 Musaad B. Alsahly1,2
Musaad B. Alsahly1,2 Madaniah O. Zakari1,3Lauren G. Koch4
Madaniah O. Zakari1,3Lauren G. Koch4 Steven Britton5
Steven Britton5 Laxmansa C. Katwa1
Laxmansa C. Katwa1 Kelsey Fisher-Wellman1,6
Kelsey Fisher-Wellman1,6 Robert M. Lust1,6*
Robert M. Lust1,6*- 1Department of Physiology, Brody School of Medicine, East Carolina University, Greenville, NC, United States
- 2East Carolina Diabetes and Obesity Center, East Carolina University, Greenville, NC, United States
- 3Department of Physiology, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 4Department of Physiology, College of Medicine, Taibah University, Medina, Saudi Arabia
- 5Department of Physiology and Pharmacology, University of Toledo, Toledo, OH, United States
- 6Departments of Anesthesiology and Molecular and Integrative Medicine, University of Michigan, Ann Arbor, MI, United States
Rationale: Regular active exercise is considered therapeutic for cardiovascular disease, in part by increasing mitochondrial respiratory capacity, but a significant amount of exercise capacity is determined genetically. Animal models, demonstrating either high capacity aerobic running (HCR) or low capacity aerobic running (LCR) phenotypes, have been developed to study the intrinsic contribution, with HCR rats subsequently characterized as “disease resistant” and the LCRs as “disease prone.” Enhanced cardioprotection in HCRs has been variable and mutifactoral, but likely includes a metabolic component. These studies were conducted to determine the influence of intrinsic aerobic phenotype on cardiac mitochondrial function before and after ischemia and reperfusion.
Methods: A total of 34 HCR and LCR rats were obtained from the parent colony at the University of Toledo, housed under sedentary conditions, and fed normal chow. LCR and HCR animals were randomly assigned to either control or ischemia-reperfusion (IR). On each study day, one HCR/LCR pair was anesthetized, and hearts were rapidly excised. In IR animals, the hearts were immediately flushed with iced hyperkalemic, hyperosmotic, cardioplegia solution, and subjected to global hypothermic ischemic arrest (80 min). Following the arrest, the hearts underwent warm reperfusion (120 min) using a Langendorff perfusion system. Following reperfusion, the heart was weighed and the left ventricle (LV) was isolated. A midventricular ring was obtained to estimate infarction size [triphenyltetrazolium chloride (TTC)] and part of the remaining tissue (~150 mg) was transferred to a homogenation buffer on ice. Isolated mitochondria (MITO) samples were prepared and used to determine respiratory capacity under different metabolic conditions. In control animals, MITO were obtained and prepared similarly immediately following anesthesia and heart removal, but without IR.
Results: In the control rats, both resting and maximally stimulated respiratory rates were higher (32 and 40%, respectively; p < 0.05) in HCR mitochondria compared to LCR. After IR, resting MITO respiratory rates were decreased to about 10% of control in both strains, and the augmented capacity in HCRs was absent. Maximally stimulated rates also were decreased more than 50% from control and were no longer different between phenotypes. Ca++ retention capacity and infarct size were not significantly different between HCR and LCR (49.2 ± 5.6 vs. 53.7 ± 4.9%), nor was average coronary flow during reperfusion or arrhythmogenesis. There was a significant loss of mitochondria following IR, which was coupled with decreased function in the remaining mitochondria in both strains.
Conclusion: Cardiac mitochondrial capacity from HCR was significantly higher than LCR in the controls under each condition. After IR insult, the cardiac mitochondrial respiratory rates were similar between phenotypes, as was Ca++ retention capacity, infarct size, and arrhythmogenicity, despite the increased mitochondrial capacity in the HCRs before ischemia. Relatively, the loss of respiratory capacity was actually greater in HCR than LCR. These data could suggest limits in the extent to which the HCR phenotype might be “protective” against acute tissue stressors. The extent to which any of these deficits could be “rescued” by adding an active exercise component to the intrinsic phenotype is unknown.
Introduction
Cardiovascular disease (CVD) remains the leading cause of morbidity and mortality in developed countries (1). The most common cause of cardiac injury is ischemic heart disease secondary to progressive coronary occlusion and is complicated frequently by complications such as ventricular arrhythmias and congestive heart failure (2). The most effective therapy, thus, far for limiting ischemic injury is early reperfusion, but reperfusion in itself has risks (3–5) to the extent that acute coronary occlusion and therapeutic intervention are collectively considered to produce an aggregate phenomenon recognized as ischemia-reperfusion (IR) injury (6–9).
Continuing research is aimed to explore the therapeutic interventions against IR injury. Although numerous pharmacological and preconditioning approaches to cardioprotection have been explored, regular exercise participation is recognized as an important, cost-effective, and safer lifestyle intervention in the prevention and treatment of IR injury (10–14). Redundant protective effects are evident in the exercised heart, namely, increased levels of heat shock proteins (15), altered nitric oxide (NO) signaling (16–18), enhanced Ca2+ handling proteins (19), improved ATP-sensitive potassium channels (20), and enhanced endogenous antioxidant (13, 21).
As an important site for ATP production via oxidative phosphorylation, mitochondria are critical in regulating normal cardiac metabolism and play a key role in the susceptibility of the heart to IR injury (22–28). The heart is an organ with high metabolic demand. Providing the myocardium with adequate coronary perfusion and oxygen delivery is crucial, but enhanced mitochondrial capacity also is essential. Mitochondrial respiratory rate and enzyme activities are the major elements that drive the oxidative phosphorylation process and structural integrity of the mitochondria. Myocardial IR can cause severe effects on mitochondrial homeostasis which dramatically affects mitochondrial function and survival (29–32). In addition to the detrimental effects of impaired mitochondrial energy production, mitochondrial ionic imbalance, and cell stress signaling can cause mitochondrial-mediated cell death (33–37).
Myocardial IR injury is an important cause of impaired heart function in the early postoperative period after cardiac surgery and acute myocardial ischemia. Growing evidence has become available supporting a crucial role of mitochondrial dysfunction in myocardial IR injury. Mitochondrial dysfunction during ischemia is a major mechanism that contributes to cardiomyocytes damage during IR (24, 26–31). Increased reactive oxygen species (ROS) generation, defects in electron transport chain activity and oxidative phosphorylation process, impaired respiratory chain complexes activity, opening of the mitochondrial permeability transition pore (mPTP), and release of cytochrome care considered contributing factors in mitochondrial dysfunction associated with heart IR (30–36).
