 Ezra B. Ketema
Ezra B. Ketema Gary D. Lopaschuk
Gary D. Lopaschuk
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 02 August 2021
Sec. Cardiovascular Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.723996
This article is part of the Research Topic Metabolic Regulation in Cardiovascular Homeostasis and Disease View all 13 articles
Perturbations in myocardial energy substrate metabolism are key contributors to the pathogenesis of heart diseases. However, the underlying causes of these metabolic alterations remain poorly understood. Recently, post-translational acetylation-mediated modification of metabolic enzymes has emerged as one of the important regulatory mechanisms for these metabolic changes. Nevertheless, despite the growing reports of a large number of acetylated cardiac mitochondrial proteins involved in energy metabolism, the functional consequences of these acetylation changes and how they correlate to metabolic alterations and myocardial dysfunction are not clearly defined. This review summarizes the evidence for a role of cardiac mitochondrial protein acetylation in altering the function of major metabolic enzymes and myocardial energy metabolism in various cardiovascular disease conditions.
Following the discovery of histone acetylation and its regulatory effect on RNA synthesis by Allfrey et al. (1), it has been established that alterations in the level of histone acetylation can modulate gene expressions via chromatin remodeling and epigenetic modifications (2, 3). Indeed, dysregulation of histone acetylation level has been strongly linked with the development and progression of cancer and other human diseases (4, 5). This was further reinforced by the identification of histone acetyltransferases (HAT) and histone deacetylases (HDACs), enzymes that mediate the addition or removal of an acetyl group to and from a lysine residue of histone proteins (6, 7), and the development of several HDAC inhibitors to treat cancer and heart diseases (8–10).
In addition to nuclear histone acetylation, the potential for non-histone protein acetylation of non-nuclear proteins has also recently generated considerable interest. The first acetylation of cytoplasmic proteins was described in microtubules (α-tubulin) in 1987 by Piperno et al. (11). The involvement of acetylation of non-nuclear proteins was further confirmed by the isolation of other acetylated proteins in both the cytosol and mitochondria, as well as the presence of deacetylase enzymes, such as SIRT2 and SIRT3 outside the nucleus (12–15). Since then, a number of acetylases and deacetylases have been identified outside the nucleus (Figure 1) (16). However, it is the advancements in protein acetylome quantitative methods, and the identification of several thousands of cytosolic and mitochondrial acetylated proteins using these techniques, that have recently helped advance our understanding of non-histone acetylation dynamics and its biological implications.
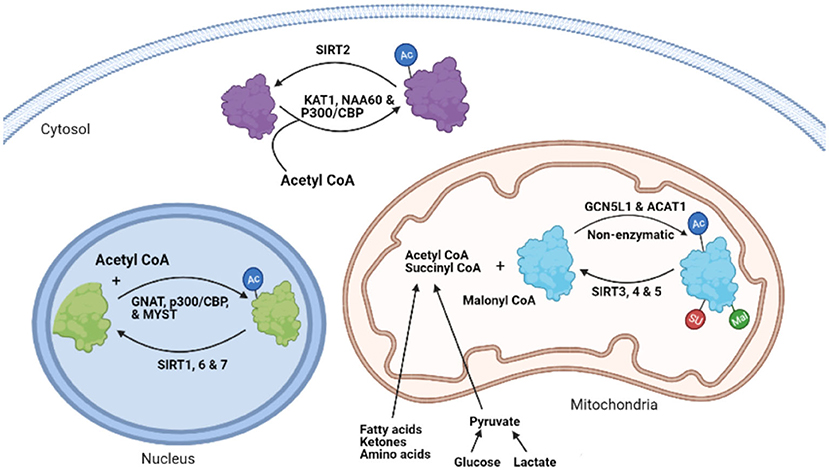
Figure 1. The process of protein lysine acetylation/acylation and deacetylation/deacylation. KAT1, lysine acetyltransferase; NAA60, N-α-acetyltransferase 60; CBP, CREB-binding protein; SIRT, sirtuin, GNAT, GCN5-related N-acetyltransferases; MYST, MYST family acetyltransferase; GCN5L1, General control of amino acid synthesis 5 (GCN5) like-1; ACAT1, acetyl-CoA acetyltransferase; AC, lysine Acetylation; SU, succinylation; MU, malonylation.
Kim et al. reported the first large acetyl proteomic data profile by identifying 388 acetylation sites in 195 proteins in HeLa cells and mouse liver using immunoprecipitation of lysine-acetylated peptides and mass spectrometry analysis (17). Of these, 133 proteins with 277 lysine acetylation sites were from mitochondria, showing for the first time the abundance of the acetylation process in mitochondria. Subsequently, several hundred acetylated proteins were also identified in other studies using either nutritional or acetylase/deacetylase enzymatic manipulations (18, 19). For example, in response to caloric restriction in mice, Schwer et al. reported acetylation of several proteins involved in metabolic pathways in the liver (19). Another study by Zhao et al. revealed the prevalence of acetylation of several metabolic enzymes and its possible regulatory role using both HDAC and non-histone acetylation inhibitors (20). Similarly, Lombard et al. demonstrated extensive mitochondrial protein acetylation in SIRT3 knockout (KO) mice (21), a finding confirmed by Hirschey et al. (22). Strikingly, these studies described the exceptional susceptibility of mitochondrial proteins to acetylation modification in response to various stressors, with up to 60% of mitochondrial proteins reported to being acetylated (23). Of importance, the majority of these acetylated proteins were enzymes catalyzing energy metabolic processes (19–21, 24). Similar to acetylation modifications, several studies have demonstrated increased succinylation and malonylation of a large number of cytosolic and mitochondrial proteins in response to SIRT5 deletion (25–27). For instance, in liver mitochondria isolated from SIRT5 KO mice, Rardin et al. identified 1190 succinylation sites, of which 386 sites on 140 proteins were seen in enzymes involved in energy metabolism, including fatty acid β-oxidation and ketogenesis (26). Despite these studies, that have revealed widespread acyl modifications on proteins in most of the metabolic pathways, its biological significance and regulation are still poorly understood. Understanding these post-translational modifications is particularly important in heart failure, where significant alterations in mitochondrial metabolism are seen, and disturbances in the metabolites used as a substrate for these post-translational modifications occur.
Impairments in myocardial energy substrate metabolism are a major contributor to the pathogenesis of heart failure (28–30). However, the underlying causes of these metabolic perturbations remain unclear. In the failing heart, changes in the transcription of genes encoding metabolic enzymes, particularly fatty acid metabolic enzymes, have been suggested as one of the mechanisms for altered energy substrate metabolism (31–34). Nevertheless, transcriptional regulation alone is not sufficient to explain all the metabolite and enzyme activity changes observed in the failing heart (35, 36). Several post-transcriptional and post-translational processes have been shown to modulate the original transcription products, and thus can significantly alter energy metabolism in the failing heart. Post-translational acetylation-mediated modification of metabolic proteins is thought to have a regulatory role in these energy metabolic changes (37–39).
In addition to acetylation, many other post-translational modifications including phosphorylation, malonylation, succinylation, glutarylation, ubiquitination, SUMOylation, O-GlcNAcylation, N-glycosylation, methylation, citrullination, and S-nitrosylation play important roles in cardiac disease pathogenesis, including metabolic perturbations (40–42). However, the focus of this review will be on the role of acetylation (and some acylation) modifications of energy metabolic enzymes and their contributions to altering cardiac energy metabolism. Although the relevance of protein acetylation changes have also been reported in other pathological processes, including inflammation, oxidative stress, and apoptosis, this review will mainly discuss the connections between acetylation imbalances and cardiac energy metabolic changes.
Lysine acetylation of proteins occurs through the covalent attachment of an acetyl group to the lysine residues of proteins. This acylation modification causes important changes to the protein at its lysine residue, which includes altering its charge status and adding an extra structural moiety (43, 44). These changes impact the proteins' native structure, its interactions with other proteins or regulatory molecules, its stability, and its function (45). Similar to acetylation, lysine succinylation, and malonylation have also emerged as functionally important acyl group modifications. These acyl modifications occur by the addition of malonyl and succinyl groups to the same or different lysine residues modified by acetylation (25, 26, 46). As discussed in the following section, cellular protein acetylation dynamics are regulated by various factors including pathological stressors, substrate availability, and the balances between acylation and deacylation enzymes (47).
While several acyltransferases have been characterized and shown to catalyze histone and other nuclear protein acetylation processes (48, 49), the involvement of these acyltransferases in the transfer of acetyl (acyl) group during cytosolic and mitochondrial protein acetylation (acylation) modifications remains to be clearly defined. A few studies suggest that some of the nuclear acetyltransferases, such as p300/CBP, may shuttle between the nucleus and cytoplasm and participate in the acetylation of cytosolic proteins (44, 50, 51). Type B lysine acetyltransferases (KATs), which include KAT1 and NAA60, are also cytoplasmic enzymes (48). The GNAT family, ATAT1 and general control of amino acid synthesis 5-like 1 (GCN5L1) acetyltransferase, also contribute to mitochondrial protein acetylation changes (Figure 1) (52, 53). However, it has also been suggested that mitochondrial protein acetylation can occur through non-enzymatic modifications (54, 55). Although widespread protein malonylation and succinylation have been described in the mitochondria (25, 56), no specific succinyltransferases or malonytransferases have been identified to date. As a result, some researchers have proposed that non-enzymatic mechanisms may be responsible for such acyl modifications (55), while others suggest that some nuclear acetyltransferases, such as histone acetyltransferase 1 (HAT1), may be involved in nuclear protein lysine succinylation (57).
Deacetylation of cytoplasmic and mitochondrial proteins mainly involves the actions of sirtuin enzymes. Sirtuins are class III NAD+-dependent protein deacetylases, which are considered as orthologs of silent information regulator 2 (SIR2) in yeast (58, 59). SIR2 regulates the transcription of silencing of mating-type loci, telomeres, and ribosomal DNA, thereby prolonging the yeast's lifespan (60, 61). Sirtuins can also regulate mammalian lifespan (62–64). This effect of sirtuins has led to the suggestion that sirtuins are mediators of the favorable effects of calorie restriction on health and aging, including metabolic reprogramming and stress tolerance. In support of this, caloric restriction is also shown to increase the expression of sirtuins (21, 22, 65, 66).
There are seven mammalian sirtuin proteins (SIRT1–SIRT7) with variation in their tissue specificity, subcellular localization, enzymatic activity, and targets (16). SIRT1, 6, and 7 are mainly localized in the nucleus (16, 67, 68), while SIRT2 is predominantly localized in the cytoplasm (12). However, SIRT1 and SIRT2 can shuttle between the nucleus and the cytoplasm and acetylate proteins in both compartments (67, 69). SIRT1 can regulate the acetylation state of diverse cellular proteins in the nucleus (70). In contrast, SIRT 3, 4, and 5 are mainly localized in the mitochondria (13, 16, 71), although some studies have reported cytosolic localization of SIRT5 (25). In terms of their enzymatic activity, SIRT 1–3 possess potent deacetylase activity, regulating protein acetylation status in the respective organelles (21, 23, 72). The other sirtuins, SIRT 4–7, have weak or no detectable deacetylase activity or either have very protein specific deacetylation activity (73) or mediate other deacylation processes (21). Of importance, SIRT5 has potent lysine demalonylation and desuccinylation activity (25, 56, 74). Additionally, SIRT4 and 6 have been shown to possess ADP-ribosyltransferase activity in the mitochondria and nucleus, respectively (71, 75). Also, SIRT7 has been described to have desuccinylase activity on nuclear histones (76). Combined, deacylation by sirtuins regulates diverse processes including, metabolism, gene expression, cell survival, and several other processes in the heart (77). In addition to sirtuins, recent studies have also suggested the participation of non-sirtuin HDACs in the regulation of mitochondrial acetylation dynamics (78, 79). In support of this, HDAC1 and HDAC2 have been detected in the mitochondrial isolates from mouse hearts (79).
Lysine acylation in the heart can be driven and affected by several factors including the altered level and function of acetyltransferases (such as GCN5L1) and deacylation enzymes (sirtuins), the levels of acetyl-CoA and short-chain acyl-CoAs, the levels of NAD+, and the underlying disease state (39, 80, 81). However, it is not yet clear how these individual factors contribute to the degree of mitochondrial protein acetylation/acylation, and whether their contribution varies according to variable conditions. As a result, despite the increased recognition of excessive protein acetylation and acylation in various forms of heart failure, there is a need to better understand the actual mechanism that is responsible for these protein post-translational modifications (PTMs).
Short-chain acyl-CoA species such as acetyl-CoA, malonyl-CoA, and succinyl-CoA are important metabolite intermediates generated during catabolism of various energy fuels. They are also donors of acetyl, malonyl, and succinyl groups for protein lysine acetylation, malonylation, and succinylation, respectively. Thus, the levels and distribution of these short acyl-CoA species can significantly affect cellular PTMs patterns.
Previous studies have suggested that increased acetylation largely arises from the non-enzymatic reaction of high levels of acetyl-CoA generated during a high-fat diet (HFD), obesity, and diabetes (82–86). Myocardial fatty acid ß-oxidation increases with a HFD, diabetes, and obesity, leading to an increase in acetyl-CoA generation (84, 87, 88). Compromised mitochondrial tricarboxylic acid (TCA) cycle activity, such as during ischemia and severe heart failure, can also increase mitochondrial acetyl-CoA levels. The mitochondrial acetyl-CoA production in these conditions may exceed the oxidative capacity of the TCA cycle and therefore increase the mitochondrial acetyl-CoA pool size. As acetyl-CoA is a substrate for acetylation, this excess acetyl-CoA has the potential to drive acetylation of mitochondrial proteins. In agreement with this, Pougovkina et al., using radioactively labeled palmitate, showed that acetyl-CoA generated by fatty acid ß-oxidation in cultured liver cells is sufficient to drive global protein hyperacetylation (89). Similarly, in a recent study, Deng et al. observed a high incorporation of fatty acid-derived 13C isotope onto acetylated peptides in failing mouse hearts. The authors also demonstrated a significant elevation in the levels of protein acetylation in H9c2 cells when incubated with palmitate, suggesting an association between fatty acid ß-oxidation and protein hyperacetylation (90). Wagner and Payne also demonstrated that widespread protein acetylation in the mitochondria may be facilitated by alkaline pH and high concentrations of reactive acyl-CoAs independent of any enzymatic action (55). Although these studies suggest that elevations in acetyl-CoA levels during increased fatty acid utilization enhances protein acetylation events, it has not yet been demonstrated whether an increased acetyl-CoA production from other fuels also contributes to protein acetylation modification in the mitochondria.
