 Xiao Li1,2Xihe Tang1,2Bo Liu2
Xiao Li1,2Xihe Tang1,2Bo Liu2 Jinghang Zhang3Yongxi Zhang4Hefan Lv2
Jinghang Zhang3Yongxi Zhang4Hefan Lv2 Dongling Liu2
Dongling Liu2 Jawahar L. Mehta5
Jawahar L. Mehta5 Xianwei Wang1,2,6,7*
Xianwei Wang1,2,6,7*- 1Department of Cardiology, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
- 2Henan Key Laboratory of Medical Tissue Regeneration, Xinxiang Medical University, Xinxiang, China
- 3Department of Pathology, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
- 4Department of Oncology, The Third Affiliated Hospital, Xinxiang Medical University, Xinxiang, China
- 5Division of Cardiology, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 6Department of Human Anatomy & Histoembryology, Xinxiang Medical University, Xinxiang, China
- 7Xinxiang Key Laboratory of Molecular Neurology, Xinxiang Medical University, Xinxiang, China
Background: Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) is a transmembrane glycoprotein that mediates uptake of oxidized low-density lipoprotein (ox-LDL) into cells. Previous studies had shown that LOX-1 deletion had a potential to inhibit cardiac fibrosis in mouse models of hypertension and myocardial infarction. Whether LOX-1 deletion also affects cardiac fibrosis associated with aging still remains unknown. The aim of this study was to investigate the effect of LOX-1 deletion on myocardial fibrosis in the aged mice.
Methods: C57BL/6 mice and LOX-1 knockout (KO) mice with C57BL/6 background were studied to the age of 60 weeks. Both genotypes of aged mice were exposed to angiotensin II (Ang II) or saline for additional 4 weeks. The mice were then sacrificed, and myocardial fibrosis, reactive oxygen species (ROS) and expression of LOX-1, fibronectin, collagens, p22phox, and gp91phox were measured.
Results: LOX-1 deletion markedly reduced Ang II-mediated rise of blood pressure in the aged mice (vs. saline-treated mice). LOX-1 deletion also limited fibrosis and decreased fibronectin and collagen-3 expression in the hearts of aged mice, but not the expression of collagen-1 and collagen-4. LOX-1 deletion also inhibited ROS production and p22phox expression. As the aged mice were exposed to Ang II for 4 weeks (resulting in hypertension), LOX-1 deletion more pronounced inhibiting myocardial fibrosis and ROS production, and decreasing expression of fibronectin, collagen-1, collagen-2, collagen-3, p22phox, and gp91phox.
Conclusion: LOX-1 deletion limited fibrosis and ROS production in the hearts of aged mice. This effect was more pronounced in the aged mice with hypertension induced by Ang II infusion.
Introduction
Heart failure associated with aging is one of the leading causes of death in the world (1). The average age of onset heart failure is 74 years (2). Myocardial fibrosis caused by aging is perhaps the most important cause of heart failure in the elderly (1). With aging, collagens are gradually deposited in myocardial interstitium, resulting in a decrease of elasticity and compliance in ventricles and leading to diastolic heart failure (3). Although many studies have provided different insights into aging-driven myocardial fibrosis, the specific mechanisms are still not completely understood.
Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), a 50-KDa type II transmembrane glycoprotein, is an important member in the class E scavenger receptor subfamily (4). The main function of LOX-1 is to recognize and bind oxidized low-density lipoprotein (ox-LDL), and thus mediate the uptake of ox-LDL into cells (5). It is generally acknowledged that LOX-1 is a critical player in atherogenesis (6, 7). Recent studies have implicated LOX-1 in cardiac remodeling following myocardial infarction and in models of hypertension (8–10). Blockade or deletion of LOX-1 has been shown to attenuate myocardial fibrosis in the rodent models (9, 10). However, whether LOX-1 also affects cardiac fibrosis related to aging still remains unknown. Recently, studies from our group and others have indicated that LOX-1 may be involved in aging of cardiovascular system (11–13). LOX-1 was found to be downregulated with aging in vascular walls, endothelial cells, and cardiac fibroblasts (11, 12). An in vitro study also showed that LOX-1 downregulation could inhibit proliferation of senescent cardiac fibroblasts by regulating cytoskeleton reorganization (12).
Based on this information, we hypothesized that LOX-1 may participate in myocardial fibrosis associated aging. This study was designed to address this hypothesis by using old wild type (WT) and LOX-1 knockout (KO) mice. Both genotypes of mice were infused with angiotensin II (Ang II) by subcutaneously implanted osmotic pumps to strengthen cardiac fibrosis.
Methods
Animal Models
The homozygous LOX-1 KO mice were backcrossed eight times with C57BL/6 strain to replace the genetic background. The genotypes of these mice were verified by genotyping assays. Male LOX-1 KO mice and C57BL/6 WT mice were housed in a conditional room for 56 weeks. Afterwards, both genotypes of mice were randomly divided into four groups (12 animals /group): WT + saline, WT + Ang II, LOX-1 KO + saline, and LOX-1 KO + Ang II. All animals were subcutaneously implanted with osmotic pumps (ALZET® International Distributors, Cupertino, CA, USA) to infuse Ang II (100ng/kg·min for 4 weeks; Abcam, Cambridge, MA, USA) or equal volumes of saline as per previously published protocols (14, 15). Blood pressure was measured every week by noninvasive tail-cuff method with a Softron BP-98A (Softron Co., Ltd., Tokyo, Japan). All experimental procedures were conducted in accordance with protocols approved by the Institutional Animal Care and Usage Committee.
