 Ye Li
Ye Li Kui-Fang Xie
Kui-Fang Xie Ya-Hong Chang
Ya-Hong Chang Cheng Wang
Cheng Wang Ying Chen
Ying Chen Ming-Jie Wang
Ming-Jie Wang Yi-Chun Zhu
Yi-Chun Zhu- 1Shanghai Key Laboratory of Bioactive Small Molecules and Shanghai Key Laboratory of Clinical Geriatric Medicine, Innovative Research Team of High-Level Local Universities in Shanghai, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, China
- 2Laboratory Animal Technical Platform, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China
Background: Endogenous hydrogen sulfide (H2S) is emerging as a key signal molecule in the development of diabetic cardiomyopathy. The aim of this study was to explore the effect and underlying mechanism of S-propargyl-cysteine (SPRC), a novel modulator of endogenous H2S, on diabetic cardiomyopathy in db/db diabetic mice.
Methods and Results: Vehicle or SPRC were orally administered to 8-month-old male db/db mice and their wild type littermate for 12 weeks. SPRC treatment ameliorated myocardial hypertrophy, fibrosis, and cardiac systolic dysfunction assessed by histopathological examinations and echocardiography. The functional improvement by SPRC was accompanied by a reduction in myocardial lipid accumulation and ameliorated plasma lipid profiles. SPRC treatment improved glucose tolerance in db/db mice, with fasting blood glucose and peripheral insulin resistance remaining unchanged. Furthermore, insulin receptor signaling involving the phosphorylation of protein kinase B (Akt/PKB) and glycogen synthase kinase 3β (GSK3β) were elevated and activated by SPRC treatment. Primary neonatal mice cardiomyocytes were cultured to explore the mechanisms of SPRC on diabetic cardiomyopathy in vitro. Consistent with the results in vivo, SPRC not only up-regulated insulin receptor signaling pathway in cardiomyocytes in dose-dependent manner in the basal state, but also relieved the suppression of insulin receptor signaling induced by high concentrations of glucose and insulin. Furthermore, SPRC also enhanced the expression of glucose transporter 4 (GLUT4) and 3H glucose uptake in cardiomyocytes.
Conclusions: In this study, we found a novel beneficial effect of SPRC on diabetic cardiomyopathy, which was associated with activation of insulin receptor signaling. SPRC may be a promising medication for diabetic cardiomyopathy in type 2 diabetes mellitus patients.
Introduction
Among adults in China, the number of diabetic patients have explosively increased. The estimated overall prevalence of diabetes was 10.9%, and that for prediabetes was 35.7% (1). Cardiovascular disease, is a leading cause of mortality in the development of diabetic complications (2). After adjusting for other risk factors including age, hypertension, obesity, dyslipidemia, the incidence of heart failure increases 2.4–5 fold in diabetic patients than non-diabetic patients (3). Diabetic cardiomyopathy (DCM) is defined as structural and functional abnormalities in the myocardium of diabetic patients independent of underlying coronary artery disease and hypertension (4). At present, treatment strategy for DCM mainly rely on conventional therapies that focus on optimizing glycemic control (5). However, meta-analysis of large clinical trials revealed that strict glycemic control had no impact on the incidence of heart failure in diabetic patients (6, 7). Therefore, it is particularly important to explore the pathogenesis mechanism and novel therapeutic drugs of DCM.
Hydrogen sulfide (H2S) has been considered toxic and odorous for a long time. However, it has been recognized as the third gasotransmitter following nitric oxide and carbon monoxide to play an important role in many physiological and pathological processes since the 1990s. Accumulating studies have shown that H2S is involved in improving DCM by multiple mechanisms (8), such as supplementation of exogenous H2S reduced endoplasmic reticulum pressure in cardiomyocytes (9, 10) and inhibited myocardial oxidative Stress, inflammation, and apoptosis (11). Endogenous H2S production by cystathionine-γ-lyase (CTH, also named CSE, the main producing enzyme of endogenous H2S in cardiovascular system) is inhibited in myocardium of DCM rats (12). Consistently, H2S levels in serum of DCM patients were significant decreased and the supplement of H2S could rescue the cardiomyopathy dysfunction induced by hyperglycemia (10, 13). However, H2S cannot be used for clinical therapy because of its instability and gaseous characteristics. S-Propargyl-Cysteine (SPRC, also named as ZYZ-802), a novel water-soluble modulator of endogenous H2S, promotes the activity of CSE and then increases H2S levels in plasma or tissue (14–16). In this study, we evaluated the effect of SPRC on DCM in db/db mice and further explored the underlying mechanism, both in vivo and in vitro.
Materials and Methods
Chemicals
S-Propargyl-Cysteine (SPRC, purity > 99%) was synthesized as described previously (17) and provided by Professor Zhu Yi-Zhun (State Key Laboratory of Quality Research in Chinese Medicine and School of Pharmacy, Macau University of Science and Technology, Macau, China). Chemical formula of SPRC is shown in Supplementary Figure 1A.
Animals and Treatments
All experimental procedures were performed in accordance with Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH) of the United States and approved by the Ethics Committee of Experimental Research, Fudan University Shanghai Medical College. Male C57BLKS/J db/db mice and their wild type littermates (7-week-old) were purchased from Nanjing Biomedical Research Institute of Nanjing University and were housed in a climate-controlled environment (22 ± 2°C, 45–75% relative humidity) with a 12 h light-dark cycle and unrestricted access to food and water. After acclimatization for 1 weeks, wild-type (WT) mice were used as a normal control group and db/db mice were randomly divided into four different groups (n = 15–16 per group): diabetic model group, low-dose SPRC treatment group (20 mg/kg per day), medium-dose SPRC treatment group (40 mg/kg per day), and high-dose SPRC treatment group (80 mg/kg per day). Both WT control group and diabetic model group were orally administered an equal volume of vehicle (ddH2O). After 12 weeks of administration, mice were anesthetized by intraperitoneal injection of 1% sodium pentobarbital and blood samples were collected. Heart weights (HW) and tibia lengths (TL) were measured and heart tissue specimens were obtained.
Transthoracic Echocardiography
At the first and last week of SPRC treatment, the hair was removed from the chest of mice using depilatory cream. The mice were then anesthetized with 1.5% isoflurane and placed in a supine position on the test bench, with ultrasound gel applied onto the chest. Mouse two-dimensional echocardiography was performed using a Vevo3100 ultrasound device (VisualSonics Inc., Canada), as previously described (18). B-mode, M-mode, and Power Doppler Mode ultrasound images of the left ventricle were recorded. All measurements were averaged for five consecutive cardiac cycles.
Fasting Blood Glucose, Body Weight, and Fasting Plasma Insulin Levels
The FBG levels and BW of mice were monitored every 2 weeks after food was removed for 12 h. Glucose measurements were performed with venous blood collected from mice tails by glucose monitors (ONETOUCH, Johnson and Johnson, USA). After food was removed overnight for 16 h, FINS were detected by enzyme-linked immunosorbent assay (ELISA) kit (Mercodia, Sweden). The formula of homeostasis model assessment for insulin resistance index (HOMA-IR) is [(HOMA-IR) = (FBG × FINS)/22.5] (19).
IPGTTs and IPITTs
Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) were performed in the morning on nonfasted mice that had their food removed 1 h prior to intraperitoneal injection (IPGTT 1 g glucose per kg body weight and IPITT 1unit recombinant human insulin (Humulin 70/30, Lilly, USA) per kg body weight), Blood glucose levels were measured before the injection (time 0) and 15, 30, 60, and 120 min after the injection. Areas under the curve (AUC) were determined using the trapezoidal rule.
Biochemical Analyses
The total cholesterol (TC), triglycerides (TG), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), non-esterified fatty acid (NEFA) in plasma were determined using commercial kits (Nanjing Jiancheng, China) according to the manufacturer's instructions. Briefly, full-wavelength microplate reader (Infinite® 200 PRO, TECAN, Switzerland) was used to detect the absorbance of plasma samples at 510 nm for TC and TG or at 546 nm for HDL-C, LDL-C and NEFA.
Histological Analysis
Heart specimens were fixed in 4% paraformaldehyde solution. After being embedded in paraffin, the specimens were cut into 5-μm-thick sections and stained with wheat germ agglutinin (WGA) and Masson's trichrome. The other heart tissue specimens were frozen and stained with Oil Red O lipid stain. The size of myocardial cells, fiber area fraction, and myocardial lipid content were determined using Image J software (Bethesda, MA, USA).
Transmission Electron Microscopy
To observe myocardial ultrastructure, heart tissues were cut into 1 mm transverse sections and immersed in 2% glutaraldehyde overnight. The sections were then immersed in 1% osmium tetroxide for 2 h, dehydrated in graded ethanol, and embedded in epoxy resin. Ultrathin sections (60–70 nm) were obtained, stained with uranyl acetate and lead citrate, and examined using a Tecnai G20 Twin transmission electron microscope (FEI, USA).