Unlike acetyl-CoA, the association between malonyl-CoA and succinyl-CoA levels and corresponding changes in lysine acylation in the heart has not been examined. However, in contrast to acetyl-CoA levels, malonyl-CoA levels are reduced under conditions of increased fatty acid ß-oxidation as a result of increased malonyl-CoA decarboxylase (MCD) enzymatic activity, the enzyme that degrades malonyl-CoA (91, 92). Others have also suggested that malonyl-CoA levels are unchanged during obesity or with a HFD (93, 94). High levels of fatty acids seen in these conditions also increase myocardial MCD expression, contributing to a decrease in malonyl-CoA levels (95). In contrast, increased malonyl-CoA levels in MCD deficient human fibroblast cells resulted in a two-fold increase in the levels malonylation, suggesting that malonyl-CoA levels may impact malonylation status (96). Although succinyl-CoA is one of the most abundant acyl-CoAs in the heart (46), it is not known if succinyl-CoA levels alter succinylation status in the heart. It is known that protein lysine succinylation is increased in mice hearts lacking SIRT5 (46), and that many of these proteins participate in metabolic pathways that include oxidative phosphorylation, fatty acid ß-oxidation, ketogenesis, branched-chain amino acid (BCAAs) catabolism, and the TCA cycle (97).
In contrast to the well-characterized role of multiple acetyltransferases for histone or nuclear protein acetylation, less is known regarding the role of acetyltransferases in cytosolic and mitochondrial lysine acetylation. Enzymatic acetylation of mitochondrial or cytosolic proteins may involve the GNAT family of acetyltransferases, including acetyl-CoA acetyltransferase (ACAT1) (98) and GCN5L1 (52, 53). Studies by Thapa et al. demonstrated a correlation between an excess nutrient (i.e., a HFD), upregulation of GCN5L1 expression, and increased mitochondrial lysine acetylation (53), although we observed no changes in GCN5L1 expression under similar experimental conditions (39). Reduced mitochondrial protein acetylation in GCN5L1 cardiac-specific KO mice subjected to a HFD has also been reported (99). We have also shown an increased expression of GCN5L1 in association with increased lysine acetylation in the newborn heart (100).
As discussed, protein lysine malonylation and succinylation modifications are highly prevalent in enzymes of mitochondrial metabolism and the TCA cycle (25, 56, 74). However, it is still unknown whether these processes are catalyzed by succinyl or malonyl transferases or whether they occur passively. Therefore, it remains unclear how these protein acylation modifications are regulated during pathological conditions.
SIRT3 is a major mitochondrial deacetylase. Studies have shown an association between SIRT3 deletion and mitochondrial protein hyperacetylation, supporting its critical deacetylating role (21, 22). Many key enzymes in fatty acid and carbohydrate metabolism are substrates for SIRT3 deacetylation (72, 101, 102). Downregulation of SIRT3 occurs in response to stressors such as a HFD or various heart diseases (81, 103). Decreased SIRT3 has been also implicated in various cardiac pathologies in association with hyperacetylation of mitochondrial proteins (54, 104). For instance, a change in the expression of SIRT3 isoforms (long and short forms) is seen in mice hearts subjected to different hypertrophic stimuli (105). However, there is a lack of understanding as to how SIRT3 gene expression is affected by altered metabolic (nutrient) state or heart failure. Moreover, most previous studies have focused on the expression levels of SIRT3, as opposed to actual SIRT3 enzymatic activity.
Cardiac SIRT1, a deacetylase enzyme in the nucleus and cytosol, is also downregulated in advanced heart failure (106). Similar findings in other heart failure studies have also been observed, which demonstrated an association between decreased SIRT1 expression and increased oxidative stress (107). Paradoxically, other researchers have shown a correlation between constitutive overexpression of SIRT1 and impaired cardiac function, as well as disturbed cardiac energy metabolism in response to acute pressure overload (108). SIRT1 protein is also negatively regulated by HFD, which induces its cleavage by the inflammation-activated caspase-1 in adipose tissue (109).
Some studies have indicated a high level of SIRT5 expression in normal hearts (16). However, the pattern of changes in SIRT5 levels under stress conditions is not well-characterized. A previous study on mouse primary hepatocytes have suggested upregulation of SIRT5 by peroxisome proliferator-activated receptor coactivator-1α (PGC-1α), and downregulation by AMP-activated protein kinase (AMPK) (110). Unlike SIRT1 and 3, the absence of SIRT5 does not affect the development of HFD-induced metabolic abnormalities and insulin resistance (111).
HDACs are known to modulate histone acetylation status and thus affect its interaction with DNA, which results in chromatin remodeling and transcriptional changes (2). However, recent studies have also implicated a role for HDAC in modifying the mitochondrial acetylome directly in a non-transcriptional manner using various HDAC inhibitors (78, 79, 112). Both hyperacetylation and hypoacetylation of mitochondrial proteins was observed in response to a pan-HDAC inhibitor in a feline model of heart failure (78). However, the effects of these acetylation modifications was not investigated in this study. Moreover, though a positive association has been made between HDAC inhibition and improved cardiac function in relation to acetylation changes, it has not yet been explored how HDAC inhibition affects sirtuin functions or whether the patterns of acetylation regulated by HDAC inhibition is different from those regulated by mitochondrial sirtuins.
NAD+ is an important cofactor for sirtuins, and as such fluctuation in NAD+ levels may be one of the contributing factors for altered protein acetylation levels (58). Through NAD+, sirtuin activity is directly linked to the energy status of the cell. NAD+ is synthesized from different biosynthetic precursors. In the salvage pathway, the major NAD+ generating pathway, nicotinamide riboside (NR) and nicotinamide (NAM) are converted into nicotinamide mononucleotide (NMN) by nicotinamide riboside kinase (NRK) and nicotinamide phosphoribosyltransferase (NAMPT) enzymes, respectively. Nicotinamide mononucleotide adenyltransferases (NMNAT) then converts NMN to NAD+. In the de novo pathway, NAD+ is generated from the amino acid tryptophan, which is ultimately converted into the biosynthetic intermediate, nicotinic acid mononucleotide (NaMN) through multiple enzymatic steps. Nicotinic acid mononucleotide is then converted to nicotinic acid dinucleotide (NaAD+) by NMN/NaMN adenylyltransferases (NMNATs) and then converted to NAD+ by NAD+ synthetase through deamination (113). Intracellular NAD+ levels can also be altered by rates of glycolytic and mitochondrial metabolic pathways using NAD+ to produce NADH, rates of mitochondrial electron transport chain activity that produce NAD+ from NADH, and by enzymes that consume or catabolize NAD+, such as the poly ADP-ribosyltransferases (PARPs) and the NAD+ cyclases (CD38) (113, 114).
Alterations in the NAD+ biosynthetic or degradation pathways may directly affect the activity of sirtuins and thus protein acetylation status (115, 116). Kinetics studies on sirtuins and NAD+ metabolites, have demonstrated the sensitivity of sirtuins to changes in nicotinamide and NAD+ levels, which inhibits and activates its enzymatic activity, respectively (58, 117, 118). While both the NADH and NAD+/NADH ratio have been previously suggested to impact lysine acetylation status (80, 119, 120), recent studies indicated that alterations in NADH have insignificant effect on sirtuin regulation (117, 121). Accordingly, NADH has a very poor binding affinity to sirtuins (117), and sirtuins are insensitive to NADH inhibition at the concentration of NADH found in the cell (121). As a result, changes in NAD+, as opposed to commonly reported changes in the NAD+/NADH ratio, should be used for assessing NAD+ regulation of sirtuin activity (122). Both NADH & NAD+, as well as NAD+/NADH ratio, also significantly varies across cellular compartments and in response to various disease or metabolic states, making it difficult to interpret the implication of NAD+/NADH ratio in controlling sirtuin activity (122). In addition, the NAD+/NADH ratio alone does not also provide specific information on the direction of changes to the individual nucleotides. Thus, measurement of free NAD+ levels is most relevant when it comes to the regulation of sirtuins and perturbations in protein acetylation.
Previous studies have demonstrated changes in the activity of sirtuins and protein acetylation levels by modulating both NAD+ synthetic and catabolic pathways (41, 115, 123). Accordingly, Lee et al. observed a significant decrease in the mitochondrial proteins acetylome in response to NAMPT overexpression or NAD+ supplementation (80). Similarly, other studies have also shown increased NAD+ levels accompanied by enhanced SIRT1 and SIRT3 activities in responses to NR supplementation, which was accompanied by an increase in oxidative metabolism and protection against HFD-induced metabolic abnormalities (123). Supporting this, producing a CD38 deficiency (which is a NAD+ degrading enzyme) protects the heart from HFD-induced oxidative stress by increasing NAD+ availability and activating SIRT3 mediated protein deacetylation (115). NAD+ depletion occurs in many cardiac pathologies, such as during ischemia-reperfusion (I/R) injury, and several therapeutic strategies to increase NAD+ levels have been proposed (124, 125). However, the mechanisms which mediate the favorable effects of increasing NAD+ levels are not completely understood, although emerging data suggests activation of sirtuins and decreasing protein acylation modifications as key effectors (126–129).
Alterations in myocardial energy metabolism, both in terms of changes in energy substrate preference, and decreased mitochondrial oxidative metabolism and ATP production, are key contributors to heart failure development (28, 130–133). Various injury or stress signals, including ischemia, hypertrophy or neurohormonal changes, are thought to mediate these metabolic derangements (130). While these disturbances result in an unbalanced use of glucose and fatty acids and a decreased contractile efficiency during heart failure, it remains controversial whether the shifts occur toward increased glucose use or increased fatty acid use, and whether these shifts are adaptive or pathological (30, 33, 134–139). Our limited understanding of the underlying mechanisms regulating these metabolic changes is a major challenge to better characterizing these alterations for potential therapeutic interventions.
Changes in metabolic gene expressions, predominantly down-regulation of fatty acid transporting and metabolizing proteins, have been described as one of the contributors to the metabolic changes seen in heart failure (31–34, 139). Recently, apart from transcription regulation, several post-transcriptional and post-translational processes have been shown to modulate transcriptional and protein products (41, 42). There is also an inconsistency in these transcriptional changes, within both the metabolic pathways and across species (31). For instance, while downregulation of fatty acid metabolic enzymes expression is observed in various animal models of heart failure, no significant alterations are observed in genes regulating glucose metabolism, or changes are largely inconsistent, in human heart failure samples (31, 35). Even within the fatty acid metabolic genes, transcriptional downregulation has been observed only in a few of them compared to the widespread post-translational modification seen in most of these enzymes (34, 35). In support to this, Sack et al. observed, a mismatch between mRNA levels and activities of some of the fatty acid metabolic enzymes in the failing heart (139), suggesting a role for post-transcriptional and post-translational changes.
Growing evidence suggests that post-translational acetylation modification may play a significant role in altering myocardial metabolism during heart failure by modifying the function and structure of major metabolic proteins (39, 104, 140). Hyperacylation of key metabolic enzymes involved in fatty acid and glucose metabolism has been shown in heart failure as well as response to excess nutrition or sirtuin deletions (81, 83, 97, 141, 142). However, despite the modification of many of these enzymes by acetylation or other acylations, the actual impact of these PTMs on individual pathways and enzymes remains poorly understood. In this section, we summarize recent evidence on the impact of hyperacetylation on selected metabolic enzyme activity in the heart as well as in other organs or cells (Figure 2).
