 Xiao-dan Xia1†
Xiao-dan Xia1† Maggie Wang
Maggie Wang Gui-qing Wang
Gui-qing Wang Da-wei Zhang
Da-wei Zhang- 1Department of Orthopedics, The Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People's Hospital, Qingyuan, China
- 2School of Economics, Management and Law, University of South China, Hengyang, China
- 3Group on the Molecular and Cell Biology of Lipids, Department of Pediatrics, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
Proprotein convertase subtilisin/kexin type 9 (PCSK9) promotes degradation of low-density lipoprotein receptor (LDLR) and plays a central role in regulating plasma levels of LDL cholesterol levels, lipoprotein(a) and triglyceride-rich lipoproteins, increasing the risk of cardiovascular disease. Additionally, PCSK9 promotes degradation of major histocompatibility protein class I and reduces intratumoral infiltration of cytotoxic T cells. Inhibition of PCSK9 increases expression of LDLR, thereby reducing plasma levels of lipoproteins and the risk of cardiovascular disease. PCSK9 inhibition also increases cell surface levels of major histocompatibility protein class I in cancer cells and suppresses tumor growth. Therefore, PCSK9 plays a vital role in the pathogenesis of cardiovascular disease and cancer, the top two causes of morbidity and mortality worldwide. Monoclonal anti-PCSK9 antibody-based therapy is currently the only available treatment that can effectively reduce plasma LDL-C levels and suppress tumor growth. However, high expenses limit their widespread use. PCSK9 promotes lysosomal degradation of its substrates, but the detailed molecular mechanism by which PCSK9 promotes degradation of its substrates is not completely understood, impeding the development of more cost-effective alternative strategies to inhibit PCSK9. Here, we review our current understanding of PCSK9 and focus on the regulation of its expression and functions.
Introduction
Plasma low-density lipoprotein cholesterol (LDL-C) levels are positively correlated to the risk of cardiovascular disease (CVD). Statins, the currently most-prescribed lipid-lowering drug, reduce cardiovascular events by 20–40%. However, there is mounting evidence that about 50% of statin-treated patients and 80% of very high-risk patients do not achieve the recommended cholesterol values even with the highest tolerated dose. Furthermore, up to 20% of statin-treated people show statin intolerance, and about 10–12% of cases exhibit maladaptive side effects (1). Thus, there is an urgent need to develop a non-statin-based cholesterol-lowering drug.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) plays a critical role in regulating plasma cholesterol homeostasis through promoting LDL receptor (LDLR) degradation (Figure 1). Gain-of-function mutations in PCSK9 cause autosomal dominant hypercholesterolemia, while loss-of-function mutations are associated with reduced plasma levels of LDL-C (2–6). PCSK9 also promotes major histocompatibility protein class I (MHCI) degradation and suppresses immune attacks to tumors (7) (Figure 1). Therefore, PCSK9 plays a central role in the pathogenesis of CVD and cancers. In addition, it has been reported that PCSK9, especially extra-hepatic PCSK9, can recruit inflammatory cells and induce local inflammation (8, 9). Here, we summarize the latest advances in PCSK9 and focus on its role in lipid metabolism and cancer immunotherapy and the molecular mechanisms for the regulation of PCSK9 expression.
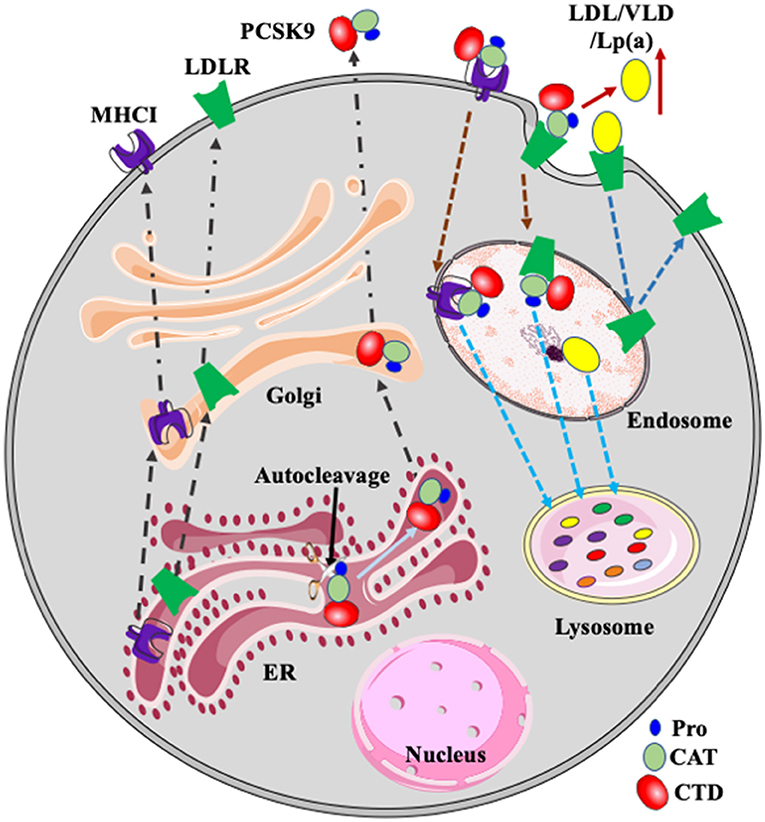
Figure 1. PCSK9, LDLR, and MHCI. PCSK9 is auto-cleaved in the ER. Mature PCSK9 is transported to the Golgi and then secreted. PCSK9 binds to LDLR and MHCI on the cell surface. After, the complex is delivered to endosomes via endocytosis and then transported to the lysosome for degradation. LDLR binds to its ligands such as LDL, VLDL, and Lp(a) and then the receptor/ligand complex enters cells via receptor-mediated endocytosis and is delivered to the endosome. In the acidic endosomal environment, the ligand, such as LDL, is released from LDLR and transported to the lysosome for degradation. LDLR is recycled to plasma membrane. PCSK9-promoted degradation of LDLR increases plasma levels of LDL and Lp(a). Pro, prodomain; CAT, catalytic domain; CTD, C-terminal domain.
PCSK9 Function
Human and mouse PCSK9 is encoded by the PCSK9/Pcsk9 gene located at chromosome 1p32.3 and 4C7, containing thirteen and twelve exons that encode a 692 and 694-amino acid PCSK9 protein, respectively (10). PCSK9 is highly conserved among mammals, including chimpanzee, monkey, camel, alpaca, rat, and mouse, with an approximate amino acid identity of 99, 96, 82, 81, 77, and 77%, respectively. The majority of identified gain-of-function and loss-of-function mutations occur in entirely conserved residues, such as gain-of-function mutations D35Y, L108R, S127R, D129G, N157K, R215H, F216L, R218S, A220T, R357H, D374Y, N425S, R468W, R496W and R499H, and loss-of-function mutations R104C, R105Q, G106R, G236S, L253F, G316C, N354I and S462P (2, 4, 11, 12). However, loss-of-function mutations R46L and R93C and gain-of-function mutations R96L, A168E, R499H, and S636R are not conserved between human and mouse or rat PCSK9. The correspondence residues of Arg46, Arg93, Arg96, Ala168, Arg499, and Ser636 in mouse/ rat PCSK9 are Pro49/Pro48, Gln93/Gln92, His99/His95, Thr211/Thr167, Trp512/Arg498, and Ser639/Ser635, respectively. Whether the difference in these residues affects PCSK9 function, however, is unclear.
PCSK9 contains a signal peptide [amino acid (aa) 1-30], a prodomain (Pro) (aa 31-152), a catalytic domain (CAT) (aa 153-425) and a Cys and His-rich C-terminal domain (CTD) (aa 426-692) (13) (Figure 1). The CAT contains a classical serine protease catalytic triad of Asp186, His226 and Ser386 and is highly conserved with the CAT of other proprotein convertases. PCSK9 is self-cleaved by the CAT at the FAQ152/SIPK site in the endoplasmic reticulum (ER). After autocleavage, the prodomain is associate with the CAT and masks the catalytic activity of PCSK9. This process is required for PCSK9 maturation and secretion. Compared to the other subtilisin-like serine protease family members, the CTD of PCSK9 is unique and contains multiple potential protein-protein interaction motifs (13). The CTD is positively charged and may interact with the negatively charged ligand-binding repeats of LDLR in the acidic endosomal environment, blocking recycling of the receptor (14, 15). In addition, partial deletion of the CTD markedly damages PCSK9 secretion, indicating its essential role in this process (16, 17). However, the underlying mechanism is unclear. Our recent study suggests that the CTD mediates PCSK9 secretion, possibly via a coat protein complex II (COPII) component Sec24 (17).
PCSK9 induces degradation of LDLR and its family members, including very low-density lipoprotein receptor (VLDLR), apolipoprotein E receptor 2 (ApoER2), and LDLR-related protein 1 (LRP1) (10, 18–20), thereby playing an essential role in lipid metabolism. However, PCSK9 at a physiological concentration can effectively degrade LDLR but not other LDLR family members in cultured cells (19, 20). Similarly, PCSK9 degrades LDLR but not LRP1 in mouse liver (21), but it can regulate visceral adipogenesis likely through promoting VLDLR degradation in mouse adipose tissues (22). Conversely, loss of functional PCSK9 in humans does not cause any known abnormality except for reduced plasma cholesterol levels (3, 6). Nevertheless, these findings indicate that PCSK9's action on its substrates is cell/tissue-type and/or species-dependent.
