 Changping Gu1,2,3†
Changping Gu1,2,3† Liang Zhao
Liang Zhao Yue-lan Wang
Yue-lan Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 13 January 2022
Sec. Cardiovascular Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.788505
This article is part of the Research Topic Mitochondrial Metabolism in Ischemic Heart Disease View all 18 articles
Mitochondrial dynamics, including continuous biogenesis, fusion, fission, and autophagy, are crucial to maintain mitochondrial integrity, distribution, size, and function, and play an important role in cardiovascular homeostasis. Cardiovascular health improves with aerobic exercise, a well-recognized non-pharmaceutical intervention for both healthy and ill individuals that reduces overall cardiovascular disease (CVD) mortality. Increasing evidence shows that aerobic exercise can effectively regulate the coordinated circulation of mitochondrial dynamics, thus inhibiting CVD development. This review aims to illustrate the benefits of aerobic exercise in prevention and treatment of cardiovascular disease by modulating mitochondrial function.
Cardiovascular diseases (CVDs) are the leading cause of global mortality and a major contributor to disability worldwide (1). CVD pathogenesis is a complex biological process, but few feasible targets exist to prevent or reverse CVD (2). The American College of Cardiology recently reported significant progress in drug treatment for CVD; however, these drugs always have negative long-term effects, causing a reduction of 10–20% in the left ventricular mass (3). Therefore, it is essential to find effective non-pharmaceutical therapies. Aerobic exercise has many benefits to the cardiovascular system, such as improving the mechanical properties of the heart and enhancing its contractility to reduce the incidence of many CVDs. Some of these improvements may arise from effects on mitochondria.
Mitochondria are the energy factories of cells, producing adenosine triphosphate (ATP), and are the main source of cellular reactive oxygen species (ROS). Dysfunctional mitochondria limit energy production, increase ROS production, and transmit apoptotic signals, leading to tissue damage and organ dysfunction (4). Mitochondrial dynamics, which include mitochondrial biogenesis, fusion, fission, and autophagy, play an important role in maintaining mitochondrial homeostasis and ensuring mitochondrial function (5). Mitochondrial dynamics are particularly important for cells with high energy requirements, such as cardiomyocytes, which continuously require ATP to support heart function (6). Therefore, regulating mitochondrial dynamics through effective interventions is crucial for preventing and treating CVDs (7). In animal models of heart failure, Campos et al. found that aerobic exercise improved the mitochondrial fusion/fission balance and restored cardiac autophagy flux, thereby improving cardiac function (8). Here, we will address the biological mechanisms of mitochondrial dynamics in CVDs and discuss the mechanism of aerobic exercise in improving cardiovascular diseases, thereby providing valuable clues for CVD prevention and treatment.
Mitochondria are highly dynamic organelles, constantly undergoing a coordinated cycle of biogenesis, fusion, fission, and degradation (Figure 1) (9), and forming a complex network to respond to dynamic changes in energy requirements (10). Maintaining healthy and functional mitochondrial networks is critical for physiological adaptation and coping with stress during development and throughout life (11). Mitochondrial dynamics consist mainly of mitochondrial fusion and fission. Mitochondria are easily damaged by various external stimuli. Damaged mitochondria are cleared by autophagy to maintain the stability of mitochondrial dynamics, which is important to ensuring a healthy mitochondrial network. Defects in this machinery cause a range of diseases especially affecting the cardiovascular system (12). Notably, mounting evidence demonstrates that some of the benefits of aerobic exercise against CVD occur through its effects on three underlying mitochondrial aspects: mitochondrial biogenesis, mitochondrial fusion and fission, and mitochondrial autophagy.