Hematoxylin-Eosin (HE) and Masson's Trichrome Staining
Hearts were collected from each group of animals (six samples /group). Following washing with PBS twice, the samples were fixed with 4% paraformaldehyde, embedded in paraffin and sliced into 5 μm sections. The ventricle sections were deparaffinized, rehydrated, and stained with HE using standard protocols and with a Masson's Trichrome Stain Kit (Sigma-Aldrich, St. Louis, MO, USA) following manufacturer's instructions. The images were viewed and captured with a digital imaging system. The collagen volume fraction (CVF) was calculated with Image J software.
Immunostaining
Left ventricular sections were blocked with 5% goat serum/1% BSA in PBS for 30 min, and then incubated with LOX-1 antibody (a gift from Dr. Tatsuya Sawamura, Shinshu University, Japan; 1:400, v/v) for 90 min at room temperature. After washing twice with PBS, the sections were incubated with TR-conjugated secondary antibody (ZSGB-Bio, Beijing, China; 1:1,000, v/v) for 30 min at room temperature. After washed with PBS and deionized water, the sections were covered with coverslips by antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI). The images were taken with a fluorescence microscope (Leica Microsystems, Bensheim, Germany). The fluorescence intensity was calculated with Image J software.
DHE Staining
Frozen ventricular tissues were sliced into 7 μm sections, and subsequently the sections were incubated with 5 μM dihydroethidium (DHE) in the dark in a humidified box at 37°C for 30 min. After washing with PBS, the sections were treated with antifade reagent with DAPI and analyzed with a fluorescent microscope. The fluorescence intensity was calculated with Image J software.
Western Blotting
Proteins were extracted from each group of left ventricular tissues with lysis buffer supplemented with protease inhibitor and phenylmethylsulfonyl fluoride (PMSF). Proteins (20 μg /sample) were separated by electrophoresis with 10% SDS/PAGE gels. After electrophoresis, proteins were transferred to nitrocellulose membranes. The blots were blocked with 5% non-fat milk in Tris-buffered saline with Tween-20 (TBS-T) for 1 h at room temperature, and then incubated with primary antibodies against mouse LOX-1 (a gift from Dr. Sawamura; 1:2,000, v/v, dilution), fibronectin (Abcam; 1:2,000, v/v, dilution), collagen-1a (Santa Cruz, Dallas, TX, USA; 1:1,000, v/v, dilution), collagen-3a (Santa Cruz; 1:1,000, v/v, dilution), collagen-4a (Santa Cruz; 1:1,000, v/v, dilution), p22phox (Santa Cruz; 1:1,000, v/v, dilution), gp91phox (Abcam; 1:2,000, v/v, dilution) or β-actin (Santa Cruz; 1:2,000, v/v, dilution) in TBS-T at 4°C overnight on a shaker. After washing with TBS-T three times, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz) for 1 h at room temperature. The blots were scanned with a ChemiDOC XRS system (Bio-Rad, Hercules, CA, USA) following exposure to Luminol Reagents (Beyotime, Shanghai, China) for 3 min.
Statistical Analysis
Statistical analysis was performed with SPSS 15.0 software (IBM, Chicago, IL, USA). Data are presented as mean ± SD from six independent experiments. Univariate comparisons of means were evaluated using one-way ANOVA with Tukey's post-hoc adjustment. P < 0.05 was considered statistical significance.
Results
Confirmation of LOX-1 Expression
Genotyping data showed that the amplified genomic LOX-1 fragments (400bp) appeared in WT mice following polymerase chain reaction (PCR), but were absent in all LOX-1 KO mice. The band was replaced by inserted neomycin resistance gene fragments (200kb) (Figure 1). Immunofluorescence staining showed that LOX-1 protein expression was increased 4-fold in the hearts derived from WT mice following Ang II infusion for 4 weeks, but was absent in LOX-1 KO mice hearts (Figures 2A,B), which was further confirmed by Western blotting data (Figure 2C).

Figure 1. Genotyping results of LOX-1 KO mice and WT mice used in this study. Neo represents the inserted neomycin resistance gene.