The CAT of PCSK9 directly binds to the epidermal growth factor precursor homology domain A (EGF-A) of LDLR on the cell surface. After endocytosis, PCSK9 remains bound to LDLR in the acidic endosome, preventing LDLR from recycling and redirecting the receptor to the lysosome for degradation (Figure 1) (19, 21, 23, 24). PCSK9 can also promote LDLR degradation via an intracellular pathway, especially when overexpressed in cultured cells (25). Of note, evolocumab and alirocumab are humanized monoclonal anti-PCSK9 antibodies targeting the catalytic domain of PCSK9. They block PCSK9 binding to LDLR on the cell surface and do not affect the intracellular pathway. On the other hand, Inclisiran is a small siRNA that targets PCSK9 mRNA and reduces its expression. It can inhibit both the intracellular and extracellular pathways. However, evolocumab, alirocumab, and Inclisiran all markedly reduce plasma LDL cholesterol levels in patients to a similar degree (26–28). Therefore, the extracellular pathway appears to be mainly responsible for the LDL-lowering effect of PCSK9 inhibition.
PCSK9 directs its substrates for lysosomal degradation (Figure 1), which does not require its proteolytic activity (29). Both caveolae-dependent and clathrin-mediated endocytosis have been reported to play an important role in the endocytosis of PCSK9/LDLR complex in HepG2 cells (30–32). Additionally, DeVay et al. reported that amyloid beta precursor-like protein 2 (APLP2) directly bound to PCSK9 and targeted the PCSK9/LDLR complex to lysosomes for degradation in HepG2 cells (33), while other studies showed that APLP2 did not affect PCSK9-promoted LDLR degradation in mice, HepG2, or Huh7 cells (34, 35). Differences in approaches and/or models used in these studies may cause these discrepancies. However, further studies are required to elucidate the underlying mechanism for PCSK9-promoted LDLR degradation.
PCSK9 and LDL
Plasma LDL is eliminated from circulation primarily via hepatic LDLR. Upon binding, the LDL-LDLR complex is internalized via the clathrin-coated pits and subsequently delivered to endosomes, where LDL is released from LDLR and then transported to lysosomes for degradation, while LDLR is recycled to the cell surface to clear more LDL. Mutations in LDLR cause familial hypercholesterolemia (FH), characterized by elevated plasma levels of cholesterol, particularly LDL cholesterol, and increased risk of CVD (36). PCSK9 promotes LDLR lysosomal degradation. Circulating PCSK9 preferentially degrades LDLR in mouse liver (37), which may be due to hepatic heparan sulfate proteoglycans (HSPG). HSPG can recruit circulating PCSK9 to hepatocytes, enhancing its action on LDLR (38). Knockout of PCSK9 increases hepatic LDLR levels, reduces plasma LDL cholesterol levels, and improves sensitivity to statin treatment in mice (37).
LDL is derived from VLDL catabolism, which is a triglyceride-enriched lipoprotein exclusively secreted by the liver. Triglycerides in VLDL are hydrolyzed by lipoprotein lipase, resulting in intermediate-density lipoprotein (IDL), which can be further metabolized to LDL (39). PCSK9 can directly interact with apoB100, the main structural lipoprotein on VLDL, and inhibit apoB100 degradation, thereby promoting its secretion. Knockout of PCSK9 reduces hepatic apoB secretion and plasma LDL cholesterol levels in Ldlr−/−/Apobec1−/− mice (40). Conversely, gain-of-function mutant PCSK9 increases apoB100 secretion in a rat hepatoma-derived cell line, McArdle-7777 cells (41). These findings indicate that, in addition to reducing the availability of hepatic LDLR, PCSK9 may promote the production of LDL through increasing secretion of VLDL. However, hepatocytes typically produce apoB100 in abundance, and the rate-limiting step in VLDL secretion is lipidation of apoB100. Therefore, the physiological role of PCSK9-promoted VLDL secretion may not be significant in vivo.
PCSK9 is also expressed in extra-hepatic cells and tissues, such as the vascular smooth muscles cells (VSMCs), macrophages, endothelial cells, the pancreas, the kidneys, the intestine and the central nervous system (10). The arterial vessel has the maximal secretion of PCSK9 at the lowest level of shear stress that occurs in the aortic branching and aorta-iliac bifurcation regions of the mouse aorta. Cultured VSMCs produce substantially more PCSK9 than endothelial cells (42). Elevated PCSK9 in VSMCs can reduce LDLR levels in VSMCs and macrophages (42, 43), which may impair LDL clearance and accelerate retention of LDL in VSMCs and macrophages in the location of arterial bifurcation. PCSK9 is also expressed in and secreted from pancreatic beta cells. However, inhibition of PCSK9 does not affect insulin secretion in the human EndoC-betaH1 beta cell line and mice even though PCSK9 promotes LDLR degradation in beta cells (44). Together, these findings indicate a cell-type-specific function of PCSK9.
PCSK9 and Triglyceride-Rich Lipoproteins
Elevated plasma levels of triglyceride-rich lipoproteins and their remnants are an independent risk factor for atherosclerosis and CVD. Hepatic LDLR binds to apoE on remnant lipoprotein particles to mediate their clearance (36). Therefore, PCSK9 can affect plasma triglyceride and remnant cholesterol levels through the LDLR pathway. Elevated plasma PCSK9 levels are positively associated with plasma TG levels in humans upon a short-term high-fructose intake (45). Treatments with PCSK9 inhibitors increase clearance of VLDL remnants in patients (46). Alirocumab, a fully human PCSK9 monoclonal antibody, reduces LDL particles by 56.3% in human patients. This reduction is partly due to an increase in the clearance rate of IDL particles, thereby decreasing the conversion of IDL to LDL (27).
PCSK9 is expressed in the intestine and can affect chylomicron metabolism. Knockout of PCSK9 in mice significantly reduces lymphatic apoB48 secretion and increases secretion of TG-rich large chylomicrons. Clearance of chylomicron remnants is also increased in Pcsk9−/− mice (47). Rashid et al. further demonstrated that PCSK9 promoted chylomicron secretion through both LDLR-dependent and -independent pathways in mice and a human enterocyte cell line, CaCo-2 cells, such as increasing the expression of apoB, microsomal triglyceride transfer protein and lipogenic genes in enterocytes (48). However, inhibition of PCSK9 by evolocumab or alirocumab does not significantly affect VLDL production or postprandial plasma levels of apoB48 and triglycerides in healthy humans or patients with hypercholesterolemia (49). Conversely, in patients with type-II diabetes mellitus, evolocumab reduces postprandial apoB48 levels even though the effect on postprandial triglyceride levels is not significant, while alirocumab can significantly reduce fasting plasma apoB48 and TG levels and postprandial TG levels (50). Plasma PCSK9 levels are also correlated with plasma apoB48-containing TG-rich lipoproteins in men with insulin resistance (51). However, the impact of PCSK9 on plasma levels of TG-rich lipoproteins, such as VLDL and chylomicrons, is much less than its effect on plasma LDL (52). This may be because VLDL and chylomicron remnants can also be effectively cleared by an LDLR-independent pathway, such as LRP1 (53). In summary, extracellular PCSK9 regulates LDLR-mediated catabolism, and intracellular PCSK9 modulates apoB secretion; the two pathways might act in a complementary fashion to regulate TG-rich lipoproteins metabolism with the extracellular pathway as the primary contributor.
PCSK9 and Lipoprotein(a)
Elevated plasma lipoprotein(a) [Lp(a)] levels are a highly prevalent risk factor for cardiovascular disease, especially for myocardial infarction, atherosclerotic stenosis and aortic valve stenosis. Lp(a) is an apoB100–containing lipoprotein particle covalently linked to the plasminogen-like glycoprotein apo(a) by a disulfide bond (54). The statin treatment and lifestyle interventions hardly affect circulating Lp(a) levels, which brings a real challenge for successfully managing elevated Lp(a) levels in patients. Conversely, PCSK9 inhibitors dramatically reduce plasma Lp(a) levels up to ~35% in patients (54, 55). Inhibition of PCSK9 reduces the risk of coronary heart disease to a much greater degree in patients with a high plasma Lp(a) level compared to patients with a low plasma Lp(a) level (23 vs. 7%) (54). However, how PCSK9 regulates Lp(a) levels is unclear. Plasma Lp(a) levels are determined by its production and clearance. Lp(a) is removed from circulation through LDLR, SR-BI, and LRP1(54). Lp(a) levels are increased in FH patients who carry loss-of-function mutant LDLR. Overexpression of LDLR enhances Lp(a) clearance in mice. PCSK9 promotes LDLR degradation and reduces Lp(a) catabolism in HepG2 cells and primary fibroblasts (31). These findings indicate that PCSK9 can regulate plasma Lp(a) levels in a LDLR-dependent pathway. However, while statin treatment increases LDLR levels, it has no significant effect on plasma Lp(a) levels in patients. Furthermore, lymphocytes from patients with homozygous FH can effectively take up Lp(a) particles, and PCSK9 inhibitors can lower circulating Lp(a) in homozygous FH patients (56), indicating a LDLR-independent pathway. Lp(a) usually cannot compete with LDL for binding to LDLR. It has been proposed that PCSK9 inhibition can promote hepatic clearance of Lp(a) through LDLR-mediated endocytosis when plasma LDL levels are low (57). Nevertheless, although the mechanism by which PCSK9 inhibitors reduce Lp(a) levels remains to be determined, the fact that PCSK9 inhibitors provide an additional beneficial effect in lowering circulating Lp(a) may confer protection against CVD from a clinical perspective. Further work is needed to understand the role of PCSK9 in the overall metabolism of apoB-containing lipoproteins, especially for Lp(a).
PCSK9 and Cancer Cell Immunity
MHCI on the cell surface presents specific antigens to T-cell receptors (TCR) on CD8+ T cells, activating CD8+ T cell-mediated cell killing. After antigen presentation, MHCI enters cells via endocytosis and is recycled to present new antigens. On the other hand, programmed cell death protein 1 receptor (PD-1) on the cell surface interacts with its ligand programmed death-ligand 1 (PD-L1) on T cells to act as an immune checkpoint, which suppresses the immune response and prevents indiscriminate attacks (58).