Stunned (reversibly injured and nonfunctioning) myocardium displays relatively excess oxygen consumption for a specified rate of contractile work and, therefore, has a declined mechanical efficiency, which may be due to a rapid recovery of the intracellular pH during reperfusion (6). Once perfusion is restored, the intracellular increased accumulation of H+ during ischemia is transported into the extracellular space to normalize the pH in exchange for Na+ via Na+/H+ exchanger, while ATP depletion inactivates Na+/K+-ATPase. The combined effect results in an increase in intracellular Na+ which, in turn, activates the sarcolemmal 2Na+/Ca2+ exchanger, resulting in the exchange of intracellular Na+ with extracellular Ca2+. A high rate of 2Na+/Ca2+exchange can finally lead to Ca2+ overload which, in turn, induce arrhythmogenesis, myocardial stunning, contracture, and ultimately apoptotic or autophagic cell death. Fluctuations in Ca2+from the sarcoplasmic reticulum during reperfusion stimulate the opening of the mPTP (29, 30). Opening of the mPTP leads to rapid dissipation of the membrane potential gradient which is essential for the synthesis of ATP, water enters through the open pore causing mitochondrial swelling and lysis triggering apoptosis and cell death (28–30).
The proposed mechanisms underlying exercise-induced cardioprotection in IR are numerous. Some are systemic (12), some are vascular (12–14, 21), some are neural (38, 39), some are structural (9), and some are energetic/metabolic (40, 41) including expression of selected mitochondrial proteins resulting in a mitochondrial phenotype that is resistant to IR-induced injury (13, 19). While these studies generally support exercise-induced adaptations that produce resistance to injury, few address the mitochondrial functional consequences following an injury.
Although regular exercise training is recognized as an important lifestyle intervention in the prevention and treatment of CVD and IR injury, not all the individuals experience the same benefits from participating in the exercise. There is a paradox that some individuals with many modifiable risk factors (hypertension, diabetes, and obesity), and who do not exercise, also do not get CVD, while others who have no risk factors, and exercise regularly, and still experience adverse cardiac outcomes, suggesting a significant intrinsic component to the exercise effect. It has been estimated that up to 70% of the variation in exercise capacity is due to the intrinsic genetic component (42). Thus, studying the differential impacts of intrinsic aerobic exercise capacity after cardiac IR injury using intrinsic aerobic phenotype rats bred for low and high aerobic running capacity would provide a better platform for understanding the influences of intrinsic aerobic capacity on cardiac metabolic capacity and mitochondrial adaptive response pre- and post-IR injury in these phenotypes.
Koch and Britton used phenotypic selection based on treadmill running time at 11 weeks of age in an outbred rat strain (NIN:N) to create divergent strains that have become known as high capacity aerobic running (HCR) and low capacity aerobic running (LCR) rats (43–47). The HCR animals generally are characterized as “disease resistant,” while the LCR animals are characterized as disease prone. Interestingly, the HCR/LCR strains demonstrate many of the same traits related to CVD that had been previously associated with active exercise (48–52) including some mitochondrial/metabolic effects (53, 54). Still, cardioprotection in the HCRs is not always observed (55–57), and while LCR did associate with a higher incidence of pump failure, it did not associate with multiorgan system failure in hemorrhagic shock (58). We have previously reported that cardioprotection in HCRs was present but limited, was likely intrinsic to the tissue, and could be overwhelmed by IR severity (57). Given the consistent metabolic differences that characterized HCR and LCR, the current studies were designed to identify the relative influence of aerobic phenotype on the impact of IR on subsequent mitochondrial function.
Materials and Methods
Animal Strains
A total of 34 HCR and LCR female rats from generation 32 were obtained from the parent colony at the University of Toledo. The protocol for generating the animal model has been described in detail previously (43–46). Briefly, starting with an outbred founder population (N: NIH stock), two-way selective breeding using run time until exhaustion on a graded treadmill exercise test as selection criteria, was used to create low capacity runner (LCR) and high capacity runner (HCR) strains. A total of 13 animals of each sex with the shortest run times and 13 animals of each sex with the highest run times were used as the founding population of LCR and HCR cohorts. Well-managed breeding strategies within each cohort were used to create subsequent generations that were increasingly divergent in total running capacity. Upon arrival at this location, animals were maintained in quarantine under standard husbandry conditions. Animal procedures were conducted following the American Physiological Society guidelines for the humans and safe use of animals and all the protocols involving animals in these experiments were approved by the East Carolina University Animal Care and Use Committee.
Cardiac Ischemic-Reperfusion Injury
Rats were anesthetized with an intraperitoneal injection of ketamine/xylazine (90/10 mg/kg ip, Patterson Veterinary Supply, Greeley, Colorado, USA). Once appropriate anesthetic depth was achieved, the thorax was opened and the heart was excised rapidly. The aortic root was cannulated and the coronary circulation was flushed immediately with warm saline. After the saline flush, the control hearts were flushed again with cold saline and transferred to buffer to being mitochondrial isolation. The IR hearts were immediately arrested with 10 ml of iced St Thomas' cardioplegic solution (NaCl 110.0 mM, NaHCO3 10.0 mM, KCl 16.0 mM, MgCl2 16.0 mM, CaCl2 1.2 mM, and pH 7.8) at 4°C and the hearts were then stored in this solution at the same temperature for 80 min. After 80 min of cold global ischemic arrest, the aorta was cannulated and the heart was immediately perfused retrogradely on a Langendorff perfusion apparatus with Krebs–Henseleit buffer (KHB) for 120 min at 37°C (KHB composition (mM): NaCl 118; KCl 4.7; MgSO4 1.2; KH2PO4 1.2; NaHCO3 25; CaCl2 1.4; glucose 11; and pH 7.3–7.4). Perfusion was gravity-fed constant pressure maintained at 80 mm Hg, established by the appropriate height of the perfusion reservoir above the heart. Coronary flow and heart rhythm were monitored throughout the reperfusion period. The Langendorff system is widely used because of the ability to control a large number of performance variables (preload, rate, and afterload), but it also has the disadvantage of not actively pumping a volume. In addition, since the system is crystalloid perfused typically, any circulating agents like pro- or anti-inflammatory cytokines are not present, nor are hormones or autonomic nervous influences. In this case, that can be an asset, since any of those can be changed by exercise and would not be confounding aspects of the study.
Tissue Isolation and Infarct Size Quantification
Following reperfusion, the IR hearts were taken off the cannula and weighed. The right ventricle and atrial tissue were removed and a midventricular ring was obtained from the left ventricle (LV) for infarct size quantification, while the balance of the LV was prepared for mitochondrial isolation, or for −80°C storage and future analysis. The midventricular ring was placed in a 0.1% triphenyltetrazolium chloride solution and incubated at 37°C for 10 min in a shaking water bath. Following incubation, both sides of the slice were photographed with a digital camera attached to a dissecting microscope. Images were quantified using Image J software where total area, lumen area, and infarcted area were measured and quantified as described previously (21, 30, 57).
