 Zhehao Dai
Zhehao Dai Seitaro Nomura
Seitaro Nomura- Department of Cardiovascular Medicine, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
Cardiovascular diseases are among the leading causes of morbidity and mortality worldwide. Although the spectrum of the heart from development to disease has long been studied, it remains largely enigmatic. The emergence of single-cell omics technologies has provided a powerful toolbox for defining cell heterogeneity, unraveling previously unknown pathways, and revealing intercellular communications, thereby boosting biomedical research and obtaining numerous novel findings over the last 7 years. Not only cell atlases of normal and developing hearts that provided substantial research resources, but also some important findings regarding cell-type-specific disease gene program, could never have been established without single-cell omics technologies. Herein, we briefly describe the latest technological advances in single-cell omics and summarize the major findings achieved by such approaches, with a focus on development and homeostasis of the heart, myocardial infarction, and heart failure.
Introduction
Cardiovascular diseases are among the major causes of morbidity and mortality worldwide. Yet, despite relentless research efforts in recent decades, the complex structure of the heart, its incessantly beating nature, and its wide range of abnormalities and disorders made us still struggle to understand the molecular mechanisms underlying the normal development, homeostasis, and diseases of this organ.
Recent technological advances have enabled researchers to understand biology at the single-cell resolution. Single-cell RNA sequencing (scRNA-seq) and single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) allow an unbiased assessment of transcriptomics and epigenomics in heterogeneous tissues and have identified not only previously unknown cell populations, but also dynamic cellular transitions and intercellular interactions in various tissues (1, 2). These cutting-edge technologies have been applied not only to healthy humans and mice to create reference maps, namely, the Human Cell Atlas and the Tabula Muris (3, 4), but also to normal development and various diseases. Recent advances in spatial transcriptomics, the molecular profiling method that compensates for the lack of spatial information in scRNA-seq, have furthered our understanding (5). The use of single-cell omics technologies in cardiovascular research has been increasing exponentially since 2015 (Figure 1) and, as expected, is providing more insight into the workings of both healthy and pathological hearts.
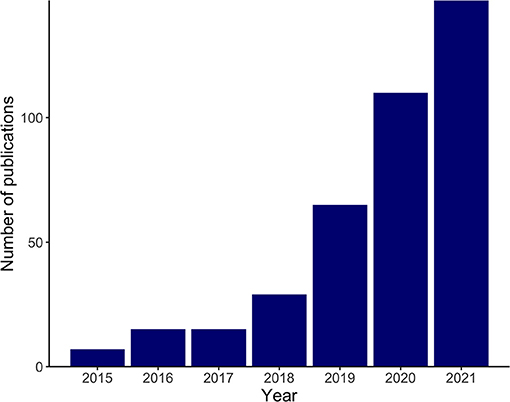
Figure 1. Exponential increase in cardiovascular research implementing single-cell omics. The bar plot shows the numbers of search results in PubMed in recent years with the following search term and the search conducted on November 2, 2021: (“RNA-Seq”[Mesh] OR “Transcriptome”[Mesh] OR “Chromatin Immunoprecipitation Sequencing”[Mesh] OR multi-omics[Title/Abstract] OR multiomics[Title/Abstract] OR omics[Title/Abstract] OR sequencing[Title/Abstract] OR seq[Title/Abstract]) AND (“Single-Cell Analysis”[Mesh] OR “single cell”[Title/Abstract] OR “single nucleus”[Title/Abstract] OR “single nuclei”[Title/Abstract]) AND (“heart”[Mesh] OR heart[Title/Abstract] OR cardiac[Title/Abstract] OR cardiomyocyte[Title/Abstract] OR myocardial[Title/Abstract]).
In this review, we briefly introduce the latest technological advances in single-cell omics and summarize recent progress in cardiovascular research that has applied these technologies, with special focus on development and homeostasis of the heart, myocardial infarction (MI) and ischemia, and heart failure (HF). We also discuss how single-cell omics can, and should support future cardiovascular research and contribute to precision cardiovascular medicine.
Technological Advances
The experimental pipelines of single-cell omics, represented by scRNA-seq, typically begin with single-cell isolation and capture, followed by sequencing and downstream analyses (6). In the early days, integrated fluidic circuit (IFC) systems, such as Fluidigm C1, were used to isolate cells into individual chambers, where they were then lysed and reverse-transcribed (plate-based method). The IFC system has been largely replaced by fluorescence-activated cell sorting (FACS), which can not only isolate cells, but also distinguish live cells through viability stains. A major drawback of plate-based systems, however, is the limited number of cells that can be analyzed. To overcome this, droplet-based methods were developed, in which single cells are encapsulated in droplets and then undergo barcoding and reverse transcription. This technology has greatly improved throughput, increasing the number of cells analyzed to up to 10,000 cells, albeit with a sacrifice in read depth.
Both IFC systems used in plate-based methods and microfluidic devices used in droplet-based methods limit the size of the cells that can be processed to < ~40 μm, which are applicable to non-cardiomyocytes, neonate cardiomyocytes (CMs), and pluripotent stem cell-derived CMs. However, adult CMs (>100 μm in length) cannot be processed by these methods. Moreover, commercially available FACS nozzles (70–130 μm in diameter) might induce terminal damage to live CMs and so are not suitable either (6, 7). Although successful isolation of viable single CMs using large-particle FACS with a nozzle of 500 μm in diameter has recently been reported (8), most published studies applying scRNA-seq in CMs have relied on either manual pick-up of CMs combined with plate-based methods (9) or the iCELL8 system, a large nozzle dispenser with a 5,184-nanowell plate and an imaging system to differentiate live cells from dead ones (10). An alternative solution is to isolate and sequence the single nuclei of CMs (11), which are smaller and can pass through FACS and droplet-based systems. Single-nucleus RNA sequencing (snRNA-seq), although is applicable in archived frozen specimens and can minimize the alteration of gene expression that may be caused by dissociation (12), should be used and its results interpreted with awareness of how it differs from scRNA-seq. A comparison of snRNA-seq and scRNA-seq showed that they both demonstrated similar cell clusters in the process of induced pluripotent stem cell (iPSC) differentiation to CMs. However, scRNA-seq captures more genes than snRNA-seq, represented by mitochondrial and ribosomal genes, while snRNA-seq detects more non-coding RNA and more reads that are mapped to intronic regions (11, 13). Importantly, the multinucleation and polyploidy of CMs also contribute to the divergence of snRNA-seq from scRNA-seq when applied to CMs, thus should be taken into consideration (14).
Besides the advances made in single-cell transcriptomics, its integration with single-cell epigenomics, namely, scATAC-seq, provides more information on both cell clustering and interactions between gene expression and open chromatin states (15, 16). Furthermore, spatial transcriptomics (i.e., technologies that recover transcriptomic information from tissue while preserving its spatial localization) is undergoing exponential development. Spatial transcriptomics uses either multiplexed RNA imaging [e.g., sequential fluorescence in situ hybridization (seqFISH)] or spatial barcoding (e.g., Visium, slide-seq) (5). Because the structure of the heart is complex and its tissues are highly heterogeneous, the addition of spatial information substantially improves our understanding of tissue architectures. It is also better able to uncover intercellular communications, thereby boosting the exploration of the underlying molecular pathways in the developing, normal, and diseased heart (5, 17).
In the following sections of this review, we focus on recent cardiovascular research that has applied single-cell omics technologies.
Development of The Heart
Early scRNA-seq studies of the heart mainly focused on its development because fetal and neonatal cardiac tissues are easier to digest and their cells, including CMs, are smaller and so can pass through various single-cell platforms. Several such studies are highlighted in Table 1.