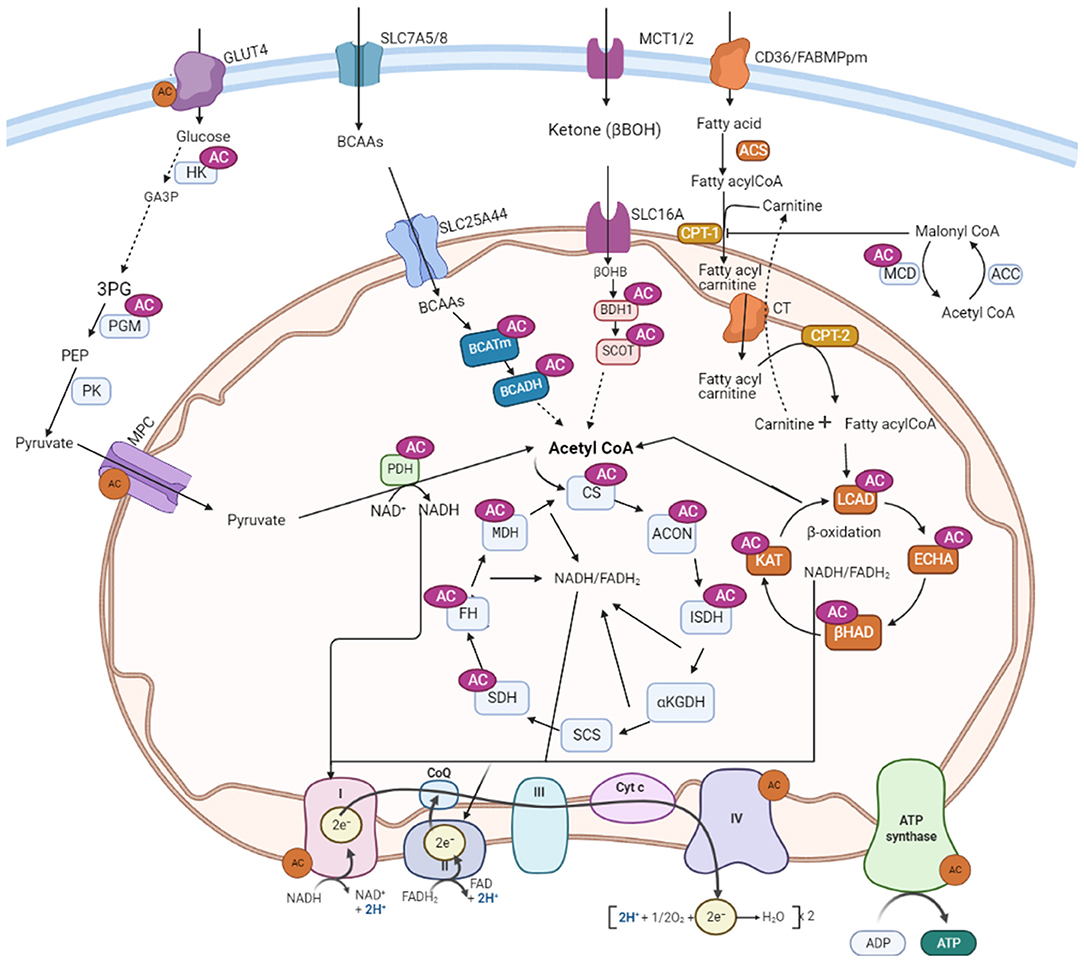
Figure 2. Metabolic proteins subjected to acetylation control in the heart. AC, lysine acetylation; GLUT4, glucose transporter isoform 4; SLC7A5/8, solute carrier family-7; SLC25A44, solute carrier family-25; SLC16, solute carrier family-16; MCT, monocarboxylate transporter 1; CD36, cluster of differentiation 36; FABPpm, plasma membrane fatty acid-binding protein; MCD, malonyl CoA decarboxylase; ACC, acetyl CoA carboxylase; PDH, pyruvate dehydrogenase; LCAD, long-chain acyl CoA dehydrogenase; β-HAD, β-hydroxyacyl CoA dehydrogenase; KAT, 3-ketoacyl-coa thiolase; ECH, enoyl-CoA hydratase; FAS, fatty acyl CoA synthase; CPT-1, carnitine palmitoyltransferase 1; CPT-2, carnitine palmitoyltransferase-2; CT, carnitine acyl translocase; BCAA, branched-chain amino acids; BCATm, mitochondrial branched-chain aminotransferase; BCADH, branched-chain amino acid dehydrogenase; β-OHB, β-hydroxybutyrate; BDH-1, 3-hydroxybutyrate dehydrogenase 1; SCOT, 3-ketoacid CoA transferase; CS, citrate synthase; ISDH, iso-citrate dehydrogenase; α-KGDH, alpha-ketoglutarate dehydrogenase; SCS, succinate CoA synthetase; MDH, malate dehydrogenase; SDH, succinate dehydrogenase; FH, fumarate hydratase; OAA, oxaloacetate; MCD, malonyl CoA decarboxylase; ACON, aconitase; HK, hexose kinase; GA3P, glyceraldehyde 3-phosphate; 3PG: 3-phosphoglycerate; PGM, Phosphoglycerate mutase; PK, pyruvate kinase; CoQ, coenzyme Q; Cytc: cytochrome C; FAD/FADH2, flavin adenine dinucleotide; NAD/NADH2, nicotinamide adenine dinucleotide.
The enzymes catalyzing the cyclic reactions of fatty acid β-oxidation (which converts fatty acid carbons into acetyl-CoA moeities) includes long-chain acyl CoA dehydrogenase (LCAD), enoyl-CoA hydratase, L-3-hydroxy acyl-CoA dehydrogenase (β-HAD), and 3-ketoacyl-CoA thiolase (KAT) (143, 144). Acetylation of these enzymes has been widely reported in various studies (Table 1) (20, 22, 83). The functional consequences of acetylation are relatively well-studied for LCAD and β-HAD. However, most of these studies were conducted in the liver and skeletal muscle, and only a few studies examined the direct impact of acetylation on fatty acid metabolism in the heart.

Table 1. Effect of lysine acetylation on major metabolic enzymes in the heart.
The exact effect of acetylation on fatty acid metabolizing enzymes' activity remains controversial, and there are two opposing views. Using both HFD and SIRT3 KO mice, we demonstrated a positive correlation between increased acetylation of myocardial LCAD and β-HAD and their enzymatic activities as well as increased fatty acid ß-oxidation rates in the heart (83). A similar relationship between acetylation and increased fatty acid ß-oxidation was also seen in newborn rabbits and human hearts. In the early newborn period, a dramatic maturational change in myocardial energy substrate metabolism occurs, accompanied by an increase in fatty acid ß-oxidation. In association with this, we have shown increased acetylation of LCAD and β-HAD, accompanied by an increase in their activities, during the maturation of fatty oxidation in neonatal rabbit hearts (100). The increased acetylation of these enzymes is accompanied by an up-regulation of the acetyltransferase, GCN5L1. In a separate study, we also found a decreased LCAD and βHAD activities and a decrease in fatty acid ß-oxidation rates in hypertrophied newborn human and rabbit hearts in association with decreased acetylation status of these enzymes (154). Similarly, Thapa et al. revealed a positive association between increased acetylation and activities of several cardiac fatty acid ß-oxidation enzymes, including LCAD, β-HAD, and short-chain acyl-CoA dehydrogenase in chronic HFD mice, along with GCN5L1 upregulation (53). Furthermore, decreasing acetylation by GCN5L1 knockdown leads to diminished fatty acid ß-oxidation in cultured H9C2 cells, supporting the idea that lysine acetylation promotes fatty acid ß-oxidation in the heart (53).
Additional evidence for a positive correlation between increased acetylation and increased activities of fatty acid ß-oxidation enzymes have been reported from studies in diabetic animals. In streptozotocin-induced type 2 diabetic rat hearts, Vazquez et al. found a significant increase in mitochondrial lysine acetylation compared to the controls (143). Increased activities of medium- and long-chain acyl-CoA dehydrogenases (MCAD, LCAD) and fatty acid ß-oxidation rates were observed in the hearts of diabetic animals. Analysis of substrate preference in these hearts also revealed an increase in state 3 respiration using palmitoylcarnitine as a substrate (146). Similarly, in type 1 diabetic mice, a 2.5-fold increase in total acetylation levels compared to age-matched controls was observed in the heart. In this study, the maximal rate of respiration remained unchanged only when palmitoylcarnitine or a fatty acid-based substrate was used (85). Furthermore, data from the two most recent studies also indicated that fatty acid utilization in the heart is either unaffected or proceed in harmony with increased acetylation state (90, 155).
Activation of fatty acid ß-oxidation by acetylation is also reported in other tissues/cells. A direct relationship between hyperacetylation and activity of enoyl-coenzyme A hydratase/3-hydroxy acyl-coenzyme A was demonstrated in the presence of high fat and deacetylase inhibitors in cultured Chang human liver cells (20). Other investigators have also found a link between HDAC3 mediated deacetylation of 3-hydroxy acyl-coenzyme A and its decreased activity in macrophages, while HDAC3 depletion reversed this effect (112). Moreover, in SIRT3 KO mice and SIRT3 deficient skeletal muscle cells, Jing et al. showed an increase in palmitate oxidation rates in the presence of excessive acetylation (155). In the presence of high palmitate, oxygen consumption rates are significantly higher in SIRT3 lacking myoblasts, which is lost in the presence of etomoxir, a fatty acid ß-oxidation inhibitor (156). Together, these studies demonstrate that increased acetylation of myocardial fatty acid ß-oxidation enzymes is associated with their enhanced activities. This is further supported by the fact that both myocardial fatty acid utilization and mitochondrial acetylation are enhanced during a HFD, obesity, and diabetes (157). The high fatty acid ß-oxidation rate seen in these conditions may also lead to the increased production of acetyl-CoA that can serve as a substrate for acetylation (82). Thus, it is reasonable to assume that increased acetylation of fatty acid oxidative enzymes can further trigger the enzyme activity and led to the continuation of fatty acid ß-oxidation in the heart in these circumstances.
In contrast to the scenarios discussed above, a study by Koentges et al., in isolated working hearts, found a negative correlation between hyperacetylation of LCAD and its activity along with reduced palmitate oxidation in SIRT3 deficient transverse aortic constriction (TAC) mice (158). However, these hearts were perfused with a buffer that contained an ultra-physiological high concentration of glucose (11 mM) and a lower fatty acid to albumin ratio (1.5%) where both conditions may contribute to decreased fatty acid ß-oxidation rates. Likewise, Chen et al. also reported an abnormal lipid accumulation and decreased palmitate ß-oxidation rates in TAC-induced hypertrophic hearts, with a further decline in SIRT3 KO mice hearts in association with hyperacetylation of LCAD (145). A recent study by Davidson et al. showed reduced expression of genes of fatty acid catabolism despite no overt abnormalities in mitochondrial respiration in mice deficient for cardiac carnitine acetyltransferase and SIRT3, despite the fact that the hearts exhibited extreme acetylation (159). In common, these three studies were done in mice with TAC-induced heart failure, while the last two did not assess directly the acetylation status of the enzymes. Overall, it is not clear if TAC alters the dynamics of acetylation on LCAD differently, such as through distinctive sites or multiple site modifications. However, several acyl modifications may likely coexist under these circumstances, which possibly compete with acetylation for the same lysine residue (104). However, experimental data are lacking regarding these interactions in heart failure.
Unlike the heart, studies conducted in the liver reported an inhibitory effect of acetylation on fatty acid metabolism. Hirschey et al. described a decreased activity of LCAD enzyme and reduced fatty acid ß-oxidation following hyperacetylation of these enzymes in SIRT3 KO mice. The authors further reported an accumulation of long-chain acylcarnitine species, fatty acid ß-oxidation intermediate products, and triacylglycerol in livers from SIRT3 KO mice that could suggest a decreased rate of fatty acid ß-oxidation. From the eight acetylated lysine residues detected on LCAD, lysine residue 42 was identified as a critical regulation site for acetylation (22). Furthermore, analysis of the rate of conversion of radiolabeled palmitate revealed lowered oxidizing capacity and ATP production in tissue homogenates from SIRT3–/– compared to wild-type tissue at a high substrate concentration. Reduced activities of fatty acid ß-oxidation enzymes in the liver by acetylation were also reported in other studies (160). In addition to LCAD and β-HAD, hyperacetylation of hydroxy acyl-CoA dehydrogenase, another important enzyme in fatty acid ß-oxidation, led to its decreased activity and decline in overall fatty acid oxidation rate in the mouse liver. Deletion of GCN5L1 acetylase enzyme or overexpression of SIRT3 reduced hydroxy acyl-CoA dehydrogenase acetylation and increased its activity as well as fatty acid ß-oxidation in the liver (161, 162). However, there is presently no agreement on the functionally significant acetylation sites or SIRT3 target sites in these studies. While Hirschey et al. noted lysine 42 residue on LCAD as an important regulation site (22), using chemically acetylated recombinant proteins, Bharathi et al. identified Lys-318 and Lys-322 residues as an important site for LCAD acetylation/sirt3 deacetylation (160). To date, a detailed analysis of lysine residue modification and functional acetylation/deacetylation target sites for LCAD and β-HAD is lacking in the heart. Not all lysine residues within LCAD that are acetylated are expected to impact LCAD activity in the same manner. Understanding the acetylation status of different acetylation sites, and their effect on the enzyme function in multiple tissues will help to characterize the tissues specific effects of acetylation dynamics.
Compared to acetylation, the effect of succinylation and malonylation modification on fatty acid ß-oxidation enzymes is not clear. Some studies have shown an impaired β-oxidation and accumulation of medium- and long-chain acylcarnitines in the liver and muscles of SIRT5 KO mice (26). Most of the acyl-CoA dehydrogenase enzymes, including very long-chain acyl-CoA dehydrogenase (VLCAD), LCAD, and MCAD, were found to be hypersuccinylated, suggesting a suppressive effect of excessive succinylation on fatty acid oxidizing enzymes (26). In contrast, cardiac ECH is desuccinylated and activated by SIRT5 (46, 97). But the effects of similar modifications on other enzymes in this pathway have yet to be determined.
Overall, from all these data it is possible to suggest that the effect of lysine acetylation on fatty acid ß-oxidative enzymes may not be similar between different tissues, at least between the liver and heart. Reasonably, these differences may account for the specialization of these tissues in regulating fatty acid ß-oxidation differently in line with their physiological functions. The liver has a high capacity for both synthesizing and oxidizing fatty acids and the two processes are regulated reciprocally during a fed or fasting state as well in disease conditions such as in obesity or diabetes. On the contrary, the heart continually uses fatty acids as a source of energy, which accounts for up 60–90% of the total energy requirements for its normal contractile function irrespective of the fed state or disease conditions. While acetylation may serve as a feedback regulation in the liver as suggested by Bharathi et al. (160), the same mechanism would potentially compromise the heart's ability to produce energy if fatty acid ß-oxidation enzymes were down-regulated by acetylation induced by excess fatty acid ß-oxidation. Thus, future studies are needed to determine the differences in acetylome between the two tissues, and if acetylation-mediated regulation of fatty acid ß-oxidation is tissue- or site-specific.