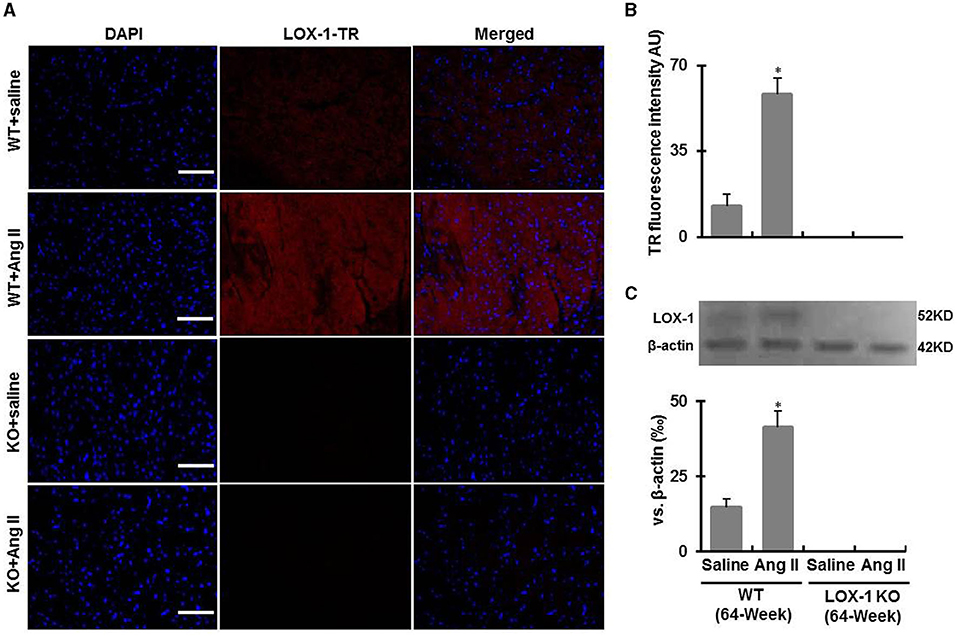
Figure 2. LOX-1 expression in the hearts of each group of mice following infusion with Ang II or saline for 4 weeks. (A) Representative immunostaining images show LOX-1 expression in the hearts of each group of mice following Ang II infusion; (B) Quantitative data of immunostaining images; (C) Western blotting data show LOX-1 expression in the hearts of each group of mice following Ang II infusion. n = 6 /group. *P < 0.05, vs. WT + saline group. Scale bars represent 50 μm.
LOX-1 Deletion Suppresses Increase of Blood Pressure in Aged Mice With Ang II Infusion
As shown in Figures 3A,B, systolic and diastolic blood pressures both were markedly increased in both WT + Ang II (vs. WT + saline) and LOX-1 KO + Ang II (vs. LOX-1 KO + saline) groups of mice (P < 0.05). LOX-1 deletion markedly suppressed Ang II-induced increase of blood pressure in the aged mice (LOX-1 KO + Ang II vs. WT + Ang II, P < 0.05), but did not significantly affect blood pressure of aged mice without Ang II infusion (LOX-1 KO + saline vs. WT + saline, P > 0.05). Notably, there were no significant differences in heart rate among four groups (P > 0.05; Figure 3C).
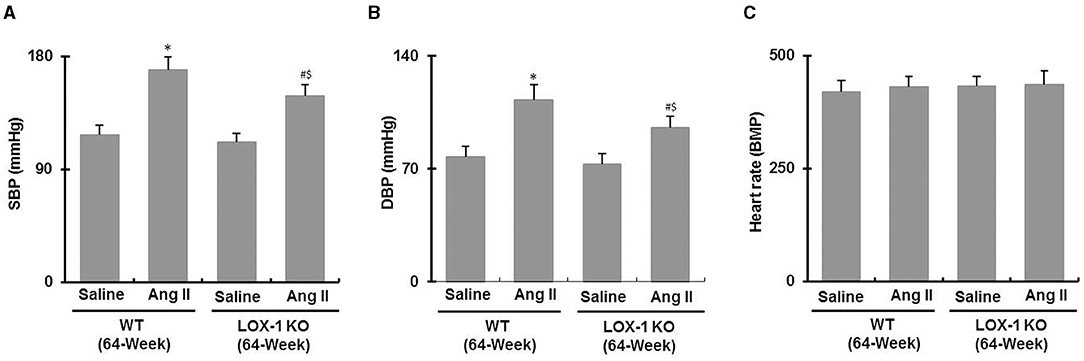
Figure 3. Blood pressure and heart rate in each group of mice following infusion with Ang II or saline for 4 weeks. (A) Systolic blood pressure (SBP); (B) diastolic blood pressure (DBP); (C) heart rate. n = 6 /group. *P < 0.05, vs. WT + saline group; #P < 0.05, vs. LOX-1 KO + saline group; $P < 0.05, vs. WT + Ang II group.
LOX-1 Deletion Attenuates Myocardial Fibrosis of Aged Mice Regardless of Infusion With Ang II or Saline
After sacrifice of mice, we measured heart weight and calculated the ratio of heart weight to body weight. As shown in Figure 4, the ratios were markedly increased in WT + Ang II and LOX-1 KO + Ang II groups of mice following infusion with Ang II for 4 weeks, as compared with WT + saline and LOX-1 KO +saline groups, respectively (P < 0.05). More importantly, this ratio was markedly smaller in LOX-1 KO + Ang II group than that in WT + saline group (P < 0.05), which indicated that LOX-1 deletion could limit cardiac hypertrophy of aged mice with hypertension. Of note, the ratio of heart weight to body weight was also slightly smaller in LOX-1 KO + saline group than that in WT + saline group, but did not reach statistical significance (P > 0.05). HE staining data showed that myocytes clearly became hypertrophied in WT + Ang II and LOX-1 KO + Ang II groups as compared with WT + saline and LOX-1 KO +saline groups, respectively (Figure 5A). More importantly, LOX-1 deletion not only limited myocyte hypertrophy in the aged mice with hypertension, but also the aging mice alone (infused with saline) (Figure 5A).
