 Daniella M. Mizurini
Daniella M. Mizurini Eugenio D. Hottz
Eugenio D. Hottz Patrícia T. Bozza
Patrícia T. Bozza Robson Q. Monteiro
Robson Q. Monteiro- 1Institute of Medical Biochemistry Leopoldo de Meis, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil
- 2Oswaldo Cruz Foundation, Laboratory of Immunopharmacology, Oswaldo Cruz Institute, Rio de Janeiro, Brazil
- 3Laboratory of Immunothrombosis, Department of Biochemistry, Federal University of Juiz de Fora (UFJF), Juiz de Fora, Brazil
The novel coronavirus disease (COVID-19) is associated with a high incidence of coagulopathy and venous thromboembolism that may contribute to the worsening of the clinical outcome in affected patients. Marked increased D-dimer levels are the most common laboratory finding and have been repeatedly reported in critically ill COVID-19 patients. The infection caused by Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) is followed by a massive release of pro-inflammatory cytokines, which mediate the activation of endothelial cells, platelets, monocytes, and neutrophils in the vasculature. In this context, COVID-19-associated thrombosis is a complex process that seems to engage vascular cells along with soluble plasma factors, including the coagulation cascade, and complement system that contribute to the establishment of the prothrombotic state. In this review, we summarize the main findings concerning the cellular mechanisms proposed for the establishment of COVID-19-associated thrombosis.
General Features of Hemostatic Imbalance in Covid-19 Progression
Since the first reports comparing mild and severe covid COVID-19 patients, it was clear that the more severe illness was strongly associated with hypercoagulability (1, 2). COVID-19-associated coagulopathy has been consistently reported in patients admitted to hospital, in particular those severely ill. The most frequently reported laboratory findings related to COVID-19 coagulopathy include increased D-dimer levels and fibrinogen concentration, decreased platelet count, and slightly prolonged clotting time (3–5). A meta-analysis identified significantly lower platelet count in COVID-19 patients, and showed an association between thrombocytopenia, disease severity and increased risk of mortality (3). Fibrinogen levels are commonly high in the early stage of COVID-19 infection; however, a decline in plasma fibrinogen levels was observed in the late stage of the disease among non-survivors (5, 6). On the subject of coagulation tests, it has been demonstrated that COVID-19 patients have a modest prolongation of prothrombin time (PT) and activated partial thromboplastin time (APTT), although a marked increase in the D-dimer levels has been repeatedly reported (5, 7). An increasing number of studies have shown that D-dimer is commonly elevated in COVID-19 patients and that increased levels of this coagulation marker are associated with poor outcomes (2, 5, 8). In this context, Tachil et al. early proposed D-dimer concentrations as essential markers of COVID-19 severity (8). Other authors have proposed progressive stages of COVID-19–associated hemostatic abnormalities that mostly rely on D-dimer levels and could define the antithrombotic therapy as well as additional patient management procedures (9, 10). As the pandemic continues, it has become clear that COVID-19 progresses with increased thrombosis propensity being a significant cause of death in hospitalized patients (11–14). In this context, both arterial and venous thrombosis have been reported (15, 16). In addition, post-mortem analyses have shown microvascular thrombi in the lung, kidney, and heart of COVID-19 patients (17). Interestingly, histology of lung and skin samples revealed generalized thrombotic microvascular injury accompanied by the presence of complement deposits, more specifically C5b-9 complement complex and C4d complement split factor (18). The complement system is an important part of the innate defense against common pathogens, and its activation has previously been described in respiratory viral infections like influenza A and SARS-CoV (19). Additional evidence of complement activation following SARS-CoV-2 infection includes the observation of increased plasma levels of complement markers, such as sC5b-9, C5a, and C3a, in COVID-19 patients compared to non-COVID-19 and healthy controls, and their association with disease severity (20–22). Considering that a crosstalk between complement activation and coagulation system has long been documented, the recent findings open debate on whether complement activation triggered by COVID-19 contributes to COVID-19-associated thrombosis. Whether the prothrombotic phenotype observed in COVID-19 patients is a combination of hypercoagulability and impaired fibrinolysis has been one of the theories of several clinical studies. Recent systematic reviews have found a decreased fibrinolytic capacity in COVID-19 patients (23, 24). Some studies reported a reduced fibrinolytic activity in COVID-19 patients in intensive care unit (ICU) compared with non-ICU COVID-19 patients, and in patients with thrombotic events among those requiring ICU care (23). Using diverse viscoelastic tests, different research groups were able to detect impaired fibrinolysis in blood samples from COVID-19 patients, despite a contradictory increase in circulating levels of D-dimer (23, 24).
In this review, we summarize the main findings concerning the mechanisms proposed for the establishment of COVID-19-associated thrombosis, focusing on the involvement of cellular components of the clotting and immune system.
Endothelium
The vascular endothelium is a highly specialized and dynamic organ that exerts a number of important functions that are critical to maintaining adequate blood supply to vital organs (25). Some of these functions include control of hemostasis as well as regulation of vascular tonus and permeability (25–28). In addition to regulating the systemic blood flow, the endothelium actively participates in both the innate and adaptive immune responses (29).
Viral pathogens, including respiratory viruses, members of the herpesvirus family, and human immunodeficiency virus (HIV) can damage the endothelium, leading to detrimental shifts in the vascular equilibrium toward inflammation, vasoconstriction and procoagulant activity (30–34). Correspondingly, the autopsy of lung samples from different cohorts of COVID-19 patients revealed the presence of viral inclusions in endothelial cells (35, 36). In addition to the evidence of direct viral infection of endothelial cells, post-mortem histology also disclosed the presence of endothelial inflammation (35, 36). Immunohistochemical analysis of lung specimens revealed staining patterns consistent with apoptotic endothelial cells and mononuclear cell infiltrates (36). Indeed, SARS-CoV-2 infected endothelial cells were also detected in several organs besides the lung, including the kidney, heart, and liver (36–38).
There is ample evidence that SARS-CoV-2 uses the functional receptor for the SARS-CoV virus, the angiotensin-converting enzyme 2 (ACE2) receptor, to infect the host cells (39–41). ACE2 is a type I integral transmembrane protein widely expressed in human tissues in addition to the lungs (42). Interestingly, ACE2 has been detected in arterial and venous endothelial cells in a variety of human tissues, including the nasal mucosa, lung, small intestine, kidney, and brain (42). Similar to what has been reported to other coronaviruses, the cell entry of SARS-CoV-2 also depends on cell surface proteases which cleave the Spike protein, allowing the fusion of the viral and cellular membranes (39, 41, 43). Recent studies suggest that both SARS-CoV and SARS-CoV-2 employ the protease transmembrane protease serine 2 (TMPRSS-2), which is also expressed in human endothelial cells (39, 42). These findings support the hypothesis that SARS-CoV-2 can directly infect blood vessel cells, which was recently confirmed by Monteil et al. (40).
Besides the direct effects of SARS-CoV-2 on endothelial integrity, evidence from recent studies points that endothelial cells can be activated by humoral factors presented in the blood of SARS-CoV-2 infected patients (44, 45). Stimulation of human umbilical vein endothelial cells (HUVECs) with plasma or sera from COVID-19 patients resulted in activation of the endothelial cells, determined by overexpression of adhesion molecules and upregulation of thromboinflammatory genes associated with downregulation of antithrombotic genes (44, 45). This shift toward a prothrombotic surface appears to be triggered by alterations in the Tie2-angiopoietin axis (44). Tie2, or angiopoietin receptor 2, is a tyrosine kinase receptor selectively expressed on vascular endothelial cells, and its activation by angiopoietin 1 maintains the endothelial quiescence and suppresses vessel inflammation (46). It has been experimentally demonstrated that pharmacological activation of Tie2 effectively reversed the expression of thromboinflammatory genes induced by COVID-19 plasma in vitro (44). Another mechanism whereby COVID-19 may induce activation of endothelial cells appears to be mediated by antiphospholipid (aPL) antibodies. aPL antibodies comprise a heterogeneous population of autoantibodies directed against phospholipids and phospholipid-binding proteins and are associated with a higher risk of thrombosis in COVID-19 (47). A high prevalence of antiphospholipid antibodies has been reported in COVID-19 patients (12, 47), and the presence of different aPL correlates with endothelial activation (45). Positive aPL sera from COVID-19 patients induced upregulation of the cell adhesion molecules E-selectin, vascular cell adhesion molecule 1 (VCAM-1), and intracellular adhesion molecule 1 (ICAM-1) in HUVECs, which was abolished by total IgG depletion from the COVID-19 sera (45).
It is well-documented that activated endothelial cells evolve from an antithrombotic phenotype to a prothrombotic state (48). Under normal circumstances, endothelial cells possess anti-coagulant functions by expressing several molecules that limit or inhibit the coagulation process, however after inflammatory stimuli (e.g., interleukin (IL)-1 and tumor necrosis factor (TNF)-α), these cells acquire pro-coagulant functions, such as shedding of microparticles with exposed phosphatidylserine (PS) that enable the assembly of clotting enzyme complexes and the generation of thrombin (49, 50). Interestingly, activation of HUVECs with plasma from severe COVID-19 patients incited externalization of PS, creating a suitable surface for the assembly of coagulation complexes on the endothelium (44). Indeed, generation of factor Xa and thrombin were detected in HUVECs following exposure to COVID-19 plasma (44). Nevertheless, IL-1 and TNF-α, cytokines that are markedly increased in COVID-19 patients, also induce endothelial cells to synthesize tissue factor (TF), the primary initiator of coagulation (51, 52). TF is a transmembrane cellular receptor for the plasma coagulation factor VIIa (FVIIa), and the binding of FVIIa to TF is the initial step of a sequential proteolytic cleavage of coagulation factors that results in thrombin generation, platelet activation, and clot formation (53).
Endothelial cell dysfunction commonly observed in COVID-19 patients may be a result of pro-inflammatory cytokines that are produced in response to viral infection (54, 55). There is strong evidence that severe SARS-CoV-2 infection is accompanied by a massive inflammatory response resulting in the release of a large number of pro-inflammatory cytokines (56–59). The resultant increase in the pro-inflammatory cytokine levels might result in the loss of the antithrombotic phenotype of the endothelium, occasioning the activation of the coagulation cascade, platelets, and complement system in the vasculature (26, 48). For example, it is well-known that IL-6 secreted by immune and endothelial cells in response to a viral infection plays an important role in activating the endothelium during the early phase of inflammation (60). This cytokine not merely increases the vascular permeability but also promotes the secretion of pro-inflammatory cytokines by endothelial cells, sustaining an amplification loop that contributes to the excessive cytokine production, one of the most prominent characteristics of COVID-19 (61, 62).
Endothelial cell activation triggered by pro-inflammatory cytokines is an initial step in the platelet plug formation. It is well-known that endothelial cells store von Willebrand factor (VWF) and surface adhesion molecules, including P-selectin in intracellular granules, called Weibel–Palade bodies, which are then released upon endothelial cell activation (48, 63–65). Elevated levels of both VWF antigen and VWF activity were detected in the plasma of COVID-19 patients (with the higher values being observed among patients requiring intensive care), corroborating the previous observations of endothelial cell activation elicited by SARS-CoV-2 infection (66, 67). However, VWF activity depends on its size, that is, the larger the length of the VWF multimer, the greater its ability to adhere to platelets and other blood cells (68). Under normal circumstances, ultra-large VWF multimeters are cleaved by ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motifs, member 13), preventing thrombus formation (69). The deficiency of ADAMTS13, which was also observed in blood samples from COVID-19 patients, results in disseminated microvascular thrombosis, characteristic of thrombotic thrombocytopenic purpura (TTP) (67, 70). There is evidence that, in COVID-19 patients, the increase in VWF levels coexist with a moderate reduction in ADAMTS13 activity (67).
