 Rebecca Hernandez
Rebecca Hernandez Changcheng Zhou
Changcheng Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 08 December 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.752337
This article is part of the Research Topic Immunometabolism, Atherosclerosis and Metabolic Diseases View all 13 articles
Cardiometabolic diseases, including cardiovascular disease, obesity, and diabetes, are the leading cause of mortality and morbidity worldwide. Cardiometabolic diseases are associated with many overlapping metabolic syndromes such as hypertension, hyperlipidemia, insulin resistance, and central adiposity. However, the underlying causes of cardiometabolic diseases and associated syndromes remain poorly understood. Within the past couple of decades, considerable progresses have been made to understand the role of inflammatory signaling in the pathogenesis of cardiometabolic diseases. The transcription factor, NF-κB, a master regulator of the innate and adaptive immune responses, is highly active in cardiometabolic diseases. IκB kinase β (IKKβ), the predominant catalytic subunit of the IKK complex, is required for canonical activation of NF-κB, and has been implicated as the critical molecular link between inflammation and cardiometabolic diseases. Recent studies have revealed that IKKβ has diverse and unexpected roles in mediating adiposity, insulin sensitivity, glucose homeostasis, vascular function, and atherogenesis through complex mechanisms. IKKβ has been demonstrated as a critical player in the development of cardiometabolic diseases and is implicated as a promising therapeutic target. This review summarizes current knowledge of the functions of IKKβ in mediating the development and progression of cardiometabolic diseases.
Cardiometabolic diseases such as atherosclerosis, obesity, and diabetes are related to several risk factors termed cardiometabolic syndromes (1, 2). Cardiometabolic syndromes encompass a group of metabolic dysfunctions like hypertension, hyperlipidemia, insulin resistance, and central adiposity (1). Chronic low-grade inflammation has been established as a major contributor to the development of cardiometabolic diseases such as type 2 diabetes and atherosclerosis (3, 4). Many inflammatory pathways that contribute to the cardiometabolic disease risk are regulated by the transcriptional factor NF-κB, a master regulator of the innate and adaptive immune responses (1, 5). In non-stimulated cells, NF-κB remains in the cytoplasm bound to specific inhibitory proteins—the inhibitors of NF-κB (IκBs). In response to various stimuli including proinflammatory cytokines, infectious agents, reactive oxygen species, and free fatty acids (FFAs), NF-κB can be rapidly activated through the IκB kinase (IKK) complex (1, 5, 6). The IKK complex is composed of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ/NEMO). Activation of IKK can lead to the phosphorylation and ubiquitination of IκB. Consequently, free NF-κB can then translocate to the nucleus and regulate the expression of many target genes (1, 7).
While IKKβ and IKKα, have a similar structure, they have different functions as IKKα contains a putative nuclear localization signal and IKKβ contains a ubiquitin binding domain. In addition, IKKβ activation is necessary for canonical NF-κB pathway activation, while IKKα is not (8, 9). The stimuli that can activate IKKβ include proinflammatory cytokines, growth factors, microbial products, stress stimuli, and the engagement of T cell receptors. These stimuli can activate membrane-bound receptors such as the Tumor necrosis factor receptor superfamily (TNFRSF), Interleukin-1 receptor (IL-1R), and Toll-like receptors (TLR), subsequently leading to the activation of the IKK complex (10, 11). IKKβ and its serine-threonine kinase activity are essential for regulating inflammatory and immune responses, and many studies have uncovered its function in chronic inflammation-associated cardiometabolic diseases such as atherosclerosis, obesity, and insulin resistance (Figure 1). In addition to regulating the NF-κB pathway, more and more new targets of IKKβ have also been identified. The known IKKβ substrates and their functions in tumorigenesis, inflammation, diabetes, hormone response, and cell survival have been discussed in detail in several comprehensive reviews (12, 13). For the purpose of this review, we focus on IKKβ, its known substrates, and their functions in the development of cardiometabolic diseases (Figure 1).
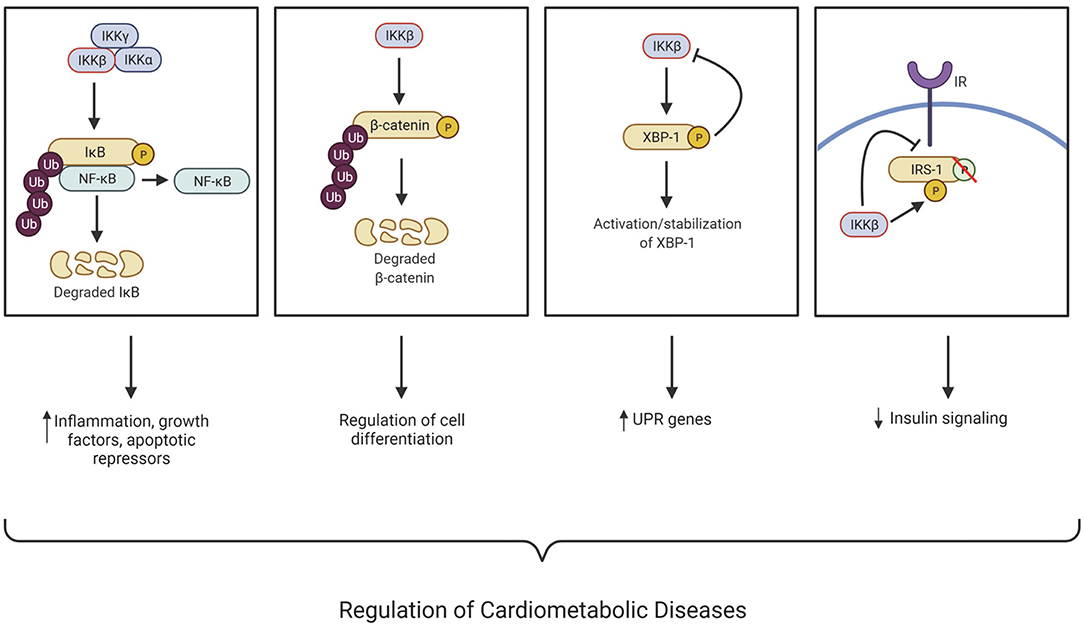
Figure 1. IKKβ regulates many cellular processes associated with the development of cardiometabolic diseases through NF-κB-dependent and -independent mechanisms. IκB kinase (IKK); Nuclear factor kappa B (NF-κB); X-box binding protein (XBP-1); Insulin receptor substate 1 (IRS-1); Insulin receptor (IR); Ubiquitination (Ub); Unfolded protein response (UPR); Phosphorylation (P). This figure was created using BioRender.com.