During tumor development, cancer cells evolve various mechanisms to escape immune attacks, such as stimulating immune checkpoint targets and reducing tumor-specific antigen (TSA) presentation. Monoclonal anti-PD1 or PDL1 antibodies, which inhibit the PD1 pathway and promote antitumor immune response, have been approved to treat various types of cancers such as melanoma, bladder cancer, non-small cell lung cancer, and renal cell carcinoma. On the other hand, MHCI on the cancer cell surface presents TSA to CD8+ cells, activating CD8+ cell-mediated cancer cell killing (58). Recently, Liu et al. reported that PCSK9 bound to MHCI on the cancer cell surface and redirected it to the lysosome for degradation, thereby reducing cell surface MHCI levels and TSA presentation. Knockout of PCSK9 or inhibition of circulating PCSK9 increased CD8+ T cell intratumoral infiltration and enhanced antitumor activity of CD8+ T cells in mice. This suppressed tumor growth of several mouse cancer cell lines, including 4T1 (breast cancer), MC38 (colon adenocarcinoma), and the PD-1 inhibitor-resistant cancer cell line, MC38R, in mice (7). In addition, knockout of PCSK9 suppressed tumor growth in Ldlr−/− mice, indicating a LDLR-independent mechanism. However, Yuan et al. reported that inhibition of PCSK9 attenuated MC38 tumor growth in a LDLR-dependent manner. They found that LDLR directly interacted with T-cell receptor complex (TCR) and increased its cell surface levels. Inhibition of PCSK9 increased LDLR and TCR levels in MC38 tumors, enhancing TCR signaling and CD8+ T cell-dependent cancer cell killing in mice. The reason for the discrepancy is unclear. Yuan et al. did not report whether MHCI levels in MC38 tumors were affected by PCSK9, but they found that PCSK9 inhibition did not alter MHCI levels in B16F10 melanoma cells (59), indicating that PCSK9 regulates MHCI levels in a tumor/cell type-specific manner. These findings suggest that PCSK9 may control tumor growth through the LDLR and the MHCI pathway independently and/or collaboratively.
PCSK9 produced locally in vascular cells and cardiomyocytes can promote inflammation via the NF-κB signaling pathway (60, 61). Chronic inflammation increases the risk of cancer. PCSK9 expression is high in various cancers, such as hepatocellular carcinoma, gastricadenocarcinoma and prostate cancer cell lines (62–64). Zhang et al. reported that PCSK9 expression is positively correlated with poor prognosis. The authors found that PCSK9 suppressed apoptosis in cultured hepatoma-derived cell lines through the Bax/Bcl-2/Caspase9/3 pathway (64). Consistently, inhibition of PCSK9 by siRNA promotes apoptosis in a human lung adenocarcinoma cell line, A549 cells, via activation of caspase-3 and stimulation of ER stress (62). On the other hand, silencing PCSK9 by siRNA reduces radiation-induced apoptosis in prostate cancer cell lines, PC-3 and LnCap and thus enhances cell viability (65). However, the authors did not investigate the potential contribution of inflammation in these studies. Nevertheless, these studies suggest that PCSK9 plays a complex role in cancer development via different mechanisms. Cancer risk analysis of subjects carrying loss-of-function and gain-of-function mutations in PCSK9 will further reveal and confirm the role of PCSK9 in cancer progression.
Regulation of PCSK9
PCSK9 plays a critical role in regulating circulating lipid homeostasis and MHCI-dependent immune responses. The complexity of PCSK9's functions indicates that its activity is strictly regulated by various mechanisms at multiple levels.
Regulation of PCSK9 Expression
Epigenetically, binding of forkhead box O (FoxO) 3 to the promoter of PCSK9 recruits Sirt6 to deacetylate histone H3, suppressing PCSK9 expression (Figure 2) (66). On the other hand, histone nuclear factor P (HINFP) binds to a HINFP motif in 20 bp upstream of the sterol regulatory element motif (SRE) in PCSK9 promoter, promoting histone H4 acetylation to activate sterol regulatory element-binding protein 2 (SREBP2)-mediated upregulation of PCSK9 transcription (67). Furthermore, PCSK9 promoter is methylated. Alcohol use disorder (AUD) causes hypomethylation in PCSK9 promoter and consequently reduces PCSK9 expression and plasma cholesterol levels in a mouse model of AUD, which may partially contribute to the protective effect on CVD risk observed in light alcohol users (68).
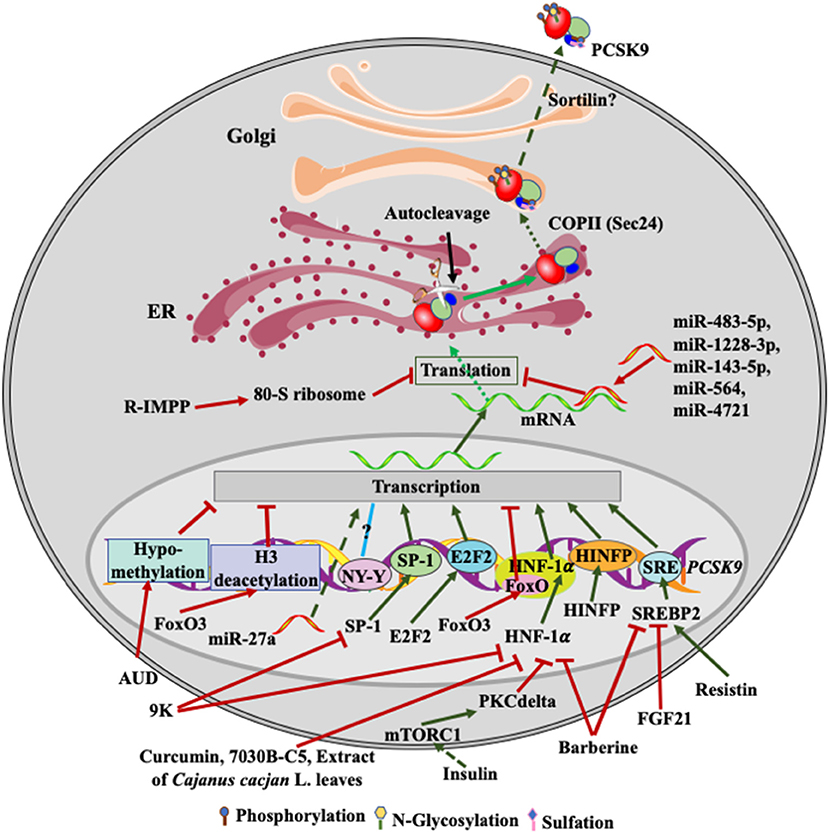
Figure 2. Regulation of PCSK9 expression. Transcriptional factors, such as SREBP2, HNF-1α, SP-1and E2F2, upregulate PCSK9 transcription. FGF21 and resistin inhibit and increase SREBP2-mediated transcription of PCSK9, respectively. Barberine reduces PCSK9 expression via suppressing the activity of SREBP2 and HNF-1α on PCSK9 transcription. 9K suppresses PCSK9 expression through SP1 and HNF-1α, while Curcumin, 7030B-C5 inhibits HNF-1α-induced transcription of PCSK9. Alcohol use causes hypomethylation of PCSK9 promoter and then reduces PCSK9 expression. Insulin activates mTROC1 and then PKCδ to suppress PCSK9 transcription via HNF-1α. miR-483-5p, miR-1228-3p, miR-143-5p, miR-564, and miR-4721 bind to the 3'UTR of PCSK9 mRNA, reducing PCSK9 expression, while miR27a somehow increases PCSK9 expression. R-IMPP inhibits 80S ribosome and reduces PCSK9 expression. After autocleavage in the ER, PCSK9 is transported to the Golgi via classical COPII vesicles. There, PCSK9 undergoes posttranslational modifications, such as phosphorylation, glycosylation, and sulfation. Mature PCSK9 is then secreted into the extra cellular environment.
At the transcriptional level, both SREBP1 and SREBP2 have been reported to bind to SRE in PCSK9 promoter and thus upregulate PCSK9 expression in cultured cells; however, PCSK9 is predominantly regulated by SREPB2 in vivo (69–71). Statin treatment activates the transcriptional activity of SREBP2 and thus increases the expression of LDLR and PCSK9 (71). However, Poirier et al. reported that the expression of PCSK9 in the rodent central nervous system was regulated in a SREBP2-independnet manner (72). PCSK9 promoter also contains a binding site of the transcription factor hepatocyte nuclear factor 1 alpha (HNF1α) (70, 73). Silencing of HNF1α but not HNF1β significantly reduced PCSK9 expression. Furthermore, insulin increases the mTORC1 signaling pathway and activates PKCδ, which reduces HNF1α-mediated expression of PCSK9 and increases hepatic LDLR levels (74). Li et al. reported that HNF1α worked cooperatively with SREBP2 to activate PCSK9 transcription since mutations in the HNF1α-binding site significantly reduced SREBP2-mediated upregulation of PCSK9 transcription (73). However, the HNF1α binding site is just 28 bp upstream of the SREBP2 bindings site. Mutations in the HNF1α binding site may affect the integrity of SRE and then indirectly impair SREBP2 binding.
The HNF1α binding site in the promoter of PCSK9 contains a consensus FoxO binding site (Figure 2). FoxO3 can inhibit PCSK9 expression competitively via inhibiting HNF1α-mediated upregulation of PCSK9 (66). In addition, Lai et al. reported that transcription factor E2F2 could bind to the PCSK9 promoter region and upregulate its expression under a condition of feeding or high cellular cholesterol levels (75). The promoter region of PCSK9 also contains NF-Y and SP1 binding sites upstream of SRE (71). The putative NF-Y binding site appears not to affect PCSK9 expression. However, the SP1 site may mediate basal transcription of PCSK9 since it is not required for the sterol-dependent regulation of PCSK9 expression, but mutations in this site reduce PCSK9 expression (69). A variant, C-332C>A, in the SP1 binding site, increases PCSK9 expression by approximately 2.5-fold independent of lovastatin treatment (76).
