. Synergistic expression of histone deacetylase 9 and matrix metalloproteinase 12 in m4 macrophages in advanced carotid plaques. Eur J Vasc Endovasc Surg. (2017) 53:632–40. doi: 10.1016/j.ejvs.2017.02.014
32. Ghosh AK. p300 in cardiac development and accelerated cardiac aging. Aging Dis. (2020) 11:916–26. doi: 10.14336/AD.2020.0401
33. Yang D, Wei G, Long F, Nie H, Tian X, Qu L, et al. Histone methyltransferase Smyd3 is a new regulator for vascular senescence. Aging Cell. (2020) 19:e13212. doi: 10.1111/acel.13212
34. Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. (2004) 6:731–40. doi: 10.1038/ncb1151
35. Xu ST, Wu J, Sun BF, Zhong C, Ding JP. Structural and biochemical studies of human lysine methyltransferase Smyd3 reveal the important functional roles of its post-SET and TPR domains and the regulation of its activity by DNA binding. Nucleic Acids Res. (2011) 39:4438–49. doi: 10.1093/nar/gkr019
36. Cencioni C, Heid J, Krepelova A, Rasa SMM, Kuenne C, Guenther S, et al. Aging triggers H3K27 trimethylation hoarding in the chromatin of nothobranchius furzeri skeletal muscle. Cells. (2019) 8:1169. doi: 10.3390/cells8101169
37. Martin-Herranz DE, Aref-Eshghi E, Bonder MJ, Stubbs TM, Choufani S, Weksberg R, et al. Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. (2019) 20:146. doi: 10.1186/s13059-019-1753-9
38. Pu M, Ni Z, Wang M, Wang X, Wood JG, Helfand SL, et al. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. (2015) 29:718–31. doi: 10.1101/gad.254144.114
39. Luo X, Yang D, Wu W, Long F, Xiao C, Qin M, et al. Critical role of histone demethylase Jumonji domain-containing protein 3 in the regulation of neointima formation following vascular injury. Cardiovasc Res. (2018) 114:1894–906. doi: 10.1093/cvr/cvy176
40. Xu C, Wang L, Fozouni P, Evjen G, Chandra V, Jiang J, et al. SIRT1 is downregulated by autophagy in senescence and aging. Nat Cell Biol. (2020) 22:1170–9. doi: 10.1038/s41556-020-00579-5
41. Guo J, Wang ZY, Wu JC, Liu M, Li MM, Sun Y, et al. Endothelial SIRT6 is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ Res. (2019) 124:1448–61. doi: 10.1161/CIRCRESAHA.118.314032
42. Xu S, Yin M, Koroleva M, Mastrangelo MA, Zhang W, Bai P, et al. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging. (2016) 8:1064–82. doi: 10.18632/aging.100975
43. Guzik TJ, Cosentino F. Epigenetics and immunometabolism in diabetes and aging. Antioxid Redox Sign. (2018) 29:257–74. doi: 10.1089/ars.2017.7299
44. Camici GG, Savarese G, Akhmedov A, Luscher TF. Molecular mechanism of endothelial and vascular aging: implications for cardiovascular disease. Eur Heart J. (2015) 36:3392–403. doi: 10.1093/eurheartj/ehv587
45. Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. (2016) 96:1297–325. doi: 10.1152/physrev.00041.2015
46. Zhang F, Zhang R, Zhang X, Wu Y, Li X, Zhang S, et al. Comprehensive analysis of circRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of atherosclerosis in rabbits. Aging. (2018) 10:2266–83. doi: 10.18632/aging.101541
47. Huang SL, Li XZ, Zheng H, Si XY, Li B, Wei GQ, et al. Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation. (2019) 139:2857–76. doi: 10.1161/CIRCULATIONAHA.118.038361
48. Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, et al. Long noncoding RNAs with snoRNA ends. Mol Cell. (2012) 48:219–30. doi: 10.1016/j.molcel.2012.07.033
49. Michel CI, Holley CL, Scruggs BS, Sidhu R, Brookheart RT, Listenberger LL, et al. Small nucleolar RNAs U32a, U33, and U35a are critical mediators of metabolic stress. Cell Metab. (2011) 14:33–44. doi: 10.1016/j.cmet.2011.04.009