Mitochondria Isolation
Left ventricular mitochondria were isolated as described in Fisher-Wellman et al. (68). Briefly, one portion of isolated LV (~40 mg) was immediately placed in ice-cold buffer A [phosphate-buffered saline (pH = 7.4), supplemented with EDTA (10 mM)]. All the tissues were minced and resuspended in buffer C (MOPS (4-Morpholinepropanesulfonic acid, 50 mM; pH = 7.1), KCl (100 mM), EGTA (egtazic acid, 1 mM), and MgSO4 (5 mM) supplemented with bovine serum albumin (BSA; 2 g/l)) and then homogenized via a Teflon pestle and borosilicate glass vessel. Tissue homogenates were centrifuged at 500 × g for 10 min at 4°C. Supernatant from each tissue was then filtered through thin layers of gauze and subjected to additional centrifugation at 10,000 × g for 10 min at 4°C. Mitochondrial pellets were washed in 1.4 ml of buffer B [MOPS (50 mM; pH = 7.1), KCl (100 mM), EGTA (1 mM), and MgSO4 (5 mM)], transferred to microcentrifuge tubes, and centrifuged at 10,000 × g for 10 min at 4°C. Buffer B was aspirated from each tube and final mitochondrial pellets were suspended in 100–200 μl of buffer B. Protein content was determined via the Pierce BCA protein assay. Functional assays involving isolated mitochondria were carried out in the following buffers: buffer D-potassium-MES (105 mM; pH = 7.2), KCl (30 mM), KH2PO4 (10 mM), MgCl2 (5 mM), EGTA (1 mM), BSA (2.5 g/l); buffer E-HEPES (20 mM; pH = 8.0), KCl (100 mM), KH2PO4 (2.5 mM), MgCl2 (2.5 mM), and glycerol (1%).
Mitochondrial Respiratory Control (JO2)
High-resolution O2 consumption measurements were conducted at 37°C in 2 ml of assay buffer by using the Oroboros Oxygraph-2K (Oroboros Instruments, Innsbruck, Austria), as previously described (67). Briefly, isolated mitochondria (0.025 mg/ml) were added to assay buffer, supplemented with creatine (Cr; 5 mM), phosphocreatine (PCr; 1 mM), and creatine kinase (CK; 20 U/ml), followed by the addition of respiratory substrates then ATP (5 mM). Respiratory control was assessed through the sequential additions of PCr to final concentrations of 6 mM, 12 mM, 15 mM, and 21 mM before additions of 5 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP). Calculation of ATP free energy of hydrolysis (ΔGATP) was determined for each PCR concentration as previously described (68) using an online tool (Bioenergetic Calculators (dmpio.github.io)).
Citrate Synthase Activity
Citrate synthase activity was measured using a standard, commercially available kit (Sigma, St Louis, Missouri, USA), according to the instructions of the manufacturer. The assay generates a colorimetric signal in proportion to the rate of conversion between acetyl-CoA and oxaloacetic acid and is read on a spectrophotometer.
Ca2+ Retention Capacity
Calcium retention protocols were modified from Sloan et al. (30) where 0.5 mg mitochondria were suspended in an assay buffer containing: 125 mM KCl, 5 mM HEPES, 2 mM KH2PO4, and 1 mM MgCl2 (25°C, pH = 7.3). The fluorescent Ca2+ indicator, calcium green 5 N salt, was utilized to track changes in extramitochondrial calcium levels. Extramitochondrial calcium fluorescence was measured using a fluorescence spectrophotometer (Photon Technology International, Birmingham, New Jersey, USA), with excitation and emission wavelengths set to 506/532 nm, respectively. Calcium-induced mPTP opening experiments were performed under state 2 respiration conditions (5 mM glutamate/5 mM malate). Mitochondrial PTP opening was induced by subjecting mitochondria to sequential 50 nmol CaCl2 pulses every 3 min, which causes a repeated decrease in the fluorescent signal as Ca2+ is taken up by the mitochondria. Induction of mPTP was denoted by a sharp increase in extramitochondrial Ca2+fluorescence, representing the release of the accumulated Ca2+ from the mitochondrial matrix. Calcium-retention capacity was quantified as the amount of calcium needed to induce PTP opening (nmol CaCl2/mg mitochondria).
Statistical Analysis
Statistical analysis was performed by using commercial software (GraphPad Prism, San Diego, California, USA). Data are expressed as means ± SD and a p < 0.05 was considered as statistically significant.
Results
Myocardial Infarct Size
Infarctions were somewhat larger than expected despite the relatively long ischemic times, considering the use of the cold cardioplegic solution. About 80 min of cold global ischemic arrest and 120 min of warm reperfusion, myocardial infarct size was not significantly different between LCRs and HCRs (47.2 ± 4.8 vs. 42.9 ± 5.2, respectively). Hearts from HCR and LCR animals showed different patterns of infarction, with HCR being more contiguous and LCR more diffuse, but relatively similar in the overall amount of tissue involved (Figure 1).
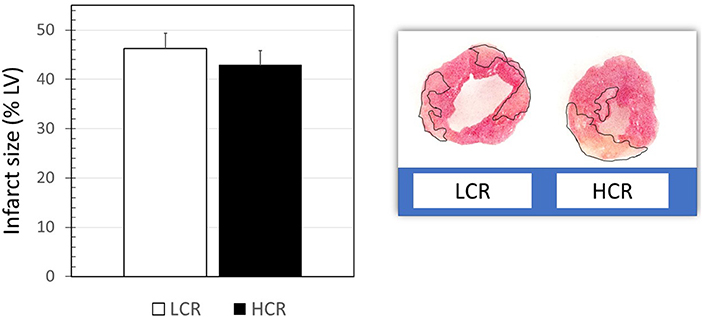
Figure 1. Examples of triphenyltetrazolium chloride (TTC)-stained myocardial sections, and graphical summary of group data for infarct size results (n = 9 per group).
Coronary Artery Flow
When normalized to heart weight, baseline coronary flow at the onset of reperfusion was not different between LCR and HCR hearts (20.5 ± 3.2 vs. 23.6 ± 2.8, respectively). However, coronary flow decreased by 68% in LCRs and by 56% in HCRs over the 2 h of reperfusion and coronary flow decreased more quickly in the LCRs compared to the HCRs, becoming significantly different at 40 min of reperfusion (Figure 2). In a constant pressure system, the decreasing flow is consistent with an increased coronary vascular resistance. Still, there was a substantial drop in coronary flow in both groups, and the relatively higher/less diminished flow, was not enough to produce any difference in overall infarction size.
