 Yanan Sun
Yanan Sun Shreya Sangam
Shreya Sangam Qiang Guo4
Qiang Guo4 Jian Wang
Jian Wang Haiyang Tang
Haiyang Tang Stephen M. Black
Stephen M. Black Ankit A. Desai
Ankit A. Desai
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 10 September 2021
Sec. Sex and Gender in Cardiovascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.719058
This article is part of the Research Topic Metabolic Hormones and Inflammation View all 8 articles
Pulmonary arterial hypertension (PAH) is a complex and devastating disease with a poor long-term prognosis. While women are at increased risk for developing PAH, they exhibit superior right heart function and higher survival rates than men. Susceptibility to disease risk in PAH has been attributed, in part, to estrogen signaling. In contrast to potential pathological influences of estrogen in patients, studies of animal models reveal estrogen demonstrates protective effects in PAH. Consistent with this latter observation, an ovariectomy in female rats appears to aggravate the condition. This discrepancy between observations from patients and animal models is often called the “estrogen paradox.” Further, the tissue-specific interactions between estrogen, its metabolites and receptors in PAH and right heart function remain complex; nonetheless, these relationships are essential to characterize to better understand PAH pathophysiology and to potentially develop novel therapeutic and curative targets. In this review, we explore estrogen-mediated mechanisms that may further explain this paradox by summarizing published literature related to: (1) the synthesis and catabolism of estrogen; (2) activity and functions of the various estrogen receptors; (3) the multiple modalities of estrogen signaling in cells; and (4) the role of estrogen and its diverse metabolites on the susceptibility to, and progression of, PAH as well as their impact on right heart function.
Pulmonary arterial hypertension (PAH) is a complex and devastating disease characterized by a progressive increase in pulmonary vascular resistance (PVR) and subsequent, right heart failure, considered the most common cause of death in patients (1). The updated hemodynamic criteria define pulmonary hypertension (PH) using a threshold for mean pulmonary artery pressure (mPAP) > 20 mm Hg, pulmonary artery wedge pressure (PAWP) ≤ 15 mm Hg, and PVR ≥ 3 Wood units (WU) (2) during a resting right heart catheterization (RHC). Obliterative pulmonary arterial (PA) remodeling, sustained vasoconstriction, reduced wall elasticity, in-situ thrombosis in small pulmonary arterioles, and formation of plexiform lesions (PXL) (3) are hallmarks of PAH pathology and contribute to the rise in PVR.
Evidence of sexual dimorphism is a distinguishing feature of developing PAH. The World Health Organization describes sex as a biologically determined characteristic, whereas gender is used to describe a characteristic that is socially constructed (4). Therefore, the term sex is used in this review to discriminate differences between male and female subjects. Furthermore, while the proportion of women with PAH is greater, their survival rate is higher than that of men (5–10). Based on these clinical observations, a significant amount of research has been carried out to investigate the potential roles of estrogen signaling in PAH. Specifically, the identification of protective or pathogenic roles of estrogen signaling provide a complex molecular model to decipher the contrasting clinical observations in both sexes. In some circumstances, published research on estrogen signaling from conventional animal models of PAH appear to directly contradict observations from human disease, often termed the “estrogen paradox.” Female rodents, for example, are less susceptible than male rodents to the development of PAH (11, 12). Indeed, these observations along with other findings provide more credence to the notion that established animal models do not fully mimic PAH in patients (11). Indeed, these contradictory findings from animal models emphasize the need for caution in interpreting the translatability as well as the importance of choosing the appropriate animal models when studying the sexual dimorphism of PAH. This review will summarize our most up to date knowledge regarding the sex differences in existing pre-clinical models of PAH, including data from novel transgenic models (13–15), that provide novel insights into the sex differences related to estrogen signaling in PAH. In the process, we will highlight the importance of selecting suitable animal models for PAH research.
Estrogen signaling is complex and requires the evaluation of both the hormone itself and its metabolites. As a sex steroid hormone, estrogen is produced through multiple reactions and has intricate catabolic pathways in vivo. Some of its metabolites have been reported to promote the development of PAH, while others have been shown to attenuate PAH development. A careful inventory of each metabolite and its function is critical to better understanding the estrogen paradox. Moreover, both PAH status and progression, themselves affect the synthesis and catabolism of estrogen. This latter observation is based on prior studies (16–18) that reported changes in estrogen-metabolizing enzymes and the proportion of estrogen metabolites during PAH development. The relationship between estrogen and PAH, therefore, appears bidirectional in terms of cause and effect. Further, beyond circulating levels, local concentrations of estrogen signaling mediators also appear to be important in PAH. For example, estrogen-metabolizing enzymes are tissue-specific, raising questions about possible discrepancies between the levels of estrogen (and its metabolites) in the circulation vs. in the heart and lungs. Whether the presence of PAH or its progression leads to changes in the local metabolism of estrogen in the heart and lungs also deserves more attention. In addition, the relationship between estrogen and gene expression is complex. Signal transduction of estrogen is influenced by various factors including, age, sex, receptor type, cell type, disease status, etc. Indeed, the three known estrogen receptors (ERs-ERα, ERβ, and GPER) exhibit different roles in regulating gene expression. Moreover, dimers formed by ERα and ERβ may manifest as hetero- or homodimers and function via individual or dimer forms, further complicating estrogen signaling (19). This review will further discuss estrogen signaling in the context of the relative content of ERs, ERs' different position (membrane or nucleus) in cells, and varying modifications of ERs, to give a full picture of the sex-specific differences and the need for the development of sex-specific treatments in PAH.
In order to better understand the estrogen paradox, this review will summarize available evidence and discuss the following: (1) potential origins of sex differences; (2) mechanisms involving estrogen anabolism and catabolism; (3) the diverse cellular roles of ERs; (4) multiple modes of estrogen signaling; (5) direct and indirect effects of estrogen hormone and its metabolites. These discussions will also shed insight into the impact of both the presence and severity of PAH on estrogen anabolism and catabolism, ERs, and estrogen metabolites.
As early as the 1950s, a predilection was observed for female patients in PAH (20, 21), a finding that has since been confirmed by multiple studies (7–9, 22). In 1987, National Institutes of Health (NIH) registry reported the ratio of male to female patients in primary PH was 1–1.7. This data, however, did not break down PH into the current sub-group classifications (5). The findings of the French National Registry of PH also suggested that women account for a greater proportion of patients with most PAH subtypes, such as idiopathic, familial, anorexia, connective tissue disease and congenital heart diseases associated PAH (6). In hereditary PAH, the incidence of PAH among carriers of bone morphogenic protein receptor type II (BMPR2) mutations also varies by sex−42% for women and 14% for men (7). Sexual dimorphism is also reflected in surveys across geographical regions and nationalities. Data from the Japan PH Registry (JAPHR) showed that 76.2% of Japanese patients with PAH were female (8). The Registry to EValuate Early And Long-term pulmonary arterial hypertension disease management (REVEAL Registry) shows a greater number of women with the disease in the US (9, 22). These and other registries, composed of mostly adult patients, consistently show a female predilection (Table 1). Differences in exact rates across these registries may reflect different inclusion/exclusion criteria, drug exposure history, lifestyle differences, among other confounders. Notably, hemodynamic criteria for PAH in these registries were based on older definitions: mPAP ≥ 25 mm Hg at rest; these data did not include any patients whose mPAP was between 20 and 24 mm Hg based on the new definition of PAH with a lower threshold of mPAP (≥20 mm Hg). Interestingly, Hoeper et al. re-analyzed the results of the Comparative Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) registry and stratified patients into two categories: old (>65 years old) and young (18–65 years old), they reported a sex difference was apparent in young but not older subjects (25).
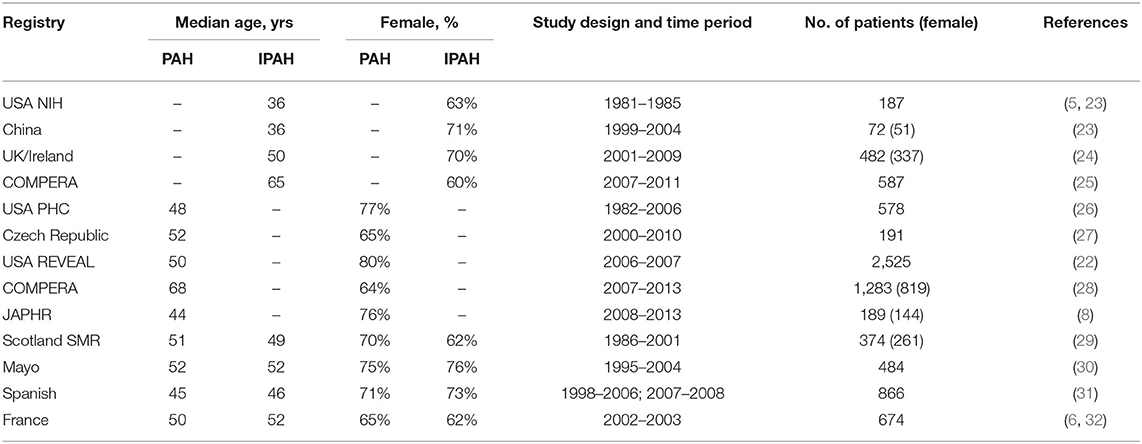
Table 1. Proportion of female patients in major PAH registries.
While female patients are more susceptible, mortality data consistently show that they exhibit better survival outcomes than males. For example, in the Spanish Registry of Pulmonary Arterial Hypertension, male sex is an independent predictor of death, associated with an increased risk of death (31). The REVEAL registry also reported a worse prognosis for men than women (33) and the French registry showed female sex was an independent predictor of survival (34). Data from the COMPERA registry showed similar survival benefits for female patients (28). These observations cumulatively establish significant differences between male and female patients with PAH including disease risk and outcomes.