50. Yang F, Chen QS, He SP, Yang M, Maguire EM, An WW, et al. miR-22 is a novel mediator of vascular smooth muscle cell phenotypic modulation and neointima formation. Circulation. (2018) 137:1824–41. doi: 10.1161/CIRCULATIONAHA.117.027799
51. Farina FM, Hall IF, Serio S, Zani S, Climent M, Salvarani N, et al. miR-128-3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ Res. (2020) 126:e120–35. doi: 10.1161/CIRCRESAHA.120.316489
52. Hadji F, Boulanger MC, Guay SP, Gaudreault N, Amellah S, Mkannez G, et al. Altered DNA methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation. (2016) 134:1848–62. doi: 10.1161/CIRCULATIONAHA.116.023116
53. Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. (2010) 6:e1001233. doi: 10.1371/journal.pgen.1001233
54. Jinn S, Brandis KA, Ren A, Chacko A, Dudley-Rucker N, Gale SE, et al. snoRNA U17 regulates cellular cholesterol trafficking. Cell Metab. (2015) 21:855–67. doi: 10.1016/j.cmet.2015.04.010
55. Ma L, Wang K, Shang J, Cao C, Zhen P, Liu X, et al. Anti-peroxynitrite treatment ameliorated vasorelaxation of resistance arteries in aging rats: involvement with NO-sGC-cGKs pathway. PLoS ONE. (2014) 9:e104788. doi: 10.1371/journal.pone.0104788
56. Dikalova AE, Pandey A, Xiao L, Arslanbaeva L, Sidorova T, Lopez MG, et al. Mitochondrial Deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ Res. (2020) 126:439–52. doi: 10.1161/CIRCRESAHA.119.315767
57. Nguyen A, Leblond F, Mamarbachi M, Geoffroy S, Thorin E. Age-dependent demethylation of Sod2 promoter in the mouse femoral artery. Oxid Med Cell Longev. (2016) 2016:8627384. doi: 10.1155/2016/8627384
58. Yu LM, Yang G, Zhang XJ, Wang P, Weng XY, Yang YY, et al. Megakaryocytic Leukemia 1 bridges epigenetic activation of NADPH oxidase in macrophages to cardiac ischemia-reperfusion injury. Circulation. (2018) 138:2820–36. doi: 10.1161/CIRCULATIONAHA.118.035377
59. Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-Dependant manner. Cell Stem Cell. (2011) 8:59–71. doi: 10.1016/j.stem.2010.11.028
60. Mathew OP, Ranganna K, Mathew J, Zhu ML, Yousefipour Z, Selvam C, et al. Cellular effects of butyrate on vascular smooth muscle cells are mediated through disparate actions on dual targets, Histone Deacetylase (HDAC) activity and PI3K/Akt signaling network. Int J Mol Sci. (2019) 20:2902. doi: 10.3390/ijms20122902
61. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. (2011) 12:21–35. doi: 10.1038/nrm3025
62. Miao L, Xin X, Xin H, Shen X, Zhu YZ. Hydrogen sulfide recruits macrophage migration by integrin beta1-Src-FAK/Pyk2-Rac pathway in myocardial infarction. Sci Rep. (2016) 6:22363. doi: 10.1038/srep22363
63. Nishimura M, Kumsta C, Kaushik G, Diop SB, Ding Y, Bisharat-Kernizan J, et al. A dual role for integrin-linked kinase and beta1-integrin in modulating cardiac aging. Aging Cell. (2014) 13:431–40. doi: 10.1111/acel.12193
64. Olmos G, Lopez-Ongil S, Ruiz Torres MP. Integrin-linked kinase: a new actor in the ageing process? Exp Gerontol. (2017) 100:87–90. doi: 10.1016/j.exger.2017.10.026
65. Kaburagi T, Kizuka Y, Kitazume S, Taniguchi N. The inhibitory role of alpha 2,6-Sialylation in adipogenesis. J Biol Chem. (2017) 292:2278–86. doi: 10.1074/jbc.M116.747667
66. Liu X, Zheng N, Shi YN, Yuan J, Li L. Thyroid hormone induced angiogenesis through the integrin αvβ3/protein kinase D/histone deacetylase 5 signaling pathway. J Mol Endocrinol. (2014) 52:245–54. doi: 10.1530/JME-13-0252
67. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. (2016) 540:579–82. doi: 10.1038/nature20602
68. Tang AT, Choi JP, Kotzin JJ, Yang Y, Hong CC, Hobson N, et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature. (2017) 545:305–10. doi: 10.1038/nature22075
69. Du P, Gao K, Cao Y, Yang S, Wang Y, Guo R, et al. RFX1 downregulation contributes to TLR4 overexpression in CD14(+) monocytes via epigenetic mechanisms in coronary artery disease. Clin Epigenetics. (2019) 11:44. doi: 10.1186/s13148-019-0646-9
70. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
71. Lim EJ, Park DW, Lee JG, Lee CH, Bae YS, Hwang YC, et al. Toll-like receptor 9-mediated inhibition of apoptosis occurs through suppression of FoxO3a activity and induction of FLIP expression. Exp Mol Med. (2010) 42:712–20. doi: 10.3858/emm.2010.42.10.070
72. Ohya S, Kito H, Hatano N, Muraki K. Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol Ther. (2016) 160:11–43. doi: 10.1016/j.pharmthera.2016.02.001
73. Krapivinsky G, Krapivinsky L, Manasian Y, Clapham DE. The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell. (2014) 157:1061–72. doi: 10.1016/j.cell.2014.03.046
74. Denda S, Takei K, Kumamoto J, Goto M, Denda M. Expression level of Orai3 correlates with aging-related changes in mechanical stimulation-induced calcium signalling in keratinocytes. Exp Dermatol. (2017) 26:276–8. doi: 10.1111/exd.13175
75. Behringer EJ, Hakim MA. Functional interaction among K(Ca) and TRP channels for cardiovascular physiology: modern perspectives on aging and chronic disease. Int J Mol Sci. (2019) 20:e77. doi: 10.3390/ijms20061380
76. Liao J, Zhang Y, Ye F, Zhang L, Chen Y, Zeng F, et al. Epigenetic regulation of L-type voltage-gated Ca(2+) channels in mesenteric arteries of aging hypertensive rats. Hypertens Res. (2017) 40:441–9. doi: 10.1038/hr.2016.167
77. Moreau KL. Modulatory influence of sex hormones on vascular aging. Am J Physiol Heart Circ Physiol. (2019) 316:H522–6. doi: 10.1152/ajpheart.00745.2017
78. Francomano D, Fattorini G, Gianfrilli D, Paoli D, Sgro P, Radicioni A, et al. Acute endothelial response to testosterone gel administration in men with severe hypogonadism and its relationship to androgen receptor polymorphism: a pilot study. J Endocrinol Invest. (2016) 39:265–71. doi: 10.1007/s40618-015-0325-4
79. Chen YQ, Zhou HM, Chen FF, Liu YP, Han L, Song M, et al. Testosterone ameliorates vascular aging via the Gas6/Axl signaling pathway. Aging. (2020) 12:16111–25. doi: 10.18632/aging.103584
80. Wang S, Long CL, Chen J, Cui WY, Zhang YF, Zhang H, et al. Pharmacological evidence: a new therapeutic approach to the treatment of chronic heart failure through SUR2B/Kir6.1 channel in endothelial cells. Acta Pharmacol Sin. (2017) 38:41–55. doi: 10.1038/aps.2016.118
81. Grossi E, Raimondi I, Goñi E, González J, Marchese FP, Chapaprieta V, et al. A lncRNA-SWI/SNF complex crosstalk controls transcriptional activation at specific promoter regions. Nat Commun. (2020) 11:936. doi: 10.1038/s41467-020-14623-3
82. Duckles SP, Miller VM. Hormonal modulation of endothelial NO production. Pflugers Arch. (2010) 459:841–51. doi: 10.1007/s00424-010-0797-1
83. Katoh N, Kuroda K, Tomikawa J, Ogata-Kawata H, Ozaki R, Ochiai A, et al. Reciprocal changes of H3K27ac and H3K27me3 at the promoter regions of the critical genes for endometrial decidualization. Epigenomics. (2018) 10:1243–57. doi: 10.2217/epi-2018-0006