Several lines of evidence demonstrate the regulation of PCSK9 transcription by small molecules (Figure 2). Curcumin and the methanol extract of Cajanus cajan L. leaves reduce HNF1α levels and downregulate PCSK9 transcription in HepG2 cells (77, 78). Epigallocatechingallate (humans, Sprague-Dawl rats, HepG2 and Huh7 cells), ascorbic acid (mice, HepG2 and Huh7 cells), Pinostrobin (HepG2 cells), and tanshinone IIA (HepG2 cells) reduce PCSK9 expression in a FoxO3a-dependent manner, probably via attenuating HNF1α-mediated activation of PCSK9 expression and/or methylation in the promoter region of PCSK9 (79–82). A small molecular, 7030B-C5, also reduces PCSK9 expression in HepG2 cells and mice mainly through the FoxO1 and HNF1α pathway (83). In addition, Berberine reduces PCSK9 expression mainly through attenuating SREBP2 and HNF1α-mediated upregulation of PCSK9 transcription in HepG2 cells (73), which may account for its cholesterol-lowering effect. Conversely, a berberine derivative, 9k, downregulates PCSK9 expression via suppressing the transcriptional activity of HNF1α and/or SP1 in HepG2 cells (84). Fibroblast growth factor 21 inhibits the transcriptional activity of SREBP2, thereby reducing PCSK9 expression in mouse liver (85). In addition, glucagon, bile acids, fibrate, and oncostatin M have been reported to inhibit PCSK9 expression at the transcriptional levels in HepG2 cells (86–88), but the underlying mechanisms are unclear. On the other hand, resistin, a small cysteine-rich protein secreted from macrophages and adipose tissues, increases PCSK9 transcription via the SREBP2 pathway in HepG2 cells and primary human hepatocytes (89). Nevertheless, these findings indicate the potential of inhibiting PCSK9 transcription as an avenue to lower plasma cholesterol levels, reducing CVD risk. However, the aforementioned transcriptional factors also regulate the transcription of many other proteins that play important roles in various physiological processes. For example, inhibition of SREBP2 reduces LDLR expression, attenuating LDL clearance. Inhibition of HNF1α activity does not affect LDLR expression. However, HNF1α can act as a tumor suppressor, and its expression is reduced in patients with liver malignancies (90). Thus, it is a big challenge to develop small molecules that can specifically modify PCSK9 expression at the transcriptional level.
Post-transcriptionally, the expression of PCSK9 is regulated by microRNAs (miRNA) (Figure 2). miR-483-5p targets the 3′-UTR of the PCSK9 mRNA, reduces PCSk9 expression and decreases plasma cholesterol levels in HepG2 cells and mice (91). Similarly, miR-224, miR-191, miR-222, miR-1228-3p, miR-564, miR-4721, miR-337-3p, and miR-143-5p can reduce PCSK9 expression through targeting its 3′-UTR in cultured cells, such as Huh7, HepG2 and BON-1 cells (92–94). A common variant, 1420C>G, decreases the inhibitory effect of miR-1228-3p and miR-143-5p on PCSK9 expression, reducing plasma levels of PCSK9 and LDL cholesterol (95). Similarly, Los et al. identified several variants in PCSK9 3′-UTR in FH patients. The variant 345C>T impairs binding of miR-4721 and miR-564 to PCSK9 3′-UTR and increases PCSK9 expression (93). Conversely, miR-27a upregulates PCSK9 expression, possibly through binding to the upstream of PCSK9 promoter in HepG2 cells (96). It is of note that a single miRNA often targets multiple genes as binding of miRNAs to their target genes requires seed pairing of as few as six nucleotides or even imperfect seed pairing (97). Thus, one of the key issues of miRNA-based therapies is their potential off-target effect.
Compared to miRNA, siRNAs bind to their complementary sequence in mRNA that completely matches their antisense strand, thereby specifically reducing the expression of their target genes. Phase III trials of Inclisiran, a chemically modified siRNA that targets PCSK9 mRNA, shows a promising lipid-lowering effect. Subcutaneous injection of Inclisiran reduces plasma LDL-C levels up to 50% in heterozygous FH patients without any major side-effects (26). Inclisiran, which requires twice-yearly administration, may reduce the cost of PCSK9 inhibitors compared to the current PCSK9 monoclonal antibody therapy that needs administration every 2–4 weeks. However, it is still a financial burden as a primary prevention measure for all eligible patients. In addition, siRNAs, particularly at a high dose, also exhibit miRNA-like off-target activity (98). Additionally, duplex siRNA can trigger an innate immune response in Toll-like receptors-dependent and independent mechanisms (99). Patients with Inclisiran treatment do show a slightly increased rate of mild-to-moderate bronchitis (4.3 vs. 0.7% for Inclisiran and placebo, respectively) (26). Therefore, possible long-term side effects of using siRNAs as a lifelong primary prevention strategy need to be assessed.
A small molecule, R-IMPP, can selectively target human 80S ribosome and inhibit PCSK9 translation. R-IMPP significantly reduces the protein level of PCSK9, thereby increasing LDLR levels and LDL uptake in Huh7 cells (100). However, the therapeutic potential of R-IMPP is uncertain because ribosomes are the core of protein translation machinery and not an ideal therapeutic target.
Most recently, Liu et al. reported that the blood flow rate regulated PCSK9 expression through the toll-like receptor 4-MyD88-NF-κB signaling pathway in the rabbit thoracic aorta. Low-flow state increased, whereas high-flow state reduced the mRNA and protein level of PCSK9 in vascular cells. Interestingly, they observed an opposite effect on the expression of LDLR (101), indicating that the impact of flow rate is independent of SREBP2. Interestingly, knockdown of PCSK9 suppressed, while overexpression of PCSK9 enhanced the toll-like receptor 4-NF-κB signaling pathway and inflammation in the atherosclerotic lesions of apoE−/− mice (60). It will be of interest to see if the increased expression of PCSK9 under the low-flow state promotes the toll-like receptor 4-NF-κB signaling pathway in the thoracic aorta.
Circulating PCSK9 is mainly produced by hepatocytes. Blood flow rate may not have a similar effect on PCSK9 expression in hepatocytes since hepatocytes, unlike aortic vascular cells, are not directly exposed to blood flow. On the other hand, blood flow rate equals blood flow divided by cross-section area. Atherosclerosis causes blood vessels to harden and to narrow, which increases blood flow rate. It would be of interest to assess whether PCSK9 expression in vascular cells near and at atherosclerotic lesion area is reduced due to the increased blood flow rate, which could lead to a beneficial outcome since vascular cell PCSK9 can promote inflammation.
Regulation of PCSK9 Secretion
Although multiple tissues express PCSK9, circulating PCSK9 is mainly secreted from the liver (102). Loss-of-function PCSK9 mutations such as G236S, S462P, and C679X reduce its secretion, while gain-of-function mutations such as E32K enhance PCSK9 secretion (17, 103). Furthermore, circulating PCSK9 is rapidly cleared from the blood with a half-life of about 5 min in mice (37), indicating that targeting PCSK9 secretion is a promising therapeutic strategy. However, the machinery system controlling PCSK9 secretion is still elusive.
PCSK9 undergoes autocleavage in the ER (104), which is essential for its maturation and secretion. However, the enzymatic activity is not required for this processing. Mutant PCSK9 that losses its catalytic activity is retained in the ER, but coexpression of prodomain with catalytic dead mutant PCSK9 rescues its secretion in HepG2 cells (29). In addition, deletion of part of the C-terminal domain of PCSK9 impairs its secretion but does not affect its autocleavage in cultured human hepatocytes, such as HepG2 and Huh7 cells (17, 105), indicating that the autocleavage is not sufficient to support PCSK9 secretion. After autocleavage, the cleaved N-terminal PRO is associated with the catalytic domain and functions as an intramolecular chaperone, guaranteeing the correct folding of the catalytic domain in the ER. This step is believed to be the rate-limiting step for PCSK9 mature and secretion (103).
PCSK9 is transported from the ER to the Golgi via the classical COPII vesicles. The lack of SEC24, one of COPII components, significantly reduces PCSK9 secretion in mice and cultured human hepatocytes, HepG2 and Huh7 cells (17, 106). However, PCSK9 is a secretory protein located in the ER lumen, while SEC24 is located in the cytosol. Therefore, a cargo receptor is required to bridge the interaction between PCSK9 and SEC24. Emmer et al. reported that a cargo receptor Surf4 facilitated secretion of PCSK9 that was overexpressed in HEK293 cells. They found that Surf4 co-immunoprecipitated with PCSK9, and knockout of Surf4 significantly reduced the amount of PCSK9 detected in culture medium (107). Surf4 is a transmembrane protein that mainly resides in the ER membrane. It contains five putative transmembrane domains, an ER lumen-exposed N-terminus that binds cargo proteins within the lumen, and a cytoplasmic domain that interacts with COPII components, facilitating cargo sorting into COPII vesicles. However, we found that knockdown of Surf4 in cultured immortalized human hepatocytes, HepG2 and Huh7 cells, did not impair endogenous PCSK9 secretion (108). This discrepancy may be caused by different types of cells used in the two studies. We investigated endogenous PCSK9 secretion from cultured hepatocytes, HepG2 and Huh7 cells, while Emmer et al. studied the effect of Surf4 on secretion of PCSK9 overexpressed in HEK293 cells that do not express endogenous PCSK9. Furthermore, we found that knockdown of Surf4 in mouse liver had no significant effect on plasma and hepatic PCSK9 levels. In liver-specific Surf4 knockout mice, the levels of PCSK9 in plasma and liver homogenate were also comparable to that in the wild-type mice (109). Therefore, Surf4 is not required for endogenous PCSK9 secretion.