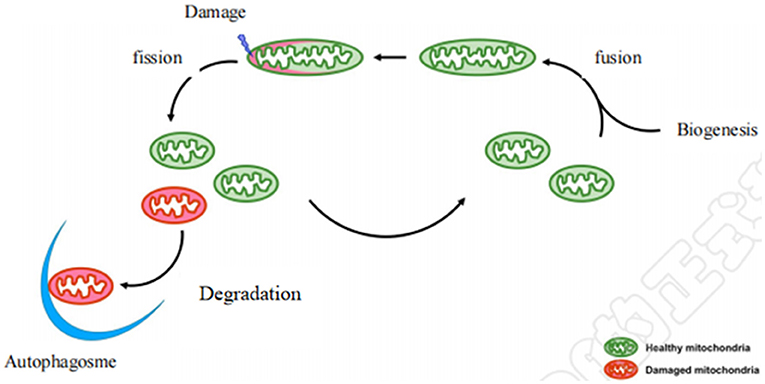
Figure 1. Diagram of mitochondrial dynamics. Mitochondrial biogenesis is the process by which new mitochondria are formed in the cell after mitochondrial fusion. Mitochondrial biogenesis is activated by numerous different signals which can lead to mitochondrial fission and damaged mitochondria are degraded by autophagy.
Mitochondrial biogenesis requires coordination of nuclear and mitochondrial DNA (12). It is critical to healthy mitochondrial dynamics and further affects myocardial function. Mitochondrial biosynthesis is regulated by the peroxisome proliferator–activated receptor gamma coactivator-1 (PGC-1) family. This includes PGC-1α, PGC-1β, and peroxisome proliferator-activated receptor gamma (PPARγ). PGC-1α plays a vital role in regulating cardiac metabolism. Activation of PGC-1 family transcription factors under conditions of cellular energy requirements, such as cell growth, hypoxia, glucose deprivation, or exercise, enhances mitochondrial remodeling or biosynthesis and restores intracellular energy balance (13). PGC-1 knockdown in mice induces cardiomyopathy via fragmentation and elongation of cardiac mitochondria, which is related to changes in mitofusin 1 (MFN1), optic atrophy 1 (OPA1), and dynamin-related protein 1 (DRP1) expression (14).
Multiple transcription factors are found downstream of PGC-1α, and peroxisome proliferator–activated receptors (PPARs), nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (TFAM) are all involved in mitochondrial biogenesis (15). PPARα is a member of the nuclear receptor superfamily of PPARs. PPARs participate in decomposition and metabolism of fatty acids and play a key role in maintaining myocardial energy metabolism (16). Specific knockout of PPARα results in cardiac hypertrophy and fat accumulation in rats, and ultimately heart failure leads to animal death (17, 18). In addition, PPARα can ameliorate cardiac hypertrophy caused by hypertension. The PPARα/NRF2 signaling pathway protects the heart from remodeling induced by stress overload, and up-regulation of PPARα protein expression can improve cardiac hypertrophy (19). NRF1/2 is a key component in regulating nuclear coding of mitochondrial proteins and is closely involved in mitochondrial biogenesis. In models of cardiac hypertrophy and heart failure, gene expression and protein levels of NRF1 and TFAM and protein levels of NRF2 are decreased in the cardiac tissue (20).
Mitochondrial biogenesis is inhibited in CVDs (21). The number and ATP production of myocardial mitochondria and mitochondrial synthesis regulatory factors increased in rats after an 8-week treadmill test, indicating that aerobic exercise could improve myocardial energy supply and cardiac function by promoting biosynthesis of myocardial mitochondria (22). Treadmill exercise helps improve CVDs; thus, aerobic exercise may exert direct benefit on mitochondrial biogenesis.
In line with this, aerobic exercise can activate the silencing regulatory protein 3 (SIRT3)/PGC-1α/phosphatidylinositol 3 kinase (PI3K)/Akt signaling pathway, resulting in improved mitochondrial biogenesis (23). Such physical activity can also activate the AMPK/PGC-1α pathway in myocardial tissue. PGC-1α can upregulate expression of mitofusin2 (MFN2) protein, which promotes mitochondrial biogenesis and mitochondrial fusion (13). Further, aerobic exercise enhances protein levels of PGC-1α and NRF2, which increases the mRNA levels of TFAM and NRF1 and increases mitochondrial DNA replication. NRF2 promotes expression of various antioxidant enzymes, thus reducing the oxidative stress level of myocardial tissue (24). Thus, aerobic exercise can promote expression of PGC-1α, NRF1, and NRF2, and expression of regulatory factors and induction of mitochondrial biogenesis improve CVDs.