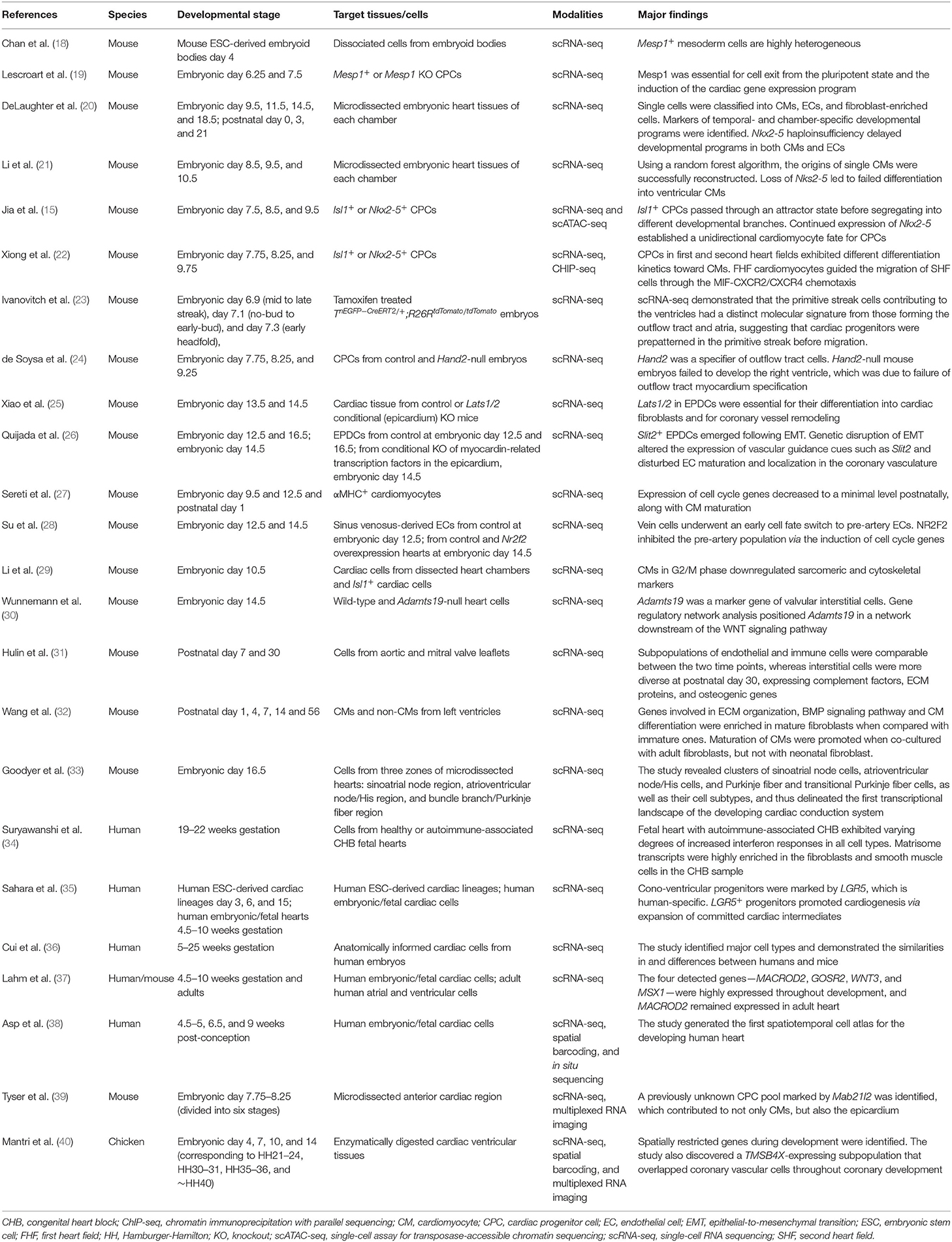
Table 1. Summary of studies of heart development.
It had been unclear whether Mesp1+ mesoderm cells were intrinsically heterogeneous or were homogeneous but capable of multiple lineage decisions. Taking advantage of scRNA-seq, Chan et al. unraveled the heterogeneity of Mesp1+ mesoderm cells from mouse embryonic stem cell (ESC)-derived embryoid bodies (18). Lescroart et al., by examining Mesp1+ cardiac progenitor cells (CPCs) as well as Mesp1-expressing cells in the Mesp1 knockout context, found that Mesp1 was essential for exit from the pluripotent state and for induction of the cardiac gene expression program (19).
Efforts to establish a transcriptomic atlas of the developing heart have been undertaken by multiple teams. By investigating >1,200 cells from murine heart chambers of different developmental stages, DeLaughter et al. not only successfully clustered CMs, endothelial cells (ECs), and fibroblast-enriched cells, but also identified markers of temporal- and chamber-specific developmental programs (e.g., genes involved in cell cycle at embryonic day 9.5–11.5 and genes involved in cellular growth and CM differentiation between embryonic day 14.5 and postnatal day 3) (20). Li et al. used a similar approach together with a random forest algorithm to reconstruct the spatial origins of single CMs (21). Both studies demonstrated that Nkx2-5 deficiency led to failure of CM maturation (20, 21). Jia et al. integrated scRNA-seq and scATAC-seq in Nkx2−5+ and Isl1+ CPCs, enabling developmental trajectories to be reconstructed and revealing that Isl1+ CPCs passed through an attractor state before segregating into different developmental branches, whereas continued expression of Nkx2-5 established a unidirectional CM fate for CPCs (15). A similar analysis of Nkx2−5+ and Isl1+ CPCs found interactions between first and second heart field CPCs, with second heart field CPCs attracted to the first heart field-populated heart tube region through MIF (macrophage migration inhibitory factor)-CXCR2/CXCR4 chemotaxis, contributing to the growth of the outflow tract and right ventricle (22). Furthermore, using genetic lineage tracing and live imaging, Ivanovitch et al. discovered the temporal order in which different cardiac lineages arose within the primitive streak: the progenitors of the left ventricle at the mid-streak stage, those of the right ventricle at the late-streak stage, and those of the outflow tract and atria at the no-bud to early-bud stage. More importantly, scRNA-seq demonstrated distinct molecular signatures of these subpopulations of primitive streak cells, indicating their prepatterned fate in the primitive streak before migration (23). Using a Boolean network-based lineage-specifier prediction method in the downstream analysis of scRNA-seq of mouse embryos as an alternative approach, de Soysa et al. identified Hand2 as a specifier of outflow tract cells. Hand2-null mouse embryos failed to specify outflow tract myocardium, whereas the right ventricle myocardium was specified but not properly differentiated and migrated, which was associated with disrupted anterior–posterior patterning of CPCs (24).
With a focus on epicardium-derived cells (EPDCs), Xiao et al. demonstrated that epicardium conditional knockout of Lats1/2, encoding LATS1 and LATS2 that are negative regulators in the Hippo signaling pathway, caused arrested differentiation into fibroblasts and defective coronary vasculature remodeling (25). Quijada et al. also investigated epicardial contributions to the growing coronary plexus by applying scRNA-seq to EPDCs and uncovered a Slit2+ subpopulation that emerged following epithelial-to-mesenchymal transition (EMT). Genetic disruption of EMT altered the expression of vascular guidance cues such as Slit2 and disturbed EC maturation and localization in the coronary vasculature (26). Su et al. analyzed sinus venosus-derived ECs, discovered that the vein cells underwent an early cell fate switch to pre-artery ECs, and identified the transcription factor NR2F2 as a vein specifier, whose overexpression prevented differentiation to the pre-artery population via the induction of cell cycle genes (28). By performing a genome-wide association study (GWAS) in humans, Wunnemann et al. detected mutations in ADAMTS19 that are responsible for valvular diseases. They then applied scRNA-seq to wild-type and Adamts19-null murine heart at embryonic day 14.5 and identified Admats19 as a marker of valve interstitial cells. Gene regulatory network analysis helped to identify the Wnt-Adamts19-Klf2 axis as being essential for valve development (30). Another study of murine postnatal valve leaflets demonstrated that, whereas subpopulations of ECs and immune cells were comparable between day 7 and 30, interstitial cells were more diverse at postnatal day 30, expressing complement factors, extracellular matrix proteins, and osteogenic genes (31). Wang et al. analyzed the dynamic change of subtypes of non-CMs along with postnatal CM maturation in murine hearts. scRNA-seq of CMs and non-CMs of murine hearts at postnatal day 1, 4, 7, 14, and 56 showed that fibroblasts increasingly expressed maturation promoting genes such as Dcn. Co-culture experiments demonstrated that maturation of CMs was promoted when co-cultured with adult fibroblasts, but not with neonatal fibroblasts. Their results indicated fibroblasts subtype switching as a crucial microenvironmental event regulating postnatal CM maturation (32).