Atherosclerosis is the major contributing risk factor for the development of cardiovascular disease (CVD). It is a very complex disease involving the development of plaques in large arteries causing narrowing of the vessel lumen leading to various clinical manifestations, including stroke, ischemic heart disease, chronic kidney disease, and peripheral artery disease. The plaques are characterized by accumulating lipids and immune cells into the sub-endothelial space (14–18). Atherosclerosis has been characterized as a chronic inflammatory disease, which may be initiated when the endothelium undergoes a phenotypic change, termed endothelial dysfunction, stimulated by modified LDL such as oxidized-LDL (oxLDL) and inflammatory stimuli. The endothelium fails to maintain vascular homeostasis during endothelial dysfunction like vasodilation, eliminating reactive oxygen species, and maintaining an appropriate inflammatory balance. Various chemotactic factors and adhesion molecules are differentially expressed by endothelial cells undergoing endothelial dysfunction, which aids in monocyte migration and infiltration. Ox-LDL is rapidly taken up by monocyte scavenger receptors upon monocyte infiltration, leading to the conversion of monocytes into lipid-filled macrophage foam cells. The lesional foam cells can release inflammatory factors to further contribute to the monocyte and lipid build-up (16–19). While acute vessel wall inflammation leads to asymptomatic fatty streaks, chronic inflammation can cause the gradual and uncontrolled accumulation of macrophage foam cells that later develop into symptomatic atheromas or plaques. For many years, the NF-κB pathway has been implicated in the pathogenesis of atherosclerosis (20). For example, NF-κB activation has been detected in atherosclerotic plaques, including macrophages, endothelial cells, and smooth muscle cells in both human and animal models (21–24). Previous studies have implicated that NF-κB activation in human atherosclerosis was IKKβ-dependent and resulted in up-regulation of proinflammatory and prothrombotic mediators (25). However, recently studies have demonstrated that the functions of IKKβ in atherosclerosis are complex and that IKKβ in different tissues or cell types may have different impact on atherosclerosis development in animal models.
As a significant player in atherosclerosis initiation and progression, studies have suggested that the inflammatory response in endothelial dysfunction can be driven by IKKβ/NF-κB signaling (14, 21, 26). Gareus et al. previously demonstrated that inhibition of NF-κB activity through the deletion of IKKγ, also known as NF-κB essential modulator (NEMO), or expression of a dominant-negative IκBα decreases atherosclerosis in atherogenic prone mice (14). They also found that inhibition of NF-κB in endothelial cells reduced the expression of proinflammatory cytokines, chemokines, and adhesion molecules, leading to decreased monocyte recruitment into the plaque (14). Consistently, inhibition of IKKβ in human umbilical vein endothelial cells has been shown to block NF-κB activation, leading to decreased adhesion molecule gene expression including E-selectin, ICAM-1, and VCAM-1 (27). These adhesion molecules are essential for the attachment and infiltration of the recruited monocytes into the intimal layer (16–18). By contrast, constitutive activation of endothelial IKKβ in mice increased monocyte infiltration into the subintimal space, which contributed to exacerbating early and late-stage atherosclerosis (28). Indeed, the rise of age-associated endothelial dysfunction is correlated with increased IKK activation in arteries while pharmacological inhibition of IKK by salicylate has been shown to improve age-related endothelial dysfunction (29). Thus, targeting endothelial cell IKKβ may have beneficial effects against atherosclerosis development.
The M1, or proinflammatory, macrophage plays a key role in atherosclerosis development, while M2, or anti-inflammatory, macrophages enhance plaque regression and stability (30). The link between macrophage polarization and IKKβ remains elusive, though evidence suggests that IKKβ/NF-κB pathway activation polarizes macrophages to the M2, anti-inflammatory phenotype through negative crosstalk with STAT1 (31, 32). To study the role of macrophage IKKβ in atherosclerosis, Kanters et al. transplanted IKKβ-deficient bone marrow-derived macrophages into atherogenic prone low-density lipoprotein receptor-deficient (LDLR−/−) mice. They found that the mice receiving IKKβ-deficient macrophages exhibited enhanced atherosclerotic lesion development and increased necrosis, which suggest a protective role of bone marrow-derived macrophage IKKβ against atherosclerosis development (33). However, the same group used a similar method to delete IκBα in myeloid cells, aimed to activate NF-κB signaling. Interestingly, those mice displayed increased atherosclerosis lesion size and leukocyte adhesion without significantly increasing NF-κB targeted genes (34), indicating pro-atherogenic effects of canonical NF-κB activation. Several other studies have also found that macrophage IKKβ/NF-κB pathway has pro-atherogenic effects (35, 36). For example, inhibition of NF-κB in macrophages through the overexpression of a trans-dominant and non-degradable form of IκBα can reduce macrophage foam cell formation in vitro (35). Further, myeloid-specific IKKβ deficiency decreased diet-induced atherosclerosis in LDLR−/− mice by diminishing macrophage inflammatory responses such as adhesion, migration and lipid uptake in macrophages (36). Collectively, these results indicate the role of macrophage IKKβ/NF-κB in atherogenesis is complex and more studies are needed to completely understand how IKKβ functions in myeloid cells to regulate atherosclerosis development.
In addition to endothelial and immune cells, vascular smooth muscle cells (VSMCs) also play an important role in atherogenesis. In the early stages of atherosclerosis, VSMCs undergo a phenotypic switch from contractile to synthetic where they gain the ability to proliferate and migrate into the intimal layer. This provides a beneficial effect as these VSMCs proliferate and migrate to the cap of the plaque and reinforces its stability, lowering the risk for plaque rupture (37). An earlier study demonstrated that IKKα and IKKβ was activated in IL-1β-induced proliferative response of human saphenous vein smooth muscle cells (38). Notably, the proliferative ability of human VSMCs were diminished in IKKα and IKKβ mutant transfected cells (38). The role of VSMC IKKβ in atherosclerosis was also investigated in LDLR−/− mice (39). Deficiency of IKKβ in VSMCs driven by a SM22Cre-IKKβ-flox system protected LDLR−/− mice from diet-induced vascular inflammation and atherosclerosis development (39). Since inhibition of NF-κB activity in endothelia cells also decreased vascular inflammation and atherosclerosis in ApoE−/− mice (14), these studies suggest that inhibiting IKKβ/NF-κB signaling in the vasculature has anti-atherogenic effects.