To better understand sex differences in patients with PAH, several animal models have been studied. Interestingly, while sex differences are also found in animal models of PH, not all are consistent with those observed in patient studies. We summarize sex differences in four conventional animal models of PH: Sugen/Hypoxia induced rodent PH (SuHx-PH), monocrotaline-induced rodent PH (MCT-PH), chronic hypoxia-induced PH (HPH), and genetically modified animal models of PH.
Female rats manifest a milder form of disease severity than male rats when exposed to HPH or MCT-PH. Ovariectomy (OVX) further exacerbates PH in female rats, and exposure to OVX-treated animals with 17β-estradiol (E2) alleviates PH in these animal models (11, 12, 35). Apart from improved right heart function and survival advantages, female mice have less PA remodeling than male mice in HPH (13). Moreover, vascular occlusion and PXL are not observed in the HPH model (11). These findings appear to be inconsistent with the suspected pathogenic role of estrogen in pulmonary blood vessels of patients. It is also worth noting that the MCT is biologically activated in the liver by cytochrome P-450 (CYP-450) into MCT pyrrole (MCTP) (36), which can damage the lung endothelium and leads to the development of PH. Studies have shown a sex-specific role for CYP-450 involvement in the metabolism of MCT in the liver (36), which may partly explain sex discrepancies between animal MCT-PH models and patients.
In the SuHx-PH model, although female rats exhibit more pronounced smooth muscle thickening of the pulmonary vasculature, they display better cardiac function and a better ability to cope with increased afterload caused by acute exercise challenge than male rats (37). The survival rate of female rats is also higher than in males. OVX treatment exacerbates the severity of SuHx-PH in female rats, and supplementing with exogenous E2 after OVX, improves right heart function and PA remodeling. The increased severity in vascular remodeling, improved right ventricular (RV) adaptability, and superior survival rate of female rats in the SuHx-PH model better mimic those observed in PAH patients. Comparing these features from HPH and MCT-PH models, SuHx-PH may represent a more suitable model to study sex differences in PAH.
Select transgenic models have also successfully imitated the increased susceptibility in women observed in patients with PAH and may be leveraged to investigate the role of a targeted pathway in PAH-related sexual dimorphism. For example, in signal transducers and activators of transcription 5 heterozygous (Stat5+/−) or homozygous (Stat5−/−) mice, female mice exhibited more severe PH than males after exposure to hypoxia (13). In addition, in mice over-expressing the calcium-binding protein S100A4/Mts1, right ventricle systolic pressure (RVSP) and pulmonary vascular remodeling were increased in female mice, while male mice were unaffected (14). In mice over-expressing serotonin transporter (SERT+), female mice showed increased RVSP and PVR, while male mice were unaffected. Moreover, OVX eliminates the effect of SERT+ in females. Also, long-term administration of E2 re-established the PAH phenotype after OVX in both normoxic and hypoxic SERT+ mice (15).
Although PAH is considered a disease of the pulmonary vasculature, the ability of the right heart to adapt to the increased PVR determines the survival rate in patients with PAH (32–34, 38). Registries of PAH show that female patients with PAH have more favorable hemodynamic characteristics and better RV function than male patients (39, 40), including higher cardiac index (CI) and lower RVSP, lower average PA pressure and PVR (41, 42). Subsequent animal studies have also shown that estrogen mainly exhibits protective effects on the RV, while in pulmonary blood vessels, its role is relatively complex. In the pulmonary vasculature of animal models, the pathogenic effect of estrogen may be greater than the protective effect, but these findings remain unclear. Nonetheless, these animal model observations may partly explain why women are more susceptible to disease risk but have a lower mortality rate in PAH.
To dissect the underpinnings of the sexual dimorphism and the estrogen paradox, significant attention has been paid to estrogen synthesis, signaling, and metabolism and their contribution to the development of PAH. Given the diversity of estrogen, its metabolites, as well as some estrogen analogs, it is important to discuss evidence of their involvement and potential as therapeutic targets in PAH.
The basic metabolic pathways of estrogen synthesis are important to briefly summarize when considering their role in PAH. The initial step in the synthesis of sex hormones (Figure 1) is the conversion of cholesterol to pregnenolone under the action of cytochrome P450 (CYP) enzymes (43). Pregnenolone is converted to progesterone by 3β-hydroxysteroid dehydrogenase (3β-HSD). Pregnenolone and progesterone are oxidized to dehydroepiandrosterone (DHEA) and androstenedione under the action of CYP17A1, respectively. They are transformed into testosterone under the action of different β-hydroxysteroid dehydrogenase 1 (β-HSD1). The final step in the production of estrogen is the conversion of androstenedione or testosterone into estrone (E1) or E2 under the action of aromatase (CYP19A1), respectively. Notably, aromatase is increased in the lungs of female patients with PAH (16), suggesting that PAH development is associated with increased estrogen production. Estrogens produced by the human body include E1, E2, and estriol (E3). Among them, E2 is the most abundant estrogen with the strongest biological activity, and it is one of the main female hormones produced by the ovaries. E1 and E3 are weak estrogens. E1 is increased in post-menopausal females. E3 is the degradation product of E2 and is increased during pregnancy.
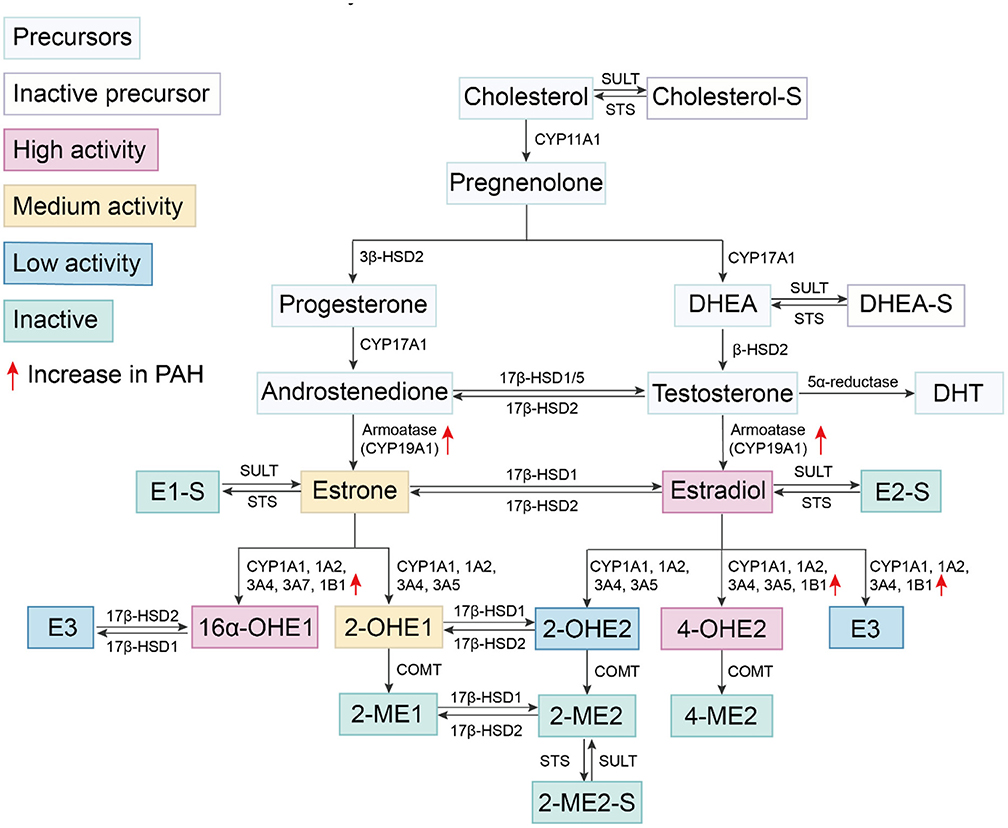
Figure 1. Estrogen synthesis and metabolism. Cholesterol is catalyzed by the cytochrome P450 enzyme and undergoes several conversions to become DHEA. DHEA is converted to testosterone under the catalysis of different β-HSDs. Androstenedione and testosterone are converted into estrone and E2 by CYP19A1, respectively. E2 is oxidized on carbon at multiple positions, including oxidation at the C17 position by 17β-HSD responsible for the reversible conversion between E1 and E2. Other oxidation sites include those at C2, C4, and C16 positions and produce different metabolites via varying cytochrome P450 enzyme. E1, E2, estrogen precursors and some metabolites can be combined with sulfonate groups by SULT to convert them into sulfuric steroids. Sulfation of steroids can be converted into active forms by STS.
E2 is oxidized at multiple carbon positions. Oxidation at the C17 position is required for the conversion between two estrogens, E1 and E2 by 17β-HSD; specifically, E1 converts to E2 via 17β-HSD1 while E2 converts back to E1 via 17β-HSD2. This process is reversible, but the rate of conversion from E1 to E2 is slower. Other sites of oxidation of E2 predominantly occur at C2, C4, and C16 positions which results in the production of different metabolites. These estrogen metabolites have previously been shown to exert diverse biological effects in PAH (44–46).
Hydroxylation at the C2 position represents one of several available pathways for E2 metabolism (47). The metabolites produced from this pathway have minimal to no estrogen activity, thereby, rapidly reducing the amount of active estrogen. CYP450 isomers CYP1A1, 1A2, 3A4, 3A5, etc. catalyze conversion of E2 to form 2-hydroxyestradiol (2-OHE2) (47). 2-OHE2 has low estrogen activity and is further converted into 2-methoxyestradiol (2-ME2) via catechol-O-methyltransferase (COMT). 2-ME2 has no detectable estrogenic activity (48, 49). Hydroxylation of E1 at this C2 position generates 2-hydroxyestrone (2-OHE1), which has a higher ability to bind ERα than ERβ (50). Conversion of both E1 and E2 into other metabolites predominantly occurs in the liver. Cardiovascular tissues also facilitate conversion of E2 into 2-OHE2 and 2-ME2, such as cardiac fibroblasts (51), vascular endothelial cells (EC) (52), and smooth muscle cells (SMC) (53).