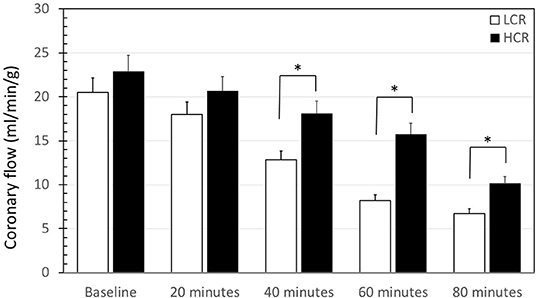
Figure 2. Summary of coronary flow results during reperfusion of low capacity aerobic running (LCR) and high capacity aerobic running (HCR) hearts. Data are expressed as mean ± SD. n = 9 per group. *p < 0.05 in designated comparison.
Reperfusion Arrhythmia
Upon harvest, the hearts were placed immediately into the cold storage solution, so there were no baseline rhythm measurements. The first perfusion on the column constituted the onset of rewarming and reperfusion. Arrhythmia was evaluated followed by the guidelines established by the Lambeth Conventions and graded by using a numerical scoring system as described previously (30). Fibrillation was a uniform finding early in reperfusion with similar frequency and severity in both LCR and HCR hearts (Figure 3).
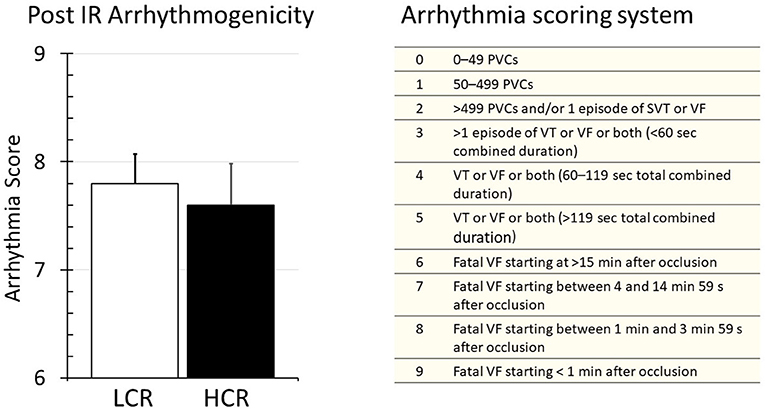
Figure 3. Summary of arrhythmia results during reperfusion of LCR and HCR hearts (left panel), and the arrhythmia scoring system that was used (right panel) to generate the data. Data are expressed as mean ± SD. n = 9 per group. *p < 0.05 in designated comparison.
Mitochondrial Respiratory Capacity
Control Comparisons
Cardiac mitochondria respiratory rates were 32% higher at rest in HCR and more than 40% higher under maximally stimulated conditions, compared to LCR (both p < 0.05). Generally, HCR mitochondria showed significantly higher mitochondria respiration with all substrates compared to LCR. The addition of the uncoupling agent, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (1 μM), at the end of the respirometry experiments rescued absolute rates of oxygen consumption (Figure 4).
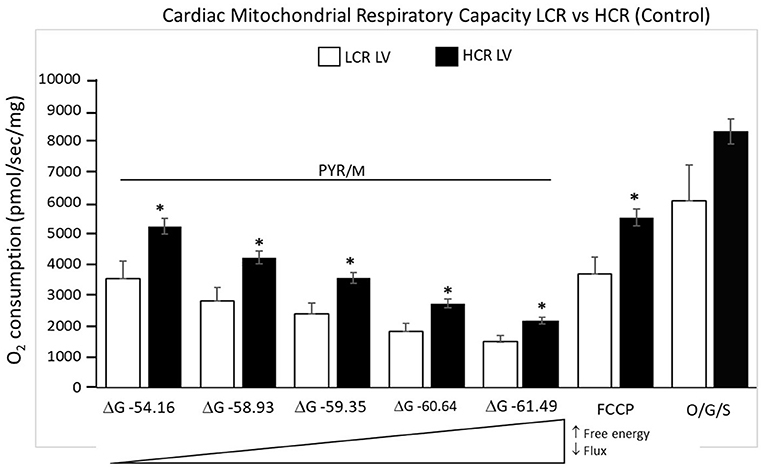
Figure 4. Summary of mitochondrial respiration results in samples isolated from HCR and LCR control ventricles. The top inset shows representative tracing, while the graphic summarizes group results. Data are expressed as mean ± SD. n = 8 per group. *p < 0.05 in designated comparison.
Post-ir Injury Comparisons
In contrast to control experiments, HCR mitochondria no longer showed relatively increased respiratory capacity compared to LCR (Figure 5). Comparing control to post-I/R under pyruvate/malate conditions, respiratory rates were reduced in both phenotypes to levels <10% of baseline (Figure 6). There was recruitable respiratory capacity in both HCR and LCR postischemic mitochondria, but the differences between phenotypes that been observed across the spectrum of substrate conditions were no longer present after ischemia. In fact, HCRs showed a greater loss of respiratory capacity in response to FCCP than the LCR (Figure 6).
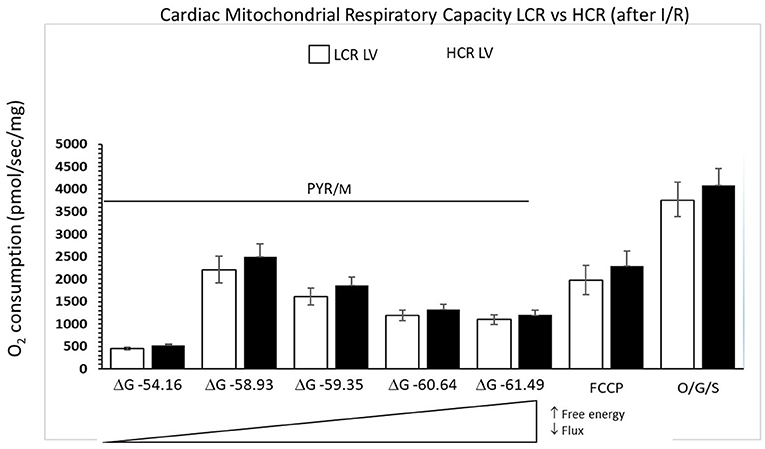
Figure 5. Summary of mitochondrial respiration results in samples isolated from HCR and LCR ventricles following IR. The top inset shows representative tracing, while the graphic summarizes group results. Data are expressed as mean ± SD. n = 9 per group. *p < 0.05 in designated comparison.