Under pathological conditions, adipose tissue is at a chronic low level of inflammation (3). The circulating inflammatory mediators secreted by adipocytes participate in vascular dysfunction, which can lead to atherosclerosis (40). However, the role of adipocyte IKKβ signaling in atherogenesis is poorly understood. A recent study found that adipocyte-specific deletion of IKKβ did not affect obesity and atherosclerosis in lean LDLR−/− mice when fed a low-fat diet (41). When fed a high-fat diet, however, IKKβ-deficient LDLR−/− mice had defective adipose remodeling, leading to increased adipose tissue and systemic inflammation (41). Deficiency of adipocyte IKKβ did not affect atherosclerotic lesion size but resulted in enhanced lesional inflammation and increased plaque vulnerability in obese IKKβ-deficient LDLR−/− mice (41). In addition to regular fat depots, adipocytes can also be found adjacent to the vascular wall called perivascular adipose tissue (PVAT). Under homeostatic conditions, PVAT holds a protective role on vascular homeostasis by secreting bioactive molecules like adiponectin, nitric oxide (NO), and IL-10 (42, 43). However, under pathological conditions, PVAT switches to a proinflammatory phenotype by secreting adipokines, cytokines, and chemokines (43). The role of PVAT in atherosclerosis and vascular injury has not been extensively investigated. However, studies have found that PVAT may contribute to endothelial dysfunction (42), macrophage migration, and VSMC proliferation and migration (44). The role of PVAT IKKβ in vascular function and atherosclerosis remains elusive. Future studies should be considered to investigate the role of PVAT IKKβ/NF-κB signaling on vascular function and atherosclerotic development under normal or pathological conditions (e.g., obesity).
Obesity is a worldwide epidemic and a risk factor for developing severe metabolic and cardiovascular diseases. According to the updated 2020 Heart Disease and Stroke Statistics, 39.6% of adults and 18.5% of youth are living with obesity in the US (45). Thus, research surrounding this field has become increasingly popular due to the financial, economic, and mental burden it carries (46). Obesity is associated with a low-grade chronic inflammation that contributes to the development of many chronic diseases including insulin resistance, diabetes, and CVD (3, 4, 47–49). Adipocytes are responsible for energy storage and respond to overnutrition by increasing adiposity and inflammation. There are three general steps to adipose chronic inflammation. First, adipocytes are introduced to a stressor, like overnutrition. The adaptive physiological response, which includes acute inflammation, aims to balance, and reduce this stressor. However, chronic exposure to this stressor creates new set basal points, which includes higher blood glucose levels and increased body weight (50). Thus, understanding the mechanistic link between inflammatory pathways in obesity, and obesity induced metabolic disorders is critical for developing essential therapeutic targets.
The IKKβ/NF-κB pathway is highly active in the adipose tissues of obese patients and in mouse models of obesity and insulin resistance (1, 51, 52). In addition to regulating inflammatory responses, IKKβ also plays important roles in regulating cell proliferation, differentiation, survival, and apoptosis (47, 53). However, the function of IKKβ during obesity in the context of adipose tissue development remain elusive. Recent studies have revealed the previously unrecognized function of IKKβ in regulating adiposity.
While deletion of IKKβ in VSMCs decreased atherosclerosis development in LDLR−/− mice (39), those mice were also protected from diet-induced obesity and insulin resistance. Interestingly, many adipocyte precursor cells express SMC markers and ablation of IKKβ blocked adipocyte differentiation in vitro and in vivo, suggesting that IKKβ functions in adipocyte precursor cells to regulate adiposity (39). Indeed, selective deletion of IKKβ in the white adipose lineage further elucidated the role of adipose progenitor cell IKKβ signaling in regulating adiposity and metabolic function (39, 54). Deficiency of adipose progenitor IKKβ decreased high-fat feeding-induced adipogenesis and systemic inflammation, resulting in decreased adiposity and insulin resistance in those mice (39, 54). The function of IKKβ in the regulation of adipogenesis was further confirmed in mesenchymal stem cells (MSCs) (55). Mechanistic studies then revealed an important crosstalk between IKKβ and Wnt/β-catenin signaling (Figure 1) (55). Interestingly, IKKβ is a β-catenin kinase that can directly phosphorylate the conserved degron motif of β-catenin to prime it for β-TrCP-mediated ubiquitination and degradation (10, 55). Wnt/β-catenin signaling has been well studied to inhibit adipocyte differentiation (56, 57) and the impact of IKKβ signaling on adipogenesis was abolished in β-catenin-deficient MSCs (10, 55). Thus, IKKβ-mediated β-catenin phosphorylation may play a critical role in regulating adipocyte differentiation and adiposity in obesity (Figure 1).
While studies have suggested a pro-obesogenic role of progenitor IKKβ, the function of IKKβ in mature adipocytes is apparently more complicated. Constitutive activation of IKKβ in adipocytes has been demonstrated to increased energy expenditure in mice, leading to protective effects against diet-induced obesity and insulin resistance (58). However, targeted deletion of IKKβ in adipocytes did not affect obesity but resulted in increased tissue inflammation, impaired adipose remodeling, and exacerbated metabolic disorders (59, 60). In addition to mediating inflammation, IKKβ can also promote cell survival by upregulating NF-κB-mediated anti-apoptotic gene expression (61–63) and by direct phosphorylation of pro-apoptotic protein, BAD (64). Previous reports have linked adipocyte death with obesity, adipocyte macrophage infiltration, and systemic insulin resistance (65). IKKβ has been shown to be a key adipocyte survival factor in obesity, and deficiency of IKKβ in adipocytes can lead to high fat feeding-elicited cell death, impaired adipose tissue remodeling and partial lipodystrophy in visceral adipose tissue (59, 60). Further studies are required to completely understand the role of adipocyte IKKβ in regulating energy expenditure, homeostasis, and adiposity.