While hydroxylation at the C4 position accounts for only a small portion of E2 hydroxylation, the resulting metabolite, 4-hydroxyestradiol (4-OHE2), has high estrogenic activity. These C4 reactions are catalyzed by enzymes such as CYP1A1, 1A2, 3A4, 3A5, and 1B1. The latter, CYP1B1, catalyzes the reaction of C4 for the most part, while the other enzymes primarily catalyze the reaction at C2, followed by the reaction at C4 (47). Notably, 4-OHE2 reportedly exhibits reduced binding affinity with ERs, is reactive oxygen species (ROS)-dependent and has carcinogenic effects (54). Furthermore, similar to the metabolic reaction at the C2 position, 4-OHE2 is converted into 4-methoxyestradiol (4-ME) via catechol-O-methyl transferase (COMT) or into 4-hydroxyestrogen (4-OHE1) via 17β-HSD.
Hydroxylation of E2 at the C16 position is also one of the main mechanisms for the formation of highly estrogenic metabolites. The enzymes involved include CYP1A1, 1A2, 3A4, 3A7, and 1B1 (55). Among them, CYP3A7 has high catalytic activity for 16α-hydroxylation of E1, but not for E2 (56). E1 and E2 are metabolized to 16α-hydroxyestrone (16α-OHE1) and 16α-hydroxyestradiol (E3, estriol), respectively (47). E3 is only produced in large amounts during pregnancy. Although the estrogenic activity of E3 is weak, it can be catalyzed by 17β-HSD2 to produce 16α-OHE1. 16α-OHE1 is a highly estrogenic metabolite, more potent than the highly abundant E2, and has a slightly higher affinity for ERβ than for ERα (50). Moreover, 16α-OHE1 also prolongs ER activation (57, 58). In patients and various experimental PAH models, the expression of CYP1B1 is also increased, especially in the pulmonary vasculature (17, 46, 59). CYP1B1 catalyzes the catabolism of estrone to 16α-OHE1 in extrahepatic tissues. Austin et al. compared the levels of estrogen metabolites in female subjects with BMPR2 mutations, and found that 2-OHE: 16α-OHE1 ratio of affected mutation carriers was 2.3 times lower than that of unaffected mutation carriers (18). The accumulation of 16α-OHE1 and 16α-OHE2 has been independently validated in idiopathic PAH patients (60). Broadly, these studies indicate dysregulation of estrogen catabolism in PAH. Dexfenfluramine (Dfen)-induced PH (59), HPH, and SuHx-induced PH (46) are all attenuated in CYP1B1 knockout (KO) mice. The selective CYP1B1 inhibitor, 2,3′,4,5′-tetramethoxystilbene (TMS), attenuates Dfen-induced PH, primarily by inhibiting estrogen-induced proliferation of human PASMCs (46). Therefore, PAH development may predispose estrogen catabolism toward 16α-OHE1 generation. By increasing 16α-OHE1, a series of PAH-promoting estrogen effects are potentially amplified. However, this hypothesis requires further validation.
Estrogen metabolism and catabolism are well-known but its characteristics across tissue-types remain poorly studied. In PAH, emerging data indicate parallel complexity and specificity based on disease-relevant tissues including EC, SMC, and cardiomyocytes. There are two obvious changes in estrogen metabolism in PAH. Aromatase is increased in the lungs of female patients (16) while CYP1B1 is increased in patients and various experimental PAH models, which in turn increases the 16α-OHE (17, 46, 59, 60). The enzymes responsible for estradiol synthesis are expressed in the liver as well as in vascular SMCs (61), cardiac fibroblasts and cardiomyocytes (62). Moreover, the CYP subtypes involved in the oxidative metabolism of estrogen have different levels of catalytic activity and unique regioselectivity (47). For example, estrogen catabolism in liver tissue is mainly mediated by CYP1A2 and 3A4, while in extrahepatic tissue, it is catalyzed by CYP1B1 and 1A1 (55). CYP1B1, 1A1, as well as COMT were all observed in cardiovascular tissues (51, 62–64). These finding demonstrate that estrogen can be locally synthesized and metabolized in the blood vessels of heart and lungs, tissues where the effects of estrogen and its metabolites likely target during PAH pathogenesis.
Combining sulfonate groups, sulfotransferase (SULT) converts E1, E2, estrogen precursors and metabolites (DHEA, 2-ME, etc.) into sulfuric acid E1, sulfuric acid E2, sulfuric acid 2-ME, etc. These sulfated steroids are considered inactive, cannot activate ERs, and also hydrophilic, which makes them easier to excrete through the kidney (65). In various cancer cell lines, increased SULT activity is related to the weakening of the anti-mitotic effect of 2-ME2 (66). Specifically, the level of activity of these steroids are reduced by sulfation. Sulfation of steroids, however, can further be converted into active forms by sulfatase (STS). Therefore, SULT and STS regulate the content of biologically active estrogen, its precursors and metabolites depending on whether they are sulfated.
Estrogen exerts most of its biological functions through ERs. ERs are composed of a DNA-binding domain, a ligand binding domain, and an amino terminal transcription control domain (AF-1). ERs interact with regulatory binding proteins through AF-1, a region with significant differences between ERα and ERβ (67). GPER, a 7-pass transmembrane G protein-coupled ER that is predominantly localized in the endoplasmic reticulum (68), binds estrogen with high affinity to activate several cellular signaling cascades (69). GPER is an orphan receptor unrelated to nuclear ERs (nERs), which mimics the binding and signaling characteristics of membrane ERs (mERs) (70). There are three receptors expressed in lung (71–74) and heart tissue (75, 76). While both, ERα and ERβ, are expressed in PAECs (77) and PASMCs (78), ERβ is primarily expressed in PAECs (79). GPER is seen in the intima and media of the aorta by immunohistochemical staining (75), and its abundance is similar in males and females. Studies on the roles and expression of ERs in the pulmonary vasculature have not been as exhaustive as those in the systemic vasculature, and much of our knowledge is still derived from animal studies.
The content and ratio of ERα and ERβ influence estrogen effects on gene expression. ER levels appear to be different in men than in women, and in women, before and after menopause. Gavin et al. described that early follicular ERα expression was 30% lower than that of late follicles in vascular ECs of premenopausal women, and ERα levels were 33% lower than that of late follicles in postmenopausal women (80). In addition, although Grohe et al. did not observe any sex differences in the expression of ERβ in cardiomyocytes, a significant change in ERα expression by sex was observed (71). GPER is also affected by sex and age. The expression of GPER in the mesenteric artery of men and elderly women is reduced by ~50% compared with young women (81). The abundance of ERα and ERβ in different tissues and cells may vary (82). In the lung, ERβ is reportedly more abundant than ERα (83, 84). The relative function of a specific ER may further depend on the clinical context, for example, men or women, premenopausal women or postmenopausal women.
In addition to age and sex, disease factors also influence ER expression levels in the cardiovascular system. Cardiac pressure load, for example, increases both, ERα and ERβ, in the human heart (85), while heart failure increases ERα (86). ERα expression is reduced in the RV of SuHx-PH when they are subject to OVX and this recovers after E2 supplementation (87). The expression of ERα in the RV is negatively correlated with RVSP and right ventricular hypertrophy (RVH) (87). Similarly, Pinzone et al. found that estrogen positively regulates ER levels (88). Ihionkhan et al. reported that chronic use of exogenous estrogen modifies transcription of ERα gene in the endothelium through ERα or ERβ, such that the expression of ERα is increased while ERβ is decreased (89). Further, increased expression of ERα in female PAH patients correlates in increased PASMC proliferation and remodeling (79) via multiple pathways including MAPK and Akt signaling. In contrast, in hypoxic male rats, increased ERβ, but not ERα, expression is observed in pulmonary vessels (90). Also, the expression of ERβ mRNA in male blood vessels is increased after vessel injury (91). Moreover, the biological functions of ERβ may depend on the presence of ERα in certain cell types and tissues (92). Therefore, when discussing the role of ERs activity in PAH, it is necessary to pay attention to the effect of PAH on ERs in the heart and lungs.
Modification events also influences ER activity. For example, the methylation of the ER promoter reduces ER expression (93) while acetylation increases the transcriptional activity of ERα (94) and enhances the activity of ERα-dependent gene regulation (95). In addition, the phosphorylation of ER exerts ER ligand-independent regulation, enhancing ER signaling (96). NO-mediated S-nitrosylation of ER has also been shown to occur at the two major DNA-binding zinc-finger domains of ER and this leads attenuated binding to specific estrogen response elements (EREs) and may potentially favor activation of non-genomic signaling pathways (97).
As mentioned above, ERs perform different functions according to both the recruited co-regulators and the bound transcription factors (TFs). Studies have also shown that ERα and ERβ may have opposing effects in the same tissue (98). For example, in vascular SMCs, ERβ up-regulates the expression of inducible nitric oxide synthase (iNOS) while ERα causes down-regulation (99). In addition, ERα has been shown to mostly up-regulate gene expression in the mouse aorta while 90% of estrogen-mediated genes were downregulated in an ERβ-dependent manner (82). Conversely, in the hearts of female mice, 122 genes were up-regulated and only 23 genes were down-regulated by ERβ. The gene ontology “muscle contraction” was down-regulated after using an ERβ-selective agonist, diarylpropionitrile (DNP), whereas ERβ upregulated immune/chemokine genes and genes involved in regulating cell death (100). Together these findings demonstrate that ERα and ERβ have divergent effects, not only in their abundance and functions, but also in the types of genes they regulate downstream as well as across different tissues. Importantly, the effect of ERs on gene expression may also be time-dependent (101). Schnoes et al. reported that estrogen recruits specific TFs in vascular tissues in a fast and temporary manner (102). Recent studies showed that the method of administering estrogen or ER agonists can be divided into acute (ranging from several minutes to several hours) or chronic (ranging from 1 week to several weeks), which may work through different estrogen signaling pathways.
