 Zeyi Cheng
Zeyi Cheng Tingting Fang2†
Tingting Fang2† Yingqiang Guo
Yingqiang Guo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 25 October 2021
Sec. General Cardiovascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.722340
This article is part of the Research Topic Cardiovascular Fibrosis and Related Diseases: Basic and clinical research advances View all 14 articles
Hypertrophic cardiomyopathy (HCM) is a very common inherited cardiovascular disease (CAD) and the incidence is about 1/500 of the common population. It is caused by more than 1,400 mutations in 11 or more genes encoding the proteins of the cardiac sarcomere. HCM presents a heterogeneous clinical profile and complex pathophysiology and HCM is the most important cause of sudden cardiac death (SCD) in young people. HCM also contributes to functional disability from heart failure and stroke (caused by atrial fibrillation). Current treatments for HCM (medication, myectomy, and alcohol septal ablation) are geared toward slowing down the disease progression and symptom relief and implanted cardiac defibrillator (ICD) to prevent SCD. HCM is, however, entering a period of tight translational research that holds promise for the major advances in disease-specific therapy. Main insights into the genetic landscape of HCM have improved our understanding of molecular pathogenesis and pointed the potential targets for the development of therapeutic agents. We reviewed the critical discoveries about the treatments, mechanism of HCM, and their implications for future research.
Hypertrophic cardiomyopathy (HCM) is a heterogeneous myocardial disease characterized by left ventricular hypertrophy and in the cases of hypertrophic obstructive cardiomyopathy (HOCM), it is characterized by asymmetric septal hypertrophy. In the majority of the cases, the average interventricular septal thickness is 26 mm, which cannot be fully explained by the loading conditions of the left ventricle (1, 2). There are several types of HCM based on the distribution of hypertrophy: symmetric, asymmetric, apical, and focal (3). In addition to the hypertrophy, the abnormalities of the mitral valve and subvalvular apparatus lead to the systolic anterior motion (SAM) and left ventricular outflow tract (LVOT) obstruction in about two-thirds of the HCM cases, the characteristic features of HOCM, as well as the microvascular dysfunction and subendocardial ischemia (3). Due to a combination of these factors, patients with HCM frequently experience reduced exercise capacity, dyspnea, and/or chest pain. HCM is mainly inherited in an autosomal dominant pattern, linked with mutations (nucleotide sequence variants) in 11 or more genes encoding the proteins of myocardial sarcomere structure (~60% of all the causes and >90% of the genetically defined patients), and with beta-myosin heavy chain and myosin-binding protein C genes most commonly involved (Table 1; Figure 1) (4–6). Patients with HCM suffer from the cardiovascular death rates of 1-2% per year including the sudden cardiac death (SCD) ~ 1%, heart failure (HF) ~ 0.5%, and thromboembolism ~ 0.1% (2, 3). In recent years, there has been tremendous development in this field with a translation of the basic science discoveries into the new therapeutic methods. In this study, we reviewed the recent development in pharmacological and gene-based therapies, which we believe will result in a comprehensive understanding of the treatment of HCM in the future.
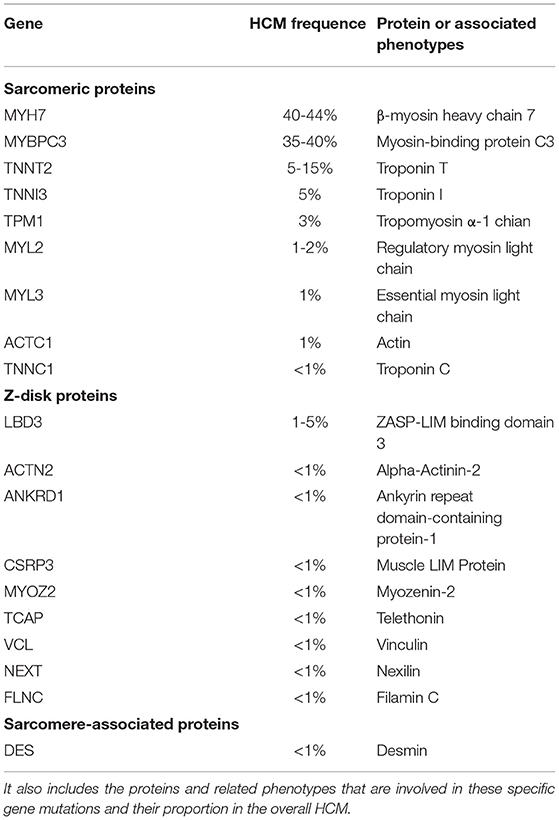
Table 1. A list of the genes in which the pathogenic mutations are associated with hypertrophic cardiomyopathy (HCM).