Insulin resistance is a very complex syndrome and IKKβ has been shown to regulate insulin resistance by directly interfering with the insulin signaling pathway (66). Once stimulated by its ligand, insulin, the insulin receptor (IR) becomes activated and phosphorylates insulin receptor substrate-1 (IRS-1) on its tyrosine residues, leading to increased glucose uptake (67). As a serine kinase, IKKβ can ectopically phosphorylate IRS-1 on multiple serine residues, which impairs insulin signaling (Figure 1) (68). Several studies have demonstrated that treatment with glucose lowering drugs and molecules such as kaempferol (69), timosaponin B-II (TB-II) (70), rosiglitazone (71), and bovine α-lactalbumin hydrolysates (α-LAH) (72) can alleviate insulin resistance by decreasing or inhibiting IKKβ levels/activity resulting in a reduction of ectopic IRS-1 serine phosphorylation.
The IKKβ/NF-κB pathway has been demonstrated to be active in both obesity-dependent and independent insulin resistance (47, 53). Inhibition of IKKβ with salicylate or other methods is associated with reduced insulin resistance and glucose intolerance (54, 73–75). Previous studies demonstrated that constitutively active hepatic IKKβ induced obesity-independent systemic insulin resistance, while inhibiting hepatic NF-κB reversed both local and systemic insulin resistance (51, 76). These findings indicate an important role of IKKβ in regulating hepatic and systemic insulin resistance. Another study utilizing hepatocyte-specific IKKβ deficient mice found improved hepatic insulin response while maintaining systemic insulin resistance during obesity (77). These results can be attributed to obesity-associated systemic inflammation that cannot be alleviated by IKKβ knockdown in the liver alone. More recently, it has been reported that hepatic IKKβ in the liver can improve glucose homeostasis by interacting with x-box binding protein 1 (XBP1) and enhancing its activity, stabilization, and nuclear translocation (Figure 1) (78). While it is generally recognized that hepatic inflammation drives the detrimental perspectives of obesity-induced insulin resistance (1, 73, 79), upregulation of certain inflammatory signaling could have positive or negative contributions to whole-body metabolism, depending on conditions of signaling activation and related physiological statuses. Therefore, the hepatic IKKβ function in insulin resistance is complex and future studies are required to define the detailed mechanisms through which hepatic IKKβ regulates insulin responsiveness under normal and pathophysiological conditions.
Inflammation is an important contributor of insulin resistance, and adipose tissue is one of the important tissues for this high-fat feeding-elicited inflammatory response (80). Adipose IKKβ signaling has been implicated in obesity-associated insulin resistance. For example, studies have found that IKKβ deficiency in adipocyte precursors or adipose lineage cells can protect mice from diet-induced obesity, systemic inflammation and insulin resistance (39, 54). Several studies demonstrated that IKKβ deficiency and XBP1 overexpression attenuates FFA-induced inflammation and impairment of insulin signaling in cultured adipocytes (81, 82). While hepatic IKKβ increases nuclear translocation of XBP1 (78), adipocyte IKKβ is inhibited by XBP1 (82), indicating a more complex role of IKKβ/XBP1 interaction in cardiometabolic disease. Overexpression of IKKβ in adipocytes also led to increased adipose tissue inflammation in mice (58). Paradoxically, those mice were resistant to diet-induced obesity and insulin resistance, likely due to increased energy expenditure (58). Deletion of adipocyte IKKβ did not affect obesity in mice but resulted in elevated adipose tissue inflammation, increased macrophage infiltration and exacerbate insulin resistance (59, 60).
Skeletal muscle is another insulin responsive tissue that is impaired in obesity and diabetes (67). Studies revealed elevated IKKβ activity in isolated skeletal muscle of obese patients with type 2 diabetes and obese mice (83, 84). By contrast, inhibition of IKKβ or NF-κB signaling can restore insulin signaling in vitro (85, 86) and systemic IKKβ inhibition can alleviate skeletal muscle and systemic insulin resistance all together (73, 74). However, under obese conditions, targeting skeletal muscle IKKβ can only alleviate local insulin resistance, but not systemic insulin responsiveness (87).
While tissue-specific inhibition of IKKβ (i.e., liver, adipose, skeletal muscle) may be able to abrogate local insulin resistance, it may not be sufficient for systemic inflammation-induced insulin resistance under obese conditions. For example, it is reported that myeloid-specific IKKβ deficiency can improve obese-dependent systemic insulin resistance (77, 87), indicating that myeloid cell IKKβ plays a role in systemic insulin resistance and inflammation in obesity. Furthermore, Cai et al. linked the IKKβ/NF-κB pathway with paracrine IL-6 signaling (51), which is associated with type 2 diabetes and insulin resistance (88, 89). IL-6 can induce the expression of suppressor of cytokine signaling 3 (SOCS-3), which inhibits autophosphorylation of IRS-1 and insulin receptor (90). The IKKβ/NF-κB/IL-6 axis was confirmed to be involved in insulin resistance when IL-6 neutralization improved insulin resistance (51).
Non-alcoholic fatty liver disease or non-alcoholic steatohepatitis is also associated with metabolic syndrome. The activation of the IKKβ/NF-κB pathway has been shown to promote fatty liver disease, or hepatic steatosis (91), whereas inhibition of IKKβ prevents the initiation of steatosis and non-alcoholic steatohepatitis (75, 92). Inhibition of IKKβ significantly reduced the expression of essential proinflammatory genes like TNFα and IL-6 in the liver (92). In line with lipid metabolism, the peroxisome proliferator-activated receptor family (PPAR) is an important regulator of lipid homeostasis in multiple organs and tissues (93). PPARα, highly expressed in the liver, can upregulate IκB, thus inhibiting the NF-κB pathway (92, 93). Interestingly, IKKβ inhibition can also lead to PPARα upregulation and reduced lipid accumulation in the liver by increasing CPT-1 and ACOX—two important molecules that decreases fatty acid accumulation through β-oxidation (92). Additionally, IKKβ inhibition attenuated hepatic inflammation, apoptosis, and collagen deposition, therefore preventing liver fibrosis (54, 92). By contrast, hepatic IKKβ activation promoted liver fibrosis by inducing chronic inflammation (94). While the mechanism behind IKKβ-mediated hepatic steatosis and fibrosis remain to be explored, these findings suggest that inhibiting IKKβ may prevent lipid and collagen accumulation in the liver, leading to decreased hepatic steatosis and fibrosis development.