Development of the conduction system has also attracted researchers' interest. Goodyer et al. performed scRNA-seq on cells from murine embryonic heart regions including the conduction system. The study revealed clusters of sinoatrial node cells, atrioventricular node/His cells, and Purkinje fiber and transitional Purkinje fiber cells, thus delineated the first transcriptional landscape of the developing cardiac conduction system (33). Suryawanshi et al. investigated single-cell transcriptomes from healthy human fetal hearts and a fetal heart with autoimmune-associated congenital heart block and showed, through a comparison with normal fetal hearts, that the fetal heart with autoimmune-associated congenital heart block exhibited increased interferon responses of varying degrees in all cell types. Additionally, matrisome transcripts were highly enriched in the fibroblasts and smooth muscle cells in the congenital heart block sample compared with healthy hearts, indicating their contribution to fibrosis (34).
Other studies of human heart development, although rare, have also been conducted to unravel the human-specific features. Cui et al. identified cell types from human fetal hearts and compared their gene expression profiles with those of mice (36). A study of human ESC-derived cardiac derivates and human embryonic/fetal hearts revealed a human-specific cono-ventricular progenitor population marked by LGR5. LGR5 (leucine rich repeat containing G protein-coupled receptor 5) is a receptor for R-spondins, which is involved in the WNT signaling pathway. LGR5+ progenitors were shown to promote cardiogenesis via expansion of committed cardiac intermediates (35). Lahm et al. identified four congenital heart disease-associated genes—MACROD2, GOSR2, WNT3, and MSX1—by GWAS and explored their expression in embryonic and adult human heart by scRNA-seq. They found that MACROD2, GOSR2, WNT3, and MSX1 were highly expressed throughout development and that MACROD2 expression persisted in adult heart (37).
More recent research implemented multiple advanced technologies along with scRNA-seq. Asp et al. generated the first spatiotemporal cell atlas for human developing heart by integrating scRNA-seq, spatial barcoding, and multiplexed RNA imaging (38). Tyser et al., by combining scRNA-seq with genetic lineage labeling and abundant imaging modalities including multiplexed RNA in situ hybridization in murine embryonic hearts, identified and located the CPC subpopulations. Via this spatial transcriptomic approach, they unraveled a previously unknown progenitor subpopulation marked by Mab21l2 that located at the rostral border of the cardiac crescent and contributed to both CMs and epicardium. Mab21l2 (Mab-21 like 2) was previously known to be required for neural and eye development, was here implied to represent the earliest progenitors of the epicardium (39). By integrating scRNA-seq and spatial transcriptomics in developing chicken hearts, Mantri et al. identified spatially restricted genes in cardiac tissue during development. They found that TBX5 was enriched in the left ventricle compared with the right but decreased from embryonic day 7 to 14 and that CHGB expression was restricted to the right ventricle from day 7 onward. In addition, they discovered a TMSB4X-expressing subpopulation that overlapped with coronary vascular cells throughout coronary development. TMSB4 (thymosin β4) participates in regulating actin polymerization, thus is involved in cell proliferation, migration, and differentiation. Its role in heart development had long been under significant debate, given the paradoxical results of several previous Tmsbx knockout and knockdown experiments in mouse models, and was provided deeper insights into by this study (40). Undoubtedly, the latest technological advances, represented by spatial transcriptomics, are furthering our understanding of the molecular mechanisms of cardiac development.
Homeostasis of The Adult Heart
Single-cell omics approaches have also been implemented in studies of adult mice and humans in homeostasis (Table 2), with more studies focusing on non-CMs than on CMs. Yekelchyk et al. performed scRNA-seq on murine CMs using the iCELL8 platform, finding that gene expression was homogeneous between mono- and multi-nucleated CMs in heathy ventricles (41). By performing scRNA-seq on non-CMs of murine heart, Skelly et al. detected major cell types and transcriptional heterogeneity, revealed communications between cell types, and showed cell type-specific sexual dimorphism of cardiac gene expression, such as male upregulation of Irf8 (interferon regulatory factor 8) and female upregulation of Tsc22d3 (TSC22 domain family member 3, a transcription factor implicated in anti-inflammatory functions) in macrophages. Their results provided molecular cues for the sexual difference of cardiac responses to the environmental insults (42). Chakarov et al. identified two distinct interstitial macrophage populations occupying different niches, which were conserved across lung, heart, fat, and dermis tissues: Lyve1lowMHCIIhighCX3CR1high and Lyve1highMHCIIlowCX3CR1low monocyte-derived interstitial macrophages. They also demonstrated that depletion of Lyve1highMHCIIlowCX3CR1low interstitial macrophages exacerbated lung and heart fibrosis (43), which was highly consistent with the results obtained using a mouse MI model (55). Macrophages also facilitate conduction in the atrioventricular node. Genes involved in cardiac conduction are enriched in macrophages from the atrioventricular node when compared with those from other tissues. Atrioventricular node macrophages highly express Cx43, contributing to the gap junctions that link them to neighboring CMs (44). In regard to the conduction system, quantitative proteomics of the murine sinus node demonstrated a remarkable abundance of ion channels responsible for the pacemaking process (e.g., HCN4), which were indicated by snRNA-seq to be predominantly expressed by sinus node myocytes (45). Liang et al. then verified the existence of a cell cluster in the sinus node marked by Hcn1 and Hcn4, where Vsnl1 was also highly expressed. VSNL1 (visinin-like 1) is a member of visinin/recoverin subfamily of neuronal calcium sensor proteins, and is involved in calcium signaling pathways. Adeno-associated virus-mediated knockdown of Vsnl1 in mice resulted in a reduced heart rate and decreased expression of Hcn4, Cacna1d, Cacna1i, and Serca2a, but not that of Ryr2 or of the genes involved in potassium channels (46).
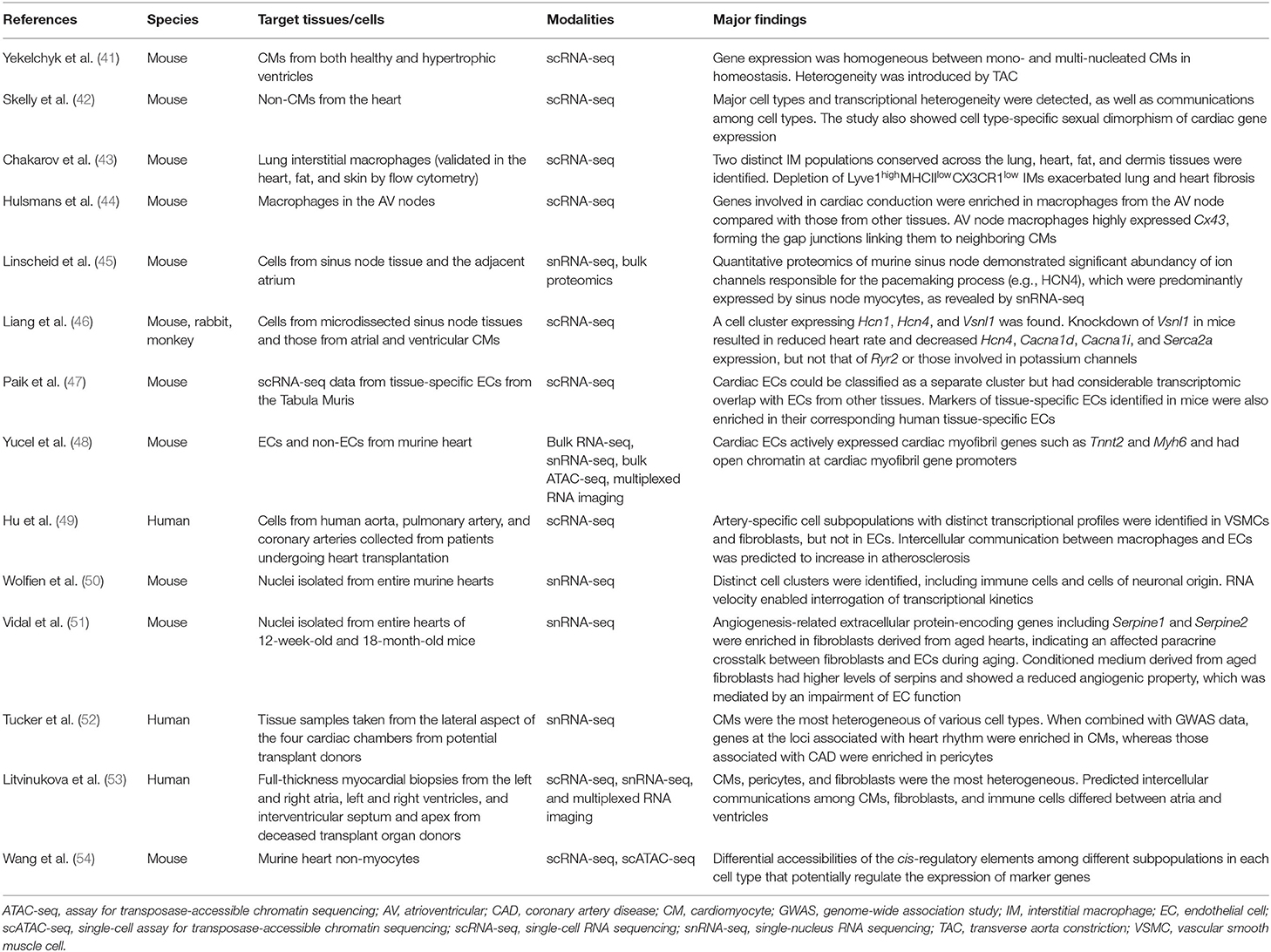
Table 2. Summary of studies of homeostasis of the adult heart.