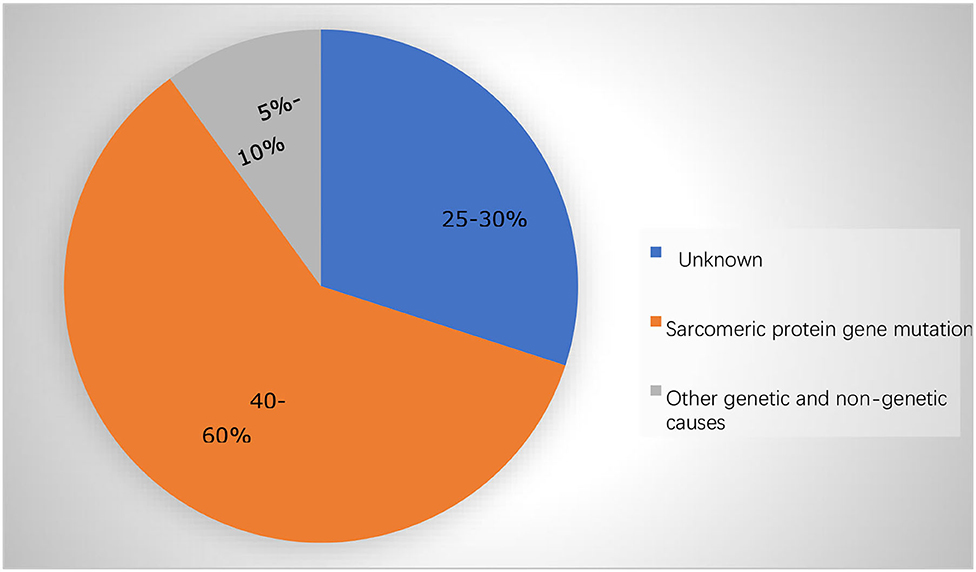
Figure 1. The majority of the cases in adolescents and adults are caused by mutations in the sarcomere protein genes.
Gene mutation is the initiating pathogenesis of HCM affecting the proteins by playing a critical role in the function of the cardiac muscle unit “sarcomeres.” The function of the sarcomere may weaken due to an abnormality in or shortage of any one of these proteins, which, in turn, affects the normal myocardial contractility. It is still not exactly described how the mutations in the sarcomere-related genes cause hypertrophy of the heart muscle (7, 8). However, there are several hypotheses that are as follows (Figure 2):
(1) Mutations in the sarcomere-related genes are associated with an increased affinity for calcium in the myofilaments, activate the calmodulin kinase II (CaMKII) pathway, and delay the downstream targets of the CaMKII sodium channels, thus increase the intracellular calcium and, thus, forming a vicious cycle (9–12). This results in the impaired relaxation and diastolic dysfunction of the myocardium.
(2) Mutations in the sarcomere-related genes in HCM can lead to inefficient contractility with a resultant increase in the ATP demand. This impairs the structure and function of the mitochondria leading to energy supply disorders (13–16). Microvascular dysfunction further exacerbates the myocardial energy deficiency of HCM and restricts the transport of the oxidative metabolites. The imbalance between the energy supply and demand leads to the myocardial cells in a state of peroxidation and then produces various reactive oxygen species (ROS), resulting in the glutathione acylation of the muscle filaments [cardiac myosin-binding protein C (cMyBP-C)] (17, 18). Functionally, this modification increases the myofilament calcium sensitivity and inhibits the kinetics of cross-bridge cycling, leading to the diastolic dysfunction and ultimately aggravating the HCM phenotype (18–21).
(3) Due to the mutations in the sarcomere-related genes, the accumulation of the harmful proteins results in a toxic effect on the myocardial contractile devices and myocardial cells (22).
(4) Sarcomeric protein transcription and posttranslational modifications, as well as the other modified genes, also promote the development of HCM. Studies have shown that polymorphism of angiotensin I can contribute to the hypertrophic phenotype (23). These modified factors stimulate non-cardiac cell proliferation such as fibroblasts (23), thereby promoting the development of HCM. In conclusion, the functional changes at the cellular and molecular levels could be target of innovative therapies.