In contrast to fatty acid metabolism, the effects of acetylation on glucose metabolism have received less attention, especially in the heart. However, recent studies reported the acetylation of several proteins involved in glucose transport, glycolysis, and glucose oxidation (83, 149, 156). The pyruvate dehydrogenase complex (PDH) is one of the most widely investigated glucose metabolizing enzymes in relation to acetylation. It is a key enzyme that catalyzes the irreversible and rate-limiting step in glucose oxidation that links glycolysis to the TCA cycle. PDH is regulated by several mechanisms, but the change in its phosphorylation status is critical to its activity. It is inhibited by phosphorylation on the E1 subunit by a specific PDH kinase (PDK), PDK4, and is activated when dephosphorylated by PDH phosphatase (163, 164).
Modification of PDH by acetylation has been demonstrated in several studies (53, 83, 156). In HFD induced obese mice subjected to a TAC, we showed a significant increase in PDH acetylation with a decrease in glucose oxidation rates (147). Similarly, Thapa et al. reported increased acetylation of the α-subunit of PDH in the heart after HFD feeding, and its hyperacetylation was shown to inhibit its activity (53). Reduced activity of PDH in association with its increased acetylation and decreased SIRT3 level has been also demonstrated in mice with angiotensin II-induced cardiac hypertrophy (165).
In addition, increased acetylation has also been implicated in a reduced transport of pyruvate into the mitochondria. Akita type 1 diabetic mice hearts exhibit a significant hyperacetylation state, along with a 70% decrease in the rate of mitochondrial pyruvate transport that occurs without any changes in the protein level of the mitochondrial pyruvate carriers 1 and 2 (MPC1 and MPC2). Mass spectrometry analysis revealed that acetylation of lysines 19 and 26 of MPC2 were increased in Akita mice heart mitochondria, and that acetylation at these sites is associated with impaired pyruvate metabolism in the heart (148).
The impact of acetylation on PDH has also been examined in skeletal muscle by Jing et al. Deletion of SIRT3, both in vivo in SIRT KO mice and in vitro in myoblasts, lead to a significant increase in acetylation associated with decreased PDH activity along with a reduced glucose oxidation rate and accumulation of pyruvate and lactate metabolites. Six acetylation sites have been identified on the PDH E1α subunit, with lysine 336 being significantly altered by SIRT3 deletion (156). Interestingly, it was also shown that hyperacetylation of the PDH E1α at lysine 336 enhances its phosphorylation leading to suppressed PDH enzymatic activity. Additionally, Ozden et al. and Fan et al. also demonstrated the inhibitory effect of acetylation at lysine 321 on PDH activity in cancer cells, which is completely reversed by SIRT3 activation (97, 164). In the latter study, it was also shown that lysine acetylation at lysine 202 inhibits PDP1 by dissociating it from PDHA1, thus promoting its phosphorylation (97).
Hypersuccinylation of PDH accompanied by a decrease in SIRT5 expression is also associated with a decrease in PDH activity as the heart maturates in postnatal rabbits (100). Unexpectedly, Park et al. observed the suppressive effect of SIRT5 catalyzed desuccinylation on PDH in SIRT5 KO mouse embryonic fibroblasts (MEFs), while SIRT5 deletion led to an increase in PDH activity (25). In contrast, Zhang et al. found significantly reduced malate/pyruvate-driven respiration in SIRT5-deficient HEK293 cells, as well as in homogenates prepared from SIRT5 KO livers (166). While these discrepancies need further investigation, tissue/cell-specific variation in these modifications may contribute to these differences.
Acetylation of glycolytic enzymes has been reported in hearts as well as various other cells. In newborn rabbit hearts, we showed a significant decline in glycolysis rates in line with hyperacetylation of its enzymes, including hexokinase (HK-1) and phosphoglycerate mutase (PGM) (100). In contrast, Hallows et al. have shown a negative regulation of PGM by SIRT1 (165). Acetylated PGM displayed enhanced activity, while Sirt1-mediated deacetylation reduced its activity in human embryonic kidney (HEK293) cells (167). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is another glycolytic enzyme subjected to acetylation modification. Similar to PGM, acetylation of GAPDH at lysine 254 increases its enzymatic activity in response to glucose in HEK293T cells (168). In contrast, GAPDH acetylation enhances its translocation from the cytoplasm to the nucleus in NIH3T3 cells, thereby inhibiting downstream glycolysis and accumulation of glycolytic intermediates (169). In another study, Xiong et al. showed an inhibitory effect of acetylation of pyruvate kinase (PK), which catalyzes the last step of glycolysis (170). In addition to acetylation, other acyl modifications can regulate glycolysis. GAPDH, PGK, and enolase are hypermalonylated in livers of db/db mice (142), although the effects of this hypermalonylation on the activities of these enzymes has not been investigated. Another study in hepatocytes demonstrated SIRT5 mediated demalonylation of GAPDH and increased activity, suggesting that malonylation decreases glycolytic flux (27). Similarly, desuccinylation of PK by SIRT5 increases its kinase activity (171). In contrast, Xiangyun et al. reported that desuccinylation of PKM2 by SIRT5 inhibits its activity in tumor cells (172). Unfortunately, there is a lack of data on the effect of malonylation and succinylation modifications on glycolysis in the heart.
At the transcriptional level, glycolysis is regulated by the level of hypoxia-inducible factor-1α (HIF-1α), a master transcriptional regulator of glycolytic enzymes (173). Studies by Geng et al. have shown a positive correlation between increased acetylation of HIF-1α by p300 at lysine 709 and its stability, or decreased polyubiquitination, in HEK293 cells (174). Similarly, other studies also showed an inhibition of HIF-1α by SIRT1 mediated deacetylation at lysine 674, in HT1080 and HEK293 cells (175). These results were also found in SIRT6 deficient embryonic stem cells and MEFs cells (174). Interestingly, these cells exhibit increased glucose uptake with up-regulation of glycolysis in association with increased HIF-1α activity.
The impact of acetylation on glucose uptake and its transporters has also been described. In both cultured cardiomyocytes and perfused hearts, Renguet et al. found that acetylation of GLUT 4 inhibits glucose uptake in adult cardiomyocytes, as well as in perfused hearts, by decreasing its translocation to the plasma membrane (173). Strikingly, treatment with inhibitors of acetyltransferases prevents the increase in protein acetylation and reverses the inhibition of glucose uptake and GLUT4 translocation (149). Unfortunately, the direct acetylation status of GLUT4 was not analyzed in this study. However, the inhibitory effect of acetylation on glucose uptake is supported by other studies. Using SIRT3 KO mice and hyperinsulinemic-euglycemic clamp experiments, Lantier et al. showed that increased acetylation leads to insulin resistance and reduced muscle glucose uptake that is associated with decreased hexokinase II (HKII) binding to the mitochondria in HFD-fed SIRT3 KO mice (176). This suggests a reduced HKII activity and translocation as a result of increased acetylation. Similar to the above study, unfortunately, there was no direct analysis of the acetylation status of the proteins involved in glucose uptake or glucose phosphorylation.
Insulin resistance in type 2 diabetes and obesity as well as in other heart conditions contributes to a number of adverse changes in the heart that includes alterations in cardiac energy metabolism, lipotoxicity, and hypertrophy, and is associated with an increased risk of heart failure (84, 177, 178). Cardiac insulin signaling is impaired in heart failure, diabetes, and obesity (147, 178, 179). Recent studies have shown that several proteins in the insulin signaling pathway are targets for acetylation modification, which therefore may impact insulin signaling. Akt is an important component of the insulin signaling pathway. Akt activation requires binding with phosphatidylinositol 3,4,5-trisphosphate [PIP (3)], which promotes its membrane localization and phosphorylation by the upstream kinase, phosphoinositide-dependent protein kinase 1 (PDK1). Previously, we have shown a negative association between acetylation of insulin signaling mediators, such as Akt and PDK1, and their decreased activation as a result of changes in their phosphorylation status due to acetylation (147). In support of this, Sundaresan et al. showed that acetylation of Akt and PDK1 occurs in their pleckstrin homology (PH) domains, which blocks PIP (3) binding, and that this is reversed by SIRT1 deacetylation (150). SIRT2 also binds and activates Akt in insulin-responsive cells, through its interaction with the PH domain, whereas SIRT2 inhibition impairs AKT activation by insulin (180).
Ketone body and BCAA oxidation can impact cardiac energy metabolism and heart failure progression (179, 181, 182). Both pathways may also contribute to mitochondrial acetylation changes. However, only a few studies have characterized the acetylation status and its impact on enzymes involved in ketone and BCCA metabolism.
In hepatic mitochondria of SIRT3 KO mice, hydroxymethyl-glutaryl (HMG)-CoA synthase (HMGCS2), the rate-limiting enzyme in ketogenesis, is hyperacetylated and its enzymatic activity reduced, leading to a decrease in β-hydroxybutyrate synthesis. Deacetylation of HMGCS2 by SIRT3 increases its enzymatic activity and β-hydroxybutyrate levels (183). Similar to the acetylation effect, loss of SIRT5 results in hypersuccinylation of HMGCS2 and reduces its activity both in vivo and in vitro in liver mitochondria, which leads to reduced β-hydroxybutyrate levels during fasting (26).
With regard to ketone oxidation, succinyl-CoA:3-ketoacid-CoA transferase (SCOT), a key enzyme of ketone oxidation, is hyperacetylated in the brain and heart at multiple sites in SIRT3 KO mice (184). In vitro biochemical analysis of recombinant SCOT demonstrates that acetylation at lysine 451 residues results in decreased enzyme activity that is reversed by SIRT3 activation. Moreover, in brain homogenates from WT and SIRT3 KO mice, acetoacetate-dependent acetyl-CoA production is decreased by three-fold in SIRT3 KO mice, suggesting decreased ketone oxidation rates upon increased acetylation (180). In contrast, a decrease in ketogenesis capacity was noted in the liver of mice lacking SIRT3 (184). However, similar studies are lacking in the heart.
Similarly, enzymes in BCAA (isoleucine, leucine, and valine) catabolic pathways are among the proteins regulated by acetylation and SIRT3 in the liver. Some acetylation sites were detected in branched-chain alpha-keto acid dehydrogenase (BCKDH), a key enzyme catalyzing the breakdown of BCAAs, in SIRT3 KO mice (181, 182). As BCAA levels were raised, the authors suggested that acetylation may have an inhibitory effect on branched-chain ketoacid dehydrogenase (BCKDH) activity (23, 185). Other investigators have also suggested that acetylation of BCAA aminotransferase (BCAT) promotes its degradation in the ubiquitin-proteasome pathway, thereby decreasing BCAA catabolism in the pancreas (186). cAMP-responsive element-binding (CREB)-binding protein (CBP) and SIRT4 were identified as the acetyltransferase and deacetylase for BCAT at lysine 44 (K44), respectively (186).
Acetyl-CoA is the final common product in the oxidative metabolism of various fuels, and is a substrate for the TCA cycle. While acetylation of all TCA cycle enzymes have been reported in the liver (20), 6 of the 8 enzymes were found to be acetylated in the heart (151). However, examination of the effect of acetylation on the TCA cycle has produced mixed results. Increased acetylation of malate dehydrogenase (MDH) in Chang liver cells enhances its enzyme activity. When cells were treated with deacetylase inhibitors, trichostatin A (TSA) and NAM, MDH acetylation doubled the endogenous MDH activity, while in vitro deacetylation of purified MDH decreased its activity (20). Similarly, significant acetylation-dependent activation of aconitase was found in both isolated heart mitochondria subjected to in vitro chemical acetylation, and in hearts of HFD fed obese mice (152). Increased aconitase acetylation at multiple sites were found, with acetylation at K144 identified as a responsible site for structural change at the active site that was reversed by increasing SIRT3 overexpression (152).
Although acetylation at multiple sites was detected on the TCA cycle enzyme isocitrate dehydrogenase (IDH), no significant effect of this acetylation on enzyme activity was found (151). In contrast, others have reported a significant loss of function of IDH when acetylated at lysine 413, which is fully restored by SIRT3 mediated deacetylation (187). Additionally, increased acetylation of succinate dehydrogenase (SDH), which functions both in the TCA cycle and electron transport chain, is associated with a decrease in activity in human and mice heart failure (81). Further investigation on its acetylation sites revealed that lysine 179 of SDH as an important site for acetylation that regulates enzyme activity by interfering in FAD+ binding to the enzyme. However, despite widespread acetylation of its enzymes, the overall TCA cycle activity appears to be unaffected by excessive acetylation (83).
Succinylation and malonylation of TCA cycle enzymes have also been reported (25, 26, 142). However, the impact of this succinylation and malonylaton has been assessed on only a few enzymes. SIRT5 mediated desuccinylation activates IDH (188). Paradoxically, SDH desuccinylation has been suggested to inhibit its activity in MEFs, while SIRT5 deletion leads to an increase in SDH activity (25).
Lysine acetylation of mitochondrial respiratory complex enzymes, NADH dehydrogenase 1, ubiquinol cytochrome c reductase core protein 1, and ATP synthase mitochondrial F1 complex assembly factor 1 is increased in mice hearts that lack SIRT3. In these hearts, functional studies demonstrated inhibition of Complex I activity (24). In another study, a similar effect was observed in neonatal rat cardiomyocytes, as well as in H9c2 cells treated with high glucose, oleate, and palmitate (153). Treatment with HDACs and sirtuin inhibitors, TSA and NAM, respectively, further increase the levels of acetylated proteins in mitochondrial complexes I, III, and V, with a concomitant decrease in ATP production. However, treatment with exogenous H2S elevates the NAD+/NADH ratio and the activity of SIRT3, both of which are decreased in the presence of high glucose and fatty acid as well as in diabetes (153). Similarly, a decrease in the activity of complex V in association with increased acetylation was shown by Kerner et al. (151).
