 Jiangen Liu
Jiangen Liu Xianjing Song
Xianjing Song Youyou Yan
Youyou Yan Bin Liu
Bin Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 30 September 2021
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.720085
This article is part of the Research Topic Extracellular Vesicles in Cardiovascular Diseases View all 17 articles
Heart function maintenance requires a large amount of energy, which is supplied by the mitochondria. In addition to providing energy to cardiomyocytes, mitochondria also play an important role in maintaining cell function and homeostasis. Although adult cardiomyocyte mitochondria appear as independent, low-static organelles, morphological changes have been observed in cardiomyocyte mitochondria under stress or pathological conditions. Indeed, cardiac mitochondrial fission and fusion are involved in the occurrence and development of heart diseases. As mitochondrial fission and fusion are primarily regulated by mitochondrial dynamins in a GTPase-dependent manner, GTPase-dependent mitochondrial fusion (MFN1, MFN2, and OPA1) and fission (DRP1) proteins, which are abundant in the adult heart, can also be regulated in heart diseases. In fact, these dynamic proteins have been shown to play important roles in specific diseases, including ischemia-reperfusion injury, heart failure, and metabolic cardiomyopathy. This article reviews the role of GTPase-dependent mitochondrial fusion and fission protein-mediated mitochondrial dynamics in the occurrence and development of heart diseases.
Over the course of a human lifetime, the heart beats ~2.5 billion times. The enormous amount of energy required for this cyclic beating process is provided by the mitochondria of myocardial cells. Mitochondria are membrane-bound organelles found in the cytoplasm of almost all eukaryotic cells (cells with clearly defined nuclei), the primary function of which is to generate large quantities of energy in the form of adenosine triphosphate (ATP). Aside from their role in energy production, mitochondria also have a vital role in maintaining cell homeostasis.
Mitochondria are organelles whose morphology and function are interconnected. Through the dynamic balance of fission and fusion, the normal morphology of mitochondria is regulated to meet the energy and metabolic needs of the cell. These dynamic changes in morphology maintain the integrity and distribution of mitochondria, thereby allowing their function to adapt to changing physiological needs (1, 2). Mitochondria are highly abundant and widely distributed throughout the cytoplasm of cardiomyocytes. In fact, more than 40% of the cytoplasmic space of adult cardiac cardiomyocytes is occupied by densely packed mitochondria (3), which are primarily located between the sarcomeres of cardiomyocytes, around the nucleus, and under the serosa (3), allowing them to continuously provide energy for myocardial contraction. Therefore, the morphology of mitochondria is essential to maintain the health of cardiomyocytes. Cardiac mitochondria can be divided into two types based on their location, namely, subserosa mitochondria and interfiber mitochondria. The cristae morphology of these two mitochondrion types differs; that is, the subserosa mitochondria are flaky, whereas interfiber mitochondria are primarily tubular. However, cristae morphology can change in response to the pathophysiological state of heart disease, suggesting that mitochondrial morphology also has an important role in cardiomyocyte homeostasis (4).
Mitochondrial fission is mediated by dynamin-related protein 1 (DRP1), a GTPase-dependent enzyme that is recruited to the outer mitochondrial membrane through a series of receptor proteins (MFF, FIS1, MID49, and MID50) (5). Together, DRP1 and FIS1 mediate fission and participate in apoptosis activation, necrosis, and autophagy (6), after which DRP1 divides the mitochondria into two in a GTP-dependent manner (See in Figure 1). In mammals, mitochondrial fusion is generally regulated by three GTPase-dependent proteins in the dynamin superfamily, namely, mitofusin-1 (MFN1), mitofusin-2 (MFN2), and optic atrophy 1 (OPA1), all of which participate in the processes of energy metabolism, mitochondrial permeability transition, and calcium homeostasis (6, 7). Generally, mitochondrial fusion requires three steps. First, MFN1 or MFN2 reversibly approach the outer mitochondrial membranes. Second, the outer membranes of the two mitochondria fuse via MFN1 and MFN2 under GTPase mediation. Third, the inner mitochondrial membranes fuse via OPA1 in a GTPase-dependent manner (Figure 2).
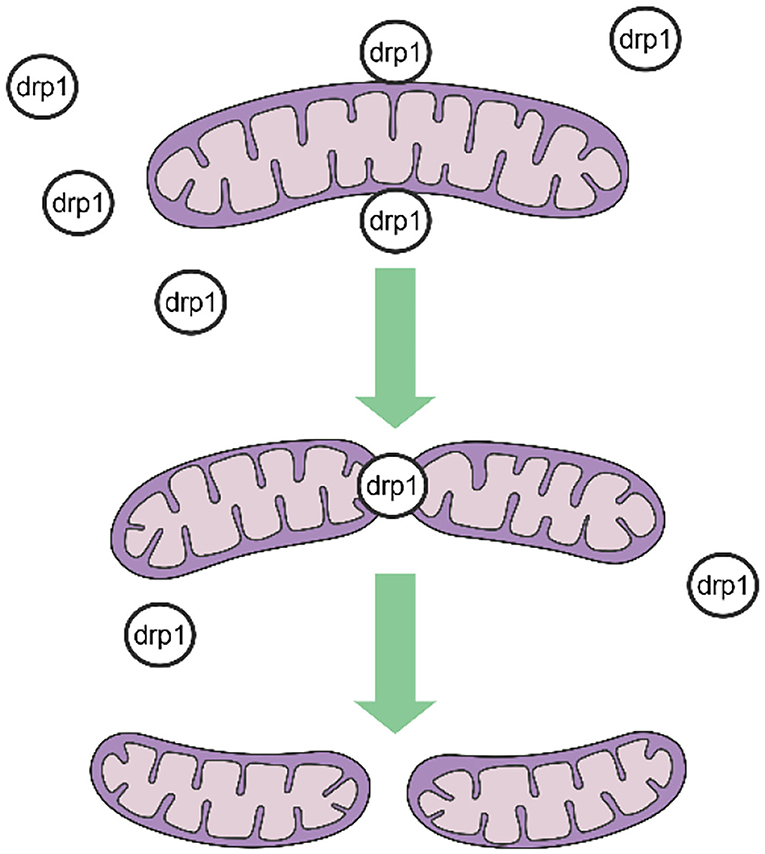
Figure 1. Procedures of mitochondrial fission.
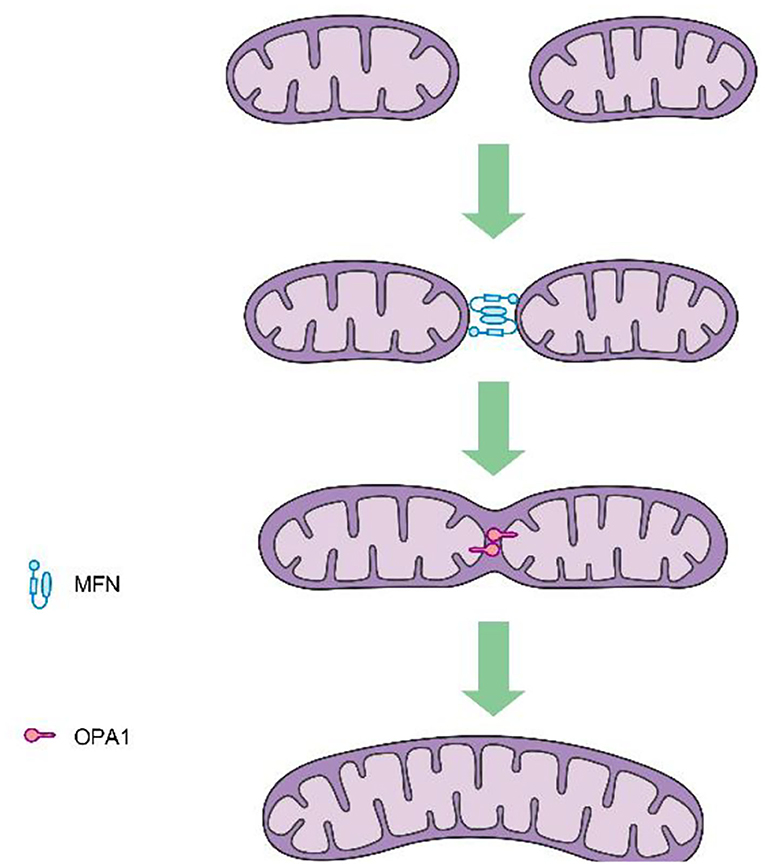
Figure 2. Procedures of mitochondrial fusion.