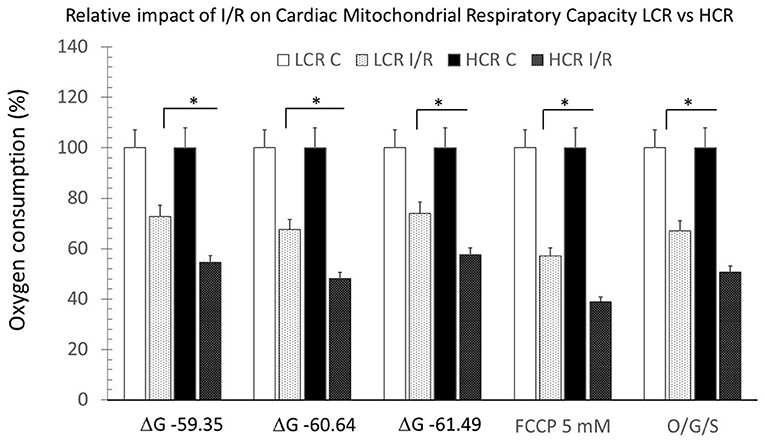
Figure 6. Summary of mitochondrial respiration, comparing results within and between phenotypes, under control and post-ischemia-reperfusion (IR) conditions. Data are expressed as mean ± SD. n = 8 per group for controls and 9 per group for IR. *p < 0.05 in designated comparison.
The amount of mitochondrial protein harvested from LV tissue samples (w/w) was not different between HCR and LCR under control conditions (Figure 7A). Following cold storage ischemia and rewarming/reperfusion, there was a significant decrease in mitochondrial protein in both strains (p < 0.05), consistent with a loss of mitochondria. However, the decrease was similar in both groups (Figure 7A). Interestingly, the citrate synthase activity of the remaining mitochondria following reperfusion also was reduced comparably in both groups, consistent with possible functional impairment in surviving mitochondria (Figure 7B).
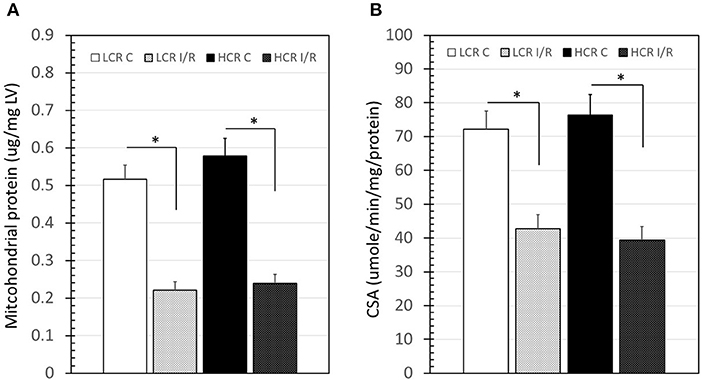
Figure 7. Summary of mitochondrial protein “yield” from left ventricle (LV) samples (left panel) and citrate synthase activity (CSA, right panel) before and after IR. Data are expressed as mean ± SD. n = 8 per group for controls and 9 per group for IR. *p < 0.05 in designated comparison.
Ca2+ Retention Capacity
There were no differences in baseline mitochondrial calcium retention. Similar to the respiratory deficits that occurred in mitochondria after I/R, the ability to retain calcium was also reduced significantly following ischemia. On average, the permeability transition pore (PTP) opened with about 75% less calcium than it did before the I/R insult but the impairments were similar in both the phenotypes (Figure 8).
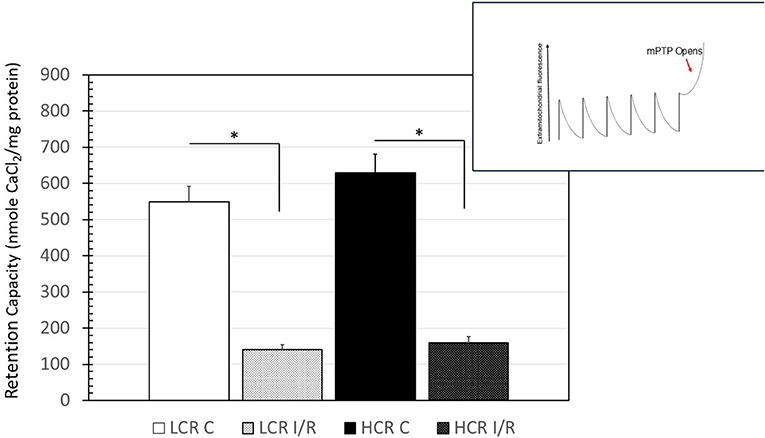
Figure 8. Summary of results for the calcium-retention experiments. Inset is a representation of the tracings obtained, while the chart is a graphical summary of the group data. Arrow in inset indicates the change in calcium uptake that occurs when the transition pore opens. Data are expressed as mean ± SD. n = 8 per group for controls and 9 per group for IR. *p < 0.05 in designated comparison.
Discussion
The results of this study showed that while there was increased mitochondrial capacity in HCRs compared to LCRs, the increased cellular respiratory capacity did not translate to reduced injury from ischemic arrest and reperfusion even with the benefit of supplemental cardioprotection by using established cardioplegia regimens. Moreover, there was accelerated relative loss of mitochondrial respiratory capacity in the HCRs compared to the LCRs, even though coronary blood flow was better preserved during reperfusion in the HCRs compared to the LCRs. The lack of benefit despite better flow was a bit unexpected, as dynamic exercise has generally associated cardioprotection with improved perfusion. A decrease in infarct size after training was seen in rats subjected to a swim training regimen after permanent occlusion which the authors attributed to increased myocardial vascularity (59). In a much-cited paper, Brown et al. demonstrated that prolonged endurance training confers a cardioprotective effect against infarction in myocardium subjected to severe ischemia and subsequent reperfusion (21). In addition, they observed that during severe ischemia, coronary flow to regions of the myocardium outside the ischemic area at risk was better maintained in hearts isolated from endurance-trained rats. Furthermore, on reperfusion of the area at risk, the increase in flow to the previously ischemic region of the heart was markedly higher in hearts isolated from trained rats (21).
We have demonstrated that the cardioprotection associated with the HCR phenotype may be limited (57). We did not have sufficient animals available to complete a preliminary dose response study to determine the optimal cold storage time, and it is possible that despite the cardioplegia, the ischemic time was simply too long and overwhelmed and benefit the HCR phenotype might have conferred. However, the results are consistent with those reported by others working with the model (55, 56). Still, there are important differences worth noting. In particular, active exercise training programs produce improvements to ischemic tolerance via a host of mechanisms (5, 6, 9, 10, 14, 15), notably including increased mitochondrial functional capacity (19, 24, 59). Gallego-Selles et al. described experiments in humans subjects indicating that, at least in skeletal muscle, nrf2 is activated rapidly by exercise to exhaustion and can dynamically modulate ROS and mitochondrial-sourced proteins such as catalase (61). Flockhart demonstrated, also in human subjects, that participating in an intense, active exercise program produced functional impairment in the mitochondria that also was associated with at least a transient decrease in glucose tolerance (62). In our hands, despite an increased resting mitochondrial capacity in the HCRs, there was no protection against the ischemic insult. Together, it seems clear that there can be substantial differences in the role of mitochondria in the response to ischemia that can be influenced by the nature and duration of the exercise training and confounded by differences dictated by the intrinsic capacity for exercise, independent of actual active exercise. The importance and role of pathways such as nrf2/keap1/catalase and others in bridging the components of an exercise effect (intrinsic vs. active) remain to be determined.