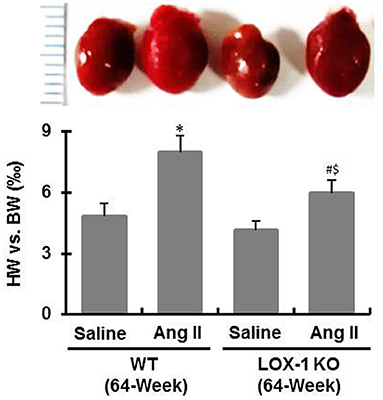
Figure 4. The ratio of heart weight to body weight of each group of mice following infusion with Ang II or saline for 4 weeks. n = 6 /group. *P < 0.05, vs. WT + saline group; #P < 0.05, vs. LOX-1 KO + saline group; $P < 0.05, vs. WT + Ang II group.
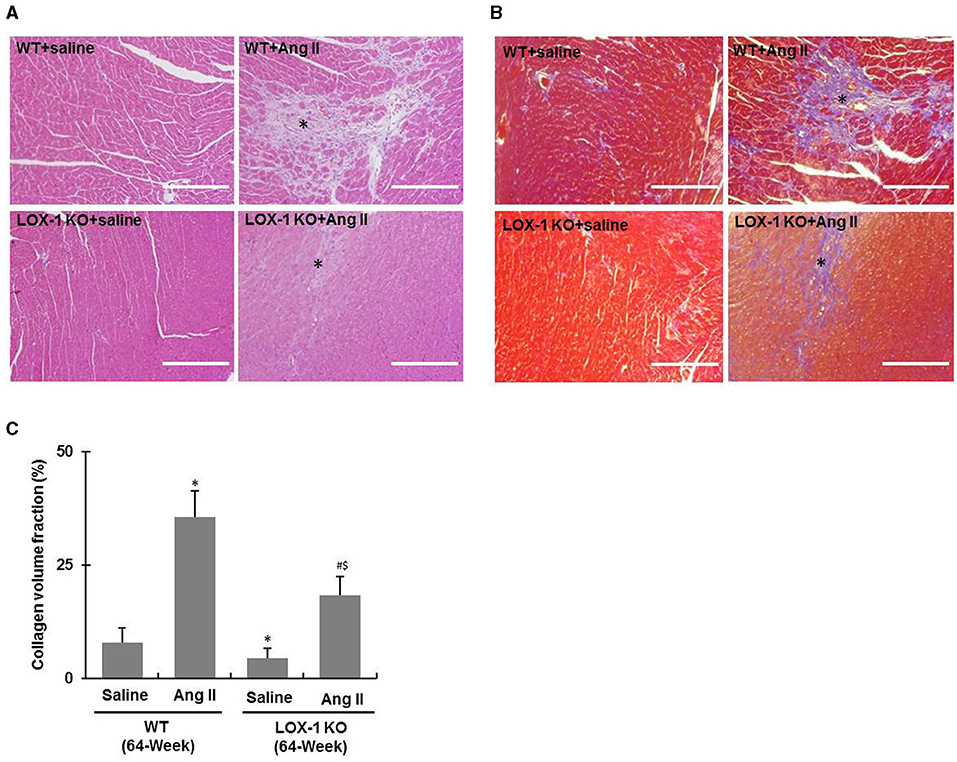
Figure 5. Cardiac fibrosis of each group of mice following infusion with Ang II or saline for 4 weeks. (A) Representative HE staining images; (B) Representative Masson's trichrome staining images. Asterisks indicating fibrotic scars. (C) Quantification of collagen volume fraction in Masson's trichrome staining images. n = 6 /group. *P < 0.05, vs. WT + saline group; #P < 0.05, vs. LOX-1 KO + saline group; $P < 0.05, vs. WT + Ang II group. Scale bars represent 200 μm.
Further, HE staining showed that the fibrotic scar areas were more intense and larger in the hearts of WT + Ang II and LOX-1 KO + Ang II groups of mice as compared with WT + saline and LOX-1 KO +saline groups, respectively (Figure 5A, asterisks indicated). LOX-1 deletion also limited fibrotic scar area in the aged mice with hypertension (Figure 5A, indicated by asterisks). Masson's trichrome staining (Figures 5B,C) further confirmed that LOX-1 deletion could inhibit cardiac fibrosis in the aged mice regardless of infusion with Ang II (aging + hypertension) or saline (aged alone).
Western blotting data showed that Ang II infusion significantly increased fibronectin, collagen-1a, collagen-3a and collagen-4 expression in left ventricles of both WT and LOX-1 KO mice (P < 0.05; Figures 6A–D), and LOX-1 deletion markedly attenuated Ang II-induced fibronectin, collagen-1 and−3 expression in the hearts of aged mice (P < 0.05; Figures 6A–C), but did not significantly affect collagen-4 expression (P > 0.05; Figure 6D). In addition, LOX-1 deletion also markedly inhibited fibronectin and collagen-3 expression in the hearts of aged mice infused with saline (P < 0.05) (Figures 6A,C), but did not significantly affect collagen-1 and collagen-4 expression (P > 0.05) (Figures 6B,D).
