
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 29 November 2021
Sec. General Cardiovascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.773473
This article is part of the Research Topic Cardiovascular Fibrosis and Related Diseases: Basic and clinical research advances View all 14 articles
 Keman Xu1
Keman Xu1 Ying Shao1
Ying Shao1 Fatma Saaoud1Aria Gillespie2Charles Drummer IV1
Fatma Saaoud1Aria Gillespie2Charles Drummer IV1 Lu Liu3Yifan Lu1
Lu Liu3Yifan Lu1 Yu Sun1Hang Xi3
Yu Sun1Hang Xi3 Çagla Tükel4
Çagla Tükel4 Domenico Pratico5
Domenico Pratico5 Xuebin Qin6
Xuebin Qin6 Jianxin Sun7Eric T. Choi8Xiaohua Jiang1,3
Jianxin Sun7Eric T. Choi8Xiaohua Jiang1,3 Hong Wang3
Hong Wang3 Xiaofeng Yang1,3*
Xiaofeng Yang1,3*To determine whether pro-inflammatory lipid lysophosphatidylinositols (LPIs) upregulate the expressions of membrane proteins for adhesion/signaling and secretory proteins in human aortic endothelial cell (HAEC) activation, we developed an EC biology knowledge-based transcriptomic formula to profile RNA-Seq data panoramically. We made the following primary findings: first, G protein-coupled receptor 55 (GPR55), the LPI receptor, is expressed in the endothelium of both human and mouse aortas, and is significantly upregulated in hyperlipidemia; second, LPIs upregulate 43 clusters of differentiation (CD) in HAECs, promoting EC activation, innate immune trans-differentiation, and immune/inflammatory responses; 72.1% of LPI-upregulated CDs are not induced in influenza virus-, MERS-CoV virus- and herpes virus-infected human endothelial cells, which hinted the specificity of LPIs in HAEC activation; third, LPIs upregulate six types of 640 secretomic genes (SGs), namely, 216 canonical SGs, 60 caspase-1-gasdermin D (GSDMD) SGs, 117 caspase-4/11-GSDMD SGs, 40 exosome SGs, 179 Human Protein Atlas (HPA)-cytokines, and 28 HPA-chemokines, which make HAECs a large secretory organ for inflammation/immune responses and other functions; fourth, LPIs activate transcriptomic remodeling by upregulating 172 transcription factors (TFs), namely, pro-inflammatory factors NR4A3, FOS, KLF3, and HIF1A; fifth, LPIs upregulate 152 nuclear DNA-encoded mitochondrial (mitoCarta) genes, which alter mitochondrial mechanisms and functions, such as mitochondrial organization, respiration, translation, and transport; sixth, LPIs activate reactive oxygen species (ROS) mechanism by upregulating 18 ROS regulators; finally, utilizing the Cytoscape software, we found that three mechanisms, namely, LPI-upregulated TFs, mitoCarta genes, and ROS regulators, are integrated to promote HAEC activation. Our results provide novel insights into aortic EC activation, formulate an EC biology knowledge-based transcriptomic profile strategy, and identify new targets for the development of therapeutics for cardiovascular diseases, inflammatory conditions, immune diseases, organ transplantation, aging, and cancers.
Atherosclerosis is a pathological process underlying the development of myocardial infarction, stroke, and peripheral arterial disease, which is a substantial cause of morbidity and mortality (1). Vascular inflammation contributes significantly to the atherosclerotic onset and the development of its complications (2–5). In addition to consistent findings across multiple mouse models (6), the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) demonstrated that the inhibition of pro-inflammatory interleukin-1β (IL-1β) reduces the atherosclerotic burden in cardiovascular disease (7–9). The activation of endothelial cells (ECs) is the earliest event and a central pathological process associated with the onset of atherosclerosis. Based on our previous findings, we propose that:(1) ECs are innate immune cells (3–5), as they display innate immune functions similar to those of prototypical innate immune cells, such as macrophages (5, 10, 11) and monocytes (12–18). (2) In addition to increased secretion of cytokines and chemokines and upregulation of adhesion molecules, activated ECs also exhibit two new hallmarks of innate immune cells, namely, upregulation of both danger-associated molecular patterns (DAMPs) receptors and major histocompatibility complex (MHC) molecules for antigen presentation (19). (3) Endogenous metabolites that bind to their intrinsic receptors, rather than classical DAMP receptors, such as toll-like receptors (TLRs) and nod-like receptors/inflammasomes, can become conditional DAMPs, for example, lysophospholipids (19–23). (4) Similar to macrophages and monocytes, ECs have innate immune memory functions (trained immunity) (2, 3, 24–26). Although many transcriptomic data have been reported, there is no standard universal framework to analyze these data. To address this knowledge gap, we applied the ontology transcriptomic formula to characterize aortic endothelial cell activation.
There are four key features for conditional DAMPs as we proposed previously: (i) acting as endogenous metabolites; (ii) elevating in pathologically conditions; (iii) contributing to physiological signaling roles; (iv) binding to their intrinsic receptors and carrying out cytokine-like signal amplification functions (27). Conditional DAMPs include lysophospholipids, hyperhomocysteinemia (14–17, 28–30), and succinate (27) among others. Lysophospholipids are a group of bioactive lipids; some of which are pro-inflammatory molecules (31, 32), such as lysophosphatidylcholines (LPCs, lysoPC) (23, 33, 34), lysophosphatidic acid (LPA) (34–36), lysophosphatidylinositols (LPIs, lysoPIs) (19), and sphingosine-1-phosphate (37). LPA, LPCs, and LPIs are the characteristics of atherosclerotic aorta plaque in apolipoprotein E deficient (ApoE−/−) and low-density lipoprotein receptor (LDLR−/−) mice. One of the sub-species of LPIs, 18:0, has been reported and is mainly localized in the necrotic core of the plaque (38). In addition to pro-inflammatory molecules, we also proposed anti-inflammatory endogenous metabolites, such as lysophosphatidylethanolamine and lysophosphatidylserine, pro-resolving mediators (39), IL-35 (40), and itaconate (41, 42) as homeostasis-associated molecular patterns (20). As we have reported, most lysophospholipids (LPLs) contribute to aortic endothelial cell (EC) activation (23, 43, 44) and the progression of atherosclerosis (22). The molecular mechanisms underlying LPC-induced aortic EC activation included calcium influx-increased proton leaks via uncoupled mitochondrial electron transport chain, increased mitochondrial reactive oxygen species (mtROS), increased histone 3 lysine 14 acetylation (H3K14ac), and transcription factor AP-1 driven ICAM-1 upregulation (23, 43–46). In addition, we also reported that LPC induces caspase-1 activation and pyroptosis (inflammatory cell death) (33, 34, 47). Moreover, by RNA-Sequencing (RNA-Seq), we reported that LPC and LPIs induce prolonged EC activation by upregulating adhesion molecule ICAM-1, additional DAMP receptors such as CD36, and MHC molecules for antigen presentation (19). However, the transcriptomic formula of aortic EC activation in a panoramic view remained poorly characterized.
Low-throughput techniques used in current cardiovascular science research laboratories limit our understanding of aortic EC activation. Thus, high-throughput computational bioinformatics screening is often introduced to provide a whole picture at the beginning of an experimental project. As an initial step, RNA-Seq data can be profiled via various databases, for example, Gene Set Enrichment Analysis (GSEA) (19). To improve our panoramic understanding of the importance of aortic EC activation induced by conditional DAMP proinflammatory lipid LPIs, we hypothesized that transcriptomic profiling using high-throughput RNA-sequencing data can be formulated on an EC biology knowledge basis. We examined this new hypothesis by massive profiling. Aortic EC phenotypic research was studied from EC adhesion and secretory functions. LPIs induce aortic EC activation by upregulating EC biomarkers and membrane adhesion molecules (159 genes), clusters of differentiation (CDs) signaling (373 genes), six types of secretomic gene sets, namely, canonical secretome (2,640 genes with signal peptide) (13), caspase-1-gasdermin D (GSDMD) non-canonical secretome (964 genes), caspase-4-GSDMD non-canonical secretome (1,223 genes), exosome non-canonical secretome (6,560 genes) (48), Human Protein Atlas (HPA) database-classified cytokines (1,176 genes), and HPA-classified chemokines (200 genes) (49). Three mechanistic studies were included in this article to identify molecular mechanisms underlying the upregulation of these key features of EC activation, such as increased endothelial cell membrane adhesion functions and secretory functions. We focused on determining the expression changes in a complete list of 165 reactive oxygen species regulators (ROS regulatome) (50) and 1,158 nuclear DNA-encoded mitochondrial genes (mitoCarta genes), and a complete list of 1,496 human genome-encoded TFs (49) (Figure 1), as have we reported for CD4+Foxp3+ regulatory T (Treg) cells (49). Our results have provided novel insights into aortic endothelial cell (EC) activation, formulated an EC biology knowledge-based transcriptomic profile strategy, and identify new targets for the future development of therapeutics for cardiovascular diseases, inflammations, immune diseases, transplantation, aging, and cancers (51).

Figure 1. Comprehensive analysis of lysophosphatidylinositol LPI)-treated human aorta endothelial cells (HAECs) at messenger RNA (mRNA) level from 11 different aspects. (A) Phenotypic and mechanistic studies. The table shows how a gene changes 18 h after HAECs were treated with 10 μM LPIs. The last column indicates that the pathways are promoted by LPI-upregulated genes. Pathways associated with endothelial cell activation are marked in red. (B) Logic flow for our knowledge-based transcriptomic profile strategy.
Eleven gene lists were generated in this manuscript for phenotypic and mechanistic studies of LPI-treated HAECs (Figure 1). One hundred fifty-nine EC biomarkers were modified from PMID: 29333215; 373 CD markers, 1,176 cytokines, 200 chemokines, and 1,496 TFs were generated from PMID: 33613572; 2,640 canonical secretomes were downloaded from the comprehensive protein database Human Protein Atlas (https://www.proteinatlas.org/); 964 non-canonical caspase-1-gasdermin D (GSDMD) secretomes were generated from PMID: 18329368;1,223 non-canonical caspase-4 (humans)/11 (mice) secretomes were extracted from PMID: 28196878; 6,560 exosome secretome downloaded from a comprehensive exosome database (http://www.exocarta.org/); 165 ROS regulators were downloaded from PMID: 33154757; 1,158 human nuclear genome DNA-encoded mitochondrion genes were downloaded from the Broad Institute at MIT (mitoCarta, https://www.broadinstitute.org/mitocarta/mitocarta30-inventory-mammalian-mitochondrial-proteins-and-pathways).
Microarray datasets were collected from the National Institutes of Health (NIH)-National Center for Biotechnology Information (NCBI)-Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/gds/) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) databases and analyzed with online software GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/), as we have reported (3, 10, 52–54). Three GEO datasets were used in this manuscript, namely, GSE59226 (influenza virus infection), GSE 79218 (MERS-CoV infection for 0, 12, 24, 36, and 48 h), and GSE 1377 (Kaposi's sarcoma-associated herpes virus).
Metascape (https://metascape.org/gp/index.html#/main/step1) was used for enrichment analysis. This website contains the core of most existing gene annotation portals. Our 11 gene lists mentioned in Figure 1A were compared with thousands of gene sets and ontology databases (KEGG, MSigDB, and GO) that were defined by their involvement in specific biological processes, pathway membership, enzymatic function, and protein localization. More details about Metascape can be found in cited references (55).
The ClueGo v2.5.8 in Cytoscape (https://cytoscape.org/) v3.8.2 was applied to identify gene connections and interactions between functional terms/pathways, as we have reported (56). Eight ClueGO databases were used for our network analysis, namely, GO-Biological Process (17,776 terms/pathways with 18,058 available unique genes), GO-Cellular Component (1,975 terms/pathways with 18,983 available unique genes), GO-Immune System Process (1,195 terms/pathways with 3,625 available unique genes), GO-Molecular Function (5,468 terms/pathways with 18,336 available unique genes), KEGG (333 terms/pathways with 8,093 available unique genes), Reactome pathways (2,474 terms/pathways with 10,855 available unique genes), Reactome reactions (13,015 terms/pathways with 10,855 available unique genes), and Wiki Pathways (667 terms/pathways with 7,633 available unique genes).
As we have reported previously, human aortic endothelial cells (HAECs) were treated with vehicle control or lysophosphatidylinositol (LPIs, 16:0) (10 μM) for 18 h. The RNA-Seq data are available in the Array Express database under accession number E-MTAB-6605 (19).
The expression changes were listed in the results with p < 0.05 (statistical significance). Genes with expression changes more than log2 (1) in our RNA-Seq data were defined as upregulated, while those with expression decrease of more than log2 (1) were defined as downregulated (Supplementary Tables).
To significantly improve our understanding of LPI-induced activation of HAECs with focus on EC activation key features, such as membrane protein adhesion and signaling and secretory function, an endothelial biology knowledge (3–5, 23, 24, 33, 34, 57–59)-based transcriptomic profile strategy was formulated, and 11 gene lists with 16,114 genes: (i) a comprehensive list of 373 cluster of differentiation (CD) markers (plasma membrane proteins) identified by specific monoclonal antibodies (https://en.wikipedia.org/wiki/List_of_human_clusters_of_differentiation); (ii) 159 updated EC-specific biomarkers (60); six types of secretomes namely, (iii) canonical secretome with 2,640 genes (encoded by all human genome-encoded proteins with a signal peptide) as we have reported (13); (iv) DAMP-sensor caspase-1 (1, 26, 33, 34, 47, 61–65)-gasdermin D (GSDMD) (66) secretome (proteins secreted extracellularly via activated caspase-1 cleaved N-terminal GSDMD protein pore) with 961 genes (48, 67); (v) caspase-4-GSDMD secretome (proteins secreted extracellularly via activated caspase-4 cleaved N-terminal GSDMD protein pore) with 1,223 genes (48, 68), (vi) exosome secretome with 6,560 genes, as we have reported (48); (vii) a complete list of 1,176 Human Protein Atlas (HPA)-classified cytokines; viii) a complete list of 200 HPA-classified chemokines, as we have reported (49); (ix) a complete list of 165 reactive oxygen species (ROS) regulators (regulatome), as we have reported (50); (x) a complete list of 1,496 human genome-encoded TFs from the Human Protein Atlas, as we have reported (3, 49); finally, (xi) a complete list of 1,158 human nuclear genome DNA-encoded mitochondria genes from the Broad Institute at MIT, were analyzed in this study (Figure 1A). As outlined in Figure 1B, all the examinations on EC membrane proteins, such as EC-specific biomarkers, CD markers, and the six types of secretomes were phenotypic studies. The three molecular mechanisms, namely TFs, mitoCarta genes, and ROS regulatome, were mechanistic studies.
As we mentioned in the introduction, G protein-coupled receptor 55 (GPR55, 319 amino acids, NIH-NCBI Protein database ID: NP_005674.2) is the specific receptor for LPIs (51, 69). The tissue RNA-Seq data from NIH-NCBI Gene database ID 9290 (https://www.ncbi.nlm.nih.gov/gene/9290) showed that significant GPR55 expressions (>0.5 reads per kilobase million, RPKM) were found in six tissues, such as the appendix, duodenum, lymph node, small intestine, spleen, and testis among 27 human tissues from 95 human individuals (Supplementary Figure 1). The expression of GPR55 was found in the human heart, although the GPR55 expression data from the vessel were not listed. However, the expressions of GPR55 in human and mouse aortic endothelial cells remained unknown. Hence, we hypothesized that GPR55 is expressed in human and mouse aortic endothelial cells. To examine this hypothesis, the human thoracic aorta single-cell RNA-Seq data were analyzed on the Single CellBeta Portal database of the Broad Institute at Massachusetts Institute of Technology (MIT) and Harvard (https://singlecell.broadinstitute.org/single_cell/study/SCP1265/deep-learning-enables-genetic-analysis-of-the-human-thoracic-aorta?genes=GPR55#study-summary). As shown in Figures 2A,B, the expressions of GPR55 were distributed in six aortic cell clusters identified in 54,092 cells, such as vascular smooth muscle cells, fibroblasts, macrophages, endothelial cells, pericytes, and lymphocytes. Of note, GPR55 expression in both subsets of EC made EC the only cell type with GPR55 expression among all subsets of the cell type (Figure 2B). The maximum GPR55 expression in EC reached 2.62 log2 (transcripts per million, TPM+1), ranking third among all the six cell types (Figure 2C). In addition, GPR55 was also expressed in ECs of the mouse aorta. Transcriptions of 24,001 aortic cells were profiled, and ten aortic cell types were identified (https://singlecell.broadinstitute.org/single_cell/study/SCP1361/single-cell-transcriptome-analysis-reveals-cellular-heterogeneity-in-the-ascending-aorta-of-normal-and-high-fat-diet-mice?genes=Gpr55#study-summary). GPR55 mRNA transcripts were found in B cells, dendritic cells, endothelial cells, fibroblasts, macrophages, mesothelial cells, and T cells of mouse aortas (Figures 2D,E). However, no significant expression of GPR55 was found in aortic neural cells, pericyte cells, and smooth muscle cells (Figure 2D). Moreover, GPR55 mRNA transcripts in aortic cells were expressed at much higher levels in the aortas of high-fat-fed mice than in the aortas of normal chow diet-fed healthy control mice (Figure 2F).