Platelets
Platelets are anucleate blood cells generated in the bone marrow from precursor cells called megakaryocytes and are well-known for their essential role in hemostasis and thrombosis [reviewed in (71)]. Low platelet count and altered morphology are often observed during viral infections (72, 73). The low number of circulating platelets following a viral infection might be a result of either decreased platelet production or increased platelet destruction (72). Usually, the reduction in platelet production is observed at later stages of infection, whereas an abrupt decrease in the platelet count in response to viral infections is mediated by enhanced platelet destruction/clearance as a consequence of their activation [reviewed in (72)]. Data from several sources have identified a reduction in the platelet count in COVID-19 patients, where it is commonly associated with disease severity and increased risk of mortality (3, 5, 74–76). It has now been shown that SARS-CoV-2 and platelet interaction triggers programmed cell death, as defined by the presence of necroptosis and apoptosis markers in platelets from COVID-19 patients (77). Indeed, markers of platelet apoptosis, such as mitochondrial inner membrane depolarization, cytosolic calcium concentration, and PS exposure, were increased in COVID-19 patients in the ICU compared with non-ICU COVID-19 patients and healthy controls, demonstrating an association between severe COVID-19 and platelet apoptosis (78). In critically ill COVID-19 patients, these markers were also positively correlated with D-dimer levels and incidence of thrombotic complications (78). These clinical findings were corroborated by in vitro studies where isolated platelets from healthy donors stimulated with sera from severe COVID-19 patients resulted in platelet apoptosis mediated by immunoglobulin G (IgG) fractions from COVID-19 (78). These findings may explain the thrombocytopenia and the thrombotic phenotype observed in COVID-19 patients.
Platelets have been shown to sense and respond to viral components as part of the immune response [reviewed in (79)]. The literature review points out that platelets express a wide range of receptors on their surface which may mediate the binding and uptake of viral pathogens during influenza, HIV, and dengue infection (79–81). For example, during influenza infection, viral particles were detected inside platelets where they colocalized with Toll-like receptor 7 (TLR7) in the lysosomes (80). In the same way, it has been demonstrated that single-stranded RNA viruses activate the TLR7 receptor in human platelets leading to α-granule release and translocation of P-selectin to the cell surface (80, 82). Interestingly, blood samples or isolated platelets from COVID-19 patients examined either by different microscopy techniques or RNA-sequencing analysis revealed that SARS-CoV-2 particles are either attached to or inside platelets (76, 77). However, whether the SARS-CoV-2 virus can enter platelets via binding ACE2 and TMPRSS2 receptors is still debated since there is no consensus if human platelets express both receptors (76, 83, 84). The fact remains that SARS-CoV-2 interacts and possibly enters platelet, in spite of the concern whether ACE2 plays a role in this interaction. Either binding of SARS-CoV-2 to platelets or the SARS-CoV-2 RNA uptake has been shown to enhance platelet activation (76, 83, 84). Uptake of SARS-CoV-2 virions leads to morphological changes in platelets with microparticles shedding and content release (77). In fact, increased levels of platelet-derived extracellular vesicles (EVs) have been previously described in the plasma of COVID-19 patients (84). Indeed, other significant laboratory findings, including increased mean platelet volume (MPV) and increased αIIbβ3 activation and P-selectin expression, corroborate that platelets from COVID-19 patients show a hyperactive phenotype (76, 85, 86). Once activated, platelets secrete a vast repertoire of bioactive molecules from their intracellular granules that play essential roles not only in thrombus formation, such as ADP and thromboxane A2 (TXA2), but also in inflammation [e.g., pro-inflammatory cytokines; reviewed in (87)]. The activation process also results in a dramatic reorganization of the platelet membrane, involving the exposure of new receptors, including glycoprotein αIIbβ3, which enables fibrinogen binding and allows platelet aggregation, and the translocation of P-selectin to the platelet surface (88, 89).
Similar to what has been observed for other viral infections, including influenza, HIV, and dengue (90–93), there is an increased P-selectin expression during clinical infection with SARS-CoV-2 (76, 83, 94). P-selectin surface expression is increased in COVID-19 patients compared to healthy controls. Whether or not this finding could be related to the disease severity has remained elusive so far (76, 83, 94). P-selectin expression was also found to be increased in platelets from COVID-19 patients upon stimulation (83). Interestingly, Spike protein was found to induce integrin activation and P-selectin expression in platelets isolated from healthy donors even in the absence of platelet agonists (76). In addition to P-selectin, increased CD63 expression was also observed in platelets from SARS-CoV-2 infected patients (83, 94). Both P-selectin and CD63 are increased on the surface of platelets after granule release and are therefore considered markers of platelet activation (95). In line with the observation that CD63 modulates platelet spreading on immobilized fibrinogen (96), it has been demonstrated that platelets from COVID-19 patients exhibit greater adhesion and spreading on fibrinogen and collagen (83). Additionally, in vitro studies where platelets from healthy donors were incubated with SARS-CoV-2 or Spike protein showed an enhanced spreading on immobilized fibrinogen and clot retraction (76).
P-selectin is also considered an essential receptor for forming platelet leukocyte aggregates since the binding to its counter-receptor (P-selectin glycoprotein ligand 1; PSGL-1) on the leukocyte surface enables the adhesion of activated platelets to leukocytes (97–99). Platelet-neutrophil and platelet-monocyte aggregates, which are considered a more sensitive indicator of in vivo platelet activation, were significantly elevated in COVID-19 patients compared to healthy individuals (83, 85, 94, 100, 101). A similar outcome was observed when the whole blood of healthy controls treated with SARS-CoV-2 and Spike protein in vitro resulted in an increased proportion of platelet-leukocyte aggregates (76). It should be mentioned that increased levels of circulating platelet-leukocyte aggregates have been considered a marker of a prothrombotic state and are associated with several thrombotic diseases (102).
Another reason to believe that SARS-CoV-2 infection induces greater platelet reactivity is that platelets from COVID-19 patients are more sensitive to aggregation in response to low concentrations of agonists, such as thrombin and collagen, compared to healthy individuals. This hyperreactivity seems to be more pronounced in severe patients than in non-severe patients (76, 83). There is evidence that increased platelet response during SARS-CoV-2 infection is, at least in part, mediated by increased MAPK signaling pathway since the phosphorylation of ERK1/2, p38, and eIF4E, was found to be upregulated in platelets from COVID-19 patients (76, 83). Phosphorylated MAPK was detected in platelets from healthy donors after stimulation with SARS-CoV-2 or Spike protein (76). It was previously demonstrated that activation of MAPK in platelets induces the activation of a cytosolic phospholipase A2 (cPLA2), which results in thromboxane synthesis (103). Interestingly, phosphorylation of cPLA2 was increased in COVID-19 patients both at baseline and following platelet activation in vitro (83). It is well-known that increases in cPLA2 phosphorylation activate cPLA2 activity, upregulating thromboxane production (103). Indeed, it was found that plasma levels of TXA2 and thromboxane B2, a metabolite from platelet TXA2 synthesis, are higher in severe COVID-19 patients (83, 94). Although TXA2 can be secreted by cell types other than platelets, such as endothelial cells (ECs) and macrophages, it was observed that platelets from severe COVID-19, but not mild/asymptomatic subjects, had increased TXA2 synthesis (94).
The platelet hyperactivation observed in COVID-19 patients may be triggered by damaged endothelium. It is well-known that exposure of platelets to components of the subendothelial matrix might lead to platelet activation and aggregation in an attempt to repair the injured tissue (104, 105). Under normal circumstances, platelets circulate in a quiescent, non-adhesive state; however, they can be rapidly activated by subendothelial matrix components and soluble agonists such as ADP, TXA2, and thrombin (104). Another important step of platelet activation is the loss of lipid asymmetry, favoring the exposure of anionic lipids such as PS to the outer membrane. This rearrangement in the membrane phospholipids creates a procoagulant surface on platelets where the clotting factors can anchor and be activated, bursting thrombin formation (89). The thrombin generated cleaves fibrinogen into fibrin, which then interacts with activated platelets in order to stabilize the aggregates (89, 106). Notably, increased factor XII activity was detected in platelets isolated from COVID-19 patients, which was accompanied by a shortening in the activated partial thromboplastin time (APTT) measured in platelet-rich plasma (85). In other words, in the study conducted by Taus et al., platelets from COVID-19 patients show a procoagulant phenotype.
One of the most prominent characteristics of severe COVID-19 is the massive release of a wide range of cytokines, such as TNF, interferon γ (IFN-γ) and IL-1, IL-6, and IL-18, which characterizes the cytokine storm observed in SARS-Cov-2 infected patients (56–59, 107, 108). It is well-established that some of these inflammatory cytokines can have direct effects on platelet function, contributing to their thrombotic propensity (109–111). In line with the observation that IL-6 and IL-1β affect platelet function, it has been demonstrated that both cytokines can enhance agonist-induced platelet aggregation (109, 110). Additionally, there is robust evidence that IL-6 and IL-1β foster platelets hyperactivation and spreading (111). Interestingly, plasma from COVID-19 patients triggered platelet activation in vitro, as demonstrated by a significant increase in platelet P-selectin and CD63 surface translocation and platelet-leukocyte aggregates formation (94, 112), which was dampened by pretreatment with IL-6 receptor inhibitor (113). The resultant platelet hyperactivation may be evoked by inflammatory mediators present in the plasma of COVID-19 patients.
Monocytes/Macrophages
Severe SARS-CoV-2 infection is characterized by an excessive inflammatory response with the release of a large number of pro-inflammatory cytokines (57–59, 107). Similar findings have been documented in patients infected with the severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS) coronaviruses, where hypercytokinemia was considered the main cause of morbidity (114). There is emerging evidence that infiltration of pro-inflammatory monocytes is the critical mediator of the hyperinflammatory response following SARS-CoV-2 infection, being responsible for the cytokine storm observed during the acute phase in severe cases (115–117). Single-cell transcriptomic analysis of bronchoalveolar fluid from COVID-19 patients revealed an increased number of mononuclear phagocytes (MNPs) in severe patients compared to those with mild symptoms or healthy controls (115). The MNP composition in severe patients showed a lower proportion of tissue-resident alveolar macrophages and a higher proportion of inflammatory monocyte-derived macrophages (115). On the other hand, single-cell RNA sequencing of peripheral blood from COVID-19 patients revealed a reduced number of non-classical and intermediate monocytes and impaired immune response by myeloid cells with reduced expression of cytokines, such as IL-6, TNF, and IL-1β (118–120). Considering that cytokine plasma levels are enhanced in SARS-CoV-2 infected patients, those more recent findings suggest a tissue origin of the plasma cytokines. Moreover, circulating levels of pro-inflammatory cytokines were found to be significantly higher in severe COVID-19 patients than in those with mild symptoms, suggesting that this intense cytokine production is positively associated with disease severity (56, 57, 59, 107, 108).
Similar to what has been reported in other respiratory viral diseases, the infection of airway epithelial cells with SARS-CoV-2 triggers the innate immune response, resulting in the activation of monocytes, macrophages, and dendritic cells (121). Several lines of evidence have suggested that monocytes/macrophages are attracted to the alveolar space in response to the viral infection elicited by SARS-CoV-2, where they secrete a wide range of pro-inflammatory cytokines and chemokines, including IL-1β and IL-6 and TNF, contributing to the hyperinflammatory and hypercoagulable phenotypes observed in severe COVID-19 patients (122, 123). In critically ill COVID-19 patients, plasma levels of pro-inflammatory cytokines, in particular the aforementioned IL-1, IL-6, and TNF-α, were found to be upregulated (57–59, 107). Higher levels of IL-6 seem to be associated with the severity of the disease (56, 107, 108), even though it has been reported that circulating IL-6 levels are lower in COVID-19 patients compared to what is observed in other pathological conditions, such as acute respiratory disease syndrome (ARDS) and cytokine release syndrome (CRS) (124).
In fact, it is well-known that activation of coagulation and intravascular coagulation that occurs during sepsis are mainly mediated by the expression of TF on monocytes in response to a number of different inflammatory extracellular stimuli (125–127). In such conditions, monocytes and macrophages respond with increased expression and release of TF (125–127). Likewise, increased TF activity was detected in EVs isolated from plasma of COVID-19 patients compared with healthy controls (128). Since Rosell et al. could not determine the origin of the TF-positive EVs, they speculate that they are derived from activated monocytes and endothelial cells.