The complexity of ER-mediated gene regulation may represent a significant reason underlying the “estrogen paradox” in PAH. The lack of comparative studies on the distribution and proportion of ER subtypes in and between humans and animal models further compound the assumptions in translation. Specifically, they raise the question of reliability of using animal models to mirror estrogen signaling in humans. In addition, the signal transduction of ERs requires the participation of co-regulators. The type, level and post-translational modification of co-regulators will affect the signal transduction of ERs. Research on these aspects remains scarce, and subsequent studies will need to define the impact of these factors on PAH development.
ERα and ERβ show contrasting effects during disease states through the regulation of different genes. Overall, ERβ appears to exhibit a protective role in cardiopulmonary diseases, and has shown to exert anti-fibrotic (103), anti-hypertrophic (104), anti-inflammatory (12) and vaso-dilatory properties (105). ERβ has been shown to promote the production of various angiogenic factors to regulate angiogenesis, and NO, exerting vasodilator properties, mediating protective effects of estrogen in PAH. ERβ agonists have also been used to treat PAH (12). Further, studies support an important role for ERα during E2 signaling in PAH. These include data demonstrating that ERα mainly mediates the effect of E2 on cardiopulmonary hemodynamic parameters in HPH (90), that there are minimal to no effects of estrogen on vascular injury in ERα KO mice that underwent OVX (106) and that ERα in ECs plays a key role in E2-mediated vascular endothelialization (107). GPER has been shown to mediate cardioprotection through the activation of the PI3K/Akt pathway (75). GPER agonist, G1, inhibits the proliferation of cardiac fibroblasts in a dose-dependent manner (76), and alleviates cardiac remodeling and diastolic dysfunction in rats (108). G1 also improves the function and reduces myocardial inflammation during ischemia/reperfusion (109). The stimulation of GPER prevents changes in intracellular calcium concentration and vascular tone caused by vasoconstrictor and inhibits the proliferation of human vascular SMC (74). Conversely, the deletion of the GPER gene in mice eliminates the vascular effects of GPER activation (74). Based on these differential responses, selective activation of specific ER subtypes may help elucidate the effects of estrogen signaling on cardiopulmonary function in PAH.
The role of estrogen signaling in regulating gene expression is well-characterized (110). Multiple modes of signaling have been previously described (19, 96, 110). Two of these pathways are considered “genomic signaling” due to the direct involvement of DNA binding (19, 96), While a third type of non-genomic signaling cascade also has been described, typically via protein kinases (110).
Binding of estrogen (and its metabolites) to ERs can directly regulate gene expression and is often referred to as the classical pathway. Specifically, this pathway is initiated when estrogen enters the cytoplasm and binds with nERs to form an estrogen-ER complex (Figure 2). The combination of estrogen-ER complex leads to dimerization [either homodimer or heterodimer (19)] leading to DNA binding in two ways. First, the estrogen-ER complex can directly bind to EREs and acts as a TF complex, resulting in either up- or down-regulation of gene expression according to the types of recruited co-regulators (co-activators or co-suppressors). Second, on genes without EREs, the estrogen-ER complex indirectly binds to DNA through other TFs to regulate gene expression. This specific mechanism is often referred to as transcriptional crosstalk. For example, estrogen interacts with other promoter binding proteins to activate or inhibit AP-1 dependent transcription (111). Notably, many estrogen-responsive genes that lack full ERE sequences contain either partial ERE sequences or the binding site of SFRE, a response element with orphan nuclear hormone receptor (SF-1) that serves as a direct ER binding site (112). To our knowledge, only ERα has been observed to bind to SFRE (113). Importantly, about one-third of human genes are estimated to be targets for ER binding indirectly through intermediate TFs (112).
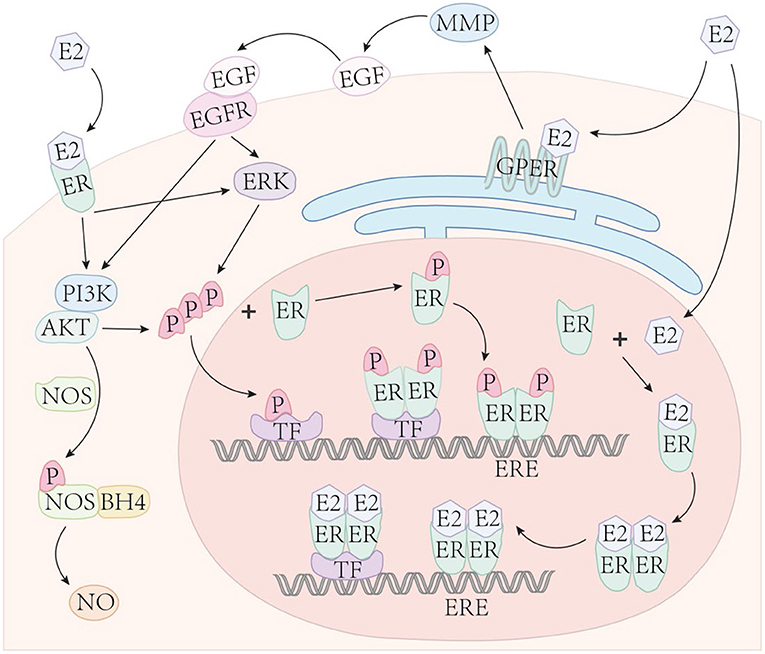
Figure 2. Estrogen signaling. Estrogen enters the cell nucleus and binds with nuclear ERs to form an estrogen-ER complex. The combination of the two estrogen-ER complexes typically lead to dimerization, then bind to DNA in two ways: (1) The estrogen-ER complex directly binds to the ERE sites on DNA and acts as a transcription factor, up- or down-regulating gene expression based on the types of recruited co-regulators; (2) In genes with absent EREs, ER indirectly binds to DNA through other TFs to influence gene expression. Phosphorylated ERs directly binds to ERE or indirectly binds to DNA through TFs, similar to estrogen-ER complexes. Estrogen binds to membrane ERα, Erβ, and GPER to mediate non-genomic (acute) estrogen signaling through the activation of various protein kinase cascades.
ERs can also independently interact with DNA without binding to estrogen and regulate gene expression, called ligand-independent transcriptional regulation. This mechanism requires ERs to be phosphorylated at specific serine sites (96). The phosphorylated ER directly combines with EREs or indirectly binds to DNA through TFs, similar to estrogen-ER complexes. This mode of ER ligand-independent transcriptional regulation also requires phosphorylation of growth factor-dependent coactivators. For example, Carascossa et al. demonstrated that although protein kinase A (PKA) is not required to participate in the direct phosphorylation of ERα, PKA assists in the phosphorylation of activator-related arginine methyltransferase 1 (CARM1), which is necessary for ERα ligand-independent transcriptional regulation (114).
The non-genomic pathway is also a common form of signal transduction of steroid hormones, generally through the activation of various protein kinase cascades (110). Both, ERα and ERβ, present on the cell membrane (115), as well as the estrogen receptor, GPER (also known as GPR30) (116), can mediate non-genomic estrogen signaling. E2 binds to the membrane-bound receptor and leads to the activation of G-protein directly coupled to the receptor, indicating that GPER is a G protein-coupled receptor. The activity of adenylate cyclase is also increased in this mode of signaling (70). Estrogen binds to the above receptors and then activates kinases or secondary messengers, including phosphoinositide 3-kinase (PI3K) (115), mitogen-activated protein kinase (MAPK) (117), etc.
Based on prior work, cardiovascular protection derived from estrogen is mediated, in part, by the activation of non-genomic effects (74, 118, 119). ERα and ERβ both have been shown to induce Akt-dependent activation of endothelial NOS (eNOS) in vascular ECs through a non-genomic mechanism, thereby, rapidly causing vasodilation (115, 118, 120–124). ERα also mediates the activation of eNOS through a MAPK-dependent mechanism (121). MAPK activation plays an important role in mediating the above non-genomic ER effects. The phosphorylation of ERα and ERβ is also mediated by the MAPK signaling pathway. Therefore, the non-genomic pathway can enhance genomic pathways by phosphorylation of ER. GPER activation also shows cardioprotection in both male and female rats (75), The GPER selective agonist, G1, effectively reverses PH-induced RV dysfunction, structural abnormalities and exercise intolerance in male rats (125).
Non-genomic pathways of estrogen conduction may indirectly affect gene expression by activating other signal transduction pathways that can act on target TFs. While the genomic effect takes several hours to manifest, non-genomic pathway is typically considered “acute” and characterized to manifest in a few seconds to minutes.
In PAH, mortality is closely related to sex and RV hemodynamic function (32, 39). Even though medical therapy reduces PVR, the prognosis of PAH patients remains poor from RV failure (38). RV ejection fraction (RVEF), assessed by magnetic resonance imaging, is an important determinant of the prognosis of patients with PAH. Increased RVEF indicates a higher survival rate (126). Male patients with PAH display lower RVEF than those of female patients (40). The response of males to heart pressure overload, cardiac contractility, and heart adaptability are also not as strong as females. Among females treated with estrogen, higher E2 levels were also associated with better RV contractile function (41, 42, 127), suggesting that estrogen may have cardioprotective effects.