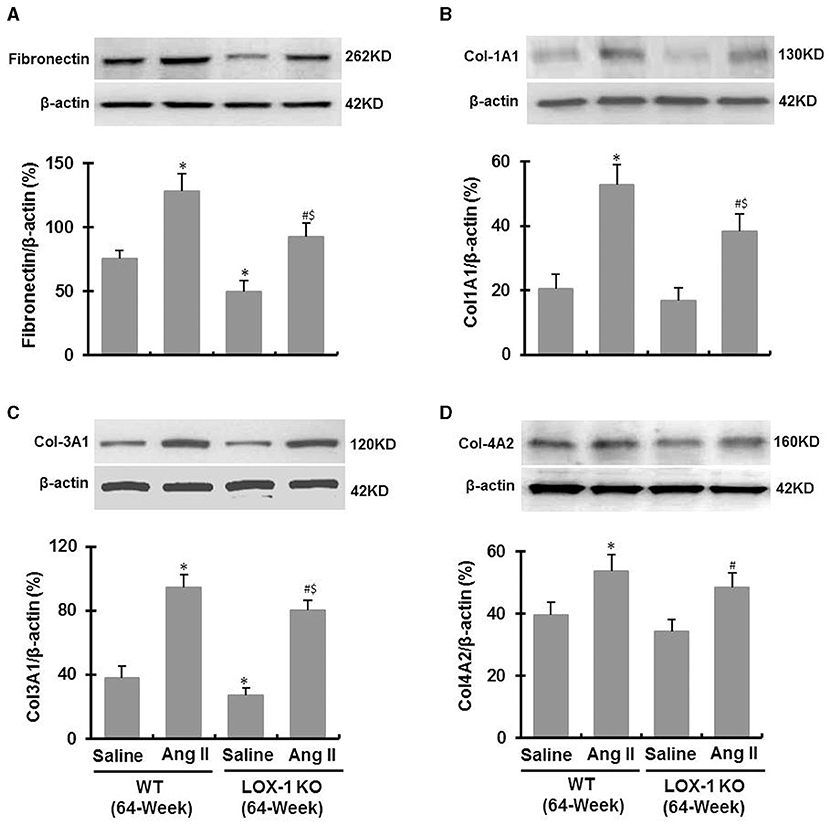
Figure 6. Expression of fibronectin and collagens in the ventricle of each group of mice following infusion with Ang II or saline for 4 weeks (Western blotting). (A) Fibronectin expression; (B) Collagen-1A1 expression; (C) Collagen-3A1 expression; (D) Collagen-4A2 expression. n = 6 /group. *P < 0.05, vs. WT + saline group; #P < 0.05, vs. LOX-1 KO + saline group; $P < 0.05, vs. WT + Ang II group.
LOX-1 Deletion Inhibits ROS Production in the Hearts of Aged Mice Regardless of Infusion With Ang II or Saline
DHE staining showed that ROS levels were markedly increased in the hearts of WT + Ang II and LOX-1 KO + Ang II groups of mice following infusion of Ang II for 4 weeks (P < 0.05; Figures 7A,B). LOX-1 deletion attenuated Ang II-induced ROS production in the hearts of aged mice as well as in the hearts aged mice with hypertension (P < 0.05; Figures 7A,B). Western blotting data showed that p22phox and gp91phox expression was also markedly increased in WT + Ang II and LOX-1 KO + Ang II groups of mice following infusion of Ang II for 4 weeks as compared with WT + saline and LOX-1 KO +saline groups, respectively (Figures 7C,D). LOX-1 deletion markedly suppressed p22phox expression in the hearts of aged mice as well as in the hearts of aged mice with hypertension (P < 0.05; Figure 7C). Of note, gp91phox expression was suppressed in the aged mice with hypertension (P < 0.05), but not the aged mice not given Ang II (P > 0.05) (Figure 7D).
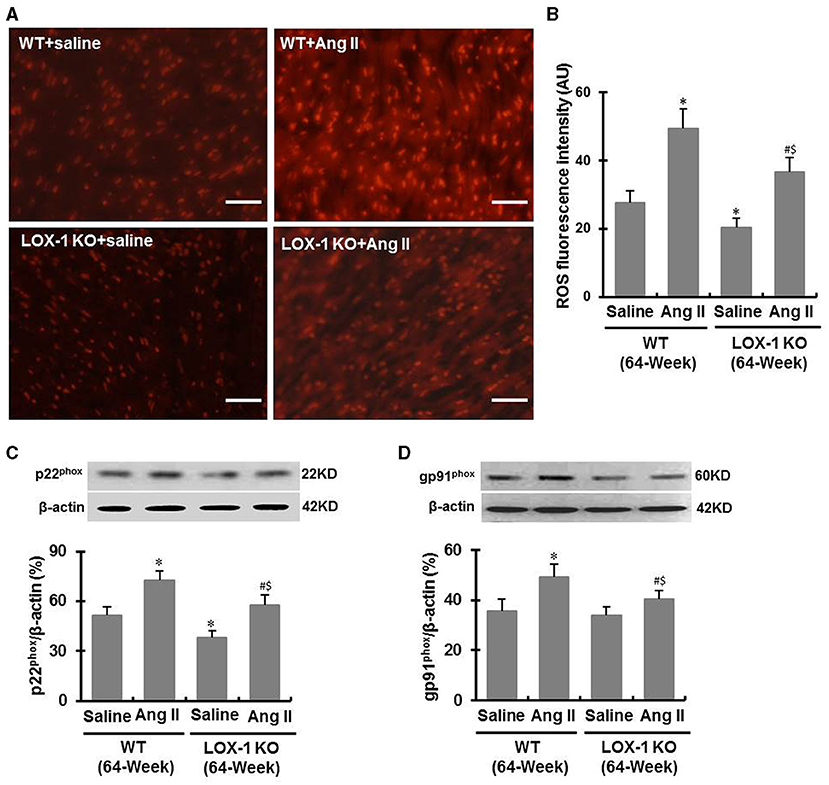
Figure 7. ROS level and expression of p22phox and gp91phox in the ventricle of each group of mice following infusion with Ang II or saline for 4 weeks. (A) Representative DHE staining images shows ROS level in the ventricle of each group of mice; (B) Quantitative data of DHE staining images; (C) Western blotting data show p22phox expression; (D) Western blotting data show gp91phox expression. n = 6 /group. *P < 0.05, vs. WT + saline group; #P < 0.05, vs. LOX-1 KO + saline group; $P < 0.05, vs. WT + Ang II group. Scale bars represent 50 μm.