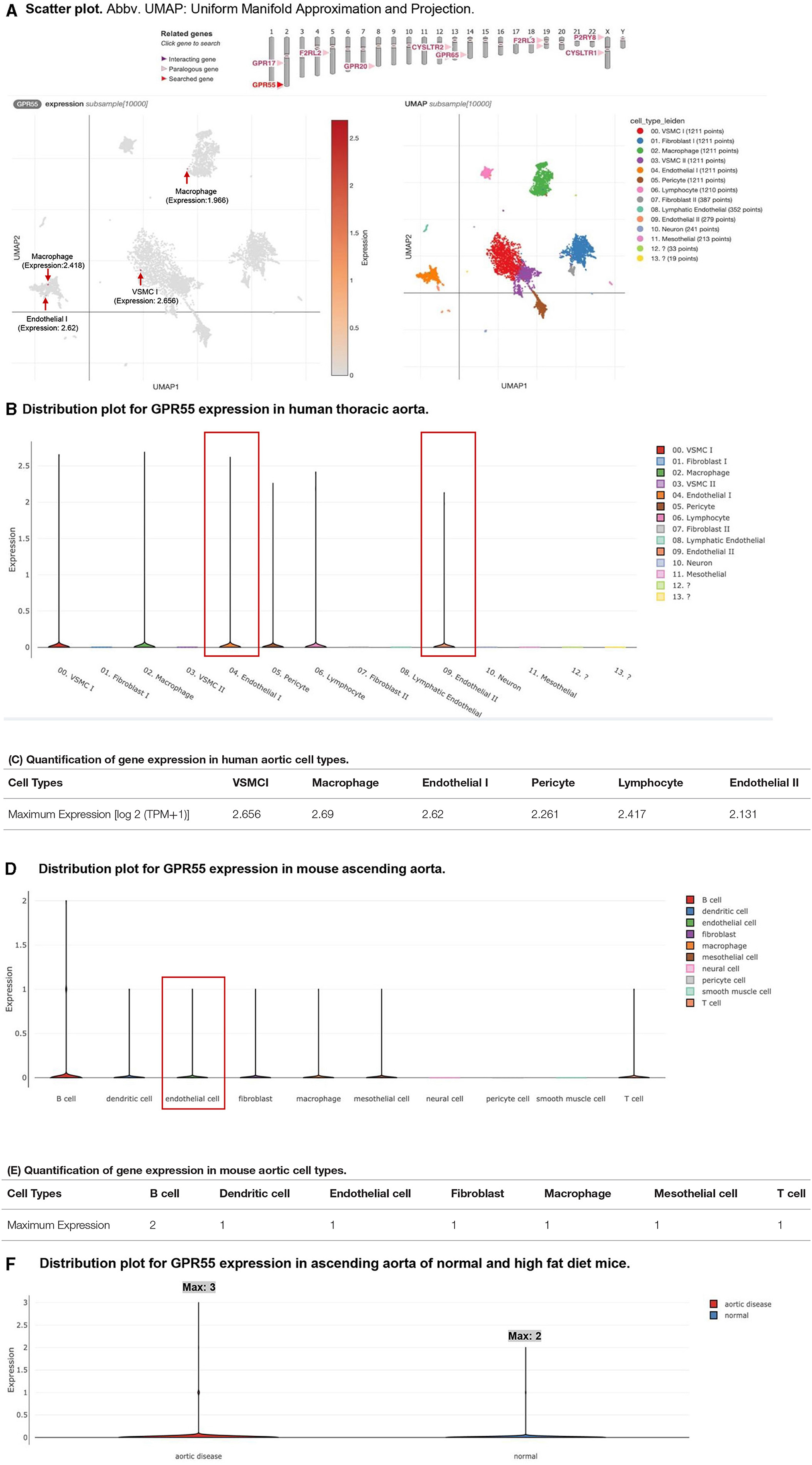
Figure 2. G protein-coupled receptor 55 (GPR55) mRNA transcripts are found in endothelial cells in both human and mouse aortas. Moreover, the expression of GPR55 is upregulated in aortic disease. (A) Single-cell RNA-seq analyses were performed on the Single Cell RNA-Seq database of the Broad Institute of MIT and Harvard (Single-Cell RNA-Seq database portal for human thoracic aorta: https://singlecell.broadinstitute.org/single_cell/study/SCP1265/deep-learning-enables-genetic-analysis-of-the-human-thoracic-aorta?genes=GPR55#study-visualize (https://doi.org/10.1101/2020.05.12.091934); GPR55 expressions in macrophage, vascular smooth muscle cells, and endothelial cells of the human aorta are labeled with red arrows in the Scatter. (B) GPR55 expressions in endothelial cells of the human aorta were boxed in red in the Distribution. (C) The table shows the maximum expressions of GPR55 in each cell type. The expression unit should be the log2 (TPM+1). Note: (A–C) show the expression of GPR55 in humans. The single-cell transcriptional profiles in (B,C) from 54092 thoracic aortic cells singled out 14 clusters representing 13 cell types. (D) Single Cell RNA-Seq database portal for mouse study: https://singlecell.broadinstitute.org/single_cell/study/SCP1361/single-cell-transcriptome-analysis-reveals-cellular-heterogeneity-in-the-ascending-aorta-of-normal-and-high-fat-diet-mice?genes=Gpr55#study-visualize). GPR55 expressions in endothelial cells of mouse aorta were boxed in red in the Distribution. (E) The table shows the maximum expression of GPR55 in each cell type. (F) The expression of GPR55 increased in the high fat-fed aortic disease mouse model compared with the normal control mice. The expression unit should be the log2 (TPM+1). Note: The transcriptional profiles in (D–F) represent 24,001 aortic cells and 10 cell types.
Taken together, these findings have demonstrated that first, LPI receptor GPR55 is expressed in human and mouse aortic endothelial cells; second, GPR55 is also expressed in human aortic vascular smooth muscle cells, fibroblasts, macrophages, pericytes, and lymphocytes, and mouse aortic B cells, dendritic cells, fibroblasts, macrophages, mesothelial cells, and T cells. Of note, the expression patterns of GPR55 in aortic endothelial cells, fibroblasts, macrophages, and lymphocytes are shared between human aortas and mouse aortas; third, high fat diet-induced hyperlipidemia upregulates aortic GPR55 expression, suggesting critical roles of GPR55 in hyperlipidemia-accelerated atherosclerosis (11, 14, 15, 33, 44, 47, 57, 70, 71). The results were well correlated with our report on LPI-induced activation of EC (19).
Our recent report showed that LPIs upregulate the expressions of membrane proteins, such as E selectin (SELE), intercellular adhesion molecule 1 (ICAM1), CD74, human leukocyte antigen (HLA) allele-DRB1, and HLA-DMA in HAECs (19). EC expresses specific clusters of differentiation (CDs), such as CD31, which includes various membrane-bound or cytoplasmic molecules on its surface, helps in easier identification of ECs from other cell types, such as CD4+ T cells (72–77), and can be defined by specific monoclonal antibodies (78). However, the overall LPI-modulated membrane protein expressions remained unknown. An excellent review summarized that 11 CDs expressed in ECs, namely, CD54 (ICAM1), CD102 (ICAM2), CD146 (MCAM), CD322 (JAM-B), CD106 (VCAM1), CD31 (PECAM1), CD155 (poliovirus receptor), CD99 (MIC2), CD62E (E-selectin), CD62P (P-selectin), and CD144 (VE-Cadherin), are involved in monocyte trafficking across the vessel wall (79). However, an important question remained whether the expression of all the other CDs is modulated in EC activation. We hypothesize that LPIs play a vital role in modulating the expressions of CDs and other EC adhesion molecules. To examine this hypothesis and study how LPIs change immunophenotyping and alter the behavior of ECs, we collected a complete list of 373 CD markers from a human protein database (https://www.proteinatlas.org/search/protein_class:CD+markers) and screened these CD markers in our LPI-treated HAEC RNA-Seq dataset (19). By comparing the RNA-Seq data of the LPI-treated HAECs with that of untreated HAEC controls, 21,252 genes were found to be significantly modulated (p < 0.05, |log 2FC| ≥ 1). As shown in Supplementary Table S1, 65 out of 373 (17.4%) CDs showed significant expression changes in LPI-treated HAECs. Among them, 43 CDs out of 373 (11.5%) were dramatically upregulated (Figure 3A). Of the 43 upregulated CD markers, we found that nine were involved in the regulation of cell adhesion, namely, selectin E (SELE, CD62E), intercellular adhesion molecule-1 (ICAM1, CD54, which are ligands for the leukocyte adhesion protein LFA-1), integrin a6 (ITGA6, CD49f, and beta4, which promote tumorigenesis where beta1 inhibits erbB2/HER2 signaling), ITGA1 (CD49a, which is involved in cell adhesion, inflammation, and fibrosis), ITGB1 (CD29, which is involved in cell adhesion and recognition in various processes such as embryogenesis, hemostasis, tissue repair, immune response, and cancer metastasis), lysosome-associated membrane protein 2 (LAMP2 and CD107b, which play an important role in chaperone-mediated autophagy), interferon-induced transmembrane protein 1 (IFITM1, which inhibits the entry of viruses, viral fusion, and release to the cytoplasm), CD164 (multi-glycosylated core protein 24, which regulates the proliferation, adhesion, and migration of hematopoietic progenitor cells), and nectin cell adhesion molecule 3 (nectin3 and CD113, which function as adhesion molecules at adherens junctions). These CDs were also functional in leukocyte recruitment, cell-cell interaction, and slow rolling (80–90). The second group of five upregulated CDs, namely CD27 (a tumor necrosis factor receptor (TNFR), a superfamily member and co-stimulation receptor), semaphoring 7A (SEMA7A), CD108, (which promotes axonal growth and T cell development), TNFSF4 (CD134, OX40 ligand, a co-stimulation receptor), and GGT1[CD224, which promotes clear cell renal cell carcinoma initiation and progression (91)], played roles in co-stimulating T cell immune responses, promoting cancer growth (92), and establishing immune memory (93–97). In addition, the third group of four inflammation-related CDs, such as MHC HLA-DR gamma chain (CD74) for MHC class II antigen presentation, interleukin-7 receptor (IL7R, which plays a critical role in the development of lymphocytes in a process called VDJ recombination), scavenger receptor class B, member 3 (CD36) for oxidized low-density lipoprotein (oxLDL) cell internalization (98), and toll-like receptor 3 (TLR3) for binding to double-stranded RNA/unmethylated CpG DNA and cooperating with scavenger receptor SREC-I to trigger inflammatory innate immune response (99), were dramatically upregulated in LPI-treated HAECs (Figure 3B) (100–103). Moreover, the fourth group included 25 CDs involved in many cellular functions, such as viral infection (IFITM1), interferon-gamma receptor signaling (IFITM1, IFNGR1), growth factor/cytokine receptors (TNFSF10, ADAM10, TFRC, FAS, CD109, IL13RA2, IL3RA, LIFR, DPP4), immune checkpoint receptors (PDCD1LG2), complement signaling (CD55, CD46), and hematopoiesis and stem cells [CD34, EVI2B, and KIT (104)] (Figure 3C).

Figure 3. LPIs upregulated EC adhesion molecules and cluster of differentiation (CD) marker-mediated signaling pathways. (A) 373 CD markers were used for database mining. Genes with p < 0.05 and log2 FC > |1| were selected as significantly changed genes. The total number of significantly changed CD markers is 65; upregulated genes account for 66.2% (43/65) and downregulated genes occupied 33.8% (22/65). Downregulated CDs can be found in Supplementary Table S1. (B) Eighteen out of 43 LPI-induced CDs mediate EC adhesion, immune cell responses, and inflammatory cell signaling. (C) Twenty-five out of 43 LPI-induced CDs mediates the other cell signaling. (D) One hundred fifty-nine endothelial cell-specific markers were generated (modified from PMID: 29333215). The LPI-upregulated EC-specific genes were compared with the upregulated CD markers. The Venn diagram indicated that five adhesion molecules showed in the overlapped area between endothelial cell-specific cell markers and LPI-stimulated CD markers. The functions of five overlapped CD markers and most of them participate in the cell adhesion process. (E) About 43 upregulated CDs were screened in seven virus-stimulated EC datasets (PMID:34248940). Sixteen out of 43 showed different changes in these seven datasets. (F) Upregulated genes of LPI-treated HAECs were analyzed by Metascape (https://metascape.org/gp/index.html#/main/step1). Pathways with high expression are related to the cell adhesion process, leukocyte migration, and inflammation. (G) A schematic presentation shows how LPIs regulate endothelial cells by mediating membrane protein interactions. *(G) was created with Biorender.com.
To better understand the alteration of endothelial cell surface markers induced by LPIs, we further gathered a list of 159 endothelial cell-specific biomarkers (60) (Supplementary Table S2). Figure 3D shows that five CD markers (11.6%) out of 43 LPI-upregulated CDs were shared with 5 out of 10 LPI-upregulated EC-specific cell biomarkers: CD36, ICAM1, CD34, ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1), and ADAM metallopeptidase domain 10 (ADAM10). Among these five CD markers, ICAM1, CD34, and ADAM10 directly mediate cell-cell adhesion. For example, the classic adhesion molecule ICAM1 on the surface of EC could interact with the molecule LFA-1 on lymphocytes, leading to a pro-inflammatory signaling cascade (82). CD34, a marker for human hematopoietic progenitor cells, exhibits E-selectin binding activity, facilitating leukocyte rolling and adhesion (105). ADAM10 showed a significantly high expression in atherosclerotic plaque, and its activity was necessary for chemotaxis/migration of monocytes and ECs (106). Of note, the LPIs upregulating 38 out of 43 CDs (88.4%) that did not overlap with EC-specific markers suggested that as high as 88.4% of the CDs upregulated by LPIs are functional in various aspects and not limited to EC-specific functions. These results have demonstrated that the LPI stimulation of aortic ECs may induce innate immune trans-differentiation of ECs, as we have reported (19), and non-EC-specific functions.
In order to identify CD markers specifically induced by LPIs, we examined the expressions of 43 LPI-induced CD markers in the microarray datasets of seven virus-infected human endothelial cells, such as influenza virus-infected human umbilical vein endothelial cells (HUVEC), middle east respiratory syndrome coronavirus (MERS-CoV, homologous to severe acute respiratory syndrome coronavirus 2, SARS-CoV2, or COVID-19)-infected human microvascular endothelial cells, and Kaposi's sarcoma-associated herpes virus(KSHV)-infected human dermal endothelial cells, as we have reported (3). As shown in Figure 3E, the 43 LPI-induced CD markers can be classified into three groups: (1) 12 LPIs were upregulated, and pathogen-associated molecular pattern (PAMP)-triggered (virus-infection) was upregulated (activated endothelial cell shared), namely, IL7R, IL3RA, ICAM1, FAS, Ifitm1, TNFSF10, CD274, CD74, CD36, SELE, ITGA6, and HMMR; (2) 4 LPIs were upregulated, but virus infection was downregulated (LPI-specific group 1), such as IFNGR1, ITGB1, TLR3, and ITGA2; 3) 27 LPIs were upregulated but virus infection was not changed (LPI-specific group 2) namely, CD27, IL13RA2, GGT1, MME, KIT (CD117, stem cell growth factor receptor), SEMA7A (CD108), CD34 (107), EVI2B, DPP4 (CD26, its inhibitors approved for treating type 2 diabetes), PDCD1LG2, CD302, CD164, TNFSF4 (OX40 ligand, CD252), ENTPD1, CD55, CD46, LAMP2, ADAM10, ABCG2 (CDw338, breast cancer resistant protein), PRNP, TFRC, CD109, NECTIN3, ITGA1, SLC44A1, LIFR, and CD82. Of note, future work is needed to determine whether LPIs upregulated CDs share with the CDs upregulated in responses to the stimulation of PAMPs/DAMPs and conditional DAMPs (20, 21).