Primary Culture of Neonatal Mice Cardiomyocytes
Primary cultures of cardiomyocytes were prepared from neonatal mice hearts. In brief, hearts were excised from 1- to 2-day-old mice pups. Ventricles were minced by small scissors and digested using 1 mg/ml collagenase type II (Worthington, USA). The digested solution was collected. The process was repeated 3–4 times until no chunks of tissue were visible. The final pooled solution was centrifuged and the cell pellet re-suspended in Dulbecco's Modified Eagle Medium (DMEM, Hyclone, USA), high Glucose containing 10% fetal bovine serum (FBS, Gibco, USA). The cells were pre-plated for 1 h to allow the attachment and removal of fibroblasts. The unattached cardiomyocytes remaining in suspension were then collected and plated in DMEM, high Glucose containing 10% FBS. After 48 h Cardiomyocytes cultures were used for subsequent experiments.
Cell Viability
Cell viability was determined by cell counting kit-8 assay according to manufacturer's instructions (DOJINDO, Japan). Cardiomyocytes were cultured in a 96-well-culture plate and treated with different concentrations of SPRC (0–1,000 μM) for 24 h. Cells were subsequently incubated with 10 ml CCK-8 solution at 37°C for 4 h. The absorbance at 450 nm was measured.
Real-Time PCR
Total RNA was extracted by Trizol reagent from heart tissue or cardiomyocytes. RNA was reverse-transcribed using a cDNA synthesis kit (Toyobo Life Science, Japan). Real-Time PCR was performed using a StepOnePlus Real-Time PCR Detection System (Applied Biosystems Inc., CA, USA). A total volume of 20 μL reaction mix containing 2 μL cDNA, 10 μL SYBR Green PCR Master Mix (Toyobo, Japan) and 1 μL each primer (10 μM). Gapdh was used for normalization and the relative expression of mRNA was calculated according to the ΔΔCt method. The specific primers were as follows: Myh7: 5′-ATCAATGCAACCCTGGAGAC-3′, 5′-CGAACATGTGGTGGTTGAAG-3′; InsR: 5′-GCTTCTGCCAAGACCTTCAC-3′, 5′-CACTCGGGGATGCACTTATT-3′; Glut4: 5′-ACCCTGGGCTCTGTATCCC-3′, 5′-CCCTGACCACTGAGTGCAAA-3′; Gapdh: 5′-TTCACCACCATGGAGAAGGC-3′, 5′-GGCATGGACTGTGGTCATGA-3′; β-actin: 5′- GACAGGATGCAGAAGGAGATTACT-3′, TGATCCACATCTGCTGGAAGGT-3′.
Western Blotting
Left ventricular tissue and cell samples were lysed with Cell Lysis Buffer for Western (P0031; Beyotime Biotechnology, China) containing a protease inhibitor cocktail (049693132001; Roche, Basel, Switzerland). The crude cell lysate was centrifuged and the supernatant was harvested. The concentration of protein in the supernatant was quantified by standard bicinchoninic acid assay (BCA, P0012, Beyotime Biotechnology, China). For western blotting, equal amounts of proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (1:10,000; A0208; Beyotime Biotechnology, China) for 2 h, and immunoreactive bands were visualized by chemiluminescence. And the gray value of the captured bands was determined by ImageJ software. The primary antibodies used included: anti-Insulin Receptor β (#3025), anti-Phospho-Insulin Receptor β Tyr1150/1151(#3024), anti-Akt (#9272), anti-phospho-Akt Ser473 (#4060), anti-GSK-3β (#9315), anti-Phospho-GSK-3β Ser9 (#5558) were purchased from Cell Signaling Technology (Beverly, MA, USA); anti-GAPDH (60004-1), anti-β-Actin (66009-1), and anti-α-Tubulin (11224-1) purchased from Proteintech Group (Chicago, USA), and anti-CSE(sc-374249) were purchased from Santa Cruz Biotechnology (Texas, USA).
2-Deoxyglucose Uptake
Primary mice cardiomyocytes were incubated with 33.3 mM glucose (HG), 100 nM insulin (HI), or 33.3 mM glucose and 100 nM insulin (HG++HI) for 48 h. Among them, the HG+HI group treated with vehicle or different doses of SPRC (25–100 μM) for 24 h. then the uptake of 2-deoxyglucose by cardiomyocytes was measured as previously described (20). The cardiomyocytes were rinsed with pre-warmed KRP buffer (128 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, 5 mM NaH2PO4, 5 mM Na2HPO4, and 10 mM HEPES, pH 7.4) for three times and then treated with 100 nM insulin in KRP buffer without glucose for 30 min in 37°C. Subsequently, the cardiomyocytes were incubated with 2-deoxy-D[3H]-glucose (1 μCi/ml) dissolved in KRP buffer with 100 nM insulin for 10 min in 37°C. After washing with pre-cold phosphate-buffered saline (PBS) containing 10 mM glucose for 3 times, the cells were lysed with 0.4 mM NaOH for 2 h. The radioactivity of 3H was measured in a liquid scintillation counter (Beckman LS6500) and the concentration of protein were detected by BCA method for standardization.
Measurement of H2S Levels
H2S levels in heart tissues and plasma were measured as previously described (21). Briefly, heart tissues were homogenized in Tris-HCl (100 mmol/L, pH 8.5), then centrifuged at 12,000 g for 20 min at 4°C. The plasma sample were directly centrifuged at 3,500 rpm for 15 min at 4°C. And 30 μl of the supernatant from heart tissues and plasma were derivatized by MBB and detected by HPLC-MS. The concentration of protein from heart tissues were detected by BCA method for standardization.
Statistical Analysis
All data were expressed as the means ± SEM from at least three independent experiments. SPSS 13.0 software (SPSS, Inc., Chicago, USA) was used for statistical analysis. Student's unpaired t-test was performed to statistically analyze individual group data. Multiple-group comparisons were evaluated by one-way analysis of variance (ANOVA) followed by Fisher's Least Significant Difference (LSD) and the non-parametric test was used when the variance was not equal. Values of P < 0.05 were considered to be statistically significant.
Results
SPRC Treatment Improves Cardiac Function, and Alleviates Myocardial Hypertrophy and Fibrosis on Diabetic Cardiomyopathy
To explore the effect of SPRC on cardiac function of db/db diabetic mice, echocardiograph images were recorded before and after 12 weeks of SPRC treatment (Figure 1A). Compared with WT littermates, db/db mice preserved cardiac function at 8-week-old, but developed diastolic and systolic dysfunction at 20-week-old characterized by elevated deceleration time (DT, Figure 1B), reduced left ventricular ejection fraction (EF, Figure 1D) and decreased fractional shortening (FS, Figure 1E, P = 0.054) with a consistent heart rate (HR, Figure 1C). All of three parameters examined were improved by SPRC treatment (Supplementary Tables 1, 2). The beneficial effect of SPRC treatment on cardiac hypertrophy was confirmed by histopathological examinations and RT-PCR (Figure 2). Heart weight to tibia length (HW/TL) ratio was significantly increased in db/db mice compared with WT mice (Figure 2C). Consistently, cardiomyocyte cross-sectional area examined in db/db mice by wheat germ agglutinin (WGA) was markedly enlarged (Figure 2D). Administration of SPRC to db/db mice decreased HW/TL ratios and cardiomyocyte cross-sectional area, reduced mRNA level of beta-myosin heavy chain (Myh7) gene, marker for cardiac hypertrophy (Figure 2B). Masson staining revealed that obvious fibrosis in the interstitial of myocardium was observed in db/db mice, but significantly alleviated by SPRC treatment (Figure 2E). These data suggest that SPRC treatment improves cardiac function, and alleviates cardiac hypertrophy and fibrosis in the db/db diabetic model.