Cardiac ECs are another research interest. Paik et al. re-explored the scRNA-seq data from tissue-specific ECs in the Tabula Muris (4) and demonstrated that cardiac ECs could be classified as a separate cluster, even though they had considerable transcriptomic overlap with ECs from other tissues (47). Yucel et al. unexpectedly observed that adult murine cardiac ECs actively expressed cardiac myofibril genes such as Tnnt2 and Myh6, which was consistent with the analytical results of multiple datasets; this result was validated by RNA in situ hybridization. They also showed that cardiac ECs had open chromatins at cardiac myofibril gene promoters, suggesting that the results were not due to technical contamination or paracrine transfer of mRNA. Interestingly, the accessibility of cardiac myofibril genes was no longer maintained in cardiac ECs upon culture. Although the significance of the expression of cardiac myofibril genes in cardiac ECs is still unclear, the authors hypothesized that those genes played a role in maintaining cardiac EC maturity and specificity, which needs further investigation (48). Hu et al. used scRNA-seq to analyze cells from human aorta, pulmonary artery, and coronary arteries collected from patients who underwent heart transplantation. Artery-specific cell subpopulations with distinct transcriptional profiles were identified in vascular smooth muscle cells and fibroblasts, but not in ECs (49).
Recently, efforts have focused on depicting the full picture of the cell atlas of the whole heart. snRNA-seq of murine hearts successfully clustered distinct cell subpopulations, including immune cells, cells of neuronal origin, and cardiac glial cells (50). With snRNA-seq, Vidal et al. investigated the transcriptomes of murine hearts of 12-week-old and 18-month-old, and found upregulated angiogenesis-related extracellular protein-encoding genes in aged hearts, indicating an affected paracrine crosstalk between fibroblasts and ECs during aging. In vitro experiments confirmed that conditioned medium derived from aged fibroblasts showed a reduced angiogenic property, which was mediated by an impairment of EC function (51). In humans, samples of myocardium from cardiac chambers, rather than of the whole heart, have been studied. By applying snRNA-seq to biopsied human cardiac chambers, Tucker et al. showed that CMs were the most spatially heterogeneous of the distinct cell types. Furthermore, by combining the snRNA-seq results with GWAS data for cardiometabolic traits, they revealed that genes at the loci associated with heart rhythm were enriched in CMs, whereas those at the loci associated with coronary artery disease were enriched in pericytes (52). Litvinukova et al. investigated full-thickness biopsy samples from deceased transplant donors and were able to provide more in-depth information, predicting different intercellular communications among immune cells, CMs, and fibroblasts between ventricles and atria (53). Wang et al. integrated scRNA-seq and scATAC-seq for non-CMs in murine hearts and revealed the heterogeneity of non-CM populations and their subpopulations at transcriptomic and epigenomic levels. Cardiac fibroblasts were classified into three distinct functional states related to response to stimuli (marked by Hsd11b1; 11β-hydroxysteroid dehydrogenase type 1), the cytoskeleton (marked by Gfpt2; glutaminefructose-6-phosphate transaminase 2, involved in the hexosamine biosynthesis pathway), and immune regulation (marked by C1qa), respectively. Importantly, they also identified differential accessibilities of the cis-regulatory elements among different subpopulations of each cell type that potentially regulate the expression of marker genes (54). The success of these studies indicates that single-cell omics represents an indispensable toolbox for broadening our insights into the homeostasis of the adult heart, particularly the heterogeneity of cells and their functions, as well as intercellular communications.
Myocardial Infarction/Ischemia-Reperfusion Injury
In addition to development and adult homeostasis, single-cell omics approaches have played a crucial role in research into diseased hearts (Table 3). MI, as a consequence of atherosclerotic disease, may have a marked effect on cardiac function and quality of life. The toolbox from single-cell omics technologies has enabled detailed investigation of not only the cells involved in the process of plaque formation and rupture, but also the ischemic myocardium that is susceptible to remodeling (71).
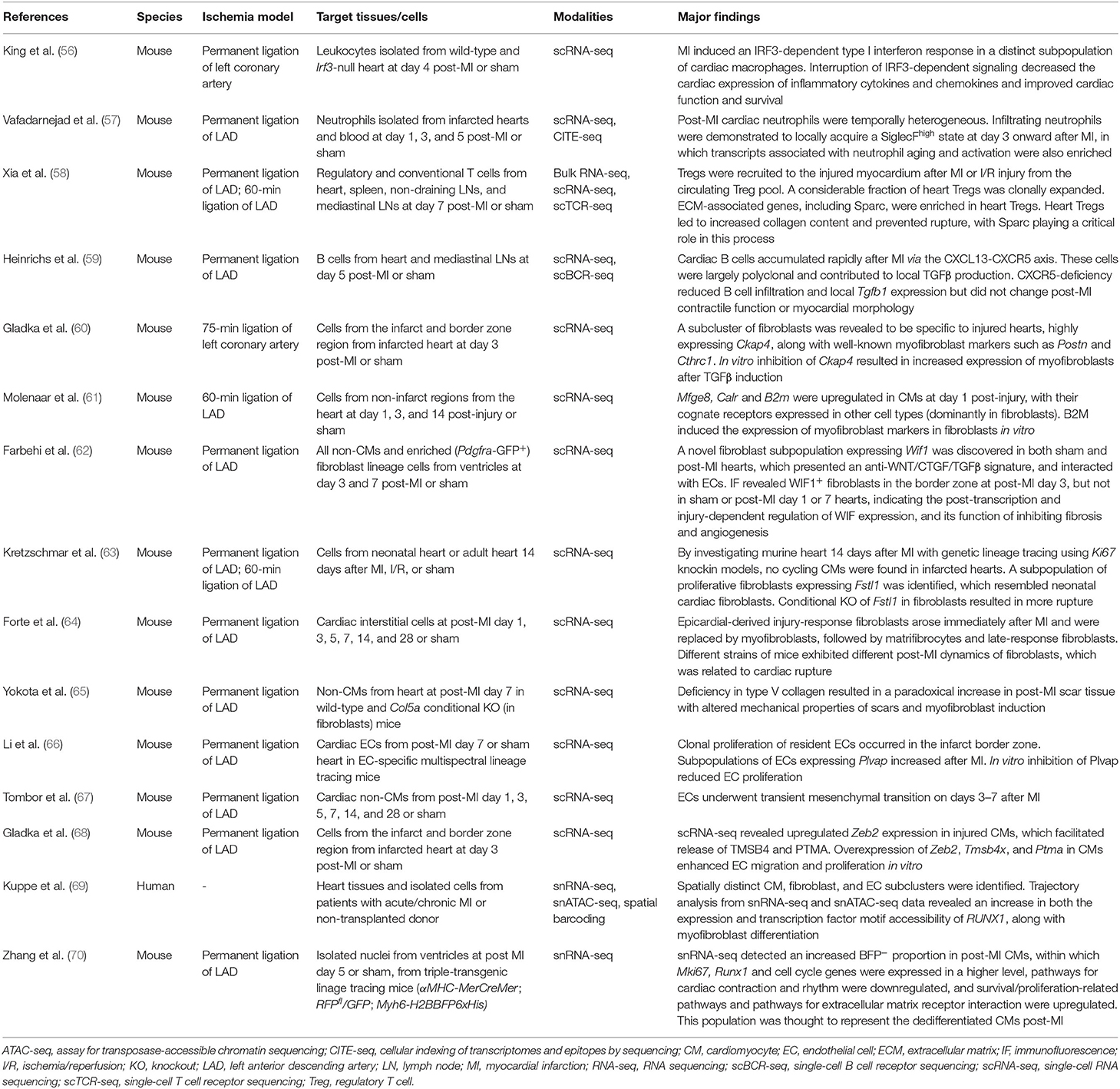
Table 3. Summary of studies of myocardial infarction or ischemia-reperfusion injury.