Mitochondria constantly undergo fission and fusion. After mitochondrial fission, offspring mitochondria with higher membrane potential enter the next round of fusion to maintain mitochondrial number and function. These processes are regulated by mitochondrial fusion and fission proteins, and are particularly important for maintaining normal mitochondrial function (25). Mitochondrial fusion proteins include MFN1, MFN2, and OPA1. MFN1 and MFN2 mediate fusion of the outer mitochondrial membrane (OMM), while OPA1 mediates fusion of the inner mitochondrial membrane. DRP1 serves as a critical effector of mitochondrial fission (26).
In CVDs, mitochondrial morphology and fusion-related proteins exhibit alterations. For example, severe mitochondrial fragmentation occurs in myocardial ischemia–reperfusion injury (I/R) models, which significantly inhibits OPA1 expression (27). Further, there is an abnormal increase in myocardial mitochondria, downregulation of mitochondrial biogenesis-related genes, and aggravation of cardiomyopathy after specific deletion of Mfn1 and Mfn2 in the mouse myocardium (28). Mitochondrial fission was caused by translocation of phosphorylated DRP1 from the cytoplasm to the mitochondria (26). The use of DRP1 inhibitor reduced DRP1 translocation to the mitochondria, improved the structure and function of the mitochondria, and alleviated myocardial hypertrophy and myocardial fibrosis (29, 30). Besides the positive changes in mitochondrial dynamics in cardiovascular diseases, inhibiting mitochondrial fission may also lead to heart impairment (31, 32). Therefore, both mitochondrial fusion and fission are essential for CVD development. A balanced state of mitochondrial fission and fusion are conducive to disease prevention and improved prognosis.
In CVDs, mitochondria exhibit a high rate of fission and a low rate of fusion, resulting in imbalanced mitochondrial dynamics (12). Notably, aerobic exercise can increase expression of PGC-1α mRNA and protein to improve the level of MFN2 protein and promote mitochondrial fusion (33). Swimming training can downregulate the mir-30B-p53-DRP1 pathway, reduce the contents of p53 and DRP1 proteins in the mouse myocardium, and inhibit myocardial mitochondrial fission (34). The swimming training lasted 8 weeks, 5 days/week. At the beginning, the swimming duration is 30 min. The swimming time was increased by 10 min ever week. Therefore, swimming duration maintained at 90 min after 7–8 weeks. Then, the mice trained twice a day with an interval of 6 h. Aerobic exercise training can induce increased mitochondrial fusion (upregulating the expression of MFN2 and OPA1 proteins) and decreased mitochondrial fission (downregulating the expression of DRP1 protein), and promote mitochondrial kinetic remodeling, effectively alleviating mitochondrial dysfunction in rats with myocardial infarction (MI) (35). After moderate aerobic exercise training, the mRNA levels of Ppargc1α, Opa1, Mfn2, and Drp1 significantly increased and the diastolic parameters improved in spontaneously hypertensive rats (SHRs) (22). These results suggest that aerobic exercise maintains the balance of mitochondrial fusion and fission in the SHR myocardium and improves mitochondrial function. A comparison of the myocardial mitochondria of animals with heart failure who exercised vs. did not exercise revealed that the number of myocardial mitochondria in the exercise group was decreased and mitochondrial size was increased. Further experiments showed that exercise increased MFN1, MFN2, and DNM1L GTPase activity in the myocardium and reversed the translocation of DNM1L to the mitochondria (8). Therefore, aerobic exercise may have protective effects on the balance of mitochondrial fusion and fission in CVDs.
Mitochondrial autophagy is a selective form of autophagy, which is specialized to remove aging or irreversibly damaged mitochondria and plays a decisive role in control of mitochondrial quality (36). Mitochondrial autophagy and fission can coordinate with each other, and mitochondrial autophagy can remove damaged mitochondrial parts formed by mitochondrial fission and keep mitochondria healthy (37). DRP1 migration and mitochondrial autophagy activation occur almost simultaneously after transverse aortic constriction (TAC) treatment, indicating that mitochondrial autophagy is closely related to mitochondrial fission (38).