In most organs, mitochondrial dynamics are highly active, with mitochondria continually undergoing fusion, fission, transportation, and degradation. These dynamic processes are necessary to maintain the normal shape, health, and number of mitochondria, thus, maintaining cellular physiological functioning. Yet, the description of mitochondria in a hyperdynamic state may not be suitable to adult cardiomyocytes, where the mitochondria appear as independent, lowly dynamic organelles. Meanwhile, under stress or pathological conditions, morphological changes have been observed in myocardial mitochondria. Moreover, the delicate balance between fission and fusion is critical to ensure a steady energy supply and cardiomyocyte hemostasis (8). To date, many studies have confirmed that the fusion and fission dynamins of cardiac mitochondria, in particular the GTPase dependent dynamins (DRP1, MFN1, MFN2, OPA1) are associated with the occurrence and development of various heart diseases. As such, this article provides a review of current literature related to the effects of these specific GTPase-dependent dynamins that mediate mitochondrial fusion and fission in myocardial tissue.
Along with mitochondrial fusion, fission is essential to maintain normal cell and tissue physiological functions (9, 10). Mitochondrial fission is mediated by the GTPase-dependent dynamin DRP1, which is composed of four fragments: the N-terminal GTPase active fragment, middle fragment, variable fragment, and C-terminal GTPase effector fragment. Before Drp1 participates in the fission of mitochondria, a series of proteins are needed to transport it to the outer mitochondrial membrane. These proteins are called Drp1 Adaptors. The adaptors include fission 1 (Fis1), mitochondrial fission factor (Mff) and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49, MiD51) (5). And MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and suggest that they provide specificity to the division of mitochondria (11). A number of reports have shown that Fis1 is not required for Drp1 recruitment, but Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells (12).
Through the action of various kinases and phosphatases, the C-terminal GTPase effector fragment is reversibly phosphorylated to achieve DRP1 aggregation on the surface of mitochondria (13, 14). This DRP1 aggregation allows for mitochondria reshaping in response to intracellular or extracellular signals. Phosphorylation of the commonly confirmed DRP1 sites, Ser616 and Ser637, represents important regulatory mechanisms following DRP1 translation, whereby Ser616 phosphorylation induces DRP1 activity, while Ser637 phosphorylation reduces DRP1 activity (15). For example, during cell mitosis, CDK1 enhances mitochondrial division through Ser616 phosphorylation (16). Meanwhile, phosphorylation of Ser637 on the DRP1 C-terminal GTPase effector fragment by cAMP-dependent protein kinase, reduces the GTPase activity of DRP1 and inhibits mitochondrial fission (17). In contrast, dephosphorylation of Ser637 mediated by calcium-dependent phosphatase and calcineurin increases DRP1 recruitment from the cytoplasm to the mitochondria (18). Additionally, the sumoylation status of DRP1 can also affect its intracellular localization; that is, when DRP1 is modified by SUMO-1, localization of DRP1 to mitochondria increases (19, 20), while DRP1 modification by SUMO-2/3 inhibits its localization to mitochondria (21) (Figure 3).
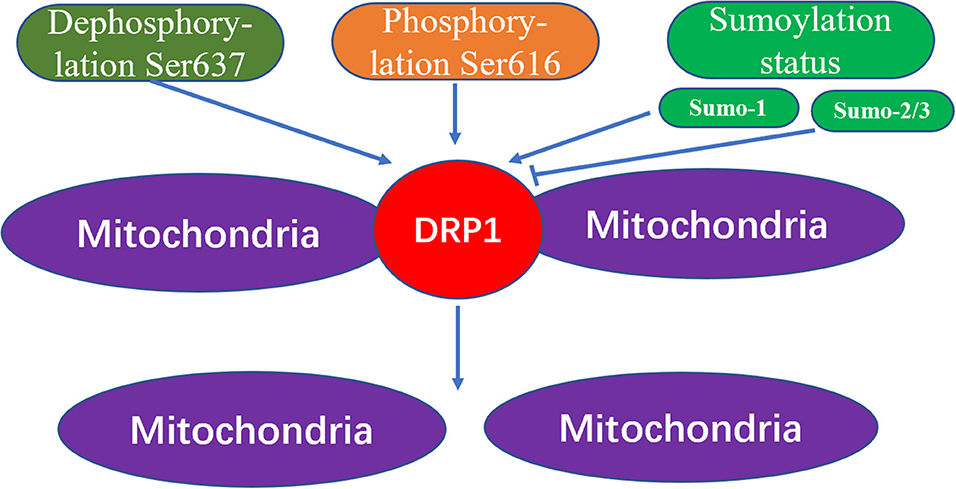
Figure 3. The mechanism of mitochondrial fission caused by Drp1 modification. The increased dephosphorylation of Ser637 on the drp1 protein or the increased phosphorylation of Ser616, and the change of the sumoylation state of DRP1 will effect the activity of DRP1, thereby increasing the fission of mitochondria.
Endogenous DRP1 plays an important role in mitochondrial quality control and cardiomyocyte survival. For example, Drp1 knock out in mice leads to embryonic lethality, where the embryos generally die within 9.5–11.5 embryonic days, a phenotype accompanied by elongated mitochondria, decreased cell differentiation, and decreased developmental regulatory apoptosis (22). Moreover, targeted knock out of Drp1 in murine heart tissue after birth caused dilated cardiomyopathy and death. Meanwhile, the cardiomyocyte mitochondria exhibited internally aggregated ubiquitin proteins accompanied by weakened cell respiration (23–25). The effects associated with DRP1 were also demonstrated with a mutation scan of N-ethyl-N-nitrosourea, in which mice with Drp1 mutations developed dilated cardiomyopathy related to reduced levels of respiratory complexes and loss of ATP (26). Additionally, three studies that specifically knocked out Drp1 in murine cardiac tissues reported that lack of DRP1 led to enlarged mitochondria, while DRP1 loss was fatal during the perinatal period or in the mature heart. Moreover, mitochondrial quality and abundance were found to be related to mitochondrial autophagy (24, 27, 28). The specific deletion of myocardial Drp1 not only increases the mitochondrial size, but also causes their abundance to progressively decrease (23).
In mice with muscle-specific Drp1 knockout, all pups died between 7 and 10 days after delivery, however, the death was not accompanied by neonatal defects or weight changes. Meanwhile, the hearts of Drp1 knockout mice were enlarged 7 days after delivery, accompanied by a thinning of the heart walls, however, the heart weight remained normal. Ultrasonography revealed clear cardiac dysfunction, including an increase in the end-systolic diameter (0.85 mm in the control compared to 1.64 mm in the Drp1 knockout) and a decrease in the shortening rate of the left ventricular short axis (46.5% in the control compared to 21.4% in the Drp1 knockout). Furthermore, heart-specific knockout of Drp1 via gene recombination after birth caused cardiomyopathy and death in young mice (28). Collectively, these results indicate that DRP1 is necessary for the maintenance of neonatal heart function.
DRP1 clearly plays an important role in embryogenesis and heart development in newborn mice. During these developmental stages, Drp1 inhibition or knock out causes enlargement and excessive fusion of mitochondria, both of which are lethal to mice. Hence, DRP1 has important roles in different stages of ontogeny. Under normal physiological conditions, inhibiting or silencing Drp1 reduces mitochondrial fission, increases fusion, and causes loss of heart function and even death. Under pathological conditions, the DRP1 expression becomes increased; its subsequent excessive phosphorylation or acetylation cause apoptosis and autophagy resulting in excessive mitochondrial fission and reduced fusion, thus further inducing cardiac dysfunction.
One study found that within 60 min of ischemia reperfusion, DRP1 expression increased with excessive mitochondrial fission following localization of DRP1 and BAX (apoptosis-related protein) to the mitochondria (29). During this process, MFN1 and MFN2 activity was inhibited, which may have led to induced activation of the mitochondrial permeability transition pore (MPTP) (30), causing subsequent mitochondrial fragmentation and permeation of the outer membrane (31, 32). Increased mitochondrial outer membrane permeability or opening of the MPTP leads to cytochrome C release, thus, further activating the caspase apoptosis pathway (33, 34). In the case of ischemia-reperfusion injury, inhibition of DRP1 activity elicits a protective effect on the heart. Thus, in different stages of the life cycle and development, as well as during physiological and pathological states, the degree of change in DRP1 activity differs, leading to different effects on cardiomyocytes and the heart.