MI has long been known to involve both pro- and anti-inflammatory cascades. King et al. were the first to apply scRNA-seq to leukocytes isolated from infarcted and non-infarcted murine hearts, finding that MI induced an IRF3-dependent type I interferon response in a distinct subpopulation of cardiac macrophages and that interruption of IRF3-dependent signaling decreased the cardiac expression of inflammatory cytokines and chemokines and improved cardiac function (56). Cardiac-infiltrating neutrophils were demonstrated to locally acquire a SiglecFhigh ICAMhighCXCR4high state from day 3 after MI, indicating local neutrophil activation and aging. Similar neutrophil subpopulation was also found in atherosclerotic aortas, suggesting their roles both in acute and chronic cardiovascular inflammation (57). Regulatory T cells (Tregs) accumulated in the injured myocardium after MI or ischemia-reperfusion injury. The majority of these heart Tregs were derived from the circulating Treg pool; a considerable fraction of them experienced clonal expansion and displayed a unique T-cell receptor repertoire. Importantly, extracellular matrix-associated genes, including Sparc, were enriched in heart Tregs compared with other Tregs. Further experiments revealed that heart Tregs contributed to increased collagen content and prevented rupture and that Sparc was critical in this process (58). After MI, cardiac B cells also accumulated rapidly via the CXCL13-CXCR5 axis but were largely polyclonal and contributed to local TGFβ production, indicating their involvement in fibroblast activation (59).
Fibroblasts are directly involved in post-infarct scar formation. Gladka et al. performed scRNA-seq on cells sorted by FACS with a 130-μm nozzle from infarcted and control murine hearts and were able to identify distinct clusters of CMs, fibroblasts, ECs, and macrophages. A subset of activated fibroblasts was revealed to be specific to injured hearts and to strongly express Ckap4. CKAP4 (cytoskeleton associated protein 4) is a transmembrane receptor whose function in cardiac fibroblasts had remained unknown. In vitro inhibition of Ckap4 resulted in increased expression of genes related to activated fibroblasts after TGFβ induction, suggesting a modulating function for CKAP4 in fibroblast activation (60). Molenaar et al. focused on the ligands upregulated in CMs 1 day after ischemia, whose cognate receptors were expressed by another cell type, and detected B2m (β2 microglobulin) in CMs and its receptors in fibroblasts. In vitro experiments demonstrated that fibroblasts expressed myofibroblast markers when cultured with B2M (61). Farbehi et al. discovered a novel fibroblast subpopulation with upregulated Wif1 expression, in sham hearts, which persisted after MI, had an anti-WNT/CTGF/TGFβ signature, and interacted with ECs. Immunofluorescence revealed WIF1+ fibroblasts in close proximity with ECs in the border zone at post-MI day 3, but not in sham or post-MI day 1 or 7 hearts, indicating the injury-dependent and post-transcriptional regulation of WIF expression, and its function of inhibiting fibrosis and angiogenesis in post-MI repair (62). By investigating the murine heart 14 days after MI, Kretzschmar et al. identified a subpopulation of proliferative fibroblasts resembling neonatal cardiac fibroblasts, which presented with upregulated Fstl1 (follistatin-like 1, which had been known to be expressed in adult cardiac fibroblasts). Conditional knockout of Fstl1 in fibroblasts induced less proliferative cells at the infarct and border zones and lead to more cardiac ruptures, indicating the importance of autocrine FSTL1 signaling in preventing rupture (63). Longitudinal scRNA-seq analysis of murine heart interstitial cells from post-MI day 1 to 28 uncovered a dynamic shift in the subpopulations. In particular, epicardial-derived injury-response fibroblasts arose immediately after MI and were replaced by myofibroblasts, followed by matrifibrocytes and late-response fibroblasts. Different strains of mice exhibited different post-MI dynamics of fibroblasts, which was related to the risk of cardiac rupture (64). Yokota et al. demonstrated that a deficiency in type V collagen resulted in a paradoxical increase of scar tissue, where mechanical properties of scars were altered, and myofibroblasts were induced (65).
ECs are involved in angiogenesis after MI and have thus been another target of single-cell studies. Li et al. discovered that clonal proliferation of resident ECs occurred in the infarct border zone, in which Plvap was enriched. PLVAP (plasmalemma vesicle associated protein) had been known as an EC-specific membrane protein involved in the formation of transendothelial channels and microvascular permeability. In vitro inhibition of Plvap reduced EC proliferation, indicating its involvement in neoangiogenesis (66). Li et al. also observed transient endothelial mesenchymal activation, but the extent to which was comparable between post-MI day 7 and sham. By contrast, Tombor et al. demonstrated that ECs underwent transient mesenchymal activation on days 1 to 7 after MI, which returned to homeostasis afterwards, indicating its involvement in facilitating regeneration of the vascular network via EC migration and clonal expansion (67). A crosstalk between injured CMs and ECs has also been identified: transcription factor Zeb2 expression was upregulated in injured CMs, which facilitated the release of TMSB4 and PTMA (prothymosin α). Overexpression and knockdown experiments of Zeb2, Tmsb4x, and Ptma in iPSC-derived CMs clearly demonstrated their involvement in enhancing EC proliferation and migration (68).
Kuppe et al. integrated snRNA-seq, single-nucleus assay for transposase-accessible chromatin sequencing (snATAC-seq), and spatial transcriptomics in human heart tissues from patients with and without a history of MI. Trajectory analysis from snRNA-seq and snATAC-seq data revealed an increase in both the expression and the transcription factor motif accessibility of RUNX1 in fibroblasts, as well as in their differentiation to myofibroblasts. In vitro experiments then validated that RUNX1 amplified TGFβ signaling and thus mediated myofibroblast differentiation (69). Zhang et al. developed a triple-transgenic mouse model (αMHC-MerCreMer; RFPfl/GFP; Myh6-H2BBFP6xHis, tamoxifen treated) for fate mapping of CMs after MI. They discovered an increase in the rare GFP+BFPlow CMs in post-MI hearts compared to sham hearts that presented with a high BrdU+ incorporation rate, which were thought to represent dedifferentiated CMs. snRNA-seq also detected an increased BFP− proportion in post-MI CMs, within which Mki67, Runx1, and cell cycle genes were expressed in a higher level. They further showed that pathways for cardiac contraction and rhythm were downregulated, and pathways for survival/proliferation and extracellular matrix receptor interaction were upregulated (70).
MI is not a local event but rather a spatiotemporally heterogeneous state of disease involving the entire heart. The conventional approach for increasing the spatial resolution in earlier studies was dissecting infarct hearts into ischemic zone, border zone, and remote zone. To this end, spatial transcriptomics adds a continuous spatial resolution to the transcriptomic information, which is being used in ongoing research into MI.