In addition to analyzing the functions of upregulated CD markers, Metascape was used for pathway analysis (https://metascape.org/gp/index.html#/main/step1) for small gene sets in comparison with that analyzed by IPA. Twenty pathways, using upregulated CD markers from LPI-treated HAECs (Figure 3F), were identified, such as the top 10 functions of hematopoietic cell lineage, regulation of cell adhesion, production of molecular mediator of the immune response, cell adhesion molecules (CAMs), cytokine-cytokine receptor interaction, leukocyte migration, leukocyte activation involved in immune response, positive regulation of cell migration, positive regulation of cytokine production, and regulation of IL-10 production. Among these 20 pathways, 11 were related to EC activation (boxed), namely, the top 2–9 functions mentioned above, and myeloid leukocyte differentiation, regulation of inflammatory response, and cell adhesion mediated by integrin. Of note, the “regulation of cell adhesion” showed extraordinarily high enrichment up to around log10 (16).
Our results on LPI-induced CD markers in HAECs demonstrated that, first, of the total 65 LPI-changed CD markers, 66.2% were significantly upregulated by LPI stimulation, and that only 33.8% were downregulated by LPIs. Among the LPI-upregulated CD markers, 23% were associated with cell adhesion; 9.3 and 9.3% were related to immune response and inflammation, respectively. These results suggest that LPIs induce aortic EC activation through the upregulation of various adhesion molecules, via which LPIs may initiate inflammation by recruiting immune cell accumulation; second, by comparing LPI-upregulated CD markers with EC-specific markers, we found that LPI stimulation upregulates CDs that are significantly different from EC-specific markers, suggesting that LPIs may induce the innate immune trans-differentiation of ECs, as we have reported (19), LPI-activated HAECs may carry out many non-EC-specific functions; third, 31 out of the 43 LPI-induced CD markers (72.1%) are LPI induction-specific CD markers that are not induced in three types of virus-infected endothelial cells, which significantly enhance our understanding of CD markers upregulated in activated ECs; fourth, in addition to inducing EC activation, LPI-induced CD markers may promote other immune cell functions and inflammatory responses via membrane protein interactions (Figure 3G).
Secretome refers to a collection of actively secreted proteins for a destination outside the nucleus and cytoplasm of the cells. Those proteins are actively transported within the secretory pathways and participate in various signaling functions, such as cytokines, chemokines, adhesion molecules (108), angiogenesis, and wound healing (109). ECs are secretory cells, and protein secretion plays a pivotal role in EC functions, as we and others have reported/reviewed (1, 3, 4, 40, 44, 50, 110, 111). Especially during EC activation, secreted proteins are responsible for cell-cell interaction, affecting vascular tone, cell adhesion, and inflammation (112). The 18 cytokines secreted from EC (110) included pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-3, IL-5, IL-6, IL-8, IL-11, IL-15, monocyte chemoattractant protein-1 (MCP-1), granulocyte-macrophage colony-stimulating factor (GM-CSF) (3, 57), CD40/CD40L, endothelin-1, regulated upon activation, normal T cell expressed and presumably secreted (RANTES, C-C motif ligand 5, CCL5) and anti-inflammatory cytokine IL1 receptor antagonist (IL1ra), IL10 (59), IL13, transforming growth factor-β (TGF-β), and IL-35 (40, 44, 58, 59, 111). We hypothesize that LPI treatment drives HAEC activation via the upregulation of inflammatory and adhesion-related secreted proteins. To gain a comprehensive understanding of how the LPI stimulation of HAECs regulates the secretory functions of ECs, we collected six secretomic gene lists:(1) 2,640 conventional secretomes (with signal peptide) were downloaded from the comprehensive protein database Human Protein Atlas (https://www.proteinatlas.org/), as we have reported (13); (2) 964 non-canonical caspase-1-gasdermin D (GSDMD) secretomes (67); (3) 1,223 non-canonical caspase-4 (humans)/11 (mice) secretomes (68), and 4) 6,560 exosome secretomes downloaded from a comprehensive exosome database (http://www.exocarta.org/) (113), as we have reported (48). As shown in Figure 4A, 216 (8.2%) out of the 2,640 canonical secretomic genes were dramatically increased in LPI-treated HAECs. Similarly, 60 (6.2%) out of the 964 caspase-1-GSDMD non-canonical secretomic genes and 117 (9.6%) out of the 1,223 caspase-4-GSDMD non-canonical secretomic genes were significantly upregulated in the LPI-activated HAECs, respectively (Figures 4B,C). In addition, 40 out of the 6,560 total exosome secretomic genes showed dramatic elevation, with >log2FC 1.5 (fold change) (Figure 4D).
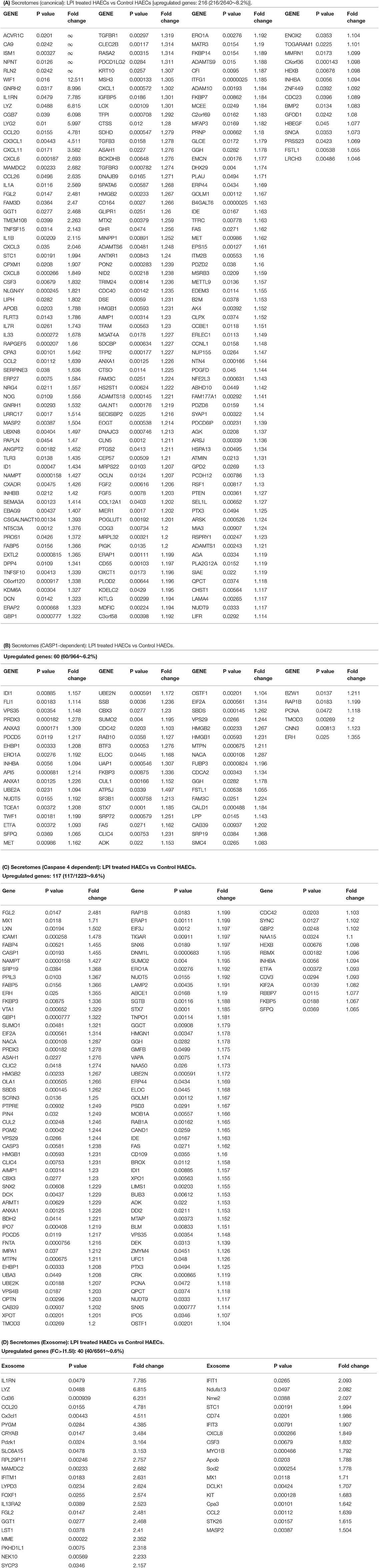
Figure 4. LPIs significantly upregulate secretomic genes of canonical secretomes, non-canonical caspase-1-Gasdermin D (GSDMD), caspase-4-GSDMD, and exosome secretomes in HAECs. (A) Among 2,640 canonical secretomes, 216 were significantly upregulated in LPI-treated HAECs. The upregulated genes accounted for 8.2% of the total canonical secretomes. (B) Sixty non-canonical caspase 1 dependent secretomes were significantly elevated in LPI-treated HAECs. (C) One hundred seventeen non-canonical caspase 4 dependent secretomes were dramatically increased in LPI-treated HAECs. (D) Forty exosomes were dramatically increased in LPI-stimulated HAECs. *Genes in (A–C) were selected with p < 0.05 and log2 FC > I1I as significantly changed genes, while genes in exosome listed p < 0.05 and log2 FC > |1.5| as significantly changed genes. The full list of 923 LPI-upregulated exosomes can be found in Supplementary Table S3. *Created with Biorender.com.
Of note, secretomes secrete a variety of biologically active molecules, such as (1) growth factors, (2) hormones, (3) cytokines [myokines/exerkines from muscle (113), adipokines from adipose tissues (114), cardiokines from the heart (115), hepatokines from the liver (116), osteokines from bones (116), nephrokines from kidney (113), and neurokines from the brain (113)]; (4) chemokines (117), and (5) many other secretory molecules with poorly characterized functions (13, 48, 49). Cytokines and chemokines released from endothelium have long been documented to be essential in promoting leukocyte recruitment, and inflammation during atherosclerosis, as we and others have reported/reviewed (1, 3, 4, 40, 44, 50, 110, 111). However, the vital question remained whether ECs secrete large pools of cytokines and chemokines during LPI-induced EC activation. Thus, we collected two lists of 1,176 cytokines and 200 chemokines (49) classified by the Human Protein Atlas (HPA, https://www.proteinatlas.org/) and examined the expression changes in the HPA-classified cytokines and chemokines, and their interactors in the LPI-treated HAEC RNA-Seq dataset. Of note, some cytokines and chemokines were overlapped with secretory proteins classified in other secretomes. As shown in Figure 5A, the expressions of 179 (15.2%) out of 1,176 HPA-classified cytokines showed a significant increase, and the expressions of 28 (14%) out of 200 HPA-classified chemokines were significantly upregulated (Figure 5B).
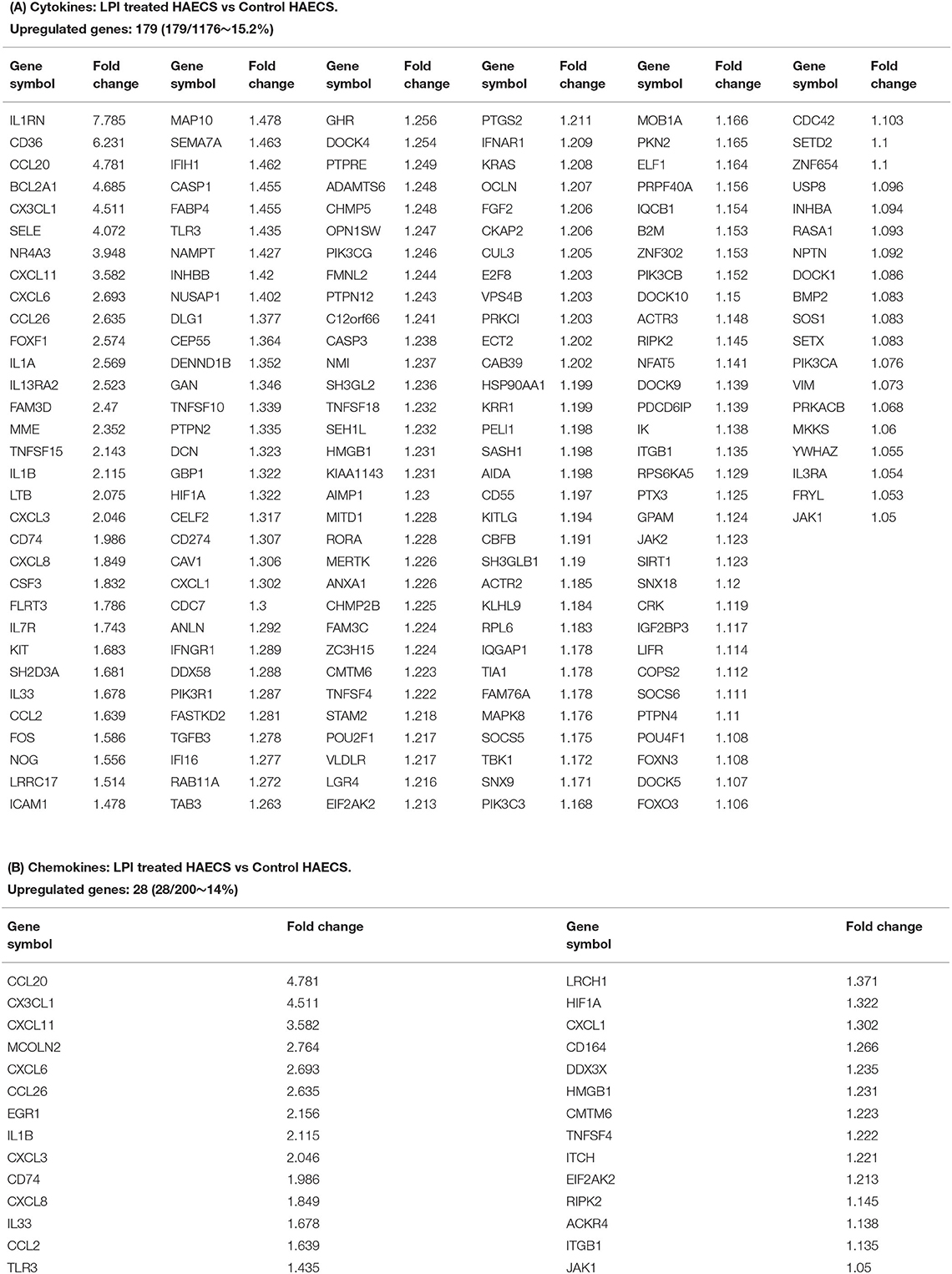
Figure 5. Cytokines and chemokines showed significant upregulation in LPI-treated HAECs compared with the control HAECs. (A) One hundred seventy-nine cytokines were significantly upregulated in LPI-treated HAECs. (B) Twenty-eight chemokines were significantly upregulated in LPI-treated HAECs.
The Metascape (https://metascape.org/gp/index.html#/main/step1) database analysis with six types of upregulated secretomes, cytokines, and chemokines in LPI-activated HAECs in Figure 6 demonstrated that the LPI-upregulated canonical secretome had top 10 functional pathways, namely, NABA matrisome associated, extracellular structure organization, glycoprotein metabolic process, vasculature development, myeloid leukocyte activation, regulation of cell adhesion, positive regulation of cell migration, IL-10 signaling, cellular response to growth factor stimulus, and VEGFA-VEGFR2 signaling (Figure 6A). The LPI-upregulated caspase-1-GSDMD non-canonical secretome had top 10 functional pathways, namely, regulation of nuclease activity, homeostasis of the number of cells, renal cell carcinoma, negative regulation of protein complex assembly, myeloid cell differentiation, CDC5L complex, cellular response to oxidative stress, 7q11.23 copy number variation syndrome, neutrophil degranulation, and nucleotide excision repair (Figure 6B). The LPI-upregulated caspase-4-GSDMD non-canonical secretome had top 10 functional pathways, namely, regulated exocytosis, autophagy, cytokine signaling in the immune system, cellular component disassembly, viral life cycle, response to an inorganic substance, response to tumor necrosis factor, apoptotic signaling pathway, cellular protein catabolic process, and regulation of nuclease activity (Figure 6C). The LPI-upregulated exosome non-canonical secretome had top 10 functional pathways, namely, membrane trafficking, endomembrane system organization, organelle localization, vesicle organization, actin filament-based process, regulated exocytosis, protein localization to the membrane, cellular protein catabolic process, endocytosis, and autophagy (Figure 6D). The LPI-upregulated cytokines had top 10 functions, namely, signaling by interleukins, regulation of cell adhesion, cytokinesis, cytokine-cytokine receptor interaction, response to molecule of bacterial origin, transmembrane receptor protein tyrosine kinase signaling, positive regulation of locomotion, Kaposi sarcoma-associated herpesvirus infection, positive regulation of cytokine production, and regulation of MAPK cascade (Figure 6E). The LPI-upregulated chemokines had top 10 functions, namely, chemokine production, response to chemokine, cellular response to interleukin-1, signaling by interleukins, positive regulation of response to external stimulus, regulation of leukocyte migration, positive regulation of vasculature development, regulation of the multi-organism process, positive regulation of cytokine biosynthetic process, and influenza A-related process (Figure 6F).