84. Perez-Cremades D, Mompeon A, Vidal-Gomez X, Hermenegildo C, Novella S. miRNA as a new regulatory mechanism of estrogen vascular action. Int J Mol Sci. (2018) 19:473. doi: 10.3390/ijms19020473
85. Bendale DS, Karpe PA, Chhabra R, Shete SP, Shah H, Tikoo K. 17-beta Oestradiol prevents cardiovascular dysfunction in post-menopausal metabolic syndrome by affecting SIRT1/AMPK/H3 acetylation. Br J Pharmacol. (2013) 170:779–95. doi: 10.1111/bph.12290
86. Wang F, Tu Y, Gao Y, Chen H, Liu J, Zheng J. Smooth muscle sirtuin 1 blocks thoracic aortic aneurysm/dissection development in mice. Cardiovasc Drugs Ther. (2020) 34:641–50. doi: 10.1007/s10557-020-07005-w
87. Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, et al. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc Res. (2015) 106:421–31. doi: 10.1093/cvr/cvv128
88. Zhou L, Ren M, Zeng T, Wang W, Wang X, Hu M, et al. TET2-interacting long noncoding RNA promotes active DNA demethylation of the MMP-9 promoter in diabetic wound healing. Cell Death Dis. (2019) 10:813. doi: 10.1038/s41419-019-2047-6
89. Labrie M, St-Pierre Y. Epigenetic regulation of mmp-9 gene expression. Cell Mol Life Sci. (2013) 70:3109–24. doi: 10.1007/s00018-012-1214-z
90. Zheng HX, Zhang XS, Sui N. Advances in the profiling of N(6)-methyladenosine (m(6)A) modifications. Biotechnol Adv. (2020) 45:107656. doi: 10.1016/j.biotechadv.2020.107656
91. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
92. Wu Z, Shi Y, Lu M, Song M, Yu Z, Wang J, et al. METTL3 counteracts premature aging via m6A-dependent stabilization of MIS12 mRNA. Nucleic Acids Res. (2020) 48:11083–96. doi: 10.1093/nar/gkaa816
93. Liu P, Li F, Lin J, Fukumoto T, Nacarelli T, Hao X, et al. m(6)A-independent genome-wide METTL3 and METTL14 redistribution drives the senescence-associated secretory phenotype. Nat Cell Biol. (2021) 23:355–65. doi: 10.1038/s41556-021-00656-3
94. Kruger N, Biwer LA, Good ME, Ruddiman CA, Wolpe AG, DeLalio LJ, et al. Loss of endothelial FTO antagonizes obesity-induced metabolic and vascular dysfunction. Circ Res. (2020) 126:232–42. doi: 10.1161/CIRCRESAHA.119.315531
95. Huypens P, Sass S, Wu M, Dyckhoff D, Tschop M, Theis F, et al. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat Genet. (2016) 48:497–9. doi: 10.1038/ng.3527
96. Chu PH, Chen G, Kuo D, Braisted J, Huang R, Wang Y, et al. Stem cell-derived endothelial cell model that responds to tobacco smoke like primary endothelial cells. Chem Res Toxicol. (2020) 33:751–63. doi: 10.1021/acs.chemrestox.9b00363
97. Zhang W, Feng Y, Guo Q, Guo W, Xu H, Li X, et al. SIRT1 modulates cell cycle progression by regulating CHK2 acetylation-phosphorylation. Cell Death Differ. (2020) 27:482–96. doi: 10.1038/s41418-019-0369-7
98. Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2'-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. (2008) 28:752–71. doi: 10.1128/MCB.01799-07
99. Ye F, Xu XC. Benzo[a]pyrene diol epoxide suppresses retinoic acid receptor-beta2 expression by recruiting DNA (cytosine-5-)-methyltransferase 3A. Mol Cancer. (2010) 9:93. doi: 10.1186/1476-4598-9-93
100. Yang D, Xiao C, Long F, Wu W, Huang M, Qu L, et al. Fra-1 plays a critical role in angiotensin II-induced vascular senescence. FASEB J. (2019) 33:7603–14. doi: 10.1096/fj.201801671RRRR
101. Caliri AW, Caceres A, Tommasi S, Besaratinia A. Hypomethylation of LINE-1 repeat elements and global loss of DNA hydroxymethylation in vapers and smokers. Epigenetics. (2020) 15:816–29. doi: 10.1080/15592294.2020.1724401
102. Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. (2019) 29:871–85.e5. doi: 10.1016/j.cmet.2019.02.014
103. Potus F, Pauciulo MW, Cook EK, Zhu N, Hsieh A, Welch CL, et al. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation. (2020) 141:1986–2000. doi: 10.1161/CIRCULATIONAHA.119.044320
104. Zhang Q, Zhao K, Shen QC, Han YM, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. (2015) 525:389–93. doi: 10.1038/nature15252
105. Badrnya S, Baumgartner R, Assinger A. Smoking alters circulating plasma microvesicle pattern and microRNA signatures. Thromb Haemost. (2014) 112:128–36. doi: 10.1160/TH13-11-0977
106. Zhang Y, Wang Y, Zhang L, Xia L, Zheng M, Zeng Z, et al. Reduced platelet miR-223 induction in kawasaki disease leads to severe coronary artery pathology through a miR-223/PDGFRbeta vascular smooth muscle cell axis. Circ Res. (2020) 127:855–73. doi: 10.1161/CIRCRESAHA.120.316951
107. Frati G, Forte M, di Nonno F, Bordin A, Chimenti I, Picchio V, et al. Inhibition of miR-155 attenuates detrimental vascular effects of tobacco cigarette smoking. J Am Heart Assoc. (2020) 9:e017000. doi: 10.1161/JAHA.120.017000
108. Holz-Schietinger C, Reich NO. RNA modulation of the human DNA methyltransferase 3A. Nucleic Acids Res. (2012) 40:8550–7. doi: 10.1093/nar/gks537
109. Zhang G, Esteve PO, Chin HG, Terragni J, Dai N, Correa IR, et al. Small RNA-mediated DNA (cytosine-5) methyltransferase 1 inhibition leads to aberrant DNA methylation. Nucleic Acids Res. (2015) 43:6112–24. doi: 10.1093/nar/gkv518
110. Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, et al. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid Redox Signal. (2010) 13:1023–32. doi: 10.1089/ars.2010.3251
111. Hwang JW, Yao H, Caito S, Sundar IK, Rahman I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med. (2013) 61:95–110. doi: 10.1016/j.freeradbiomed.2013.03.015
112. Feng Y, Liu T, Dong SY, Guo YJ, Jankovic J, Xu H, et al. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J Neurochem. (2015) 134:668–76. doi: 10.1111/jnc.13172
113. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. (2009) 136:62–74. doi: 10.1016/j.cell.2008.10.052
114. Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. (2010) 115:2971–9. doi: 10.1182/blood-2009-05-224824
115. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
116. Khan SS, Singer BD, Vaughan DE. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell. (2017) 16:624–33. doi: 10.1111/acel.12601
117. Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. (2017) 31:101–26. doi: 10.1101/gad.291518.116
118. Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol Sci. (2018) 39:424–36. doi: 10.1016/j.tips.2018.02.001
119. Gao R, Ma Z, Hu Y, Chen J, Shetty S, Fu J. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L847–53. doi: 10.1152/ajplung.00274.2014
120. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. COVID-19 cytokines and the hyperactive immune response: synergism of TNF-alpha and IFN-gamma in triggering inflammation, tissue damage, and death. bioRxiv. 2020:361048. doi: 10.1101/2020.10.29.361048
121. Costantino S, Camici GG, Mohammed SA, Volpe M, Luscher TF, Paneni F. Epigenetics and cardiovascular regenerative medicine in the elderly. Int J Cardiol. (2018) 250:207–14. doi: 10.1016/j.ijcard.2017.09.188
122. Nosalski R, Siedlinski M, Denby L, McGinnigle E, Nowak M, Cat AND, et al. T-Cell-Derived miRNA-214 mediates perivascular fibrosis in hypertension. Circ Res. (2020) 126:988–1003. doi: 10.1161/CIRCRESAHA.119.315428