Heart Failure and Miscellaneous
HF presents with various underlying causes and culminates in cardiac dysfunction and pump insufficiency, with the most often used experimental animal model of HF being transverse aorta constriction (TAC)-induced pressure overload in mice (Table 4). See et al. investigated single nuclei from CMs in the mouse pressure overload model, in which CMs accurately segregated into clusters specific to sham or TAC groups. Weighted gene co-expression network analysis (WGCNA) revealed a disease module in which the long non-coding RNAs (lncRNAs) Gas5 and Sghrt were identified as key nodal regulators and were correlated with cell cycle genes and the fetal reprogramming marker Nppa, providing early evidence for lncRNA-regulated state alteration in stress-response CMs (72). Nomura et al. investigated CMs at various time points after TAC and revealed that activated p53 signaling in the late stage of hypertrophy facilitated the HF gene program (73). They also analyzed single-cardiomyocyte transcriptomes from patients with HF and confirmed the conservation of pathological gene programs. Ren et al. used the iCELL8 platform for scRNA-seq of both CMs and non-CMs in murine hearts after TAC and revealed that fibroblasts underwent a subtype switching away from protective features at the initial stage of hypertrophy. The study also showed that the macrophages switched toward a pro-inflammatory state and that their considerable interaction with CMs was associated with deterioration of cardiac function (74). With the iCELL8 platform, Wang et al. constructed a comprehensive resource of single-cell transcriptomes of both CMs and non-CMs from normal, failed, and recovered human heart biopsy samples. In addition to revealing the inter- and intra-compartmental CM heterogeneity, a major finding of this study was that ACKR1+ ECs, which had a protective function and possessed the highest counts of interactions with other cell types, decreased in failed hearts. This observation was validated by in vivo experiments where injection of ACKR1+ ECs rescued cardiac function after MI by increasing vessel density in both infarct and border regions in mice (10). Zaman et al. demonstrated that cardiac macrophages in the acute and chronic phase after angiotensin II-induced hypertensive stress were enriched in Igf1-containing reparative pathways related to wound healing and angiogenesis and that conditional knockout of Igf1 in tissue-resident macrophages inhibited adaptive CM hypertrophy and led to cardiac dysfunction after long-term hypertensive stress (75). Besides the interaction with CMs, cardiac macrophages have also been reported to activate fibroblasts through paracrine signaling in HF. Ramanujam et al. applied ligand–receptor pair analysis to scRNA-seq data from non-CMs in pressure-overloaded murine hearts by analyzing the transcripts of secreted protein ligands in one cell type and their receptors in another cell type, and predicted that M1 like macrophages interacted with activated fibroblasts via secreted proteins such as TGFβ and fibronectin. These interactions were demonstrated to be mediated by macrophage miR-21, depletion of which rescued hypertrophic phenotype after TAC (76). Martini et al., by sequencing cardiac CD45+ cells, showed increases in Osm+ pro-inflammatory macrophages, as well as Pdcd1+ Tregs, in pressure-overloaded murine hearts when compared with healthy hearts (77).
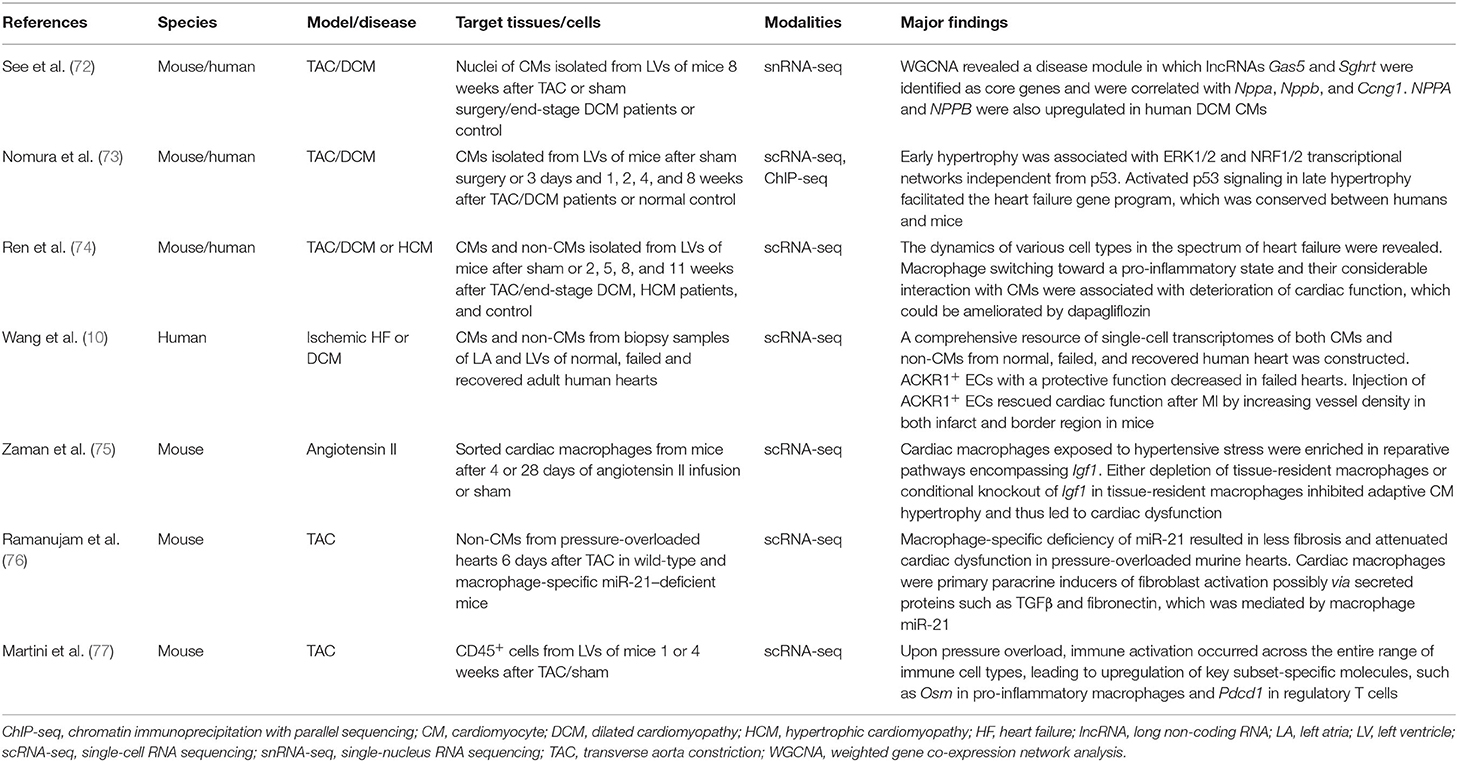
Table 4. Summary of studies of heart failure.
Single-cell omics approaches have also played important roles in recent studies investigating SARS-CoV-2–associated cardiac injury. ACE2 (the cellular receptor for virus spike protein) is expressed in CMs and pericytes and is upregulated at both mRNA and protein levels in failing hearts, indicating a vulnerability of these cell states to infection (78, 79). By combining phosphoproteomics and snRNA-seq, Mills et al. reported that the inflammatory response in human cardiac organoids was mediated by STAT1 and epigenetic activation, including BRD4 (bromodomain containing 4, a member of bromodomain and extraterminal protein family). They then validated the results in the hearts of SARS-CoV-2–infected K18-hACE2 mice. Bromodomain and extraterminal family inhibitors were identified as drug candidates that could prevent COVID-19–mediated cardiac damage (80).
Future Perspectives
Single-cell omics technologies are advancing rapidly and are being applied to a wide range of research subjects. Meanwhile, enabled by progress in bioinformatic analysis, the integration of multi-omics data, as well as spatial transcriptomics, is becoming more common and is providing us with more comprehensive information. These technologies offer deeper insights into the molecular pathways involved in heart development, homeostasis, and specifically in cardiovascular diseases and are thus helping to identify therapeutic candidates.
Besides elucidating pathophysiology, single-cell omics can also contribute to precision medicine. The response to treatment in cardiovascular diseases is non-uniform, as in other organ systems, and this might be associated with interindividual differences in not only genetic backgrounds but also cell populations and microenvironment. Single-cell omics enables assessment of cell populations, functional states, and intercellular communications and thus can enhance patient-specific understanding of diseases alongside the use of existing diagnostic tools. Kim et al. exemplified how single-cell analysis can facilitate a personalized therapeutic approach in a patient with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms (81). Such an application to cardiovascular diseases is expected in the near future.
Author Contributions
ZD: writing—original draft preparation. SN: writing—review and editing, supervision, and funding acquisition. Both authors have approved the submitted version and agreed to be accountable for the content of the work.