The exposition of TF to the blood allows it to bind plasma factor VIIa forming a complex that initiates blood coagulation. The sequential proteolytic cleavage of coagulation factors culminates in the generation of thrombin, which can promote fibrin clot formation and platelet activation and enhance the pro-inflammatory response (53). Although monocytes are considered the primary source of TF in inflammatory states, especially during bacterial sepsis (127), the role of monocytes in virus-induced hypercoagulability remains limited. In vitro studies have shown that cytomegalovirus and influenza virus can upregulate TF expression on infected human monocytes, giving these cells a procoagulant phenotype (129). In vivo studies have correspondingly demonstrated a correlation between TF expression on monocytes and coagulopathy in HIV infection (130). In COVID-19, EV-TF activity was positively correlated with D-dimer, prothrombin time, and von Willebrand factor levels, indicating a link between TF-positive EVs, coagulation, and endothelial activation, which may contribute to thrombosis in COVID-19 patients (128).
It was demonstrated that TF expression by monocytes in COVID-19 is mediated by the crosstalk between monocytes and platelets (94). The platelet-monocyte interaction is also observed in other viral infections, including influenza (131), and during SARS-CoV-2 infection, it is mediated by P-selectin (83, 94). Of note, there is evidence of a positive and significant correlation between platelet-monocyte aggregates and P-selectin expression on the platelet surface in COVID-19 patients, suggesting that activated platelets interact with monocytes and, thereby, induce the expression of TF (85, 94). Indeed, monocyte TF expression appears to be higher in critically ill COVID-19 patients compared to non-infected individuals (94). Interestingly, monocytes from healthy volunteers exhibited increased TF expression after incubation with platelets from COVID-19 patients (94). Not surprisingly, severe COVID-19 patients showed a higher level of platelet-monocyte aggregates than asymptomatic/mild infected subjects and healthy controls (94).
Research over the past decade has linked anti-viral responses and virus-associated illnesses to the NLRP3 inflammasome activation (132–134). It has been recently reported that the SARS-CoV-2 virus can elicit such response. However, to date, the exact mechanisms by which SARS-CoV-2 activates NLRP3 inflammasome remain unclear (135–137). NLRP3 inflammasome activation mediates the autocatalytic activation of caspase-1, leading to maturation and secretion of IL-1β and IL-18, and has recently been implicated in the pathogenesis of thrombosis (138–140). Previous research has observed that IL-1β elicits tissue factor expression in monocytes-macrophages (141) and enhances the production of plasminogen activation inhibitor (142), resulting in a hypercoagulable state. High concentrations of active caspase-1 were detected in the sera of COVID-19 patients, and this increase was found to be more prominent in patients with the severe form (137). Additionally, active intracellular caspase-1 was detected in PBMCs obtained from COVID-19 patients, and the maintenance of these cells in culture resulted in increased active caspase-1 and IL-1β in the supernatant (137). In vitro studies have shown that infection of human monocytes with SARS-CoV-2 leads to an increase in procaspase-1 cleavage and IL-1β production, which can be impaired by NLRP3 inhibitors such as MCC950 and glyburide (136, 137). Together, these findings strongly support the participation of NLRP3 inflammasome in the innate immune responses to SARS-CoV-2. Upon activation, the NLRP3 recruits and interacts with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) which in turn interacts with procaspase-1 (138). During this event, ASC forms aggregates known as “specks” in the cytosol, which are considered a readout for inflammasome activation (143). Interestingly, it was recently reported that SARS-CoV-2 induces ASC speck formation in human monocytes, which can be inhibited by MCC950 (137). In addition, NLRP3 and ASC specks were visualized by fluorescence microscopy in PBMCs from COVID-19 patients, indicating active inflammasomes in cells from patients infected with SARS-CoV-2 (137). Indeed, NLRP3 and ASC specks were also observed in postmortem lung tissues from COVID-19 patients (137). Immunofluorescence analysis of the autopsy lung samples revealed higher numbers of NLRP3 and ASC speck in samples from COVID-19 patients compared to controls (137).
Another feature of inflammasome activation is the cleavage of Gasdermin D (GSDMD) mediated by caspases, which triggers a form of programmed cell death termed pyroptosis (138). Human monocytes infected with viable SARS-CoV-2 showed enhanced cleaved GSDMD, which suggests that SARS-CoV-2 induces pyroptotic cell death in human monocytes (136). Interestingly, it was demonstrated that following inflammasome activation, pyroptotic macrophages release TF that is required for coagulation activation (144).
There is evidence that cells undergoing a lytic cell death release their intracellular content, including lactate dehydrogenase (LDH). Flow cytometry analysis revealed that SARS-CoV-2 induced lytic cell death in human monocytes, which culminated in an increased concentration of LDH in the supernatant compared to non-infected cells (136). Both LDH and IL-1β are released as a result of inflammasome activation (59, 145, 146). A large number of clinical data collected from COVID-19 patients revealed high levels of circulating lactate dehydrogenase (LDH) and IL-1β in critically ill patients (56–58, 100, 147).
Neutrophils
Neutrophil count was found to be higher in severe COVID-19 patients than in patients with less severe symptoms (2, 57, 75, 100, 148, 149). The increase in blood neutrophil levels is concomitant to the disease progression and severity (1), and it has been described as an indicator of poor outcome (56, 150). In particular diseases, including some types of cancer and myeloproliferative disorders, neutrophilia is considered a reliable marker of a prothrombotic state (151–153). Several prospective cohort studies have highlighted the association between the high number of circulating neutrophils and the occurrence of venous thromboembolism (VTE) in cancer patients (154, 155). Whereas, the number of neutrophils is increased, severe COVID-19 patients often experience a drop in the lymphocyte levels, which results in a high neutrophil to lymphocyte ratio (NLR) (100, 150, 156). High NLR, a recognized marker of systemic inflammation that has been considered a predictor of VTE in cancer patients, was also found to be an independent risk factor predicting COVID-19 severity (148, 157–160).
It has been experimentally demonstrated that, in addition to their increased number in the circulation, neutrophils from COVID-19 patients show decreased granularity, which suggests a pre-activated state, and increased ability to spontaneously form NETs (100, 101, 147, 149, 161). Previous studies have shown that upon activation or during NETosis, neutrophils release calprotectin (162, 163). Noteworthy, increased levels of circulating calprotectin have been observed in COVID-19 patients with either severe or moderate disease (164, 165). Serum and plasma levels of calprotectin were found to be positively correlated with disease severity and negatively correlated with oxygenation efficiency (164, 165). Recent evidence suggests that increased levels of calprotectin during COVID-19 are not only associated with a worse outcome but also with thrombogenic and pro-inflammatory phenotypes following SARS-CoV-2 infection. Indeed, it was demonstrated that calprotectin secreted by platelets from COVID-19 patients promotes endothelial activation, as defined by upregulation of coagulation-related and pro-inflammatory genes by ECs in vitro (166).
NETs are formed when neutrophils, upon activation, expel their nuclear content decorated with granule proteins [e.g., myeloperoxidase (MPO) and neutrophil elastase (NE)] to the extracellular milieu (167, 168). They were first recognized for their role in bacterial clearance, but there is recent evidence that NETs are part of the innate immune response during a viral infection, such as those caused by influenza and the respiratory syncytial viruses (167, 169, 170). Similar to the phenomena observed in pneumonia-associated ARDS, neutrophils from COVID-19 patients are more prone to form NETs (171–175). In fact, there is evidence that neutrophils infected with a virus respond to it by releasing NETs (170, 176, 177). Not surprisingly, neutrophils containing SARS-CoV-2 antigens were found in blood samples from a cohort of COVID-19 patients, where they were more efficient in forming NETs compared to healthy neutrophils (149). Additionally, it was demonstrated that viable SARS-CoV-2 could induce NET formation in neutrophils from healthy donors in vitro (149). In line with the observation that aPL antibodies stimulate neutrophils to release NETs (178) it was demonstrated that IgG isolated from COVID-19 patient serum enriched for aPL induces healthy neutrophils to form NETs to a similar extent compared to IgG samples obtained from patients with antiphospholipid syndrome (APS) (47).
An increasing number of studies have detected high circulating levels of NETs in COVID-19 patients (101, 147, 149, 161). As compared with samples from healthy individuals, the COVID-19 patient samples showed higher levels of cell-free DNA (100, 147), MPO-DNA complexes (100, 101, 147, 149, 161), and citrullinated histone H3 (100, 147), well-known markers of NET formation. These markers have been positively correlated with circulating D-dimer levels in SARS-CoV-2 infected patients sera, indicating an association between NETosis and a higher risk of thromboembolic events in COVID-19 (179). The excessive NET formation is also observed in several pathological conditions such as autoimmune and pulmonary diseases and thrombosis (180). It has been recognized by the literature that NETs exhibit a number of thrombogenic properties, including the ability to initiate the intrinsic pathway of coagulation, serving as a scaffold for the adherence of platelets and red blood cells, to degrade natural coagulation inhibitors, and finally, to exert antifibrinolytic effects (181). There is a growing body of literature that provides mechanistic insights into how NETs propagate inflammation and thrombosis. For example, DNA released from activated neutrophils, like other polyanionic molecules, elicits blood coagulation by amplifying factors XI and XII activation (182). In severe sepsis patients, the cell-free DNA levels in circulation correlate with increased thrombin generation (182). Histones, another main component of NETs, exert prothrombotic activity by enhancing thrombin generation either by activating platelets (183) or by impairing protein C activation (184). At the same time, the serine protease neutrophil elastase (NE) acts, together with extracellular nucleosomes, to inactivate the tissue factor pathway inhibitor (TFPI), thus resulting in increased procoagulant activity (185). Together, these studies support the hypothesis that the hypercoagulable state observed in COVID-19 patients is, at least partially, mediated by neutrophil activation and NET formation. This notion is further supported by the evidence that MPO-DNA levels were positively correlated with thrombin-antithrombin (TAT) levels in SARS-CoV-2 infected patients (161).
Neutrophils can also interact with activated platelets at sites of inflammation, thereby facilitating NET formation. The interactions between neutrophils and platelets are mainly mediated by the binding of platelet adhesion molecules or glycoproteins to their ligands on the neutrophil surface (99, 186). The formation of platelet-neutrophil aggregates not only contributes to NET-mediated virus clearance (187), but also propagates thrombus formation (188). Interestingly, neutrophils from COVID-19 patients were found to be decorated with platelets, especially in severe patients (100). Indeed, higher levels of circulating platelet-neutrophil aggregates were detected in COVID-19 patients compared with healthy adults (100, 101). The crosstalk between neutrophils and platelets relies not only on cell-to-cell communication but also on secreted substances. Upon activation, platelets secrete a number of molecules, such as high mobility group box 1 (HMGB1) and platelet factor 4 (PF4), capable of modulating the activation of neutrophils, thus triggering NET formation (189, 190). In vitro studies revealed that platelet-rich plasma from COVID-19 patients stimulates neutrophils from healthy donors to increase the expression of TF and to release NETs (161). These TF-bearing NETs also showed a procoagulant activity indicated by the high levels of TAT complex, a parameter that reflex a hypercoagulable state (161).
Another prominent characteristic of COVID-19 is the extensive neutrophil infiltration in the pulmonary capillaries. Different groups have been describing a robust neutrophil infiltration in the pulmonary capillaries in COVID-19 revealed by autopsy of lung samples from patients infected with SARS-CoV-2 (101, 156, 191–193). Other respiratory viruses, including SARS-CoV and MERS-CoV, are also associated with neutrophil infiltration at sites of infection and development of ARDS (194, 195). Indeed, the ARDS is characterized by increased neutrophil infiltration and accumulation in the alveoli in response to pro-inflammatory cytokines and chemokines produced by an exuberant immune response (196). The neutrophil-attracting chemokines CXCL2 and CXCL8 were shown to be overexpressed by human epithelial cells infected with SARS-CoV-2 in vitro (197). Immunofluorescence analysis of lung sections from COVID-19 patients revealed not only a robust neutrophil infiltration but also typical NETs structures (101, 149). Neutrophils within the lung microvasculature were found to be trapped, associated with platelets, in fibrin meshworks (101, 156) and these clots may contribute to the disease severity. In this context, microvascular thrombi containing NETs associated with platelets and fibrin have been found in the lung, kidney, and heart of COVID-19 patients, as demonstrated by post-mortem histopathological analysis (17).