As detailed above, the excessive mitochondrial fission observed within 60 min of reperfusion after transient ischemia, led to mitochondrial dysfunction and a decrease in myocardial contractility. However, P110, an inhibitor of DRP1, can reverse the decrease in myocardial contractility, thereby improving heart function (35). During reperfusion, dephosphorylation of Ser637 on DRP1 induces the transfer of DRP1 to the mitochondrial membrane and enhances mitochondrial fission (36). This dephosphorylation event in ischemia-reperfusion injury can be mediated by calcineurin (36, 37). Thus, because FK506 is an inhibitor of calcineurin, it can inhibit Ser637 dephosphorylation and ultimately preserve cardiac function during ischemia-reperfusion injury (36). In addition, AKT (protein kinase B) can activate DRP1 (38). For instance, in diabetes, the level of SIRT1 (sirtuin 1) in cardiomyocytes decreases, causing increased phosphorylation of Ser473 on AKT, leading to increased dephosphorylation of Ser637 on DRP1, thereby enhancing DRP1 activity, which subsequently increases mitochondrial fission and aggravates cardiac ischemia-reperfusion injury (39). Meanwhile, mammalian STE20-like kinase 1 (MST1) is upregulated after myocardial infarction and increases mitochondrial fission through the JNK-DRP1 (DJ-1) cell signal transduction pathway, leading to cardiomyocyte fibrosis and aggravating myocardial damage (40). Moreover, DJ-1 activation can alter the sumoylation state of DRP1, thereby inhibiting mitochondrial division, and playing a protective role in ischemia-reperfusion injury. A potential mechanism underlying this process may involve the interaction of DJ-1 with SENP5 to enhance DRP1 sumoylation mediated by SUMO-2/3, which is modified to inhibit mitochondrial division (41).
By inhibiting the DRP1 expression mitochondrial fission is also inhibited, which can alleviate cardiac ischemia-reperfusion injury. At the same time, increased mitochondrial fusion occurs in these DRP1-inhibited cardiomyocytes (36, 42, 43). These studies assessed the effect of mitochondrial fission in cultured cardiomyocytes by RNA interference and via expression of the main negative form of DRP1. That is, adenovirus-transfected cells overexpress the negative regulator of DRP1, K38A in vivo and in vitro, which subsequently protects myocardial cells from the damage associated with ischemia-reperfusion (43). In cardiac ischemia, endogenous levels of the microRNA miR-499 are downregulated, while calcineurin is activated, and Ser637 on DRP1 is dephosphorylated, thus, further increasing mitochondrial fragmentation, and in turn leading to increased apoptosis of cardiomyocytes. Thus, miR-499 overexpression inhibits calcineurin-mediated DRP1 dephosphorylation, thereby reducing cardiomyocyte apoptosis (44). miR-499 can also inhibit DRP1 through the cAMP-PKA cell signal transduction pathway, thereby prolonging the mitochondrial lifespan (17). SIRT3 can further reduce the phosphorylation level of DRP1 by normalizing AMPK cell signal transduction pathways, thus, inhibiting mitochondrial fission and ultimately reducing myocardial damage following myocardial infarction (45). Similarly, treatment of mice with the DRP1 inhibitor Mdivi-1 leads to decreased mitochondrial fission, thus imparting a protective effect against hypoxia and ischemia reperfusion (36, 42). Moreover, in vitro, a simulated ischemia-reperfusion model with cultured HL-1 cells pretreated with Mdivi-1 improved cell survival and delayed the opening of MPTP (42). In Mdivi-1 pretreated mice, reperfusion in the occluded artery and the area of myocardial infarction were found to decrease (42). In fact, emergency inhibition of mitochondrial division after acute myocardial infarction can improve long-term cardiac function (35, 36). Meanwhile, in a mouse cardiac arrest model, inhibition of myocardial mitochondria fission improves the survival rate of mice (46). In pressure load-induced heart failure, Mdivi-1 can also improve left ventricular function and reduce myocardial fibrosis (47). Furthermore, as an inhibitor of DRP1, P110 can inhibit the upregulation of the autophagy marker LC3-II whether it is applied before or after cardiac reperfusion, however, it also inhibits the increase in phosphorylation of cleaved caspase 3 and JNK when applied after reperfusion, both of which are associated with cell apoptosis and death (35, 48, 49). Indeed, treatment of the heart with Mdivi-1 and P110 elicited a protective effect against ischemia-reperfusion injury (35, 42, 50), as DRP1 inhibitors not only maintain mitochondrial morphology but also reduce calcium ions in the cytoplasm, inhibit the opening of MPTP, and inhibit cardiomyocyte apoptosis, thereby reducing myocardial damage. In fact, a recent study showed that injecting Mdivi-1 prior to myocardial infarction in ischemic mice can increase the length of mitochondria in myocardial cells (51). However, other studies that injected Mdivi-1 into the coronary arteries of larger animals, including pigs, as models for cardiac reperfusion after acute myocardial infarction, reported no significant changes in mitochondrial morphology between the blank control and experimental animals. Moreover, a protective cardiac effect was not detected, which may be related to the dose of Mdivi-1, timing of application, method of administration, etc. (52). Further studies are required to resolve these discrepancies in in vivo findings; hence, much work is still required before inhibitors of DRP1 can be recommended for the clinical treatment of heart diseases.
Diabetic cardiomyopathy refers to the abnormal structure and function of the heart caused by diabetes after excluding dangerous factors, including hypertension, coronary heart disease, and valvular disease, which may cause cardiac insufficiency. Mitochondrial dysfunction plays an important role in diabetic cardiomyopathy (53). Within minutes, acute hyperglycemia conditions can induce formation of shortened mitochondria mediated by the fission protein DRP1 (54). The acute response depends on the post-translational modification mechanism, which may involve glucose-induced intracellular calcium transients and ERK1/2 activation, causing phosphorylation of Ser616 on DRP1 (55). Specifically, PKCδ, CDK1, and CDK5 phosphorylate Ser616 to increase the fission activity of DLP1 (16, 56, 57). Melatonin regulates the expression of DRP1 through the SIRT1-PGC1α cell signal transduction pathway to reduce mitochondrial fission and ultimately alleviate diabetes-induced cardiac insufficiency (58). The rapid formation of small mitochondria under acute metabolic stress leads to an increase in mitochondrial reactive oxygen species (ROS). When the blood sugar continues to rise, the pathological fragmentation of mitochondria is related to the chronic increase in ROS levels, apoptosis, and necrosis (54, 59).
In the hearts of rats with chronic hypertension and hypertrophy, the level of cardiac DRP1 is reduced (60); however, myocardial DRP1 levels are elevated in the hypertrophic heart induced by neurohormone-norepinephrine (61). Studies have confirmed that mitochondria play an important role in cardiac hypertrophy (26, 62). For example, disorganized and fragmented mitochondria have been shown to cause cardiac hypertrophy (61, 63). Meanwhile, in vitro, within the NE (Norepinephrine)-induced cardiomyocyte hypertrophy model, an increase in accumulated DRP1 on the surface of mitochondria, as well as an increase in mitochondrial fragmentation, and a decrease in mitochondrial capacity are observed. However, if adenovirus-transfected cells are used to inhibit the expression of DRP1, mitochondrial fission is reduced, alleviating cardiomyocyte hypertrophy (61). Angiotensin-(1–9) can regulated mitochondria morphology through the AT2R/miR-129-3p/PKA cell signal transduction pathway, thereby inhibiting mitochondria fission and further reducing the occurrence of cardiomyocyte hypertrophy (64). Moreover, leptin can induce cardiac hypertrophy and activate calcineurin (65). The dephosphorylation of Ser637 on DRP1 mediated by calcineurin increases the recruitment of DRP1 from the cytoplasm to the mitochondria (18). It is, therefore, thought that leptin-induced cardiomyocyte hypertrophy is related to enhanced mitochondrial fission. In the cardiac hypertrophy cell model, fission of mitochondria has also been shown to participate in the calcium-dependent calcineurin cell signal transduction pathway (61). Mitochondrial fission is also crucial for the maintenance of heart function. Targeted Drp1 knock out in the adult mouse heart induced development of cardiomyopathy after 6–13 weeks (27, 28). Moreover, mitochondrial fission was significantly inhibited; however, this failed to inhibit the occurrence of heart failure. Meanwhile, in a murine model of pressure overload, Mdivi-1 reduced myocardial hypertrophy and fibrosis without affecting blood pressure (66). Similarly, in an aortic constriction model in which Drp1 was specifically knocked out of cardiac tissue cells via persistent heterozygous, excessive hypertrophy of cardiomyocytes and decreased heart function were observed. At the same time, it was confirmed that DRP1-dependent mitochondrial autophagy plays a role in resisting pressure load. With this important role, DRP1-dependent mitochondrial autophagy can ensure normal abundance of mitochondria and ultimately, contribute to amelioration of decreased heart function (67). Moreover, during heart failure, IGF-IIR can enhance the phosphorylation of Ser616 of DRP1 through extracellular signal-regulated kinase (ERK), thereby enhancing mitochondrial fission and causing mitochondrial dysfunction (68).
To make a summary of the whole section, DRP1 plays an important role in both the embryonic development period and the development of the newborn heart. DRP1 is elevated in ischemia-reperfusion injury causing excessive mitochondrial fission, thereby, further aggravating ischemia-reperfusion injury. DRP1 also plays an important role in the occurrence and development of heart failure, hence, inhibiting DRP1 can reduce ischemia-reperfusion injury and heart failure (Table 1). Specifically, DRP1 can affect mitochondrial morphology and function through phosphorylation, acetylation, sumoylation, and other modifications (Figure 4). Thus, DRP1 affects the occurrence and development of heart disease, which may be related to the imbalance of mitochondrial fission and fusion; however, further research is required to understand the specific mechanisms involved.