Funding
This work was supported by grants from a Grant-in-Aid for Young Scientists (to SN), the Japan Foundation for Applied Enzymology (to SN), the SENSHIN Medical Research Foundation (to SN), the Kanae Foundation for the Promotion of Medical Science (to SN), MSD Life Science Foundation (to SN), the Tokyo Biomedical Research Foundation (to SN), Astellas Foundation for Research on Metabolic Disorders (to SN), the Novartis Foundation (Japan) for the Promotion of Science (to SN), the Japanese Circulation Society (to SN), the Takeda Science Foundation (to SN), the Cell Science Research Foundation (to SN), a Grant-in-Aid for Scientific Research (B) (to SN), and AMED (JP21ek0210152, JP21gm6210010, JP20bm0704026, JP21ek0210141, JP21ek0109440, JP21ek0109487, JP21gm0810013, JP21km0405209, JP21ek0210118, JP21ek0109406, JP21ek0109543, JP21ek0109569, and JP21tm0724601).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. (2015) 523:486–90. doi: 10.1038/nature14590
2. Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. (2014) 11:163–6. doi: 10.1038/nmeth.2772
3. Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The human cell atlas. Elife. (2017) 6:e27041. doi: 10.7554/eLife.27041
4. Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. (2018) 562:367–72. doi: 10.1038/s41586-018-0590-4
5. Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet. (2021) 22:627–44. doi: 10.1038/s41576-021-00370-8
6. Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. (2020) 17:457–73. doi: 10.1038/s41569-020-0359-y
7. Yamada S, Nomura S. Review of single-cell RNA sequencing in the heart. Int J Mol Sci. (2020) 21:218345. doi: 10.3390/ijms21218345
8. Kannan S, Miyamoto M, Lin BL, Zhu R, Murphy S, Kass DA, et al. Large particle fluorescence-activated cell sorting enables high-quality single-cell RNA sequencing and functional analysis of adult cardiomyocytes. Circ Res. (2019) 125:567–9. doi: 10.1161/CIRCRESAHA.119.315493
9. Katoh M, Nomura S, Yamada S, Aburatani H, Komuro I. Single-cardiomyocyte RNA sequencing to dissect the molecular pathophysiology of the heart. Methods Mol Biol. (2021) 2320:183–92. doi: 10.1007/978-1-0716-1484-6_18
10. Wang L, Yu P, Zhou B, Song J, Li Z, Zhang M, et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. (2020) 22:108–19. doi: 10.1038/s41556-019-0446-7
11. Selewa A, Dohn R, Eckart H, Lozano S, Xie B, Gauchat E, et al. Systematic comparison of high-throughput single-cell and single-nucleus transcriptomes during cardiomyocyte differentiation. Sci Rep. (2020) 10:1535. doi: 10.1038/s41598-020-58327-6
12. Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun. (2016) 7:11022. doi: 10.1038/ncomms11022
13. Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE. (2018) 13:e0209648. doi: 10.1371/journal.pone.0209648
14. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell. (2015) 161:1566–75. doi: 10.1016/j.cell.2015.05.026
15. Jia G, Preussner J, Chen X, Guenther S, Yuan X, Yekelchyk M, et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat Commun. (2018) 9:4877. doi: 10.1038/s41467-018-07307-6
16. Wang Z, Cui M, Shah AM, Tan W, Liu N, Bassel-Duby R, et al. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. (2020) 33:108472. doi: 10.1016/j.celrep.2020.108472
17. Rao A, Barkley D, Franca GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. (2021) 596:211–20. doi: 10.1038/s41586-021-03634-9
18. Chan SS, Chan HHW, Kyba M. Heterogeneity of Mesp1+ mesoderm revealed by single-cell RNA-seq. Biochem Biophys Res Commun. (2016) 474:469–75. doi: 10.1016/j.bbrc.2016.04.139
19. Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sanchez-Danes A, et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. (2018) 359:1177–81. doi: 10.1126/science.aao4174
20. DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, et al. Single-cell resolution of temporal gene expression during heart development. Dev Cell. (2016) 39:480–90. doi: 10.1016/j.devcel.2016.10.001
21. Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell. (2016) 39:491–507. doi: 10.1016/j.devcel.2016.10.014
22. Xiong H, Luo Y, Yue Y, Zhang J, Ai S, Li X, et al. Single-cell transcriptomics reveals chemotaxis-mediated intraorgan crosstalk during cardiogenesis. Circ Res. (2019) 125:398–410. doi: 10.1161/CIRCRESAHA.119.315243
23. Ivanovitch K, Soro-Barrio P, Chakravarty P, Jones RA, Bell DM, Mousavy Gharavy SN, et al. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. (2021) 19:e3001200. doi: 10.1371/journal.pbio.3001200
24. de Soysa TY, Ranade SS, Okawa S, Ravichandran S, Huang Y, Salunga HT, et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature. (2019) 572:120–4. doi: 10.1038/s41586-019-1414-x
25. Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Wang S, et al. Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev Cell. (2018) 45:153–69 e6. doi: 10.1016/j.devcel.2018.03.019
26. Quijada P, Trembley MA, Misra A, Myers JA, Baker CD, Perez-Hernandez M, et al. Coordination of endothelial cell positioning and fate specification by the epicardium. Nat Commun. (2021) 12:4155. doi: 10.1038/s41467-021-24414-z
27. Sereti KI, Nguyen NB, Kamran P, Zhao P, Ranjbarvaziri S, Park S, et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat Commun. (2018) 9:754. doi: 10.1038/s41467-018-02891-z
28. Su T, Stanley G, Sinha R, D'Amato G, Das S, Rhee S, et al. Single-cell analysis of early progenitor cells that build coronary arteries. Nature. (2018) 559:356–62. doi: 10.1038/s41586-018-0288-7
29. Li G, Tian L, Goodyer W, Kort EJ, Buikema JW, Xu A, et al. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development. (2019) 146:173476. doi: 10.1242/dev.173476
30. Wunnemann F, Ta-Shma A, Preuss C, Leclerc S, van Vliet PP, Oneglia A, et al. Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat Genet. (2020) 52:40–7. doi: 10.1038/s41588-019-0536-2
31. Hulin A, Hortells L, Gomez-Stallons MV, O'Donnell A, Chetal K, Adam M, et al. Maturation of heart valve cell populations during postnatal remodeling. Development. (2019) 146:173047. doi: 10.1242/dev.173047
32. Wang Y, Yao F, Wang L, Li Z, Ren Z, Li D, et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat Commun. (2020) 11:2585. doi: 10.1038/s41467-020-16204-w
33. Goodyer WR, Beyersdorf BM, Paik DT, Tian L, Li G, Buikema JW, et al. Transcriptomic profiling of the developing cardiac conduction system at single-cell resolution. Circ Res. (2019) 125:379–97. doi: 10.1161/CIRCRESAHA.118.314578
34. Suryawanshi H, Clancy R, Morozov P, Halushka MK, Buyon JP, Tuschl T. Cell atlas of the foetal human heart and implications for autoimmune-mediated congenital heart block. Cardiovasc Res. (2020) 116:1446–57. doi: 10.1093/cvr/cvz257
35. Sahara M, Santoro F, Sohlmer J, Zhou C, Witman N, Leung CY, et al. Population and single-cell analysis of human cardiogenesis reveals unique LGR5 ventricular progenitors in embryonic outflow tract. Dev Cell. (2019) 48:475–90 e7. doi: 10.1016/j.devcel.2019.01.005
36. Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. (2019) 26:1934–50 e5. doi: 10.1016/j.celrep.2019.01.079
37. Lahm H, Jia M, Dressen M, Wirth F, Puluca N, Gilsbach R, et al. Congenital heart disease risk loci identified by genome-wide association study in European patients. J Clin Invest. (2021) 131. doi: 10.1172/JCI141837
38. Asp M, Giacomello S, Larsson L, Wu C, Furth D, Qian X, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. (2019) 179:1647–60 e19. doi: 10.1016/j.cell.2019.11.025
39. Tyser RCV, Ibarra-Soria X, McDole K, Arcot Jayaram S, Godwin J, van den Brand TAH, et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science. (2021) 371:abb2986. doi: 10.1126/science.abb2986
40. Mantri M, Scuderi GJ, Abedini-Nassab R, Wang MFZ, McKellar D, Shi H, et al. Spatiotemporal single-cell RNA sequencing of developing chicken hearts identifies interplay between cellular differentiation and morphogenesis. Nat Commun. (2021) 12:1771. doi: 10.1038/s41467-021-21892-z