Mitochondrial autophagy may occur through Parkin-dependent or Parkin-independent mechanisms (12). In Parkin-mediated mitotic phagocytosis, PTEN-induced putative kinase 1 (PINK1) recruits Parkin to the outer membrane of mitochondria. Once recruited to the mitochondria, Parkin ubiquitinates several mitochondrial outer membrane proteins, including the mitochondrial fusion proteins MFN1 and MFN2, Miro, Translocase of OMM 20, and voltage-dependent anion channel. Then, the selective autophagy adapter protein P62/Sequestosome 1 is recruited to the mitochondria to interact with LC3 and initiate mitochondrial autophagy (36, 37, 39). Meanwhile, ubiquitination and proteasome degradation of MFN1 and MFN2 results in mitochondrial fission and fragmentation, which further induces mitochondrial autophagy (40). PINK1 and Parkin protein levels are significantly reduced in heart failure models (41). PINK1 knockout mice have a significantly increased number of atherosclerotic lesions (42). Also, Parkin knockout mice show excessive myocardial hypertrophy, myocardial fibrosis, and left ventricular systolic dysfunction in response to TAC (43).
Parkin-independent mitochondrial autophagy occurs independently of Parkin; some autophagy receptor proteins are located in mitochondria and interact with LC3 to recruit autophagosomes to damaged mitochondria. These autophagy receptor proteins include BCL-2/adenovirus E1B19-kDa interacting protein 3 (BNIP3), NIX (also called BNIP3L), and Fun14 Domain–containing 1 (FUNDC1) (36, 44, 45). Lowering of BNIP3 protein levels impairs mitochondrial function, which in turn leads to impaired myocardial cell function (46); also, FUNDC1 gene knockout exacerbates I/R injury (47).
Mitochondrial autophagy has a significant protective effect on the heart, and autophagy activation is observed in the boundary region of the subacute stage of MI (1 week after MI) (48). Enhancement of autophagy with rapamycin 2 weeks after coronary artery ligation ameliorates cardiac dysfunction and maladaptive remodeling, whereas inhibition of autophagy by buffamycin A1 worsens cardiac dysfunction (49). In conclusion, insufficient mitochondrial autophagy is closely related to CVD development.
CVD pathology is associated with weakening of mitochondrial autophagy. Yet, 8 weeks of aerobic exercise on a treadmill can increase the LC3II/LC3I ratio, and thus upregulate Beclin1, LC3, and BNIP3 in the rat myocardium to promote myocardial autophagy (50). For treadmill excise study, rats were trained on a treadmill with an exercise intensity and schedule set at 70% of maximum aerobic capacity. The training group run for 60 min/day, 5 days per week at 10 m/min speed for 8 weeks. After the treadmill excise training, PINK1, Parkin, ubiquitin, P62, and LC3 levels were significantly increased in the rat skeletal muscle (51). Consistent with this finding, exercise improves the oxidative capacity of myocardial mitochondria in animals with heart failure, and this improvement in oxidative capacity is related to reconstruction of autophagy flux. Exercise stimulates mitochondrial autophagy flux by increasing Parkin recruitment in myocardial mitochondria (8). Notably, aerobic exercise training enhances the PINK1/Parkin signaling pathway, thus inducing mitochondrial autophagy (52). This physical activity also upregulates SIRT3 expression, enhances the antioxidant capacity of the body, improves the quality of mitochondria, and helps alleviate cardiac dysfunction in mice after MI (53). These studies suggested that aerobic exercise induces mitochondrial autophagy and improves CVDs by increasing the expression of autophagy-related and mitochondrial autophagy–related proteins.
Abundant evidence illustrates the key role of mitochondrial dynamics in cardiac homeostasis. Imbalanced mitochondrial fusion and fission, insufficient mitochondrial autophagy, and weakened mitochondrial biogenesis are the pathological factors leading to CVD occurrence and development. Regular aerobic exercise is a simple and effective way to balance mitochondrial dynamics and improve mitochondrial function, which can build human health. MFN2 and OPA1, which are beneficial to mitochondrial fusion and biogenesis, can be elevated by aerobic exercise. DRP1-induced mitochondrial fission can be inhibited by aerobic exercise in cardiovascular diseases. PINK1/parkin signaling is still the main targeting pathway that induces mitochondrial autophagy following aerobic exercise in cardiovascular diseases. However, one study indicated that acute exercise had no effect on the proteins involved in mitochondrial dynamics. Indeed, after exercise training, the proteins involved in mitochondrial dynamics changed significantly in normotensive control rats, but remained unchanged in spontaneously hypertensive rats (22). This finding aligned with another study finding that myocardial mitochondrial dynamics-related proteins did not change after acute exercise in rats (54).