Although there have been strong links between IKKβ and metabolic diseases within the periphery, more recently, inflammatory activation has been seen within the central nervous system (CNS). Specifically, IKKβ in the hypothalamus can be activated in obesity and obesity-related metabolic dysregulation such as energy, body weight, and glucose dysregulation (95–98). A study found that FFAs induce TLR4-mediated hypothalamic cytokine production and anorexigenic signal resistance which may lead to obesity (99). Signaling between the gut and brain (gut-brain-axis) is a major influencer in developing obesity. Obese mice and mice stimulated with overnutrition display overall higher levels of IKKβ within the hypothalamic neurons, which is consistent with the systemic trend (95, 100). However, it was observed that overnutrition-mediated activation of IKKβ/NF-κB was activated intracellularly by ER stress and prompted both hypothalamic leptin and insulin resistance through the induction of suppressor of cytokine signaling 3 (SOCS3), an inhibitor of leptin and insulin signaling (95, 101). ER stress can also lead to impaired hepatic insulin signaling, which was improved upon ER stress inhibition (102). TLR-dependent IKKβ activation in the CNS was also involved in obesity and leptin resistance (96). Deficiency of IKKβ in hypothalamic AGRP neurons displayed anti-obese phenotype along with preserved leptin and insulin signaling and reduced SOCS3 gene expression, and overexpression of SOCS3 reversed the protective effects of IKKβ knockout in mice (95). By contrast, activation of IKKβ in AGRP neurons resulted in impaired glucose homeostasis, without affecting body weight and leptin signaling (103).
While it is critical to study the effects of hypothalamic inflammation on obesity and metabolic syndromes, it is also important to investigate the upstream targets mediating hypothalamic inflammation. For example, astrocytes play essential roles in neuronal development; regulation of blood flow; fluid, ion, pH, and transmitter homeostasis; the regulation of synaptic transmission; and regulate immune response (104). Under pathological conditions or external stressors, astrocytes and other glial cells undergo gliosis, or astrogliosis, which is characterized by proliferation and accumulation of astrocytes (104, 105). Zhang et al. demonstrated an important role of astrocyte IKKβ in stimulating glucose intolerance, hypertension, and weight gain (106). While overnutrition and IKKβ overexpression inhibited proper astrocytic plasticity, inhibition of IKKβ prevented overnutrition-induced metabolic diseases and impaired astrocytic plasticity (106). Mechanistically, IKKβ-induced shortening of astrocyte processes led to increased extracellular GABA, an inhibitory neurotransmitter, and lower brain derived neurotrophic factor (BDNF) levels through inhibition of BDNF secreting neurons in the hypothalamus (106). Low levels of BDNF have been associated with metabolic disorders such as obesity, energy metabolism, and hyperglycemia (107). The protective role of IKKβ deficiency in astrocytes were reversed by BDNF inhibition, suggesting that the GABA-BDNF axis is important in regulating energy homeostasis and metabolic syndromes (106). In addition to developed cells within the CNS, the hypothalamic neural stem cells are important mediators for metabolic syndrome. IKKβ/NF-κB activation in the mediobasal hypothalamus can lead to obesity and insulin resistance, along with loss of neuronal development including POMC neurons (108).
Hypertension, a chronic elevation in arterial blood pressure, is one of the major risk factors for developing CVD such as myocardial infarction, stroke, and heart failure. Although there are therapeutic interventions aimed to target and treat hypertension, it is still a prevalent contributor to cardiometabolic disease burden (109). IKKβ in the CNS, mainly in the hypothalamus, can regulate blood pressure. Overexpression of a constitutively active form of IKKβ in the mediobasal hypothalamus induces hypertension in mice, while NF-κB inhibition attenuated high-fat feeding induced hypertension in mice (110). Additionally, astrocyte-specific IKKβ overexpression in mice led to higher daytime blood pressure, while NF-κB inhibition reversed obesity-induced hypertension in mice (106). In line with the previous discussion linking ER stress to insulin resistance, thapsigargin-induced ER stress increased blood pressure and phosphorylated IκB, but inhibition of NF-κB alleviated these effects (102).
Recent research advancements have expanded our knowledge on the function of IKKβ in cardiometabolic diseases. A summary of the role of IKKβ in cardiometabolic diseases is listed in Table 1. By exploring various mechanisms of chronic inflammation-associated diseases, such atherosclerosis, obesity, and insulin resistance, IKKβ and its regulated main canonical NF-κB pathway in various cell types have been found to play diverse roles in cardiometabolic disease development. In addition, new discoveries revealed that NF-κB-independent mechanisms may also contribute to the impact of IKKβ on the development of cardiometabolic diseases. For example, IKKβ can interact with several important signaling molecules such as β-catenin, BAD, and IRS-1 that are essential for regulating cell survival, differentiation and insulin signaling. With more new molecular targets of IKKβ being discovered, there will be more opportunities for fully understanding the complex function of IKKβ in cardiometabolic diseases and for developing new and effective therapeutic approaches.

Table 1. Overview of IKKβ modulation and mechanism in cardiometabolic diseases.