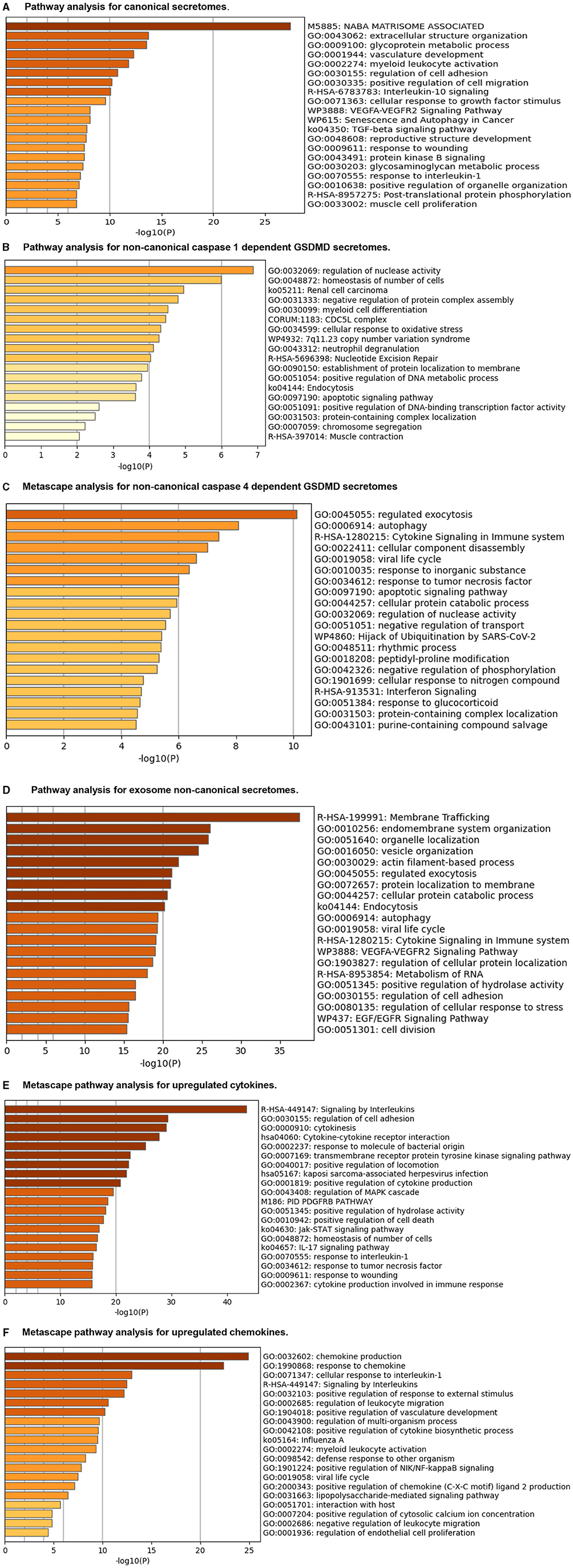
Figure 6. Metascape pathway analysis for upregulated six secretomes and cytokine and chemokines in LPI-treated HAECs. (A) Pathway analysis for canonical secretomes. (B) Pathway analysis for caspase-1-dependent non-canonical GSDMD secretomes. (C) Pathway analysis for caspase-4-dependent non-canonical GSDMD secretomes. (D) Pathway analysis for exosomes non-canonical secretomes. (E) Metascape pathway analysis for upregulated cytokines. (F) Metascape pathway analysis for upregulated chemokines.
To find potential connections among the LPI-treated HAEC secretory protein molecules, we created a Venn diagram for the pathways of canonical secretome, caspase-1 secretome, caspase-4 secretome, exosome secretome, HPA cytokines, and HPA chemokines. As shown in Figure 7A, among the 118 secretomic gene pathways identified by the Metascape analysis, the majority of the pathways were secretome-specific; and 12 pathways were shared among the six types of secretomic genes. The caspase-1-GSDMD secretome shared homeostasis of numbers of cells with HPA-cytokines; the caspase-1-GSDMD secretome shared endocytosis with the exosome secretome; the exosome secretome shared positive regulation of hydrolase activity with the HPA cytokines, and three types of secretomes, canonical, exosome, HPA cytokines, shared regulation of cell adhesion; the canonical secretome and HPA chemokines shared myeloid leukocyte activation; the canonical secretome and HPA cytokines shared response to wounding and response to interleukin-1; the caspase-4-GSDMD and exosome secretomes shared three pathways, regulated exocytosis, autophagy, and cytokine signaling in the immune system; three types of secretomes, namely, caspase-4-GSDMD, exosome, and HPA chemokines, shared viral life cycle; caspase-4-GSDMD and HPA cytokines shared response to tumor necrosis factor.
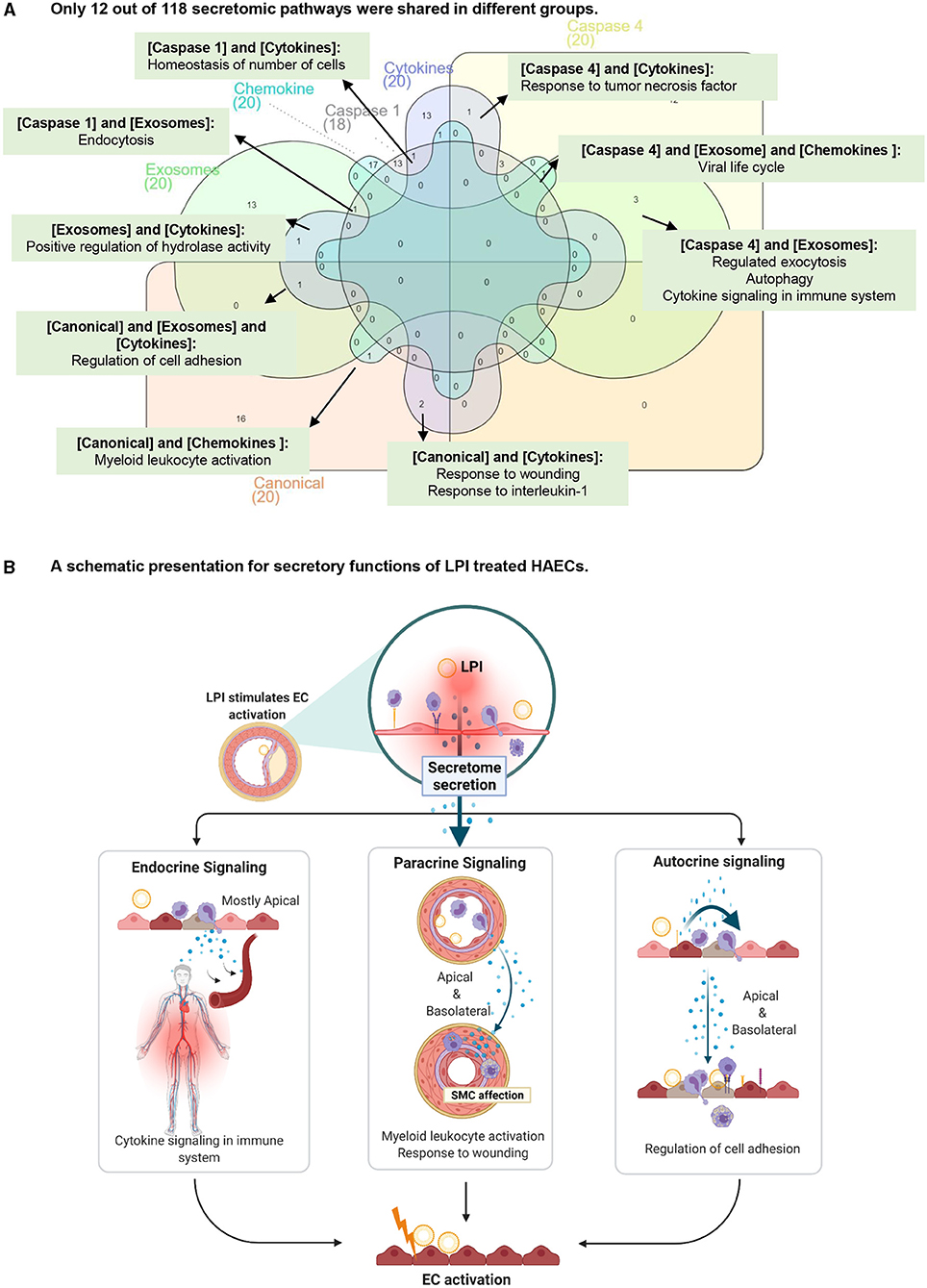
Figure 7. The majority of secretory pathways are mutually exclusive in LPI-stimulated HAECs. (A) Venn diagram was used to check the overlapped pathways among six secretomic groups. *(B) was created with Biorender.com.
The results have demonstrated for the first time that first, in contrast to the 20 cytokines reported to be secreted from ECs as mentioned above (110), activated aortic ECs are a large secretory organ that can upregulate the transcripts of large numbers (640 genes) of secretory proteins, potentially secrete six long lists of cytokines (179 genes), chemokines (28 genes), and 433 secretomic genes (216 + 60 + 117 + 40 = 433) and modulate the functions of innate and adaptive immune cells, inflammatory cells, other vascular cells, and non-vascular cells via three manners, such as autocrine, paracrine, and endocrine (Figure 7B); second, around 10% of secretomes in each category (canonical, caspase-1-GSDMD, caspase-4-GSDMD, exosome, HPA-cytokines, HPA-chemokines) showed significant upregulation after LPI stimulation. The percentages of LPI-upregulated cytokines and chemokines are higher than that of the four types of secretomes, around 15% in each. A similar percentage also occurred in the LPI-stimulated exosome secretomic genes, 923 (923/6,560 in total, ~14.1%) with p < 0.05 and log2FC >1; third, based on the comparison of top 10 functional pathways related to the LPI-upregulated secretomic genes, canonical secretome, caspase-1-GSDMD non-canonical secretome, caspase-4-GSDMD non-canonical secretome, and exosome non-canonical secretome in LPI-activated aortic EC may carry out different functions in EC adhesion, immune and inflammatory cell activation, regulation of leukocyte migration, regulation of cellular response to stress, and many other functions; fourth, a previous report has suggested that pools of human coronary artery ECs and human umbilical vein ECs have polarized secretomes, such as apical secretome and basolateral secretome. The majority of EC secretomes with 840 proteins and extracellular vesicles (EVs), such as exosome (53)) secretome, are polarized to the apical surface (112). A future proteomic study is needed to determine the polarized secretomes of LPI-activated aortic ECs (Figure 7B).
To identify molecular mechanisms underlying LPI-induced transcriptomic changes in CDs and EC-specific biomarkers, and six types of secretomic genes, we first examined the LPI-induced transcriptomic remodeling of the master gene transcription factors. We previously reported that three transcription factors (TFs), GATA-binding protein 3 (GATA3), B cell lymphoma 6 (Bcl-6), and histone deacetylase 6 (HDAC6), regulate CD4+Foxp3+ regulatory T cell (Treg) plasticity and determine Treg conversion into either novel antigen-presenting cell-like Treg or Th1-Treg (118). This result suggests that other T helper cell subsets, such as type 2 CD4+ T helper cell (Th2), and TFs such as GATA3, follicular T helper cell (Tfh) TF Bcl-6, and HDAC6, cooperate with Foxp3 to determine Treg transcriptomes and functions. Moreover, three upregulated TFs, Jun (AP-1 transcription factor subunit), hypoxia-inducible factor-1α (HIF1A), and endothelial PAS domain protein 1 (EPAS1, HIF-2α), collaborate with other pathways and membrane receptors to potentially trans-differentiate CD14+ thrombus leukocytes into angiogenic endothelial cells (12). The expressions of 232 transcription regulators are differentially regulated in 28 sets of endothelial cell microarrays in response to the stimulation of a broad spectrum of pathophysiologically relevant pathogen-associated molecular patterns (PAMPs)/danger-associated molecular patterns (DAMPs) (3). We hypothesized that LPIs activate HAECs by upregulating a set of specific TFs. To test this hypothesis, we collected 1,496 TFs from the comprehensive protein database Human Protein Atlas (HPA, https://www.proteinatlas.org/search/cytokine), as we reported recently (13). As shown in Figure 8A, 172 out of the total 1,496 TFs (11.5%, log2FC >1, p < 0.05) were significantly upregulated in LPI-activated HAECs. In addition, the numbers of LPI-induced upregulation for more than log2FC 2 folds, more than log2FC 1.5-fold, more than log2FC 1.4-fold, more than log2FC 1.3-fold, and more than log2FC 1.2-fold were 5, 3, 8, 15, and 49 TFs, respectively. Among the highly LPI-upregulated TFs, nuclear receptor subfamily 4 group A member 3 (NR4A3) was a novel target of p53 contributing to apoptosis (119); FoxF1 was a therapy target of Hedgehog-related cancers (120); FOS (AP-1 TF subunit) was one of the TFs linked to ERK/MAPK activation (121), inflammation, and atherosclerosis (122); Kruppel-Like Factor 3 (KLF3) was one of the key mechanosensitive master switches in gene expression in promoting atherosclerosis (123); hypoxia-inducible factor-1α (HIF1A) was a master regulator of EC biology for diabetic atherosclerosis (124).
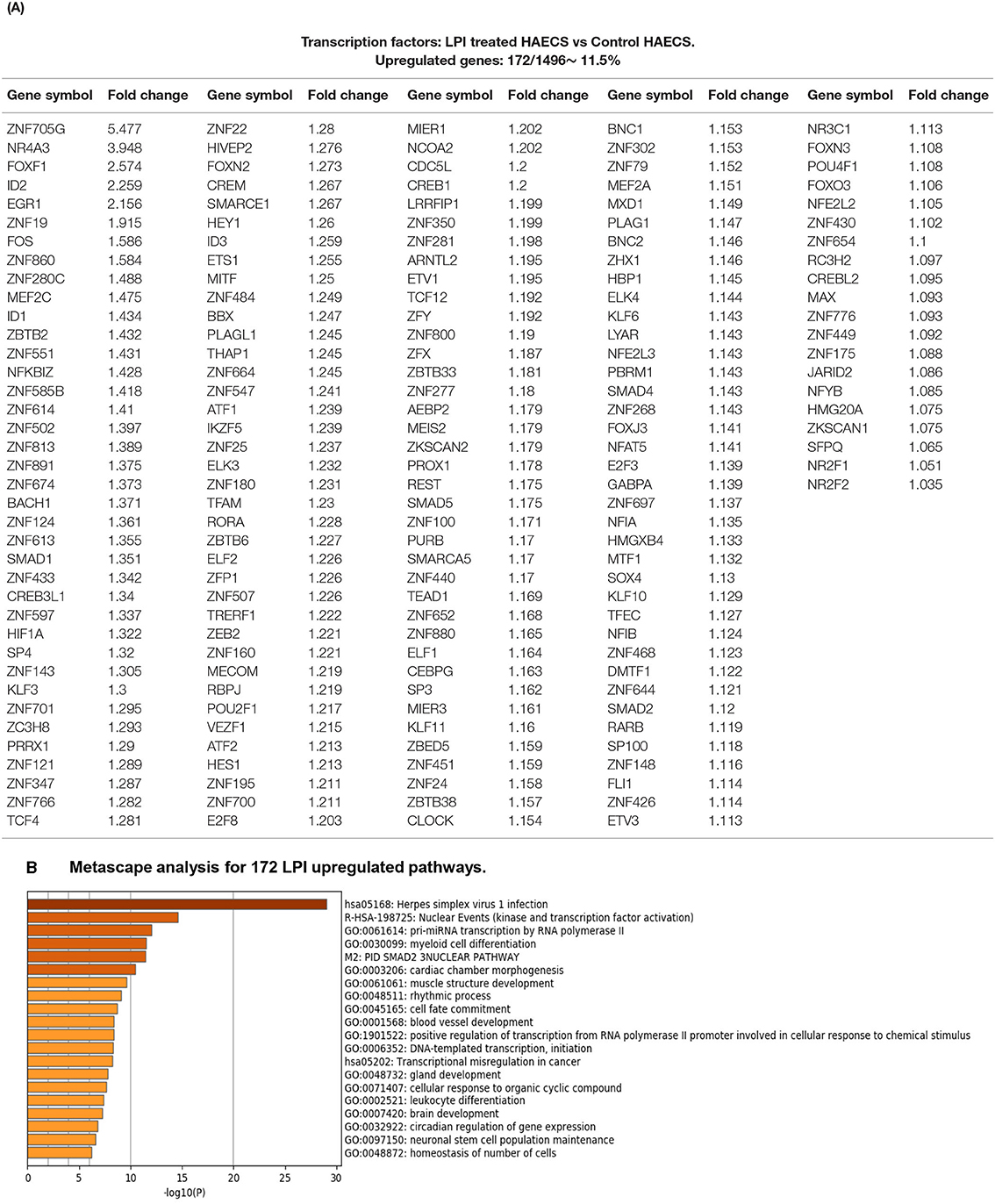
Figure 8. One hundred seventy-two LPI-upregulated transcription factors (TFs) were identified, which mediates 20 pathways after screening for a total of 1,496 TFs. (A) Around 11.5% of the genes (172/1496) showed significant upregulation among total TFs. (B) Metascape pathways and biological process enrichment analysis for upregulated genes in the LPI group.