Therapeutic Perspectives
COVID-19 is generally a respiratory infection; however, some patients develop with non-respiratory symptoms. Research groups around the world have observed a hypercoagulability and an increased risk for venous thromboembolism and arterial thrombosis in SARS-CoV-2 infected patients (4–7, 15, 16, 57). There is, indeed, an association between thromboembolic events and higher mortality among COVID-19 positive patients (11–14). In this review, we summarized the main findings regarding the cellular mechanism of COVID-19-associated coagulopathy that may provide evidence for the use of conventional therapy for thrombosis. In fact, some studies have been conducted to validate the impact of prophylactic or treatment dose anticoagulation, as well as antiplatelet therapy on both thromboembolism incidence and mortality in COVID-19 patients; nevertheless, data are still controversial.
Data from the HEP-COVID study showed that therapeutic anticoagulation with low molecular weight heparin (LMWH) or unfractionated heparin (UFH) was associated with a reduction in venous thromboembolism (VTE), arterial thromboembolism (ATE) in a cohort of non-severe or non-ICU COVID-19 patients (198). In addition, therapeutic heparins doses significantly reduced the mortality from all cause in non-ICU patients, which was not observed in patients admitted to the ICU (198). However, data from the INSPIRATION trial showed no benefit of an intermediate dose of LMWH in preventing VTE or ATE compared with the prophylactic dose of LMWH in ICU patients (199). Similarly, another randomized clinical trial (RAPIDTrial) did not detect any significant reduction in the primary composite of death, mechanical ventilation, or ICU admission in patients receiving a therapeutic dose of LMWH or UFH compared with a prophylactic dose of heparins, although it was associated with a lower incidence of death at 28 days (200). Early initiation of anticoagulation with prophylactic doses of heparin or enoxaparin within 24 h of hospital admission was associated with a decreased risk of 30-day mortality compared with no anticoagulation (201).
A recent retrospective study found that therapeutic anticoagulation either in combination with antiplatelet therapy or alone was associated with improved outcomes and decreased mortality in hospitalized COVID-19 patients in comparison to patients receiving prophylactic anticoagulation. The concomitant prophylactic anticoagulation and antiplatelet therapy were associated with a significantly lower rate of invasive mechanical ventilation compared with prophylactic anticoagulation alone (202). In line with these observations, a single-center retrospective study demonstrated an association between prophylactic anticoagulation and decreased in-hospital mortality (203).
An observational cohort study using an online multicenter international registry [Health Outcome Predictive Evaluation Registry (HOPE-COVID-19)] for patients with laboratory-confirmed SARS-CoV-2 observed no difference in embolic events and the need for mechanical ventilation between patients receiving antiplatelet therapy and those without; however, the duration of mechanical ventilation for those using antiplatelet therapy was significantly shorter (204). Multivariable regression analysis revealed that antiplatelet therapy during hospitalization was associated with a lower risk of mortality, even among critically ill COVID-19 patients (204). Comparably, a recent retrospective, observational cohort study analyzed the data from COVID-19 patients who were taking daily aspirin prior to hospitalization and concluded that pre-admission aspirin therapy was associated with a better in-hospital outcome, in spite of no difference in mortality rate compared to patients who had not received aspirin (205). A multicenter retrospective study observed that aspirin, administered within 24 h or in the 7 days before hospitalization, did not decrease the rate of thrombosis; however, it was associated with a lower risk of ICU admission, mechanical ventilation, and in-hospital death (206).
Research over the past year has provided evidence that the innate immune system plays a critical role in patients' response to SARS-CoV-2 infection. It is becoming increasingly clear that some COVID-19 patients develop a hyperinflammatory syndrome resembling cytokine storm syndromes, which in turn drives the ARDS observed in these patients (56–59, 107, 108). The massive cytokine release in response to SARS-CoV-2 infection raises the possibility that pro-inflammatory cytokines may be a therapeutic target in COVID-19. Thus, immunomodulatory drugs have been proposed as potential therapies for the treatment of COVID-19 since they may mitigate the effects of the hyperinflammatory response. Indeed, several clinical trials are currently in progress to evaluate the benefits of using immunomodulators in COVID-19, and preliminary studies have been already published for anakinra, an IL-1 receptor antagonist that blocks the activity of IL-1α and IL-1β, canakinumab, a monoclonal antibody targeting IL-1β and tocilizumab, a monoclonal antibody that specifically targets IL-6 receptor (207–210). The findings of these studies are summarized in Table 1.
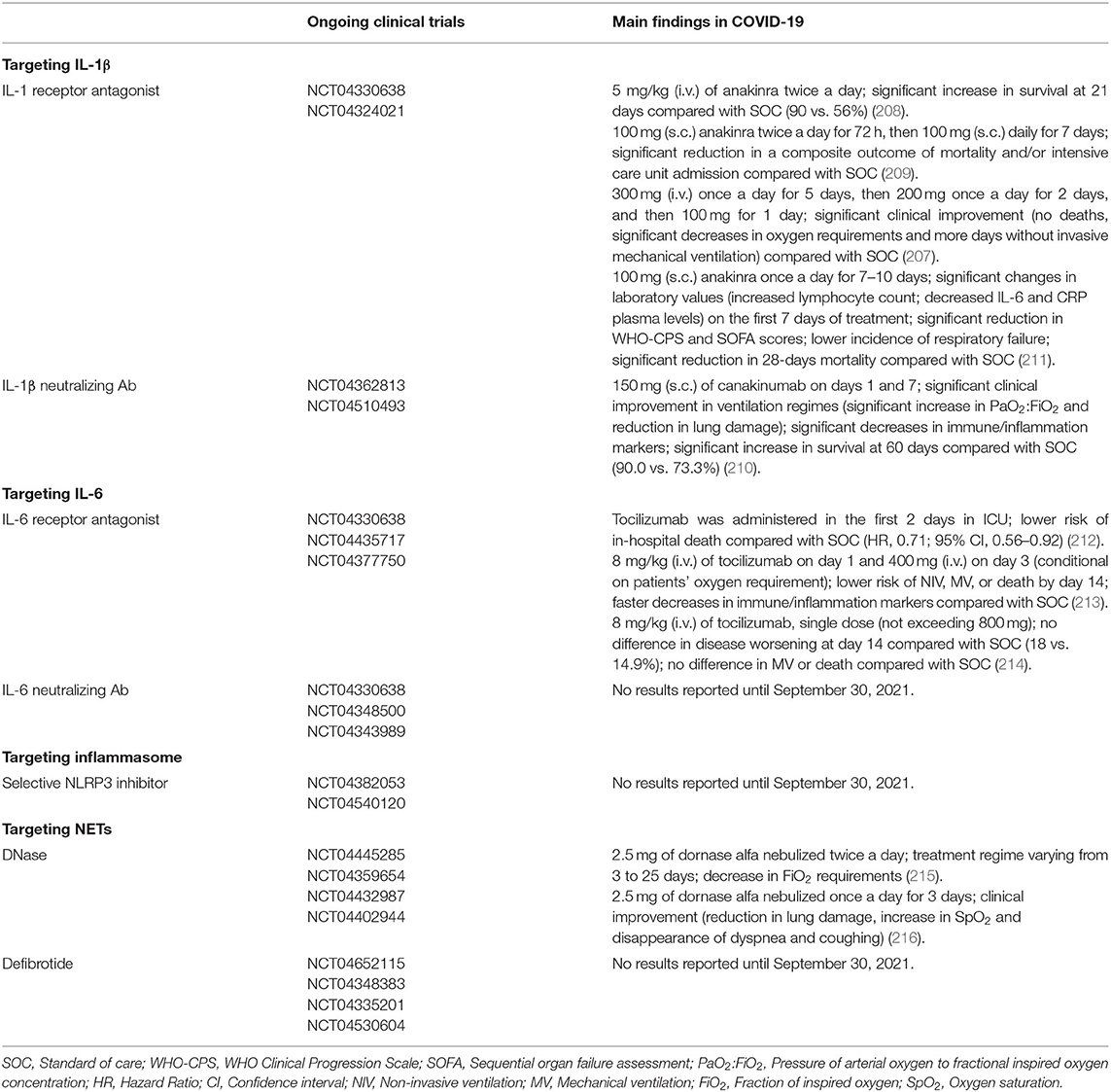
Table 1. Ongoing clinical trials for immunomodulators in COVID-19.
Given that pro-inflammatory cytokines, inflammasome and NETs may contribute to the thromboembolic events observed in critically ill COVID-19 patients. It is, therefore, reasonable to speculate that these aforementioned immunomodulators can also ameliorate the COVID-19–related hypercoagulable state. Actually, immunomodulators have been used not only to treat autoimmune and autoinflammatory diseases but also to improve cardiovascular outcomes in patients with cardiovascular diseases. In the CANTOS trial (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study), the treatment of patients with previous myocardial infarct with the monoclonal IL-1β-neutralizing antibody canakinumab reduced the risk of recurrent cardiovascular events (217). The IL-1β blockade has also been proven to be effective in reducing thrombosis under hypoxic conditions (139). In their experimental model, mice treated with specific antibodies against active IL-1β showed a reduction in coagulation markers, including D-dimer and prothrombin fragment 1+2, and developed smaller venous thrombi than IgG treated mice (139). Neutralizing IL-1 resulted in a prominent decrease in venous thrombogenesis in CD39-deficient mice (140). The deficiency of CD39 in mice results in a higher incidence of thrombosis, and the treatment with either a neutralizing IL-1β antibody or the IL-1 receptor antagonist anakinra resulted in reduced occurrence or size of thrombus in inferior vena cava (IVC) stenosis mice (140). In a 4T1 murine breast cancer model, the treatment with anakinra decreased not only the tumor growth but also the thrombosis occurrence in tumor-bearing mice (218). Given that IL-1β can be secreted as a result of NLRP3 inflammasome activation (138), it is rational to think that NLRP3 inhibitors can dampen the pro-inflammatory and prothrombotic effects elicited by IL-1β. In fact, genetic ablation of NLRP3 was able to curtail venous thrombosis in animals exposed to hypoxia (139). In vitro studies revealed that pretreatment of human platelets with a direct NLRP3 inhibitor significantly reduces platelet aggregation in response to low concentrations of collagen and ADP and impairs clot retraction (219). Of note, a recent phase 3 clinical trial showed that early initiation of anakinra treatment in hospitalized patients with moderate or severe COVID-19 resulted in a significant increase in the lymphocyte count concurrent to a decrease in circulating IL-6 and CRP levels on the first 7 days of treatment. In this study, the 10-day treatment with anakinra was associated with a lower incidence of respiratory failure and a significant reduction in 28-days mortality (211). Further assessment of thrombosis-related parameters in this or similar trials may help to support a role for IL1/ILR in COVID-19-associated hyperthrombotic state.
The humanized monoclonal anti-IL-6 receptor antibody tocilizumab (TCZ) is used to treat rheumatoid arthritis (RA) and recent clinical trials have been proposed its use, in combination with standard of care, in the treatment of COVID-19 patients. Apart from the anti-inflammatory activity in RA, some trials also showed significant benefits with TCZ in terms of reducing hemostatic parameters associated with thrombosis (220, 221). Patients with moderately to severely active RA treated with TCZ intravenous reported fast and sustained reductions in fibrinogen and D-dimer (220). Additionally, another cohort of RA patients receiving subcutaneous TCZ treatment experienced an improvement in endothelial function and decreased NETs formation (221).
It is known that released NET components contribute to thrombus formation as a result of their function on blood coagulation, platelet activation and/or endothelial activation (182–185, 222). One of the major components of NETs are histones, and they are known to promote endothelial cell activation (222). It has recently been shown that endothelial cell activation induced by histones can be inhibited by defibrotide, a mixture of oligonucleotides currently used in the treatment of hepatic veno-occlusive disease (223, 224). Interestingly, a recent study showed that endothelial cell activation induced by COVID-19 patients' serum could be partially inhibited by defibrotide in vitro (224). NETs, which play an important role in thrombus propagation and stabilization, can be degraded by nucleases, such as DNase 1 (225). Studies in experimental models of thrombosis have shown that intravenous administration of DNase 1 protected mice from DVT after IVC flow restriction (226). Markedly, DNase 1-treated mice were less susceptible to DVT than vehicle-treated mice (226). Similarly, DNase 1 has also been shown to protect mice from ischemia-reperfusion injury (227). A combination of intravenous and intraperitoneal injections of DNase 1 in mice subjected to transient middle cerebral artery occlusion (tMCAO) reduced the infarct size by about 40% and significantly improved stroke outcome (227). Tumor-bearing mice also benefit from DNase 1 treatment. Mice bearing 4T1 breast tumors and treated with DNase 1 prior to thrombosis induction were protected from the increased thrombus formation characteristic of this experimental model (228). However, it has been demonstrated that long-term systemic treatment with DNase 1 may be deleterious in mice models of sepsis and cancer-associated thrombosis (229, 230).