Aerobic exercise can prevent or alleviate CVD development by regulating proteins involved in mitochondrial dynamics. Exercise can reduce mitochondrial fission, enhance mitochondrial fusion and improve mitochondrial autophagy or biogenesis. Regarding to the type of exercise, we compared some excise programs of treadmill training with swimming, and also summarized the influence of different exercise programs on mitochondrial dynamics (Table 1). Treadmill training seems to have a greater effect on modulation of the cardiac proteome than swimming (55). While we can control the intensity and duration of treadmill training to calculate the amount of exercise, swimming training is more difficult to control. Some animals do not exhibit continuous swimming behavior, but rather tend to dive or swing, which results in intermittent hypoxia and may affect interpretation of the results.
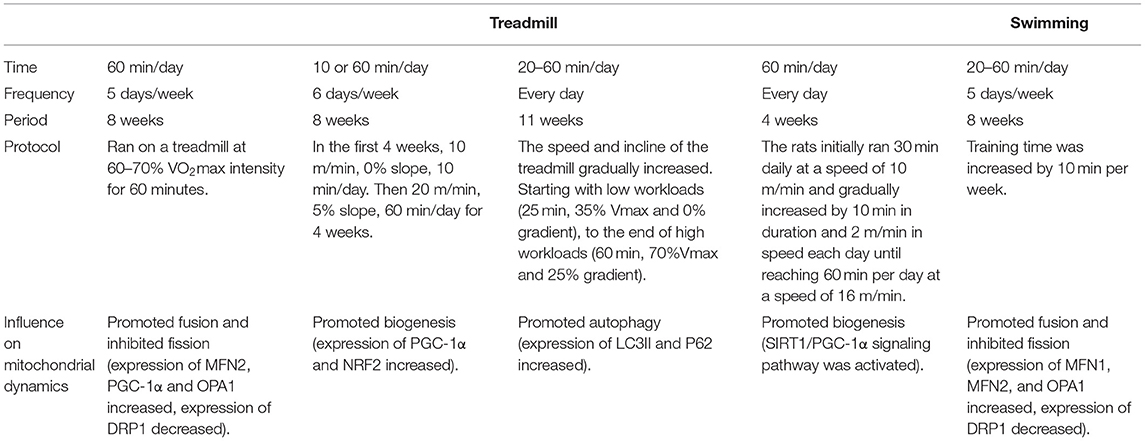
Table 1. Comparison of different exercise training programs.
Mitochondrial dynamics, including mitochondrial biogenesis, mitochondrial fusion or fission, and mitochondrial autophagy, can be used as disease prevention and treatment targets under exercise intervention. Moreover, the balance between mitochondrial dynamics may be a key factor in disease occurrence or development. However, the mechanism of aerobic exercise intervention in mitochondrial dynamics is complex and needs further long-term research due to its gradual effects and individual differences. Improper exercise can also lead to arthritis, decreased immunity, etc. Therefore, verified and reasonable individualized exercise prescription has important guiding significance for health. It is important to carry out long-term and proper exercise intervention.
The concept of aerobic exercise for disease prevention is increasingly popular, while the number of subhealthy people and older adults is increasing every year. Reasonable exercise prescription intervention for these populations or people with a tendency for organ disease development can significantly reduce disease occurrence. Further studies are needed to explore this idea.