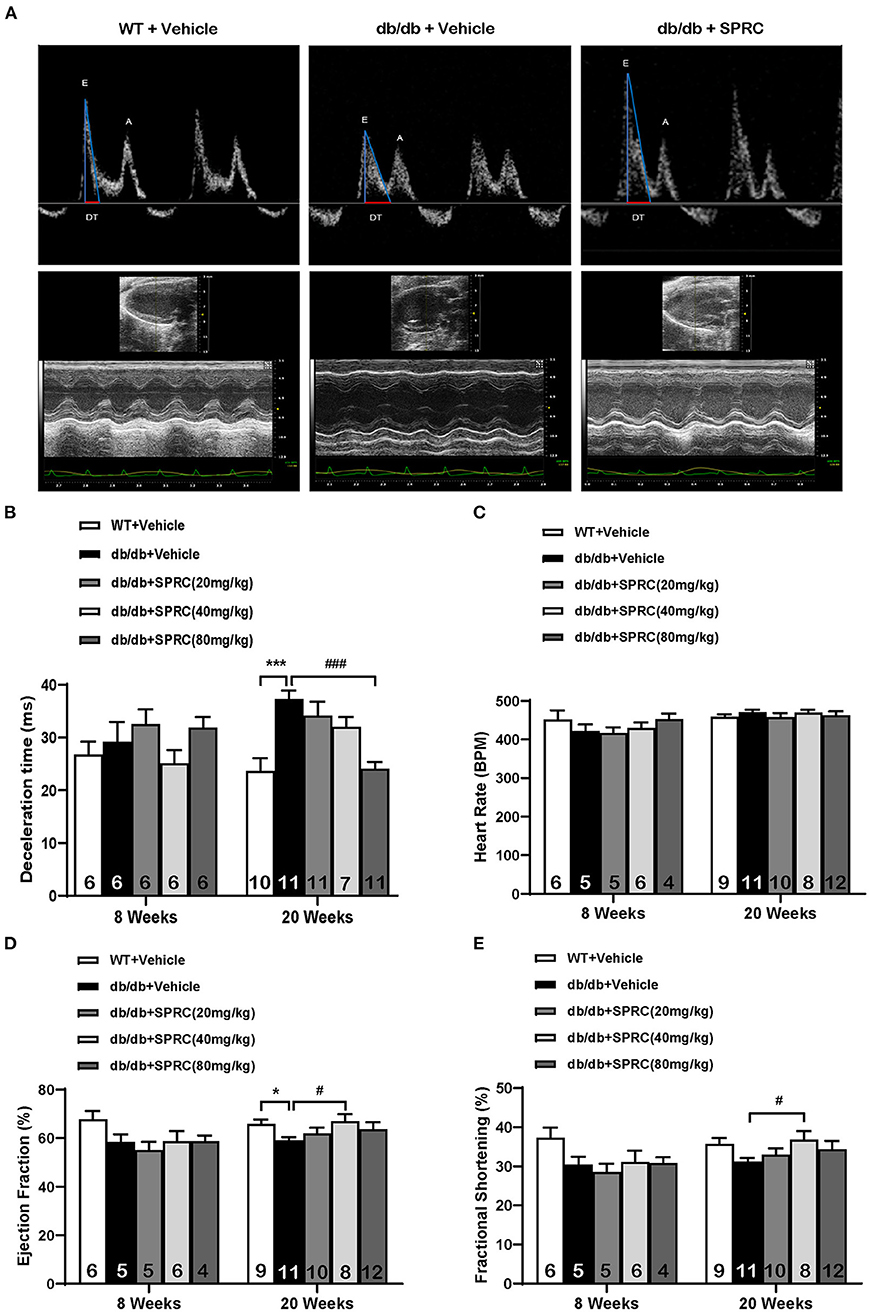
Figure 1. SPRC treatment improves cardiac function in db/db mice. Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. Echocardiograph images were recorded before and after 12 weeks of SPRC treatment. (A) Representative Power Doppler Mode (upper) and M-Mode (lower) echocardiograph images. (B–E) Echocardiographic assessment of deceleration time (DT), heart rate (HR), left ventricular ejection fraction (EF), and fractional shortening (FS). Values are presented as means ± SEM (n = 4–12). *P < 0.05, ***P < 0.001 vs. WT + Vehicle group, #P < 0.05, ###P < 0.001 vs. db/db + Vehicle mice.
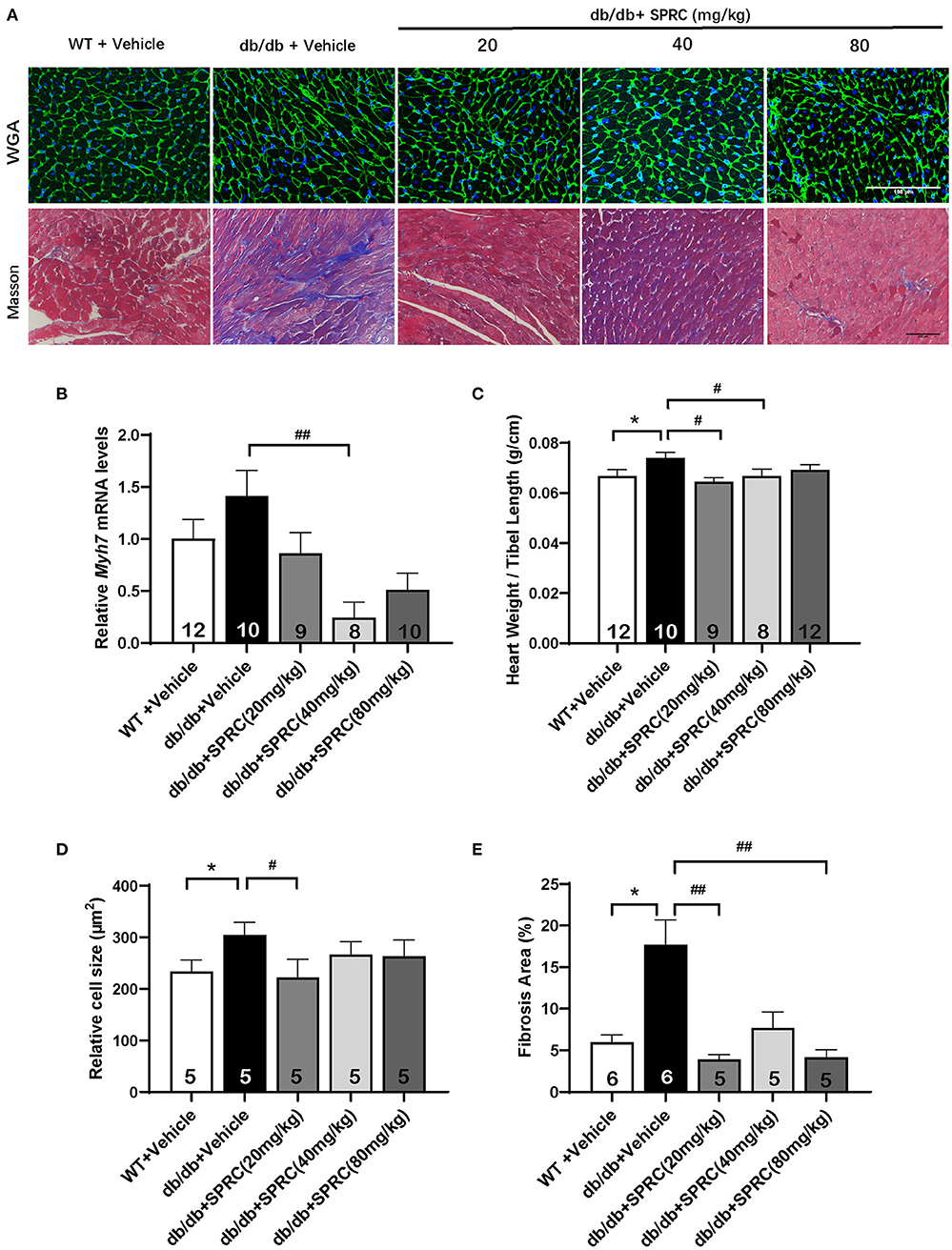
Figure 2. SPRC treatment improves myocardial hypertrophy and fibrosis in db/db mice. Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. Heart tissue specimens were collected. (A) Wheat germ agglutinin (WGA) and masson staining of myocardium were performed. (B) mRNA levels of Myh7 in heart tissue were determined by real-time qPCR. mRNA levels of Gapdh was used as reference for normalization (n = 8–12). (C) The ratio of heart weights (HW) and tibia lengths (TL) was calculated (n = 8–12). Average cell area (n = 5) (D) and fibrosis area (n = 5–6) (E) were shown. Values are presented as means ± SEM. *P < 0.05 vs. WT + Vehicle group, #P < 0.05, ##P < 0.01 vs. db/db + Vehicle mice.
SPRC Treatment Alleviated Abnormal Myocardial Ultrastructure on Diabetic Cardiomyopathy
SPRC treatment protected db/db mice from abnormalities in the ultrastructure of cardiomyocytes. The mitochondria in the cardiomyocytes of WT mice were regularly arranged and exerted integral cristae, and the myofilaments were arranged tightly, while a portion of mitochondria in the myocardium of db/db mice were swollen, disorderly arranged with broken cristae, and the myofilaments were loose. After SPRC treatment, fewer abnormal ultrastructure of mitochondria and myofilaments were observed in diabetic mice (Figure 3).

Figure 3. SPRC treatment improves abnormal ultrastructure of cardiomyocytes in db/db mice. Vehicle or SPRC (20,40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. Heart tissue specimens were collected and representative images of the ultrastructural morphology of cardiomyocytes were shown. The red arrow indicates myofilament and the blue arrow indicates mitochondria (n = 2).
SPRC Treatment Reduces Myocardial Lipid Accumulation and Dyslipidemia on Diabetic Cardiomyopathy
Oil red O staining showed a predictable and significantly increase in lipid droplets of db/db hearts, which was markedly decreased in response to SPRC (Figures 4A,B). Meanwhile, systemic dyslipidemia was observed in db/db mice characterized by markedly increased TC, TG, LDL-C, and NEFA. SPRC treatment inhibited the increased plasma TC levels, indicating that SPRC has a certain lipid-lowering effect (Figures 4C–G).