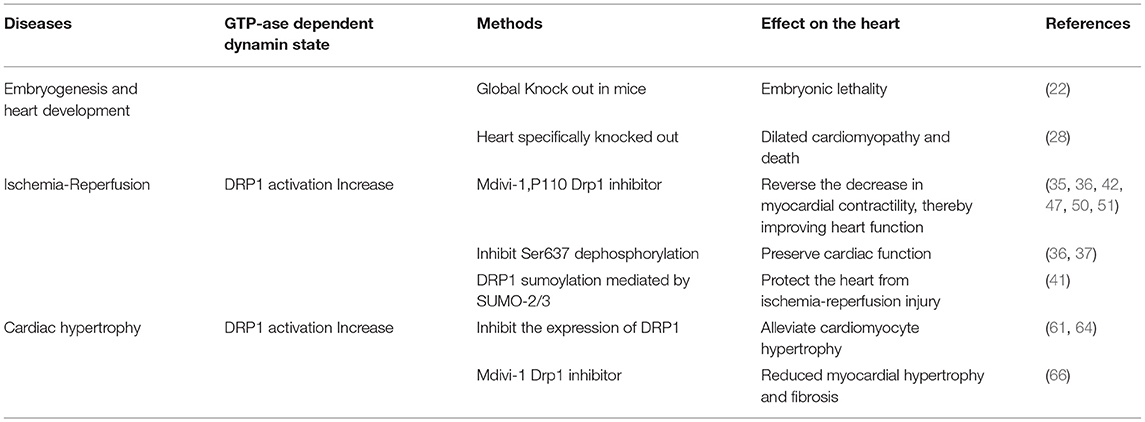
Table 1. Drp1 the role of Drp1 in heart development and heart disease.
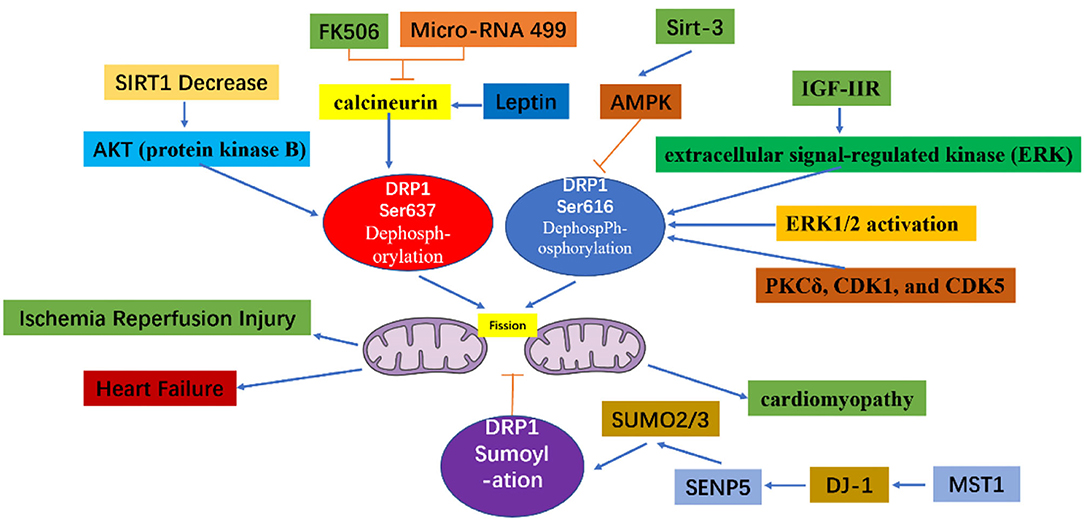
Figure 4. Cell signal transduction pathway acting on DRP1 and its role in heart disease.
Mitochondrial fusion is regulated by GTPase-dependent proteins, including MFN1, MFN2, and OPA1. Mitochondrial fusion plays an important role in the maintenance of mitochondrial DNA homeostasis. In cultured cells and skeletal muscle cells lacking mitochondrial fusion factors, mtDNA decreased and mtDNA accumulation mutation increased (69).
Mammalian mitochondrial fusion protein has been identified as a human homolog of the Drosophila fuzzy onion protein (Fzo) (70). MFN1 is composed of 741 amino acids, whereas MFN2 is composed of 757 amino acids. Both are transmembrane GTPases located on the outer mitochondrial membrane, with functional domains that share 63% homology. The conserved domain of mitofusins consists of an amino-terminal GTP binding domain, a coiled-coil domain (heptapeptide repeat HR1), a carboxy-terminal with a two-part transmembrane domain and a second coiled-coil domain (heptapeptide repeat sequence HR2). Both the GTPase and coiled-coil domains are exposed to the cytoplasm. In addition, MFN2 possesses an N-terminal Ras binding domain that does not exist in MFN1, implying that MFN2 has unique functions that are not shared with MFN1 (71). Although both MFN1 and MFN2 support mitochondrial fusion, MFN2 also mediates the coupling of the endoplasmic reticulum and mitochondria (72). Therefore, MFN2 plays a critical role in maintaining intercellular calcium regulation, which is of great significance to the excitation-contraction-metabolic coupling of cardiomyocytes. Unsurprisingly, MFN2 is highly abundant in the heart compared to other tissues (73).
Cardiac-specific Mfn1 and Mfn2 knockout mice exhibit death due to heart failure, respiratory dysfunction, and mitochondrial fragmentation and swelling. In dual Mfn1 and Mfn2 cardiac knockout mice, no births were observed with embryos dying on embryonic days 9.5 and 10.5. However, the expression of Mfn1 or Mfn2 alone is sufficient for murine survival, thus only the simultaneous absence of both is incompatible with life (74). In single Mfn1 or Mfn2 knockout mice, mitochondrial outer membrane fusion and aggregation proceed normally, while double knockout result in loss of fusion induced by mitofusins (75). In 8-week-old mice, when Mfn1 and Mfn2 were specifically and simultaneously knocked out in cardiac tissue, the cardiomyocytes showed mitochondrial fragmentation and respiratory dysfunction, rapidly developing into lethal dilated cardiomyopathy (76). Additionally, MFN1 levels have been found to decrease in myocardial hypertrophy (77), while MFN2 levels increase under oxidative stress (78), however, MFN2 levels in the late stage after myocardial infarction and diabetis late stageare significantly reduced (79, 80). A recent study showed that microRNA-20b can downregulate Mfn2 and promote cytoplasmic Ca2+ overload, thus, weakening the buffering capacity of mitochondria and increasing cardiac hypertrophy (81).
In mice with Mfn1 only knockout, cardiac function, mitochondrial respiratory function, and respiratory function are maintained; however, an increase in spherical mitochondria can still be observed (82, 83). In contrast, cardiac Mfn2 ablation causes early-dilated cardiomyopathy in mice. Indeed, MFN1 loss is better tolerated than MFN2 loss in cardiomyocytes (30, 84), which may account for why single Mfn1 knockout elicit minimal effects on heart function. These data imply that when either MFN1 or MFN2 are missing, the other protein may compensate for the lost function, however, this requires further verification.
In ischemia-reperfusion injury, DRP1 and BAX are transferred to mitochondria (29). In this process, the activities of MFN1 and MFN2 are inhibited, resulting in mitochondrial fragmentation and outer membrane permeability (31, 32). MFN inhibition can induce the activation of MTPT (30). When either mitochondrial outer membrane permeability is increased or the mitochondrial permeability transition pore opens, cytochrome C is released, further activating the caspase cell apoptosis pathway (33, 34). In the simulated ischemia-reperfusion injury model, the overexpression of MFN1 or MFN2 in the HL-1 cardiomyocyte cell line reduces cell death (42). Meanwhile, following siRNA silencing of Mfn2 expression in a rat neonatal cardiomyocyte ischemia-reperfusion injury model, the cell survival rate was significantly decreased (77). Acute knockout of Mfn1 and Mfn2 in the heart showed a short-term protective effect against acute myocardial infarction, related to the decreased contact between mitochondria and endoplasmic reticulum, thereby reducing the calcium overload in the mitochondria (76). In response to ischemia-reperfusion injury, mitochondria transport calcium ions from the endoplasmic reticulum to the mitochondria through a calcium ion unidirectional transport pump. Upregulation of calcium ions in the mitochondria can induce the opening of the MPTP, which is a determinant of cardiomyocyte death (85). In ischemia-reperfusion injury, inhibiting calcium overload in mitochondria can reduce myocardial cell death and the myocardial infarction area (86–88). Moreover, MFN2 reportedly has an important role in the connection between mitochondria and the endoplasmic reticulum, ensuring sufficient calcium transfer for the production of bioenergy (72). After its activation, βII protein kinase C can phosphorylate Ser86 on MFN1, effectively inhibiting its GTPase activity and leading to fragmentation and dysfunctional mitochondrial accumulation, which in turn causes deterioration of heart function (89).