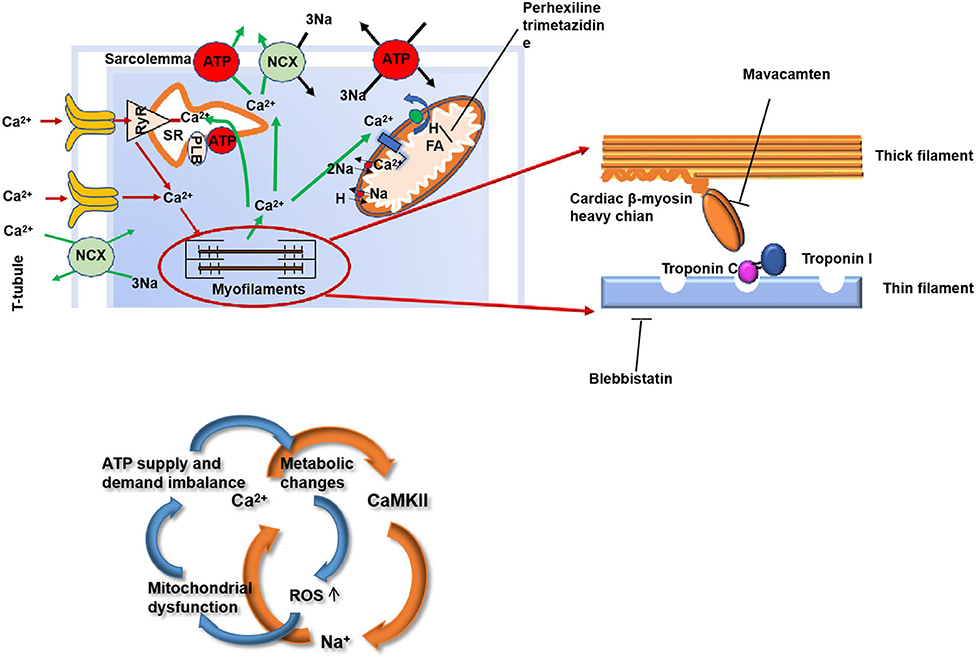
Figure 2. The structure of the myocardium and the mechanism of myocardial contraction are the potential targets for HCM therapy. HCM, hypertrophic cardiomyopathy; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; NCX, sodium/calcium exchange pump; PLB, phospholamban; ROS, reactive oxygen species; FA, fatty acid.
The structural abnormalities of HCM include the following:
(1) Abnormal myofibrils and abnormal arrangement of the cardiomyocytes (24).
(2) Coronary artery microvascular dysfunction. Thickening of the blood vessel wall leading to asymptomatic myocardial ischemia, further inducing myocardial injury and fibrosis (25, 26). Interstitial connective tissue increases significantly. Fibrosis is patchy or widely distributed around the cells and poor remodeling of the myocardial tissue ultimately leads to irreversible dysfunction such as severe HF and SCD (27, 28).
Ca2+ overload, CaMKII, and increased INaL play a very important role that drive the myocardial remodeling from the earliest stage of the development of hypertrophy, diastolic dysfunction, and the arrhythmogenic substrate (5, 6). There are many studies aimed at an increased calcium sensitivity (Figure 2).
Blebbistatin is an inhibitor of actin-myosin interaction functioning independently of Ca2+ influx (29). Studies have shown that blebbistatin, in a mouse model of HCM caused by troponin T mutation, can reduce the sensitivity of Ca2+ to myofilaments and the incidence of arrhythmias; meanwhile, several studies Grillo et al. also reported that reducing the sensitivity of Ca2+ to myofilaments can be a target for the HCM treatment (29–32).
Parvalbumin is a Ca2+ buffering molecule not expressed in the cardiomyocytes; when the concentration of Ca2+ increases, parvalbumin will release Mg2+ and binds to Ca2+. Piguet et al. and Coutu et al. found that parvalbumin can correct the myocardial diastolic dysfunction in the rat and mouse HCM models (33, 34).
In a mouse model of HCM caused by a tropomyosin mutation, SERCA2a, an SR protein, was transported by the adenovirus to 1-day-old mice. After several weeks, The sarco/endoplasmic reticulum calcium ATPase 2a isoform (SERCA2a) protein expression increased in the heart of the mouse and significantly improved the morphology of the heart. In HCM mice knocked out with SERCA2a inhibitory protein, phosphoprotein gene [phospholamban (PLN)] can also increase the absorption of Ca2+ by the sarcoplasmic reticulum (SR) and improve the phenotype (35).
Diltiazem can inhibit the L-type calcium channels (34). Early application of diltiazem caused the upregulation of the SR protein and eased the development of the HCM phenotype (36). In recent years, a study by Ho CY et al. found that diltiazem may relieve left ventricular remodeling in the asymptomatic sarcomere mutation carriers (NCT00319982) (37). The summarized novel therapies can be seen in Table 2.

Table 2. Table for the novel therapies.
In HCM, the mutations in the sarcomere gene result in reduced contractile efficiency of the sarcomere and an increase in ATP consumption. The characteristic of the HCM substrate metabolism is the preferential use of fatty acid (FA) oxidation, but in order to adapt to the consumption of more ATP, energy metabolism transfers to glucose metabolism to produce more ATP. This increased glucose metabolism, however, results in the accumulation of pyruvate and lactate produced by glycolysis, which is harmful to the myocardium (Figure 2).