The Metascape analysis in Figure 8B shows that LPI-upregulated TFs had 20 significant pathways, namely, herpes simplex virus 1 infection, nuclear events (kinase and transcription factor activation), pri-miRNA transcription by RNA polymerase II, myeloid cell differentiation, SMAD2-3 nuclear pathway [main signal transducers for transforming growth factor-β (TGF-β)], cardiac chamber morphogenesis, muscle structure development, rhythmic process, cell fate commitment, blood vessel development, positive regulation of transcription in response to chemical stimulus, DNA-template transcription-initiation, transcription misregulation in cancer, gland development, cellular response to organic cyclic compound, leukocyte differentiation, brain development, circadian regulation of gene expression, neuronal stem cell regulation maintenance, and homeostasis of the number of cells.
Taken together, the results have demonstrated that first, LPIs upregulate 172 (11.5%) out of 1,496 TFs and 80 (5.3%) TFs (log2FC > 1.2, p < 0.05), suggesting that LPIs have a broad effect on aortic EC transcriptome; second, some LPI-upregulated TFs, such as NR4A3, FOS, KLF3, and HIF1A, play significant roles in promoting inflammation and atherosclerosis; third, other Metascape analysis-identified inflammatory pathways include myeloid cell differentiation, positive regulation of transcription in response to chemical stimulus, cellular response to organic cyclic compound, and leukocyte differentiation.
Our previous reports showed that LPC induces aortic EC activation by increasing mitochondrial reactive oxygen species (mtROS) and proton leaks uncoupled from ATP synthesis (23, 44–46, 125) and that similar to LPC, LPIs also induces the upregulation of ICAM1 and aortic EC activation (19). We hypothesized that LPIs activate aortic ECs via a mitochondrion-dependent mechanism and modulate the transcription of genomic (nuclear) DNA-encoded mitochondrial genes (mitoCarta genes). To test this hypothesis, we collected the mitoCarta gene list from the Broad Institute at MIT (https://www.broadinstitute.org/mitocarta/mitocarta30-inventory-mammalian-mitochondrial-proteins-and-pathways). Figure 9A shows that LPIs upregulated 152 (13.1%) out of 1,158 mitoCarta genes. In addition, the Metascape analysis showed that the LPI-upregulated mitoCarta genes had functions of mitochondrion organization, cellular respiration, mitochondrial translation, mitochondrial gene expression, mitochondrial transport, propanoate metabolism, small-molecule catabolic process, ribose phosphate metabolic process, mitochondrial membrane organization, regulation of cellular respiration, mitochondrial biogenesis, metabolism of lipids, tRNA aminoacylation for protein translation, citric acid cycle (TCA cycle), ribosome disassembly, glycerol-3-phosphate metabolic process, protein depalmitoylation, mitochondrial iron-sulfur cluster biogenesis, protein complex oligomerization, and regulation of mitochondrial membrane potential (Figure 9B). Taken together, the results have demonstrated that LPI-activated aortic ECs activate a mitochondrial mechanism by upregulating 152 nuclear DNA-encoded mitochondrial genes (MitoCarta) and promote the mitochondrial organization, cellular respiration, translation, transport, and membrane organization.
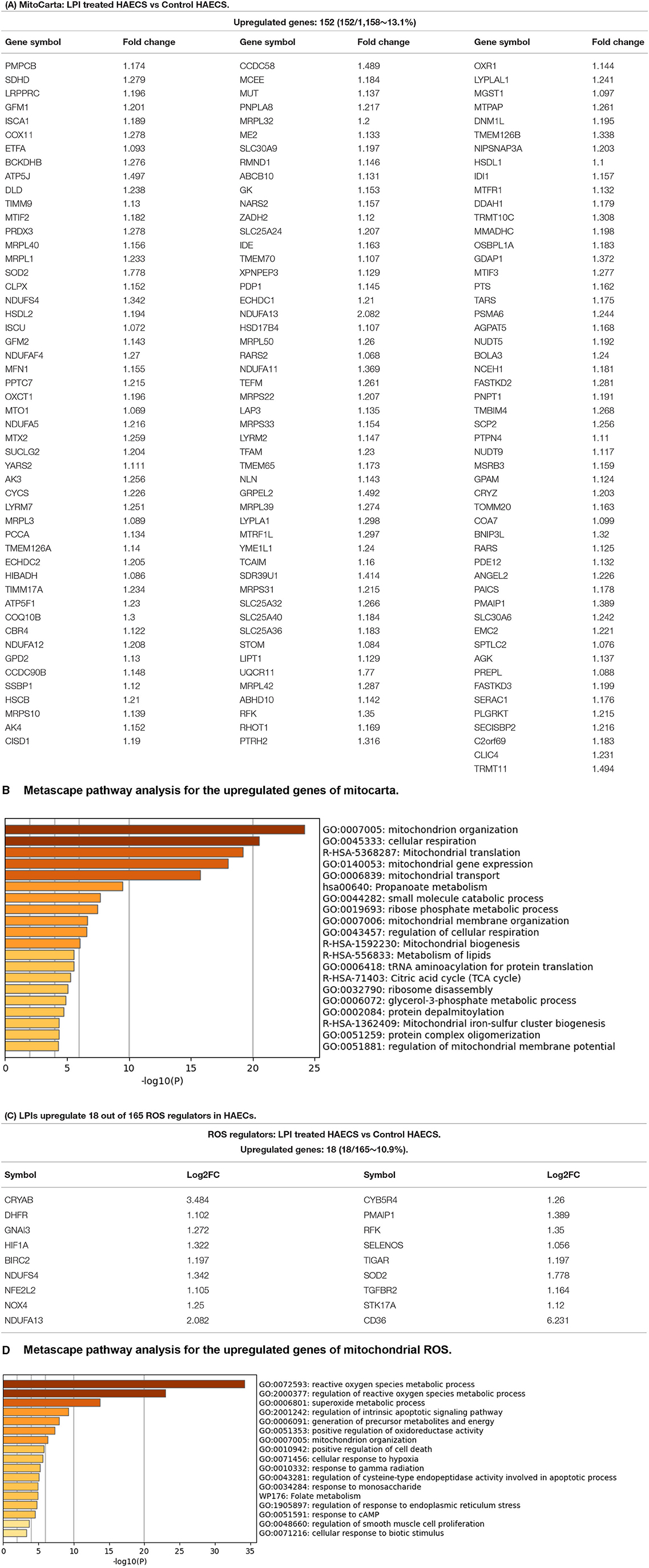
Figure 9. Mitochondrion-related genes showed significant upregulation in LPI-treated HAECs compared with the control HAECs. (A) One hundred fifty-two out of 1,158 mitocarta genes were significantly upregulated in LPI-treated HAECs. (B) Metascape pathway analysis for the upregulated genes of mitocarta. (C) Eighteen mitochondrial reactive oxygen species (ROS) regulators were significantly upregulated in LPI-treated HAECs. (D) Metascape pathway analysis for the upregulated genes of mitochondrial ROS.
It has been reported that ROS plays a key role in regulating pathophysiological signaling in endothelial cell activation (126) and cardiovascular diseases (127). We also reported that mitochondrial ROS plays a significant role in mediating EC activation (23, 44, 59). In addition, we recently proposed a new working model in which ROS is an integrated cellular network for sensing homeostasis and alarming DAMPs (128). We hypothesized that LPIs modulate the expressions of ROS regulators in HAECs. We collected 165 ROS regulators classified in the Gene Set Enrichment Analysis (GSEA) (https://www.gsea-msigdb.org/gsea/index.jsp) database, as we have reported (50). Figure 9C shows that LPIs upregulated 18 (10.9%) out of 165 ROS regulators in activated HAECs. In addition, the Metascape analysis showed that LPIs upregulated ROS regulators and promoted the functions of ROS metabolic process, regulation of ROS metabolic process, superoxide metabolic process, regulation of intrinsic apoptotic signaling, generation of precursor metabolites and energy, positive regulation of oxidoreductase activity, mitochondrion organization, positive regulation of cell death, cellular response to hypoxia, response to gamma radiation, regulation of cysteine-type endopeptidase activity involved in apoptosis, response to monosaccharide, folate metabolism, regulation of response to endoplasmic reticulum stress, response to cyclic adenosine 3', 5'-monophosphate (cAMP), regulation of smooth muscle cell proliferation, and cellular response to biotic stimulus (Figure 9D). Taken together, the results have demonstrated that first, LPIs upregulate 18 (10.9%) out of 165 ROS regulators in activated HAECs, suggesting that LPIs activate human aortic endothelial cells potentially via ROS-mediated mechanisms; second, LPIs upregulate many pathways in regulating ROS metabolic process, mitochondrial metabolism, and cell death.
We further hypothesized that three molecular mechanisms underlying the LPI activation of human aortic endothelial cells can be connected. To examine this hypothesis, we used the Cytoscape (https://cytoscape.org/) database to visualize and integrate the complex network among 172 LPI-upregulated TFs, 152 LPI-upregulated mitoCarta genes, and 18 LPI-upregulated ROS regulators. As shown in Figure 10A, three groups of genes are loaded in the function ClueGO of the Cytoscape database, and the visual style is set as the clusters with assigned colors. The three groups of genes included 172 LPI-upregulated TFs (shown in cluster 1, red), 152 LPI-elevated Mitocarta genes (shown in cluster 2, blue), and 18 LPI-increased ROS regulators (shown in cluster 3, purple). In the search for potential connections between three color clusters, two selection criteria were used. First, the GO tree interval was set between GO levels 4–10 to identify the representative and specific pathways, meaning mapped genes represent 4 to 50% of the total associated genes. When the pathways were selected to be only presented when the p-value of the pathway was less than .05, 185 terms/pathways were identified. The second criteria/step were to find potential connections among the lists of LPIs stimulated TF (Red Cluster), MitoCarta genes (Blue Cluster), and ROS regulators (Purple Cluster). Thus, the genes in all three clusters (Red, Blue, Purple colors) were selected for further analysis. After the first and second screening, five terms/pathways were chosen that genes associated with the term/pathways were from different, overlapping clusters (all clusters <60%). The representative genes are shown in Figure 10B, and include: (i) mitochondrial biogenesis (13% associated genes to the term, 41% for cluster 1, 50% for cluster 2, and 9% for cluster3); (ii) regulation of cellular response to oxidative stress (13% associated genes to the term, 38% for cluster 1, 20% for cluster 2, and 42% for cluster3); (iii) regulation of oxidative stress-induced cell death (11% associated genes to the term, 50% for cluster 1, 17% for cluster 2, and 33% for cluster3); (iv) transcriptional activation of mitochondrial biogenesis (16% associated genes to the term, 52% for cluster 1, 36% for cluster 2, and 11% for cluster3); and (v) mitochondrion localization (12% associated genes to the term, 27% for cluster 1, 56% for cluster 2, and 17% for cluster 3). Figure 10C shows the overlapped genes between each term. Taken together, the Cytoscape results have demonstrated that three molecular mechanisms, such as 172 LPI-upregulated TFs, 152 LPI-upregulated mitoCarta genes, and 18 LPI-upregulated ROS regulators, are integrated to promote HAEC activation.
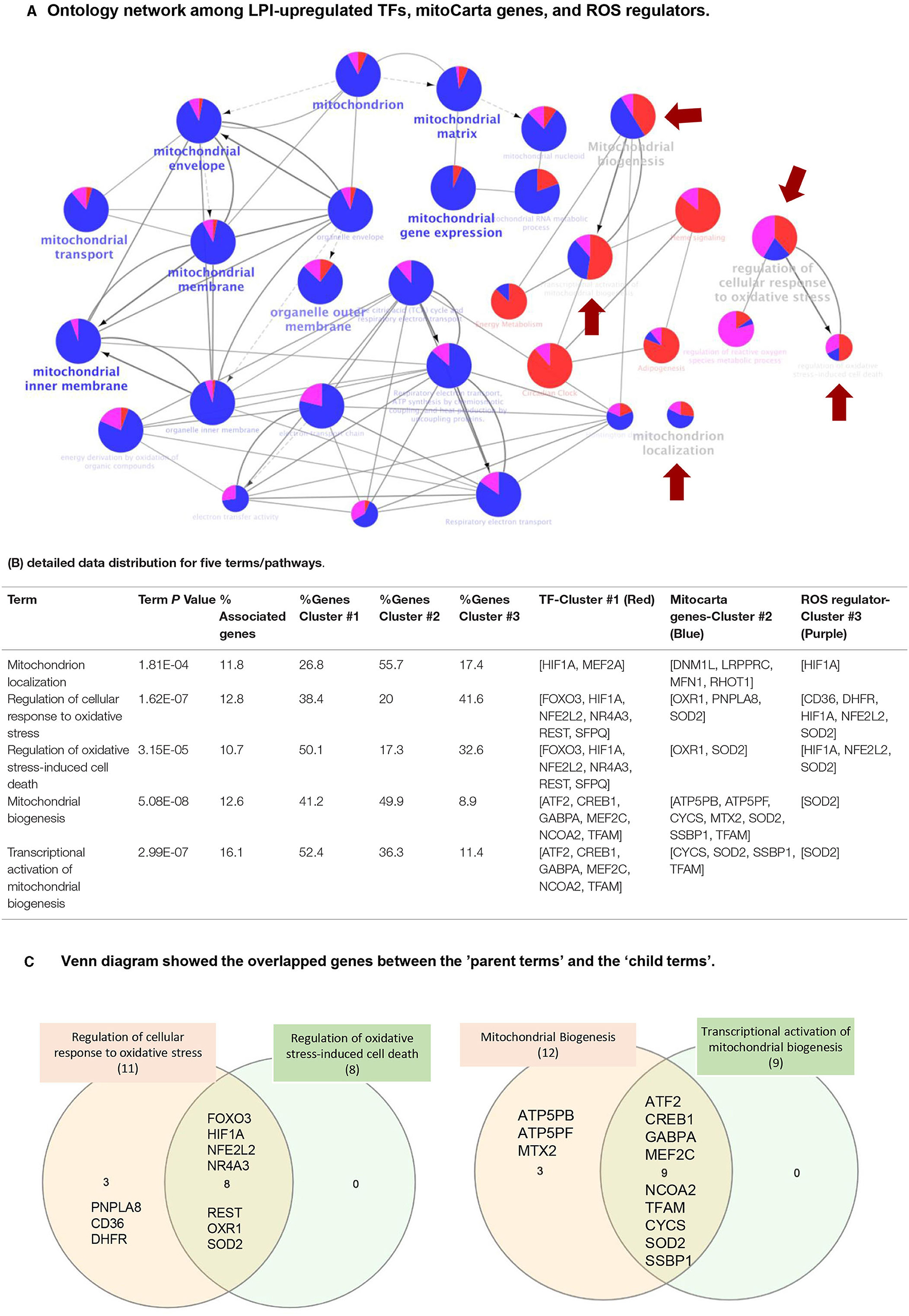
Figure 10. Network data integration database Cytoscape analysis (https://cytoscape.org/) was performed to analyze the network connection among 172 upregulated LPI TFs, LPI-increased mitocarta genes, and LPI-upregulated ROS regulators. (A) Ontology network among LPI-upregulated TFs, mitoCarta genes, and ROS regulators. The cluster of 172 LPI-increased TFs is shown in red, the cluster of 152 LPI-upregulated mitoCarta genes is shown in blue, and the cluster of 18 LPI-elevated ROS regulators is shown in purple. One node represents a term/pathway, and the node size stands for significance. The bigger the node size, the higher the significance. (the smallest node in this figure is p < 0.05). The proportion of the colors in each node indicates the percentage of genes that contribute to this term/pathway from each cluster. The red arrow highlights five terms/pathways, indicating that the three clusters of genes equally contribute to a term/pathway. We focus on these five pathways, and the other 25 shown in this figure are closely related to the five. The gray lines indicate the connection between each term. The thicker the line, the stronger the correlation. The gray arrow starts from “parent term” to “child term.” The child term is more specialized than its parent term. (B) Detailed data distribution for five terms/pathways. The associated genes% represent how many genes from our three clusters matched to the genes of terms in the database. The gene clusters #1, #2, #3 indicate how many percentages of each cluster of genes contribute to the term/pathway. (C) Venn diagram shows the overlapped genes between the “parent terms” and the “child terms.”