Discussion
Cardiac fibrosis is a chronic process characterized by abnormal collagen accumulation, resulting in myocardium stiffening, reduced contractility, impaired cardiac function and clinical heart failure (16, 17). It is known that aging is an independent risk factor for cardiac fibrosis in the elderly (3). However, the mechanisms of myocardial fibrosis induced by aging needs further studies. LOX-1 has been shown to be involved in myocardial fibrosis following myocardial infarction and sustained hypertension (10, 18). Whether LOX-1 also participate in the aging-induced cardiac fibrosis has still not been elucidated. This is the first study to define the role of LOX-1 in myocardial fibrosis using the natural aging mouse model with or without hypertension. We found that LOX-1 deletion could significantly inhibit myocardial fibrosis and fibronectin and collagen-3 expression in the hearts of aged mice, but not collagen-1 and collagen-4. Furthermore, LOX-1 deletion could also inhibit ROS production and NADPH oxidase subunit p22phox expression in the hearts of aged mice, but not gp91phox. In the aged mice with hypertension, LOX-1 deletion attenuated myocardial fibrosis and ROS production, and decreased expression of fibronectin, collagen-1, collagen-2, collagen-3, p22phox, and gp91phox. These data indicate that LOX-1 deletion has the potential to limit aging-induced myocardial fibrosis, and this process is more pronounced in pathological conditions such as hypertension.
Previous studies indicated that LOX-1 deletion attenuated the evolution of hypertension in young and adult mice or rats presumably by blocking the crosstalk between LOX-1 and renin angiotensin system (RAS) (4, 10, 19). Blockade and deletion of LOX-1 also reduced Ang II type 1 receptor (AT1R) expression in cardiovascular system (18, 20). We postulated that deletion of LOX-1 should reduce evolution of blood pressure in the aged mice. However, in this study we observed that LOX-1 deletion only inhibited blood pressure in the aged mice with Ang II-induced hypertension, but did not affect basal blood pressure of in the aged mice not given Ang II (Figure 3A). It has been reported that there is no significant difference in blood pressure between the aged and young mice (21). Our findings suggest that LOX-1 contributes potency to the evolution of hypertension induced by Ang II, but not to the alterations of blood pressure that occur with aging.
It is known that aging-dependent stimulation to RAS in myocardium induces an increase of NADPH oxidase activity, promotes ROS production, and activates transforming growth factor-β (TGF-β) signaling, which leads to pathological myocardial fibrosis (22, 23). Ang II stimulates collagen synthesis in fibroblasts through activating AT1R (17). In this study, we also observed that cardiac fibrosis and collagen accumulation were markedly enhanced in the hearts of the aged mice given Ang II for 4 weeks (Figures 5, 6). Previous studies demonstrated that Ang II upregulates LOX-1 expression in the heart and other tissues (19, 24). Consistent with previous reports, LOX-1 expression in the hearts of WT aged mice was markedly increased by Ang II infusion, but in the LOX-1 KO mice (Figure 2). LOX-1 deletion inhibited Ang II-induced myocardial fibrosis in the aged mice (Figure 5). LOX-1 deletion also limited myocardial fibrosis of the aged mice infused with saline. These data indicate that LOX-1 deletion has the potential to inhibit cardiac fibrosis of aged mice. This phenomenon is prominent in the aged mice concurrently with Ang II-induced hypertension.
Fibronectin is an important mediator of collagen formation, and collagen-1 and−3 are the major players, and−4 a relatively minor player in matrix accumulation in the hearts under pathological conditions (25). Quantitative protein detection by Western blotting showed that LOX-1 deletion inhibited fibronectin and collagen-3 expression, but not collagen-1 and−4 in the hearts of aged mice infused with saline (Figure 6). Of note, LOX-1 deletion also inhibited different matrix proteins including fibronectin, collagen-1,−3, and−4 in the hearts of aged mice infused with Ang II (Figure 6). This may be the reason why the inhibitory effect of LOX-1 deletion on myocardial fibrosis in the aged mice given Ang II is more prominent than in the aged mice not given Ang II.