OPA1 is a GTPase-dependent protein that mediates the fusion of mitochondria, while organizing the morphological regeneration of mitochondrial cristae, and resisting apoptosis for physiological needs (7, 90, 91). Evidencing the important role of OPA1 in the maintenance of mitochondrial morphology, knocking out Opa1 during embryogenesis is fatal (92). In fact, Opa1 or Mfn2 knockout, via gene trapping, prevents embryonic stem cells (ESCs) from differentiating into cardiomyocytes (93). Mitochondrial fusion disrupts and increases the entry of capacitive calcium, thereby enhancing Notch1 signaling through calcium-induced calcineurin activation. Meanwhile, inhibition of calcium and calcineurin signals is sufficient to reverse the non-differentiated phenotype of gene-trapped ESCs without affecting mitochondrial morphology. This places mitochondrial dynamics upstream of Notch signaling ESC differentiation in cardiomyocytes, thereby supporting the regulation of mitochondrial dynamics in heart development.
Wai et al. (94) observed morphological changes in the mitochondria of hearts from Yme1l knockout mice, finding that the smaller mitochondria aggregated, while their cristae structure was normal. This indicates that mitochondrial dynamics were damaged due to YME1L loss. Furthermore, in cardiomyocytes lacking YME1L, the D form of s-OPA1 disappears, while the C and E forms of s-OPA1 aggregate as a result of OMA1 activation. Fragmentation of the mitochondrial network was also found in fibroblasts with Yme1l knocked out in vitro, and cell damage caused by s-OPA1 aggravation was also observed. The absence of YME1L in cardiomyocytes activates OMA1, which further increases the processing level of OPA1, increases the fragmentation of mitochondria, and ultimately leads to dilated cardiomyopathy and heart failure. In ischemia-induced heart failure, OPA1 expression decreases, indicating that OPA1 plays an important role in ischemic cardiomyopathy (95). In Drosophila and mice, inhibiting Opa1 expression may cause abnormal mitochondrial morphology and cardiac hypertrophy (96, 97). Therefore, the inhibition of mitochondrial fusion-related proteins can increase mitochondrial fragmentation, which further affects the arrangement of cardiomyocytes and sarcoplasmic reticulum, thus, impacting cardiac function.
The change in OPA1 form represents a key step in mitochondrial cooperative fusion and fission regulation (98, 99). The balance between long and short forms of OPA1 maintains the normal shape of mitochondria: fusion depends on long OPA1, while short OPA1 is related to mitochondrial fission (100, 101). Cell pressure, mitochondrial dysfunction, or genetic intervention (such as Yme1l deletion) can activate OMA1, thereby increasing the conversion of long OPA1 to short OPA1, and increasing the fragmentation of mitochondria (100, 102–104). Thus, GTPase OPA1 located on the inner mitochondrial membrane is a key enzyme that regulates mitochondrial fission-fusion. Mitochondrial protease OMA1 and AAA protease (AAA proteases comprise a conserved family of membrane bound ATP-dependent proteases that ensures the quality control of mitochondrial inner-membrane proteins) YME1L can cleave OPA1 from its long to short form and, while short OPA1 does not play a role in mitochondrial fusion, the aggregation of short OPA1 may cause accelerated fission. In cultured mammalian cells, OMA1 is activated under pressure, which leads to increased mitochondrial fission (94, 105, 106). Sadhana et al. (107) confirmed that SIRT3 can regulate the enzymatic activity of OPA1 through lysine deacetylation. Mitochondrial respiration depends on the acetylation state of the C-terminal K926 and K931 lysine fragments in the GED region of the OPA1 protein. Cardiac stress causes hyperacetylation of OPA1, thereby reducing its GTPase activity, however, SIRT3 can directly bind to OPA1, thereby enhancing mitochondrial function and networking (107). Studies have found that OPA1 is acetylated in the states of cardiac hypertrophy and SIRT3 deficiency. Overexpression of SIRT3 can play a protective role in the process of adriamycin-mediated mitochondrial fragmentation and cell death. These mechanisms are all related to the acetylation status of OPA1. SIRT3 exerts its activity by directly binding to OPA1 on the inner mitochondrial membrane and modifying it by deacetylation. SIRT3 maintains or strengthens the mitochondrial respiratory complex by activating OPA1. SIRT3 can inhibit mitochondria-mediated apoptosis by mediating OPA1. Meanwhile, hyperacetylation reduces the GTPase activity of OPA1, indicating that the decreased activity of OPA1 may partially account for mitochondrial defects in hypertrophy of the heart.
Normal expression of OPA1 is necessary for the maintenance of mitochondrial autophagy. The expression of OPA1 is decreased in infarcted hearts in vivo or in cardiomyocytes cultured in vitro. Meanwhile, within cardiomyocytes, irisin can activate OPA1-induced mitochondrial autophagy (108). In myocardial ischemia-reperfusion injury, decreased expression of OPA1 leads to increased mitochondrial fragmentation, and thus mitochondrial dysfunction and increased mitochondrial apoptosis, further aggravating ischemia-reperfusion injury (109). The increase or overexpression of OPA1 maintains mitochondrial function and cardiac function during myocardial ischemia and reperfusion (110). In contrast, in H9C2 cells cultured in vitro, overexpression of OPA1 leads to mitochondrial lengthening with no detectable reduction in apoptosis under ischemic stimulation (95). OPA1 levels decrease in failing hearts and in late stages after myocardial infarction (80, 95), while increasing in hypertensive hypertrophic hearts and cardiomyocytes treated with insulin (60, 111). MFN1, MFN2, and OPA1 levels showed a consistent decrease in the genetically induced lipid-overloaded heart (112). The stability of OPA1 in cardiomyocytes is regulated by ERK, AMPK, and YAP cell signal transduction pathways (113).
Melatonin can activate OPA1-mediated mitochondrial autophagy and fusion in an AMPK-dependent manner, thereby reducing cardiac ischemia-reperfusion injury (110). Coenzyme Q10 can induce the AMPK-YAP-OPA1 cell signal transduction pathway, thus, improving mitochondrial function and reducing the degree of arteriosclerosis (114). Calenduloside E can reduce cardiac ischemia-reperfusion injury by regulating OPA1-related mitochondrial fusion mediated by AMPK activation (115). In the cardiomyocyte model of ischemia-reperfusion injury, increased expression of OPA1 can reduce the oxidative stress of cells through the Ca+2/calmodulin-dependent protein kinase II (CaMKII signaling) pathway (116).
Studies in murine diabetes models have shown that O-GlcNAc of OPA1 and DLP1 increase mitochondrial fragmentation (117, 118). O-GlcNAc modification of OPA1 can inhibit its function, leading to fragmentation of mitochondria, reduction of mitochondrial membrane potential, and reduction of complex IV activity (118). It is reported that enhanced O-GlcNAc modification of diabetic cardiomyopathy (DCM) is associated with disease progression in type 2 diabetes models, and reversal of this modification can restore cardiac function (119). Studies have shown that the occurrence of heart failure may be related to the fragmentation and dysfunction of mitochondria caused by the short-form aggregation of OPA1. The dysfunction of mitochondrial function causes the energy utilization mode of cardiomyocytes to change from lipid to sugar intake (94), which further causes cardiac function insufficiency (See in Figure 5).
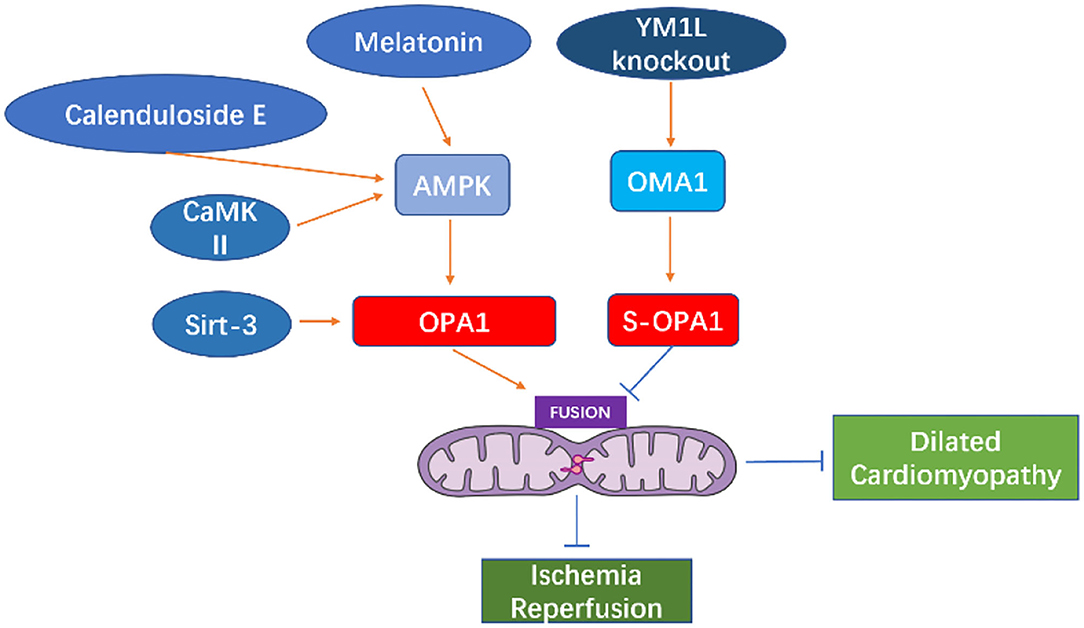
Figure 5. Cell signal transduction pathway acting on OPA1 and its role in heart disease.