RH: conceptualized, wrote, and edited the manuscript. CZ: reviewed, edited, and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported in part by National Institutes of Health grants (R01HL131925 and R01ES023470) and American Heart Association grant (19TPA34890065) to CZ. RH was supported by an NIH T32 training grant (T32ES018827).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Baker RG, Hayden MS, Ghosh S. NF-κB, Inflammation, and metabolic disease. Cell Metab. (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
2. Zhou C. Novel functions of PXR in cardiometabolic disease. Biochim Biophys Acta. (2016) 1859:1112–20. doi: 10.1016/j.bbagrm.2016.02.015
3. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
4. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. (2008) 8:923–34. doi: 10.1038/nri2449
5. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
6. Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A, et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. (2006) 116:2280–9. doi: 10.1172/JCI26283
7. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
8. Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. (2010) 2:a000158. doi: 10.1101/cshperspect.a000158
9. Page A, Navarro M, Suárez-Cabrera C, Bravo A, Ramirez A. Context-dependent role of IKKβ in cancer. Genes. (2017) 8:376. doi: 10.3390/genes8120376
10. Ma B, Hottiger MO. Crosstalk between Wnt/β-Catenin and NF-κB signaling pathway during inflammation. Front Immunol. (2016) 7:378. doi: 10.3389/fimmu.2016.00378
11. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. (2009) 1:a001651–a. doi: 10.1101/cshperspect.a001651
12. Hinz M, Scheidereit C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. (2014) 15:46–61. doi: 10.1002/embr.201337983
13. Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. (2012) 246:239–53. doi: 10.1111/j.1600-065X.2012.01107.x
14. Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJJ, Kardakaris R, et al. Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metab. (2008) 8:372–83. doi: 10.1016/j.cmet.2008.08.016
15. Hansson GK. Inflammation and atherosclerosis: the end of a controversy. Circulation. (2017) 136:1875–7. doi: 10.1161/CIRCULATIONAHA.117.030484
17. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. (2012) 32:2045–51. doi: 10.1161/ATVBAHA.108.179705
19. Libby P, Bornfeldt KE. How far we have come, how far we have yet to go in atherosclerosis research. Circ Res. (2020) 126:1107–11. doi: 10.1161/CIRCRESAHA.120.316994
20. Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, et al. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. (1996) 97:1715–22. doi: 10.1172/JCI118598
21. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. (2000) 97:9052–7. doi: 10.1073/pnas.97.16.9052
22. Brand K, Eisele T, Kreusel U, Page M, Page S, Haas M, et al. Dysregulation of monocytic nuclear factor-kappa B by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. (1997) 17:1901–9. doi: 10.1161/01.ATV.17.10.1901
23. Bennett BL, Lacson RG, Chen CC, Cruz R, Wheeler JS, Kletzien RF, et al. Identification of signal-induced IkappaB-alpha kinases in human endothelial cells. J Biol Chem. (1996) 271:19680–8. doi: 10.1074/jbc.271.33.19680
24. Bourcier T, Sukhova G, Libby P. The nuclear factor kappa-B signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. J Biol Chem. (1997) 272:15817–24. doi: 10.1074/jbc.272.25.15817
25. Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci USA. (2004) 101:5634–9. doi: 10.1073/pnas.0401060101
26. de Winther Menno PJ, Kanters E, Kraal G, Hofker Marten H. Nuclear Factor κB Signaling in Atherogenesis. Arterioscler Thromb Vasc Biol. (2005) 25:904–14. doi: 10.1161/01.ATV.0000160340.72641.87
27. Meiler SE, Hung RR, Gerszten RE, Gianetti J, Li L, Matsui T, et al. Endothelial IKK β Signaling is required for monocyte adhesion under laminar flow conditions. J Mol Cell Cardiol. (2002) 34:349–59. doi: 10.1006/jmcc.2001.1519
28. Mussbacher M, Salzmann M, Haigl B, Basílio J, Hochreiter B, Gleitsmann V, et al. Ikk2-mediated inflammatory activation of arterial endothelial cells promotes the development and progression of atherosclerosis. Atherosclerosis. (2020) 307:21–31. doi: 10.1016/j.atherosclerosis.2020.06.005
29. Lesniewski LA, Durrant JR, Connell ML, Folian BJ, Donato AJ, Seals DR. Salicylate treatment improves age-associated vascular endothelial dysfunction: potential role of nuclear factor kappaB and forkhead Box O phosphorylation. J Gerontol A Biol Sci Med Sci. (2011) 66:409–18. doi: 10.1093/gerona/glq233
30. Yang S, Yuan HQ, Hao YM, Ren Z, Qu SL, Liu LS, et al. Macrophage polarization in atherosclerosis. Clin Chim Acta. (2020) 501:142–6. doi: 10.1016/j.cca.2019.10.034
31. Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, et al. An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J Exp Med. (2008) 205:1269–76. doi: 10.1084/jem.20080124
32. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci USA. (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
33. Kanters E, Pasparakis M, Gijbels MJJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJA, et al. Inhibition of NF-κB activation in macrophages increases atherosclerosis in LDL receptor–deficient mice. J Clin Invest. (2003) 112:1176–85. doi: 10.1172/JCI200318580
34. Goossens P, Vergouwe MN, Gijbels MJJ, Curfs DMJ, van Woezik JHG, Hoeksema MA, et al. Myeloid IκBα deficiency promotes atherogenesis by enhancing leukocyte recruitment to the plaques. PLoS ONE. 2011;6:e22327–e. doi: 10.1371/journal.pone.0022327
35. Ferreira V, van Dijk KW, Groen AK, Vos RM, van der Kaa J, Gijbels MJJ, et al. Macrophage-specific inhibition of NF-κB activation reduces foam-cell formation. Atherosclerosis. (2007) 192:283–90. doi: 10.1016/j.atherosclerosis.2006.07.018
36. Park S-H, Sui Y, Gizard F, Xu J, Rios-Pilier J, Helsley Robert N, et al. Myeloid-specific IκB kinase β deficiency decreases atherosclerosis in low-density lipoprotein receptor–deficient mice. Arterioscler Thromb Vasc Biol. (2012) 32:2869–76. doi: 10.1161/ATVBAHA.112.254573
37. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
38. Sasu S, Beasley D. Essential roles of IκB kinases α and β in serum- and IL-1-induced human VSMC proliferation. Am J Physiol Heart Circ Physiol. (2000) 278:H1823–H31. doi: 10.1152/ajpheart.2000.278.6.H1823