E2 protection in the heart has been reported to signal through various pathways (Figure 3). In one study, the conversion of cardiac fibroblasts to myofibroblasts is inhibited by estrogen through ERβ, thereby preventing myocardial fibrosis (103). The expression of fibrosis markers and metalloproteinases in cardiac fibroblasts were directly inhibited by E2. Reversing fibrosis and up-regulating the expression of new extracellular matrix enzymes such as ADAM15, ADAM17 and OPN was associated with E2's ability to reverse the adverse RV remodeling associated with PH. These effects also may be mediated by ERβ (128). ERβ not only weakens fibrosis, but also inhibits the development of apoptosis, thereby slowing the progression of heart failure, which was more obvious in females (129). In addition, estrogen exhibits an inhibitory effect on the hypertrophic response caused by stress overload through the ERβ genomic effects (104, 130, 131). Estrogen also protects the heart through ERβ-mediated cardioprotective protein S-nitrosation (SNO), which requires the involvement of NOS (132, 133). Umar et al. found that recovery after removing estrogen still improves the structure and function of RV in MCT rats and rescues the original severity of PH. The beneficial effects of estrogen in PH appear to come predominantly from its protection of the heart, including stimulation of cardiopulmonary neovascularization, inhibition of fibrosis, RVH (12) and stimulating RV contractility (134).
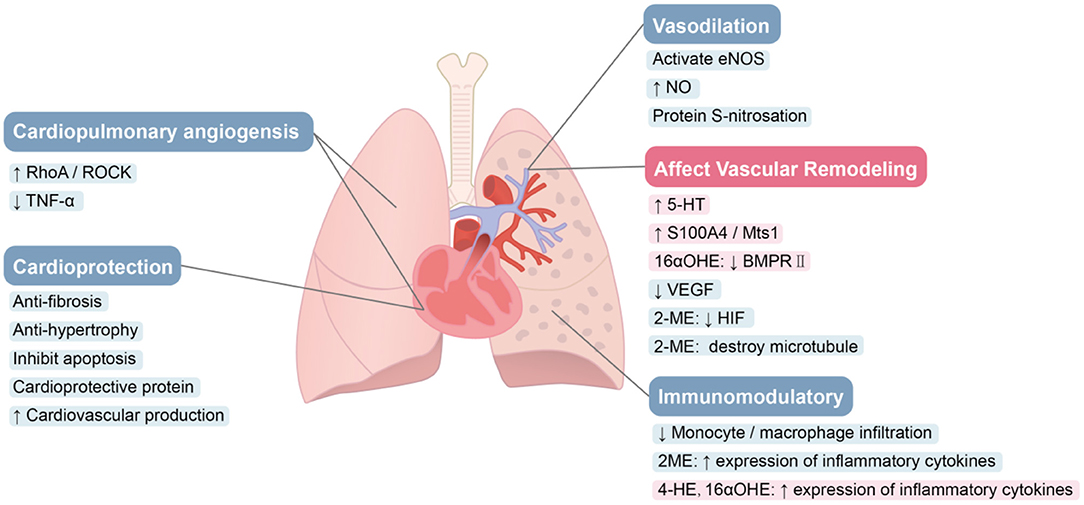
Figure 3. Role of estrogen and its metabolites in PAH. E2 protects the heart through various pathways. Estrogen can promote blood vessel remodeling. E2 improves cardiac structure and function via augmentation of angiogenesis. Estrogen can improve pulmonary hemodynamics. Estrogen and 2-ME appear to suppress inflammation in PAH, while 4-OHE and 16α-OHE1 have significant pro-inflammatory effects.
The protective effects of estrogen on the heart are considered to be partly mediated by ERβ (12). Many protective genes such as GADD45β and COX-2 are up-regulated by ERβ selective agonist, DPN (100). ERβ-mediated PI3K/Akt signal transduction was shown to improve myocardial function (135). Calcineurin-related hypertrophy is also inhibited by ERβ (130). Furthermore, activating ERβ reduces cardiac hypertrophy and fibrosis caused by AngII signaling (104). This finding was confirmed in ERβ KO mice (136). However, ER-α-mediated acute myocardial protection in female has also been reported, and differential activation of MAPK may mediate this protection (137). GPER has also shown cardioprotective effects in both male and female rats (75, 109, 138).
Vascular remodeling in PAH leads to occlusion of the lumen and a resulting rise in PVR. Abnormal proliferation of PASMCs plays a pivotal role in this remodeling (139). Estrogen and its metabolites have key roles in the pathological process. E2 promotes PA remodeling (Figure 3). In PASMCs, E2 induces increased expression of the calcium binding protein, S100A4/Mts1, and activates its endogenous receptor (receptor for advanced glycosylation end products; RAGE), thereby stimulating cell proliferation (14). Serotonin-induced human PASMC proliferation is mediated by SERT and 5-HT1B receptors (140, 141), and E2 mediates the upregulation of SERT and 5-HT1B receptors (15). Increased SERT expression causes increased 5-hydroxytryptamine (5-HT) in cells, and both E2 and 5-HT increase CYP1B1 expression in PAH-derived PASMCs (59). Thus, the vascular remodeling in PAH may be associated with dysregulation of estrogen metabolic pathways through increased CYP1B1 activity and the formation of pathogenic metabolites that promote PASMC proliferation. E2 also promotes PASMC hyperproliferation by inhibiting BMPR2 expression (142–144). Even in normal subjects, the expression and activity of BMPR2 is sex-specific. In PASMCs, the mRNA and protein expression of BMPR2 in females was observed to be lower than that of males in control subjects, and in mice (142), which may contribute to the proliferation phenotype of female PASMCs, and may partly explain why women are more susceptible to PAH (145). BMPR2 signal transduction is reduced in the lungs of SuHx-PH rats. Inhibition of estrogen synthesis with aromatase inhibitors not only reverses SuHx-PH in female rats, but also restores BMPR2 signaling (16), indicating that endogenous estrogen has an inhibitory effect on BMPR2 signaling. Moreover, there is direct binding of ERα to EREs on the promoter of the BMPR2 gene, resulting in estrogen-mediated reductions in BMPR2 expression (142). In addition, changes in oxygen concentration can alter E2 effects on BMP signaling in human PAECs (146).
Metabolites of estrogen also play a significant role in the abnormal proliferation of PASMCs. The highly estrogenic metabolite, 16α-OHE1, induces the proliferation of human PASMCs isolated from PAH patients (18, 46). 16α-OHE1 has also been shown to stimulate nicotinamide adenine dinucleotide phosphate oxidase (NOX)-induced ROS generation and cause abnormal proliferation, via Erα, in human PASMCs (44). In vivo, chronic 16α-OHE1 exposure in BMPR2 mutant male mice doubles the prevalence of PAH and decreased cardiac output. In the whole lungs of control mice, 16α-OHE1 inhibits BMPR2 protein and BMP signaling, but not in BMPR2 mutant mice, indicating a role for BMPR2 inhibition in 16α-OHE1-mediated proliferative effects.
2-ME2 has been shown to significantly reduce angiogenesis and remodeling. Using COMT KO mice, Lefteris et al. demonstrated the vascular anti-mitotic effects of E2 derived from catabolism to 2-ME2, which was ER-independent (147). SMC mitosis is inhibited by 2-ME2 through inhibition of key cell cycle regulatory proteins, up-regulating COX-2, reducing HIF protein expression, interfering with the polymerization of tubulin and disrupting the microtubule network (45, 148–150). The synthetic analog of 2-ME, 2-ethoxyestradiol (2-EE), can also inhibit vascular remodeling in PH in a dose-dependent manner. Moderate concentrations inhibit the growth of human PASMCs in vitro, and have a biphasic effect on the growth of ECs (low stimulation and high concentration inhibitory effect) (151). Furthermore, in primary rat aortic SMCs (152) and rat liver epithelial cell lines (153), the expression of angiotensin type 1 receptor (AT1R) was down-regulated by 2-ME2, a response mediated by GPER. Up-regulation of AT1R expression and signal transduction has been shown to be important in pulmonary vascular remodeling in PAH (154). Notably, E2 protection in HPH does not require conversion of E2 to downstream metabolites such as 2-ME2. E2 also directly reduces the secretion of vascular endothelial growth factor (VEGF) in hypoxic rat PAECs in an ER-dependent manner. E2 can inhibit the cell cycle, and reduce vascular remodeling and hemodynamic parameters in HPH through direct antiproliferative effects (90). As 2-ME2 does not appear to interact with nuclear ERα and ERβ in the cardiovascular system the anti-mitotic effects of 2-ME2 may be ER-independent (64).
The effects of E2 on vascular remodeling may be influenced by several factors. Oxygen concentrations can impact E2 signaling and there is a dichotomous effect of E2 in cultured PAECs, with E2 inhibiting hyperplasia pathways during hypoxia but promoting cell proliferation under normal oxygen levels (90). It should be noted that studies that have observed estrogen-promoting effects on angiogenesis and remodeling have all been conducted during normal oxygen levels. Dose-dependence is also important in E2 signaling (151, 155). Metabolite specificity also drives directionality in estrogen signaling. E2 metabolites exert both, proliferative and anti-proliferative, actions. For example, 2-ME is known to have anti-proliferative effects on cells, while 16α-OHE1 stimulates cell proliferation (156). Tissue specificity can influence estrogen signaling. ERα has pro-proliferative properties in some tissues and cancers, while ERβ has anti-proliferative effects (157). The activity of each ER subtype in tissues may also influence the relationship between estrogen and vascular remodeling.
The diverse array of E2 metabolites and their varying biological activities as well as signaling pathways combined may contribute to the diverging effects of estrogen (158). They may explain some of the opposing actions of estrogen metabolites on angiogenesis and remodeling across different studies. Equally important, disruption in the balance of estrogen metabolites may also explain the differential effects of estrogen in animal models of PH (15).