41. Yekelchyk M, Guenther S, Preussner J, Braun T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res Cardiol. (2019) 114:36. doi: 10.1007/s00395-019-0744-z
42. Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, et al. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. (2018) 22:600–10. doi: 10.1016/j.celrep.2017.12.072
43. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. (2019) 363:aau0964. doi: 10.1126/science.aau0964
44. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169:510–22 e20. doi: 10.1016/j.cell.2017.03.050
45. Linscheid N, Logantha S, Poulsen PC, Zhang S, Schrolkamp M, Egerod KL, et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat Commun. (2019) 10:2889. doi: 10.1038/s41467-019-10709-9
46. Liang D, Xue J, Geng L, Zhou L, Lv B, Zeng Q, et al. Cellular and molecular landscape of mammalian sinoatrial node revealed by single-cell RNA sequencing. Nat Commun. (2021) 12:287. doi: 10.1038/s41467-020-20448-x
47. Paik DT, Tian L, Williams IM, Rhee S, Zhang H, Liu C, et al. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. (2020) 142:1848–62. doi: 10.1161/CIRCULATIONAHA.119.041433
48. Yucel N, Axsom J, Yang Y, Li L, Rhoades JH, Arany Z. Cardiac endothelial cells maintain open chromatin and expression of cardiomyocyte myofibrillar genes. Elife. (2020) 9:e55730. doi: 10.7554/eLife.55730
49. Hu Z, Liu W, Hua X, Chen X, Chang Y, Hu Y, et al. Single-cell transcriptomic atlas of different human cardiac arteries identifies cell types associated with vascular physiology. Arterioscler Thromb Vasc Biol. (2021) 41:1408–27. doi: 10.1161/ATVBAHA.120.315373
50. Wolfien M, Galow AM, Muller P, Bartsch M, Brunner RM, Goldammer T, et al. Single-nucleus sequencing of an entire mammalian heart: cell type composition and velocity. Cells. (2020) 9:20318. doi: 10.3390/cells9020318
51. Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight. (2019) 4:131092. doi: 10.1172/jci.insight.131092
52. Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, et al. Transcriptional and cellular diversity of the human heart. Circulation. (2020) 142:466–82. doi: 10.1161/CIRCULATIONAHA.119.045401
53. Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. (2020) 588:466–72. doi: 10.1038/s41586-020-2797-4
54. Wang L, Yang Y, Ma H, Xie Y, Xu J, Near D, et al. Single cell dual-omics reveals the transcriptomic and epigenomic diversity of cardiac non-myocytes. Cardiovasc Res. (2021). doi: 10.1093/cvr/cvab134
55. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. (2019) 124:263–78. doi: 10.1161/CIRCRESAHA.118.314028
56. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. (2017) 23:1481–7. doi: 10.1038/nm.4428
57. Vafadarnejad E, Rizzo G, Krampert L, Arampatzi P, Arias-Loza AP, Nazzal Y, et al. Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circ Res. (2020) 127:e232–49. doi: 10.1161/CIRCRESAHA.120.317200
58. Xia N, Lu Y, Gu M, Li N, Liu M, Jiao J, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. (2020) 142:1956–73. doi: 10.1161/CIRCULATIONAHA.120.046789
59. Heinrichs M, Ashour D, Siegel J, Buchner L, Wedekind G, Heinze KG, et al. The healing myocardium mobilises a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism. Cardiovasc Res. (2021). doi: 10.1093/cvr/cvab181
60. Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, et al. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation. (2018) 138:166–80. doi: 10.1161/CIRCULATIONAHA.117.030742
61. Molenaar B, Timmer LT, Droog M, Perini I, Versteeg D, Kooijman L, et al. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun Biol. (2021) 4:146. doi: 10.1038/s42003-020-01636-3
62. Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. (2019) 8:e43882. doi: 10.7554/eLife.43882
63. Kretzschmar K, Post Y, Bannier-Helaouet M, Mattiotti A, Drost J, Basak O, et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci USA. (2018) 115:E12245–54. doi: 10.1073/pnas.1805829115
64. Forte E, Skelly DA, Chen M, Daigle S, Morelli KA, Hon O, et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. (2020) 30:3149–63 e6. doi: 10.1016/j.celrep.2020.02.008
65. Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, et al. Type V collagen in scar tissue regulates the size of scar after heart injury. Cell. (2020) 182:545–62 e23. doi: 10.1016/j.cell.2020.06.030
66. Li Z, Solomonidis EG, Meloni M, Taylor RS, Duffin R, Dobie R, et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur Heart J. (2019) 40:2507–20. doi: 10.1093/eurheartj/ehz305
67. Tombor LS, John D, Glaser SF, Luxan G, Forte E, Furtado M, et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. (2021) 12:681. doi: 10.1038/s41467-021-20905-1
68. Gladka MM, Kohela A, Molenaar B, Versteeg D, Kooijman L, Monshouwer-Kloots J, et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat Commun. (2021) 12:84. doi: 10.1038/s41467-020-20361-3
69. Kuppe C, Ramirez Flores RO Li Z, Hannani M, Tanevski J, Halder M, et al. Spatial multi-omic map of human myocardial infarction. bioRxiv. (2020). doi: 10.1101/2020.12.08.411686
70. Zhang Y, Gago-Lopez N, Li N, Zhang Z, Alver N, Liu Y, et al. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. (2019) 5:30. doi: 10.1038/s41421-019-0095-9
71. van Blokland IV, Groot HE, Franke LH, van der Wijst MGP, van der Harst P. Translational insights from single-cell technologies across the cardiovascular disease continuum. Trends Cardiovasc Med. (2021). doi: 10.1016/j.tcm.2021.02.009
72. See K, Tan WLW, Lim EH, Tiang Z, Lee LT, Li PYQ, et al. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat Commun. (2017) 8:225. doi: 10.1038/s41467-017-00319-8
73. Nomura S, Satoh M, Fujita T, Higo T, Sumida T, Ko T, et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat Commun. (2018) 9:4435. doi: 10.1038/s41467-018-06639-7
74. Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, et al. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation. (2020) 141:1704–19. doi: 10.1161/CIRCULATIONAHA.119.043053
75. Zaman R, Hamidzada H, Kantores C, Wong A, Dick SA, Wang Y, et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity. (2021). doi: 10.1016/j.immuni.2021.07.006
76. Ramanujam D, Schon AP, Beck C, Vaccarello P, Felician G, Dueck A, et al. MicroRNA-21-dependent macrophage-to-fibroblast signaling determines the cardiac response to pressure overload. Circulation. (2021) 143:1513–25. doi: 10.1161/CIRCULATIONAHA.120.050682
77. Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation. (2019) 140:2089–107. doi: 10.1161/CIRCULATIONAHA.119.041694
78. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. (2020) 116:1097–100. doi: 10.1093/cvr/cvaa078
79. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe A, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. (2021) 595:107–13. doi: 10.1038/s41586-021-03570-8
80. Mills RJ, Humphrey SJ, Fortuna PRJ, Lor M, Foster SR, Quaife-Ryan GA, et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell. (2021) 184:2167–82 e22. doi: 10.1016/j.cell.2021.03.026
Keywords: single-cell omics, cardiovascular research, heart development, cardiac homeostasis, myocardial infarction, heart failure
Citation: Dai Z and Nomura S (2021) Recent Progress in Cardiovascular Research Involving Single-Cell Omics Approaches. Front. Cardiovasc. Med. 8:783398. doi: 10.3389/fcvm.2021.783398
Received: 26 September 2021; Accepted: 22 November 2021;
Published: 16 December 2021.
Edited by:
Christoph Dieterich, Heidelberg University, GermanyReviewed by:
Robert Kelly, UMR7288 Institut de Biologie du Développement de Marseille (IBDM), FranceMonika Gladka, Academic Medical Center, Netherlands
Copyright © 2021 Dai and Nomura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seitaro Nomura, c2Vub211cmEtY2liQHVtaW4uYWMuanA=