DNase is currently used in the treatment of cystic fibrosis, where it is administered to these patients by nebulization (231). Clinical trials currently in progress also propose the use of nebulized DNase 1 for the treatment of respiratory failure in COVID-19. Although it has been disclosed that aerosolized DNase 1 promotes a reduction in systemic inflammatory markers (232), it is not yet clear whether nebulized DNase 1 will have any effect on circulating levels of NET or NET-mediated prothrombotic state in COVID-19.
In addition to the drugs mentioned in this section, current approaches to COVID-19 therapeutics also include other compounds with the potential to ameliorate either inflammation or thrombosis triggered by SARS-CoV-2, such as colchicine, Bruton tyrosine kinase inhibitors and activated protein C. However, while some studies are in a more advanced stage of analysis, others are still in the theoretical field and thus are not discussed in detail (191, 233, 234).
Concluding Remarks
Severe COVID-19 elicits an inflammatory-related hyperthrombotic state that significantly contributes to the fatal outcome. COVID-19-associated thrombosis is a complex process that seems to engage different vascular cells, including endothelial cells, platelets, monocytes, and neutrophils, that contribute to the establishment of the prothrombotic state (Figure 1). Remarkably, heterotypic cell-cell associations seem to play a pivotal role in amplifying the inflammatory/prothrombotic response not only during SARS-CoV-2 infection but also after recovering. Recent studies have shown a persistent prothrombotic phenotype in convalescent COVID-19 patients (235–237). It has been discussed that endothelial cell activation triggered by SARS-CoV-2 is sustained up to 10 weeks following acute infection (236). This may be responsible for the procoagulant state observed in convalescent COVID-19 patients (236, 237). Indeed, a higher level of D-dimer was detected in patients recovering from COVID-19, while other coagulation and inflammation markers were at basal levels (237). Besides the aforementioned factors, the assessment of NET markers may also be beneficial in the management of long-term post-COVID-19 patients since it was demonstrated that circulating NET remnants are increased in individuals up to 2 years after the diagnosis of venous thromboembolism (238). Although less is known about the long-term consequences, there is evidence that endothelial activation, low-grade inflammation, and hypercoagulability may persist in post-acute COVID-19. Thus, it is crucial to establish strategies to identify and monitor patients with a high risk of developing post-acute COVID-19 syndrome.
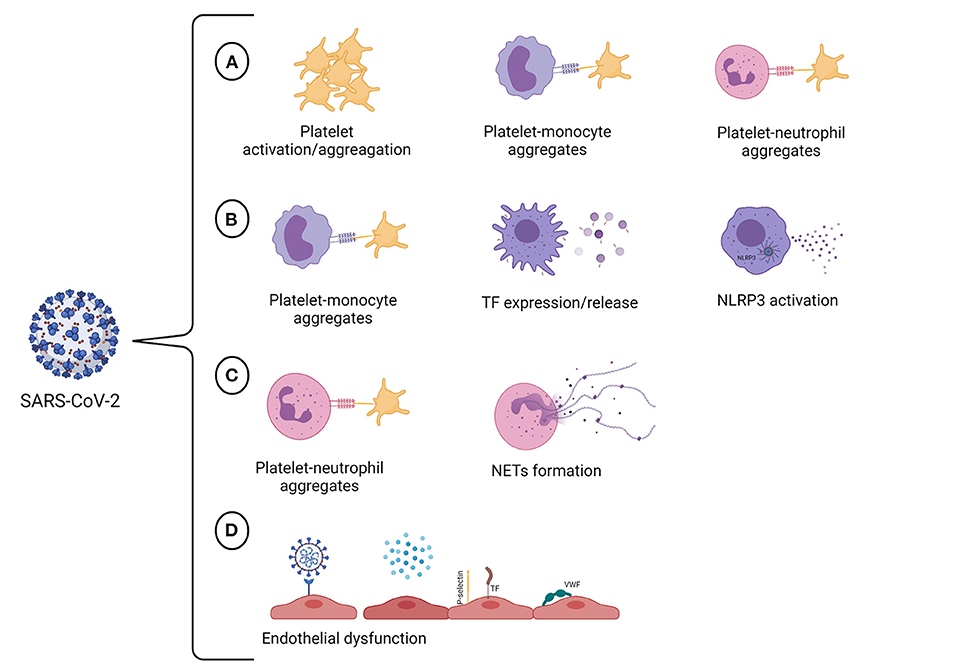
Figure 1. SARS-CoV-2 targets multiple vascular cells. (A) SARS-CoV-2 infection promotes hyperactivation of platelets. The consequences of platelet activation include reorganization of the platelet membrane, which enables fibrinogen binding and allows platelet aggregation, and the translocation of P-selectin to the platelet surface, enabling the formation of platelet-monocyte and platelet-neutrophil aggregates. (B) The crosstalk between platelets and monocytes can upregulate TF expression and release by monocytes, giving these cells a procoagulant phenotype. The infection of human monocytes with SARS-CoV-2 may activate the NLRP3 inflammasome, leading to an increase in procaspase-1 cleavage and IL-1β production and, therefore, resulting in a hypercoagulable state. (C) The interactions between neutrophils and platelets at sites of inflammation facilitate NET formation. It has also been proved that SARS-CoV-2 can induce NET formation in healthy neutrophils in vitro. NETs can propagate thrombus formation due to their thrombogenic properties, including the ability to initiate the intrinsic pathway of coagulation, to degrade natural coagulation inhibitors, and exert antifibrinolytic effects. (D) COVID-19 may trigger endothelial cell dysfunction either due to SARS-CoV-2 infection of endothelial cells or by pro-inflammatory cytokines that are produced in response to viral infection. The activation of endothelial cells results in the loss of the antithrombotic phenotype of the endothelium, occasioning the activation of the coagulation cascade, platelets and complement system in the vasculature. Created with BioRender.com.
Taken together, we conclude that, as seen with other pathological states such as cancer, the prothrombotic condition observed in COVID-19 is a multifactorial and complex process (Figure 2). Further evaluation of additional mechanisms may help delineate novel pharmacological strategies and determine how COVID-19-associated thrombosis can contribute to the multiorgan dysfunction that characterizes this disease (239).
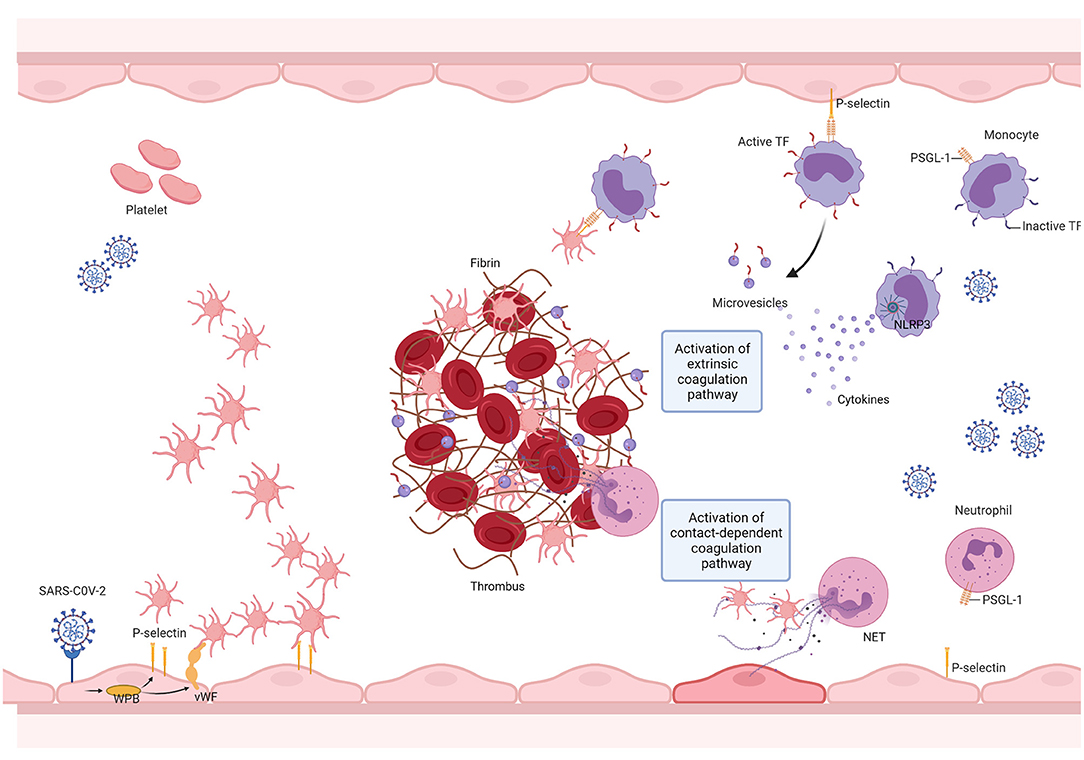
Figure 2. SARS-CoV-2 promotes thrombus formation by engaging multiple vascular cells. The SARS-CoV-2 infection triggers the innate immune response, resulting in the activation of monocytes, which in turn elicit a pro-inflammatory cytokine storm. This results in neutrophil recruitment, endothelial cell, and platelet activation. Endothelial cell activation is followed by the release of von Willebrand factor (VWF) and adhesion molecules, leading to platelet recruitment and activation. Platelet activation results in the exposure of new receptors, including glycoprotein αIIbβ3, which enables fibrinogen binding and allows platelet aggregation, and the translocation of P-selectin to the platelet surface, enabling the formation of platelet-monocyte and platelet-neutrophil aggregates. The interaction between platelets and monocytes and pro-inflammatory cytokines upregulate tissue factor (TF) expression and release it into microvesicles. TF then binds to coagulation factor VII (FVII), activating coagulation. SARS-CoV-2 infection may activate the NLRP3 inflammasome, leading to an increase in IL-1β production and, therefore, resulting in a hypercoagulable state. SARS-CoV-2 mediated cytokine storm promotes sustained neutrophil recruitment and activation, culminating in neutrophil extracellular trap (NET) formation, which fosters thrombus formation. Created with BioRender.com.
Author Contributions
DM: writing—original draft preparation and editing. DM and RM: figure design. EH, PB, and RM: writing—review and editing and funding acquisition. All authors read the final draft and approved the manuscript for submission.