Loss of pulmonary microvessels and impaired regeneration leads to the progression of PAH. The loss of RV myocardial microvessels causes RV ischemia, which is directly related to RV dysfunction (159). Promoting the regeneration of “lost” distal vasculature is considered to be an important future treatment direction (160). Experimental studies have shown that estrogen plays a role in these phenotypes. For example, in MCT-PH, “loss” or pruning of blood vessels can be reversed by E2 treatment. E2 also stimulates the growth of new capillaries in healthy control rats beyond normal levels (12). Furthermore, E2 enhances EC activity during neovascularization (161), enhances EC motility through ERα (162), and promotes functional endothelial recovery after traumatic de-endothelialization injury (163). Tumor necrosis factor-α (TNF-α)-induced EC apoptosis is also inhibited by E2 in a dose-dependent (155). E2 enhances RhoA/ROCK signaling through ERs and increases protein expression related to the cell cycle, promoting EC migration and proliferation (164). The effect of E2 on RV capillary vascularization and RV ERK1/2 activation is only weakened in non-selective ER retardation, and is not effective in ERα or ERβ selective arrest (90). Estrogen binds to EREs by forming an estrogen-ER complex and regulates VEGF gene transcription (165). E2 enhances the mobilization of endothelial progenitor cells from bone matrix, and promotes its entry into sites of neovascularization through eNOS-mediated matrix metalloproteinase-9 expression enhancement in the bone matrix (166). Therefore, E2 may lead to improvements in cardiopulmonary structure and function by increasing myocardial blood vessels and angiogenesis, which may represent an additional mechanism to alleviate PH. This is supported by data showing that E2 cannot rescue PH in the presence of the angiogenesis inhibitor, TNP (12).
Exogenous E2 rapidly reduces PA vascular reactivity and acute hypoxic pulmonary vasoconstriction (HPV) in a dose-dependent fashion (167). Changes in endogenous E2 abundance in females can also affect the vasoconstrictor response (168). Recent research suggests that low levels of NO in PAH may not be solely due to reduced NOS expression, but rather, may be influenced by factors that regulate NOS activity (169). The beneficial effects of estrogen in PAH partly reflect its effects on eNOS induction via both expression and activation, resulting in NO production, improved pulmonary hemodynamics and vascular remodeling. Although this is likely and predominantly mediated by ERs at the level of gene transcription, estrogen also appears to have non-genomic effects on ERα-mediated eNOS activity in ECs. The combination of these genomic and non-genomic pathways is critical to the vascular protective properties of estrogen (121, 170).
Lahm et al. showed that even physiological changes in endogenous estrogen, such as the menstrual cycle, can affect pulmonary artery vasoreactivity and pulmonary vasoconstriction during acute hypoxia (168). Chronic E2 treatment in vivo increases the expression of eNOS, resulting in enhanced endothelium-dependent dilation in aorta and lungs (171). Long-term E2 infusion causes abnormal vasodilation patterns in the lungs of fetal sheep (172). The pulsatile load of the mechanical proximal PA is reduced after E2 supplementation, thereby improving ventricular-vascular coupling (173). High-dose exogenous E2 acutely attenuates PA vasoreactivity and acute HPV in a rapid and dose-dependent manner (167). This immediate effect suggests the role of non-genomic estrogen mechanisms. E2 also rapidly activates eNOS through Src kinase in human EC, inducing the formation of a complex containing ERs, c-Src and p85 (the regulatory subunit of PI3K). The formation of this complex leads to the continuous activation of PI3K, Akt, and eNOS, thereby enhancing the release of NO via eNOS (174). NO induced post-translational protein modification protects blood vessels through S-nitrosated cell proteins (175, 176). Indeed, physiologically relevant doses of E2 has been shown to increase protein S-nitrosation in vascular EC through ERα and eNOS. Although both ERα and ERβ specific agonists increase the expression of eNOS protein, only ERα specific agonists activate eNOS through phosphorylation (177). To sum up, E2 increases the protein level of eNOS through the genomic pathway, and activates eNOS in a rapid non-genomic manner in vascular. A similar effect of estrogen on eNOS was also observed in the heart. E2 stimulated the expression of iNOS and eNOS in cardiomyocytes (178). Both, ERα and ERβ, reduce PA vasoconstriction, and the contribution of specific ERs seems to be stimulus specific. ERα appears to mainly regulate phenylephrine-induced vasoconstriction, while ERβ was shown to inhibits HPV (123). ERα also affects women's vascular endothelial function by affecting protein levels and activating eNOS (80). While the anti-atherosclerotic effect of estrogen may be partially mediated by ERα-induced up-regulation of eNOS gene expression and maintenance of EC function and integrity (179).
A large body of evidence shows that inflammation and autoimmunity play important roles in the pathogenesis of PAH. At the beginning of the disease, the inflammatory response, caused by injured small pulmonary vascular endothelium, is believed to protect the body. However, as the disease progresses, Dorfmüller proposed that severe PH results in immune system disorders (180). This ineffective immune regulation (immune imbalance) may be related to the pathology and biology of PAH and leads to aggravation of the disease (181). In and around the reconstructed lung resistance vessel wall and near the plexiform lesions of PAH patients and PH animal models, varying degrees of perivascular inflammatory infiltration have been found, including T lymphocytes and B lymphocytes, macrophages, dendritic cells and mast cells (182), and elevated levels of proinflammatory cytokines (such as IL-1β and IL-6) in serum (183). Perivascular inflammation plays an important role in the process of vascular remodeling (184). Pulmonary vascular remodeling in HPH requires the recruitment of circulating mesenchymal precursors of the monocyte/macrophage lineage (185), which is essential for the late onset of HPH (186). Studies investigating links between estrogen and inflammation in other diseases have revealed that proinflammatory cytokines such as TNFα, IL-1 and IL-6 stimulate the activity of fibroblast aromatase and increase the level of estrogen (187, 188). Up-regulation of CYP1B1 leads to increased estrogen catabolism to 4-OHE and 16α-OHE1, which have significant angiogenic, pro-inflammatory and mitogenic properties, and thus promote the development of PAH (46). However, estrogen-induced PAH recovery is related to the inhibition of inflammation, and E2 limits the infiltration of lung monocytes/macrophages associated with PAH (12). E2 mediated inhibition of inflammation in the mouse lung is associated with modified levels of vascular cell adhesion molecules and proinflammatory mediators (189). Further, E2 has been shown to regulate several genes involved in inflammation during hypoxia in an ER-dependent manner. The anti-inflammatory effect of E2 appears to be predominantly mediated by ERβ (190, 191).
Another metabolite of estrogen, 2-ME2, also suppresses inflammation in PAH. 2-ME has significant anti-inflammatory and immunomodulatory effects (192). In several cardiovascular and kidney injury models, this effect is manifested by inhibiting macrophage influx/activation (193). Moreover, contrary to 16α-OHE1, 2-ME2 is found to inhibit the expression of inflammatory cytokines (TNF-α, IL-6 and PGE2) in other disease models, reducing local inflammation and preventing new angiogenesis (192). By blocking inflammatory cytokines, 2-ME reduces its stimulation of aromatase activity (194), which in turn may reduce E2 levels in local tissues. E2 appears to regulate lung inflammation in a sex-independent and age-restricted manner via ERα (195). Although the specific effect of inflammation on E2 metabolism in PAH is unclear, spontaneous inflammation is known to increase the production and utilization of E2 and its proinflammatory metabolites, which influences the pathology of severe PAH.
Pregnancy further complicates this complex estrogen signaling process. PAH is considered a significant risk factor during pregnancy (196). The levels of progesterone and estrogen continue to increase significantly throughout pregnancy (197–200). Other sex hormones such as DHEA and testosterone also increase during pregnancy (198, 201). These hormones mediate vasodilation (123, 167, 168, 202–204), considered a major driver for the increase in plasma volume caused by pregnancy (204, 205). In addition, elevated E2 and DHEA during pregnancy are thought to have a beneficial effect on RV function (206, 207). However, during pregnancy, especially during the perinatal period, the increase in RV pressure caused by hemodynamic changes and volume changes may far exceed the protective effects mediated by E2 and DHEA, resulting in RV failure. Moreover, while estrogen and some of its metabolites can promote pulmonary vascular remodeling in PAH (14, 18, 46), it remains uncertain whether the worsening of PAH observed in patients after pregnancy is due to, in part, to the direct impact of sex hormones on the cardiopulmonary system. Rapid deterioration of PAH often occurs in the postpartum period (208, 209), when levels of sex hormones drop sharply (210). The protective effects of sex hormones on PAH are likely reduced as their levels decrease, which at least partly contributes to postpartum RV failure. Broadly, the increase in sex hormones caused by pregnancy and their sharp decline during postpartum have complex effects on the pulmonary vascular system and RV in PAH. There are a paucity of studies in this field with significant need to fill this gap.
Although significant progress has been made in our understanding of estrogen synthesis and catabolism, earlier research efforts primarily focused on circulating levels of estrogen and/or its metabolism in the liver. Measuring circulating estrogen concentration or its concentration in urine, however, may not provide an adequate window into comprehensively studying the role of estrogen biology in PAH. A better understanding of the local concentrations of estrogen and its metabolites in the heart and lungs, enzymes involved in estrogen metabolism, and their activity, is required to further clarify the role of estrogen in PAH.