Acetylation can also affect transcription factors that regulate energy metabolism. Peroxisome proliferators-activated receptors (PPARs) are a family of nuclear receptors that have a critical role in regulating the expression of proteins involved in fatty acid metabolism. The PPAR transcription factors, comprised of PPARα, PPARδ, and PPARγ, form a complex with retinoid X receptor (RXR) and bind the peroxisome proliferator response element (PPRE) in the promoter region of target genes, thereby initiating their transcription (189, 190). Similarly, estrogen receptor-related receptors (ERRs) including ERR α, β, and γ affect the expression of enzymes in the glucose and fatty acid metabolic pathways after binding with ERR responsive elements (191). Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) is an inducible cofactor of both PPARs and ERRs (192). Activation of PGC-1α together with PPAR and ERR promotes fatty acid utilization while suppressing glucose metabolism. All of these transcription factors have been shown to be subject to acetylation/acylation.
SIRT1 interaction with PPARα positively affects PPARα activity. While SIRT1 deficiency impairs PPARα signaling and decreases fatty acid ß-oxidation, while its overexpression upregulates PPARα targets (193). On the contrary, SIRT4 decreases PPARα activity and consequently the expression of PPARα target genes (194). Acetylation of PPARγ at different lysine residues has been shown in various tissues (195, 196). Repression of PPARγ by SIRT1 is seen in 3T3-L1 adipocytes (197). Activation of PPARγ has been seen with acetylation and its suppression by histone deacetylase 3 (HDAC3) (198). In contrast, PGC-1α is deacetylated and activated by SIRT1 (199–201). ERRα is another transcription regulator that is modified by acetylation with suppressed sensitivity after acetylation (202).
Altered energy metabolism may also influence the expression pattern of metabolic genes through chromatin modification by post-translational histone acetylation. Accordingly, increased glucose supply leads to increased histone acetylation with a corresponding activation of glucose metabolism genes in vitro (203). Similarly, upregulation of several lipid metabolism-related genes is observed in response to fatty acid-derived acetyl-CoA-induced histone hyperacetylation (204). While these data support substrate-dependent induction of specific metabolic genes, it unclear how cells respond differently to the acetyl-CoA derived from either glucose and fatty acid ß-oxidation. Additionally, the contribution of histone acetylation modifications as a result of metabolite imbalances has not been determined in the transcription dysregulation seen in pathological states.
Studies in both animal models and humans have shown increases in acetylation of mitochondrial proteins in the failing heart compared to healthy control hearts (Figure 3) (80, 81). Davidson et al. compared the acetyl proteomics profile between dual KO mice for SIRT3 and carnitine acetyltransferase (which causes extreme mitochondrial acetylation) and TAC induced heart failure mice (159). These authors found an approximately 86% overlap in acetylated peptides between the double KO mice and the experimental heart failure mice. Furthermore, in rat models of hypertensive heart failure, the Dahl salt-sensitive (SS) and spontaneously hypertensive heart failure-prone (SHHF) rats, a large number of proteins were found exclusively hyperacetylated in the failing hearts compared to control hearts (140). Increased acetylation was accompanied by a reduced level of SIRT3 in these pressure-overload-induced failing hearts (140). Similarly, in failing hearts of obese patients, Castillo et al. found a 46% decline in SIRT3 expression and increased acetylation profiles in heart failure patients with a BMI >30, as well as in obese rat hearts (205). Increased cardiac acetylation was also observed in obesity-related left ventricular remodeling and cardiac fibrosis (206). A dramatic increase in protein lysine acetylation was also seen in the heart and mitochondria from diabetic mice, along with decreased deacetylation reactions (146). Several other studies have also revealed the abundance of hyperacetylation of different proteins in various forms of heart failure (207–209). Although these data have consistently shown the increased protein acetylation in heart failure settings, the specific impact, regulations, and the mechanisms of how these changes are linked to heart failure remain incompletely understood. Moreover, the mass spectrometry-based acetylome proteomics used in these studies have inherent limitations to measure acetylation changes at the protein level and individual acetylation sites within each protein, which would be necessary to understand the biological significance of acetylation (210–213).
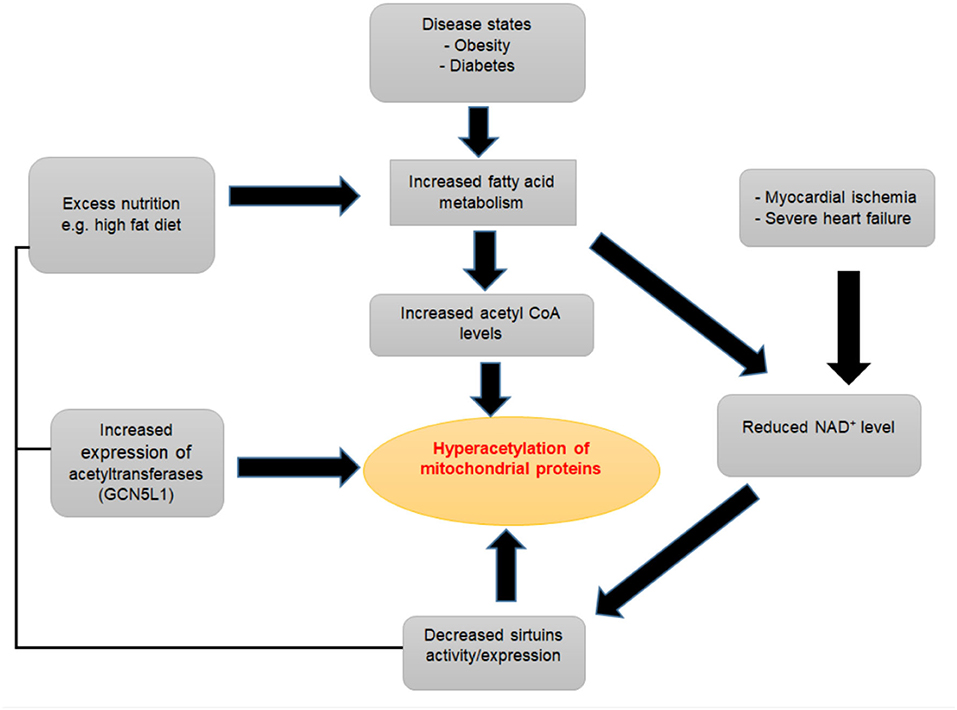
Figure 3. Contributing factors to mitochondrial protein hyperacetylation.
Numerous studies have demonstrated altered substrate preferences and metabolism in the failing heart [see (28) for review]. However, the actual direction of these shifts in energy substrate utilization remains controversial. While the shift toward increased oxidation of fatty acid is widely observed in ischemic and diabetic heart failure, other studies have suggested a shift in myocardial metabolism away from fatty ß-oxidation and its association with the progression to ventricular dysfunction in hypertrophic hearts (214). According to the principles outlined by Randle (215), glucose and fatty acid metabolism are regulated reciprocally. As highlighted in the preceding sections, lysine acetylation affects the main enzymes of both fatty acid and glucose oxidation inversely in the heart. In fact, various animal studies, as described below, have demonstrated that acetylation may be sufficient to cause shifts in myocardial substrate preference in the presence of pathological stressors.
In type 1 diabetic mice hearts, increased acetylation induces mitochondria metabolic inflexibility accompanied by decreased activities of PDH and complex II enzyme activities (85). A dramatic decrease in mitochondrial respiration in the presence of non-fatty acid substrates was observed in contrast to minimal inhibition in palmitoylcarnitine-supported respiration (85). Similarly, other studies have shown a switching in cardiac energy metabolic substrate preference toward fatty utilization by lysine hyperacetylation in type 2 diabetes mice (216). In db/db mice, and in cardiomyocytes in culture, increased acetylation of enzymes involved in mitochondrial fatty acid ß-oxidation and glucose oxidation were found, with a concomitant decrease in the expression and activity of SIRT3. While LCAD acetylation is accompanied by a significant upregulation in its activity, the hyperacetylation of PDH is associated with a decrease in its activity. In support of this, decreasing acetylation by increasing the expression and activities of SIRT3 (through exogenous hydrogen sulfide administration) switches cardiac substrate utilization from fatty acid ß-oxidation to glucose oxidation in diabetic mice hearts (216).
Additionally, Romanick et al. observed marked changes in lysine acetylation in cardiac tissues with obesity (206). Of those significantly impacted by increased acetylation due to obesity were very long-chain specific acyl-CoA dehydrogenase, aconitate hydratase 2, and dihydrolipoyl dehydrogenase. Interestingly, increased transcriptional activation of KLF15 and PPARα, with increased expression of downstream target genes and their interaction with these significantly acetylated proteins, was observed in diet-induced obesity. In addition, the authors of this study found enhanced expression of PDK4 and MCD in the heart (206). PDK4 is known to inhibit glucose oxidation by inhibiting pyruvate dehydrogenase, while malonyl CoA decarboxylase promotes fatty acid ß-oxidation via activation of carnitine palmitoyltransferase 1 (CPT1). Together this suggests that shifting toward fatty acid ß-oxidation, at the expense of glucose oxidation, occurs in the presence of hyperacetylation (206). We and others have also observed that hyperacetylation on PDH, LCAD, and β-HAD, accompanied by a decrease in glucose oxidation and an increase in fatty acid oxidation, is seen in the heart in response to HFD feeding (28, 53). Likewise, as discussed in the preceding sections, in skeletal muscle a switch in substrate utilization from glucose oxidation toward fatty acid utilization is also induced by SIRT3 KO, as a result of PDH inhibition by hyperacetylation of its E1α subunit (156). Thus, acetylation could contribute to the metabolic inflexibility seen in heart failure by regulating metabolic enzyme activities differently.
Myocardial I/R is another pathology where shifts in energy substrate utilization are implicated in heart injury. Increased fatty acid ß-oxidation rates following ischemia result in suppression of glucose oxidation and a subsequent uncoupling of glucose oxidation from glycolysis, which contributes to ischemic damage (217, 218). As discussed in the preceding section, the increased acetylation of proteins following enhanced fatty acid ß-oxidation and increased acetyl-CoA generation may contribute to the metabolic phenotype observed in I/R. In addition, as NAD+ availability is a critical determinant for Sirtuin activity, ischemia-induced decreases in the NAD+/NADH redox couple during I/R may inactivate SIRT3 and lead to the hyperacetylation of mitochondrial proteins (80). Furthermore, the mRNA and protein levels of NAMPT, the rate-limiting enzyme that converts NAM to NMN in the NAD+ salvage synthesis pathway is downregulated in the heart during I/R injury, further reducing NAD+ levels and thus Sirtuin activity (219). Several studies have demonstrated the cardioprotective role of NAD+ in I/R injury either by exogenous NAD+ supplementation or enzymatic manipulation (80, 125, 219, 220). However, this mechanism has not yet been investigated in relation to changes in metabolic enzymes activity or metabolic alterations as a result of acetylation suppression by NAD+ boosting. There is little data that support the idea that this effect may be related to increased glucose oxidation rates. Accordingly, in rats subjected to I/R after prolonged caloric restriction, Shinmura et al. showed a decreased level of acetylated mitochondrial proteins associated with enhanced Sirtuin activity and attenuated myocardial oxidative damage. Interestingly, caloric restriction increases respiratory control index and oxygen consumption in the presence of pyruvate/malate substrates in mitochondria isolated from I/R hearts (221). However, the hearts in this study were reperfused for only 3–5 min after ischemia and the proteomic analysis was not robust enough to analyze the acetylome changes in metabolic enzymes.
As discussed above, hyperacetylation of myocardial proteins is common in heart failure. In addition to its regulatory role in myocardial energy metabolism, several studies have also analyzed the impact of hyperacetylation of myocardial proteins on heart failure development and progression. While the majority of the studies suggested a link between hyperacetylation and worsening of heart failure (80, 81, 105, 146, 206, 222, 223), others found no association between myocardial dysfunction and hyperacetylation (159, 224). The cause for the discrepancies in these studies awaits further studies.
Hypertrophy is one of the pathological processes where increased acetylation is implicated in disease progression. In response to various hypertrophic stimuli, SIRT3-deficient mice appear to be more sensitive to injuries and manifested various abnormalities compared to their wild-type counterparts (105). On the other hand, SIRT3 transgenic mice are protected from hypertrophic injuries (105). Activation of forkhead box O3a-dependent (Foxo3a) and manganese superoxide dismutase and catalase are also seen in response to SIRT3 activation. Activation of SIRT3 has been also implicated in lessening the severity of cardiac hypertrophy by blocking interstitial fibrosis, as well as fibroblast proliferation and differentiation (222). Furthermore, in SIRT5 KO mice subjected to TAC, Herschberger et al. observed a reduced survival of SIRT5 KO mice compared with wild-type mice (225). The increased pathological hypertrophy and mortality in these mice is associated with several biochemical abnormalities including reduced fatty acid ß-oxidation and glucose oxidation, suggesting that SIRT5-mediated desuccinylation plays an important role in regulating cardiac metabolism during stress.
Changes in protein acetylation have been also shown in myocardial I/R injury in association with decreased SIRT3 protein levels (158, 223, 226). In support of this, increased mitochondrial protein acetylation in SIRT3 KO mice is associated with increased sensitivity to injury, as shown by larger infarct size, less functional recovery, and low O2 consumption rates (223). In contrast to these findings, the study by Koentges et al. found no additional susceptibility to I/R-specific injury in SIRT3 KO mice that underwent permanent ligation of the left anterior descending coronary artery (LAD) (125). On the other hand, Boylston et al. found an increase in infarct size and impaired recovery during I/R in SIRT5 KO hearts compared to WT littermates (97). This injury was decreased by pretreatment with dimethyl malonate, a competitive inhibitor of SDH, suggesting that the enhanced activity of SDH by hypersuccinylation is an important cause for increased ischemic injury (which inconsistent with other reports) (25). Similarly, using exogenous NAD administration, Liu et al. demonstrated that SIRT5-mediated SDH desuccinylation decreased the activity of SDH, which attenuated the succinate accumulation during I/R and alleviated reactive oxygen species generation (227). In a separate study, accumulation of succinate during ischemia and its rapid oxidation by SDH during reperfusion can drive extensive ROS generation in a murine I/R injury model (228).