Funding
The Brazilian National Council for Scientific and Technological Development (CNPq) under Grants 309946/2018-2 and 311686/2019-2, The State of Rio de Janeiro Research Foundation (FAPERJ) under Grants E-26/202.871 and E26.200.992/2021, The State of Minas Gerais Research Foundation (FAPEMIG) Grant APQ-02720-21, and the Coordination for the Improvement of Higher Education Personnel (CAPES) under Grants 88887.506989/2020-00 and 23038.009431/2021-42 supported this work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
2. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
3. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
4. Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E, et al. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost. (2020) 120:998–1000. doi: 10.1055/s-0040-1714350
5. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. (2020) 18:844–7. doi: 10.1111/jth.14768
6. Han H, Yang L, Liu R, Liu F, Wu KL, Li J, et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. (2020) 58:1116–20. doi: 10.1515/cclm-2020-0188
7. Xiong M, Liang X, Wei YD. Changes in blood coagulation in patients with severe coronavirus disease 2019 (COVID-19): a meta-analysis. Br J Haematol. (2020) 189:1050–2. doi: 10.1111/bjh.16725
8. Thachil J, Cushman M, Srivastava A. A proposal for staging COVID-19 coagulopathy. Res Pract Thromb Haemost. (2020) 4:731–6. doi: 10.1002/rth2.12372
9. Bristogiannis S, Swan D, Thachil J. Thromboprophylaxis in COVID-19 - rationale and considerations. Adv Biol Regul. (2021) 81:100819. doi: 10.1016/j.jbior.2021.100819
10. Iba T, Warkentin TE, Thachil J, Levi M, Levy JH. Proposal of the definition for COVID-19-associated coagulopathy. J Clin Med. (2021) 10:20191. doi: 10.3390/jcm10020191
11. Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. (2020) 18:1421–4. doi: 10.1111/jth.14830
12. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. (2020) 46:1089–98. doi: 10.1007/s00134-020-06062-x
13. Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Muller MCA, et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. (2020) 18:1995–2002. doi: 10.1111/jth.14888
14. Shah A, Donovan K, McHugh A, Pandey M, Aaron L, Bradbury CA, et al. Thrombotic and haemorrhagic complications in critically ill patients with COVID-19: a multicentre observational study. Crit Care. (2020) 24:561. doi: 10.1186/s13054-020-03260-3
15. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
16. Cheruiyot I, Kipkorir V, Ngure B, Misiani M, Munguti J, Ogeng'o J. Arterial thrombosis in coronavirus disease 2019 patients: a rapid systematic review. Ann Vasc Surg. (2021) 70:273–81. doi: 10.1016/j.avsg.2020.08.087
17. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. (2020) 142:1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
18. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
19. Wang R, Xiao H, Guo R, Li Y, Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infect. (2015) 4:e28. doi: 10.1038/emi.2015.28
20. Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement activation in patients with COVID-19: a novel therapeutic target. J Allergy Clin Immunol. (2020) 146:215–7. doi: 10.1016/j.jaci.2020.05.006
21. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci USA. (2020) 117:25018–25. doi: 10.1073/pnas.2010540117
22. Ma L, Sahu SK, Cano M, Kuppuswamy V, Bajwa J, McPhatter J, et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci Immunol. (2021) 6:abh2259. doi: 10.1126/sciimmunol.abh2259
23. Bareille M, Hardy M, Douxfils J, Roullet S, Lasne D, Levy JH, et al. Viscoelastometric testing to assess hemostasis of COVID-19: a systematic review. J Clin Med. (2021) 10:81740. doi: 10.3390/jcm10081740
24. Slomka A, Kowalewski M, Zekanowska E. Hemostasis in coronavirus disease 2019-lesson from viscoelastic methods: a systematic review. Thromb Haemost. (2021) 121:1181–92. doi: 10.1055/a-1346-3178
25. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. (2007) 100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a
26. Wu KK, Thiagarajan P. Role of endothelium in thrombosis and hemostasis. Annu Rev Med. (1996) 47:315–31. doi: 10.1146/annurev.med.47.1.315
27. Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesth. (2004) 93:105–13. doi: 10.1093/bja/aeh163
28. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. (2010) 4:302–12. doi: 10.2174/1874192401004010302
29. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol. (2020) 40:e138–52. doi: 10.1161/ATVBAHA.120.314330
30. Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, et al. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci USA. (1990) 87:7095–9. doi: 10.1073/pnas.87.18.7095
31. Toulon P, Lamine M, Ledjev I, Guez T, Holleman ME, Sereni D, et al. Heparin cofactor II deficiency in patients infected with the human immunodeficiency virus. Thromb Haemost. (1993) 70:730–5. doi: 10.1055/s-0038-1649660
32. Chi D, Henry J, Kelley J, Thorpe R, Smith JK, Krishnaswamy G. The effects of HIV infection on endothelial function. Endothelium. (2000) 7:223–42. doi: 10.3109/10623320009072210
33. Visseren FL, Bouwman JJ, Bouter KP, Diepersloot RJ, de Groot PH, Erkelens DW. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb Haemost. (2000) 84:319–24. doi: 10.1055/s-0037-1614014
34. Armstrong SM, Darwish I, Lee WL. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence. (2013) 4:537–42. doi: 10.4161/viru.25779
35. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
36. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
37. Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet. (2020) 396:320–32. doi: 10.1016/S0140-6736(20)31305-2
38. Fox SE, Lameira FS, Rinker EB, Vander Heide RS. Cardiac endotheliitis and multisystem inflammatory syndrome after COVID-19. Ann Intern Med. (2020) 173:1025–7. doi: 10.7326/L20-0882
39. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80 e278. doi: 10.1016/j.cell.2020.02.052
40. Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. (2020) 181:905–13 e907. doi: 10.1016/j.cell.2020.04.004
41. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. (2020) 117:11727–34. doi: 10.1073/pnas.2003138117
42. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203:631–7. doi: 10.1002/path.1570
43. Hofmann H, Pohlmann S. Cellular entry of the SARS coronavirus. Trends Microbiol. (2004) 12:466–72. doi: 10.1016/j.tim.2004.08.008
44. Schmaier AA, Hurtado GP, Manickas-Hill ZJ, Sack KD, Chen SM, Bhambhani V, et al. Tie2 activation protects against prothrombotic endothelial dysfunction in COVID-19. medRxiv. (2021). doi: 10.1172/jci.insight.151527
45. Shi H, Zuo Y, Navaz S, Harbaugh A, Hoy C, Gandhi AA, et al. Endothelial cell-activating antibodies in COVID-19. medRxiv. (2021). doi: 10.1101/2021.01.18.21250041
46. Parikh SM. Angiopoietins and Tie2 in vascular inflammation. Curr Opin Hematol. (2017) 24:432–8. doi: 10.1097/MOH.0000000000000361
47. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.abd3876
48. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. (2015) 15:130. doi: 10.1186/s12872-015-0124-z
49. Bombeli T, Karsan A, Tait JF, Harlan JM. Apoptotic vascular endothelial cells become procoagulant. Blood. (1997) 89:2429–42. doi: 10.1182/blood.V89.7.2429
50. Leroyer AS, Anfosso F, Lacroix R, Sabatier F, Simoncini S, Njock SM, et al. Endothelial-derived microparticles: Biological conveyors at the crossroad of inflammation, thrombosis and angiogenesis. Thromb Haemost. (2010) 104:456–63. doi: 10.1160/TH10-02-0111
51. Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MAJr. Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci USA. (1986) 83:4533–7. doi: 10.1073/pnas.83.12.4533
52. Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res. (2005) 96:1233–9. doi: 10.1161/01.RES.0000171805.24799.fa
53. Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. (2004) 24:1015–22. doi: 10.1161/01.ATV.0000130465.23430.74
54. Pons S, Fodil S, Azoulay E, Zafrani L. The vascular endothelium: the cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit Care. (2020) 24:353. doi: 10.1186/s13054-020-03062-7
55. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. (2020) 20:389–91. doi: 10.1038/s41577-020-0343-0
56. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1172/JCI137244
57. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
58. Li S, Jiang L, Li X, Lin F, Wang Y, Li B, et al. Clinical and pathological investigation of patients with severe COVID-19. JCI Insight. (2020) 5:138070. doi: 10.1172/jci.insight.138070
59. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the inflammatory response to severe COVID-19 illness. Am J Respir Crit Care Med. (2020) 202:812–21. doi: 10.1164/rccm.202005-1583OC
60. Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol. (2019) 10:1057. doi: 10.3389/fmicb.2019.01057
61. Bhaskar S, Sinha A, Banach M, Mittoo S, Weissert R, Kass JS, et al. Cytokine storm in COVID-19-immunopathological mechanisms, clinical considerations, and therapeutic approaches: the REPROGRAM consortium position paper. Front Immunol. (2020) 11:1648. doi: 10.3389/fimmu.2020.01648
62. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. (2020) 368:473–4. doi: 10.1126/science.abb8925
63. Wagner DD. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb Haemost. (1993) 70:105–10. doi: 10.1055/s-0038-1646169
64. Gawaz M, Brand K, Dickfeld T, Pogatsa-Murray G, Page S, Bogner C, et al. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis. (2000) 148:75–85. doi: 10.1016/S0021-9150(99)00241-5
65. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. (2004) 104:100–6. doi: 10.1182/blood-2004-01-0107
66. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. (2020) 7:e575–82. doi: 10.1016/S2352-3026(20)30216-7
67. Mancini I, Baronciani L, Artoni A, Colpani P, Biganzoli M, Cozzi G, et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J Thromb Haemost. (2021) 19:513–21. doi: 10.1111/jth.15191
68. Furlan M. Von Willebrand factor: molecular size and functional activity. Ann Hematol. (1996) 72:341–8. doi: 10.1007/s002770050184
69. Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. (2002) 100:4033–9. doi: 10.1182/blood-2002-05-1401
70. Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program. (2004) 2004:407–23. doi: 10.1182/asheducation-2004.1.407
71. Machlus KR, Italiano JE Jr. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. (2013) 201:785–96. doi: 10.1083/jcb.201304054
72. Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front Immunol. (2014) 5:649. doi: 10.3389/fimmu.2014.00649
73. Raadsen M, Du Toit J, Langerak T, van Bussel B, van Gorp E, Goeijenbier M. Thrombocytopenia in virus infections. J Clin Med. (2021) 10:40877. doi: 10.3390/jcm10040877
74. Jiang SQ, Huang QF, Xie WM, Lv C, Quan XQ. The association between severe COVID-19 and low platelet count: evidence from 31 observational studies involving 7613 participants. Br J Haematol. (2020) 190:e29–33. doi: 10.1111/bjh.16817
75. Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. (2020) 18:1469–72. doi: 10.1111/jth.14848
76. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13:120. doi: 10.1186/s13045-020-00954-7
77. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res. (2021) 129:631–46. doi: 10.1161/CIRCRESAHA.121.319117
78. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Haberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. (2021) 137:1061–71. doi: 10.1182/blood.2020008762
79. Hottz ED, Bozza FA, Bozza PT. Platelets in immune response to virus and immunopathology of viral infections. Front Med. (2018) 5:121. doi: 10.3389/fmed.2018.00121
80. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. (2019) 10:1780. doi: 10.1038/s41467-019-09607-x
81. Dib PRB, Quirino-Teixeira AC, Merij LB, Pinheiro MBM, Rozini SV, Andrade FB, et al. Innate immune receptors in platelets and platelet-leukocyte interactions. J Leukoc Biol. (2020) 108:1157–82. doi: 10.1002/JLB.4MR0620-701R
82. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. (2014) 124:791–802. doi: 10.1182/blood-2013-11-536003
83. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136:1317–29. doi: 10.1182/blood.2020007214
84. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. (2020) 120:317703. doi: 10.1161/CIRCRESAHA.120.317703
85. Taus F, Salvagno G, Cane S, Fava C, Mazzaferri F, Carrara E, et al. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol. (2020) 40:2975–89. doi: 10.1161/ATVBAHA.120.315175
86. Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. (2021) 19:e3001109. doi: 10.1371/journal.pbio.3001109
87. Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. (2015) 29:153–62. doi: 10.1016/j.blre.2014.10.003
88. Furie B, Furie BC, Flaumenhaft R. A journey with platelet P-selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost. (2001) 86:214–21. doi: 10.1055/s-0037-1616219
89. Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. (2002) 88:186–93. doi: 10.1055/s-0037-1613209