The C-terminal domain of PCSK9 has been implicated in its secretion. Loss-of-function mutations such as E498K and S462P located in the C-terminus of PCSK9 damage its secretion (12, 103). Deletion of the entire C-terminal PCSK9 from amino acids 456 to 692 does not impair PCSK9 secretion. Conversely, removing part of the C-terminal region (from amino acids 457 to 528 or 608 to 692) damages PCSK9 secretion (16, 17, 105). Furthermore, the deletion of part of or the entire hinge region that connects the catalytic domain and the C-terminal domain also significantly reduces PCSK9 secretion, indicating the important role of this region. SEC24 silencing significantly reduces the secretion of the wild-type but not mutant PCSK9 without the C-terminal domain, indicating that the C-terminal region of PCSK9 may be involved in SEC24-facilitated PCSK9 secretion (17). Further studies are required to elucidate how PCSK9 is sorted into COPII vesicles. Most recently, Rogers et al. reported that dynamin-related protein1 (DRP1)-mediated ER remodeling involved in PCSK9 secretion (110). Inhibition of DRP1 by mitochondrial division inhibitor 1 or knockout hepatic DRP1 markedly reduced PCSK9 secretion in HepG2 cells and mice.
After delivery to the Golgi apparatus, PCSK9 undergoes various posttranslational modifications and is then packed into secretory vesicles. The vesicles are delivered to and fused with the plasma membrane, releasing PCSK9 into the extracellular milieu. Gustafsen et al. reported that sortilin co-immunoprecipitated with PCSK9, and the two proteins were colocalized in the trans-Golgi network in HepG2 cells. Knockout of sortilin significantly reduced plasma PCSK9 levels in mice and reduced PCSK9 secretion from mouse primary hepatocytes. The author further showed that PCSK9 levels were positively correlated with sortilin levels in human serum. Thus, they concluded that sortilin interacted with PCSK9 in the trans-Golgi network, facilitating PCSK9 secretion (111). On the other hand, Butkinaree et al. reported that plasma levels of PCSK9 were comparable in sortilin knockout mice and wild-type littermates, and sortilin had no effect on PCSK9-promoted LDLR degradation in HepG2 and Huh7 cells. Instead, PCSK9 induced sortilin degradation (35). The reason for this discrepancy is unclear. The mice used in the Gustafsen study were C57BL/6J background, while Butkinaree et al. did not report their mouse background. Nevertheless, these findings reveal the complexity of the molecular mechanisms of PCSK9 secretion.
Posttranslational Modifications of PCSK9
PCSK9 is predicted to be phosphorylated on serine, threonine, asparagine, and lysine residues by PhosphoSitePlus (www.phosphosite.org). Mass spectrometry analysis of plasma samples confirms this prediction. Furthermore, Dewpura et al. reported that PCSK9 was partially phosphorylated on serine residues at positions 47 and 688 by a Golgi casein kinase-like kinase in a cell-type dependent manner, with the highest phosphorylation in HepG2 cell (~70%), followed by Huh 7 cells (~54%), HEK293 cells (~23%), and CHO-K1cells (none). Phosphorylation may protect PCSK9 against proteolysis and increase its stability in Huh 7 cells (112). Meanwhile, this finding also indicates that serine phosphorylation is not required for PCSK9's action on LDLR since PCSK9 purified from CHO-K1 cells is unphosphorylated and can effectively promote LDLR degradation (38). It has also been reported that PCSK9 was phosphorylated on serine residues at positions 47, 666, 668, and 688 by family with sequence similarity 20, member C (FAM20C), which increased PCSK9 secretion and enhanced its ability to promote LDLR degradation in HepG2 cells (113). However, phosphorylation at these sites was also not required for PCSK9-promoted LDLR degradation since mutant PCSK9 that lost phosphorylation at the four serine residues still could stimulate LDLR degradation. In addition, FAM20C phosphorylates serine residue in a consensus Ser-x-Glu motif present in many secreted proteins (114). Thus, it cannot be ruled out that FAM20C may indirectly affect PCSK9 secretion and function via phosphorylation of other proteins.
PCSK9 is N-glycosylated on asparagine residue at position 533 and sulfated on tyrosine residues (104). Detailed analysis revealed that the N-glycosylation at Asn533 and sulfation at Tyr38 were not required for PCSK9 processing, secretion and function in HepG2 and Huh 7 cells (115). Treatment of cells with tunicamycin that inhibits N-glycosylation or chlorate that inhibits tyrosine sulfation had no effect on PCSK9 expression and secretion in the human hepatocyte cell line, Huh7 cells. When overexpressed, mutant PCSK9 that lost the N-glycosylation and Tyr sulfation sites alone or together could be efficiently secreted and promote LDLR degradation in Huh7 and HepG2 cells (116). However, the secreted mutant PCSK9 appeared to promote LDLR degradation less effectively than the wild-type protein, suggesting that N-glycosylation may enhance the ability of PCSK9 to stimulate LDLR degradation.
Plasma levels of PCSK9 in subjects carrying phosphomannose mutase 2 (PMM2) variants (p.R141H and p.P69S) are significantly reduced by approximately 42% compared to the controls, which might contribute to hypolipidemia observed in these patients (116). The PMM2 variants may affect N-glycosylation of PCSK9 and its secretion in vivo, even though removal or inhibition of N-glycosylation in PCSK9 does not affect its secretion in cultured cells. Alternatively, these variants may impair PCSK9 secretion indirectly by affecting unknown factors that are important for PCSK9 secretion since PMM2 is required for the synthesis of GDP-mannose, a mannose donor for N-glycosylation. Analysis of secretion of N-glycosylation defective PCSK9 mutant in Pcsk9−/− mice might provide a clue for the potential role of N-glycosylation in PCSK9 secretion in vivo. Nevertheless, these studies indicate that posttranslational modifications, including phosphorylation, sulfation and N-glycosylation, may affect but are not required for PCSK9 processing, secretion, stability and function.
Conclusion and Perspectives
PCSK9 regulates plasma cholesterol levels and tumor-specific antigen presentation primarily through promoting LDLR and MHCI degradation, respectively. Of note, the lack of PCSK9 in humans does not cause any known notable side effects (3, 6). Therefore, PCSK9 is a promising therapeutic target to reduce the risk of the top two leading causes of mortality worldwide, cardiovascular disease and cancer. Various strategies have been or are being developed to inhibit PCSK9 (117–120) (Table 1). Current PCSK9 inhibitors, evolocumab and alirocumab, are humanized monoclonal anti-PCSK9 antibodies. They can significantly reduce plasma LDL cholesterol levels and cardiovascular events in patients with hypercholesterolemia and suppress tumor growth in mice. Inclisiran, a chemically synthesized siRNA targeting PCSK9 mRNA, also significantly reduces plasma LDL cholesterol levels by about 30–50% with only two injections each year. However, these strategies are expensive, limiting their widespread use. Other strategies, such as CRISPR-Cas9 gene editing and PCSK9 vaccine, are only in preclinical studies or phase I clinical trials (119, 120). Therefore, there is an urgent need for further research to elucidate the underlying mechanisms for PCSK9's impact on lipid metabolism and cancer growth. For example, (1) PCSK9 binds to LDLR with a much higher affinity at the acidic endosomal environment to block LDLR recycling. Does PCSK9 bind to MHCI in a pH-dependent manner? (2) PCSK9, LDLR and MHCI do not contain a lysosomal targeting signal; how is PCSK9/LDLR and PCSK9/MHCI complex redirected from the endosome to the lysosome for degradation? (3) Circulating PCSK9 is mainly secreted from hepatocytes and then promotes LDLR and MHCI degradation. What machinery system assists PCSK9 secretion? (4) HSPG facilitates PCSK9-promoted hepatic LDLR degradation. Is there a cofactor assisting PCSK9's action on MHCI? Answering these questions is critical to the development of innovative and more cost-effective treatment options to inhibit PCSK9-promoted degradation of LDLR and MHCI.
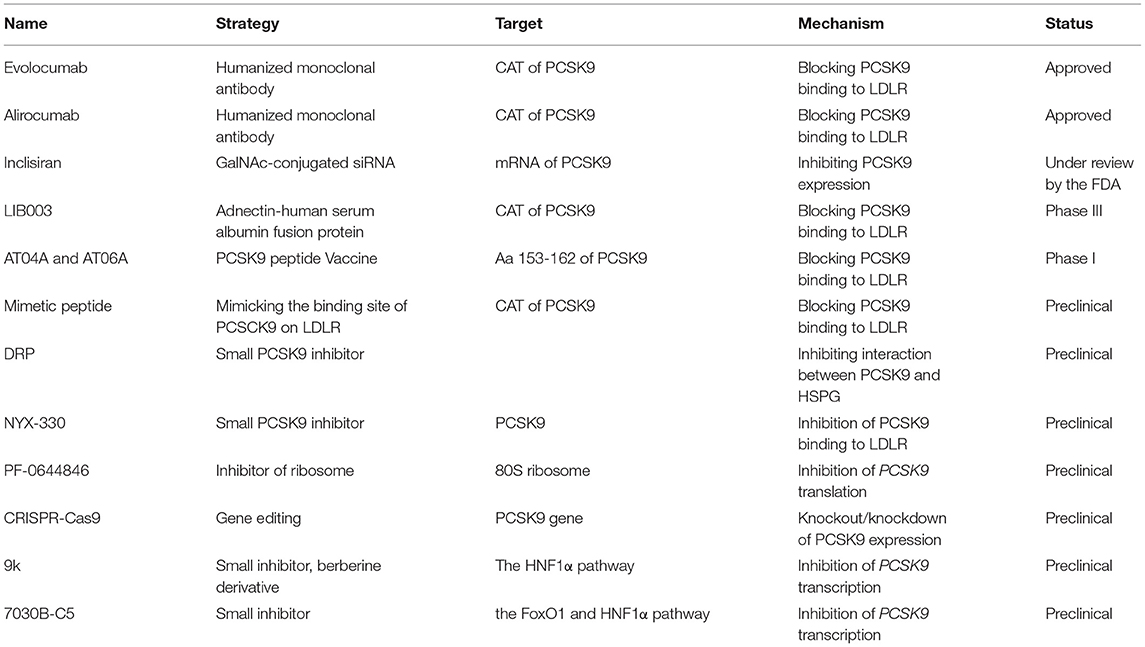
Table 1. PSCK9 inhibitors.