Perhexiline improves the energy production efficiency by transferring the substrate utilization from the free fatty acids (FFAs) to glucose and improves the symptoms, exercise capacity (VO2max), and function of heart in the patients with systolic heart failure caused by the ischemic and non-ischemic factors that are very effective (38). Perhexiline promotes the use of carbohydrates as the substrate for the myocardial energy by inhibiting carnitine palmitoyltransferase 1 (CPT1); meanwhile, CPT2 resulting in increased efficiency of the myocardial oxygen utilization (39). Perhexiline would be likely to induce an increase of at least 13% efficiency of the myocardial oxygen utilization (40). Perhexiline appears to exert the important anti-inflammatory (in part via nicotinamide adenine dinucleotide phosphate oxidase inhibition) and nitric oxide-potentiating effects that may occur independently of CPT inhibition (NCT00500552) (41).
During the period of myosin force production, there is an autoinhibited state, also referred to as a super-relaxed state. With certain myosin mutations, the HCM sarcomere spends lesser time in this state, resulting in the hyperactivation and excess utilization of ATP. The small molecule mavacamten stabilizes this inhibited state, effectively extend the time that myosin is inhibited. Mavacamten is specific for β-myosin heavy chain. Many studies in the mouse HCM models have pointed out that the early treatment of phenotype-negative HCM mice can prevent HCM hypertrophy and other features (42). Administration to the HCM mice reduced the hypertrophic phenotype and reduced the expression of the fibrotic genes (43). The gradient of 8 out of 21 participating patients had significantly reduced LVOT to <30 mm Hg. It also resulted in the reduced serum N-terminal pro B-type natriuretic peptide (NT-proBNP) levels in patients with HCM. This is the biomarker associated with increased wall stress and myocardial injury (NCT03470545) (43). The trial was designed to evaluate the dose of mavacamten in the non-obstructive HCM. This study found that the mavacamten treatment group had no significant toxicity compared to the placebo group, proving that the drug was well-tolerated (NCT03442764) (44).
Omecamtiv mecarbil (OM) is being tested in treating hypercontractility by accelerating ATP generation, thus increasing myosin head binding to actin, resulting in an enhanced force-producing situation (45). The effects of OM are dependent on the intracellular Ca2+ levels (46). OM has shown promising clinical practical values, progressing to phase III trials (NCT02929329). The summarized metabolic therapy can be seen in Table 3.

Table 3. Table for metabolic therapy.
In the past decades, gene therapy got tremendous development in the field of HCM. From the current evidence, gene therapy seems a very promising treatment in HCM caused by the mutations in the genes that encode the sarcomeric proteins.
The key problem for gene therapy is the effective and safe delivery of the gene therapy drugs into the body of the patient. It has been shown that the adeno-associated virus serotype 9 (AAV9) is a very promising candidate for cardiac gene transfer after systemic delivery in mouse and large animal HCM models (47). The SERCA2a gene therapy phase II trial also showed a very exciting result of the safety and feasibility of AAV1-mediated gene transfer (48). However, this investigation has not shown significant positive outcomes in the treated patients (49, 50). The defect of AAV-mediated gene therapy is that the human body easily generates neutralizing antibodies against AAV. These neutralizing antibodies seriously impact the outcomes of gene therapy; another question is an increased readministration rate. This could be avoided by the pharmacological modulation of the immune response and/or use of another AAV serotype (51, 52).
Fortunately, the difficulty of delivering the gene therapy medicinal product into the body of the patient has been resolved to some extent. Various methods were developed to suppress the expression of the genetic defects on the DNA or RNA levels as well as genome editing (42, 53–58), exon skipping (59, 60), allele-specific silencing, (61–63) spliceosome-mediated RNA trans-splicing (61–64), and gene replacement therapy (49, 65, 66). Due to the advancement in genome modification technologies, antisense oligonucleotides, clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9, wild-type complementary DNA (cDNA) (wild-type MYBPC3 cDNA) sequences, and RNA interference molecules are clustered regularly interspaced. Specific editing that promotes the genetic mutations of an individual may lead to the individual-based pharmacological approaches in HCM. The summarized gene therapy can be seen in Table 4.