Y-lW: conceptualization and methodology. CG: data curation and writing—original draft preparation. JY: writing—reviewing and editing. LZ: visualization and investigation. GW: software. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 Study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. Sedding DG, Boyle EC, Demandt J, Sluimer JC, Dutzmann J, Haverich A, et al. Vasa vasorum angiogenesis: key player in the initiation and progression of atherosclerosis and potential target for the treatment of cardiovascular disease. Front Immunol. (2018) 9:706. doi: 10.3389/fimmu.2018.00706
3. Whelton PK, Carey RM, Aronow WS, Casey DE, Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension (Dallas, Tex.: 1979). (2018) 71:1269–324. doi: 10.1161/HYP.0000000000000075
4. Sorrentino V, Menzies KJ, Auwerx J. Repairing mitochondrial dysfunction in disease. Annu Rev Pharmacol Toxicol. (2018) 58:353–89. doi: 10.1146/annurev-pharmtox-010716-104908
5. Dorn GW, III. Evolving concepts of mitochondrial dynamics. Ann Rev Physiol. (2019) 81:1–17. doi: 10.1146/annurev-physiol-020518-114358
6. Tahrir FG, Langford D, Amini S, Mohseni Ahooyi T, Khalili K. Mitochondrial quality control in cardiac cells: mechanisms and role in cardiac cell injury and disease. J Cell Physiol. (2019) 234:8122–33. doi: 10.1002/jcp.27597
7. Crisafulli A, Pagliaro P, Roberto S, Cugusi L, Mercuro G, Lazou A, et al. Diabetic Cardiomyopathy and ischemic heart disease: prevention and therapy by exercise and conditioning. Int J Mol Sci. (2020) 21:2896. doi: 10.3390/ijms21082896
8. Campos JC, Queliconi BB, Bozi L, Bechara L, Dourado P, Andres AM, et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy. (2017) 13:1304–17. doi: 10.1080/15548627.2017.1325062
9. Kiriyama Y, Nochi H. Intra- and intercellular quality control mechanisms of mitochondria. Cells. (2017) 7:1. doi: 10.3390/cells7010001
10. Xie LL, Shi F, Tan Z, Li Y, Bode AM, Cao Y. Mitochondrial network structure homeostasis and cell death. Cancer Sci. (2018) 109:3686–94. doi: 10.1111/cas.13830
11. Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. (2018) 28:R170–85. doi: 10.1016/j.cub.2018.01.004
12. Forte M, Schirone L, Ameri P, Basso C, Catalucci D, Modica J, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. (2021) 178:2060–76. doi: 10.1111/bph.15068
13. Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, et al. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. (2014) 114:626–36. doi: 10.1161/CIRCRESAHA.114.302562
14. Kulikova TG, Stepanova OV, Voronova AD, Valikhov MP, Sirotkin VN, Zhirov IV, et al. Pathological remodeling of the myocardium in chronic heart failure: role of PGC-1α. Bull Exp Biol Med. (2018) 164:794–7. doi: 10.1007/s10517-018-4082-1
15. Jiang Q, Ji A, Li D, Shi L, Gao M, Lv N, et al. Mitochondria damage in ambient particulate matter induced cardiotoxicity: roles of PPAR alpha/PGC-1 alpha signaling. Environ Poll (Barking, Essex: 1987). (2021) 288:117792. doi: 10.1016/j.envpol.2021.117792
16. Yin Z, Zhao Y, He M, Li H, Fan J, Nie X, et al. MiR-30c/PGC-1β protects against diabetic cardiomyopathy via PPARα. Cardiovasc Diabetol. (2019) 18:7. doi: 10.1186/s12933-019-0811-7
17. Zhu ZY, Gao T, Huang Y, Xue J, Xie ML. Apigenin ameliorates hypertension-induced cardiac hypertrophy and down-regulates cardiac hypoxia inducible factor-lα in rats. Food Funct. (2016) 7:1992–8. doi: 10.1039/C5FO01464F
18. Warren JS, Tracy CM, Miller MR, Makaju A, Szulik MW, Oka SI, et al. Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc Natl Acad Sci USA. (2018) 115:E7871–80. doi: 10.1073/pnas.1800680115
19. Wu QQ, Deng W, Xiao Y, Chen JJ, Liu C, Wang J, et al. The 5-lipoxygenase inhibitor zileuton protects pressure overload-induced cardiac remodeling via activating PPARα. Oxid Med Cell Longev. (2019) 2019:7536803. doi: 10.1155/2019/7536803