39. Sui Y, Park S-H, Xu J, Monette S, Helsley RN, Han S-S, et al. IKKβ links vascular inflammation to obesity and atherosclerosis. J Exp Med. (2014) 211:869–86. doi: 10.1084/jem.20131281
40. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. (2005) 96:939–49. doi: 10.1161/01.RES.0000163635.62927.34
41. Lu W, Park SH, Meng Z, Wang F, Zhou C. Deficiency of adipocyte IKKbeta affects atherosclerotic plaque vulnerability in obese LDLR deficient mice. J Am Heart Assoc. (2019) 8:e012009. doi: 10.1161/JAHA.119.012009
42. Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM. Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res. (2015) 52:299–305. doi: 10.1159/000443885
43. Qi X-Y, Qu S-L, Xiong W-H, Rom O, Chang L, Jiang Z-S. Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword. Cardiovasc Diabetol. (2018) 17:134. doi: 10.1186/s12933-018-0777-x
44. Manka D, Chatterjee TK, Stoll LL, Basford JE, Konaniah ES, Srinivasan R, et al. Transplanted perivascular adipose tissue accelerates injury-induced neointimal hyperplasia: role of monocyte chemoattractant protein-1. Arterioscler Thromb Vasc Biol. (2014) 34:1723–30. doi: 10.1161/ATVBAHA.114.303983
45. Virani Salim S, Alonso A, Benjamin Emelia J, Bittencourt Marcio S, Callaway Clifton W, Carson April P, et al. Heart disease and stroke statistics −2020 update: a report from the American Heart Association. Circulation. (2020) 141:e139–596. doi: 10.1161/CIR.0000000000000757
46. Seidell JC, Halberstadt J. The global burden of obesity and the challenges of prevention. Ann Nutr Metab. (2015) 66:7–12. doi: 10.1159/000375143
47. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. (2006) 444:840–6. doi: 10.1038/nature05482
48. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI200319451
49. Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. (2006) 444:875–80. doi: 10.1038/nature05487
50. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. (2017) 13:633–43. doi: 10.1038/nrendo.2017.90
51. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. (2005) 11:183–90. doi: 10.1038/nm1166
52. Solinas G, Karin M. JNK1 and IKKβ: molecular links between obesity and metabolic dysfunction. FASEB J. (2010) 24:2596–611. doi: 10.1096/fj.09-151340
53. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. (2005) 115:1111–9. doi: 10.1172/JCI25102
54. Helsley RN, Sui Y, Park S-H, Liu Z, Lee RG, Zhu B, et al. Targeting IκB kinase β in adipocyte lineage cells for treatment of obesity and metabolic dysfunctions. Stem Cells. (2016) 34:1883–95. doi: 10.1002/stem.2358
55. Sui Y, Liu Z, Park S-H, Thatcher SE, Zhu B, Fernandez JP, et al. IKKβ is a β-catenin kinase that regulates mesenchymal stem cell differentiation. JCI Insight. (2018) 3:e96660. doi: 10.1172/jci.insight.96660
56. Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science. (2000) 289:950–3. doi: 10.1126/science.289.5481.950
57. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. (2006) 7:885–96. doi: 10.1038/nrm2066
58. Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, et al. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice. Endocrinology. (2012) 153:154–65. doi: 10.1210/en.2011-1346
59. Park S-H, Liu Z, Sui Y, Helsley RN, Zhu B, Powell DK, et al. IKKβ is essential for adipocyte survival and adaptive adipose remodeling in obesity. Diabetes. (2016) 65:1616. doi: 10.2337/db15-1156
60. Kwon H, Laurent S, Tang Y, Zong H, Vemulapalli P, Pessin JE. Adipocyte-specific IKKβ signaling suppresses adipose tissue inflammation through an IL-13-dependent paracrine feedback pathway. Cell Rep. (2014) 9:1574–83. doi: 10.1016/j.celrep.2014.10.068
61. Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. (1999) 189:1839–45. doi: 10.1084/jem.189.11.1839
62. Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, et al. Inhibition of JNK activation through NF-κB target genes. Nature. (2001) 414:313–7. doi: 10.1038/35104568
63. De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, et al. Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature. (2001) 414:308–13. doi: 10.1038/35104560
64. Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, et al. Inactivation of BAD by IKK inhibits TNFα-induced apoptosis independently of NF-κB activation. Cell. (2013) 152:304–15. doi: 10.1016/j.cell.2012.12.021
65. Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, et al. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem. (2010) 285:3428–38. doi: 10.1074/jbc.M109.074252
66. Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem. (2017) 426:27–45. doi: 10.1007/s11010-016-2878-8
67. Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. (2005) 87:99–109. doi: 10.1016/j.biochi.2004.10.019
68. Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. (2002) 277:48115–21. doi: 10.1074/jbc.M209459200
69. Luo C, Yang H, Tang C, Yao G, Kong L, He H, et al. Kaempferol alleviates insulin resistance via hepatic IKK/NF-κB signal in type 2 diabetic rats. Int Immunopharmacol. (2015) 28:744–50. doi: 10.1016/j.intimp.2015.07.018
70. Yuan YL, Lin BQ, Zhang CF, Cui LL, Ruan SX, Yang ZL, et al. Timosaponin B-II Ameliorates palmitate-induced insulin resistance and inflammation via IRS-1/PI3K/Akt and IKK/NF-[formula: see text]B pathways. Am J Chin Med. (2016) 44:755–69. doi: 10.1142/S0192415X16500415
71. Zhou X, You S. Rosiglitazone inhibits hepatic insulin resistance induced by chronic pancreatitis and IKK-β/NF-κB expression in liver. Pancreas. (2014) 43:1291–8. doi: 10.1097/MPA.0000000000000173
72. Gao J, Song J, Du M, Mao X. Bovine α-lactalbumin hydrolysates (α-LAH) ameliorate adipose insulin resistance and inflammation in high-fat diet-fed C57BL/6J mice. Nutrients. (2018) 10:242. doi: 10.3390/nu10020242
73. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li Z-W, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science. (2001) 293:1673. doi: 10.1126/science.1061620
74. Kim JK, Kim Y-J, Fillmore JJ, Chen Y, Moore I, Lee J, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. (2001) 108:437–46. doi: 10.1172/JCI11559
75. Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T, et al. Interferon regulatory factor 3 constrains IKKβ/NF-κB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology. (2014) 59:870–85. doi: 10.1002/hep.26751