The GTPase-dependent mitochondrial fusion proteins MFN1, MFN2, and OPA1 play important roles in maintaining cell homeostasis, cell pressure, calcium regulation, apoptosis, and autophagy. Downregulation or post-translational modification of these fusion proteins have been confirmed as related to a variety of heart diseases (Table 2), however, the specific mechanisms through which MFN1, MFN2, and OPA1 function in cardiomyocytes require further research.
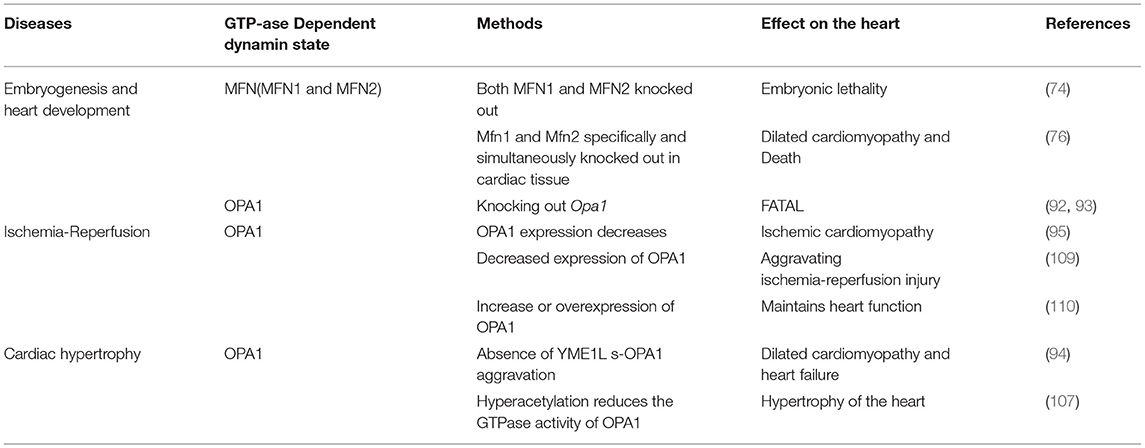
Table 2. The role of GTPase-dependent fusion proteins in heart development and heart disease.
The morphology of mitochondria in cardiomyocytes is a highly connected network structure that regulates the key functions of mitochondria through the balance of fission and fusion. Under normal physiological conditions, the fission and fusion of mitochondria are balanced to meet the normal physiological cell functions. In response to cellular stress, mitochondrial fragmentation and excessive fusion have been observed in various diseases, including ischemia-reperfusion injury and heart failure. Under normal physiological conditions, mitochondrial dynamin-related proteins maintain the shape and function of mitochondria to meet the organ needs through their own expression and post-translational modifications. However, under pathological conditions, these fission or fusion proteins can cause mitochondria morphological changes and dysfunction. Post-translational modifications of these proteins, such as acetylation and phosphorylation, can cause excessive fusion or fission of mitochondria; however, this process can be reversed by inhibiting the related proteins.
Therefore, GTP-ase-dependent mitochondrial dynamic-related proteins may represent target protein for the treatment of heart diseases. However, it remains unclear whether it is more beneficial to inhibit mitochondrial fission or promote its fusion to improve heart disease pathology. For instance, although there is a significant body of evidence suggesting that inhibition of DRP1 activity can inhibit mitochondrial fission, while improving heart function and inhibiting the progression of heart disease, a recent study reported that inhibiting mitochondrial fission by enhancing fusion leads to different cardiac outcomes. Meanwhile, up-regulation of MFN2 in the heart can correct excessive mitochondrial fragmentation compared to down-regulation of DRP1. Additionally, the safest strategies will maintain cardiac mitochondrial function (120). Maneechote's team used M1 (2 mg/kg) to enhance mitochondrial fusion to interfere with ischemia-reperfusion in rats and found that the application of M1 before ischemia can reduce infarct size and cardiac apoptosis, exerting the greatest cardioprotective effect. This indicates that myocardial ischemia-reperfusion injury can be reduced by increasing mitochondrial fusion (121). Similarly, the mitochondrial fusion enhancing substance M1 can effectively restore mitochondrial balance and improve diabetic cardiomyopathy in an OPA1-dependent manner (122).
Meanwhile, in different animals and different administration methods, the same type of DRP1 inhibition elicited differing effects. For instance, a recent study showed that injection of Mdivi-1 in advance of myocardial infarction can increase the length of mitochondria in myocardial cells and reduce the area of myocardial infarction in ischemic mice (51). However, another study using pigs as a model for cardiac reperfusion after acute myocardial infarction, reported that injection of Mdivi-1 directly into the coronary artery did not impact mitochondrial morphology nor elicit a protective effect on the heart, which may be related to the dose of Mdivi-1, timing of application, method of administration, etc. (52).Therefore, the timing and methods of intervention in mitochondrial dynamics still need further research.
Collectively, the relevant data has indicated that regulating the fission and fusion balance in mitochondria represents an important strategy for maintaining heart health and function. However, prior to clinical applications for the treatment of heart disease, long-term studies are needed to further investigate the effect of inhibiting mitochondrial motility-related proteins, as well as the appropriate timing and doses for administration.
This review demonstrates that the balance of mitochondrial fission and fusion plays a critical role in maintaining cell and heart function. Meanwhile, the imbalance in these processes likely contributes to the pathophysiological process of heart diseases, thereby providing potential new drug targets for heart diseases. Therefore, mitochondrial fission and fusion balance related proteins warrant further research.
BL proposed that mitochondrial-related proteins may play an important role in heart disease based on his previous research and organized the writing of this paper. JL wrote this paper. XS and YY provided guidance, help in the writing process, and the illustrations of the thesis. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the Finance Department Project of Jilin Province (No. 2019SCZT004), Education Department Project of Jilin Province (No. JJKH20190055KJ) and Science and Technology Department Project of Jilin Province (No. 2019090502SF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Friedman JR, Nunnari J. Mitochondrial form and function. Nature. (2014) 505:335–43. doi: 10.1038/nature12985
2. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. (2014) 15:634–46. doi: 10.1038/nrm3877
3. Hom J, Sheu SS. Morphological dynamics of mitochondria–a special emphasis on cardiac muscle cells. J Mol Cell Cardiol. (2009) 46:811–20. doi: 10.1016/j.yjmcc.2009.02.023
4. Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol. (2009) 41:1949–56. doi: 10.1016/j.biocel.2009.05.004
5. Atkins K, Dasgupta A, Chen KH, Mewburn J, Archer SL. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease. Clin Sci. (2016) 130:1861–74. doi: 10.1042/CS20160030
6. Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. (2010) 88:16–29. doi: 10.1093/cvr/cvq237
7. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
8. Li A, Gao M, Jiang W, Qin Y, Gong G. Mitochondrial dynamics in adult cardiomyocytes and heart diseases. Front Cell Dev Biol. (2020) 8:584800. doi: 10.3389/fcell.2020.584800
9. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. (2016) 212:379–87. doi: 10.1083/jcb.201511036
10. Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Ann Rev Cell Dev Biol. (2014) 30:357–91. doi: 10.1146/annurev-cellbio-101011-155756
11. Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem. (2013) 288:27584–93. doi: 10.1074/jbc.M113.479873
12. Otera Hidenori Chunxin Wang Cleland Megan . Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. (2010) 191:1141–58. doi: 10.1083/jcb.201007152
13. Pitts KR, McNiven MA, Yoon Y. Mitochondria-specific function of the dynamin family protein DLP1 is mediated by its C-terminal domains. J Biol Chem. (2004) 279:50286–94. doi: 10.1074/jbc.M405531200
14. Chang CR, Blackstone C. Drp1 phosphorylation and mitochondrial regulation. EMBO Rep. (2007) 8:1088–9; author reply 9–90. doi: 10.1038/sj.embor.7401118
15. Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res. (2017) 1864:1260–73. doi: 10.1016/j.bbamcr.2017.03.006
16. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
17. Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. (2007) 282:21583–7. doi: 10.1074/jbc.C700083200
18. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
19. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. (2007) 177:439–50. doi: 10.1083/jcb.200610042
20. Figueroa-Romero C, Iniguez-Lluhi JA, Stadler J, Chang CR, Arnoult D, Keller PJ, et al. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. Faseb J. (2009) 23:3917–27. doi: 10.1096/fj.09-136630
21. Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. (2013) 32:1514–28. doi: 10.1038/emboj.2013.65
22. Wakabayashi J, Zhang ZY, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. (2009) 186:805–16. doi: 10.1083/jcb.200903065
23. Song M, Dorn GWJCM. Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. (2015) 21:195–205. doi: 10.1016/j.cmet.2014.12.019
24. Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. (2014) 33:2798–813. doi: 10.15252/embj.201488658
25. Yoshiyuki I, Akihiro S, Yasuhiro M, Peiyong Z, Sebastiano S, Jessica T, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circul Res. (2014) 116:264.
26. Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, et al. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. (2010) 6:e1001000. doi: 10.1371/journal.pgen.1001000
27. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. (2015) 116:264–78. doi: 10.1161/CIRCRESAHA.116.303356
28. Song M, Mihara K, Chen Y, Scorrano L, Dorn GW II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. (2015) 21:273–86. doi: 10.1016/j.cmet.2014.12.011
29. Karbowski M. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. (2003) 159:931–8. doi: 10.1083/jcb.200209124
30. Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. (2011) 31:1309–28. doi: 10.1128/MCB.00911-10
31. Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. (2005) 280:25060–70. doi: 10.1074/jbc.M501599200
32. Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. (2007) 104:11649–54. doi: 10.1073/pnas.0703976104
33. Jason K, Onur K, Brody MJ, Sargent MA, Michael DM, Molkentin JD, et al. Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS ONE. (2015) 10:e0130520. doi: 10.1371/journal.pone.0130520
34. Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA. (2012) 109:6566–71. doi: 10.1073/pnas.1201608109
35. Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, et al. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. (2013) 2:e000461. doi: 10.1161/JAHA.113.000461
36. Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. (2014) 28:316–26. doi: 10.1096/fj.12-226225
37. Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. (2008) 105:15803–8. doi: 10.1073/pnas.0808249105
38. Yang YL, Li J, Liu K, Zhang L, Liu Q, Liu BL, et al. Ginsenoside Rg5 increases cardiomyocyte resistance to ischemic injury through regulation of mitochondrial hexokinase-II and dynamin-related protein 1. Cell Death Dis. (2017) 8:e2625. doi: 10.1038/cddis.2017.43
39. Tao A, Xu X, Kvietys P, Kao R, Martin C, Rui T. Experimental diabetes mellitus exacerbates ischemia/reperfusion-induced myocardial injury by promoting mitochondrial fission: role of down-regulation of myocardial Sirt1 and subsequent Akt/Drp1 interaction. Int J Biochem Cell Biol. (2018) 105:94–103. doi: 10.1016/j.biocel.2018.10.011
40. Wang XS, Song Q. Mst1 regulates post-infarction cardiac injury through the JNK-Drp1-mitochondrial fission pathway. Cell Mol Biol Lett. (2018) 23:21. doi: 10.1186/s11658-018-0085-1
41. Shimizu Y, Lambert JP, Nicholson CK, Kim JJ, Wolfson DW, Cho HC, et al. DJ-1 protects the heart against ischemia–reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol. (2016) 97:56–66. doi: 10.1016/j.yjmcc.2016.04.008
42. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. (2010) 121:2012–22. doi: 10.1161/CIRCULATIONAHA.109.906610
43. Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, Riquelme JA, et al. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J Cardiovasc Pharmacol. (2014) 63:477–87. doi: 10.1097/FJC.0000000000000071
44. Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. (2011) 17:71–8. doi: 10.1038/nm.2282
45. Jixuan L, Wei Y, Xiaojing Z, Qian J, Jinda W, Huawei Z, et al. Sirt3 attenuates post-infarction cardiac injury via inhibiting mitochondrial fission and normalization of AMPK-Drp1 pathways. Cell Signal. (2018) 53:1–13. doi: 10.1016/j.cellsig.2018.09.009
46. Sharp WW, Beiser DG, Fang YH, Han M, Piao L, Varughese J, et al. Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Crit Care Med. (2015) 43:e38–47. doi: 10.1097/CCM.0000000000000817
47. Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS ONE. (2012) 7:e32388. doi: 10.1371/journal.pone.0032388
48. Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. (2003) 35:615–21. doi: 10.1016/S0022-2828(03)00084-1
49. El-Mowafy AM, White RE. Resveratrol inhibits MAPK activity and nuclear translocation in coronary artery smooth muscle: reversal of endothelin-1 stimulatory effects. FEBS Lett. (1999) 451:63–7. doi: 10.1016/S0014-5793(99)00541-4
50. Ding M, Dong Q, Liu Z, Liu Z, Qu Y, Li X, et al. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc Diabetol. (2017) 16:1–11. doi: 10.1186/s12933-017-0540-8
51. Ra A, Sa B, Csa B, Mm B, Ssn B, Mp C, et al. Pleiotropic effects of mdivi-1 in altering mitochondrial dynamics, respiration, and autophagy in cardiomyocytes. Redox Biol. (2020) 36:101660. doi: 10.1016/j.redox.2020.101660
52. Ong SB, Kwek XY, Katwadi K, Hernandez-Resendiz S, Crespo-Avilan GE, Ismail NI, et al. Targeting mitochondrial fission using Mdivi-1 in A clinically relevant large animal model of acute myocardial infarction: a pilot study. Int J Mol Sci. (2019) 20:3972. doi: 10.3390/ijms20163972
53. Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. (2015) 22:1545–62. doi: 10.1089/ars.2015.6293
54. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. (2006) 103:2653–8. doi: 10.1073/pnas.0511154103
55. Yu TZ, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal. (2011) 14:425–37. doi: 10.1089/ars.2010.3284
56. Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol Biol Cell. (2011) 22:256–65. doi: 10.1091/mbc.E10-06-0551
57. Strack S, Wilson TJ, Cribbs JT. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol. (2013) 201:1037–51. doi: 10.1083/jcb.201210045
58. Ding M, Feng N, Tang D, Feng J, Li Z, Jia M, et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J Pineal Res. (2018) 65:e12491. doi: 10.1111/jpi.12491
59. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. (2008) 79:341–51. doi: 10.1093/cvr/cvn104
60. Tang Y, Mi C, Liu J, Gao F, Long J. Compromised mitochondrial remodeling in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Cardiovasc Pathol. (2014) 23:101–6. doi: 10.1016/j.carpath.2013.11.002
61. Pennanen C, Parra V, López-Crisosto C, Morales PE, del Campo A, Gutierrez T, et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci. (2014) 127:2659–71. doi: 10.1242/jcs.139394
62. Watanabe T, Saotome M, Nobuhara M, Sakamoto A, Urushida T, Katoh H, et al. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res. (2014) 323:314–25. doi: 10.1016/j.yexcr.2014.02.027
63. Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. (2009) 58:2303–15. doi: 10.2337/db07-1781
64. Sotomayor-Flores C, Rivera-Mejías P, Vásquez-Trincado C, López-Crisosto C, Lavandero S. Angiotensin-(1–9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Different. (2020) 27:2586–2604. doi: 10.1038/s41418-020-0522-3
65. Jong CJ, Yeung J, Tseung E, Karmazyn M. Leptin-induced cardiomyocyte hypertrophy is associated with enhanced mitochondrial fission. Mol Cell Biochemy. (2018) 454:33–44. doi: 10.1007/s11010-018-3450-5
66. Prottoy H, Masao S, Takenori I, Keisuke I, Hideya K, Toshihide I, et al. Mitochondrial fission protein, dynamin-related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl-salt sensitive rats. J Mol Cell Cardiol. (2018) 121:103–6. doi: 10.1016/j.yjmcc.2018.07.004
67. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure-overload-induced mitochondrial dysfunction and heart failure. Circulation. (2016) 134:e75–6. doi: 10.1161/CIRCULATIONAHA.116.023667
68. Huang CY, Lai CH, Kuo CH, Chiang SF, Pai PY, Lin JY, et al. Inhibition of ERK-Drp1 signaling and mitochondria fragmentation alleviates IGF-IIR-induced mitochondria dysfunction during heart failure. J Mol Cell Cardiol. (2018) 122:58–68. doi: 10.1016/j.yjmcc.2018.08.006
69. Chen HC, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA Stability in skeletal muscle and tolerance of mtDNA mutations. Cell. (2010) 141:280–9. doi: 10.1016/j.cell.2010.02.026
70. Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. (1997) 90:121–9. doi: 10.1016/S0092-8674(00)80319-0
71. Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. (2004) 6:872–83. doi: 10.1038/ncb1161
72. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456:605–10. doi: 10.1038/nature07534
73. Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. (2003) 116(Pt 13):2763–74. doi: 10.1242/jcs.00479
74. Chen Y, Liu YQ, Dorn GW. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circulat Res. (2011) 109:1327–U36. doi: 10.1161/CIRCRESAHA.111.258723
75. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
76. Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. (2016) 7:e2238. doi: 10.1038/cddis.2016.139
77. Fang L, Moore XL, Gao XM, Dart AM, Lim YL, Du XJ. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. (2007) 80:2154–60. doi: 10.1016/j.lfs.2007.04.003
78. Shen T, Zheng M, Cao CM, Chen CL, Tang J, Zhang WR, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chemi. (2007) 282:23354–61. doi: 10.1074/jbc.M702657200
79. Gao Q, Wang XM, Ye HW, Yu Y, Kang PF, Wang HJ, et al. Changes in the expression of cardiac mitofusin-2 in different stages of diabetes in rats. Mol Med Rep. (2012) 6:811–4. doi: 10.3892/mmr.2012.1002
80. Javadov S, Rajapurohitam V, Kilić A, Hunter JC, Zeidan A, Faruq NS, et al. Expression of mitochondrial fusion–fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol. (2011) 106:99–109. doi: 10.1007/s00395-010-0122-3
81. Qiu Y, Cheng R, Liang C, Yao Y, Zhang W, Zhang J, et al. MicroRNA-20b promotes cardiac hypertrophy by the inhibition of mitofusin 2-Mediated Inter-organelle Ca2+ Cross-Talk - ScienceDirect. Mol Ther Nucleic Acids. (2020) 19:1343–56. doi: 10.1016/j.omtn.2020.01.017
82. Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. doi: 10.1126/science.1231031
83. Papanicolaou KN, Ngoh GA, Dabkowski ER, O'Connell KA, Ribeiro RF Jr, et al. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. (2012) 302:H167–79. doi: 10.1152/ajpheart.00833.2011
84. Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circul Res. (2012) 111:863–75. doi: 10.1161/CIRCRESAHA.112.266585
85. Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol. (2009) 297:H1281–9. doi: 10.1152/ajpheart.00435.2009
86. Figueredo VM, Dresdner KP Jr, Wolney AC, Keller AM. Postischaemic reperfusion injury in the isolated rat heart: effect of ruthenium red. Cardiovasc Res. (1991) 25:337–42. doi: 10.1093/cvr/25.4.337
87. Miyamae M, Camacho SA, Weiner MW, Figueredo VM. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am J Physiol. (1996) 271:H2145–53. doi: 10.1152/ajpheart.1996.271.5.H2145
88. Murata M, Akao M, O'Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. (2001) 89:891–8. doi: 10.1161/hh2201.100205
89. Ferreira JCB, Campos JC, Qvit N, Qi X, Bozi LHM, Bechara LRG, et al. A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats. Nat Commun. (2019) 10:329. doi: 10.1038/s41467-018-08276-6
90. Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. (2003) 278:7743–6. doi: 10.1074/jbc.C200677200
91. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. (2006) 126:177–89. doi: 10.1016/j.cell.2006.06.025
92. Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. (2007) 16:1307–18. doi: 10.1093/hmg/ddm079
93. Kasahara A, Cipolat S, Chen Y, Dorn GW II, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. (2013) 342:734–7. doi: 10.1126/science.1241359
94. Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. (2015) 350:aad0116. doi: 10.1126/science.aad0116
95. Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. (2009) 84:91–9. doi: 10.1093/cvr/cvp181
96. Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. (2012) 94:408–17. doi: 10.1093/cvr/cvs117
97. Le C, Liu T, Tran A, Lu X, Association AAKJJotAH. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc. (2012) 1:e003012. doi: 10.1161/JAHA.112.003012
98. Quiros PM, Langer T, Lopez-Otin C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. (2015) 16:345–59. doi: 10.1038/nrm3984
99. Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol. (2015) 33:111–8. doi: 10.1016/j.ceb.2015.02.001
100. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. (2014) 204:919–29. doi: 10.1083/jcb.201308006
101. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. Embo J. (2006) 25:2966–77. doi: 10.1038/sj.emboj.7601184
102. Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, et al. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. (2014) 33:578–93. doi: 10.1002/embj.201386474
103. Zhang K, Li H, Song Z. Membrane depolarization activates the mitochondrial protease OMA1 By stimulating self-cleavage. EMBO Rep. (2014) 15:576–85. doi: 10.1002/embr.201338240
104. Jiang X, Jiang H, Shen Z, Wang X. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc Natl Acad Sci USA. (2014) 111:14782–7. doi: 10.1073/pnas.1417253111
105. Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. (2007) 178:757–64. doi: 10.1083/jcb.200704112
106. Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. (2009) 187:1023–36. doi: 10.1083/jcb.200906084
107. Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. (2014) 34:807–19. doi: 10.1128/MCB.01483-13
108. Xin T, Lu C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging. (2020) 12:4474–88. doi: 10.18632/aging.102899
109. Wang K, Liu Z, Zhao M, Zhang F, Wang K, Feng N, et al. kappa-opioid receptor activation promotes mitochondrial fusion and enhances myocardial resistance to ischemia and reperfusion injury via STAT3-OPA1 pathway. Europ J Pharmacol. (2020) 874:172987. doi: 10.1016/j.ejphar.2020.172987
110. Zhang Y, Wang Y, Xu J, Tian F, Hu S, Chen Y, et al. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res. (2019) 66:e12542. doi: 10.1111/jpi.12542
111. Parra V, Verdejo HE, Iglewski M, Campo AD, Lavandero SJD. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NF B-Opa-1 signaling pathway. Diabetes. (2014) 63:75. doi: 10.2337/db13-0340
112. Elezaby A, Sverdlov AL, Tu VH, Soni K, Luptak I, Qin F, et al. Mitochondrial remodeling in mice with cardiomyocyte-specific lipid overload. J Mol Cell Cardiol. (2015) 79:275–83. doi: 10.1016/j.yjmcc.2014.12.001
113. Xin T, Lv W, Liu D, Jing Y, Hu F. Opa1 reduces hypoxia-induced cardiomyocyte death by improving mitochondrial quality control. Front Cell Dev Biol. (2020) 8:853. doi: 10.3389/fcell.2020.00853
114. Xie T, Wang C, Jin Y, Meng Q, Liu Q, Wu J, et al. CoenzymeQ10-induced activation of AMPK-YAP-OPA1 pathway alleviates atherosclerosis by improving mitochondrial function, inhibiting oxidative stress and promoting energy metabolism. Front Pharmacol. (2020) 11:1034. doi: 10.3389/fphar.2020.01034
115. Wang M, Wang RY, Zhou JH, Xie XH, Sun GB, Sun XB. Calenduloside E ameliorates myocardial ischemia-reperfusion injury through regulation of AMPK and mitochondrial OPA1. Oxidative Med Cell Longevity. (2020) 2020:1–12. doi: 10.1155/2020/2415269
116. Luo H, Song S, Chen Y, Xu M, Zhang W. Inhibitor 1 of protein phosphatase 1 regulates Ca 2+ /calmodulin-dependent protein kinase II to alleviate oxidative stress in hypoxia-reoxygenation injury of cardiomyocytes. Oxidative Med Cell Longevity. (2019) 2019:1–19. doi: 10.1155/2019/2193019
117. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. (2012) 287:30024. doi: 10.1074/jbc.M112.390682
118. Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, et al. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol. (2011) 300:R1296–302. doi: 10.1152/ajpregu.00437.2010
119. Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R689–99. doi: 10.1152/ajpregu.00548.2011
120. Qin Y, Li A, Liu B, Jiang W, Gao M, Tian X, et al. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. FASEB J. (2020) 34:663–75. doi: 10.1096/fj.201901671R
121. Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn S, Chattipakorn N. Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin Sci. (2019) 133:497–513. doi: 10.1016/S0735-1097(19)30659-X
Keywords: heart disease, DRP1, mfn1, MFN2, OPA1
Citation: Liu J, Song X, Yan Y and Liu B (2021) Role of GTPase-Dependent Mitochondrial Dynamins in Heart Diseases. Front. Cardiovasc. Med. 8:720085. doi: 10.3389/fcvm.2021.720085
Received: 03 June 2021; Accepted: 06 September 2021;
Published: 30 September 2021.
Edited by:
Junjie Yang, University of Maryland, Baltimore, United StatesReviewed by:
Shiyue Xu, The First Affiliated Hospital of Sun Yat-sen University, ChinaCopyright © 2021 Liu, Song, Yan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Liu, bGl1YmluMzMzM0B2aXAuc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.