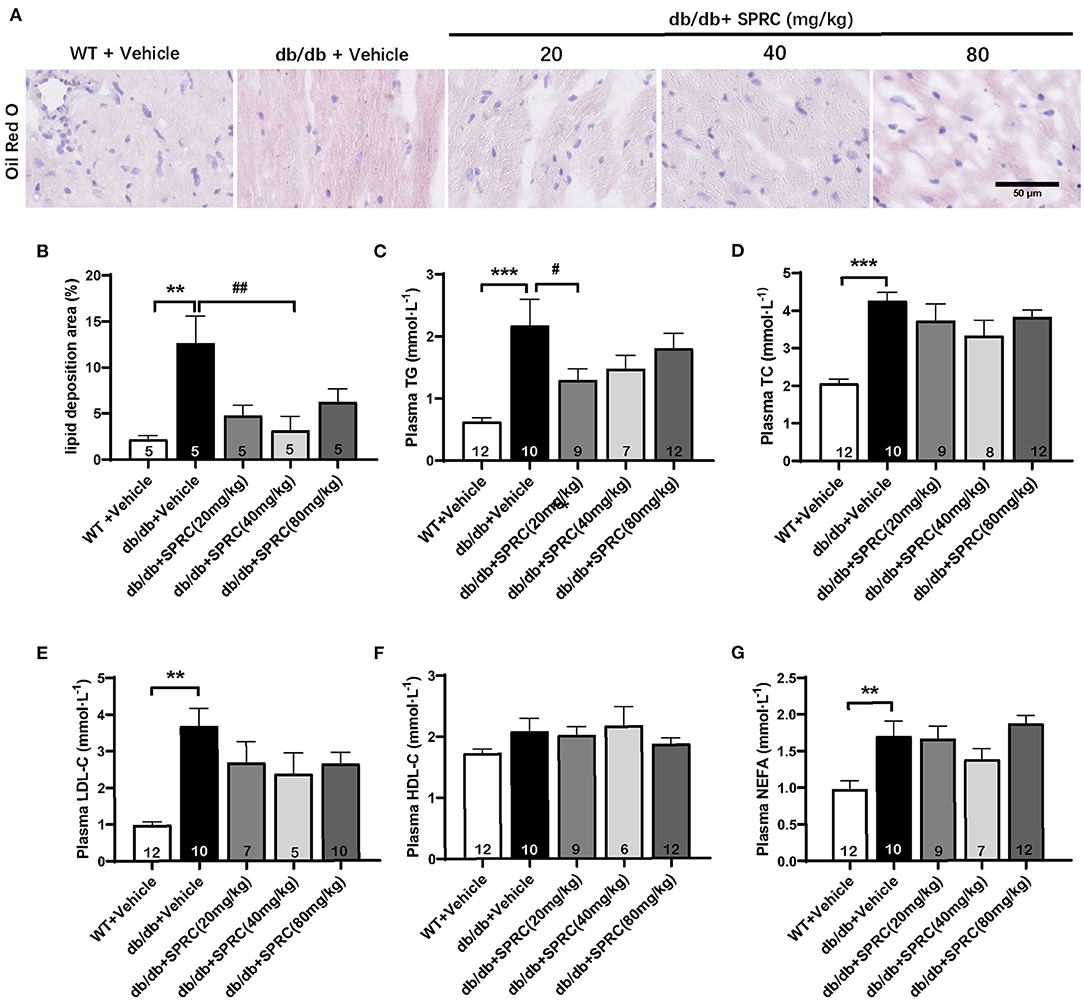
Figure 4. SPRC treatment reduces myocardial lipid accumulation and dyslipidemia in db/db mice. Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. Heart tissue specimens and plasma were collected. (A) Oil Red O staining of myocardium were performed (n = 5) lipid deposition area (B) was shown. The levels of TC (C), TG (D), LDL-C (E), HDL-C (F), NEFA (G) were measured (n = 5–12). Values are presented as means ± SEM. **P < 0.01, and ***P < 0.001 vs. WT + Vehicle group, #P < 0.05, ##P < 0.01 vs. db/db + Vehicle mice.
SPRC Treatment Improves Glucose Tolerance in db/db Mice, With Fasting Blood Glucose and Systemic Insulin Resistance Remaining Unchanged
Throughout the experiment, the body weight and fasting blood glucose of db/db mice were significantly increased compared to their WT littermates. However, SPRC treatment showed no significant impact on body weight and blood glucose (Figures 5A,B). To investigate the effect of SPRC on systemic insulin resistance, IPGTT and IPITT were preformed and the results demonstrated SPRC treatment at a dosing of 80 mg·kg−1·day−1 improved glucose tolerance to a certain extent, but has no significant impact on IPITT and its area under curve (Figures 5C–F). Furthermore, fasting plasma insulin levels and HOMA-IR index were remarkably elevated, with SPRC treatment failed to alleviate (Figures 5G,H).
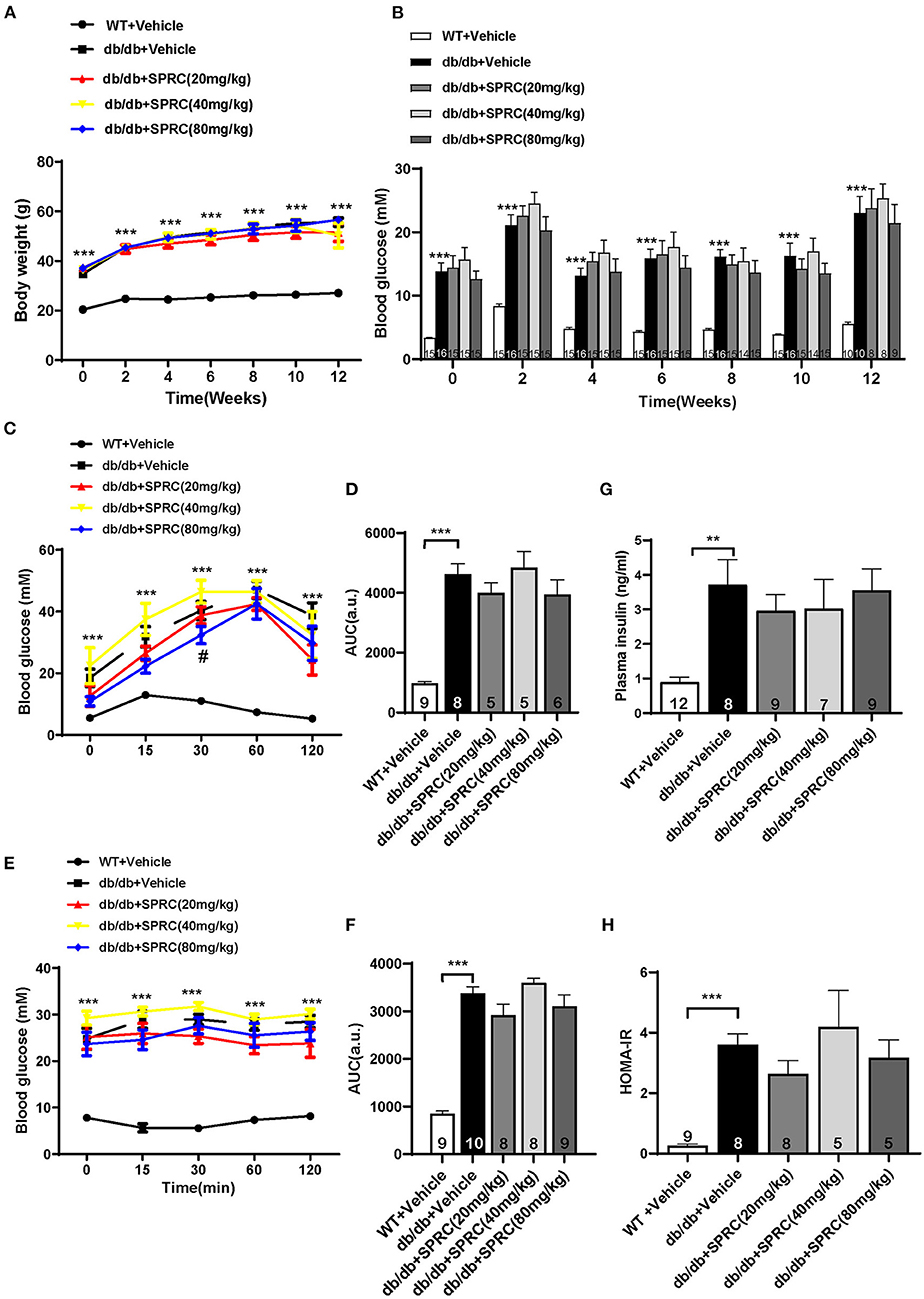
Figure 5. SPRC treatment improves glucose tolerance in db/db mice, with fasting blood glucose and systemic insulin resistance remaining unchanged. Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. (A) Body weight (n = 8–16), (B) fasting blood glucose (n = 8–16), (C) intraperitoneal glucose tolerance test (IPGTT) and (D) area under curve (AUC) of IPGTT (n = 5–9), (E) intraperitoneal insulin tolerance test (IPITT) and (F) area under curve (AUC) of IPITT (n = 8–10), (G) fasting plasma insulin (n = 7–12), and (H) HOMA-IR index were determined (n = 5–9). Values are presented as means ± SEM. **P < 0.01, and ***P < 0.001 vs. WT + Vehicle group, #P < 0.05 vs. db/db + Vehicle mice.
SPRC Treatment Activated Insulin Receptor Signaling in Both Primary Mice Cardiomyocytes and Myocardium of db/db Mice
Primary neonatal mice cardiomyocytes were cultured and identified to explore the mechanisms of SPRC on diabetic cardiomyopathy in vitro (Supplementary Figure 1B). Incubation of cardiomyocytes with SPRC (0.1–1,000 μM) for 24 h had no detectable cytotoxicity. Moreover, the cell viability was significantly increased at doses of 0.1 and 10 μM of SPRC treatment (Figure 6A). SPRC improved both mRNA and protein expression level of IR in cardiomyocytes, with the classical downstream pathway protein kinase B (Akt/PKB) and glycogen synthase kinase 3β (GSK3β) activated in the dose-dependently manner (Figures 6B–F).