Author Contributions
X-dX and Z-sP wrote the initial draft. G-qW and D-wZ supervised the final version. H-mG participated in the discussion and the preparation of the manuscript. MW reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-06479) and Canadian Institutes of Health Research (PS 155994). X-dX and G-qW were supported by funding from Qingyuan People's Hospital.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fitchett DH, Hegele RA, Verma S. Cardiology patient page. Statin intolerance. Circulation. (2015) 131:e389–91. doi: 10.1161/CIRCULATIONAHA.114.013189
2. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. (2003) 34:154–6. doi: 10.1038/ng1161
3. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. (2005) 37:161–5. doi: 10.1038/ng1509
4. Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. (2006) 79:514–23. doi: 10.1086/507488
5. Guo S, Xia XD, Gu HM, Zhang DW. Proprotein convertase subtilisin/Kexin-Type 9 and lipid metabolism. Adv Exp Med Biol. (2020) 1276:137–56. doi: 10.1007/978-981-15-6082-8_9
6. Lebeau P, Platko K, Al-Hashimi AA, Byun JH, Lhotak S, Holzapfel N, et al. Loss-of-function PCSK9 mutants evade the unfolded protein response sensor GRP78 and fail to induce endoplasmic reticulum stress when retained. J Biol Chem. (2018) 293:7329–43. doi: 10.1074/jbc.RA117.001049
7. Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. (2020) 588:693–8. doi: 10.1038/s41586-020-2911-7
8. Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci Rep. (2018) 8:2267. doi: 10.1038/s41598-018-20425-x
9. Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, et al. Local effects of human PCSK9 on the atherosclerotic lesion. J Pathol. (2016) 238:52–62. doi: 10.1002/path.4630
10. Seidah NG. The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. Glob Cardiol Sci Pract. (2017) 2017:e201702. doi: 10.21542/gcsp.2017.2
11. Guo Q, Feng X, Zhou Y. PCSK9 variants in familial hypercholesterolemia: a comprehensive synopsis. Front Genet. (2020) 11:1020. doi: 10.3389/fgene.2020.01020
12. Benjannet S, Hamelin J, Chretien M, Seidah NG. Loss- and gain-of-function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J Biol Chem. (2012) 287:33745–55. doi: 10.1074/jbc.M112.399725
13. Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. (2007) 14:413–9. doi: 10.1038/nsmb1235
14. Tveten K, Holla OL, Cameron J, Strom TB, Berge KE, Laerdahl JK, et al. Interaction between the ligand-binding domain of the LDL receptor and the C-terminal domain of PCSK9 is required for PCSK9 to remain bound to the LDL receptor during endosomal acidification. Hum Mol Genet. (2012) 21:1402–9. doi: 10.1093/hmg/ddr578
15. Yamamoto T, Lu C, Ryan RO. A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J Biol Chem. (2011) 286:5464–70. doi: 10.1074/jbc.M110.199042
16. Saavedra YG, Day R, Seidah NG. The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low-density lipoprotein receptor (LDLR) degradation pathway. J Biol Chem. (2012) 287:43492–501. doi: 10.1074/jbc.M112.394023
17. Deng SJ, Shen Y, Gu HM, Guo S, Wu SR, Zhang DW. The role of the C-terminal domain of PCSK9 and SEC24 isoforms in PCSK9 secretion. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158660. doi: 10.1016/j.bbalip.2020.158660
18. Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA. (2005) 102:5374–9. doi: 10.1073/pnas.0501652102
19. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. (2007) 282:18602–12. doi: 10.1074/jbc.M702027200
20. Gu HM, Adijiang A, Mah M, Zhang DW. Characterization of the role of EGF-A of low-density lipoprotein receptor in PCSK9 binding. J Lipid Res. (2013) 54:3345–57. doi: 10.1194/jlr.M041129
21. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. (2006) 116:2995–3005. doi: 10.1172/JCI29383
22. Roubtsova A, Munkonda MN, Awan Z, Marcinkiewicz J, Chamberland A, Lazure C, et al. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler Thromb Vasc Biol. (2011) 31:785–91. doi: 10.1161/ATVBAHA.110.220988
23. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. (2008) 105:1820–5. doi: 10.1073/pnas.0712064105
24. Zhang DW, Garuti R, Tang WJ, Cohen JC, Hobbs HH. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci USA. (2008) 105:13045–50. doi: 10.1073/pnas.0806312105
25. Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci USA. (2005) 102:2069–74. doi: 10.1073/pnas.0409736102
26. Wright RS, Ray KK, Raal FJ, Kallend DG, Jaros M, Koenig W, et al. Pooled patient-level analysis of inclisiran trials in patients with familial hypercholesterolemia or atherosclerosis. J Am Coll Cardiol. (2021) 77:1182–93. doi: 10.1016/j.jacc.2020.12.058
27. Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. (2017) 135:352–62. doi: 10.1161/CIRCULATIONAHA.116.025253
28. Bonaca MP, Nault P, Giugliano RP, Keech AC, Pineda AL, Kanevsky E, et al. Low-density lipoprotein cholesterol lowering with evolocumab and outcomes in patients with peripheral artery disease: insights from the FOURIER trial (further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk). Circulation. (2018) 137:338–50. doi: 10.1161/CIRCULATIONAHA.117.032235
29. McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. (2007) 282:20799–803. doi: 10.1074/jbc.C700095200
30. Wang Y, Huang Y, Hobbs HH, Cohen JC. Molecular characterization of proprotein convertase subtilisin/kexin type 9-mediated degradation of the LDLR. J Lipid Res. (2012) 53:1932–43. doi: 10.1194/jlr.M028563
31. Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem. (2015) 290:11649–62. doi: 10.1074/jbc.M114.611988
32. Jang H-D, Lee SE, Yang J, Lee H-C, Shin D, Lee H, et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur Heart J. (2020) 41:239–52. doi: 10.1093/eurheartj/ehz566
33. DeVay RM, Shelton DL, Liang H. Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J Biol Chem. (2013) 288:10805–18. doi: 10.1074/jbc.M113.453373
34. Fu T, Guan Y, Xu J, Wang Y. APP, APLP2 and LRP1 interact with PCSK9 but are not required for PCSK9-mediated degradation of the LDLR in vivo. Biochim Biophys Acta. (2017) 1862:883–9. doi: 10.1016/j.bbalip.2017.05.002
35. Butkinaree C, Canuel M, Essalmani R, Poirier S, Benjannet S, Asselin MC, et al. Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9 convertase-mediated low density lipoprotein receptor degradation but interact with each other. J Biol Chem. (2015) 290:18609–20. doi: 10.1074/jbc.M115.647180
36. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. (2015) 161:161–72. doi: 10.1016/j.cell.2015.01.036
37. Grefhorst A, McNutt MC, Lagace TA, Horton JD. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res. (2008) 49:1303–11. doi: 10.1194/jlr.M800027-JLR200
38. Gustafsen C, Olsen D, Vilstrup J, Lund S, Reinhardt A, Wellner N, et al. Heparan sulfate proteoglycans present PCSK9 to the LDL receptor. Nat Commun. (2017) 8:503. doi: 10.1038/s41467-017-00568-7
39. Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. (2009) 297:E271–88. doi: 10.1152/ajpendo.90920.2008
40. Sun H, Krauss RM, Chang JT, Teng BB. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J Lipid Res. (2018) 59:207–23. doi: 10.1194/jlr.M078360
41. Sun XM, Eden ER, Tosi I, Neuwirth CK, Wile D, Naoumova RP, et al. Evidence for effect of mutant PCSK9 on apolipoprotein B secretion as the cause of unusually severe dominant hypercholesterolaemia. Hum Mol Genet. (2005) 14:1161–9. doi: 10.1093/hmg/ddi128
42. Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal. (2015) 22:760–71. doi: 10.1089/ars.2014.6054
43. Ferri N, Tibolla G, Pirillo A, Cipollone F, Mezzetti A, Pacia S, et al. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis. (2012) 220:381–6. doi: 10.1016/j.atherosclerosis.2011.11.026
44. Peyot ML, Roubtsova A, Lussier R, Chamberland A, Essalmani R, Murthy Madiraju SR, et al. Substantial PCSK9 inactivation in beta-cells does not modify glucose homeostasis or insulin secretion in mice. Biochim Biophys Acta Mol Cell Biol Lipids. (2021) 1866:158968. doi: 10.1016/j.bbalip.2021.158968
45. Cariou B, Langhi C, Le Bras M, Bortolotti M, Le KA, Theytaz F, et al. Plasma PCSK9 concentrations during an oral fat load and after short term high-fat, high-fat high-protein and high-fructose diets. Nutr Metab. (2013) 10:4. doi: 10.1186/1743-7075-10-4