ROS are potent stimulators for collagen synthesis and secretion from cardiac fibroblasts and inducers for myocardial fibrosis (26). Aging and LOX-1 expression both cause ROS production in the hearts (18, 27). Our data showed that LOX-1 deletion inhibited ROS production in the hearts of both the aged mice and the aged mice concurrently with hypertension. NADPH oxidative components p22phox and gp91phox are two key regulators of ROS production (28). Previous studies showed that LOX-1 activation influenced NADPH oxidase complex including the subunits p22phox, p47phox, gp91phox, and Rac1, leading to ROS production (5, 29). In this study, we found that LOX-1 deletion suppressed p22phox and gp91phox expression in the hearts of aged mice with hypertension, but only p22phox in the aged mice without hypertension (Figure 7). We suggest that LOX-1 deletion resulting in inhibition of ROS production may partially result from the modulation of p22phox and gp91phox. The effect of LOX-1 deletion on the expression of NADPH oxidative components and ROS was at least partially responsible for more reduction in myocardial fibrosis, especially in the aged mice with hypertension.
In conclusion, we demonstrate for the first time that LOX-1 is an important player in aging-induced cardiac fibrosis, and LOX-1 deletion has the potential to attenuate myocardial fibrosis and inhibit ROS production in the hearts of aging mice. Further, the inhibitory effects LOX-1 deletion on myocardial fibrosis and ROS production appear more prominent in the aged mice that have hypertension. These findings indicate that LOX-1 may be a therapeutic target for the treatment of cardiac fibrosis with aging. Of note, there is a limitation in this study that the extent of myocardial fibrosis was not compared between young and aged mice at baseline and after Ang II infusion.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee of Xinxiang Medical University.
Author Contributions
XW: designed the experiments. XL, XT, BL, JZ, YZ, HL, and DL: performed the experiments. XW and JM: wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Natural Science Foundation of China (Nos: 81873459, U1804166, and 81370428 to XW); Henan Outstanding Young Scholars Fund (No: 212300410012 to XW); and the Supporting Plan for Scientific and Technological Innovative Talents in Universities of Henan Province (No: 19HASTIT004 to XW), and the fund from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, Washington, DC (No: BX000282-09A2 to JM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Triposkiadis F, Xanthopoulos A, Parissis J, Butler J, Farmakis D. Pathogenesis of chronic heart failure: cardiovascular aging, risk factors, comorbidities, and disease modifiers. Heart Fail Rev. (2020). doi: 10.1007/s10741-020-09987-z. [Epub ahead of print].
3. Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J heart Fail. (2017) 19:177–91. doi: 10.1002/ejhf.696
4. Wang X, Phillips MI, Mehta JL. LOX-1 and angiotensin receptors, and their interplay. Cardiovasc Drugs Ther. (2011) 25:401–17. doi: 10.1007/s10557-011-6331-7
5. Lu J, Mitra S, Wang X, Khaidakov M, Mehta JL. Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid Redox Signal. (2011) 15:2301–33. doi: 10.1089/ars.2010.3792
6. Kattoor AJ, Goel A, Mehta JL. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants (Basel). (2019) 8:218. doi: 10.3390/antiox8070218
7. Jin P, Cong S. LOX-1 and atherosclerotic-related diseases. Clin Chim Acta. (2019) 491:24–9. doi: 10.1016/j.cca.2019.01.006
8. Pothineni NV, Karathanasis SK, Ding Z, Arulandu A, Varughese KI, Mehta JL. LOX-1 in atherosclerosis and myocardial ischemia: biology, genetics, and modulation. J Am Coll Cardiol. (2017) 69:2759–68. doi: 10.1016/j.jacc.2017.04.010
9. Hu C, Dandapat A, Sun L, Marwali MR, Inoue N, Sugawara F, et al. Modulation of angiotensin II-mediated hypertension and cardiac remodeling by lectin-like oxidized low-density lipoprotein receptor-1 deletion. Hypertension. (2008) 52:556–62. doi: 10.1161/HYPERTENSIONAHA.108.115287
10. Wang X, Khaidakov M, Ding Z, Mitra S, Lu J, Dai Y, et al. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) and cardiac fibroblast growth. Hypertension. (2012) 60:1437–42. doi: 10.1161/HYPERTENSIONAHA.112.200659
11. Khaidakov M, Mitra S, Wang X, Ding Z, Bora N, Lyzogubov V, et al. Large impact of low concentration oxidized LDL on angiogenic potential of human endothelial cells: a microarray study. PLoS ONE. (2012) 7:e47421. doi: 10.1371/journal.pone.0047421