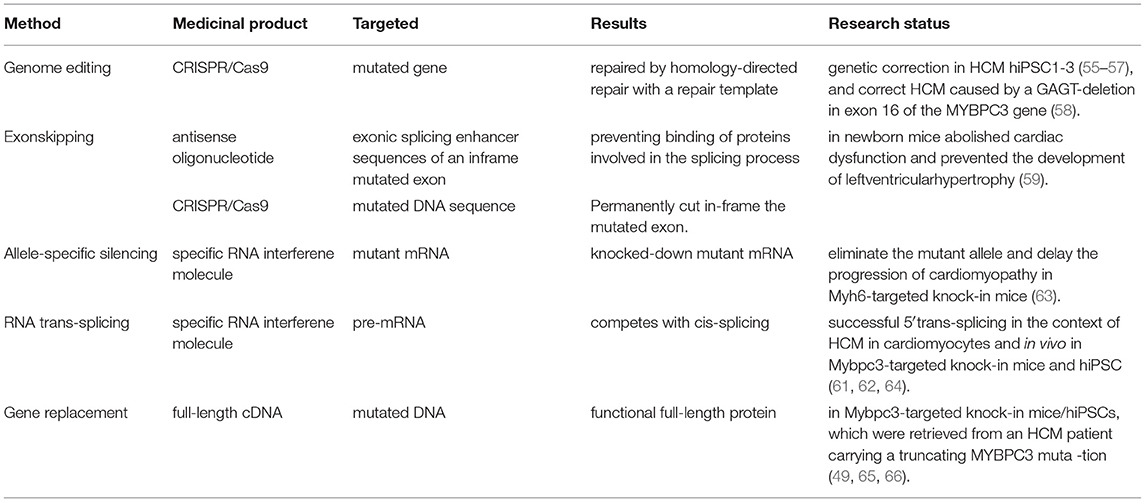
Table 4. Table for the gene therapy.
Since there are many promising treatments for HCM, it is still a complex disease that requires further study based on pathophysiology and genetics. It is necessary to further study the mechanism of gene mutations and the secondary events caused by HCM. Thus, we need to develop new therapies based on gene editing or molecular regulatory pathways. Meanwhile, a large amount of basic medical research on the pathogenesis and treatment of HCM needs to be further transformed into clinical application. In conclusion, HCM is the main hereditary disease of the heart and the sarcomeric protein gene mutation is the most common cause of HCM. HCM is hereditary cardiomyopathy. Continued study and improved understanding of the genetic mediators of HCM will help to guide the development of effective targeted therapies, small molecules that can target the key molecular pathways or events in the heart to prevent the natural course of HCM. Increasing the treatment options for HCM may block the progression of the HCM disease, but it is not possible to completely correct the mutant gene and there are still genetic risks. Finally, a better understanding of the structural and metabolic disorders caused by the gene mutations is very helpful for developing the new therapies of HCM.
YG: conception. MA and ZC: administrative support. HQ: study design. TF: collection and assembly of data. JH: data analysis and drawing. All authors: manuscript writing and final approval of manuscript.
This work was supported by the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (grant number: ZY 2017306) and Sichuan Provincial Scientific Grant: The effect and mechanism of HIPPO-YAP pathway on pulmonary artery remodeling in left heart disease-related pulmonary hypertension (2021YFS0247).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Thanks to Prof. Alam for his guidance and polishing of the English language in this article.
1. Maron BJ, Maron MS. The 20 advances that have defined contemporary hypertrophic cardiomyopathy. Trends Cardiovasc Med. (2015) 25:54–64. doi: 10.1016/j.tcm.2014.09.004
2. Masarone D, Kaski JP, Pacileo G, Elliott PM, Bossone E, Day SM, et al. Epidemiology and clinical aspects of genetic cardiomyopathies. Heart Fail Clin. (2018) 14:119–28. doi: 10.1016/j.hfc.2017.12.007
3. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. (2011) 124:e783–831. doi: 10.1161/CIR.0b013e318223e2bd
4. Santos Mateo JJ, Sabater Molina M, Gimeno Blanes JR. Hypertrophic cardiomyopathy. Med Clin. (2018) 150:434–42. doi: 10.1016/j.medcle.2017.09.029
5. Hensley N, Dietrich J, Nyhan D, Mitter N, Yee MS, Brady M. Hypertrophic cardiomyopathy: a review. Anesth Analg. (2015) 120:554–69. doi: 10.1213/ANE.0000000000000538
6. Makavos G, Kappaairis C, Tselegkidi ME, Karamitsos T, Rigopoulos AG, Noutsias M, et al. Hypertrophic cardiomyopathy: an updated review on the diagnosis, prognosis, and treatment. Heart Fail Rev. (2019) 24:439–59. doi: 10.1007/s10741-019-09775-4
7. Wolf CM. Hypertrophic cardiomyopathy: genetics and clinical perspectives. Cardiovasc Diagn Ther. (2019) 9(Suppl 2):S388–415. doi: 10.21037/cdt.2019.02.01
8. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121:749–70. doi: 10.1161/CIRCRESAHA.117.311059
9. Coppini R, Ferrantini C, Del Lungo M, Stillitano F, Sartiani L, Tosi B, et al. Response to letter regarding article, late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. (2013) 128:e157. doi: 10.1161/CIRCULATIONAHA.113.004016
10. Coppini R, Mazzoni L, Ferrantini C, Gentile F, Pioner JM, Laurino A, et al. Ranolazine prevents phenotype development in a mouse model of hypertrophic cardiomyopathy. Circ Heart Fail. (2017) 10:e003565. doi: 10.1161/CIRCHEARTFAILURE.116.003565