90. Mayne E, Funderburg NT, Sieg SF, Asaad R, Kalinowska M, Rodriguez B, et al. Increased platelet and microparticle activation in HIV infection: upregulation of P-selectin and tissue factor expression. J Acquir Immune Defic Syndr. (2012) 59:340–6. doi: 10.1097/QAI.0b013e3182439355
91. Hottz ED, Oliveira MF, Nunes PC, Nogueira RM, Valls-de-Souza R, Da Poian AT, et al. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost. (2013) 11:951–62. doi: 10.1111/jth.12178
92. Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, et al. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood. (2014) 123:2854–63. doi: 10.1182/blood-2013-07-515536
93. Le VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. (2015) 191:804–19. doi: 10.1164/rccm.201406-1031OC
94. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. (2020) 136:1330–41. doi: 10.1182/blood.2020007252
95. Michelson AD, Furman MI. Laboratory markers of platelet activation and their clinical significance. Curr Opin Hematol. (1999) 6:342–8. doi: 10.1097/00062752-199909000-00012
96. Israels SJ, McMillan-Ward EM. CD63 modulates spreading and tyrosine phosphorylation of platelets on immobilized fibrinogen. Thromb Haemost. (2005) 93:311–8. doi: 10.1160/TH04-08-0503
97. Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. (2005) 106:2334–9. doi: 10.1182/blood-2005-04-1530
98. Yokoyama S, Ikeda H, Haramaki N, Yasukawa H, Murohara T, Imaizumi T. Platelet P-selectin plays an important role in arterial thrombogenesis by forming large stable platelet-leukocyte aggregates. J Am Coll Cardiol. (2005) 45:1280–6. doi: 10.1016/j.jacc.2004.12.071
99. Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets. (2015) 26:507–20. doi: 10.3109/09537104.2015.1064881
100. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine. (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
101. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
102. Finsterbusch M, Schrottmaier WC, Kral-Pointner JB, Salzmann M, Assinger A. Measuring and interpreting platelet-leukocyte aggregates. Platelets. (2018) 29:677–85. doi: 10.1080/09537104.2018.1430358
103. Manne BK, Munzer P, Badolia R, Walker-Allgaier B, Campbell RA, Middleton E, et al. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J Thromb Haemost. (2018) 16:1211–25. doi: 10.1111/jth.14005
104. Bergmeier W, Hynes RO. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb Perspect Biol. (2012) 4:a005132. doi: 10.1101/cshperspect.a005132
105. Tomaiuolo M, Brass LF, Stalker TJ. Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Interv Cardiol Clin. (2017) 6:1–12. doi: 10.1016/j.iccl.2016.08.001
106. Swieringa F, Spronk HMH, Heemskerk JWM, van der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–60. doi: 10.1002/rth2.12107
107. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, et al. Detectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically ill patients with coronavirus disease 2019. Clin Infect Dis. (2020) 71:1937–42. doi: 10.1093/cid/ciaa449
108. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
109. Oleksowicz L, Mrowiec Z, Zuckerman D, Isaacs R, Dutcher J, Puszkin E. Platelet activation induced by interleukin-6: evidence for a mechanism involving arachidonic acid metabolism. Thromb Haemost. (1994) 72:302–8. doi: 10.1055/s-0038-1648857
110. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol. (2014) 34:552–64. doi: 10.1161/ATVBAHA.113.302700
111. Bester J, Pretorius E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep. (2016) 6:32188. doi: 10.1038/srep32188
112. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Association of neutrophil activation, more than platelet activation, with thrombotic complications in coronavirus disease 2019. J Infect Dis. (2021) 223:933–44. doi: 10.1093/infdis/jiaa756
113. Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl Sci. (2021) 6:202–18. doi: 10.1016/j.jacbts.2020.12.009
114. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
115. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
116. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. (2020) 19:102537. doi: 10.1016/j.autrev.2020.102537
117. Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, et al. Frontline Science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J Leukoc Biol. (2021) 109:13–22. doi: 10.1002/JLB.4HI0720-470R
118. Arunachalam PS, Wimmers F, Mok CKP, Perera R, Scott M, Hagan T, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. (2020) 369:1210–20. doi: 10.1126/science.abc6261
119. Gatti A, Radrizzani D, Vigano P, Mazzone A, Brando B. Decrease of non-classical and intermediate monocyte subsets in severe acute SARS-CoV-2 infection. Cytometry A. (2020) 97:887–90. doi: 10.1002/cyto.a.24188
120. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martinez-Colon GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
121. Zheng J, Wang Y, Li K, Meyerholz DK, Allamargot C, Perlman S. Severe acute respiratory syndrome coronavirus 2-induced immune activation and death of monocyte-derived human macrophages and dendritic cells. J Infect Dis. (2021) 223:785–95. doi: 10.1093/infdis/jiaa753
122. Bhattacharyya R, Iyer P, Phua GC, Lee JH. The interplay between coagulation and inflammation pathways in COVID-19-associated respiratory failure: a narrative review. Pulm Ther. (2020) 6:215–31. doi: 10.1007/s41030-020-00126-5
123. Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. (2020) 8:e46–7. doi: 10.1016/S2213-2600(20)30216-2
124. Leisman DE, Deutschman CS, Legrand M. Facing COVID-19 in the ICU: vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. (2020) 46:1105–8. doi: 10.1007/s00134-020-06059-6
125. Osterud B. Tissue factor expression by monocytes: regulation and pathophysiological roles. Blood Coagul Fibrinolysis. (1998) 9(Suppl.1):S9–14.
126. Levi M, van der Poll T, ten Cate H. Tissue factor in infection and severe inflammation. Semin Thromb Hemost. (2006) 32:33–9. doi: 10.1055/s-2006-933338
127. Pawlinski R, Mackman N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb Res. (2010) 125(Suppl.1):S70–73. doi: 10.1016/j.thromres.2010.01.042
128. Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, et al. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler Thromb Vasc Biol. (2021) 41:878–82. doi: 10.1161/ATVBAHA.120.315547
129. Bouwman JJ, Visseren FL, Bosch MC, Bouter KP, Diepersloot RJ. Procoagulant and inflammatory response of virus-infected monocytes. Eur J Clin Invest. (2002) 32:759–66. doi: 10.1046/j.1365-2362.2002.01041.x
130. Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. (2010) 115:161–7. doi: 10.1182/blood-2009-03-210179
131. Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest. (2012) 141:1490–5. doi: 10.1378/chest.11-2860
132. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
133. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. (2013) 122:3405–14. doi: 10.1182/blood-2013-05-504449
134. He Z, Chen J, Zhu X, An S, Dong X, Yu J, et al. NLRP3 inflammasome activation mediates zika virus-associated inflammation. J Infect Dis. (2018) 217:1942–51. doi: 10.1093/infdis/jiy129
135. Courjon J, Dufies O, Robert A, Bailly L, Torre C, Chirio D, et al. Heterogeneous NLRP3 inflammasome signature in circulating myeloid cells as a biomarker of COVID-19 severity. Blood Adv. (2021) 5:1523–34. doi: 10.1182/bloodadvances.2020003918
136. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. (2021) 7:43. doi: 10.1038/s41420-021-00477-1
137. Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. (2021) 218. doi: 10.1084/jem.20201707
138. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
139. Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc Natl Acad Sci USA. (2017) 114:4763–8. doi: 10.1073/pnas.1620458114
140. Yadav V, Chi L, Zhao R, Tourdot BE, Yalavarthi S, Jacobs BN, et al. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1beta-driven venous thrombosis. J Clin Invest. (2019) 129:2872–7. doi: 10.1172/JCI124804
141. Grignani G, Maiolo A. Cytokines and hemostasis. Haematologica. (2000) 85:967–72. doi: 10.3324/%25x
142. Dong J, Fujii S, Imagawa S, Matsumoto S, Matsushita M, Todo S, et al. IL-1 and IL-6 induce hepatocyte plasminogen activator inhibitor-1 expression through independent signaling pathways converging on C/EBPdelta. Am J Physiol Cell Physiol. (2007) 292:C209–215. doi: 10.1152/ajpcell.00157.2006
143. Stutz A, Horvath GL, Monks BG, Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol. (2013) 1040:91–101. doi: 10.1007/978-1-62703-523-1_8
144. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. (2019) 50:1401–11 e1404. doi: 10.1016/j.immuni.2019.04.003
145. Han Y, Zhang H, Mu S, Wei W, Jin C, Tong C, et al. Lactate dehydrogenase, an independent risk factor of severe COVID-19 patients: a retrospective and observational study. Aging. (2020) 12:11245–58. doi: 10.18632/aging.103372
146. Henry BM, Aggarwal G, Wong J, Benoit S, Vikse J, Plebani M, et al. Lactate dehydrogenase levels predict coronavirus disease 2019 (COVID-19) severity and mortality: a pooled analysis. Am J Emerg Med. (2020) 38:1722–6. doi: 10.1016/j.ajem.2020.05.073
147. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. (2020) 5:138999. doi: 10.1172/jci.insight.138999
148. Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. (2020) 18:206. doi: 10.1186/s12967-020-02374-0
149. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217. doi: 10.1084/jem.20201129
150. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J Am Med Assoc. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
151. Campbell PJ, MacLean C, Beer PA, Buck G, Wheatley K, Kiladjian JJ, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood. (2012) 120:1409–11. doi: 10.1182/blood-2012-04-424911
152. Khorana AA. Risk assessment for cancer-associated thrombosis: what is the best approach? Thromb Res. (2012) 129(Suppl.1):S10–15. doi: 10.1016/S0049-3848(12)70009-9
153. Barbui T, Masciulli A, Marfisi MR, Tognoni G, Finazzi G, Rambaldi A, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. (2015) 126:560–1. doi: 10.1182/blood-2015-04-638593
154. Blix K, Jensvoll H, Braekkan SK, Hansen JB. White blood cell count measured prior to cancer development is associated with future risk of venous thromboembolism–the Tromso study. PLoS ONE. (2013) 8:e73447. doi: 10.1371/journal.pone.0073447
155. Pabinger I, Posch F. Flamethrowers: blood cells and cancer thrombosis risk. Hematology Am Soc Hematol Educ Program. (2014) 2014:410–7. doi: 10.1182/asheducation-2014.1.410
156. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med. (2020) 8:681–6. doi: 10.1016/S2213-2600(20)30243-5
157. Ferroni P, Riondino S, Formica V, Cereda V, Tosetto L, La Farina F, et al. Venous thromboembolism risk prediction in ambulatory cancer patients: clinical significance of neutrophil/lymphocyte ratio and platelet/lymphocyte ratio. Int J Cancer. (2015) 136:1234–40. doi: 10.1002/ijc.29076
158. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. (2020) 71:762–8. doi: 10.1093/cid/ciaa248
159. Song CY, Xu J, He JQ, Lu YQ. Immune dysfunction following COVID-19, especially in severe patients. Sci Rep. (2020) 10:15838. doi: 10.1038/s41598-020-72718-9
160. Zhang B, Zhou X, Zhu C, Song Y, Feng F, Qiu Y, et al. Immune phenotyping based on the neutrophil-to-lymphocyte ratio and IgG level predicts disease severity and outcome for patients with covid-19. Front Mol Biosci. (2020) 7:157. doi: 10.3389/fmolb.2020.00157
161. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. (2020) 130:6151–7. doi: 10.1172/JCI141374
162. Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of extracellular release of human neutrophil calprotectin complex. J Leukoc Biol. (2001) 70:130–4. doi: 10.1189/jlb.70.1.130
163. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
164. Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. (2020) 182:1401–18 e1418. doi: 10.1016/j.cell.2020.08.002
165. Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA, et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol. (2021) 109:67–72. doi: 10.1002/JLB.3COVCRA0720-359R
166. Barrett TJ, Cornwell M, Myndzar K, Rolling CC, Xia Y, Drenkova K, et al. Platelets amplify endotheliopathy in COVID-19. Sci Adv. (2021) 7:eabh2434. doi: 10.1126/sciadv.abh2434
167. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
168. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. (2007) 5:577–82. doi: 10.1038/nrmicro1710
169. Tripathi S, Verma A, Kim EJ, White MR, Hartshorn KL. LL-37 modulates human neutrophil responses to influenza A virus. J Leukoc Biol. (2014) 96:931–8. doi: 10.1189/jlb.4A1113-604RR
170. Funchal GA, Jaeger N, Czepielewski RS, Machado MS, Muraro SP, Stein RT, et al. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE. (2015) 10:e0124082. doi: 10.1371/journal.pone.0124082