12. Wang X, Khaidakov M, Ding Z, Dai Y, Mercanti F, Mehta JL. LOX-1 in the maintenance of cytoskeleton and proliferation in senescent cardiac fibroblasts. J Mol Cell Cardiol. (2013) 60:184–90. doi: 10.1016/j.yjmcc.2013.04.024
13. Hsu PY, Lin WY, Lin RT, Juo SH. MicroRNA let-7g inhibits angiotensin II-induced endothelial senescence via the LOX-1-independent mechanism. Int J Mol Med. (2018) 41:2243–51. doi: 10.3892/ijmm.2018.3416
14. Ahmari N, Santisteban MM, Miller DR, Geis NM, Larkin R, Redler T, et al. Elevated bone marrow sympathetic drive precedes systemic inflammation in angiotensin II hypertension. Am J Physiol Heart Circ Physiol. (2019) 317:H279–89. doi: 10.1152/ajpheart.00510.2018
15. Ohki K, Wakui H, Kishio N, Azushima K, Uneda K, Haku S, et al. Angiotensin II type 1 receptor-associated protein inhibits angiotensin II-induced insulin resistance with suppression of oxidative stress in skeletal muscle tissue. Sci Rep. (2018) 8:2846. doi: 10.1038/s41598-018-21270-8
16. Ma ZG, Yuan YP, Wu HM, Zhang Z, Tang QZ. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci. (2018) 14:1645–57. doi: 10.7150/ijbs.28103
17. Chen P, Yang F, Wang W, Li X, Liu D, Zhang Y, et al. Liraglutide attenuates myocardial fibrosis via inhibition of AT1R-mediated ROS production in hypertensive mice. J Cardiovasc Pharmacol Ther. (2021) 26:179–88. doi: 10.1177/1074248420942007
18. Lu J, Wang X, Wang W, Muniyappa H, Hu C, Mitra S, et al. LOX-1 abrogation reduces cardiac hypertrophy and collagen accumulation following chronic ischemia in the mouse. Gene Ther. (2012) 19:522–31. doi: 10.1038/gt.2011.133
19. Liang YQ, Kakino A, Matsuzaka Y, Mashimo T, Isono M, Akamatsu T, et al. LOX-1 (lectin-like oxidized low-density lipoprotein receptor-1) deletion has protective effects on stroke in the genetic background of stroke-prone spontaneously hypertensive rat. Stroke. (2020) 51:1835–43. doi: 10.1161/STROKEAHA.120.029421
20. Yamamoto K, Kakino A, Takeshita H, Hayashi N, Li L, Nakano A, et al. Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin-like oxLDL receptor. FASEB J. (2015) 29:3342–56. doi: 10.1096/fj.15-271627
21. Toth P, Tarantini S, Springo Z, Tucsek Z, Gautam T, Giles CB, et al. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell. (2015) 14:400–8. doi: 10.1111/acel.12315
22. Ito N, Ohishi M, Yamamoto K, Tatara Y, Shiota A, Hayashi N, et al. Renin-angiotensin inhibition reverses advanced cardiac remodeling in aging spontaneously hypertensive rats. Am J Hypertens. (2007) 20:792–9. doi: 10.1016/j.amjhyper.2007.02.004
23. Conti S, Cassis P, Benigni A. Aging and the renin-angiotensin system. Hypertension. (2012) 60:878–83. doi: 10.1161/HYPERTENSIONAHA.110.155895
24. Huang D, Lu H, Liu H, Yao K, Sun A, Zou Y, et al. Losartan attenuates human monocyte-derived dendritic cell immune maturation via downregulation of lectin-like oxidized low-density lipoprotein receptor-1. J Cardiovasc Pharmacol. (2012) 60:133–9. doi: 10.1097/FJC.0b013e318258f336
25. Wang X, Guo Z, Ding Z, Khaidakov M, Lin J, Xu Z, et al. Endothelin-1 upregulation mediates aging-related cardiac fibrosis. J Mol Cell Cardiol. (2015) 80:101–9. doi: 10.1016/j.yjmcc.2015.01.001
26. Lu L, Guo J, Hua Y, Huang K, Magaye R, Cornell J, et al. Cardiac fibrosis in the ageing heart: contributors and mechanisms. Clin Exp Pharmacol Physiol. (2017) 44:55–63. doi: 10.1111/1440-1681.12753
27. Morin D, Long R, Panel M, Laure L, Taranu A, Gueguen C, et al. Hsp22 overexpression induces myocardial hypertrophy, senescence and reduced life span through enhanced oxidative stress. Free Radic Biol Med. (2019) 137:194–200. doi: 10.1016/j.freeradbiomed.2019.04.035
28. Chen S, Wang Q, Zhu J, Xiao Q, Zhang L. Reactive oxygen species: key regulators in vascular health and diseases. Br J Pharmacol. (2018) 175:1279–92. doi: 10.1111/bph.13828
Keywords: myocardial fibrosis, aging, hypertension, collagens, lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1)
Citation: Li X, Tang X, Liu B, Zhang J, Zhang Y, Lv H, Liu D, Mehta JL and Wang X (2021) LOX-1 Deletion Attenuates Myocardial Fibrosis in the Aged Mice, Particularly Those With Hypertension. Front. Cardiovasc. Med. 8:736215. doi: 10.3389/fcvm.2021.736215
Received: 04 July 2021; Accepted: 14 September 2021;
Published: 12 October 2021.
Edited by:
Jingyan Han, Boston University, United StatesReviewed by:
Henning Morawietz, Technical University of Dresden, GermanyJie Du, Capital Medical University, China
Copyright © 2021 Li, Tang, Liu, Zhang, Zhang, Lv, Liu, Mehta and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianwei Wang, d2FuZ3hpYW53ZWkxMTE2QDEyNi5jb20=