We proposed a novel concept that ECs are innate immune cells. Inflammatory mechanisms and endothelial cell activation play essential roles in promoting the progression of cardiovascular diseases, inflammatory diseases, autoimmune diseases, transplantation immune responses, cancer metastasis, and aging diseases (1, 3–6, 23, 33, 34, 44, 47, 57, 58, 61, 70, 92, 129). Significant progress has been made in elucidating molecular mechanisms underlying endothelial cell activation. However, several important issues remain to be addressed: (1) whether aortic endothelial cell activation induces conditional DAMP (20, 21). LPIs upregulate additional membrane proteins for signaling in addition to mediating inflammatory cell adhesion to EC and trans-EC migration; (2) how many secretory proteins can be upregulated during aortic EC activation, and whether aortic ECs are equipped to upregulate various secretomes during EC activation induced by LPIs; (3) whether LPIs activate aortic ECs via remodeling ROS regulatome, mitochondrial reprogramming, and TF machinery reshaping. To address these questions, we developed an EC biology knowledge-based transcriptomic formula to analyze RNA-Seq data in a panoramic manner. We made the following important findings: first, GPR55, a specific receptor for LPIs, is expressed in the endothelium of both human and mouse aortas, and is significantly upregulated in hyperlipidemia; second, LPIs upregulate 43 out of 373 clusters of differentiation (CDs) markers in HAECs, promoting EC activation, innate immune trans-differentiation, and immune and inflammatory responses; and 72.1% of LPI-upregulated CD markers are not induced in three types of virus-infected human endothelial cells; third, LPI-activated aortic ECs upregulate six types of secretomic genes, canonical secretome, caspase-1-gasdermin D (GSDMD) non-canonical secretome, caspase-4/11-GSDMD non-canonical secretome, exosome non-canonical secretome, HPA-classified cytokines, and HPA-classified chemokines, which makes HAECs a large secretory organ for inflammation, immune responses, and other functions; fourth, LPIs activate a transcription mechanism by upregulating 172 TFs, some of which, namely, NR4A3, FOS, KLF3, and HIF1A, play significant roles in promoting inflammation and atherosclerosis; fifth, LPIs activate a mitochondrial mechanism in aortic ECs by upregulating 152 nuclear DNA-encoded mitochondrial genes (MitoCarta) and promote mitochondrial organization, cellular respiration, translation, and transport, and membrane organization; sixth, LPIs activate reactive oxygen species (ROS) mechanism in activated HAECs by upregulating 18 out of 165 ROS regulators; seventh, the Cytoscape analysis results have demonstrated that three novel molecular mechanisms, namely, 172 LPI-upregulated TFs, 152 LPI-upregulated mitoCarta genes, and 18 LPI-upregulated ROS regulators, are integrated to regulate HAEC activation.
Our findings on hyperlipidemia-increased GPR55 expression in mouse aortas were correlated with several reports: (1) patients with Crohn's disease (a type of inflammatory bowel disease) manifest higher (12.5-fold) GPR55 mRNA expression in inflamed compared with non-inflamed colonic tissues (p < 0.0001) (130); (2) circulating LPIs and the liver expression of GPR55 are upregulated in patients with nonalcoholic steatohepatitis (NASH); the in vivo knockdown of GPR55 is sufficient to improve liver damage in mice fed with a high-fat diet and in mice fed with a methionine-choline-deficient diet (131); and 3) O-1602, the most potent agonist of GPR55, induces lipid accumulation in hepatocytes, which is reversed by treatment with CID16020046, an antagonist of GPR55 (132). Our findings on the LPI upregulation of 640 secretomic genes in activating HAECs and promoting inflammation were well correlated with several reviews (69) and reports: GPR55 antagonist CID16020046 protects oxLDL-induced inflammation in HAECs (133); LPIs, especially the albumin-bound form, induce pro-inflammatory cytokines TNF-a and IL-6 in macrophages via the GPR55/MAPKP38 pathway (134); GPR55 antagonist has anti-inflammatory effects in LPS-activated primary microglial cells (135); GPR55 knockout mice show reduced inflammation scores as compared with wild-type mice in an intestinal inflammation model (2.5% dextran sulfate sodium model) (136). Our findings on the LPI upregulation of 172 transcription factors in activated HAECs were well-correlated with the previous report that LPIs induce the activation of several TFs, such as nuclear factor of activated T-cells (NFAT), nuclear factor κ of activated B cells (NF-κB), and serum response element, translocation of NFAT and NF-κB, and GPR55 internalization (137). Of note, GPR55 is a non-cannabinoid receptor 1 or 2 (CB1/CB2) receptor that exhibits affinity for endogenous plant and synthetic cannabinoids. It was reported that LPI-mediated calcium release and mitogen-activated protein kinase (MAPK)-extracellular signal-regulated kinase (ERK) activation depend on the stable expression of GPR55 and that LPIs cannot have the above-mentioned calcium release and MAPK/ERK activation when CB1 or CB2 is expressed in the cells (137), suggesting the contexture (cannabinoid receptor 1 or 2 expression levels) dependence of LPI pro-inflammatory functions.
As shown in Figure 11, we proposed a novel working model to integrate all the findings. First, LPI receptor GPR55 is expressed in human and mouse aortic endothelial cells as well as other aortic cell types and is upregulated in hyperlipidemic conditions, suggesting that LPIs/GPR55 signaling is increased in aortic endothelial cells in cardiovascular diseases such as hyperlipidemia. In addition, LPI pro-inflammatory functions may depend on the contexture (cannabinoid receptor 1 or 2 expression levels). Second, by screening 12,763 secretory protein genes in six types of secretomes, we have demonstrated for the first time that human aortic endothelial cells are a large secretory organ. Under stimulation by LPIs, a prototypic conditional DAMP, pro-inflammatory lipid, and human aortic endothelial cells can upregulate as many as 640 secretomic genes via six types of secretomic mechanisms, namely, canonical secretome with all human proteins having a signal peptide via exocytic direction along the endoplasmic reticulum-Golgi-plasma membrane route, caspase-1-GSDMD non-canonical secretome without a signal peptide but secreted via the N-terminal Gasdermin D protein pore/channel, caspase-4(humans)/11 (mice)-GSDMD non-canonical secretome without a signal peptide but secreted via the N-terminal Gasdermin D protein pore/channel, exosome non-canonical secretome without a signal peptide but secreted via exosomes and docking on target cells with exosome docking mechanism but not cytokine/chemokine receptors, and HPA-classified cytokines and chemokines. In contrast to 18 traditional EC-secreted cytokines and chemokines (110), such as TNF-α, IL-1, IL-3, IL-5, IL-6, IL-8, IL-11, IL-15, MCP-1, GM-CSF (3, 57), CD40/CD40L, endothelin-1, RANTES, IL1ra, IL10 (59), IL13 and TGF-β, and IL-35 (40, 44, 58, 59, 111), these large numbers of secretomic proteins play significant roles in promoting EC activation, inflammatory cell and immune cell recruitment, cancer cell metastasis, immune cell development and regulation, vascular smooth muscle cell function regulation, and many other functions via autocrine, paracrine, and endocrine manners, either by apical secretion and/or basolateral secretion. Third, by screening 373 clusters of differentiation markers and 159 EC-specific biomarkers, we have demonstrated for the first time that LPIs upregulate 43 CD markers, five of which are shared with 159 EC-specific biomarkers, and 12 of which are shared with other human endothelial cell activation induced by an influenza virus infection, MERS-CoV infection, and KSHV infection, respectively. In contrast to traditional EC adhesion molecules, such as ICAM1, VCAM1, and SELE, as we and others have reported (33, 58), the 43 LPI-upregulated CD markers not only play significant roles in endothelial cell adhesion and inflammatory and immune cell recruitment but also promote inflammatory cell and immune cell activation, proliferation, differentiation, and immune tolerance. Fourth, three novel molecular mechanisms, namely, 172 LPI-upregulated transcription factors, 152 LPI-upregulated mitoCarta genes, and 18 LPI-upregulated ROS regulators, are integrated to promote HAEC activation.
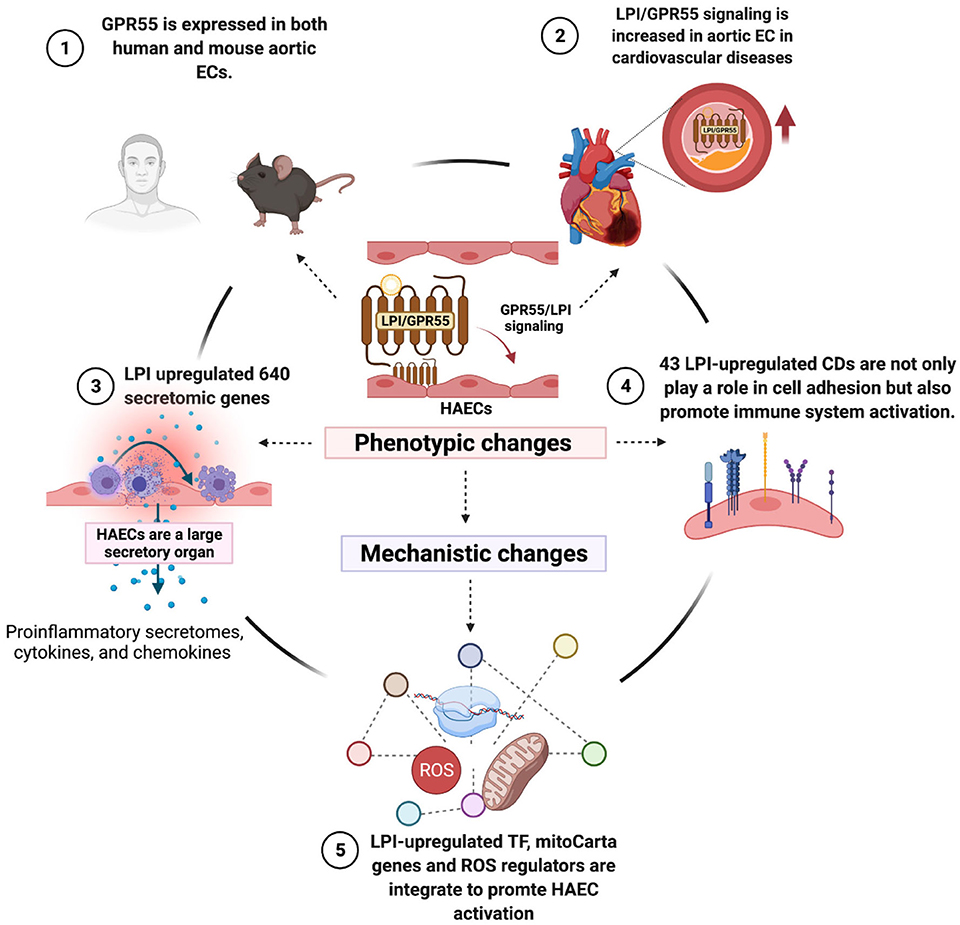
Figure 11. Working model. LPI receptor GPR55 is expressed in both human and mouse aortic endothelial cells. LPIs/GPR55 signaling is upregulated in aortic endothelial cells in hyperlipidemia. We first proposed human aortic endothelial cells as a large secretory organ, which can mediate up to 640 secretomic genes upon LPI stimulation. A large number of secretory proteins contribute a significant role in mediating EC activation and inflammation. LPI-stimulated specific CD markers not only participate in cell adhesion but also play an essential role in immune cell activation, proliferation, and differentiation. All these phenotypic changes may be caused by the mechanisms of synergy among LPI-increased TFs, mitoCarta genes, and ROS regulators. *This figure was created with Biorender.com.
Our results have provided novel insights into aortic endothelial cell (EC) activation, formulated an EC biology knowledge-based transcriptomic profile strategy, and identified new targets for the future development of therapeutics for cardiovascular diseases, inflammations, immune diseases, transplantation, aging, and cancers. One limitation of all the RNA-Seq data analyses is that due to the low-throughput nature of verification techniques in every laboratory, including ours, we could not verify every result we found with the analyses of high-throughput data, which are similar to all the studies with RNA-Seq (19, 59), single-cell RNA-Seq, metabolomics (23), chromatin immunoprecipitation (CHIP)-Seq (24, 44), and other-omics data (11, 138, 139). We acknowledge that carefully designed in vitro and in vivo experimental models will be needed in the future to verify the LPI-upregulated genes further and the underlying mechanisms we report here (9, 140).
The datasets presented in this study can be found in online repositories. The names of the repository and accession numbers can be found below: National Institutes of Health (NIH), National Center for Biotechnology Information (NCBI), Gene Expression Omnibus (GEO) DataSets database (https://www.ncbi.nlm.nih.gov/gds), GSE 59226 (Influenza virus infection), GSE 79218 (MERS-CoV infection for 0, 12, 24, 36, 48 h), and GSE 1377 (Kaposi's Sarcoma associated herpes virus).
KX carried out data gathering and data analysis and prepared the tables and figures. YSh, FS, AG, CD, LL, YL, YSu, HX, DP, XQ, JS, EC, XJ, and HW aided in the analysis of data. XY supervised the experimental design, data analysis, and manuscript writing. All the authors read and approved the final manuscript.