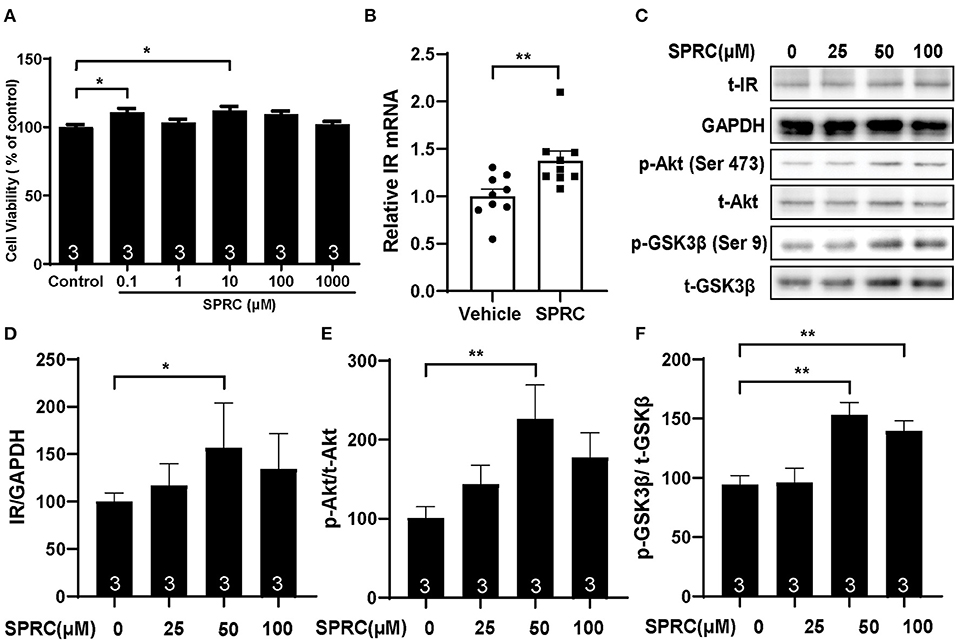
Figure 6. SPRC treatment activated cardiac insulin receptor signaling in primary cardiomyocytes. (A) Primary mice cardiomyocytes were exposed to various concentrations of SPRC (0.1–1,000 μM) for 24 h. Cell viability was determined (n = 3). *P < 0.05 vs. control group. (B) Primary mice cardiomyocytes were treated with 50 μM SPRC for 24 h. mRNA levels of insulin receptor (Insr) were determined by real-time qPCR. mRNA levels of Gapdh was used as reference for normalization (n = 3). **P < 0.01 vs. Vehicle group. (C–F) Primary mice cardiomyocytes were treated with vehicle or SPRC (25, 50, and 100 μM) for 24 h. The expression level of protein in insulin receptor signaling were determined by western blotting (n = 3). *P < 0.05, **P < 0.01 vs. Vehicle. Values are presented as means ± SEM.
The cardiac inulin receptor signaling in vivo were also determined. As shown in Figures 7A–D, SPRC treatment increased the mRNA of IR in myocardium, and it also improved protein expression of IR to a certain extent (P = 0.095). It is noteworthy that IR in the myocardium of diabetic mice was significantly phosphorylated, which is exacerbated by SPRC treatment. The phosphorylation levels of Akt and GSK3β in myocardium of db/db mice were significantly decreased, SPRC (80 mg/kg) significantly increased the phosphorylation levels of Akt and GSK3β in myocardium of DCM mice (Figures 7E,F).
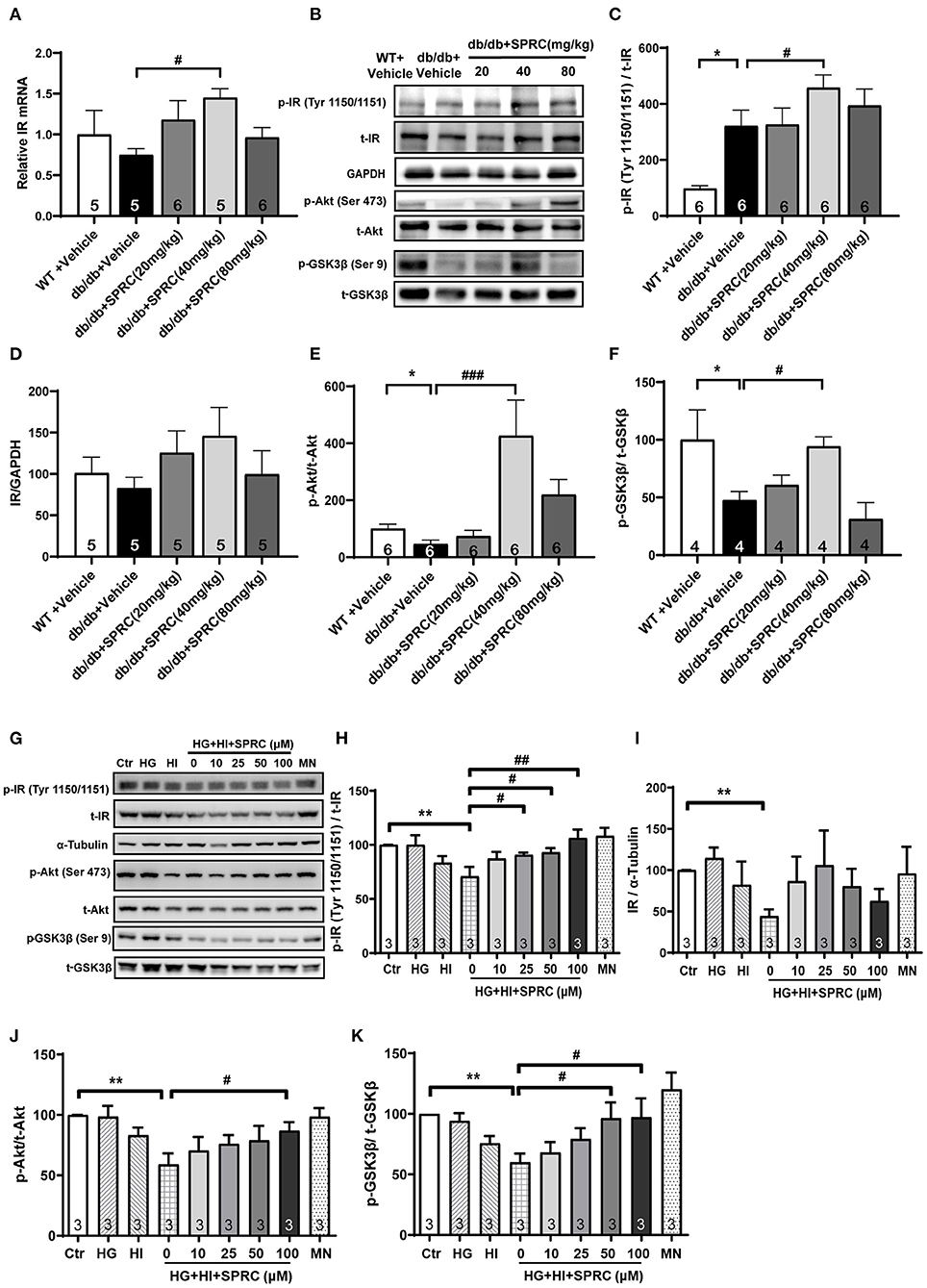
Figure 7. SPRC treatment activated insulin receptor signaling in db/db mice. Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. (A) The mRNA levels of insulin receptor (Insr) were determined by real-time qPCR. mRNA levels of Gapdh was used as reference for normalization (n = 5–6). #P < 0.05 vs. db/db + Vehicle mice. (B–F) the expression level of protein in insulin receptor signaling were determined by western blotting. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT + Vehicle group, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. db/db + Vehicle mice. (G–K) Primary mice cardiomyocytes were incubated with 33.3 mM glucose (HG), 100 nM insulin (HI), or 33.3 mM glucose + 100 nM insulin (HG + HI) for 48 h. The cardiomyocytes were then incubation with SPRC (50 μM) for 24 h. Expression of insulin receptor signaling were determined (n = 3). *P < 0.05, **P < 0.01 < 0.001 vs. control group, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. HG + HI group. Values are presented as means ± SEM.
We then exposed neonatal mice cardiomyocytes to 33.3 mM glucose and 100 nM insulin to mimic hyperglycemia and hyperinsulinemia in cardiomyocyte of diabetic heart. High concentrations of glucose and insulin stimulation significantly reduced the phosphorylation levels of Akt and GSK3β and the expression level of IR. However, after incubation with 50 μM SPRC for 24 h, the phosphorylation level of IR was preserved, and the expression level of IR was partly increased without statistical significance (P = 0.077). Consistently, the downstream of IR signaling including the phosphorylation levels of Akt and GSK3β were preserved (Figures 7G–K).
SPRC Treatment Increased GLUT4 Expression in Myocardium and Enhanced 3H Glucose Uptake in Cultured Primary Cardiomyocytes
GLUT4 transporter is the most abundant glucose transporter in the heart, which is also the major glucose transporter translocating to the plasma membrane in response to insulin (22). As shown in Figures 8A,B, the protein expression level of GLUT4 were significantly decreased in diabetic heart, and SPRC treatment increased the mRNA and protein expression level of GLUT4. SPRC also enhanced the glucose uptake in vitro (Figure 8C).