46. Hollstein T, Vogt A, Grenkowitz T, Stojakovic T, Marz W, Laufs U, et al. Treatment with PCSK9 inhibitors reduces atherogenic VLDL remnants in a real-world study. Vascul Pharmacol. (2019) 116:8–15. doi: 10.1016/j.vph.2019.03.002
47. Le May C, Kourimate S, Langhi C, Chetiveaux M, Jarry A, Comera C, et al. Proprotein convertase subtilisin kexin type 9 null mice are protected from postprandial triglyceridemia. Arterioscler Thromb Vasc Biol. (2009) 29:684–90. doi: 10.1161/ATVBAHA.108.181586
48. Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, et al. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation. (2014) 130:431–41. doi: 10.1161/CIRCULATIONAHA.113.006720
49. Watts GF, Gidding SS, Mata P, Pang J, Sullivan DR, Yamashita S, et al. Familial hypercholesterolaemia: evolving knowledge for designing adaptive models of care. Nat Rev Cardiol. (2020) 17:360–77. doi: 10.1038/s41569-019-0325-8
50. Taskinen MR, Bjornson E, Andersson L, Kahri J, Porthan K, Matikainen N, et al. Impact of proprotein convertase subtilisin/kexin type 9 inhibition with evolocumab on the postprandial responses of triglyceride-rich lipoproteins in type II diabetic subjects. J Clin Lipidol. (2020) 14:77–87. doi: 10.1016/j.jacl.2019.12.003
51. Drouin-Chartier JP, Tremblay AJ, Hogue JC, Lemelin V, Lamarche B, Couture P. Plasma PCSK9 correlates with apoB-48-containing triglyceride-rich lipoprotein production in men with insulin resistance. J Lipid Res. (2018) 59:1501–9. doi: 10.1194/jlr.M086264
52. Taskinen MR, Bjornson E, Kahri J, Soderlund S, Matikainen N, Porthan K, et al. Effects of Evolocumab on the Postprandial Kinetics of Apo (Apolipoprotein) B100- and B48-Containing Lipoproteins in Subjects With Type 2 Diabetes. Arterioscler Thromb Vasc Biol. (2020) 41:962–75 doi: 10.1161/ATVBAHA.120.315446
53. Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. (1998) 101:689–95. doi: 10.1172/JCI1240
54. O'Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. (2019) 139:1483–92. doi: 10.1161/CIRCULATIONAHA.118.037184
55. Farmakis I, Doundoulakis I, Pagiantza A, Zafeiropoulos S, Antza C, Karvounis H, et al. Lipoprotein(a) reduction with proprotein convertase subtilisin/kexin type 9 inhibitors: a systematic review and meta-analysis. J Cardiovasc Pharmacol. (2021) 77:397–407. doi: 10.1097/FJC.0000000000000963
56. Blom DJ, Harada-Shiba M, Rubba P, Gaudet D, Kastelein JJP, Charng MJ, et al. Efficacy and safety of alirocumab in adults with homozygous familial hypercholesterolemia: the ODYSSEY HoFH trial. J Am Coll Cardiol. (2020) 76:131–42. doi: 10.1016/j.jacc.2020.05.027
57. Stoekenbroek RM, Lambert G, Cariou B, Hovingh GK. Inhibiting PCSK9 - biology beyond LDL control. Nat Rev Endocrinol. (2018) 15:52–62. doi: 10.1038/s41574-018-0110-5
58. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. (2021) 21:298–312. doi: 10.1038/s41568-021-00339-z
59. Yuan J, Cai T, Zheng X, Ren Y, Qi J, Lu X, et al. Potentiating CD8(+) T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell. (2021) 12:240–60. doi: 10.1007/s13238-021-00821-2
60. Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-kappaB pathway. Atherosclerosis. (2017) 262:113–22. doi: 10.1016/j.atherosclerosis.2017.04.023
61. Guo Y, Yan B, Tai S, Zhou S, Zheng XL. PCSK9: associated with cardiac diseases and their risk factors? Arch Biochem Biophys. (2021) 704:108717. doi: 10.1016/j.abb.2020.108717
62. Xu X, Cui Y, Cao L, Zhang Y, Yin Y, Hu X. PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Exp Ther Med. (2017) 13:1993–9. doi: 10.3892/etm.2017.4218
63. Marimuthu A, Subbannayya Y, Sahasrabuddhe NA, Balakrishnan L, Syed N, Sekhar NR, et al. SILAC-based quantitative proteomic analysis of gastric cancer secretome. Proteomics Clin Appl. (2013) 7:355–66. doi: 10.1002/prca.201200069
64. Zhang SZ, Zhu XD, Feng LH, Li XL, Liu XF, Sun HC, et al. PCSK9 promotes tumor growth by inhibiting tumor cell apoptosis in hepatocellular carcinoma. Exp Hematol Oncol. (2021) 10:25. doi: 10.1186/s40164-021-00218-1
65. Gan SS, Ye JQ, Wang L, Qu FJ, Chu CM, Tian YJ, et al. Inhibition of PCSK9 protects against radiation-induced damage of prostate cancer cells. Onco Targets Ther. (2017) 10:2139–46. doi: 10.2147/OTT.S129413
66. Tao R, Xiong X, DePinho RA, Deng CX, Dong XC. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem. (2013) 288:29252–9. doi: 10.1074/jbc.M113.481473
67. Li H, Liu J. The novel function of HINFP as a co-activator in sterol-regulated transcription of PCSK9 in HepG2 cells. Biochem J. (2012) 443:757–68. doi: 10.1042/BJ20111645
68. Lohoff FW, Sorcher JL, Rosen AD, Mauro KL, Fanelli RR, Momenan R, et al. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol Psychiatry. (2018) 23:1900–10. doi: 10.1038/mp.2017.168
69. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. (2008) 49:399–409. doi: 10.1194/jlr.M700443-JLR200
70. Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, et al. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. (2010) 51:1486–95. doi: 10.1194/jlr.M003566
71. Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. (2004) 24:1454–9. doi: 10.1161/01.ATV.0000134621.14315.43
72. Poirier S, Prat A, Marcinkiewicz E, Paquin J, Chitramuthu BP, Baranowski D, et al. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem. (2006) 98:838–50. doi: 10.1111/j.1471-4159.2006.03928.x
73. Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. (2009) 284:28885–95. doi: 10.1074/jbc.M109.052407
74. Ai D, Chen C, Han S, Ganda A, Murphy AJ, Haeusler R, et al. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest. (2012) 122:1262–70. doi: 10.1172/JCI61919
75. Lai Q, Giralt A, Le May C, Zhang L, Cariou B, Denechaud PD, et al. E2F1 inhibits circulating cholesterol clearance by regulating Pcsk9 expression in the liver. JCI Insight. (2017) 2:e89729. doi: 10.1172/jci.insight.89729
76. Blesa S, Vernia S, Garcia-Garcia AB, Martinez-Hervas S, Ivorra C, Gonzalez-Albert V, et al. A new PCSK9 gene promoter variant affects gene expression and causes autosomal dominant hypercholesterolemia. J Clin Endocrinol Metab. (2008) 93:3577–83. doi: 10.1210/jc.2008-0269
77. Tai MH, Chen PK, Chen PY, Wu MJ, Ho CT, Yen JH. Curcumin enhances cell-surface LDLR level and promotes LDL uptake through downregulation of PCSK9 gene expression in HepG2 cells. Mol Nutr Food Res. (2014) 58:2133–45. doi: 10.1002/mnfr.201400366
78. Chang HY, Wu JR, Gao WY, Lin HR, Chen PY, Chen CI, et al. The cholesterol-modulating effect of methanol extract of pigeon pea (Cajanus cajan (L.) Millsp.) leaves on regulating LDLR and PCSK9 expression in HepG2 cells. Molecules. (2019) 24:493. doi: 10.3390/molecules24030493
79. Cui CJ, Jin JL, Guo LN, Sun J, Wu NQ, Guo YL, et al. Beneficial impact of epigallocatechingallate on LDL-C through PCSK9/LDLR pathway by blocking HNF1alpha and activating FoxO3a. J Transl Med. (2020) 18:195. doi: 10.1186/s12967-020-02362-4
80. Wang D, Yang X, Chen Y, Gong K, Yu M, Gao Y, et al. Ascorbic acid enhances low-density lipoprotein receptor expression by suppressing proprotein convertase subtilisin/kexin 9 expression. J Biol Chem. (2020) 295:15870–15882. doi: 10.1074/jbc.RA120.015623
81. Gao WY, Chen PY, Chen SF, Wu MJ, Chang HY, Yen JH. Pinostrobin inhibits proprotein convertase subtilisin/kexin-type 9 (PCSK9) gene expression through the modulation of FoxO3a protein in HepG2 cells. J Agric Food Chem. (2018) 66:6083–93. doi: 10.1021/acs.jafc.8b02559
82. Chen HC, Chen PY, Wu MJ, Tai MH, Yen JH. Tanshinone IIA modulates low density lipoprotein uptake via down-regulation of PCSK9 gene expression in HepG2 cells. PLoS ONE. (2016) 11:e0162414. doi: 10.1371/journal.pone.0162414
83. Wang X, Chen X, Zhang X, Su C, Yang M, He W, et al. A small-molecule inhibitor of PCSK9 transcription ameliorates atherosclerosis through the modulation of FoxO1/3 and HNF1alpha. EBioMedicine. (2020) 52:102650. doi: 10.1016/j.ebiom.2020.102650
84. Fan TY, Yang YX, Zeng QX, Wang XL, Wei W, Guo XX, et al. Structure-activity relationship and biological evaluation of berberine derivatives as PCSK9 down-regulating agents. Bioorg Chem. (2021) 113:104994. doi: 10.1016/j.bioorg.2021.104994
85. Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y, et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation. (2015) 131:1861–71. doi: 10.1161/CIRCULATIONAHA.115.015308