Though complete data are unavailable on the acetylation status of metabolic enzymes and its impact specifically in I/R, several studies have investigated this modification in other proteins or pathways. The permeability transition pore (PTP) is an important inner membrane channel that has a role in I/R injury (229). Permeability transition pore opening is facilitated by the translocation of cyclophilin D (CyPD) from matrix protein to the inner mitochondrial membrane (230). The study by Bochaton et al. in SIRT3 KO mice demonstrated that increased acetylation of CyPD following myocardial I/R facilitates PTP opening and subsequent cell death, which was prevented by attenuation of CyPD acetylation at reperfusion (231). A similar result was found in mice subjected to renal I/R where dexmedetomidine induced SIRT3 overexpression and significantly reduced I/R related mitochondrial damage by decreasing cyclophilin D acetylation (232).
Although excessive mitochondrial metabolic enzyme acetylation occurs in heart failure, its contribution to cardiac metabolic alterations remains incompletely defined. However, accumulating evidence supports the hypothesis that acetylation alters energy substrate utilization in heart failure by activating fatty acid ß-oxidation and inhibiting glucose oxidation. While the inhibitory effect of acetylation on glucose oxidation is widely accepted, there are still disagreements as to the relationship between acetylation and fatty acid ß-oxidation. While most of the studies done in the heart and skeletal muscle tissues illustrate a positive association between acetylation and fatty acid ß-oxidation, others studies suggest a suppressive effect of acetylation on fatty acid ß-oxidation in the liver. Tissue or site-specific variation in acetylation of these enzymes, as well differences in underlying pathologies, could contribute to such discrepant data. Available data also suggests that acetylation does not always has an inhibitory effect on metabolic enzymes.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funded by a Foundation Grant from the Canadian Institutes for Health Research to GL. EK is supported by an Alberta Diabetes Institute Graduate Studentship Award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. (1964) 51:786–94. doi: 10.1073/pnas.51.5.786
2. Verdone L, Caserta M, Mauro ED. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. (2005) 83:344–53. doi: 10.1139/o05-041
3. Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. (2017) 18:90–101. doi: 10.1038/nrm.2016.140
4. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. (2016). 8:a019521. doi: 10.1101/cshperspect.a019521
5. Wang R, Xin M, Li Y, Zhang P, Zhang M. The functions of histone modification enzymes in cancer. Curr Protein Pept Sci. (2016) 17:438–45. doi: 10.2174/1389203717666160122120521
6. Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, et al. Tetrahymena histone acetyltransferase a: a homolog to yeast GCN5P linking histone acetylation to gene activation. Cell. (1996) 84:843–51. doi: 10.1016/S0092-8674(00)81063-6
7. Taunton J, Hassig CA, Schreiber SL, A mammalian histone deacetylase related to the yeast transcriptional regulator RPD3P. Science. (1996) 272:408–11. doi: 10.1126/science.272.5260.408
8. Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J. (2016) 52:1–11. doi: 10.4068/cmj.2016.52.1.1
9. Zhao L, Duan YT, Lu P, Zhang ZJ, Zheng XK, Wang JL, et al. Epigenetic targets and their inhibitors in cancer therapy. Curr Top Med Chem. (2018) 18:2395–419. doi: 10.2174/1568026619666181224095449
10. Bagchi RA, Weeks KL. Histone deacetylases in cardiovascular and metabolic diseases. J Mol Cell Cardiol. (2019) 130:151–9. doi: 10.1016/j.yjmcc.2019.04.003
11. Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. (1987) 104:289–302. doi: 10.1083/jcb.104.2.289
12. North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. (2003) 11:437–44. doi: 10.1016/S1097-2765(03)00038-8
13. Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci USA. (2002) 99:13653–8. doi: 10.1073/pnas.222538099
14. Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. (2006) 103:10230–5. doi: 10.1073/pnas.0604392103
15. Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. (2006) 103:10224–9. doi: 10.1073/pnas.0603968103
16. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human sirt proteins. Mol Biol Cell. (2005) 16:4623–35. doi: 10.1091/mbc.e05-01-0033
17. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. (2006) 23:607–18. doi: 10.1016/j.molcel.2006.06.026
18. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. (2009) 325:834–40. doi: 10.1126/science.1175371
19. Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, et al. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. (2009) 8:604–6. doi: 10.1111/j.1474-9726.2009.00503.x
20. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. (2010) 327:1000–4. doi: 10.1126/science.1179689
21. Lombard DB, Alt FW, Cheng H-L, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. (2007) 27:8807–14. doi: 10.1128/MCB.01636-07
22. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. (2010) 464:121–5. doi: 10.1038/nature08778
23. Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. (2013) 49:186–99. doi: 10.1016/j.molcel.2012.10.024
24. Ahn B-H, Kim H-S, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. (2008) 105:14447–52. doi: 10.1073/pnas.0803790105
25. Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. (2013) 50:919–30. doi: 10.1016/j.molcel.2013.06.001
26. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. (2013) 18:920–33. doi: 10.1016/j.cmet.2013.11.013
27. Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell. (2015) 59:321–32. doi: 10.1016/j.molcel.2015.05.022
28. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. (2018). 5:68. doi: 10.3389/fcvm.2018.00068
29. Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure : from pathways to therapeutic targets. Curr Pharm Des. (2015) 21:3654–64. doi: 10.2174/1381612821666150710150445
30. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15:457–70. doi: 10.1038/s41569-018-0044-6
31. Barth AS, Kumordzie A, Frangakis C, Margulies KB, Cappola TP, Tomaselli GF. Reciprocal transcriptional regulation of metabolic and signaling pathways correlates with disease severity in heart failure. Circ Cardiovasc Genet. (2011) 4:475–83. doi: 10.1161/CIRCGENETICS.110.957571
32. Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid x receptor-α in pacing-induced heart failure. Circulation. (2002) 106:606–12. doi: 10.1161/01.CIR.0000023531.22727.C1
33. Dávila-Román VG, Vedala G, Herrero P., de las Fuentes L, Rogers JG, Kelly DP, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. (2002) 40:271–7. doi: 10.1016/S0735-1097(02)01967-8
34. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. (2001) 104:2923–31. doi: 10.1161/hc4901.100526
35. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. (2014) 7:1022–31. doi: 10.1161/CIRCHEARTFAILURE.114.001469
36. van Bilsen M, van der Vusse GJ, Reneman RS. Transcriptional regulation of metabolic processes: implications for cardiac metabolism. Pflugers Arch. (1998) 437:2–14. doi: 10.1007/s004240050739
37. Lu Z, Scott I, Webster BR, Sack MN. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ Res. (2009) 105:830–41. doi: 10.1161/CIRCRESAHA.109.204974
38. Hirschey MD, Shimazu T, Huang JY, Schwer B, Verdin E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb Symp Quant Biol. (2011) 76:267–77. doi: 10.1101/sqb.2011.76.010850
39. Fukushima A, Lopaschuk GD. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim Biophys Acta Mol Basis Dis. (2016) 1862:2211–20. doi: 10.1016/j.bbadis.2016.07.020
40. Yan K, Wang K, Li P. The role of post-translational modifications in cardiac hypertrophy. J Cell Mol Med. (2019) 23:3795–807. doi: 10.1111/jcmm.14330
41. Lee A, Oh JG, Gorski PA, Hajjar RJ, Kho C. Post-translational modifications in heart failure: small changes, big impact. Heart Lung Circ. (2016) 25:319–24. doi: 10.1016/j.hlc.2015.11.008
42. Liddy KA, White MY, Cordwell SJ. Functional decorations: post-translational modifications and heart disease delineated by targeted proteomics. Genome Med. (2013) 5:20. doi: 10.1186/gm424
43. Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. (2011) 7:58–63. doi: 10.1038/nchembio.495
44. Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta Proteins Proteom. (2016) 1864:1372–401. doi: 10.1016/j.bbapap.2016.06.007
45. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
46. Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci USA. (2016) 113:4320–5. doi: 10.1073/pnas.1519858113
47. Hirschey MD, Zhao Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics. (2015) 14:2308–15. doi: 10.1074/mcp.R114.046664
48. Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. (2020) 17:96–115. doi: 10.1038/s41569-019-0235-9
49. Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol. (2008) 18:682–9. doi: 10.1016/j.sbi.2008.11.004
50. Hu A, Britton L, Garcia B editors. Investigating the specificity of histone acetyltransferase activity for producing rare modifications on histones using mass spectrometry. In: The 62nd Annual American Society for Mass Spectrometry Conference on Mass Spectrometry and Allied Topics. Baltimore, MD (2014).
51. Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, et al. Lysine glutarylation is a protein posttranslational modification regulated by sirt5. Cell Metab. (2014) 19:605–17. doi: 10.1016/j.cmet.2014.03.014
52. Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5l1. Biochem J. (2012) 443:655–61. doi: 10.1042/BJ20120118
53. Thapa D, Zhang M, Manning JR, Guimarães DA, Stoner MW, O'Doherty RM, et al. Acetylation of mitochondrial proteins by GCN5l1 promotes enhanced fatty acid oxidation in the heart. Am J Physiol Heart Circ Physiol. (2017) 313:H265–74. doi: 10.1152/ajpheart.00752.2016
54. Baeza J, Smallegan MJ. Denu JM. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem Biol. (2015) 10:122–8. doi: 10.1021/cb500848p
55. Wagner GR, Payne RM. Widespread and enzyme-independent nϵ-acetylation and nϵ-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. (2013) 288:29036–45. doi: 10.1074/jbc.M113.486753
56. Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. (2011) 10:9. doi: 10.1074/mcp.M111.012658
57. Yang G, Yuan Y, Yuan H, Wang J, Yun H, Geng Y, et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. (2021) 22:29. doi: 10.15252/embr.202050967
58. Imai S-i, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. (2000) 403:795–800. doi: 10.1038/35001622
59. Greiss S, Gartner A. Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol Cells. (2009) 28:407–15. doi: 10.1007/s10059-009-0169-x
60. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. (1999) 13:2570–80. doi: 10.1101/gad.13.19.2570
61. Klar AJ, Fogel S, Macleod K. MAR1-a regulator of the HMA and HMα loci in Saccharomyces cerevisiae. Genetics. (1979) 93:37–50. doi: 10.1093/genetics/93.1.37
62. Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. (2013) 18:416–30. doi: 10.1016/j.cmet.2013.07.013
63. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. (2012) 483:218–21. doi: 10.1038/nature10815
64. Albani D, Ateri E, Mazzuco S, Ghilardi A, Rodilossi S, Biella G, et al. Modulation of human longevity by SIRT3 single nucleotide polymorphisms in the prospective study “treviso longeva (trelong)”. Age. (2014) 36:469–78. doi: 10.1007/s11357-013-9559-2
65. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. (2004) 305:390–2. doi: 10.1126/science.1099196
66. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. (2010) 143:802–12. doi: 10.1016/j.cell.2010.10.002
67. Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. (2007) 282:6823–32. doi: 10.1074/jbc.M609554200
68. Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. (2006) 20:1075–80. doi: 10.1101/gad.1399706
69. North BJ, Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE. (2007) 2:0000784. doi: 10.1371/journal.pone.0000784
70. Chen Y, Zhao W, Yang JS, Cheng Z, Luo H, Lu Z, et al. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol Cell Proteomics. (2012) 11:1048–62. doi: 10.1074/mcp.M112.019547
71. Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. (2007) 282:33583–92. doi: 10.1074/jbc.M705488200
72. Sol EM, Wagner SA, Weinert BT, Kumar A, Kim H-S, Deng C-X, et al. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase Sirt3. PLoS ONE. (2012). 7:e50545. doi: 10.1371/journal.pone.0050545
73. Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. (2013) 50:686–98. doi: 10.1016/j.molcel.2013.05.012
74. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. (2011) 334:806–9. doi: 10.1126/science.1207861
75. Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear adp-ribosyltransferase. J Biol Chem. (2005) 280:21313–20. doi: 10.1074/jbc.M413296200
76. Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun. (2016). 7:12235. doi: 10.1038/ncomms12235
77. Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. (2015) 309:31. doi: 10.1152/ajpheart.00053.2015
78. Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med. (2020). 12:eaay7205. doi: 10.1126/scitranslmed.aay7205
79. Herr DJ, Baarine M, Aune SE, Li X, Ball LE, Lemasters JJ, et al. HDAC1 localizes to the mitochondria of cardiac myocytes and contributes to early cardiac reperfusion injury. J Mol Cell Cardiol. (2018) 114:309–19. doi: 10.1016/j.yjmcc.2017.12.004
80. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. (2016) 134:883–94. doi: 10.1161/CIRCULATIONAHA.116.022495
81. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, et al. Mitochondrial protein hyperacetylation in the failing heart. Jci Insight. (2016) 2:25. doi: 10.1172/jci.insight.84897
82. Hansen BK, Gupta R, Baldus L, Lyon D, Narita T, Lammers M, et al. Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nat Commun. (2019) 10:1055. doi: 10.1038/s41467-019-09024-0
83. Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res. (2014) 103:485–97. doi: 10.1093/cvr/cvu156
84. Fukushima A, Lopaschuk GD. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim Biophys Acta. (2016) 10:18. doi: 10.1016/j.bbalip.2016.03.020
85. Vadvalkar SS, Baily CN, Matsuzaki S, West M, Tesiram YA, Humphries KM. Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochem J. (2013) 449:253–61. doi: 10.1042/BJ20121038
86. Meyer JG, Softic S, Basisty N, Rardin MJ, Verdin E, Gibson BW, et al. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS ONE. (2018) 13:e0208973. doi: 10.1371/journal.pone.0208973
87. Lopaschuk GD, Tsang H. Metabolism of palmitate in isolated working hearts from spontaneously diabetic “BB” wistar rats. Circ Res. (1987) 61:853–8. doi: 10.1161/01.RES.61.6.853
88. Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. (2006) 47:598–604. doi: 10.1016/j.jacc.2005.09.030
89. Pougovkina O., te Brinke H, Ofman R, van Cruchten AG, Kulik W, Wanders RJA, et al. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum Mol Genet. (2014) 23:3513–22. doi: 10.1093/hmg/ddu059
90. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ Res. (2020) 12:317933. doi: 10.1161/CIRCRESAHA.120.317933
91. Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5'-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. (1995) 270:17513–20. doi: 10.1074/jbc.270.29.17513
92. Sakamoto J, Barr RL, Kavanagh KM, Lopaschuk GD. Contribution of malonyl-CoA decarboxylase to the high fatty acid oxidation rates seen in the diabetic heart. Am J Physiol Heart Circ Physiol. (2000). 278:H1196–204. doi: 10.1152/ajpheart.2000.278.4.H1196
93. Zhang L, Ussher JR, Oka T, Cadete VJJ, Wagg C, Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res. (2010) 89:148–56. doi: 10.1093/cvr/cvq266
94. Keung W, Cadete VJJ, Palaniyappan A, Jablonski A, Fischer M, Lopaschuk GD. Intracerebroventricular leptin administration differentially alters cardiac energy metabolism in mice fed a low-fat and high-fat diet. J Cardiovasc Pharmacol. (2011) 57:103–13. doi: 10.1097/FJC.0b013e31820014f9
95. Young ME, Goodwin GW, Ying J, Guthrie P, Wilson CR, Laws FA, et al. Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am J Physiol Endocrinol Metab. (2001). 280:E471–9. doi: 10.1152/ajpendo.2001.280.3.E471
96. Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, et al. Proteomic and biochemical studies of lysine malonylation suggest its malonic aciduria-associated regulatory role in mitochondrial function and fatty acid oxidation. Mol Cell Proteomics. (2015) 14:3056–71. doi: 10.1074/mcp.M115.048850
97. Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. (2015) 88:73–81. doi: 10.1016/j.yjmcc.2015.09.005
98. Fan J, Shan C, Kang H-B, Elf S, Xie J, Tucker M, et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. (2014) 53:534–48. doi: 10.1016/j.molcel.2013.12.026
99. Thapa D, Manning JR, Stoner MW, Zhang M, Xie B, Scott I. Cardiomyocyte-specific deletion of GCN5L1 in mice restricts mitochondrial protein hyperacetylation in response to a high fat diet. Sci Rep. (2020). 10:10665. doi: 10.1038/s41598-020-67812-x
100. Fukushima A, Alrob OA, Zhang L, Wagg CS, Altamimi T, Rawat S, et al. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol. (2016) 311:3. doi: 10.1152/ajpheart.00900.2015
101. Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, Choudhary C. Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. (2015) 34:2620–32. doi: 10.15252/embj.201591271
102. Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. (2014) 54:5. doi: 10.1016/j.molcel.2014.03.027
103. Carrer A, Parris JL, Trefely S, Henry RA, Montgomery DC, Torres A, et al. Impact of a high-fat diet on tissue acyl-CoA and histone acetylation levels. J Biol Chem. (2017) 292:3312–22. doi: 10.1074/jbc.M116.750620
104. Parodi-Rullán RM, Chapa-Dubocq XR, Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front Physiol. (2018). 9:1094. doi: 10.3389/fphys.2018.01094
105. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. (2009) 119:2758–71. doi: 10.1172/JCI39162
106. Lu T-M, Tsai J-Y, Chen Y-C, Huang C-Y, Hsu H-L, Weng C-F, et al. Downregulation of SIRT1 as aging change in advanced heart failure. J Biomed Sci. (2014) 21:57. doi: 10.1186/1423-0127-21-57
107. Akkafa F, Halil Altiparmak I, Erkus ME, Aksoy N, Kaya C, Ozer A, et al. Reduced SIRT1 expression correlates with enhanced oxidative stress in compensated and decompensated heart failure. Redox Biol. (2015) 6:169–73. doi: 10.1016/j.redox.2015.07.011
108. Kawashima T, Inuzuka Y, Okuda J, Kato T, Niizuma S, Tamaki Y, et al. Constitutive SIRT1 overexpression impairs mitochondria and reduces cardiac function in mice. J Mol Cell Cardiol. (2011) 51:1026–36. doi: 10.1016/j.yjmcc.2011.09.013
109. Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. (2012) 16:180–8. doi: 10.1016/j.cmet.2012.07.003
110. Buler M, Aatsinki SM, Izzi V, Uusimaa J, Hakkola J. SIRT5 is under the control of PGC-1α and ampk and is involved in regulation of mitochondrial energy metabolism. FASEB J. (2014) 28:3225–37. doi: 10.1096/fj.13-245241
111. Yu J, Sadhukhan S, Noriega LG, Moullan N, He B, Weiss RS, et al. Metabolic characterization of a Sirt5 deficient mouse model. Sci Rep. (2013). 3:2806. doi: 10.1038/srep02806
112. Chi Z, Chen S, Xu T, Zhen W, Yu W, Jiang D, et al. Histone deacetylase 3 couples mitochondria to drive IL-1β-dependent inflammation by configuring fatty acid oxidation. Mol Cell. (2020). 80:43.e7–58.e7. doi: 10.1016/j.molcel.2020.08.015
113. Hershberger KA, Martin AS, Hirschey MD. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. (2017) 13:213–25. doi: 10.1038/nrneph.2017.5
114. Cantó C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. (2015) 22:31–53. doi: 10.1016/j.cmet.2015.05.023
115. Wang LF, Huang CC, Xiao YF, Guan XH, Wang XN, Cao Q, et al. CD38 deficiency protects heart from high fat diet-induced oxidative stress via activating Sirt3/FOXO3 pathway. Cell Physiol Biochem. (2018) 48:2350–63. doi: 10.1159/000492651
116. Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, et al. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech Ageing Dev. (2010) 131:21–8. doi: 10.1016/j.mad.2009.11.002
117. Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation*. J Biol Chem. (2004) 279:40122–9. doi: 10.1074/jbc.M407484200
118. Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. J Biol Chem. (2002) 277:45099–107. doi: 10.1074/jbc.M205670200
119. Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. (2004) 18:12–6. doi: 10.1101/gad.1164804
120. Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. (2003) 12:51–62. doi: 10.1016/S1097-2765(03)00226-0
121. Madsen AS, Andersen C, Daoud M, Anderson KA, Laursen JS, Chakladar S, et al. Investigating the sensitivity of NAD+-dependent sirtuin deacylation activities to NADH. J Biol Chem. (2016) 291:7128–41. doi: 10.1074/jbc.M115.668699
122. Anderson KA, Madsen AS, Olsen CA, Hirschey MD. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim Biophys Acta Bioenerg. (2017) 1858:991–8. doi: 10.1016/j.bbabio.2017.09.005
123. Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. (2012) 15:838–47. doi: 10.1016/j.cmet.2012.04.022
124. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. (2018) 137:2256–73. doi: 10.1161/CIRCULATIONAHA.116.026099
125. Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE. (2014) 9:e98972. doi: 10.1371/journal.pone.0098972
126. de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. (2016) 15:522–30. doi: 10.1111/acel.12461
127. Kane AE, Sinclair DA. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ Res. (2018) 123:868–85. doi: 10.1161/CIRCRESAHA.118.312498
128. Nelson MM, Efird JT, Kew KA, Katunga LA, Monroe TB, Doorn JA, et al. Enhanced catecholamine flux and impaired carbonyl metabolism disrupt cardiac mitochondrial oxidative phosphorylation in diabetes patients. Antioxid Redox Signal. (2020) 25. doi: 10.1089/ars.2020.8122
129. Martin AS, Abraham DM, Hershberger KA, Bhatt DP, Mao L, Cui H, et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a friedreich's ataxia cardiomyopathy model. JCI Insight. (2017) 2:20. doi: 10.1172/jci.insight.93885
130. Ardehali H, Sabbah HN, Burke MA, Sarma S, Liu PP, Cleland JGF, et al. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur J Heart Fail. (2012) 14:120–9. doi: 10.1093/eurjhf/hfr173
131. Neubauer S. The failing heart — an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
132. Fillmore N, Lopaschuk GD. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim Biophys Acta Mol Cell Res. (2013) 1833:857–65. doi: 10.1016/j.bbamcr.2012.08.014
133. Glatz JFC, Nabben M, Young ME, Schulze PC, Taegtmeyer H, Luiken JJFP. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165579. doi: 10.1016/j.bbadis.2019.165579
134. Lopaschuk GD, Ussher JR. Evolving concepts of myocardial energy metabolism. Circ Res. (2016) 119:1173–6. doi: 10.1161/CIRCRESAHA.116.310078
135. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85:1093–129. doi: 10.1152/physrev.00006.2004
136. Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to atp production in hypertrophied hearts. Am J Physiol. (1994). 267(2 Pt 2):H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
137. Degens H, de Brouwer KF, Gilde AJ, Lindhout M, Willemsen PH, Janssen BJ, et al. Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res Cardiol. (2006) 101:17–26. doi: 10.1007/s00395-005-0549-0
138. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
139. Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. (1996) 94:2837–42. doi: 10.1161/01.CIR.94.11.2837
140. Grillon JM, Johnson KR, Kotlo K, Danziger RS. Non-histone lysine acetylated proteins in heart failure. Biochim Biophys Acta. (2012) 4:607–14. doi: 10.1016/j.bbadis.2011.11.016
141. Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, et al. Mitochondrial complex i deficiency increases protein acetylation and accelerates heart failure. Cell Metab. (2013) 18:239–50. doi: 10.1016/j.cmet.2013.07.002
142. Du Y, Cai T, Li T, Xue P, Zhou B, He X, et al. Lysine malonylation is elevated in type 2 diabetic mouse models and enriched in metabolic associated proteins. Mol Cell Proteomics. (2015) 14:227–36. doi: 10.1074/mcp.M114.041947
143. Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
144. Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. (2010) 33:469–77. doi: 10.1007/s10545-010-9061-2
145. Chen T, Liu J, Li N, Wang S, Liu H, Li J, et al. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS ONE. (2015). 10:e0118909. doi: 10.1371/journal.pone.0118909
146. Vazquez EJ, Berthiaume JM, Kamath V, Achike O, Buchanan E, Montano MM, et al. Mitochondrial complex i defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc Res. (2015) 107:453–65. doi: 10.1093/cvr/cvv183
147. Karwi QG, Zhang L, Altamimi TR, Wagg CS, Patel V, Uddin GM, et al. Weight loss enhances cardiac energy metabolism and function in heart failure associated with obesity. Diabetes Obes Metab. (2019) 21:1944–55. doi: 10.1111/dom.13762
148. Vadvalkar SS, Matsuzaki S, Eyster CA, Giorgione JR, Bockus LB, Kinter CS, et al. Decreased mitochondrial pyruvate transport activity in the diabetic heart: role of mitochondrial pyruvate carrier 2 (MPC2) acetylation. J Biol Chem. (2017) 292:4423–33. doi: 10.1074/jbc.M116.753509
149. Renguet E, Ginion A, Gélinas R, Bultot L, Auquier J, Frayne IR, et al. Metabolism and acetylation contribute to leucine-mediated inhibition of cardiac glucose uptake. Am J Physiol Heart Circ Physiol. (2017) 313:H432–45. doi: 10.1152/ajpheart.00738.2016
150. Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. (2011) 4:2001465. doi: 10.1126/scisignal.2001465
151. Kerner J, Yohannes E, Lee K, Virmani A, Koverech A, Cavazza C, et al. Acetyl-l-carnitine increases mitochondrial protein acetylation in the aged rat heart. Mech Ageing Dev. (2015) 145:39–50. doi: 10.1016/j.mad.2015.01.003
152. Fernandes J, Weddle A, Kinter CS, Humphries KM, Mather T, Szweda LI, et al. Lysine acetylation activates mitochondrial aconitase in the heart. Biochemistry (Mosc). (2015) 54:4008–18. doi: 10.1021/acs.biochem.5b00375
153. Sun Y, Teng Z, Sun X, Zhang L, Chen J, Wang B, et al. Exogenous H(2)S reduces the acetylation levels of mitochondrial respiratory enzymes via regulating the NAD(+)-SIRT3 pathway in cardiac tissues of db/db mice. Am J Physiol Endocrinol Metab. (2019) 317:E284–97. doi: 10.1152/ajpendo.00326.2018
154. Fukushima A, Zhang L, Huqi A, Lam VH, Rawat S, Altamimi T, et al. Acetylation contributes to hypertrophy-caused maturational delay of cardiac energy metabolism. JCI Insight. (2018) 3:e99239. doi: 10.1172/jci.insight.99239
155. Kerr M, Miller JJ, Thapa D, Stiewe S, Timm KN, Aparicio CNM, et al. Rescue of myocardial energetic dysfunction in diabetes through the correction of mitochondrial hyperacetylation by honokiol. Jci Insight. (2020) 5:140326. doi: 10.1172/jci.insight.140326
156. Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. (2013) 62:3404–17. doi: 10.2337/db12-1650
157. Lopaschuk GD, Folmes CDL, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. (2007) 101:335–47. doi: 10.1161/CIRCRESAHA.107.150417
158. Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol. (2015) 110:015–0493. doi: 10.1007/s00395-015-0493-6