Several previous experimental studies of myocardial ischemia generally have indicated a good correlation between disruption of complex I through a variety of mechanisms (16–18, 33, 41), as well as opening of the permeability transition pore, with outcomes in cardiac ischemia and reperfusion (23, 26, 28–30). Some have suggested that complex I is considered the main site of damage to the respiratory chain in IR, while downstream electron transport chains are relatively resistant to IR injury (60, 63, 64). Furthermore, Veitch and his colleagues (63) found a major decline in complex I activity in perfused rat hearts subjected to 20 min of global ischemia, and Cairns and his group (64) demonstrated that this damage was exacerbated by reperfusion.
In contrast to these studies, our results suggest a more generalized loss of mitochondrial respiratory capacity following IR. Perhaps that should not be surprising. Animals that have been selectively inbred using aerobic running capacity as the selection criteria create a strain optimized for performance when aerobic conditions exist. Ischemia certainly is not an aerobic condition. While it is possible that the HCR phenotype augments several factors that could delay the onset of ischemia and which would be protective (57), they may be maladapted if the transition to anaerobic conditions still occurs. Structural adaptations like increased capillary density or increased vasodilator capacity, which might have been beneficial in delaying the onset of ischemia, would also be factors that could aggravate the potential for reperfusion injury once perfusion was reestablished after an acute ischemic episode. When ample oxygen is present, cardiac mitochondria have greater substrate flexibility than many other tissues, meaning that cardiac mitochondria can respire effectively using a wide variety of substrates (67). Tolerance for IR may not be related so much to the capacity to respire using a given substrate, but more to the ability to switch from one substrate to another. With the newest approaches that have been developed (68), and the energy hypothesis that is being advanced by the HCR LCR work (46, 53), the HCR LCR phenotypes could provide interesting and valuable perspective on factors contributing to substrate preference. To the extent that HCR complex I respiratory capacity was impacted strongly by IR, and that there was a similarity between PTP function and infraction size in HCR and LCR, suggest that a more complex assessment of specific mitochondria adaptations in these strains might be warranted.
Mitochondria comprise a large fraction of the heart mass and are critical for the normal mechanical and electrophysiological function of the cardiomyocyte, playing roles that extend beyond bioenergetics and metabolism. Proper function is required to meet the high energetic demand of the cardiomyocyte and playing an essential role in controlling oxidative stress and Ca2+ handling (34). Ischemia-reperfusion injury increases the production of ROS and induces calcium overload into mitochondria (35) which can interact together to induce opening of the mPTP and, therefore, triggering apoptosis by promoting the release of proapoptotic proteins (i.e., cytochromec) and subsequent activation of the programmed cell (65, 66).
Our results indicate that intrinsic aerobic capacity must be tied to mitochondrial performance at a subcellular level. It also is clear that the dynamic role of mitochondria in cardiac ischemia, perfusion, and heart failure is only now beginning to be appreciated, but remains confused, perhaps in part because the underlying intrinsic elements have not been adequately considered in the assessment of the active exercise response. Models such as these HCR and LCR phenotypes should provide a unique window on how that background influences the extent to which inducible adaptive responses can be accomplished best using an active exercise regimen.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee (IACUC) East Carolina University.
Author Contributions
RL participated in designing, conducting, analyzing, and writing. MA and MZ participated in conducting, analyzing, and writing. KF-W participated in designing, conducting, and analyzing. LCK participated in conducting and analyzing. LGK and SB provided animals and insight on study design. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. American Heart Association/American Stroke Association. Cardiovascular disease: a costly burden for america projections through 2035. Available online at: https://healthmetrics.heart.org/wp-content/uploads/2017/10/Cardiovascular-Disease-ACostly-Burden.pdf
2. Burke AP, Virmani R. Pathophysiology of acute myocardial infarction. Med Clinics North America. (2007) 91:553–72. doi: 10.1016/j.mcna.2007.03.005
3. Buja LM, Weerasinghe P. Unresolved issues in myocardial reperfusion injury. Cardiovascular Pathology. (2010) 19:29–35. doi: 10.1016/j.carpath.2008.10.001
4. Vetterlein F, Schrader C, Volkmann R, Neckel M, Ochs M, Schmidt G, et al. Extent of damage in ischemic, nonreperfused, and reperfused myocardium of anesthetized rats. Am J Physiol Circ Physiol. (2003) 285:H755–65. doi: 10.1152/ajpheart.00269.2002
5. Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Sem in Cardiothor Vasc Anes. (2012) (16):123–32. doi: 10.1177/1089253211436350
6. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Eng J Med. (2007) 357:1121. doi: 10.1056/NEJMra071667
7. Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardiol. (2005) 100:179–90. doi: 10.1016/j.ijcard.2004.04.013
8. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell Biology of Ischemia/Reperfusion Injury. Int Rev Cell Mol Biol. (2012) 229–317. doi: 10.1016/B978-0-12-394309-5.00006-7
9. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J Clin Invest. (2013) 123:92–100. doi: 10.1172/JCI62874
10. Quindry J, Hamilton K. Exercise and cardiac preconditioning against ischemia reperfusion injury. Curr Cardiol Rev. (2013) 9:220–9. doi: 10.2174/1573403X113099990033