11. Ferrantini C, Coppini R, Pioner JM, Gentile F, Tosi B, Mazzoni L, et al. Pathogenesis of hypertrophic cardiomyopathy is mutation rather than disease specific: a comparison of the cardiac troponin T E163R and R92Q mouse models. J Am Heart Assoc. (2017) 6:e005407. doi: 10.1161/JAHA.116.005407
12. Ferrantini C, Pioner JM, Mazzoni L, Gentile F, Tosi B, Rossi A, et al. Late sodium current inhibitors to treat exercise-induced obstruction in hypertrophic cardiomyopathy: an in vitro study in human myocardium. Br J Pharmacol. (2018) 175:2635–52. doi: 10.1111/bph.14223
13. Witjas-Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen GJ, et al. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol. (2014) 592:3257–72. doi: 10.1113/jphysiol.2014.274571
14. Belus A, Piroddi N, Scellini B, Tesi C, D'Amati G, Girolami F, et al. The familial hypertrophic cardiomyopathy- associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. (2008) 586:3639–44. doi: 10.1113/jphysiol.2008.155952
15. Nediani A, Borchi E, Giordano C, Baruzzo S, Ponziani V, Sebastiani M, et al. NADPH oxidase-dependent redox signaling in human heart failure: relationship between the left and right ventricle, J Mol Cell Cardiol. (2007) 42:826–34. doi: 10.1016/j.yjmcc.2007.01.009
16. Sebastiani M, Giordano C, Nediani C, Travaglini C, Borchi E, Zani M, et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J Am Coll Cardiol. (2007) 50:1362–9. doi: 10.1016/j.jacc.2007.06.035
17. Becker RC, Owens AP 3rd, Sadayappan S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J Thromb Thrombolysis. (2020) 49:177–83. doi: 10.1007/s11239-019-02026-1
18. Miceli C, Santin Y, Manzella N, Coppini R, Berti A, Stefani M, et al. Oleuropein aglycone protects against MAO-A-Induced autophagy impairment and cardiomyocyte death through activation of TFEB. Oxid Med Cell Longev. (2018) 2018:8067592. doi: 10.1155/2018/8067592
19. Santin Y, Sicard P, Vigneron F, Guilbeau-Frugier C, Dutaur M, Lairez O, et al. Oxidative stress by monoamine Oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid Redox Signal. (2016) 25:10–27. doi: 10.1089/ars.2015.6522
20. Singh SR, Zech ATL, Geertz B, Reischmann-Dusener S, Osinska H, Prondzynski M, et al. Carrier, activation of autophagy ameliorates cardiomyopathy in Mybpc3-targeted knockin mice. Circ Heart Fail. (2017) 10:e004140. doi: 10.1161/CIRCHEARTFAILURE.117.004140
21. Xu X, Roe ND, Weiser-Evans MC, Ren J. Inhibition of mammalian target of rapamycin with rapamycin reverses hypertrophic cardiomyopathy in mice with cardiomyocyte-specific knockout of PTEN. Hypertension. (2014) 63:729–39. doi: 10.1161/HYPERTENSIONAHA.113.02526
22. Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. (2009) 105:219–22. doi: 10.1161/CIRCRESAHA.109.202440
23. Geske JB, Ommen SR, Gersh BJ. Hypertrophic cardiomyopathy: clinical update. JACC Heart Fail. (2018) 6:364–75. doi: 10.1016/j.jchf.2018.02.010
24. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. (2018) 379:655–68. doi: 10.1056/NEJMra1710575
25. Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. (2017) 389:1253–67. doi: 10.1016/S0140-6736(16)31321-6
26. Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic cardiomyopathy: an overview of genetics and management. Biomolecules. (2019) 9:878. doi: 10.3390/biom9120878
27. Daubert C, Gadler F, Mabo P, Linde C. Pacing for hypertrophic obstructive cardiomyopathy: an update and future directions. Europace. (2018) 20:908–20. doi: 10.1093/europace/eux131
28. Maron BJ, Rowin EJ, Maron MS, Braunwald E. Nonobstructive hypertrophic cardiomyopathy out of the shadows: known from the beginning but largely ignored … until now. Am J Med. (2017) 130:119–23. doi: 10.1016/j.amjmed.2016.09.015
29. Huke S, Venkataraman R, Faggioni M, Bennuri S, Hwang HS, Baudenbacher F, et al. Focal energy deprivation underlies arrhythmia susceptibility in mice with calcium-sensitized myofilaments. Circ Res. (2013) 112:1334–44. doi: 10.1161/CIRCRESAHA.113.301055
30. Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. (2008) 118:3893–903. doi: 10.1172/JCI36642