86. Langhi C, Le May C, Kourimate S, Caron S, Staels B, Krempf M, et al. Activation of the farnesoid X receptor represses PCSK9 expression in human hepatocytes. FEBS Lett. (2008) 582:949–55. doi: 10.1016/j.febslet.2008.02.038
87. Kourimate S, Le May C, Langhi C, Jarnoux AL, Ouguerram K, Zair Y, et al. Dual mechanisms for the fibrate-mediated repression of proprotein convertase subtilisin/kexin type 9. J Biol Chem. (2008) 283:9666–73. doi: 10.1074/jbc.M705831200
88. Momtazi AA, Banach M, Pirro M, Katsiki N, Sahebkar A. Regulation of PCSK9 by nutraceuticals. Pharmacol Res. (2017) 120:157–69. doi: 10.1016/j.phrs.2017.03.023
89. Melone M, Wilsie L, Palyha O, Strack A, Rashid S. Discovery of a new role of human resistin in hepatocyte low-density lipoprotein receptor suppression mediated in part by proprotein convertase subtilisin/kexin type 9. J Am Coll Cardiol. (2012) 59:1697–705. doi: 10.1016/j.jacc.2011.11.064
90. Patitucci C, Couchy G, Bagattin A, Caneque T, de Reynies A, Scoazec JY, et al. Hepatocyte nuclear factor 1alpha suppresses steatosis-associated liver cancer by inhibiting PPARgamma transcription. J Clin Invest. (2017) 127:1873–88. doi: 10.1172/JCI90327
91. Dong J, He M, Li J, Pessentheiner AR, Wang C, Zhang J, et al. MicroRNA-483 ameliorates hypercholesterolemia by inhibiting PCSK9 production. JCI Insight. (2020) 5:e143812. doi: 10.1172/jci.insight.143812
92. Naeli P, Mirzadeh Azad F, Malakootian M, Seidah NG, Mowla SJ. Post-transcriptional regulation of PCSK9 by miR-191, miR-222, and miR-224. Front Genet. (2017) 8:189. doi: 10.3389/fgene.2017.00189
93. Los B, Borges JB, Oliveira VF, Freitas RC, Dagli-Hernandez C, Bortolin RH, et al. Functional analysis of PCSK9 3'UTR variants and mRNA-miRNA interactions in patients with familial hypercholesterolemia. Epigenomics. (2021) 13:779–791. doi: 10.2217/epi-2020-0462
94. Xu X, Dong Y, Ma N, Kong W, Yu C, Gong L, et al. MiR-337-3p lowers serum LDL-C level through targeting PCSK9 in hyperlipidemic mice. Metabolism. (2021) 119:154768. doi: 10.1016/j.metabol.2021.154768
95. Decourt C, Janin A, Moindrot M, Chatron N, Nony S, Muntaner M, et al. PCSK9 post-transcriptional regulation: role of a 3'UTR microRNA-binding site variant in linkage disequilibrium with c.1420G. Atherosclerosis. (2020) 314:63–70. doi: 10.1016/j.atherosclerosis.2020.10.010
96. Alvarez ML, Khosroheidari M, Eddy E, Done SC. MicroRNA-27a decreases the level and efficiency of the LDL receptor and contributes to the dysregulation of cholesterol homeostasis. Atherosclerosis. (2015) 242:595–604. doi: 10.1016/j.atherosclerosis.2015.08.023
97. Broughton JP, Lovci MT, Huang JL, Yeo GW, Pasquinelli AE. Pairing beyond the seed supports MicroRNA targeting specificity. Mol Cell. (2016) 64:320–33. doi: 10.1016/j.molcel.2016.09.004
98. Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. (2003) 21:635–7. doi: 10.1038/nbt831
99. Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. (2011) 11:59–67. doi: 10.1038/nrc2966
100. Petersen DN, Hawkins J, Ruangsiriluk W, Stevens KA, Maguire BA, O'Connell TN, et al. A small-molecule anti-secretagogue of PCSK9 targets the 80S ribosome to inhibit PCSK9 protein translation. Cell Chem Biol. (2016) 23:1362–71. doi: 10.1016/j.chembiol.2016.08.016
101. Liu S, Deng X, Zhang P, Wang X, Fan Y, Zhou S, et al. Blood flow patterns regulate PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines. Cardiovasc Res. (2020) 116:1721–32. doi: 10.1093/cvr/cvz262
102. Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. (2008) 48:646–54. doi: 10.1002/hep.22354
103. Chorba JS, Galvan AM, Shokat KM. Stepwise processing analyses of the single-turnover PCSK9 protease reveal its substrate sequence specificity and link clinical genotype to lipid phenotype. J Biol Chem. (2018) 293:1875–86. doi: 10.1074/jbc.RA117.000754
104. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. (2004) 279:48865–75. doi: 10.1074/jbc.M409699200
105. Du F, Hui Y, Zhang M, Linton MF, Fazio S, Fan D. Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem. (2011) 286:43054–61. doi: 10.1074/jbc.M111.273474
106. Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife. (2013) 2:e00444. doi: 10.7554/eLife.00444.017
107. Emmer BT, Hesketh GG, Kotnik E, Tang VT, Lascuna PJ, Xiang J, et al. The cargo receptor SURF4 promotes the efficient cellular secretion of PCSK9. Elife. (2018) 7:e38839. doi: 10.7554/eLife.38839.026
108. Shen Y, Wang B, Deng S, Zhai L, Gu HM, Alabi A, et al. Surf4 regulates expression of proprotein convertase subtilisin/kexin type 9 (PCSK9) but is not required for PCSK9 secretion in cultured human hepatocytes. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158555. doi: 10.1016/j.bbalip.2019.158555
109. Wang B, Shen Y, Zhai L, Xia X, Gu HM, Wang M, et al. Atherosclerosis-associated hepatic secretion of VLDL but not PCSK9 is dependent on cargo receptor protein Surf4. J Lipid Res. (2021) 62:100091. doi: 10.1016/j.jlr.2021.100091
110. Rogers MA, Hutcheson JD, Okui T, Goettsch C, Singh SA, Halu A, et al. Dynamin-related protein 1 inhibition reduces hepatic PCSK9 secretion. Cardiovasc Res. (2021) 117:2340–53. doi: 10.1093/cvr/cvab034
111. Gustafsen C, Kjolby M, Nyegaard M, Mattheisen M, Lundhede J, Buttenschon H, et al. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. (2014) 19:310–8. doi: 10.1016/j.cmet.2013.12.006
112. Dewpura T, Raymond A, Hamelin J, Seidah NG, Mbikay M, Chretien M, et al. PCSK9 is phosphorylated by a Golgi casein kinase-like kinase ex vivo and circulates as a phosphoprotein in humans. FEBS J. (2008) 275:3480–93. doi: 10.1111/j.1742-4658.2008.06495.x
113. Ben Djoudi Ouadda A, Gauthier MS, Susan-Resiga D, Girard E, Essalmani R, Black M, et al. Ser-phosphorylation of PCSK9 (proprotein convertase subtilisin-kexin 9) by Fam20C (family with sequence similarity 20, member C) kinase enhances its ability to degrade the LDLR (low-density lipoprotein receptor). Arterioscler Thromb Vasc Biol. (2019) 39:1996–2013. doi: 10.1161/ATVBAHA.119.313247
114. Zhang H, Zhu Q, Cui J, Wang Y, Chen MJ, Guo X, et al. Structure and evolution of the Fam20 kinases. Nat Commun. (2018) 9:1218. doi: 10.1038/s41467-018-03615-z
115. Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. (2006) 281:30561–72. doi: 10.1074/jbc.M606495200
116. Chong M, Yoon G, Susan-Resiga D, Chamberland A, Cheillan D, Pare G, et al. Hypolipidaemia among patients with PMM2-CDG is associated with low circulating PCSK9 levels: a case report followed by observational and experimental studies. J Med Genet. (2020) 57:11–17. doi: 10.1136/jmedgenet-2019-106102
117. Catapano AL, Pirillo A, Norata GD. New pharmacological approaches to target PCSK9. Curr Atheroscler Rep. (2020) 22:24. doi: 10.1007/s11883-020-00847-7
118. Katzmann JL, Gouni-Berthold I, Laufs U. PCSK9 inhibition: insights from clinical trials and future prospects. Front Physiol. (2020) 11:595819. doi: 10.3389/fphys.2020.595819
119. Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. (2021) 593:429–34. doi: 10.1038/s41586-021-03534-y
120. Zeitlinger M, Bauer M, Reindl-Schwaighofer R, Stoekenbroek RM, Lambert G, Berger-Sieczkowski E, et al. A phase I study assessing the safety, tolerability, immunogenicity, and low-density lipoprotein cholesterol-lowering activity of immunotherapeutics targeting PCSK9. Eur J Clin Pharmacol. (2021) 77:1473–1484. doi: 10.1007/s00228-021-03149-2
Keywords: lipid metabolism, cardiovascular disease, atherosclerosis, cancer immunotherapy, PCSK9, LDL receptor, major histocompatibility protein class I
Citation: Xia X-d, Peng Z-s, Gu H-m, Wang M, Wang G-q and Zhang D-w (2021) Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 8:764038. doi: 10.3389/fcvm.2021.764038
Received: 24 August 2021; Accepted: 16 September 2021;
Published: 15 October 2021.
Edited by:
Yiliang Chen, Medical College of Wisconsin, United StatesReviewed by:
Fang Li, Columbia University Irving Medical Center, United StatesDaisy Sahoo, Medical College of Wisconsin, United States
Copyright © 2021 Xia, Peng, Gu, Wang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gui-qing Wang, ZHpoYW5nQHVhbGJlcnRhLmNh; Da-wei Zhang, MTkxODYzNTM0NEBxcS5jb20=
†These authors have contributed equally to this work