11. Borges JP, da Silva Verdoorn K. Cardiac ischemia/reperfusion injury: The beneficial effects of exercise. In: Advances in Experimental Medicine and Biology. New York LLC: Springer (2017). pp. 155–79. doi: 10.1007/978-981-10-4307-9_10
12. Kavazis AN. Exercise preconditioning of the myocardium. Sports Med. (2009) 39:923–35. doi: 10.2165/11317870-000000000-00000
13. Powers SK, Quindry JC, Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic Biol Med. (2008) 44:193–201. doi: 10.1016/j.freeradbiomed.2007.02.006
14. Frasier CR, Moore RL, Brown DA. Exercise-induced cardiac preconditioning: How exercise protects your achy-breaky heart. J Appl Physiol. (2011) 111:905–15. doi: 10.1152/japplphysiol.00004.2011
15. Hamilton KL, Staib JL, Phillips T, Hess A, Lennon SL, Powers SK. Exercise, antioxidants, and HSP72: protection against myocardial ischemia/reperfusion. Free Radic Biol Med. (2003) 34:800–9. doi: 10.1016/S0891-5849(02)01431-4
16. Abe K, Hayashi N, Terada H. Effect of endogenous nitric oxide on energy metabolism of rat heart mitochondria during ischemia and reperfusion. Free Radic Biol Med. (1999) 26:379–87. doi: 10.1016/S0891-5849(98)00222-6
17. Riobó NA, Clementi E, Melani M, Boveris A, Cadenas E, Moncada S, et al. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem J. (2001) 359:139–45. doi: 10.1042/bj3590139
18. Jekabsone A, Ivanoviene L, Brown GC, Borutaite V. Nitric oxide and calcium together inactivate mitochondrial complex I and induce cytochrome c release. J Mol Cell Cardiol. (2003) 35:803–9. doi: 10.1016/S0022-2828(03)00137-8
19. Kavazis AN, Alvarez S, Talbert E, Lee Y, Powers SK. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol - Hear Circ Physiol. (2009) 297:H144–52. doi: 10.1152/ajpheart.01278.2008
20. Hausenloy DJ, Tsang A, Yellon DM. The Reperfusion Injury Salvage Kinase Pathway: A Common Target for Both Ischemic Preconditioning and Postconditioning. Trends Cardiovasc Med. (2005) 15:69–75. doi: 10.1016/j.tcm.2005.03.001
21. Brown DA, Jew KN, Sparagna GC, Musch TI, Moore RL. Exercise training preserves coronary flow and reduces infarct size after ischemia-reperfusion in rat heart. J Appl Physiol. (2003) 95:2510–8. doi: 10.1152/japplphysiol.00487.2003
22. Pell VR, Chouchani ET, Murphy MP, Brookes PS, Krieg T. Moving forwards by blocking back-flow the yin and yang of MI therapy. Circ Res. (2016) 118:898–906. doi: 10.1161/CIRCRESAHA.115.306569
23. Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochimica et Biophysica Acta - Bioenergetics. (2009) 1787:1402–15. doi: 10.1016/j.bbabio.2008.12.017
24. Machado NG, Alves MG, Carvalho RA, Oliveira PJ. Mitochondrial involvement in cardiac apoptosis during ischemia and reperfusion: Can we close the box? Cardiovasc Toxicol. (2009) 9:211–27. doi: 10.1007/s12012-009-9055-1
25. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. (2014) 114:524–37. doi: 10.1161/CIRCRESAHA.114.300559
26. Honda HM, Ping P. Mitochondrial permeability transition in cardiac cell injury and death. Cardiovasc Drugs Ther. (2006) 20:425–32. doi: 10.1007/s10557-006-0642-0
27. Paradies G, Paradies V, Ruggiero FM, Petrodillo G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. Am J Physiol Heart Circ Physiol. (2018) 315:H1341–52. doi: 10.1152/ajpheart.00028.2018
28. Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and Is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. (2001) 276:2571–5. doi: 10.1074/jbc.M006825200
29. Abdallah Y, Kasseckert SA, Iraqi W, Said M, Shahzad T, Erdogan A, et al. Interplay between Ca 2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med. (2011) 15:2478–85. doi: 10.1111/j.1582-4934.2010.01249.x
30. Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, et al. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. (2012) 52:1009–18. doi: 10.1016/j.yjmcc.2012.02.009
31. Heather LC, Carr CA, Stuckey DJ, Pope S, Morten KJ, Carter EE, et al. Critical role of complex III in the early metabolic changes following myocardial infarction. Cardiovasc Res. (2010) 85:127–36. doi: 10.1093/cvr/cvp276
32. Halestrap AP, Richardson AP. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J Mol and Cell Cardiol. (2015) 78:129–41. doi: 10.1016/j.yjmcc.2014.08.018
33. Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. (2002) 286:135–41. doi: 10.1016/S0378-1119(01)00814-9
34. Dedkova EN, Blatter LA. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol. (2013) 58:125–33. doi: 10.1016/j.yjmcc.2012.12.021
35. Vercesi AE, Kowaltowski AJ, Oliveira HCF, Castilho RF. Mitochondrial Ca2+ transport, permeability transition and oxidative stress in cell death: Implications in cardiotoxicity, neurodegeneration and dyslipidemias. Frontiers Biosci. (2006) 11:2554–64. doi: 10.2741/1990
36. Raedschelders K, Ansley DM, Chen DDY. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol Therapeut. (2012) 133:230–55. doi: 10.1016/j.pharmthera.2011.11.004
37. Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic Biol Med. (1999) 27:42–50. doi: 10.1016/S0891-5849(99)00032-5
38. Dickson EW, Hogrefe CP, Ludwig PS, Ackermann LW, Stoll LL, Denning GM. Exercise enhances myocardial ischemic tolerance via an opioid receptor-dependent mechanism. Am J Physiol - Hear Circ Physiol. (2008) 294:H402–8. doi: 10.1152/ajpheart.00280.2007
39. La Rovere MT, Bersano C, Gnemmi M, Specchia G, Schwartz PJ. Exercise-induced increase in baroreflex sensitivity predicts improved prognosis after myocardial infarction. Circulation. (2002) 106:945–9. doi: 10.1161/01.CIR.0000027565.12764.E1
40. Demirel HA, Powers SK, Caillaud C, Coombes JS, Naito H, Fletcher LA, et al. Exercise training reduces myocardial lipid peroxidation following short- term ischemia-reperfusion. Med Sci Sports Exerc. (1998) 30:1211–6. doi: 10.1097/00005768-199808000-00005
41. Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochimica et Biophysica Acta - Bioenergetics. (2014) 1837:408–17. doi: 10.1016/j.bbabio.2013.10.006
42. Bouchard C, Lesage R, Lortie G, Simoneau JA, Hamel P, Boulay MR, et al. Aerobic performance in brothers, dizygotic and monozygotic twins. Med Sci Sports Exerc. 10.