20. Nomura S, Satoh M, Fujita T, Higo T, Sumida T, Ko T, et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat Commun. (2018) 9:4435. doi: 10.1038/s41467-018-06639-7
21. Tan Y, Lei C, Tang H, Zhu X, Yi G. The hippo pathway orchestrates mitochondrial quality control: a novel focus on cardiovascular diseases. DNA Cell Biol. (2020) 39:1494–505. doi: 10.1089/dna.2019.5348
22. Quiroga C, Mancilla G, Oyarzun I, Tapia A, Caballero M, Gabrielli LA, et al. Moderate exercise in spontaneously hypertensive rats is unable to activate the expression of genes linked to mitochondrial dynamics and biogenesis in cardiomyocytes. Front Endocrinol. (2020) 11:546. doi: 10.3389/fendo.2020.00546
23. Jia D, Hou L, Lv Y, Xi L, Tian Z. Postinfarction exercise training alleviates cardiac dysfunction and adverse remodeling via mitochondrial biogenesis and SIRT1/PGC-1α/PI3K/Akt signaling. J Cell Physiol. (2019) 234:23705–18. doi: 10.1002/jcp.28939
24. Sun M, Huang C, Wang C, Zheng J, Zhang P, Xu Y, et al. Ginsenoside Rg3 improves cardiac mitochondrial population quality: mimetic exercise training. Biochem Biophys Res Commun. (2013) 441:169–74. doi: 10.1016/j.bbrc.2013.10.039
25. Sabouny R, Shutt TE. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem Sci. (2020) 45:564–77. doi: 10.1016/j.tibs.2020.03.009
26. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. (2021) 42:655–64. doi: 10.1038/s41401-020-00518-y
27. Zhang Y, Wang Y, Xu J, Tian F, Hu S, Chen Y, et al. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res. (2019) 66:e12542. doi: 10.1111/jpi.12542
28. Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, et al. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. (2012) 111:1012–26. doi: 10.1161/CIRCRESAHA.112.274142
29. Tian L, Neuber-Hess M, Mewburn J, Dasgupta A, Dunham-Snary K, Wu D, et al. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J Mol Med (Berlin, Germany). (2017) 95:381–93. doi: 10.1007/s00109-017-1522-8
30. Hasan P, Saotome M, Ikoma T, Iguchi K, Kawasaki H, Iwashita T, et al. Mitochondrial fission protein, dynamin-related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl-salt sensitive rats. J Mol Cell Cardiol. (2018) 121:103–6. doi: 10.1016/j.yjmcc.2018.07.004
31. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. (2015) 116:264–78. doi: 10.1161/CIRCRESAHA.116.303356
32. Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker K, Jung G, et al. Physiological mitochondrial fragmentation is a normal cardiac adaptation to increased energy demand. Circ Res. (2018) 122:282–95. doi: 10.1161/CIRCRESAHA.117.310725
33. Moore TM, Zhou Z, Cohn W, Norheim F, Lin AJ, Kalajian N, et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol Metab. (2019) 21:51–67. doi: 10.1016/j.molmet.2018.11.012
34. Zhao Y, Ma Z. Swimming training affects apoptosis-related microRNAs and reduces cardiac apoptosis in mice. Gen Physiol Biophys. (2016) 35:443–50. doi: 10.4149/gpb_2016012
35. Jiang HK, Wang YH, Sun L, He X, Zhao M, Feng ZH, et al. Aerobic interval training attenuates mitochondrial dysfunction in rats post-myocardial infarction: roles of mitochondrial network dynamics. Int J Mol Sci. (2014) 15:5304–22. doi: 10.3390/ijms15045304
36. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. (2015) 4:6–13. doi: 10.1016/j.redox.2014.11.006
37. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. (2018) 80:1–26. doi: 10.1146/annurev-physiol-021317-121427
38. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Response by shirakabe et al. to letter regarding article, “Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure”. Circulation. (2016) 134:e75–6. doi: 10.1161/CIRCULATIONAHA.116.023667
39. Dikic I, Bremm A. DUBs counteract parkin for efficient mitophagy. EMBO J. (2014) 33:2442–3. doi: 10.15252/embj.201490101