This study was partially supported by NIH grants to HW and XY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.773473/full#supplementary-material
1. Yang XF, Yin Y, Wang H. Vascular inflammation and atherogenesis are activated via receptors for pamps and suppressed by regulatory T cells. Drug Discov Today Ther Strateg. (2008) 5:125–42. doi: 10.1016/j.ddstr.2008.11.003
2. Drummer C. 4th, Saaoud F, Shao (邵颖) Y, Sun (孙宇) Y, Xu (徐克曼) K, Lu (路一凡) Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arteriosclerosis, Thrombosis, Vascular Biology. (2021) 41:1032–46. doi: 10.1161/ATVBAHA.120.315452
3. Shao Y, Saredy J, Xu K, Sun Y, Saaoud F, Drummer C, et al. Endothelial immunity trained by coronavirus infections, DAMP stimulations and regulated by anti-oxidant NRF2 may contribute to inflammations, myelopoiesis, covid-19 cytokine storms and thromboembolism. Front Immunol. (2021) 12:653110. doi: 10.3389/fimmu.2021.653110
4. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, et al. Vascular endothelial cells and innate immunity. Arteriosclerosis, Thrombosis, Vascular Biology. (2020) 40:e138–52. doi: 10.1161/ATVBAHA.120.314330
5. Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol Oncol. (2013) 6:61. doi: 10.1186/1756-8722-6-61
6. Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction–a novel mechanism for maintaining vascular function. J Hematol Oncol. (2014) 7:80. doi: 10.1186/s13045-014-0080-6
7. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
8. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. CANTOS trial group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. The New England journal of medicine. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
9. Liu M, Saredy J, Zhang R, Shao Y, Sun Y, Yang WY, et al. Approaching inflammation paradoxes-proinflammatory cytokine blockages induce inflammatory regulators. Front Immunol. (2020) 11:554301. doi: 10.3389/fimmu.2020.554301
10. Lai B, Wang J, Fagenson A, Sun Y, Saredy J, Lu Y, et al. Twenty novel disease group-specific and 12 new shared macrophage pathways in eight groups of 34 diseases including 24 inflammatory organ diseases and 10 types of tumors. Front Immunology. (2019) 10:2612. doi: 10.3389/fimmu.2019.02612
11. Johnson C, Drummer Iv C, Shan H, Shao Y, Sun Y, Lu Y, et al. A novel subset of CD95+ pro-inflammatory macrophages overcome miR155 deficiency and may serve as a switch from metabolically healthy obesity to metabolically unhealthy obesity. Front Immunol. (2021) 11:619951. doi: 10.3389/fimmu.2020.619951
12. Fu H, Vadalia N, Xue ER, Johnson C, Wang L, Yang WY, et al. Thrombus leukocytes exhibit more endothelial cell-specific angiogenic markers than peripheral blood leukocytes do in acute coronary syndrome patients, suggesting a possibility of trans-differentiation: a comprehensive database mining study. J Hematol Oncol. (2017) 10:74. doi: 10.1186/s13045-017-0440-0
13. Zhang R, Saredy J, Shao Y, Yao T, Liu L, Saaoud F, et al. End-stage renal disease is different from chronic kidney disease in upregulating ROS-modulated proinflammatory secretome in PBMCs - A novel multiple-hit model for disease progression. Redox Biol. (2020) 34:101460. doi: 10.1016/j.redox.2020.101460
14. Zhang D, Fang P, Jiang X, Nelson J, Moore JK, Kruger WD, et al. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ Res. (2012) 111:37–49. doi: 10.1161/CIRCRESAHA.112.269472
15. Zhang D, Jiang X, Fang P, Yan Y, Song J, Gupta S, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation. (2009) 120:1893–902. doi: 10.1161/CIRCULATIONAHA.109.866889
16. Fang P, Li X, Shan H, Saredy JJ, Cueto R, Xia J, et al. Ly6C+ inflammatory monocyte differentiation partially mediates hyperhomocysteinemia-induced vascular dysfunction in type 2 diabetic db/db Mice. Arteriosclerosis, Thrombosis, Vascular Biology. (2019) 39:2097–119. doi: 10.1161/ATVBAHA.119.313138
17. Fang P, Zhang D, Cheng Z, Yan C, Jiang X, Kruger WD, et al. Hyperhomocysteinemia potentiates hyperglycemia-induced inflammatory monocyte differentiation and atherosclerosis. Diabetes. (2014) 63:4275–90. doi: 10.2337/db14-0809
18. Yang J, Fang P, Yu D, Zhang L, Zhang D, Jiang X, et al. Chronic kidney disease induces inflammatory CD40+ monocyte differentiation via homocysteine elevation and DNA hypomethylation. Circ Res. (2016) 119:1226–41. doi: 10.1161/CIRCRESAHA.116.308750
19. Li X, Wang L, Fang P, Sun Y, Jiang X, Wang H, et al. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J Biol Chem. (2018) 293:11033–45. doi: 10.1074/jbc.RA118.002752
20. Wang X, Li YF, Nanayakkara G, Shao Y, Liang B, Cole L, et al. Lysophospholipid receptors, as novel conditional danger receptors and homeostatic receptors modulate inflammation-novel paradigm and therapeutic potential. J Cardiovasc Transl Res. (2016) 9:343–59. doi: 10.1007/s12265-016-9700-6
21. Shao Y, Nanayakkara G, Cheng J, Cueto R, Yang WY, Park JY, et al. Lysophospholipids and their receptors serve as conditional DAMPs and DAMP receptors in tissue oxidative and inflammatory injury. Antioxid Redox Signal. (2018) 28:973–86. doi: 10.1089/ars.2017.7069
22. Li YF Li RS, Samuel SB, Cueto R, Li XY, Wang H, et al. Lysophospholipids and their G protein-coupled receptors in atherosclerosis. Frontiers in Bioscience (Landmark edition). (2016) 21:70–88. doi: 10.2741/4377
23. Li X, Fang P, Li Y, Kuo YM, Andrews AJ, Nanayakkara G, et al. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arteriosclerosis, Thrombosis, Vascular Biology. (2016) 36:1090–100. doi: 10.1161/ATVBAHA.115.306964
24. Lu Y, Sun Y, Drummer C. 4th, Nanayakkara GK, Shao Y, Saaoud F, et al. jabb Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells - Novel qualification markers for chronic disease risk factors and conditional DAMPs redox. Biology. (2019) 24:101221. doi: 10.1016/j.redox.2019.101221
25. Zhong C, Yang X, Feng Y, Yu J. Trained immunity: an underlying driver of inflammatory atherosclerosis. Front Immunol. (2020) 11:284. doi: 10.3389/fimmu.2020.00284
26. Fagenson AM, Xu K, Saaoud F, Nanayakkara G, Jhala NC, Liu L, et al. Liver ischemia reperfusion injury, enhanced by trained immunity, is attenuated in caspase 1/caspase 11 double gene knockout mice. Pathogens (Basel, Switzerland). (2020) 9:879. doi: 10.3390/pathogens9110879
27. Zasłona Z, O'Neill L. Cytokine-like roles for metabolites in immunity. Mol Cell. (2020) 78:814–23. doi: 10.1016/j.molcel.2020.04.002
28. Wang H, Jiang X, Yang F, Gaubatz JW, Ma L, Magera MJ, et al. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood. (2003) 101:3901–7. doi: 10.1182/blood-2002-08-2606
29. Jiang X, Yang F, Tan H, Liao D, Bryan RM. Jr, et al. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arteriosclerosis, Thrombosis, Vascular Biology. (2005) 25:2515–21. doi: 10.1161/01.ATV.0000189559.87328.e4
30. Jamaluddin MD, Chen I, Yang F, Jiang X, Jan M, Liu X, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood. (2007) 110:3648–55. doi: 10.1182/blood-2007-06-096701
31. Chiurchiù V, Leuti A, Maccarrone M. Bioactive lipids and chronic inflammation: managing the fire within. Front Immunol. (2018) 9:38. doi: 10.3389/fimmu.2018.00038
32. Chapman MJ, Orsoni A, Tan R, Mellett NA, Nguyen A, Robillard P, et al. LDL subclass lipidomics in atherogenic dyslipidemia: effect of statin therapy on bioactive lipids and dense LDL. J Lipid Res. (2020) 61:911–32. doi: 10.1194/jlr.P119000543
33. Yin Y, Li X, Sha X, Xi H, Li YF, Shao Y, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arteriosclerosis, Thrombosis, Vascular Biology. (2015) 35:804–16. doi: 10.1161/ATVBAHA.115.305282
34. Lopez-Pastrana J, Ferrer LM Li YF, Xiong X, Xi H, Cueto R, et al. Inhibition of caspase-1 activation in endothelial cells improves angiogenesis: a novel therapeutic potential for ischemia. J Biol Chem. (2015) 290:17485–94. doi: 10.1074/jbc.M115.641191
35. An D, Hao F, Zhang F, Kong W, Chun J, Xu X, et al. CD14 is a key mediator of both lysophosphatidic acid and lipopolysaccharide induction of foam cell formation. J Biol Chem. (2017) 292:14391–400. doi: 10.1074/jbc.M117.781807
36. Smyth SS, Kraemer M, Yang L, Van Hoose P, Morris AJ. Roles for lysophosphatidic acid signaling in vascular development and disease. Biochimica et biophysica acta Molecular and cell biology of lipids. (2020) 1865:158734. doi: 10.1016/j.bbalip.2020.158734
37. Green CD, Maceyka M, Cowart LA, Spiegel S. Sphingolipids in metabolic disease: The good, the bad, the unknown. Cell Metab. (2021) 33:1293–306. doi: 10.1016/j.cmet.2021.06.006
38. Cao J, Goossens P, Martin-Lorenzo M, Dewez F, Claes B, Biessen E, et al. Atheroma-specific lipids in ldlr−/− and apoe−/− mice using 2D and 3D Matrix-assisted laser desorption/ionization mass spectrometry imaging. J Am Soc Mass Spectrom. (2020) 31:1825–32. doi: 10.1021/jasms.0c00070
39. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. (2018) 128:2657–69. doi: 10.1172/JCI97943
40. Li X, Fang P, Yang WY, Wang H, Yang X. IL-35, as a newly proposed homeostasis-associated molecular pattern, plays three major functions including anti-inflammatory initiator, effector, and blocker in cardiovascular diseases. Cytokine. (2019) 122:154076. doi: 10.1016/j.cyto.2017.06.003
41. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
42. Hooftman A, O'Neill L. The Immunomodulatory Potential of the Metabolite Itaconate. Trends Immunol. (2019) 40:687–98. doi: 10.1016/j.it.2019.05.007
43. Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K, et al. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can J Physiol Pharmacol. (2017) 95:247–52. doi: 10.1139/cjpp-2016-0515
44. Li X, Shao Y, Sha X, Fang P, Kuo YM, Andrews AJ, et al. IL-35 (Interleukin-35) suppresses endothelial cell activation by inhibiting mitochondrial reactive oxygen species-mediated site-specific acetylation of H3K14 (Histone 3 Lysine 14). Arteriosclerosis, Thrombosis, Vascular Biology. (2018) 38:599–609. doi: 10.1161/ATVBAHA.117.310626
45. Cheng J, Nanayakkara G, Shao Y, Cueto R, Wang L, Yang WY, et al. Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv Exp Med Biol. (2017) 982:359–70. doi: 10.1007/978-3-319-55330-6_20
46. Nanayakkara GK, Wang H, Yang X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation - A novel concept. Arch Biochem Biophys. (2019) 662:68–74. doi: 10.1016/j.abb.2018.12.002
47. Yin Y, Pastrana JL Li X, Huang X, Mallilankaraman K, Choi ET, et al. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Frontiers in Bioscience (Landmark edition). (2013) 18:638–49. doi: 10.2741/4127
48. Ni D, Tang T, Lu Y, Xu K, Shao Y, Saaoud F, et al. Canonical secretomes, innate immune caspase-1-, 4/11-gasdermin D non-canonical secretomes and exosomes may contribute to maintain treg-ness for treg immunosuppression, tissue repair and modulate anti-tumor immunity via ROS pathways. Front Immunol. (2021) 12:678201. doi: 10.3389/fimmu.2021.678201
49. Zhang R, Xu K, Shao Y, Sun Y, Saredy J, Cutler E, et al. Tissue treg secretomes and transcription factors shared with stem cells contribute to a treg niche to maintain treg-ness with 80% innate immune pathways, and functions of immunosuppression and tissue repair. Front Immunol. (2021) 11:632239. doi: 10.3389/fimmu.2020.632239
50. Fu H, Sun Y, Shao Y, Saredy J, Cueto R, Liu L, et al. Interleukin 35 delays hindlimb ischemia-induced angiogenesis through regulating ros-extracellular matrix but spares later regenerative angiogenesis. Front Immunol. (2020) 11:595813. doi: 10.3389/fimmu.2020.595813
51. Falasca M, Ferro R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv Biol Regul. (2016) 60:88–93. doi: 10.1016/j.jbior.2015.10.003
52. Yang Q, Nanayakkara GK, Drummer C, Sun Y, Johnson C, Cueto R, et al. Low-intensity ultrasound-induced anti-inflammatory effects are mediated by several new mechanisms including gene induction, immunosuppressor cell promotion, and enhancement of exosome biogenesis and docking. Front Physiol. (2017) 8:818. doi: 10.3389/fphys.2017.00818
53. Yang Q, Zhang R, Tang P, Sun Y, Johnson C, Saredy J, et al. Ultrasound may suppress tumor growth, inhibit inflammation, and establish tolerogenesis by remodeling innatome via pathways of ROS. immune checkpoints, cytokines, trained immunity/tolerance. Journal Of Immunology Research. (2021)2021:6664453. doi: 10.1155/2021/6664453
54. Wang J, Lai B, Nanayakkara G, Yang Q, Sun Y, Lu Y, et al. Experimental data-mining analyses reveal new roles of low-intensity ultrasound in differentiating cell death regulatome in cancer and non-cancer cells via potential modulation of chromatin long-range interactions. Front Oncol. (2019) 9:600. doi: 10.3389/fonc.2019.00600
55. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature Communications. (2019) 10:1523. doi: 10.1038/s41467-019-09234-6
56. Jan M, Cueto R, Jiang X, Lu L, Sardy J, Xiong X, et al. Molecular processes mediating hyperhomocysteinemia-induced metabolic reprogramming, redox regulation and growth inhibition in endothelial cells. Redox Biol. (2021) 45:102018. doi: 10.1016/j.redox.2021.102018
57. Mai J, Nanayakkara G, Lopez-Pastrana J, Li X, Li YF, Wang X, et al. Interleukin-17A promotes aortic endothelial cell activation via transcriptionally and post-translationally activating p38 mitogen-activated protein kinase (MAPK) pathway. J Biol Chem. (2016) 291:4939–54. doi: 10.1074/jbc.M115.690081
58. Sha X, Meng S, Li X, Xi H, Maddaloni M, Pascual DW, et al. Interleukin-35 inhibits endothelial cell activation by suppressing MAPK-AP-1 pathway. J Biol Chem. (2015) 290:19307–18. doi: 10.1074/jbc.M115.663286
59. Li X, Fang P, Sun Y, Shao Y, Yang WY, Jiang X, et al. Anti-inflammatory cytokines IL-35 and IL-10 block atherogenic lysophosphatidylcholine-induced, mitochondrial ROS-mediated innate immune activation, but spare innate immune memory signature in endothelial cells. Redox Biol. (2020) 28:101373. doi: 10.1016/j.redox.2019.101373
60. Goncharov NV, Nadeev AD, Jenkins RO, & Avdonin PV. (2017). Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxidative Medicine And Cellular Longevity. (2017). 9759735. doi: 10.1155/2017/9759735
61. Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, et al. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol. (2009) 22:311–22. doi: 10.1177/039463200902200208
62. Li YF, Huang X, Li X, Gong R, Yin Y, Nelson J, et al. Caspase-1 mediates hyperlipidemia-weakened progenitor cell vessel repair. Frontiers in Bioscience (Landmark edition). (2016) 21:178–91. doi: 10.2741/4383
63. Ferrer LM, Monroy AM, Lopez-Pastrana J, Nanayakkara G, Cueto R, Li YF, et al. Caspase-1 plays a critical role in accelerating chronic kidney disease-promoted neointimal hyperplasia in the carotid artery. J Cardiovasc Transl Res. (2016) 9:135–44. doi: 10.1007/s12265-016-9683-3
64. Li YF, Nanayakkara G, Sun Y, Li X, Wang L, Cueto R, et al. Analyses of caspase-1-regulated transcriptomes in various tissues lead to identification of novel IL-1β-, IL-18- and sirtuin-1-independent pathways. J Hematol Oncol. (2017) 10:40. doi: 10.1186/s13045-017-0406-2
65. Wang L, Fu H, Nanayakkara G, Li Y, Shao Y, Johnson C, et al. Novel extracellular and nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: a comprehensive database mining study. J Hematol Oncol. (2016) 9:122. doi: 10.1186/s13045-016-0351-5
66. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nature Reviews Immunology. (2020) 20:143–57. doi: 10.1038/s41577-019-0228-2
67. Keller M, Rüegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. (2008) 132:818–31. doi: 10.1016/j.cell.2007.12.040
68. Lorey MB, Rossi K, Eklund KK, Nyman TA, Matikainen S. Global characterization of protein secretion from human macrophages following non-canonical caspase-4/5 inflammasome activation. Molecular & Cellular Proteomics: MCP. (2017) 16:S187–S199. doi: 10.1074/mcp.M116.064840
69. Alhouayek M, Masquelier J, Muccioli GG. Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends Pharmacol Sci. (2018) 39:586–604. doi: 10.1016/j.tips.2018.02.011