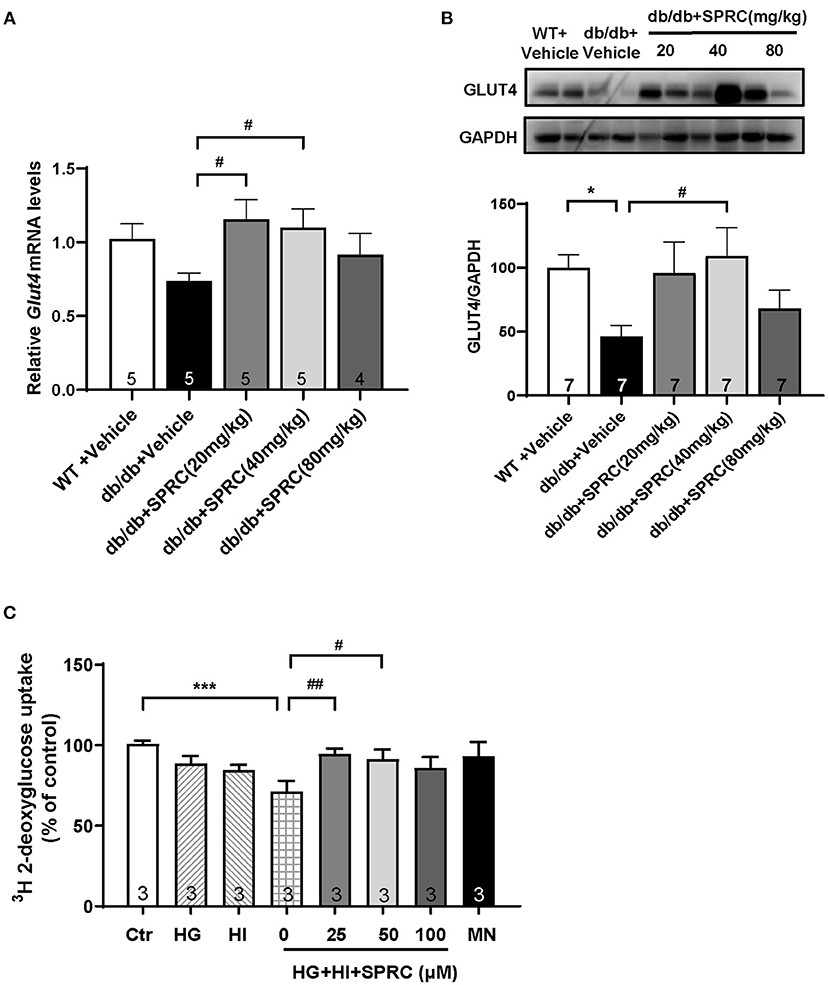
Figure 8. SPRC treatment increased GLUT4 expression and enhanced 3H glucose uptake. Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. (A) The mRNA levels of Glut4 were determined by real-time qPCR, and the mRNA levels of Gapdh was used as reference for normalization (n = 4–5). #P < 0.05 vs. db/db + Vehicle mice. (B) The expression level of Glut4 in the myocardium were determined by western blotting (n = 7). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT + Vehicle group, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. db/db + Vehicle mice. (C) Primary mice cardiomyocytes were incubated with 33.3 mM glucose (HG), 100 nM insulin (HI), or 33.3 mM glucose + 100 nM insulin (HG + HI) for 48 h. The cardiomyocytes were then incubation with SPRC (50 μM) for 24 h. 3H glucose uptake was measured in a liquid scintillation counter and the concentration of protein were detected by BCA method for standardization (n = 3). *P < 0.05 and ***P < 0.001 vs. control group, #P < 0.05, ##P < 0.01 vs. HG+HI group. Values are presented as means ± SEM.
SPRC Treatment Increased CSE Expression and H2S Content in the Myocardium
CSE is the primary H2S-generating enzyme in the cardiovascular system, catalyzing the synthesis of endogenous H2S from L-cysteine, and SPRC is an activator of CSE. We confirmed that SPRC increases the protein expression of CSE in primary cardiomyocytes (Figure 9A). Moreover, SPRC enhanced CSE expression in the left ventricular tissues of db/db mice (Figure 9B). Furthermore, H2S concentration in plasma and myocardium were detected. SPRC treatment at the doses of 40 and 80 mg·kg−1·day−1 improved H2S levels in the myocardium of db/db mice (Figure 9C). And H2S levels in plasma of db/db mice was significantly lower than that of WT mice, with SPRC treatment failed to rescue (Figure 9D).
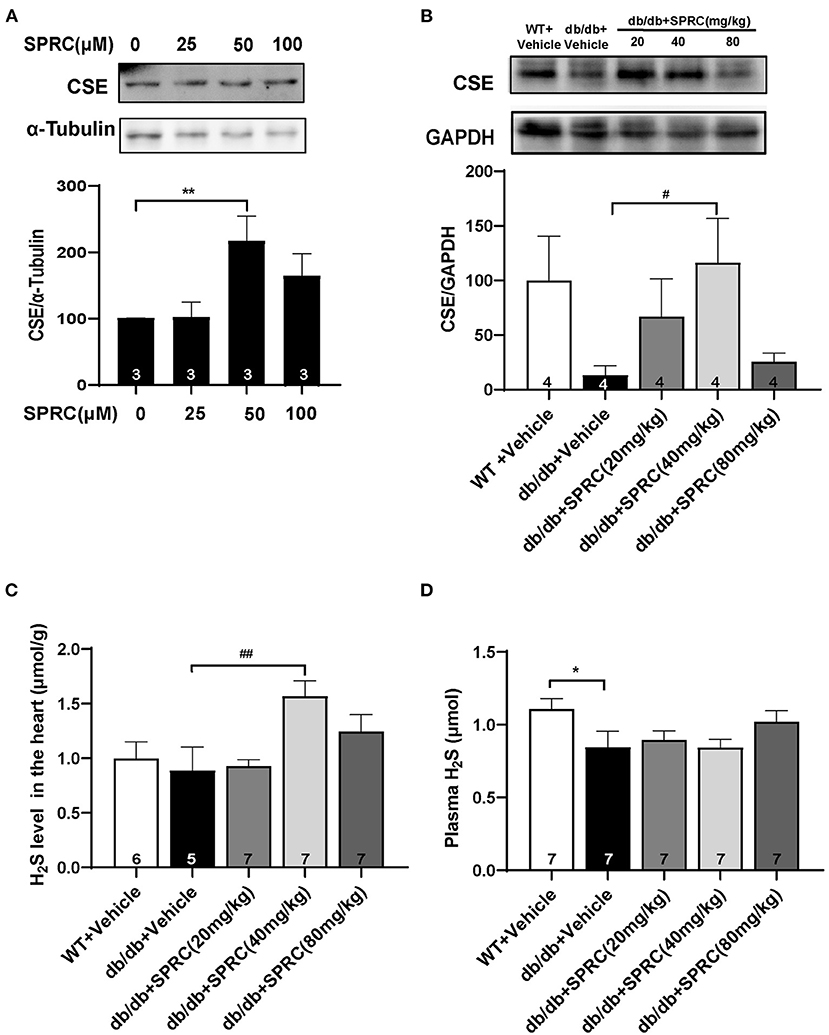
Figure 9. SPRC treatment increased CSE expression and H2S content in the myocardium. (A) Primary mice cardiomyocytes were treated with vehicle or SPRC (25, 50, and 100 μM) for 24 h. The expression level of CSE were determined by western blotting (n = 3). **P < 0.01 vs. Vehicle group. (B–D) Vehicle or SPRC (20, 40, or 80 mg/kg/day) was orally administered to WT mice or db/db mice for 12 weeks. The expression level of CSE were determined by western blotting (n = 4), H2S levels in the myocardium (n = 5–7), and plasma (n = 7) were detected. *P < 0.05, **P < 0.01 vs. WT + Vehicle group, #P < 0.05, ##P < 0.01 vs. db/db + Vehicle mice. Values are presented as means ± SEM.
Discussion
In this study, we investigated the effects of SPRC on diabetic cardiomyopathy. Our results suggested that SPRC treatment could improve the myocardial function in diabetic mice with diabetic cardiomyopathy by attenuating myocardial hypertrophy, myocardial fibrosis, myocardial lipid accumulation, abnormalities in the ultrastructure of cardiomyocytes. Importantly, we unveiled the molecular mechanism underlying SPRC protected myocardium, which involves the activation of cardiac insulin receptor signaling.
The global prevalence of type 2 diabetes mellitus has been gradually increasing over half a century especially in developing countries (23). The systemic metabolic disorder in diabetic patients exposes myocardium to hyperglycemia, hyperinsulinemia, hyperlipidemia, and insulin resistance, all of which trigger the development of myocardial dysfunction. It is widely accepted that morphological phenotypes of diabetic cardiomyopathy include cardiac hypertrophy, cardiac fibrosis, increased intramyocardial lipids, while the functional phenotypes include left ventricular diastolic dysfunction which usually precede systolic dysfunction (24–26). In this study, we used spontaneously diabetic db/db mice as type 2 diabetes model which basically reflects all the manifestation of diabetic cardiomyopathy (18, 27). Moreover, 12 weeks of SPRC treatment, especially in the medium-dose group (40 mg·kg−1·day−1), alleviated almost all the morphological and functional phenotype of diabetic cardiomyopathy in db/db mice, which indicated the protective effect of SPRC on diabetic cardiomyopathy. Consistently, only the medium-dose group (40 mg·kg−1·day−1) had a significant increase in H2S levels in the myocardium (Figure 9C), while high-dose (80 mg·kg−1·day−1) SPRC only raised H2S levels in the myocardium a little and has no statistical significance when compared with vehicle. It has been reported that the mechanisms of SPRC releasing H2S include up-regulating CSE gene and protein expression (28, 29), as well as binding and activating CSE (16). Detailed mechanism of effective dose range of SPRC in vivo needs further study.