31. Pohlmann L, Kröger I, Vignier N, Schlossarek S, Krämer E, Coirault C, et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res. (2007) 101:928–38. doi: 10.1161/CIRCRESAHA.107.158774
32. Fiset C, Giles WR. Cardiac troponin T mutations promote life-threatening arrhythmias. J Clin Invest. (2008) 118:3845–7. doi: 10.1172/JCI37787
33. Piguet F, de Montigny C, Vaucamps N, Reutenauer L, Eisenmann A, Puccio H. Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of Friedreich Ataxia. Mol Ther. (2018) 26:1940–52. doi: 10.1016/j.ymthe.2018.05.006
34. Coutu P, Bennett CN, Favre EG, Day SM, Metzger JM. Parvalbumin corrects slowed relaxation in adult cardiac myocytes expressing hypertrophic cardiomyopathylinked alpha-tropomyosin mutations. Circ Res. (2004) 94:1235–41. doi: 10.1161/01.RES.0000126923.46786.FD
35. Peña JR, Szkudlarek AC, Warren CM, Heinrich LS, Gaffin RD, Jagatheesan G, et al. Neonatal gene transfer of Serca2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. (2010) 49:993–1002. doi: 10.1016/j.yjmcc.2010.09.010
36. Flenner F, Geertz B, Reischmann-Düsener S, Weinberger F, Eschenhagen T, Carrier L, et al. Diltiazem prevents stress-induced contractile deficits in cardiomyocytes, but does not reverse the cardiomyopathy phenotype in Mybpc3knock-in mice. J Physiol. (2017) 595:3987–99. doi: 10.1113/JP273769
37. Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail. (2015) 3:180–8. doi: 10.1016/j.jchf.2014.08.003
38. Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. (2010) 122:1562–9. doi: 10.1161/CIRCULATIONAHA.109.934059
39. Horowitz JD, Chirkov YY. Perhexiline and hypertrophic cardiomyopathy: a new horizon for metabolic modulation. Circulation. (2010) 122:1547–9. doi: 10.1161/CIRCULATIONAHA.110.981464
40. Gehmlich K, Dodd MS, Allwood JW, Kelly M, Bellahcene M, Lad HV, et al. Changes in the cardiac metabolome caused by perhexiline treatment in a mouse model of hypertrophic cardiomyopathy. Mol Biosyst. (2015) 11:564–73. doi: 10.1039/C4MB00594E
41. Drury NE, Licari G, Chong CR, Howell NJ, Frenneaux MP, Horowitz JD, et al. Relationship between plasma, atrial and ventricular perhexiline concentrations in humans: insights into factors affecting myocardial uptake. Br J Clin Pharmacol. (2014) 77:789–95. doi: 10.1111/bcp.12254
42. Carroll KJ, Makarewich CA, McAnally J, Anderson DM, Zentilin L, Liu N, et al. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci USA. (2016) 113:338–43. doi: 10.1073/pnas.1523918113
43. Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2020) 396:759–69. doi: 10.1016/S0140-6736(20)31792-X
44. Ho CY, Mealiffe ME, Bach RG, Bhattacharya M, Choudhury L, Edelberg JM, et al. evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. (2020) 75:2649–60. doi: 10.1016/j.jacc.2020.03.064
45. Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. (2011) 331:1439–43. doi: 10.1126/science.1200113
46. Kampourakis T, Zhang X, Sun YB, Irving M. Omecamtiv mercabil and blebbistatin modulate cardiac contractility by perturbing the regulatory state of the myosin filament. J Physiol. (2018) 596:31–46. doi: 10.1113/JP275050
47. Ishikawa K, Fish KM, Tilemann L, Rapti K, Aguero J, SantosGallego CG, et al. Cardiac I1c overexpression with reengineered AAV improves cardiac function in swine ischemic heart failure. Mol Ther. (2014) 22:2038–45. doi: 10.1038/mt.2014.127
48. Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. (2011) 124:304–13. doi: 10.1161/CIRCULATIONAHA.111.022889
49. Mearini G, Stimpel D, Geertz B, Weinberger F, Krämer E, Schlossarek S, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long term disease prevention in mice. Nat Commun. (2014) 5:5515. doi: 10.1038/ncomms6515
50. Murrey DA, Naughton BJ, Duncan FJ, Meadows AS, Ware TA, Campbell KJ, et al. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: toxicology, biodistribution, and immunological assessments in primates. Hum Gene Ther Clin Dev. (2014) 25:72–84. doi: 10.1089/humc.2013.208
51. Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med. (2011) 3:92ra64. doi: 10.1126/scitranslmed.3002097
52. Ziegler T, Kraus M, Husada W, Gesenhues F, Jiang Q, Pinkenburg O, et al. Steerable induction of the Thymosin beta4/MRTF-A pathway via AAV-based over expression induces therapeutic neo-vascularization. Hum Gene Ther. (2018) 29:1407–15. doi: 10.1089/hum.2017.013
53. Guo Y, Van Dusen NJ, Zhang L, Gu W, Sethi I, Guatimosim S, et al. Analysis of cardiac myocyte maturation using CASAA V, a platform for rapid dissection of cardiac myocyte gene function in vivo. Circ Res. (2017) 120:1874–88. doi: 10.1161/CIRCRESAHA.116.310283
54. Johansen AK, Molenaar B, Versteeg D, Leitoguinho AR, Demkes C, Spanjaard B, et al. Postnatal cardiac gene editing using CRISPR/Cas9 with AA V9-mediated delivery of short guide RNAs results in mosaic gene disruption. Circ Res. (2017) 121:1168–81. doi: 10.1161/CIRCRESAHA.116.310370
55. Ben Jehuda R, Eisen B, Shemer Y, Mekies LN, Szantai A, Reiter I, et al. CRISPR correction of the PRKAG2 gene mutation in the patient's induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm. (2018) 15:267–76. doi: 10.1016/j.hrthm.2017.09.024
56. Mosqueira D, Mannhardt I, Bhagwan JR, Slimak KL, Katili P, Scott E, et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J. (2018) 39:3879–92. doi: 10.1093/eurheartj/ehy249
57. Wang L, Kim K, Parikh S, Gabriel Cader A, Bersell KR, He H, et al. Hypertrophic cardiomyopathy-linked mutation in troponin T causes myofibrillar disarray and pro-arrhythmic action potential changes in human iPSC cardiomyocytes. J Mol Cell Cardiol. (2018) 114:320–7. doi: 10.1016/j.yjmcc.2017.12.002
58. Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature. (2017) 548:413–9. doi: 10.1038/nature23305
59. Gedicke-Hornung C, Behrens-Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, et al. Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol Med. (2013) 5:1128–45. doi: 10.1002/emmm.201202168
60. Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. (2009) 105:239–48. doi: 10.1161/CIRCRESAHA.109.201251
61. Mearini G, Stimpel D, Kramer E, Geertz B, Braren I, Gedicke-Hornung C, et al. Repair of Mybpc3 mRNA by 5′-trans-splicing in a mouse model of hypertrophic cardiomyopathy. Mol Ther Nucleic Acids. (2013) 2:e102. doi: 10.1038/mtna.2013.31
62. Prondzynski M, Kramer E, Laufer SD, Shibamiya A, Pless O, Flenner F, et al. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol Ther Nucleic Acids. (2017) 7:475–86. doi: 10.1016/j.omtn.2017.05.008
63. Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. (2013) 342:111–4. doi: 10.1126/science.1236921
64. Azibani F, Brull A, Arandel L, Beuvin M, Nelson I, Jollet A, et al. Gene therapy via trans-splicing for LMNA-related congenital muscular dystrophy. Mol Ther Nucleic Acids. (2018) 10:376–86. doi: 10.1016/j.omtn.2017.12.012
65. Monteiro da Rocha A, Guerrero-Serna G, Helms A, Luzod C, Mironov S, Russell M, et al. Deficient cMyBP-C protein expression during cardiomyocyte differentiation underlies human hypertrophic cardiomyopathy cellular phenotypes in disease specific human ES cell derived cardiomyocytes. J Mol Cell Cardiol. (2016) 99:197–206. doi: 10.1016/j.yjmcc.2016.09.004
Keywords: hypertrophic cardiomyopathy, phenotype, pathogenesis, treatment, review
Citation: Cheng Z, Fang T, Huang J, Guo Y, Alam M and Qian H (2021) Hypertrophic Cardiomyopathy: From Phenotype and Pathogenesis to Treatment. Front. Cardiovasc. Med. 8:722340. doi: 10.3389/fcvm.2021.722340
Received: 08 June 2021; Accepted: 17 September 2021;
Published: 25 October 2021.
Edited by:
Yihua Bei, Shanghai University, ChinaReviewed by:
Kazufumi Nakamura, Okayama University, JapanCopyright © 2021 Cheng, Fang, Huang, Guo, Alam and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Qian, cWlhbmhvbmcyMjIyQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.