171. Ebrahimi F, Giaglis S, Hahn S, Blum CA, Baumgartner C, Kutz A, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur Respir J. (2018) 51:2017. doi: 10.1183/13993003.01389-2017
172. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. (2018) 3:98178. doi: 10.1172/jci.insight.98178
173. Mikacenic C, Moore R, Dmyterko V, West TE, Altemeier WA, Liles WC, et al. Neutrophil extracellular traps (NETs) are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit Care. (2018) 22:358. doi: 10.1186/s13054-018-2290-8
174. Bendib I, de Chaisemartin L, Granger V, Schlemmer F, Maitre B, Hue S, et al. Neutrophil extracellular traps are elevated in patients with pneumonia-related acute respiratory distress syndrome. Anesthesiology. (2019) 130:581–91. doi: 10.1097/ALN.0000000000002619
175. Adrover JM, Aroca-Crevillen A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed 'disarming' of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol. (2020) 21:135–44. doi: 10.1038/s41590-019-0571-2
176. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
177. Hiroki CH, Toller-Kawahisa JE, Fumagalli MJ, Colon DF, Figueiredo LTM, Fonseca B, et al. Neutrophil extracellular traps effectively control acute chikungunya virus infection. Front Immunol. (2019) 10:3108. doi: 10.3389/fimmu.2019.03108
178. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. (2015) 67:2990–3003. doi: 10.1002/art.39247
179. Zuo Y, Zuo M, Yalavarthi S, Gockman K, Madison JA, Shi H, et al. Neutrophil extracellular traps and thrombosis in COVID-19. J Thromb Thrombolysis. (2021) 51:446–53. doi: 10.1007/s11239-020-02324-z
180. Sorvillo N, Cherpokova D, Martinod K, Wagner DD. Extracellular DNA NET-works with dire consequences for health. Circ Res. (2019) 125:470–88. doi: 10.1161/CIRCRESAHA.119.314581
181. Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. J Thromb Haemost. (2013) 11(Suppl.1):233–41. doi: 10.1111/jth.12261
182. Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. (2014) 34:1977–84. doi: 10.1161/ATVBAHA.114.304114
183. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
184. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost. (2011) 9:1795–803. doi: 10.1111/j.1538-7836.2011.04422.x
185. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
186. Zucoloto AZ, Jenne CN. Platelet-neutrophil interplay: insights into neutrophil extracellular trap (NET)-driven coagulation in infection. Front Cardiovasc Med. (2019) 6:85. doi: 10.3389/fcvm.2019.00085
187. Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. (2013) 13:169–80. doi: 10.1016/j.chom.2013.01.005
188. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
189. Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. (2016) 7:271. doi: 10.3389/fimmu.2016.00271
190. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. (2016) 128:2435–49. doi: 10.1182/blood-2016-04-710632
191. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. (2020) 217:652. doi: 10.1084/jem.20200652
192. Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, da Silva LFF, de Oliveira EP, Saldiva PHN, et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. (2020) 18:1517–9. doi: 10.1111/jth.14844
193. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
194. Perlman S, Dandekar AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol. (2005) 5:917–27. doi: 10.1038/nri1732
195. Blondonnet R, Constantin JM, Sapin V, Jabaudon M. A pathophysiologic approach to biomarkers in acute respiratory distress syndrome. Dis Markers. (2016) 2016:3501373. doi: 10.1155/2016/3501373
196. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. (2019) 5:18. doi: 10.1038/s41572-019-0069-0
197. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45 e1039. doi: 10.1016/j.cell.2020.04.026
198. Spyropoulos AC, Goldin M, Giannis D, Diab W, Wang J, Khanijo S, et al. Efficacy and safety of therapeutic-dose heparin vs. standard prophylactic or intermediate-dose heparins for thromboprophylaxis in high-risk hospitalized patients with covid-19: the HEP-COVID randomized clinical trial. J Am Med Assoc Intern Med. (2021) 2021:6203. doi: 10.1001/jamainternmed.2021.6203
199. Investigators I, Sadeghipour P, Talasaz AH, Rashidi F, Sharif-Kashani B, Beigmohammadi MT, et al. Effect of intermediate-dose vs standard-dose prophylactic anticoagulation on thrombotic events, extracorporeal membrane oxygenation treatment, or mortality among patients with COVID-19 admitted to the intensive care unit: the INSPIRATION randomized clinical trial. J Am Med Assoc. (2021) 325:1620–30. doi: 10.1001/jama.2021.4152
200. Sholzberg M, Tang GH, Rahhal H, AlHamzah M, Kreuziger LB, Ainle FN, et al. Effectiveness of therapeutic heparin vs. prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with covid-19 admitted to hospital: RAPID randomised clinical trial. BMJ. (2021) 375:n2400. doi: 10.1136/bmj.n2400
201. Rentsch CT, Beckman JA, Tomlinson L, Gellad WF, Alcorn C, Kidwai-Khan F, et al. Early initiation of prophylactic anticoagulation for prevention of coronavirus disease 2019 mortality in patients admitted to hospital in the United States: cohort study. BMJ. (2021) 372:n311. doi: 10.1136/bmj.n311
202. Matli K, Chamoun N, Fares A, Zibara V, Al-Osta S, Nasrallah R, et al. Combined anticoagulant and antiplatelet therapy is associated with an improved outcome in hospitalised patients with COVID-19: a propensity matched cohort study. Open Heart. (2021) 8:1785. doi: 10.1136/openhrt-2021-001785
203. Roomi SS, Saddique M, Ullah W, Haq S, Ashfaq A, Madara J, et al. Anticoagulation in COVID-19: a single-center retrospective study. J Community Hosp Intern Med Perspect. (2021) 11:17–22. doi: 10.1080/20009666.2020.1835297
204. Santoro F, Nunez-Gil IJ, Vitale E, Viana-Llamas MC, Reche-Martinez B, Romero-Pareja R, et al. Antiplatelet therapy and outcome in COVID-19: the Health Outcome Predictive Evaluation Registry. Heart. (2021) 2021:319552. doi: 10.1136/heartjnl-2021-319552
205. Sisinni A, Rossi L, Battista A, Poletti E, Battista F, Battista RA, et al. Pre-admission acetylsalicylic acid therapy and impact on in-hospital outcome in COVID-19 patients: the ASA-CARE study. Int J Cardiol. (2021) 344:240–5. doi: 10.1016/j.ijcard.2021.09.058
206. Chow JH, Khanna AK, Kethireddy S, Yamane D, Levine A, Jackson AM, et al. Aspirin use is associated with decreased mechanical ventilation, intensive care unit admission, and in-hospital mortality in hospitalized patients with coronavirus disease 2019. Anesth Analg. (2021) 132:930–41. doi: 10.1213/ANE.0000000000005292
207. Cauchois R, Koubi M, Delarbre D, Manet C, Carvelli J, Blasco VB, et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc Natl Acad Sci USA. (2020) 117:18951–3. doi: 10.1073/pnas.2009017117
208. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. (2020) 2:e325–31. doi: 10.1016/S2665-9913(20)30127-2
209. Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. (2020) 2:e393–400. doi: 10.1016/S2665-9913(20)30164-8
210. Generali D, Bosio G, Malberti F, Cuzzoli A, Testa S, Romanini L, et al. Canakinumab as treatment for COVID-19-related pneumonia: a prospective case-control study. Int J Infect Dis. (2021) 104:433–40. doi: 10.1016/j.ijid.2020.12.073
211. Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med. (2021) 27:1752–60. doi: 10.1038/s41591-021-01499-z
212. Gupta S, Wang W, Hayek SS, Chan L, Mathews KS, Melamed ML, et al. Association between early treatment with tocilizumab and mortality among critically ill patients with COVID-19. J Am Med Assoc Intern Med. (2021) 181:41–51. doi: 10.1001/jamainternmed.2020.6252
213. Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P, et al. Effect of tocilizumab vs. usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: a randomized clinical trial. J Am Med Assoc Intern Med. (2021) 181:32–40. doi: 10.1001/jamainternmed.2020.6820
214. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of tocilizumab in patients hospitalized with covid-19. N Engl J Med. (2020) 383:2333–44. doi: 10.1056/NEJMoa2028836
215. Weber AG, Chau AS, Egeblad M, Barnes BJ, Janowitz T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: a case series. Mol Med. (2020) 26:91. doi: 10.1186/s10020-020-00215-w
216. Okur HK, Yalcin K, Tastan C, Demir S, Yurtsever B, Karakus GS, et al. Preliminary report of in vitro and in vivo effectiveness of dornase alfa on SARS-CoV-2 infection. New Microbes New Infect. (2020) 37:100756. doi: 10.1016/j.nmni.2020.100756
217. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
218. Gomes T, Varady CBS, Lourenco AL, Mizurini DM, Rondon AMR, Leal AC, et al. IL-1beta blockade attenuates thrombosis in a neutrophil extracellular trap-dependent breast cancer model. Front Immunol. (2019) 10:2088. doi: 10.3389/fimmu.2019.02088
219. Qiao J, Wu X, Luo Q, Wei G, Xu M, Wu Y, et al. NLRP3 regulates platelet integrin alphaIIbbeta3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica. (2018) 103:1568–76. doi: 10.3324/haematol.2018.191700
220. McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD, et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis. (2015) 74:694–702. doi: 10.1136/annrheumdis-2013-204345
221. Ruiz-Limon P, Ortega R, Arias de la Rosa I, Abalos-Aguilera MDC, Perez-Sanchez C, Jimenez-Gomez Y, et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl Res. (2017) 183:87–103. doi: 10.1016/j.trsl.2016.12.003
222. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
223. Pescador R, Capuzzi L, Mantovani M, Fulgenzi A, Ferrero ME. Defibrotide: properties and clinical use of an old/new drug. Vascul Pharmacol. (2013) 59:1–10. doi: 10.1016/j.vph.2013.05.001
224. Shi H, Gandhi AA, Smith SA, Wang Q, Chiang D, Yalavarthi S, et al. Endothelium-protective, histone-neutralizing properties of the polyanionic agent defibrotide. medRxiv. (2021). doi: 10.1101/2021.02.21.21252160
225. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
226. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
227. De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. (2012) 32:1884–91. doi: 10.1161/ATVBAHA.112.250993
228. Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. (2017) 7:6438. doi: 10.1038/s41598-017-06893-7
229. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS ONE. (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
230. Varady CBS, Oliveira AC, Monteiro RQ, Gomes T. Recombinant human DNase I for the treatment of cancer-associated thrombosis: a pre-clinical study. Thromb Res. (2021) 203:131–7. doi: 10.1016/j.thromres.2021.04.028
231. Pressler T. Review of recombinant human deoxyribonuclease (rhDNase) in the management of patients with cystic fibrosis. Biologics. (2008) 2:611–7. doi: 10.2147/BTT.S3052
232. Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. (2018) 9:CD001127. doi: 10.1002/14651858.CD001127.pub4
233. Pestka SB. Old drug, new Trick? The rationale for the treatment of COVID-19 with activated protein C. Med Hypotheses. (2021) 149:110537. doi: 10.1016/j.mehy.2021.110537
234. Stack M, Sacco K, Castagnoli R, Livinski AA, Notarangelo LD, Lionakis MS. BTK inhibitors for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): a systematic review. Res Sq. (2021) 2021:108816. doi: 10.1016/j.clim.2021.108816
235. Fan BE, Umapathi T, Chua K, Chia YW, Wong SW, Tan GWL, et al. Delayed catastrophic thrombotic events in young and asymptomatic post COVID-19 patients. J Thromb Thrombolysis. (2021) 51:971–7. doi: 10.1007/s11239-020-02332-z
236. Fogarty H, Townsend L, Morrin H, Ahmad A, Comerford C, Karampini E, et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J Thromb Haemost. (2021) 19:2546–53. doi: 10.1111/jth.15490
237. Townsend L, Fogarty H, Dyer A, Martin-Loeches I, Bannan C, Nadarajan P, et al. Prolonged elevation of D-dimer levels in convalescent COVID-19 patients is independent of the acute phase response. J Thromb Haemost. (2021) 19:1064–70. doi: 10.1111/jth.15267
238. Zapponi KCS, Orsi FA, Cunha JLR, de Brito IR, Romano AVC, Bittar LF, et al. Neutrophil activation and circulating neutrophil extracellular traps are increased in venous thromboembolism patients for at least one year after the clinical event. J Thromb Thrombolysis. (2021) 21:2526. doi: 10.1007/s11239-021-02526-z
Keywords: COVID-19, thrombosis, platelets, monocytes, neutrophil extracellular trap, endothelium, blood coagulation
Citation: Mizurini DM, Hottz ED, Bozza PT and Monteiro RQ (2021) Fundamentals in Covid-19-Associated Thrombosis: Molecular and Cellular Aspects. Front. Cardiovasc. Med. 8:785738. doi: 10.3389/fcvm.2021.785738
Received: 29 September 2021; Accepted: 23 November 2021;
Published: 17 December 2021.
Edited by:
Paola van der Meijden, Maastricht University, NetherlandsReviewed by:
Yogendra Kanthi, Division of Intramural Research (NHLBI), United StatesElena Campello, University of Padua, Italy
Copyright © 2021 Mizurini, Hottz, Bozza and Monteiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robson Q. Monteiro, cm9ic29ucW1AYmlvcW1lZC51ZnJqLmJy