Next, we explored the mechanism of SPRC to protect diabetic cardiomyopathy. Abnormal mitochondrial morphology is common in the progression of diabetic cardiomyopathy, and suggests the possibility of impaired mitochondrial dynamics, metabolic substrate imbalance and increased oxidative stress (5, 30). Under transmission electron microscopy, we observed obvious damage of myocardial ultrastructure in db/db mice, especially alterations of mitochondria. SPRC treatment markedly restored mitochondrial and myocardial ultrastructure. This result suggests that the effect of SPRC on diabetic cardiomyopathy may associated with improvement of myocardial mitochondria. The effect of SPRC treatment on mitochondrial function, myocardial oxidative stress and underlying signaling pathway is worth of further study.
Under conventional conditions in heart, glucose and lactic acid only supply 30% of ATP generation, with the majority of ATP generated by fatty acid (FA) oxidation. Whereas, in the case of type 2 diabetes mellitus, hyperglycemia maintains cardiac glucose uptake, but the proportion of FA as a metabolic substrate further increases in consequence of plasma FA availability (31–35). Within the heart, lipid droplet accumulation generally only appears in patients with diabetes or metabolic syndrome, indicating an imbalance of FA uptake/synthesis and consumption (36, 37). In this study, both hyperlipidaemia and cardiac lipid droplet accumulation were observed in db/db mice, and the effect of SPRC treatment on improving lipid content in myocardium seems superior to the improvement of lipid content in circulation. Similarly, SPRC treatment protected diabetic mice from cardiac insulin resistance by activating the AKT/GLUT4 signaling, with no obvious improvement on systemic insulin resistance, which were consistent with the improvement of H2S levels in myocardium but not in plasma. This may be because db/db mice as transgenic rodent is completely resistant to leptin receptor, developed morbid obesity and severe systemic metabolic phenotype at 20 weeks of age which is irreversible by SPRC treatment (23). Besides, this also demonstrates that the protective effect of SPRC treatment is targeted to the myocardium, rather than the consequence of alleviating the metabolic phenotype of whole body. In addition, SPRC releases H2S via the up-regulating of CSE, thus the organ distribution of CSE is critical to the effect of SPRC. Next, we focus on the effect of SPRC on cardiac molecular signaling pathway in the following experiments.
Insulin resistance is defined as impaired insulin signaling together with diminution in glucose transport, which promotes the development of diabetic cardiomyopathy (38). Myocardial insulin signaling plays an important role in myocardial metabolic remodeling in response to myocardial metabolic disorders by regulating glucose uptake, long-chain fatty acid uptake, and protein synthesis, wherein insulin receptor (IR) is a key protein for signal transduction (39–41). Cardiomyocyte-selective insulin receptor knockout mice revealed decreased insulin signaling in cardiomyocytes without systemic metabolic disturbances, and showed worsen cardiac remodeling in respond to stress (42) and accelerated cardiac mitochondrial dysfunction after myocardial infarction (43).
IR is a polymer transmembrane glycoprotein composed of α2β2 heterotetramer and belongs to the receptor tyrosine kinases (RTK) family Type Ò subfamily activated by ligands such as insulin and insulin-like growth factor (IGF) (44). The binding of insulin triggers autophosphorylation in kinase activation region of IR at Tyr-1146 and either Tyr-1150 or Tyr-1151 (45). The activated IR recruits and phosphorylates scaffold proteins such as insulin receptor substrates (IRS) to activate the classical PI3K/AKT pathway, with other downstream pathway of insulin signaling (including Ras/MAPK-dependent pathways) unaffected. The activation of AKT subsequently promotes the translocation of GLUT4 to the membrane, and enhances glucose uptake. The phosphorylation of AKT also inactivates GSK-3β, an essential negative regulator of cardiac hypertrophy and cardiomyopathy (46, 47). Consistent with these studies, our results showed inhibited insulin signaling such as decreased phosphorylation levels of Akt and GSK-3β and reduced protein expression of GLUT4 in the cardiomyocyte of diabetic hearts, which was notably activated by SPRC treatment. Moreover, we speculate that SPRC enhanced the glucose uptake to regulate metabolic substrate proportion, thereby protecting the heart caused by metabolic disorders. Interestingly, although the effect of SPRC on IR downstream signaling in vivo and in vitro were consistent, the phosphorylation status of IR differs. The phosphorylation level of IR was reduced by acute high concentrations of glucose and insulin in vitro, but tends to increase in chronic diabetic myocardium which may due to compensation. This suggests that in the myocardium of type 2 diabetes, the inhibition of IR signaling is not due to the decrease of phosphorylation level of IR, but the decrease of IR expression and downstream signaling transduction.
In summary, we provided evidences that SPRC protects against cardiac fibrosis and improves myocardial function in diabetic mice. This mechanism involves increased expression and activity of IR and activated Akt/ GSK-3β signaling, which subsequently enhanced glucose uptake in cardiomyocyte, resulting in improved diabetic cardiomyopathy. Thereby, SPRC may be a promising medication for diabetic cardiomyopathy in type 2 diabetes mellitus patients.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by The Ethics Committee of Experimental Research, Fudan University, Shanghai Medical College.
Author Contributions
Y-CZ and YL: designed experiments. YL, K-FX, Y-HC, and CW: performed experiments. YL and K-FX: analyzed data. Y-CZ, YC, and M-JW: provided laboratory space, reagents, and technical support. YL: wrote the manuscript. M-JW: revised the manuscript. Y-CZ: supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (31830042 and 81870212 to Y-CZ, 81970361 to M-JW), the National Key Science and Technology Project of China (2018YFC2000200/02), Macau Science and Technology Development fund (FDCT 0007/2019/AKP), and the funding of Innovative research team of high-level local universities in Shanghai and a key laboratory program of the Education Commission of Shanghai Municipality (ZDSYS14005 to Y-CZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.737191/full#supplementary-material
Abbreviations
Akt/PKB, protein kinase B; AUC, area under the curve; CSE (CTH), cystathionine-γ-lyase; DCM, diabetic cardiomyopathy; DM, diabetes mellitus; DT, deceleration time; ECL, enhanced chemiluminescence; EF, ejection fraction; FBG, fasting blood glucose; FINS, fasting plasma insulin; FS, fraction shortening; H2S, hydrogen sulfide; GLUT4, glucose transporter 4; GSK3β, glycogen synthase kinase 3β; HE, hematoxylin-eosin; HDL-C, high-density lipoprotein cholesterol; HOMA-IR, homeostasis model assessment-insulin resistance index; IPGTT, Intraperitoneal glucose tolerance test; IPITT, Intraperitoneal Insulin Tolerance Test; LDL-C, low-density lipoprotein cholesterol; SPRC, S-propargyl-cysteine; TEM, transmission electron microscope; TC, total cholesterol; TG, triglycerides.