76. Ke B, Zhao Z, Ye X, Gao Z, Manganiello V, Wu B, et al. Inactivation of NF-κB p65 (RelA) in liver improves insulin sensitivity and inhibits cAMP/PKA pathway. Diabetes. (2015) 64:3355–62. doi: 10.2337/db15-0242
77. Arkan MC, Hevener AL, Greten FR, Maeda S, Li Z-W, Long JM, et al. IKK-β links inflammation to obesity-induced insulin resistance. Nat Med. (2005) 11:191–8. doi: 10.1038/nm1185
78. Liu J, Ibi D, Taniguchi K, Lee J, Herrema H, Akosman B, et al. Inflammation improves glucose homeostasis through IKKβ-XBP1s interaction. Cell. (2016) 167:1052–66.e18. doi: 10.1016/j.cell.2016.10.015
79. Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. (2009) 6:399–409. doi: 10.1038/nrcardio.2009.55
80. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
81. Jiao P, Ma J, Feng B, Zhang H, Alan-Diehl J, Eugene-Chin Y, et al. FFA-Induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity. (2011) 19:483–91. doi: 10.1038/oby.2010.200
82. Wang M, Chen X, Zheng Z, Yu S, Zhou B, Liu Y, et al. Beneficial effect of ER stress preconditioning in protection against FFA-induced adipocyte inflammation via XBP1 in 3T3-L1 adipocytes. Mol Cell Biochem. (2020) 463:45–55. doi: 10.1007/s11010-019-03627-3
83. Green CJ, Pedersen M, Pedersen BK, Scheele C. Elevated NF-κB activation is conserved in human myocytes cultured from obese type 2 diabetic patients and attenuated by AMP-activated protein kinase. Diabetes. (2011) 60:2810–9. doi: 10.2337/db11-0263
84. Bhatt BA, Dube JJ, Dedousis N, Reider JA, O'Doherty RM. Diet-induced obesity and acute hyperlipidemia reduce IkappaBalpha levels in rat skeletal muscle in a fiber-type dependent manner. Am J Physiol Regul Integr Comp Physiol. (2006) 290:R233–40. doi: 10.1152/ajpregu.00097.2005
85. Jové M, Planavila A, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology. (2006) 147:552–61. doi: 10.1210/en.2005-0440
86. Radin MS, Sinha S, Bhatt BA, Dedousis N, O'Doherty RM. Inhibition or deletion of the lipopolysaccharide receptor Toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia. (2008) 51:336–46. doi: 10.1007/s00125-007-0861-3
87. Rohl M, Pasparakis M, Baudler S, Baumgartl J, Gautam D, Huth M, et al. Conditional disruption of IkappaB kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest. (2004) 113:474–81. doi: 10.1172/JCI200418712
88. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. (2001) 280:E745–51. doi: 10.1152/ajpendo.2001.280.5.E745
89. Rehman K, Akash MSH, Liaqat A, Kamal S, Qadir MI, Rasul A. Role of interleukin-6 in Development of insulin resistance and type 2 diabetes mellitus. Crit Rev Eukaryot Gene Expr. (2017) 27:229–36. doi: 10.1615/CritRevEukaryotGeneExpr.2017019712
90. Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. (2003) 278:13740–6. doi: 10.1074/jbc.M210689200
91. Liu Y, Sheng L, Xiong Y, Shen H, Rui L. Liver NF-κB-Inducing kinase promotes liver steatosis and glucose counterregulation in male mice with obesity. Endocrinology. (2017) 158:1207–16. doi: 10.1210/en.2016-1582
92. Beraza N, Malato Y, Vander Borght S, Liedtke C, Wasmuth HE, Dreano M, et al. Pharmacological IKK2 inhibition blocks liver steatosis and initiation of non-alcoholic steatohepatitis. Gut. (2008) 57:655–63. doi: 10.1136/gut.2007.134288
93. Gervois P, Torra IP, Fruchart JC, Staels B. Regulation of lipid and lipoprotein metabolism by PPAR activators. Clin Chem Lab Med. (2000) 38:3–11. doi: 10.1515/CCLM.2000.002
94. Sunami Y, Leithäuser F, Gul S, Fiedler K, Güldiken N, Espenlaub S, et al. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology. (2012) 56:1117–28. doi: 10.1002/hep.25711
95. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell. (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
96. Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. (2009) 10:249–59. doi: 10.1016/j.cmet.2009.08.013
97. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. (2009) 296:E1003–12. doi: 10.1152/ajpendo.90377.2008
98. Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. (2011) 286:32324–32. doi: 10.1074/jbc.M111.254417
99. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. (2009) 29:359–70. doi: 10.1523/JNEUROSCI.2760-08.2009
100. Lee CH, Suk K, Yu R, Kim MS. Cellular contributors to hypothalamic inflammation in obesity. Mol Cells. (2020) 43:431–7. doi: 10.14348/molcells.2020.0055
101. Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. (2006) 17:365–71. doi: 10.1016/j.tem.2006.09.007
102. Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci USA. (2011) 108:2939–44. doi: 10.1073/pnas.1006875108
103. Tsaousidou E, Paeger L, Belgardt BF, Pal M, Wunderlich CM, Brönneke H, et al. Distinct roles for JNK and IKK activation in agouti-related peptide neurons in the development of obesity and insulin resistance. Cell Rep. (2014) 9:1495–506. doi: 10.1016/j.celrep.2014.10.045
104. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
105. Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol Metab. (2017) 6:366–73. doi: 10.1016/j.molmet.2017.01.010
106. Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D. Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure, and body weight. Cell Metab. (2017) 25:1091–102.e4. doi: 10.1016/j.cmet.2017.04.002
107. Miranda M, Morici JF, Zanoni MB, Bekinschtein P. Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci. (2019) 13:363. doi: 10.3389/fncel.2019.00363
108. Li J, Tang Y, Cai D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. (2012) 14:999–1012. doi: 10.1038/ncb2562
109. Khor S, Cai D. Hypothalamic and inflammatory basis of hypertension. Clin Sci (Lond). (2017) 131:211–23. doi: 10.1042/CS20160001
Keywords: cardiometabolic diseases, atherosclerosis, insulin resistance, obesity, IKK-beta, NF-kB
Citation: Hernandez R and Zhou C (2021) Recent Advances in Understanding the Role of IKKβ in Cardiometabolic Diseases. Front. Cardiovasc. Med. 8:752337. doi: 10.3389/fcvm.2021.752337
Received: 02 August 2021; Accepted: 12 November 2021;
Published: 08 December 2021.
Edited by:
Rainer de Martin, Medical University of Vienna, AustriaReviewed by:
Dongsheng Cai, Albert Einstein College of Medicine, United StatesCopyright © 2021 Hernandez and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changcheng Zhou, Y2hhbmdjaGVuZy56aG91QG1lZHNjaC51Y3IuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.