159. Davidson MT, Grimsrud PA, Lai L, Draper JA, Fisher-Wellman KH, Narowski TM, et al. Extreme acetylation of the cardiac mitochondrial proteome does not promote heart failure. Circ Res. (2020) 127:1094–108. doi: 10.1161/CIRCRESAHA.120.317293
160. Bharathi SS, Zhang Y, Mohsen A-W, Uppala R, Balasubramani M, Schreiber E, et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-coa dehydrogenase by deacetylating conserved lysines near the active site. J Biol Chem. (2013) 288:33837–47. doi: 10.1074/jbc.M113.510354
161. Thapa D, Wu K, Stoner MW, Xie B, Zhang M, Manning JR, et al. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial β-oxidation enzyme HADHA. J Biol Chem. (2018) 293:17676–84. doi: 10.1074/jbc.AC118.005462
162. Nassir F, Arndt JJ, Johnson SA, Ibdah JA. Regulation of mitochondrial trifunctional protein modulates nonalcoholic fatty liver disease in mice. J Lipid Res. (2018) 59:967–73. doi: 10.1194/jlr.M080952
163. Denton RM, Randle PJ, Bridges BJ, Cooper RH, Kerbey AL, Pask HT, et al. Regulation of mammalian pyruvate dehydrogenase. Mol Cell Biochem. (1975) 9:27–53. doi: 10.1007/BF01731731
164. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. (2014) 71:2577–604. doi: 10.1007/s00018-013-1539-2
165. Mori J, Alrob OA, Wagg CS, Harris RA, Lopaschuk GD, Oudit GY. Ang II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am J Physiol Heart Circ Physiol. (2013) 304:H1103–13. doi: 10.1152/ajpheart.00636.2012
166. Zhang Y, Bharathi SS, Rardin MJ, Lu J, Maringer KV, Sims-Lucas S, et al. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem. (2017) 292:10239–49. doi: 10.1074/jbc.M117.785022
167. Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by SIRT1 protein-mediated deacetylation. J Biol Chem. (2012) 287:3850–8. doi: 10.1074/jbc.M111.317404
168. Li T, Liu M, Feng X, Wang Z, Das I, Xu Y, et al. Glyceraldehyde-3-phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J Biol Chem. (2014) 289:3775–85. doi: 10.1074/jbc.M113.531640
169. Ventura M, Mateo F, Serratosa J, Salaet I, Carujo S, Bachs O, et al. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int J Biochem Cell Biol. (2010) 42:1672–80. doi: 10.1016/j.biocel.2010.06.014
170. Xiong Y, Lei QY, Zhao S, Guan KL. Regulation of glycolysis and gluconeogenesis by acetylation of PKM and PEPCK. Cold Spring Harb Symp Quant Biol. (2011) 76:285–9. doi: 10.1101/sqb.2011.76.010942
171. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β production and to prevent dss-induced colitis in mice. Cell Rep. (2017) 19:2331–44. doi: 10.1016/j.celrep.2017.05.065
172. Xiangyun Y, Xiaomin N, Linping G, Yunhua X, Ziming L, Yongfeng Y, et al. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget. (2017) 8:6984–93. doi: 10.18632/oncotarget.14346
173. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. (2021) 599:23–37. doi: 10.1113/JP280572
174. Geng H, Liu Q, Xue C, David LL, Beer TM, Thomas GV, et al. HIF1α protein stability is increased by acetylation at lysine 709. J Biol Chem. (2012) 287:35496–505. doi: 10.1074/jbc.M112.400697
175. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol Cell. (2010) 38:864–78. doi: 10.1016/j.molcel.2010.05.023
176. Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, et al. Sirt3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat–fed mice. Diabetes. (2015) 64:3081–92. doi: 10.2337/db14-1810
177. Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res. (2016) 118:1151–69. doi: 10.1161/CIRCRESAHA.116.306206
178. Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, et al. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail. (2013) 6:1039–48. doi: 10.1161/CIRCHEARTFAILURE.112.000228
179. Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol. (2019) 18:019–0892. doi: 10.1186/s12933-019-0892-3
180. Ramakrishnan G, Davaakhuu G, Kaplun L, Chung W-C, Rana A, Atfi A, et al. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem. (2014) 289:6054–66. doi: 10.1074/jbc.M113.537266
181. Fillmore N, Wagg CS, Zhang L, Fukushima A, Lopaschuk GD. Cardiac branched-chain amino acid oxidation is reduced during insulin resistance in the heart. Am J Physiol Endocrinol Metab. (2018) 315:E1046–52. doi: 10.1152/ajpendo.00097.2018
182. Lopaschuk GD, Karwi QG, Ho KL, Pherwani S, Ketema EB. Ketone metabolism in the failing heart. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 12:158813. doi: 10.1016/j.bbalip.2020.158813
183. Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, et al. Sirt3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. (2010) 12:654–61. doi: 10.1016/j.cmet.2010.11.003
184. Dittenhafer-Reed Kristin E, Richards Alicia L, Fan J, Smallegan Michael J, Fotuhi Siahpirani A, Kemmerer Zachary A, et al. Sirt3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. (2015) 21:637–46. doi: 10.1016/j.cmet.2015.03.007
185. Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of Sirt3 in metabolic pathways. Proc Natl Acad Sci USA. (2013) 110:6601–6. doi: 10.1073/pnas.1302961110
186. Lei M-Z, Li X-X, Zhang Y, Li J-T, Zhang F, Wang Y-P, et al. Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal Transduct Target Ther. (2020) 5:1–9. doi: 10.1038/s41392-020-0168-0
187. Yu W, Dittenhafer-Reed KE, Denu JM. Sirt3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. (2012) 287:14078–86. doi: 10.1074/jbc.M112.355206
188. Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. (2016) 17:811–22. doi: 10.15252/embr.201541643
189. Azhar S. Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease. Future Cardiol. (2010) 6:657–91. doi: 10.2217/fca.10.86
190. Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications - a review. Nutr J. (2014) 13:17. doi: 10.1186/1475-2891-13-17
191. Audet-walsh É, Giguére V. The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol Sin. (2015). 36:51–61. doi: 10.1038/aps.2014.121
192. Finck BN, Kelly DP. Peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. (2007) 115:2540–8. doi: 10.1161/CIRCULATIONAHA.107.670588
193. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. (2009) 9:327–38. doi: 10.1016/j.cmet.2009.02.006
194. Laurent G, de Boer VCJ, Finley LWS, Sweeney M, Lu H, Schug TT, et al. Sirt4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol Cell Biol. (2013) 33:4552–61. doi: 10.1128/MCB.00087-13
195. Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, et al. Brown remodeling of white adipose tissue by SIRT1-dependent deacetylation of pparγ. Cell. (2012) 150:620–32. doi: 10.1016/j.cell.2012.06.027
196. Lin W, Zhang Q, Liu L, Yin S, Liu Z, Cao W. Klotho restoration via acetylation of peroxisome proliferation–activated receptor γ reduces the progression of chronic kidney disease. Kidney Int. (2017) 92:669–79. doi: 10.1016/j.kint.2017.02.023
197. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, et al. SIRT1 promotes fat mobilization in white adipocytes by repressing ppar-gamma. Nature. (2004) 429:771–6. doi: 10.1038/nature02583
198. Jiang X, Ye X, Guo W, Lu H, Gao Z. Inhibition of HDAC3 promotes ligand-independent pparγ activation by protein acetylation. J Mol Endocrinol. (2014) 53:191–200. doi: 10.1530/JME-14-0066
199. Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem. (2005) 280:16456–60. doi: 10.1074/jbc.M501485200
200. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. (2007) 26:1913–23. doi: 10.1038/sj.emboj.7601633
201. Lerin C, Rodgers JT, Kalume DE. Kim S-h, Pandey A, Puigserver P. Gcn5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab. (2006) 3:429–38. doi: 10.1016/j.cmet.2006.04.013
202. Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, et al. Direct acetylation of the estrogen receptor alpha hinge region by P300 regulates transactivation and hormone sensitivity. J Biol Chem. (2001) 276:18375–83. doi: 10.1074/jbc.M100800200
203. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. (2009) 324:1076–80. doi: 10.1126/science.1164097
204. McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. (2016) 17:1463–72. doi: 10.1016/j.celrep.2016.10.012
205. Castillo EC, Morales JA, Chapoy-Villanueva H, Silva-Platas C, Treviño-Saldaña N, Guerrero-Beltrán CE, et al. Mitochondrial hyperacetylation in the failing hearts of obese patients mediated partly by a reduction in SIRT3: the involvement of the mitochondrial permeability transition pore. Cell Physiol Biochem. (2019) 53:465–79. doi: 10.33594/000000151
206. Romanick SS, Ulrich C, Schlauch K, Hostler A, Payne J, Woolsey R, et al. Obesity-mediated regulation of cardiac protein acetylation: parallel analysis of total and acetylated proteins via TMT-tagged mass spectrometry. Biosci Rep. (2018) 38:31. doi: 10.1042/BSR20180721
207. Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca2+-atpase in heart failure. Circ Res. (2019) 124:e63–80. doi: 10.1161/RES.0000000000000277
208. Hu Q, Zhang H, Cortés NG, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res. (2020) 126:456–70. doi: 10.1161/CIRCRESAHA.119.315252
209. Stram AR, Wagner GR, Fogler BD, Pride PM, Hirschey MD, Payne RM. Progressive mitochondrial protein lysine acetylation and heart failure in a model of Friedreich's ataxia cardiomyopathy. PLoS ONE. (2017). 12:e0178354. doi: 10.1371/journal.pone.0178354
210. Olsen JV, Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. (2013) 12:3444–52. doi: 10.1074/mcp.O113.034181
211. Xiong Y, Guan K-L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. (2012) 198:155–64. doi: 10.1083/jcb.201202056
212. Baeza J, Dowell JA, Smallegan MJ, Fan J, Amador-Noguez D, Khan Z, et al. Stoichiometry of site-specific lysine acetylation in an entire proteome. J Biol Chem. (2014) 289:21326. doi: 10.1074/jbc.M114.581843
213. Weinert BT, Iesmantavicius V, Moustafa T, Schölz C, Wagner SA, Magnes C, et al. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol Syst Biol. (2014). 10:716. doi: 10.1002/msb.134766
214. Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev. (2002) 7:161–73. doi: 10.1023/A:1015380609464
215. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. (1963) 1:785–9. doi: 10.1016/S0140-6736(63)91500-9
216. Sun Y, Tian Z, Liu N, Zhang L, Gao Z, Sun X, et al. Exogenous H(2)S switches cardiac energy substrate metabolism by regulating Sirt3 expression in db/db mice. J Mol Med. (2018) 96:281–99. doi: 10.1007/s00109-017-1616-3
217. Liu B, Clanachan AS, Schulz R, Lopaschuk GD. Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ Res. (1996) 79:940–8. doi: 10.1161/01.RES.79.5.940
218. Kantor PF, Dyck JRB, Lopaschuk GD. Fatty acid oxidation in the reperfused ischemic heart. Am J Med Sci. (1999) 318:3–14. doi: 10.1016/S0002-9629(15)40566-X
219. Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. (2009) 105:481–91. doi: 10.1161/CIRCRESAHA.109.203703
220. Liu L, Wang P, Liu X, He D, Liang C, Yu Y. Exogenous NAD(+) supplementation protects H9C2 cardiac myoblasts against hypoxia/reoxygenation injury via SIRT1-p53 pathway. Fundam Clin Pharmacol. (2014) 28:180–9. doi: 10.1111/fcp.12016
221. Shinmura K, Tamaki K, Sano M, Nakashima-Kamimura N, Wolf AM, Amo T, et al. Caloric restriction primes mitochondria for ischemic stress by deacetylating specific mitochondrial proteins of the electron transport chain. Circ Res. (2011) 109:396–406. doi: 10.1161/CIRCRESAHA.111.243097
222. Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. (2015). 6:6656. doi: 10.1038/ncomms7656
223. Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. Sirt3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. (2014) 306:18. doi: 10.1152/ajpheart.00027.2014
224. Koentges C, Pfeil K, Meyer-Steenbuck M, Lother A, Hoffmann MM, Odening KE, et al. Preserved recovery of cardiac function following ischemia-reperfusion in mice lacking Sirt3. Can J Physiol Pharmacol. (2016) 94:72–80. doi: 10.1139/cjpp-2015-0152
225. Hershberger KA, Abraham DM, Martin AS, Mao L, Liu J, Gu H, et al. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J Biol Chem. (2017) 292:19767–81. doi: 10.1074/jbc.M117.809897
226. Klishadi MS, Zarei F, Hejazian SH, Moradi A, Hemati M, Safari F. Losartan protects the heart against ischemia reperfusion injury: Sirtuin3 involvement. J Pharm Pharm Sci. (2015) 18:112–23. doi: 10.18433/J3XG7T
227. Liu L, Wang Q, Zhao B, Wu Q, Wang P. Exogenous nicotinamide adenine dinucleotide administration alleviates ischemia/reperfusion-induced oxidative injury in isolated rat hearts via Sirt5-SDH-succinate pathway. Eur J Pharmacol. (2019) 858:3. doi: 10.1016/j.ejphar.2019.172520
228. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
229. Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. (1995) 307:93–8. doi: 10.1042/bj3070093
230. Di Lisa F, Bernardi P. A capful of mechanisms regulating the mitochondrial permeability transition. J Mol Cell Cardiol. (2009) 46:775–80. doi: 10.1016/j.yjmcc.2009.03.006
231. Bochaton T, Crola-Da-Silva C, Pillot B, Villedieu C, Ferreras L, Alam MR, et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J Mol Cell Cardiol. (2015) 84:61–9. doi: 10.1016/j.yjmcc.2015.03.017
Keywords: mitochondria, fatty acid oxidation, succinylation, sirtuins, lysine acetylation, glucose oxidation
Citation: Ketema EB and Lopaschuk GD (2021) Post-translational Acetylation Control of Cardiac Energy Metabolism. Front. Cardiovasc. Med. 8:723996. doi: 10.3389/fcvm.2021.723996
Received: 11 June 2021; Accepted: 30 June 2021;
Published: 02 August 2021.
Edited by:
Kunhua Song, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Bradley S. Ferguson, University of Nevada, Reno, United StatesCopyright © 2021 Ketema and Lopaschuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary D. Lopaschuk, Z2FyeS5sb3Bhc2NodWtAdWFsYmVydGEuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.