References
1. Wang L, Gao P, Zhang M, Huang Z, Zhang D, Deng Q, et al. Prevalence and ethnic pattern of diabetes and prediabetes in China in 2013. JAMA. (2017) 317:2515–23. doi: 10.1001/jama.2017.7596
2. Dal Canto E, Ceriello A, Rydén L, Ferrini M, Hansen TB, Schnell O, et al. Diabetes as a cardiovascular risk factor: an overview of global trends of macro and micro vascular complications. Eur J Prev Cardiol. (2019) 26:25–32. doi: 10.1177/2047487319878371
3. Kannel WB, McGee DL. Diabetes and cardiovascular disease: the Framingham Study. JAMA. (1979) 241:2035–8. doi: 10.1001/jama.241.19.2035
4. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. (1972) 30:595–602. doi: 10.1016/0002-9149(72)90595-4
5. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
6. Castagno D, Baird-Gunning J, Jhund PS, Biondi-Zoccai G, MacDonald MR, Petrie MC, et al. Intensive glycemic control has no impact on the risk of heart failure in type 2 diabetic patients: Evidence from a 37,229 patient meta-analysis. Am Heart J. (2011) 162:938–48.e2. doi: 10.1016/j.ahj.2011.07.030
7. Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, Duckworth WC, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. (2009) 52:2288–98. doi: 10.1007/s00125-009-1470-0
8. Zhao S, Li X, Li X, Wei X, Wang H. Hydrogen sulfide plays an important role in diabetic cardiomyopathy. Front Cell Dev Biol. (2021) 9:627336. doi: 10.3389/fcell.2021.627336
9. Sun X, Zhao D, Lu F, Peng S, Yu M, Liu N, et al. Hydrogen sulfide regulates muscle RING finger-1 protein S-sulfhydration at Cys44 to prevent cardiac structural damage in diabetic cardiomyopathy. Br J Pharmacol. (2020) 177:836–56. doi: 10.1111/bph.14601
10. Guo R, Wu Z, Jiang J, Liu C, Wu B, Li X, et al. New mechanism of lipotoxicity in diabetic cardiomyopathy: deficiency of endogenous H2S production and ER stress. Mech Ageing Dev. (2017) 162:46–52. doi: 10.1016/j.mad.2016.11.005
11. Ansari M, Kurian GA. Mechanism of hydrogen sulfide preconditioning-associated protection against ischemia–reperfusion injury differs in diabetic heart that develops myopathy. Cardiovasc Toxicol. (2020) 20:155–67. doi: 10.1007/s12012-019-09542-9
12. Mard SA, Ahmadi I, Ahangarpour A, Gharib-Naseri MK, Badavi M. Delayed gastric emptying in diabetic rats caused by decreased expression of cystathionine gamma lyase and H2S synthesis: in vitro and in vivo studies. Neurogastroenterol Motil. (2016) 28:1677–89. doi: 10.1111/nmo.12867
13. Liang W, Chen J, Mo L, Ke X, Zhang W, Zheng D, et al. ATP-sensitive K+ channels contribute to the protective effects of exogenous hydrogen sulfide against high glucose-induced injury in H9c2 cardiac cells. Int J Mol Med. (2016) 37:763–72. doi: 10.3892/ijmm.2016.2467
14. Wang M, Tang W, Xin H, Zhu YZ. S-propargyl-cysteine, a novel hydrogen sulfide donor, inhibits inflammatory hepcidin and relieves anemia of inflammation by inhibiting IL-6/STAT3 pathway. PLoS One. (2016) 11:e0163289. doi: 10.1371/journal.pone.0163289
15. Yang H, Mao Y, Tan B, Luo S, Zhu Y. The protective effects of endogenous hydrogen sulfide modulator, S-propargyl-cysteine, on high glucose-induced apoptosis in cardiomyocytes: a novel mechanism mediated by the activation of Nrf2. Eur J Pharmacol. (2015) 761:135–43. doi: 10.1016/j.ejphar.2015.05.001
16. Kan J, Guo W, Huang C, Bao G, Zhu Y, Zhu YZ. S-propargyl-cysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxid Redox Signal. (2014) 20:2303–16. doi: 10.1089/ars.2013.5449
17. Wang Q, Liu HR, Mu Q, Rose P, Zhu YZ. S-propargyl-cysteine protects both adult rat hearts and neonatal cardiomyocytes from ischemia/hypoxia injury: the contribution of the hydrogen sulfide-mediated pathway. J Cardiovasc Pharmacol. (2009) 54:139–46. doi: 10.1097/FJC.0b013e3181ac8e12
18. Mori J, Patel Vaibhav B, Abo Alrob O, Basu R, Altamimi T, DesAulniers J, et al. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart Failure. (2014) 7:327–39. doi: 10.1161/CIRCHEARTFAILURE.113.000672
19. Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab. (2015) 21:334–47. doi: 10.1016/j.cmet.2015.01.002
20. Xue R, Hao DD, Sun JP, Li WW, Zhao MM, Li XH, et al. Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid Redox Signal. (2013) 19:5–23. doi: 10.1089/ars.2012.5024
21. Shen X, Pattillo CB, Pardue S, Bir SC, Wang R, Kevil CG. Measurement of plasma hydrogen sulfide in vivo and in vitro. Free Radic Biol Med. (2011) 50:1021–31. doi: 10.1016/j.freeradbiomed.2011.01.025
22. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. (2015) 6:331–51. doi: 10.1002/cphy.c150016
23. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
24. Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy. Circ Res. (2006) 98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2
25. Lewis GA, Schelbert EB, Williams SG, Cunnington C, Ahmed F, McDonagh TA, et al. Biological phenotypes of heart failure with preserved ejection fraction. J Am Coll Cardiol. (2017) 70:2186–200. doi: 10.1016/j.jacc.2017.09.006
26. Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. (2015) 36:1718–27. doi: 10.1093/eurheartj/ehv134
27. Waldman M, Cohen K, Yadin D, Nudelman V, Gorfil D, Laniado-Schwartzman M, et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving 'SIRT1 and PGC-1α'. Cardiovasc Diabetol. (2018) 17:111. doi: 10.1186/s12933-018-0757-1
28. Li W, Ma F, Zhang L, Huang Y, Li X, Zhang A, et al. S-Propargyl-cysteine exerts a novel protective effect on methionine and choline deficient diet-induced fatty liver via Akt/Nrf2/HO-1 pathway. Oxid Med Cell Longev. (2016) 2016:4690857. doi: 10.1155/2016/4690857
29. Gong QH, Wang Q, Pan LL, Liu XH, Xin H, Zhu YZ. S-propargyl-cysteine, a novel hydrogen sulfide-modulated agent, attenuates lipopolysaccharide-induced spatial learning and memory impairment: involvement of TNF signaling and NF-κB pathway in rats. Brain Behav Immun. (2011) 25:110–9. doi: 10.1016/j.bbi.2010.09.001
30. Kubli DA, Gustafsson ÃB. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal. (2015) 22:1527–544. doi: 10.1089/ars.2015.6322
31. Wan A, Rodrigues B. Endothelial cell and cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc Res. (2016) 111:172–83. doi: 10.1093/cvr/cvw159
32. Larsen TS, Aasum E. Metabolic (In)flexibility of the diabetic heart. Cardiovasc Drugs Ther. (2008) 22:91–5. doi: 10.1007/s10557-008-6083-1
33. Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
34. van den Brom CE, Huisman MC, Vlasblom R, Boontje NM, Duijst S, Lubberink M, et al. Altered myocardial substrate metabolism is associated with myocardial dysfunction in early diabetic cardiomyopathy in rats: studies using positron emission tomography. Cardiovasc Diabetol. (2009) 8:39. doi: 10.1186/1475-2840-8-39
35. van de Weijer T, Schrauwen-Hinderling VB, Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. (2011) 92:10–18. doi: 10.1093/cvr/cvr212
36. Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, et al. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome[S]. J Lipid Res. (2009) 50:2314–23. doi: 10.1194/jlr.P900032-JLR200
37. Gao Y, Ren Y, Guo Y-K, Liu X, Xie L-J, Jiang L, et al. Metabolic syndrome and myocardium steatosis in subclinical type 2 diabetes mellitus: a (1)H-magnetic resonance spectroscopy study. Cardiovasc Diabetol. (2020) 19:70. doi: 10.1186/s12933-020-01044-1
38. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
39. Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res. (2016) 118:1151–69. doi: 10.1161/CIRCRESAHA.116.306206
40. Zamora Mn, Villena JA. Contribution of impaired insulin signaling to the pathogenesis of diabetic cardiomyopathy. Int J Mol Sci. (2019) 20:2833. doi: 10.3390/ijms20112833
41. Fu F, Zhao K, Li J, Xu J, Zhang Y, Liu C, et al. Direct evidence that myocardial insulin resistance following myocardial ischemia contributes to post-ischemic heart failure. Sci Rep. (2015) 5:17927. doi: 10.1038/srep17927
42. McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, et al. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol. (2005) 39:882–92. doi: 10.1016/j.yjmcc.2005.07.017
43. Sena S, Hu P, Zhang D, Wang X, Wayment B, Olsen C, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol. (2009) 46:910–8. doi: 10.1016/j.yjmcc.2009.02.014
44. Escribano O, Beneit N, Rubio-Longás C, López-Pastor AR, Gómez-Hernández A. The role of insulin receptor isoforms in diabetes and its metabolic and vascular complications. J Diabetes Res. (2017) 2017:1403206. doi: 10.1155/2017/1403206
45. White MF, Shoelson SE, Keutmann H, Kahn CR. A cascade of tyrosine autophosphorylation in the beta-subunit activates the phosphotransferase of the insulin receptor. J Biol Chem. (1988) 263:2969–80. doi: 10.1016/S0021-9258(18)69163-X
46. Hardt Stefan E, Sadoshima J. Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ Res. (2002) 90:1055–63. doi: 10.1161/01.RES.0000018952.70505.F1
Keywords: S-propargyl-cysteine, hydrogen sulfide, diabetic cardiomyopathy, insulin receptor signaling, glucose uptake
Citation: Li Y, Xie K-F, Chang Y-H, Wang C, Chen Y, Wang M-J and Zhu Y-C (2021) S-Propargyl-Cysteine Attenuates Diabetic Cardiomyopathy in db/db Mice Through Activation of Cardiac Insulin Receptor Signaling. Front. Cardiovasc. Med. 8:737191. doi: 10.3389/fcvm.2021.737191
Received: 06 July 2021; Accepted: 24 August 2021;
Published: 17 September 2021.
Edited by:
Hongmei Tan, Sun Yat-sen University, ChinaReviewed by:
Fenghua Yang, Guangdong Laboratory Animals Monitoring Institute, ChinaYing Shao, Temple University, United States
Copyright © 2021 Li, Xie, Chang, Wang, Chen, Wang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Jie Wang, bWp3YW5nQHNobXUuZWR1LmNu; Yi-Chun Zhu, eWN6aHVAc2htdS5lZHUuY24=
†These authors have contributed equally to this work