43. Koch LG. Britton Sl. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol Genomics. (2001) 5:45–52. doi: 10.1152/physiolgenomics.2001.5.1.45
44. Britton SL, Koch LG. Animal genetic models for complex traits of physical capacity. Exerc Sport Sci Rev. (2001) 29:7–14. doi: 10.1097/00003677-200101000-00003
45. Koch LG, Britton SL, Wisløff U. A rat model system to study complex disease risks, fitness, aging, and longevity. Trends Cardiovasc Med. (2012) 22:29–34. doi: 10.1016/j.tcm.2012.06.007
46. Koch LG, Britton SL. Theoretical and Biological Evaluation of the Link between Low Exercise Capacity and Disease Risk. Cold Spring Harb Perspect Med. (2018) 8:639–46. doi: 10.1101/cshperspect.a029868
47. Koch LG, Britton SL. Aerobic metabolism underlies complexity and capacity. J Physiol. (2008) 586:83–95. doi: 10.1113/jphysiol.2007.144709
48. Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. (2005) 307:418–20. doi: 10.1126/science.1108177
49. de Godoy MRC, Padilla J, Swanson KS, Vieira-Potter VJ. Soy improves cardiometabolic health and cecal microbiota in female low-fit rats. Sci Rep. (2017) 7:9261. doi: 10.1038/s41598-017-08965-0
50. Rosenblat M, Volkova N, Abassi Z, Britton SL, Koch LG, Aviram M. High intrinsic aerobic capacity and pomegranate juice are protective against macrophage atherogenecity: studies in high- vs. low-capacity runner (HCR vs LCR) rats J Nutr Biochem. (2015) 26:1015–21. doi: 10.1016/j.jnutbio.2015.04.001
51. Høydal MA, Stølen TO, Johnsen AB, Alvez M, Catalucci D, Condorelli G, et al. Reduced aerobic capacity causes leaky ryanodine receptors that trigger arrhythmia in a rat strain artificially selected and bred for low aerobic running capacity. Acta Physiol (Oxf). (2014) 210:854–64. doi: 10.1111/apha.12238
52. Hussain SO, Barbato JC, Koch LG, Metting PJ, Britton SL. Cardiac function in rats selectively bred for low- and high-capacity running. Am J Physiol Regul Integr Comp Physiol. (2001) 281:R1787–91. doi: 10.1152/ajpregu.2001.281.6.R1787
53. Aon MA, Cortassa S, Juhaszova M, González-Reyes JA, Calvo-Rubio M, Villalba JM, et al. Mitochondrial health is enhanced in rats with higher vs. lower intrinsic exercise capacity and extended lifespan. NPJ Aging Mech Dis. (2021) 7:1. doi: 10.1038/s41514-020-00054-3
54. Johnsen VL, Belke DD, Hughey CC, Hittel DS, Hepple RT, Koch LG, et al. Enhanced cardiac protein glycosylation (O-GlcNAc) of selected mitochondrial proteins in rats artificially selected for low running capacity. Physiol Genomics. (2013) 45:17–25. doi: 10.1152/physiolgenomics.00111.2012
55. Hjortbak MV, Grønnebæk TS, Jespersen NR, Lassen TR, Seefeldt JM, Tonnesen PT, et al. Differences in intrinsic aerobic capacity alters sensitivity to ischemia-reperfusion injury but not cardioprotective capacity by ischemic preconditioning in rats. PLoS ONE. (2020) 15:e0240866. doi: 10.1371/journal.pone.0240866
56. Høydal MA, Kaurstad G, Rolim NP, Johnsen AB, Alves M, Koch LG, et al. High inborn aerobic capacity does not protect the heart following myocardial infarction. J Appl Physiol. (1985) 115:1788–95. doi: 10.1152/japplphysiol.00312.2013
57. Alsahly MB, Zakari MO, Koch LG, Britton S, Katwa LC, Lust RM. Influence of intrinsic aerobic capacity and sex on cardiac injury following acute myocardial ischemia and reperfusion. Front Cardiovasc Med. (in press).
58. Palpant NJ, Szatkowski ML, Wang W, Townsend D, Bedada FB, Koch LG, et al. Artificial selection for whole animal low intrinsic aerobic capacity co-segregates with hypoxia-induced cardiac pump failure. PLoS ONE. (2009) 4:e6117. doi: 10.1371/journal.pone.0006117
59. McElroy CL, Gissen SA, Fishbein MC. Exercise-induced reduction in myocardial infarct size after coronary artery occlusion in the rat. Circulation. (1978) 57:958–62. doi: 10.1161/01.CIR.57.5.958
60. Solaini G, Harris DA. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem J. (2005) 390:377–94. doi: 10.1042/BJ20042006
61. Gallego-Selles A, Martin-Rincon M, Martinez-Canton M, Perez-Valera M, Martín-Rodríguez S, Gelabert-Rebato M, et al. Regulation of Nrf2/Keap1 signalling in human skeletal muscle during exercise to exhaustion in normoxia, severe acute hypoxia and post-exercise ischaemia: Influence of metabolite accumulation and oxygenation. Redox Biol. (2020) 36:101627. doi: 10.1016/j.redox.2020.101627
62. Flockhart M, Nilsson LC, Tais S, Ekblom B, Apró W, Larsen FJ. Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell Metab. (2021) 33:957–70. doi: 10.1016/j.cmet.2021.02.017
63. Veitch K, Hombroeckx A, Caucheteux D, Pouleur H, Hue L. Global ischaemia induces a biphasic response of the mitochondrial respiratory chain: Anoxic pre-perfusion protects against ischaemic damage. Biochem J. (1992) 281:709–15. doi: 10.1042/bj2810709
64. Cairns CB, Ferroggiaro AA, Walther JM, Harken AH, Banerjee A. Postischemic administration of succinate reverses the impairment of oxidative phosphorylation after cardiac ischemia and reperfusion injury. Circ. (1997) 96:260–5.
65. Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. (2000) 14:729–39. doi: 10.1096/fasebj.14.5.729
66. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. (1997) 91:479–89. doi: 10.1016/S0092-8674(00)80434-1
67. McLaughlin KL, Hagen JT, Coalson HS, Nelson MAM, Kew KA, Wooten AR, Fisher-Wellman KH. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci Rep. (2020) 10:17599.
Keywords: aerobic capacity, HCR, LCR, intrinsic, coronary occlusion, mitochondria, energetics
Citation: Alsahly MB, Zakari MO, Koch LG, Britton S, Katwa LC, Fisher-Wellman K and Lust RM (2021) Augmented Cardiac Mitochondrial Capacity in High Capacity Aerobic Running “Disease-Resistant” Phenotype at Rest Is Lost Following Ischemia Reperfusion. Front. Cardiovasc. Med. 8:752640. doi: 10.3389/fcvm.2021.752640
Received: 03 August 2021; Accepted: 30 September 2021;
Published: 03 November 2021.
Edited by:
Jian Yang, Fudan University, ChinaReviewed by:
Gentaro Ikeda, Stanford University, United StatesPongpan Tanajak, Apinop Wetchakham Hospital (Keang Khoi Medical Center), Thailand
Krekwit Shinlapawittayatorn, Chiang Mai University, Thailand
Copyright © 2021 Alsahly, Zakari, Koch, Britton, Katwa, Fisher-Wellman and Lust. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert M. Lust, bHVzdHJAZWN1LmVkdQ==