123. Li H, Fan J, Zhao Y, Zhang X, Dai B, Zhan J, et al. Nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ Res. (2019) 125:1106–20. doi: 10.1161/CIRCRESAHA.119.314898
124. Xu XJ, Su SY, Barnes VA, De Miguel C, Pollock J, Ownby D, et al. A genome-wide methylation study on obesity differential variability and differential methylation. Epigenetics. (2013) 8:522–33. doi: 10.4161/epi.24506
125. Gao Q, Wei A, Chen F, Chen X, Ding W, Ding Z, et al. Enhancing PPARgamma by HDAC inhibition reduces foam cell formation and atherosclerosis in ApoE deficient mice. Pharmacol Res. (2020) 160:105059. doi: 10.1016/j.phrs.2020.105059
126. Thounaojam MC, Bartoli M. MicroRNA-34a and vascular senescence in diabetes. Aging. (2019) 11:11799–800. doi: 10.18632/aging.102625
127. Shan K, Liu C, Liu BH, Chen X, Dong R, Liu X, et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation. (2017) 136:1629–42. doi: 10.1161/CIRCULATIONAHA.117.029004
128. Monahan KD, Dinenno FA, Seals DR, Clevenger CM, Desouza CA, Tanaka H. Age-associated changes in cardiovagal baroreflex sensitivity are related to central arterial compliance. Am J Physiol Heart Circ Physiol. (2001) 281:H284–9. doi: 10.1152/ajpheart.2001.281.1.H284
129. Duca L, Blaise S, Romier B, Laffargue M, Gayral S, El Btaouri H, et al. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res. (2016) 110:298–308. doi: 10.1093/cvr/cvw061
130. Butts B, Butler J, Dunbar SB, Corwin E, Gary RA. Effects of exercise on ASC Methylation and IL-1 Cytokines in heart failure. Med Sci Sports Exerc. (2018) 50:1757–66. doi: 10.1249/MSS.0000000000001641
131. Liang Q, Cai M, Zhang J, Song W, Zhu W, Xi L, et al. Role of Muscle-Specific Histone Methyltransferase (Smyd1) in exercise-induced cardioprotection against pathological remodeling after myocardial infarction. Int J Mol Sci. (2020) 21:7010. doi: 10.3390/ijms21197010
132. Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, et al. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech Ageing Dev. (2010) 131:21–8. doi: 10.1016/j.mad.2009.11.002
133. VerPlank JJS, Lokireddy S, Zhao J, Goldberg AL. 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc Natl Acad Sci USA. (2019). 116:4228–37. doi: 10.1073/pnas.1809254116
134. Recchioni R, Marcheselli F, Antonicelli R, Mensa E, Lazzarini R, Procopio AD, et al. Epigenetic effects of physical activity in elderly patients with cardiovascular disease. Exp Gerontol. (2017) 100:17–27. doi: 10.1016/j.exger.2017.10.016
135. Foo RS, Anene-Nzelu CG, Rosa-Garrido M, Vondriska TM. Dissecting chromatin architecture for novel cardiovascular disease targets. Circulation. (2019) 140:446–8. doi: 10.1161/CIRCULATIONAHA.119.039287
136. Cedernaes J, Schonke M, Westholm JO, Mi J, Chibalin A, Voisin S, et al. Acute sleep loss results in tissue-specific alterations in genome-wide DNA methylation state and metabolic fuel utilization in humans. Sci Adv. (2018) 4:eaar8590. doi: 10.1126/sciadv.aar8590
137. Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. (2015) 16:593–610. doi: 10.1038/nrm4048
138. Glier MB, Green TJ, Devlin AM. Methyl nutrients, DNA methylation, and cardiovascular disease. Mol Nutr Food Res. (2014) 58:172–82. doi: 10.1002/mnfr.201200636
139. Gensous N, Franceschi C, Santoro A, Milazzo M, Garagnani P, Bacalini MG. The impact of caloric restriction on the epigenetic signatures of aging. Int J Mol Sci. (2019) 20:2022. doi: 10.3390/ijms20082022
140. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. (2010) 2:247–57. doi: 10.1002/emmm.201000080
141. Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T, Bennett M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res. (2008) 103:717–25. doi: 10.1161/CIRCRESAHA.108.182899
142. Zhu YZ, Wu W, Zhu Q, Liu X. Discovery of Leonuri and therapeutical applications: from bench to bedside. Pharmacol Ther. (2018) 188:26–35. doi: 10.1016/j.pharmthera.2018.01.006
143. Jin M, Li Q, Gu Y, Wan B, Huang J, Xu X, et al. Leonurine suppresses neuroinflammation through promoting oligodendrocyte maturation. J Cell Mol Med. (2019) 23:1470–85. doi: 10.1111/jcmm.14053
144. Liao J, Suguro R, Zhao X, Yu Y, Cui Y, Zhu YZ. Leonurine affected homocysteine-methionine metabolism based on metabolomics and gut microbiota studies of clinical trial samples. Clin Transl Med. (2021) 11:e535. doi: 10.1002/ctm2.535
145. Liu XH, Pan LL, Deng HY, Xiong QH, Wu D, Huang GY, et al. Leonurine (SCM-198) attenuates myocardial fibrotic response via inhibition of NADPH oxidase 4. Free Radic Biol Med. (2013) 54:93–104. doi: 10.1016/j.freeradbiomed.2012.10.555
146. Zhang Y, Guo W, Wen Y, Xiong Q, Liu H, Wu J, et al. SCM-198 attenuates early atherosclerotic lesions in hypercholesterolemic rabbits via modulation of the inflammatory and oxidative stress pathways. Atherosclerosis. (2012) 224:43–50. doi: 10.1016/j.atherosclerosis.2012.06.066
147. Bannister CA, Holden SE, Jenkins-Jones S, Morgan CL, Halcox JP, Schernthaner G, et al. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes Metab. (2014) 16:1165–73. doi: 10.1111/dom.12354
148. de Kreutzenberg SV, Ceolotto G, Cattelan A, Pagnin E, Mazzucato M, Garagnani P, et al. Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr Metab Cardiovasc Dis. (2015) 25:686–93. doi: 10.1016/j.numecd.2015.03.007
149. Pal S, Tyler JK. Epigenetics and aging. Sci Adv. (2016) 2:e1600584. doi: 10.1126/sciadv.1600584
150. Choksomngam Y, Pattanakuhar S, Chattipakorn N, Chattipakorn SC. The metabolic role of spermidine in obesity: evidence from cells to community. Obes Res Clin Pract. (2021) 15:315–26. doi: 10.1016/j.orcp.2021.06.009
151. Pekar T, Bruckner K, Pauschenwein-Frantsich S, Gschaider A, Oppliger M, Willesberger J, et al. The positive effect of spermidine in older adults suffering from dementia: first results of a 3-month trial. Wien Klin Wochenschr. (2021) 133:484–91. doi: 10.1007/s00508-020-01758-y
152. Yang X, Yu D, Xue L, Li H, Du J. Probiotics modulate the microbiota-gut-brain axis and improve memory deficits in aged SAMP8 mice. Acta Pharm Sin B. (2020) 10:475–87. doi: 10.1016/j.apsb.2019.07.001
153. Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med. (2016) 22:657–65. doi: 10.1038/nm.4109
Keywords: vascular aging, epigenetics regulation, DNA methylation, histone modifications, chromatin architecture
Citation: Lin Z, Ding Q, Li X, Feng Y, He H, Huang C and Zhu Y (2022) Targeting Epigenetic Mechanisms in Vascular Aging. Front. Cardiovasc. Med. 8:806988. doi: 10.3389/fcvm.2021.806988
Received: 01 November 2021; Accepted: 30 November 2021;
Published: 04 January 2022.
Edited by:
Yuling Zhang, Sun Yat-sen Memorial Hospital, ChinaReviewed by:
Yuan Jiang, Sun Yat-sen University, ChinaCopyright © 2022 Lin, Ding, Li, Feng, He, Huang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: YiZhun Zhu, eXp6aHVAbXVzdC5lZHUubW8=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.