40. Basso V, Marchesan E, Peggion C, Chakraborty J, von Stockum S, Giacomello M, et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol Res. (2018) 138:43–56. doi: 10.1016/j.phrs.2018.09.006
41. Wang B, Xu M, Li W, Li X, Zheng Q, Niu X. Aerobic exercise protects against pressure overload-induced cardiac dysfunction and hypertrophy via β3-AR-nNOS-NO activation. PLoS ONE. (2017) 12:e0179648. doi: 10.1371/journal.pone.0179648
42. He L, Zhou Q, Huang Z, Xu J, Zhou H, Lv D, et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J Cell Physiol. (2019) 234:8668–82. doi: 10.1002/jcp.27527
43. Han K, Hassanzadeh S, Singh K, Menazza S, Nguyen TT, Stevens MV, et al. Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress. Sci Rep. (2017) 7:2093. doi: 10.1038/s41598-017-02339-2
44. Fu R, Deng Q, Zhang H, Hu X, Li Y, Liu Y, et al. A novel autophagy inhibitor berbamine blocks SNARE-mediated autophagosome-lysosome fusion through upregulation of BNIP3. Cell Death Dis. (2018) 9:243. doi: 10.1038/s41419-018-0276-8
45. Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. (2019) 15:1182–98. doi: 10.1080/15548627.2019.1580095
46. Li E, Li X, Huang J, Xu C, Liang Q, Ren K, et al. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell. (2020) 11:661–79. doi: 10.1007/s13238-020-00713-x
47. Zhang W, Siraj S, Zhang R, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy. (2017) 13:1080–1. doi: 10.1080/15548627.2017.1300224
48. Yu W, Sun S, Xu H, Li C, Ren J, Zhang Y. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics. (2020) 10:11244–63. doi: 10.7150/thno.46883
49. Wang Z, Zhang H, Liu Z, Ma Z, An D, Xu D. Apigenin attenuates myocardial infarction-induced cardiomyocyte injury by modulating Parkin-mediated mitochondrial autophagy. J Biosci. (2020) 45:75. doi: 10.1007/s12038-020-00047-0
50. Li H, Miao W, Ma J, Xv Z, Bo H, Li J, et al. Acute exercise-induced mitochondrial stress triggers an inflammatory response in the myocardium via NLRP3 inflammasome activation with mitophagy. Oxid Med Cell Longev. (2016) 2016:1987149. doi: 10.1155/2016/1987149
51. Shang H, Xia Z, Bai S, Zhang HE, Gu B, Wang R. Downhill running acutely elicits mitophagy in rat soleus muscle. Med Sci Sports Exerc. (2019) 51:1396–403. doi: 10.1249/MSS.0000000000001906
52. Fiuza-Luces C, Delmiro A, Soares-Miranda L, González-Murillo Á, Martínez-Palacios J, Ramírez M, et al. Exercise training can induce cardiac autophagy at end-stage chronic conditions: insights from a graft-versus-host-disease mouse model. Brain Behav Immun. (2014) 39:56–60. doi: 10.1016/j.bbi.2013.11.007
53. Zhao D, Sun Y, Tan Y, Zhang Z, Hou Z, Gao C, et al. Short-duration swimming exercise after myocardial infarction attenuates cardiac dysfunction and regulates mitochondrial quality control in aged mice. Oxid Med Cell Longev. (2018) 2018:4079041. doi: 10.1155/2018/4079041
54. Yoo SZ, No MH, Heo JW, Park DH, Kang JH, Kim JH, et al. Effects of acute exercise on mitochondrial function, dynamics, and mitophagy in rat cardiac and skeletal muscles. Int Neurourol J. (2019) 23(Suppl. 1):S22–31. doi: 10.5213/inj.1938038.019
Keywords: aerobic exercise, mitochondrial dynamics, myocardial mitochondria, cardiovascular disease, mitochondrial fusion, mitochondrial fission
Citation: Gu C, Yan J, Zhao L, Wu G and Wang Y-l (2022) Regulation of Mitochondrial Dynamics by Aerobic Exercise in Cardiovascular Diseases. Front. Cardiovasc. Med. 8:788505. doi: 10.3389/fcvm.2021.788505
Received: 02 October 2021; Accepted: 06 December 2021;
Published: 13 January 2022.
Edited by:
Lei-Lei Ma, Fudan University, ChinaReviewed by:
Stephen C. Kolwicz Jr., Ursinus College, United StatesCopyright © 2022 Gu, Yan, Zhao, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue-lan Wang, d3lsZGdmQDE2My5jb20=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.