70. Virtue A, Johnson C, Lopez-Pastraña J, Shao Y, Fu H, Li X, et al. MicroRNA-155 deficiency leads to decreased atherosclerosis, increased white adipose tissue obesity, and non-alcoholic fatty liver disease: a novel mouse model of obesity paradox. J Biol Chem. (2017) 292:1267–87. doi: 10.1074/jbc.M116.739839
71. Saaoud F, Wang J, Iwanowycz S, Wang Y, Altomare D, Shao Y, et al. Bone marrow deficiency of mRNA decaying protein Tristetraprolin increases inflammation and mitochondrial ROS but reduces hepatic lipoprotein production in LDLR knockout mice. Redox Biol. (2020) 37:101609. doi: 10.1016/j.redox.2020.101609
72. Yang XF, Weber GF, Cantor H A. novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity. (1997) 7:629–39. doi: 10.1016/S1074-7613(00)80384-2
73. Yang Y, Yang F, Xiong Z, Yan Y, Wang X, Nishino M, et al. An N-terminal region of translationally controlled tumor protein is required for its antiapoptotic activity. Oncogene. (2005) 24:4778–88. doi: 10.1038/sj.onc.1208666
74. Yang Y, Xiong Z, Zhang S, Yan Y, Nguyen J, Ng B, et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. The Biochemical Journal. (2005) 392:135–143. doi: 10.1042/BJ20050698
75. Yan Y, Xiong Z, Zhang S, Song J, Huang Y, Thornton AM, et al. CD25high T cells with a prolonged survival inhibit development of diabetes. Int J Immunopathol Pharmacol. (2008) 21:767–80. doi: 10.1177/039463200802100401
76. Xiong Z, Song J, Yan Y, Huang Y, Cowan A, Wang H, et al. Higher expression of Bax in regulatory T cells increases vascular inflammation. Frontiers in bioscience: a journal and virtual library. (2008) 13:7143–55. doi: 10.2741/3217
77. Xiong Z, Yan Y, Song J, Fang P, Yin Y, Yang Y, et al. Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis. (2009) 203:401–8. doi: 10.1016/j.atherosclerosis.2008.07.041
78. Rakocevic J, Orlic D, Mitrovic-Ajtic O, Tomasevic M, Dobric M, Zlatic N, et al. Endothelial cell markers from clinician's perspective. Exp Mol Pathol. (2017) 102:303–13. doi: 10.1016/j.yexmp.2017.02.005
79. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. (2015) 107:321–30. doi: 10.1093/cvr/cvv147
80. Kappelmayer J, & Nagy B. Jr (2017). The Interaction of Selectins and PSGL-1 as a Key Component in Thrombus Formation and Cancer Progression. BioMed research international, (2017). 6138145. doi: 10.1155/2017/6138145
81. Hu Y, Kiely JM, Szente BE, Rosenzweig A, & Gimbrone MA, Jr. (2000)E-selectin-dependent signaling via the mitogen-activated protein kinase pathway in vascular endothelial cells. Journal of immunology (Baltimore, Md.: 1950), 165: 2142–2148. doi: 10.4049/jimmunol.165.4.2142
82. Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacological reports: PR. (2009) 61:22–32. doi: 10.1016/S1734-1140(09)70004-0
83. Chistiakov DA, Bobryshev YV, Orekhov AN. Changes in transcriptome of macrophages in atherosclerosis. J Cell Mol Med. (2015) 19:1163–73. doi: 10.1111/jcmm.12591
84. Hu T, Zhou R, Zhao Y, Wu G. Integrin α6/Akt/Erk signaling is essential for human breast cancer resistance to radiotherapy. Sci Rep. (2016) 6:33376. doi: 10.1038/srep33376
85. Abair TD, Bulus N, Borza C, Sundaramoorthy M, Zent R, Pozzi A. Functional analysis of the cytoplasmic domain of the integrin {alpha}1 subunit in endothelial cells. Blood. (2008) 112:3242–54. doi: 10.1182/blood-2007-12-126433
86. Kannan K, Stewart RM, Bounds W, Carlsson SR, Fukuda M, Betzing KW, et al. Lysosome-associated membrane proteins h-LAMP1 (CD107a) and h-LAMP2 (CD107b) are activation-dependent cell surface glycoproteins in human peripheral blood mononuclear cells which mediate cell adhesion to vascular endothelium. Cell Immunol. (1996) 171:10–9. doi: 10.1006/cimm.1996.0167
87. Popson SA, Ziegler ME, Chen X, Holderfield MT, Shaaban CI, Fong AH, et al. Interferon-induced transmembrane protein 1 regulates endothelial lumen formation during angiogenesis. Arteriosclerosis, Thrombosis, Vascular Biology. (2014) 34:1011–9. doi: 10.1161/ATVBAHA.114.303352
88. Watt SM, Chan JY. CD164–a novel sialomucin on CD34+ cells. Leuk Lymphoma. (2000) 37:1–25. doi: 10.3109/10428190009057625
89. Kleemann R, Verschuren L, van Erk MJ, Nikolsky Y, Cnubben NH, Verheij ER, et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biol. (2007) 8:R200. doi: 10.1186/gb-2007-8-9-r200
90. Devilard E, Xerri L, Dubreuil P, Lopez M, Reymond N. Nectin-3 (CD113) interacts with Nectin-2 (CD112) to promote lymphocyte transendothelial migration. PLoS ONE. (2013) 8:e77424. doi: 10.1371/journal.pone.0077424
91. Bansal A, Sanchez DJ, Nimgaonkar V, Sanchez D, Riscal R, Skuli N, et al. Gamma-Glutamyltransferase 1 Promotes Clear Cell Renal Cell Carcinoma Initiation and Progression. Molecular Cancer Research: MCR. (2019) 17:1881–92. doi: 10.1158/1541-7786.MCR-18-1204
92. Shen H, Wu N, Nanayakkara G, Fu H, Yang Q, Yang WY, et al. Co-signaling receptors regulate T-cell plasticity and immune tolerance. Frontiers in bioscience (Landmark edition). (2019) 24:96–132. doi: 10.2741/4710
93. Winkels H, Meiler S, Lievens D, Engel D, Spitz C, Bürger C, et al. CD27 co-stimulation increases the abundance of regulatory T cells and reduces atherosclerosis in hyperlipidaemic mice. Eur Heart J. (2017) 38:3590–9. doi: 10.1093/eurheartj/ehx517
94. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. (2007) 117:2974–82. doi: 10.1172/JCI31344
95. van Wanrooij EJ, van Puijvelde GH, de Vos P, Yagita H, van Berkel TJ, Kuiper J. Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arteriosclerosis, thrombosis, vascular biology. (2007) 27:204–10. doi: 10.1161/01.ATV.0000251007.07648.81
96. Franzini M, Corti A, Martinelli B, Del Corso A, Emdin M, Parenti GF, et al. Gamma-glutamyltransferase activity in human atherosclerotic plaques–biochemical similarities with the circulating enzyme. Atherosclerosis. (2009) 202:119–27. doi: 10.1016/j.atherosclerosis.2008.03.023
97. Karp DR, Carlisle ML, Mobley AB, Nichols TC, Oppenheimer-Marks N, Brezinschek RI, et al. Gamma-glutamyl transpeptidase is up-regulated on memory T lymphocytes. Int Immunol. (1999) 11:1791–800. doi: 10.1093/intimm/11.11.1791
98. Sommariva E, Stadiotti I, Casella M, Catto V, Dello Russo A, Carbucicchio C, et al. Oxidized LDL-dependent pathway as new pathogenic trigger in arrhythmogenic cardiomyopathy. EMBO Mol Med. (2021) 13:e14365. doi: 10.15252/emmm.202114365
99. Murshid A, Borges TJ, Lang BJ, Calderwood SK. The Scavenger Receptor SREC-I Cooperates with Toll-Like Receptors to Trigger Inflammatory Innate Immune Responses. Front Immunol. (2016) 7:226. doi: 10.3389/fimmu.2016.00226
100. Martín-Ventura JL, Madrigal-Matute J, Muñoz-Garcia B, Blanco-Colio LM, Van Oostrom M, Zalba G, et al. Increased CD74 expression in human atherosclerotic plaques: contribution to inflammatory responses in vascular cells. Cardiovasc Res. (2009) 83:586–94. doi: 10.1093/cvr/cvp141
101. Damås JK, Waehre T, Yndestad A, Otterdal K, Hognestad A, Solum NO, et al. Interleukin-7-mediated inflammation in unstable angina: possible role of chemokines and platelets. Circulation. (2003) 107:2670–6. doi: 10.1161/01.CIR.0000070542.18001.87
102. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. (2014) 46:e99. doi: 10.1038/emm.2014.38
103. Zimmer S, Steinmetz M, Asdonk T, Motz I, Coch C, Hartmann E, et al. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ Res. (2011) 108:1358–66. doi: 10.1161/CIRCRESAHA.111.243246
105. AbuSamra DB, Aleisa FA, Al-Amoodi AS, Jalal Ahmed HM, Chin CJ, Abuelela AF, et al. Not just a marker: CD34 on human hematopoietic stem/progenitor cells dominates vascular selectin binding along with CD44. Blood advances. (2017) 1:2799–816. doi: 10.1182/bloodadvances.2017004317
106. Donners MM, Wolfs IM, Olieslagers S, Mohammadi-Motahhari Z, Tchaikovski V, Heeneman S, et al. A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor-induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arteriosclerosis, Thrombosis, Vascular Biology. (2010) 30:2188–95. doi: 10.1161/ATVBAHA.110.213124
107. Nelson J, Wu Y, Jiang X, Berretta R, Houser S, Choi E, et al. Hyperhomocysteinemia suppresses bone marrow CD34+/VEGF receptor 2+ cells and inhibits progenitor cell mobilization and homing to injured vasculature-a role of β1-integrin in progenitor cell migration and adhesion. FASEB Journal. (2015) 29:3085–99. doi: 10.1096/fj.14-267989
108. Uhlén M, Karlsson MJ, Hober A, Svensson AS, Scheffel J, Kotol D, et al. The human secretome. Science Signaling. (2019) 12: eaaz0274. doi: 10.1126/scisignal.aaz0274
109. Fromer MW, Chang S, Hagaman A, Koko KR, Nolan RS, Zhang P, et al. The endothelial cell secretome as a novel treatment to prime adipose-derived stem cells for improved wound healing in diabetes. J Vasc Surg. (2018) 68:234–44. doi: 10.1016/j.jvs.2017.05.094
110. Kofler S, Nickel T, & Weis M. Role of cytokines in cardiovascular diseases: a focus on endothelial responses to inflammation. Clinical Science (London, England: 1979). (2005) 108:205–213. doi: 10.1042/CS20040174
111. Li X, Mai J, Virtue A, Yin Y, Gong R, Sha X, et al. IL-35 is a novel responsive anti-inflammatory cytokine–a new system of categorizing anti-inflammatory cytokines. PLoS One. (2012) 7:e33628. doi: 10.1371/journal.pone.0033628
112. Wei H, Sundararaman A, Dickson E, Rennie-Campbell L, Cross E, Heesom KJ, et al. Characterization of the polarized endothelial secretome. FASEB Journal. (2019) 33:12277–87. doi: 10.1096/fj.201900262R
113. Safdar A, Tarnopolsky MA. Exosomes as Mediators of the Systemic Adaptations to Endurance Exercise. Cold Spring Harb Perspect Med. (2018) 8:a029827. doi: 10.1101/cshperspect.a029827
114. Scheja L, Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nature Reviews Endocrinology. (2019) 15:507–24. doi: 10.1038/s41574-019-0230-6
115. Planavila A, Fernández-Solà J, Villarroya F. Cardiokines as modulators of stress-induced cardiac disorders. Adv Protein Chem Struct Biol. (2017) 108:227–56. doi: 10.1016/bs.apcsb.2017.01.002
116. Gonzalez-Gil AM, Elizondo-Montemayor L. The Role of Exercise in the Interplay between Myokines, Hepatokines, Osteokines, Adipokines, and Modulation of Inflammation for Energy Substrate Redistribution and Fat Mass Loss: A Review. Nutrients. (2020) 12:1899. doi: 10.3390/nu12061899
117. L PK, Kandoi S, Misra R, S V, K R, Verma RS. The mesenchymal stem cell secretome: A new paradigm towards cell-free therapeutic mode in regenerative medicine. Cytokine & Growth Factor Reviews. (2019) 46:1–9. doi: 10.1016/j.cytogfr.2019.04.002
118. Xu K, Yang WY, Nanayakkara GK, Shao Y, Yang F, Hu W, et al. GATA3, HDAC6, and BCL6 regulate FOXP3+ treg plasticity and determine treg conversion into either novel antigen-presenting cell-like treg or Th1-treg. Front Immunol. (2018) 9:45. doi: 10.3389/fimmu.2018.00045
119. Fedorova O, Petukhov A, Daks A, Shuvalov O, Leonova T, Vasileva E, et al. Orphan receptor NR4A3 is a novel target of p53 that contributes to apoptosis. Oncogene. (2019) 38:2108–22. doi: 10.1038/s41388-018-0566-8
120. Katoh M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clinical Science (London, England: 1979). (2019) 133:953–970. doi: 10.1042/CS20180845
121. Lu N, Malemud CJ. Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. Int J Mol Sci. (2019) 20:3792. doi: 10.3390/ijms20153792
122. Nailwal NP, Doshi GM. Role of intracellular signaling pathways and their inhibitors in the treatment of inflammation. Inflammopharmacology. (2021) 29:617–40. doi: 10.1007/s10787-021-00813-y
123. Dunn J, Simmons R, Thabet S, Jo H. The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int J Biochem Cell Biol. (2015) 67:167–76. doi: 10.1016/j.biocel.2015.05.001
124. Pirri D, Fragiadaki M, & Evans PC. Diabetic atherosclerosis: is there a role for the hypoxia-inducible factors? Bioscience Reports. (2020) 40: BSR20200026. doi: 10.1042/BSR20200026
125. Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. (2013) 6:19. doi: 10.1186/1756-8722-6-19
126. Pattillo CB, Pardue S, Shen X, Fang K, Langston W., Jourd'heuil D, et al. ICAM-1 cytoplasmic tail regulates endothelial glutathione synthesis through a NOX4/PI3-kinase-dependent pathway. Free Radical Biology & Medicine. (2010) 49:1119–28. doi: 10.1016/j.freeradbiomed.2010.06.030
127. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
128. Sun Y, Lu Y, Saredy J, Wang X, Drummer Iv C, Shao Y, et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. (2020) 37:101696. doi: 10.1016/j.redox.2020.101696
129. Shao Y, Chernaya V, Johnson C, Yang WY, Cueto R, Sha X, et al. Metabolic Diseases Downregulate the Majority of Histone Modification Enzymes, Making a Few Upregulated Enzymes Novel Therapeutic Targets—“Sand Out and Gold Stays.” J Cardiovasc Transl Res. (2016) 9:49–66. doi: 10.1007/s12265-015-9664-y
130. Włodarczyk M, Sobolewska-Włodarczyk A, Cygankiewicz AI, Jacenik D, Krajewska WM, Stec-Michalska K, et al. G protein-coupled receptor 55 (GPR55) expresses differently in patients with Crohn's disease and ulcerative colitis. Scand J Gastroenterol. (2017) 52:711–5. doi: 10.1080/00365521.2017.1298834
131. Fondevila MF, Fernandez U, Gonzalez-Rellan MJ, Da Silva Lima N, Buque X, Gonzalez-Rodriguez A, et al. The L-α-Lysophosphatidylinositol/G Protein-Coupled Receptor 55 System Induces the Development of Nonalcoholic Steatosis and Steatohepatitis. Hepatology (Baltimore, Md). (2021) 73:606–24. doi: 10.1002/hep.31290
132. Kang S, Lee AY, Park SY, Liu KH, Im DS. O-1602 Promotes Hepatic Steatosis through GPR55 and PI3 Kinase/Akt/SREBP-1c Signaling in Mice. Int J Mol Sci. (2021) 22:3091. doi: 10.3390/ijms22063091
133. Wang Y, Pan W, Wang Y, Yin Y. The GPR55 antagonist CID16020046 protects against ox-LDL-induced inflammation in human aortic endothelial cells (HAECs). Archives of biochemistry and biophysics. (2020) 681:108254. doi: 10.1016/j.abb.2020.108254
134. Kurano M, Kobayashi T, Sakai E, Tsukamoto K, Yatomi Y. Lysophosphatidylinositol, especially albumin-bound form, induces inflammatory cytokines in macrophages. FASEB Journal. (2021) 35:e21673. doi: 10.1096/fj.202100245R
135. Saliba SW, Jauch H, Gargouri B, Keil A, Hurrle T, Volz N, et al. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J Neuroinflammation. (2018) 15:322. doi: 10.1186/s12974-018-1362-7
136. Stančić A, Jandl K, Hasenöhrl C, Reichmann F, Marsche G, Schuligoi R, et al. The GPR55 antagonist CID16020046 protects against intestinal inflammation. Neurogastroenterology Motility. (2015) 27:1432–45. doi: 10.1111/nmo.12639
137. Kargl J, Brown AJ, Andersen L, Dorn G, Schicho R, Waldhoer M, et al. A selective antagonist reveals a potential role of G protein-coupled receptor 55 in platelet and endothelial cell function. J Pharmacol Exp Ther. (2013) 346:54–66. doi: 10.1124/jpet.113.204180
138. Li A, Sun Y, Drummer C. 4th, Lu Y, Yu D, Zhou Y, et al. Increasing upstream chromatin long-range interactions may favor induction of circular RNAs in LysoPC-activated human aortic endothelial cells. Front physiology. (2019) 10:433. doi: 10.3389/fphys.2019.00433
139. Johnson C, Drummer C 4th, Virtue A, Gao T, Wu S, Hernandez M, et al. increased expression of resistin in microrna-155-deficient white adipose tissues may be a possible driver of metabolically healthy obesity transition to classical obesity. Frontiers in physiology. (2018) 9:1297. doi: 10.3389/fphys.2018.01297
Keywords: transcriptomic analysis, inflammation, secretomes, RNA-Seq analysis, aortic endothelial cell
Citation: Xu K, Shao Y, Saaoud F, Gillespie A, Drummer C IV, Liu L, Lu Y, Sun Y, Xi H, Tükel Ç, Pratico D, Qin X, Sun J, Choi ET, Jiang X, Wang H and Yang X (2021) Novel Knowledge-Based Transcriptomic Profiling of Lipid Lysophosphatidylinositol-Induced Endothelial Cell Activation. Front. Cardiovasc. Med. 8:773473. doi: 10.3389/fcvm.2021.773473
Received: 10 September 2021; Accepted: 04 October 2021;
Published: 29 November 2021.
Edited by:
Hong S. Lu, University of Kentucky, United StatesReviewed by:
Ting Zhou, University of Wisconsin-Madison, United StatesCopyright © 2021 Xu, Shao, Saaoud, Gillespie, Drummer, Liu, Lu, Sun, Xi, Tükel, Pratico, Qin, Sun, Choi, Jiang, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Yang, eGZ5YW5nQHRlbXBsZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.