Beyond estrogen, testosterone- as a precursor of estradiol- also deserves consideration when discussing mechanisms of sex differences. Studies show that testosterone, largely, has an anti-inflammatory effect, resulting in more pronounced pulmonary artery relaxation than estrogen (202, 203, 211). However, observations from rodent PH models, where female rodents with ovariectomy demonstrate greater severity of disease than male rats with female rats considered the least severe, suggest that the overall contribution of testosterone in the development of PAH may not be as significant as estrogen. In contrast, E2 metabolism in PAH has also begun to be better characterized. In general, increased 16α-OHE1 appears to promote the development of PAH, while 2-ME2 likely contributes to reversal of disease-related phenotypes. E2 appears to be protective in the RV but its role in the pulmonary circulation is less clear. Inhibition of the major estrogen metabolizing enzymes, aromatase (estrogen synthesizing enzyme) and CYP1B1 (catalyzes the catabolism of estrogen to “PH-damaging” metabolites), represent novel potential therapeutic targets in PAH. However, factors that influence estrogen synthesis and catabolism remain poorly characterized in PAH. For example, can increased production of 2-ME2 promote a therapeutic role in PAH? Estrogen and its metabolites play diverging roles in different cells, and in particular cases the same cells. Comprehensive studies of their activity in various cells related to PAH, remain essential for increasing our understanding of the role of estrogen in PAH and these are still lacking.
Finally, establishing reproducible and translatable observations from animal models of PAH remains a top priority. For studies that focus on understanding the “estrogen paradox,” selecting an animal model that better mirrors features of the human phenotype is critical. Importantly, recognizing that the estrous cycle and hormone levels of commonly used rats and mice are not similar to those of human patients is equally as important. Moreover, there is a significant knowledge gap in the changes across the full spectrum of estrogen synthesis, catabolism and signaling in PAH animal models during disease development, further highlighting limitations of animal models. Although none of the animal models can fully mimic the phenotype of patients with PAH, researchers should methodically select animal model(s) that are most suitable for the hypothesis and aims of the study.
Future studies will need to address a variety of topics in this field including: (1) changes in estrogen and its metabolic activity in patients and animal models during the progression of PAH; (2) the role of estrogen and its metabolites in a tissue- and temporal-specific manner in PAH; (3) defining the spectrum of estrogen synthesis-enzymes and their contributions to the estrogen content in the blood. (4) the distribution and ratio of ERα, ERβ, and GPER in cardiopulmonary tissues and their function, as well as changes in their expression and modifications with age and PAH development; (5) transcriptional programming regulated by ERα, ERβ, or/and GPER as well as estrogen metabolites; (6) the bi-directional relationship between estrogen signaling and the presence/progression of disease including elucidating any potential positive or negative feedback loops.
In conclusion, estrogen influences cardiopulmonary function in health and disease in several ways. Differences in estrogen-related enzyme activity and estrogen signaling can be observed in normal and PAH lungs, indicating that there is a correlation between PAH and estrogen catabolism, metabolism and signaling. The functions and activities of specific receptors, the concentration and ratio of estrogen and its metabolites, as well as their local concentration relative to the circulatory system, the interaction between various sex hormones, age, comorbidity, genetics, and other factors are involved in the relationship between PAH and estrogen. By analyzing and comparing existing studies, the joint contribution of specific ERs or signaling pathways and specific metabolites may partially clarify the controversy about the role of estrogen in PAH. The knowledge gained from these future investigations will provide the necessary understanding on mechanisms and better prepare us to tackle the sex gap in PAH. Ultimately, it is hoped that these studies will fuel the discovery of novel therapeutic targets for this uncurable and devastating disease.
YS, HT, SB, and AD wrote the manuscript with input from SS, QG, and JW. All authors contributed to the article and approved the submitted version.
This work was funded by National Key Research and Development Program of China (2019YFE0119400), Natural Science Foundation of China (81970052 and 81770059), Natural Science Basic Research Program of Shaanxi Province (2018JC-012), and National Institutes of Health grants (P01HL134610 and P01HL146369 to SB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tonelli AR, Arelli V, Minai OA, Newman J, Bair N, Heresi GA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2013) 188:365–9. doi: 10.1164/rccm.201209-1640OC
2. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Euro Respir J. (2019) 53:1801913. doi: 10.1183/13993003.01913-2018
3. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. (2011) 8:443–55. doi: 10.1038/nrcardio.2011.87
4. Europe WHOROf: Gender: Definitions. Available online at: https://www.euro.who.int/en/health-topics/health-determinants/gender/gender-definitions Date unknown (accessed July 26, 2021).
5. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Internal Med. (1987) 107:216–23. doi: 10.7326/0003-4819-107-2-216
6. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. (2006) 173:1023–30. doi: 10.1164/rccm.200510-1668OC
7. Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, Robbins IM, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:892–6. doi: 10.1164/rccm.201205-0886OC
8. Tamura Y, Kumamaru H, Satoh T, Miyata H, Ogawa A, Tanabe N, et al. Effectiveness and outcome of pulmonary arterial hypertension-specific therapy in japanese patients with pulmonary arterial hypertension. Circul J. (2017) 82:275–82. doi: 10.1253/circj.CJ-17-0139
9. Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US Contemporary Registries. Chest. (2011) 139:128–37. doi: 10.1378/chest.10-0075
10. Pauwaa S, Machado RF, Desai AA. Survival in pulmonary arterial hypertension: a brief review of registry data. Pulm Circ. (2011) 1:430–1. doi: 10.4103/2045-8932.87314
11. Earley S, Resta TC. Estradiol attenuates hypoxia-induced pulmonary endothelin-1 gene expression. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L86–93. doi: 10.1152/ajplung.00476.2001
12. Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. (2011) 184:715–23. doi: 10.1164/rccm.201101-0078OC
13. Yang YM, Yuan H, Edwards JG, Skayian Y, Ochani K, Miller EJ, et al. Deletion of STAT5a/b in vascular smooth muscle abrogates the male bias in hypoxic pulmonary hypertension in mice: implications in the human disease. Mol Med. (2015) 20:625–38. doi: 10.2119/molmed.2014.00180
14. Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, et al. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res. (2011) 12:159. doi: 10.1186/1465-9921-12-159
15. White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res. (2011) 90:373–82. doi: 10.1093/cvr/cvq408
16. Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. (2014) 190:456–67. doi: 10.1164/rccm.201403-0483OC
17. Yuan P, Wu WH, Gao L, Zheng ZQ, Liu D, Mei HY, et al. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, prostacyclin and endothelin-1 pathways. Euro Respir J. (2013) 41:1116–25. doi: 10.1183/09031936.00044112
18. Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, et al. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Euro Respir J. (2009) 34:1093–9. doi: 10.1183/09031936.00010409
19. Cotnoir-White D, El Ezzy M, Boulay PL, Rozendaal M, Bouvier M, Gagnon E, et al. Monitoring ligand-dependent assembly of receptor ternary complexes in live cells by BRETFect. Proc Natl Acad Sci USA. (2018) 115:E2653–e62. doi: 10.1073/pnas.1716224115
20. Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med. (1951) 11:686–705. doi: 10.1016/0002-9343(51)90020-4
21. Wood P. Pulmonary hypertension. Br Med Bull. (1952) 8:348–53. doi: 10.1093/oxfordjournals.bmb.a074201
22. Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. (2010) 137:376–87. doi: 10.1378/chest.09-1140
23. Jing ZC, Xu XQ, Han ZY, Wu Y, Deng KW, Wang H, et al. Registry and survival study in chinese patients with idiopathic and familial pulmonary arterial hypertension. Chest. (2007) 132:373–9. doi: 10.1378/chest.06-2913
24. Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. (2012) 186:790–6. doi: 10.1164/rccm.201203-0383OC
25. Hoeper MM, Huscher D, Ghofrani HA, Delcroix M, Distler O, Schweiger C, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol. (2013) 168:871–80. doi: 10.1016/j.ijcard.2012.10.026
26. Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Euro Respir J. (2007) 30:1103–10. doi: 10.1183/09031936.00042107
27. Jansa P, Jarkovsky J, Al-Hiti H, Popelova J, Ambroz D, Zatocil T, et al. Epidemiology and long-term survival of pulmonary arterial hypertension in the Czech Republic: a retrospective analysis of a nationwide registry. BMC Pulmonary Med. (2014) 14:45. doi: 10.1186/1471-2466-14-45
28. Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation. (2014) 129:57–65. doi: 10.1161/CIRCULATIONAHA.113.004526
29. Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Euro Respir J. (2007) 30:104–9. doi: 10.1183/09031936.00092306
30. Kane GC, Maradit-Kremers H, Slusser JP, Scott CG, Frantz RP, McGoon MD. Integration of clinical and hemodynamic parameters in the prediction of long-term survival in patients with pulmonary arterial hypertension. Chest. (2011) 139:1285–93. doi: 10.1378/chest.10-1293
31. Escribano-Subias P, Blanco I, López-Meseguer M, Lopez-Guarch CJ, Roman A, Morales P, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Euro Respir J. (2012) 40:596–603. doi: 10.1183/09031936.00101211
32. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. (2010) 122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818
33. Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. (2010) 122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122
34. Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Euro Respir J. (2010) 36:549–55. doi: 10.1183/09031936.00057010
35. Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol. (1993) 110:719–23. doi: 10.1111/j.1476-5381.1993.tb13871.x
36. Luo J, Yang X, Qiu S, Li X, Xiang E, Fang Y, et al. Sex difference in monocrotaline-induced developmental toxicity and fetal hepatotoxicity in rats. Toxicology. (2019) 418:32–40. doi: 10.1016/j.tox.2019.02.014
37. Lahm T, Frump AL, Albrecht ME, Fisher AJ, Cook TG, Jones TJ, et al. 17β-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L375–88. doi: 10.1152/ajplung.00132.2016
38. van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. (2011) 58:2511–9. doi: 10.1016/j.jacc.2011.06.068
39. Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest. (2014) 145:1230–6. doi: 10.1378/chest.13-1291
40. Kawut SM, Al-Naamani N, Agerstrand C, Berman Rosenzweig E, Rowan C, Barst RJ, et al. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest. (2009) 135:752–9. doi: 10.1378/chest.08-1758
41. Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD, Kawut SM. Sex and haemodynamics in pulmonary arterial hypertension. Euro Respir J. (2014) 43:523–30. doi: 10.1183/09031936.00027613
42. Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. (2012) 141:363–73. doi: 10.1378/chest.10-3114
43. Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocrine Rev. (2004) 25:947–70. doi: 10.1210/er.2003-0030
44. Hood KY, Montezano AC, Harvey AP, Nilsen M, MacLean MR, Touyz RM. Nicotinamide adenine dinucleotide phosphate oxidase-mediated redox signaling and vascular remodeling by 16α-hydroxyestrone in human pulmonary artery cells: implications in pulmonary arterial hypertension. Hypertension. (2016) 68:796–808. doi: 10.1161/HYPERTENSIONAHA.116.07668
45. Docherty CK, Nilsen M, MacLean MR. Influence of 2-methoxyestradiol and sex on hypoxia-induced pulmonary hypertension and hypoxia-inducible factor-1-α. J Am Heart Assoc. (2019) 8:e011628. doi: 10.1161/JAHA.118.011628
46. White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. (2012) 126:1087–98. doi: 10.1161/CIRCULATIONAHA.111.062927
47. Lee AJ, Cai MX, Thomas PE, Conney AH, Zhu BT. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology. (2003) 144:3382–98. doi: 10.1210/en.2003-0192
48. Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy. (2003) 23:165–72. doi: 10.1592/phco.23.2.165.32088
49. Liu D, Bachmann KA. An investigation of the relationship between estrogen, estrogen metabolites and blood cholesterol levels in ovariectomized rats. J Pharmacol Exp Ther. (1998) 286:561–8.
50. Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: insights into the structural determinants favoring a differential subtype binding. Endocrinology. (2006) 147:4132–50. doi: 10.1210/en.2006-0113
51. Dubey RK, Gillespie DG, Zacharia LC, Rosselli M, Imthurn B, Jackson EK. Methoxyestradiols mediate the antimitogenic effects of locally applied estradiol on cardiac fibroblast growth. Hypertension. (2002) 39:412–7. doi: 10.1161/hy0202.102837
52. Farin FM, Pohlman TH, Omiecinski CJ. Expression of cytochrome P450s and microsomal epoxide hydrolase in primary cultures of human umbilical vein endothelial cells. Toxicol Appl Pharmacol. (1994) 124:1–9. doi: 10.1006/taap.1994.1001
53. Zacharia LC, Jackson EK, Gillespie DG, Dubey RK. Increased 2-methoxyestradiol production in human coronary versus aortic vascular cells. Hypertension. (2001) 37:658–62. doi: 10.1161/01.HYP.37.2.658
54. Chen ZH, Na HK, Hurh YJ, Surh YJ. 4-Hydroxyestradiol induces oxidative stress apoptosis in human mammary epithelial cells: possible protection by NF-kappaB ERK/MAPK. Toxicol Appl Pharmacol. (2005) 208:46-56. doi: 10.1016/j.taap.2005.01.010
55. Badawi AF, Cavalieri EL, Rogan EG. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16alpha-hydroxylation of 17beta-estradiol. Metab Clin Exp. (2001) 50:1001–3. doi: 10.1053/meta.2001.25592
56. Lee AJ, Conney AH, Zhu BT. Human cytochrome P450 3A7 has a distinct high catalytic activity for the 16alpha-hydroxylation of estrone but not 17beta-estradiol. Cancer Res. (2003) 63:6532–6.
57. Swaneck GE, Fishman J. Covalent binding of the endogenous estrogen 16 alpha-hydroxyestrone to estradiol receptor in human breast cancer cells: characterization and intranuclear localization. Proc Natl Acad Sci USA. (1988) 85:7831–5. doi: 10.1073/pnas.85.21.7831
58. Fishman J, Martucci C. Biological properties of 16 alpha-hydroxyestrone: implications in estrogen physiology and pathophysiology. J Clin Endocrinol Metab. (1980) 51:611–5. doi: 10.1210/jcem-51-3-611
59. Dempsie Y, MacRitchie NA, White K, Morecroft I, Wright AF, Nilsen M, et al. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res. (2013) 99:24–34. doi: 10.1093/cvr/cvt064
60. Denver N, Homer NZM, Andrew R, Harvey KY, Morrell N, Austin ED, et al. Estrogen metabolites in a small cohort of patients with idiopathic pulmonary arterial hypertension. Pulmonary Circul. (2020) 10:2045894020908783. doi: 10.1177/2045894020908783
61. Harada N, Sasano H, Murakami H, Ohkuma T, Nagura H, Takagi Y. Localized expression of aromatase in human vascular tissues. Circul Res. (1999) 84:1285–91. doi: 10.1161/01.RES.84.11.1285
62. Park BK. Cytochrome P450 enzymes in the heart. Lancet. (2000) 355:945–6. doi: 10.1016/S0140-6736(00)90008-4
63. Dubey RK, Gillespie DG, Zacharia LC, Barchiesi F, Imthurn B, Jackson EK. CYP450- and COMT-derived estradiol metabolites inhibit activity of human coronary artery SMCs. Hypertension. (2003) 41:807–13. doi: 10.1161/01.HYP.0000048862.28501.72
64. Barchiesi F, Jackson EK, Gillespie DG, Zacharia LC, Fingerle J, Dubey RK. Methoxyestradiols mediate estradiol-induced antimitogenesis in human aortic SMCs. Hypertension. (2002) 39:874–9. doi: 10.1161/01.HYP.0000013863.25970.BA
65. Sánchez-Guijo A, Neunzig J, Gerber A, Oji V, Hartmann MF, Schuppe HC, et al. Role of steroid sulfatase in steroid homeostasis and characterization of the sulfated steroid pathway: evidence from steroid sulfatase deficiency. Mol Cell Endocrinol. (2016) 437:142–53. doi: 10.1016/j.mce.2016.08.019
66. Spink BC, Katz BH, Hussain MM, Pang S, Connor SP, Aldous KM, et al. SULT1A1 catalyzes 2-methoxyestradiol sulfonation in MCF-7 breast cancer cells. Carcinogenesis. (2000) 21:1947–57. doi: 10.1093/carcin/21.11.1947
67. Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. (1999) 140:5566–78.
68. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
69. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. (2006) 2:207–12. doi: 10.1038/nchembio775
70. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. (2005) 146:624–32. doi: 10.1210/en.2004-1064
71. Grohé C, Kahlert S, Löbbert K, Vetter H. Expression of oestrogen receptor alpha and beta in rat heart: role of local oestrogen synthesis. J Endocrinol. (1998) 156:R1–7. doi: 10.1677/joe.0.156r001
72. Ding Q, Gros R, Limbird LE, Chorazyczewski J, Feldman RD. Estradiol-mediated ERK phosphorylation and apoptosis in vascular smooth muscle cells requires GPR 30. Am J Phys Cell Physiol. (2009) 297:C1178–87. doi: 10.1152/ajpcell.00185.2009
73. Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, et al. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension. (2007) 49:1358–63. doi: 10.1161/HYPERTENSIONAHA.107.089995
74. Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circul Res. (2009) 104:288–91. doi: 10.1161/CIRCRESAHA.108.190892
75. Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Phys Heart Circul Physiol. (2009) 297:H1806–13. doi: 10.1152/ajpheart.00283.2009
76. Wang H, Zhao Z, Lin M, Groban L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol Cell Biochem. (2015) 405:135–48. doi: 10.1007/s11010-015-2405-3
77. Venkov CD, Rankin AB, Vaughan DE. Identification of authentic estrogen receptor in cultured endothelial cells. A potential mechanism for steroid hormone regulation of endothelial function. Circulation. (1996) 94:727–33. doi: 10.1161/01.CIR.94.4.727
78. Karas RH, Patterson BL, Mendelsohn ME. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation. (1994) 89:1943–50. doi: 10.1161/01.CIR.89.5.1943
79. Wright AF, Ewart MA, Mair K, Nilsen M, Dempsie Y, Loughlin L, et al. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res. (2015) 106:206–16. doi: 10.1093/cvr/cvv106
80. Gavin KM, Seals DR, Silver AE, Moreau KL. Vascular endothelial estrogen receptor alpha is modulated by estrogen status and related to endothelial function and endothelial nitric oxide synthase in healthy women. J Clin Endocrinol Metab. (2009) 94:3513–20. doi: 10.1210/jc.2009-0278
81. Lindsey SH, da Silva AS, Silva MS, Chappell MC. Reduced vasorelaxation to estradiol and G-1 in aged female and adult male rats is associated with GPR30 downregulation. Am J Physiol Endocrinol Metab. (2013) 305:E113–8. doi: 10.1152/ajpendo.00649.2012
82. O'Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, et al. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol. (2007) 21:1281–96. doi: 10.1210/me.2006-0497
83. Mollerup S, Jørgensen K, Berge G, Haugen A. Expression of estrogen receptors alpha and beta in human lung tissue and cell lines. Lung Cancer. (2002) 37:153–9. doi: 10.1016/S0169-5002(02)00039-9
84. Andersson C, Lydrup ML, Fernö M, Idvall I, Gustafsson J, Nilsson BO. Immunocytochemical demonstration of oestrogen receptor beta in blood vessels of the female rat. J Endocrinol. (2001) 169:241–7. doi: 10.1677/joe.0.1690241
85. Nordmeyer J, Eder S, Mahmoodzadeh S, Martus P, Fielitz J, Bass J, et al. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation. (2004) 110:3270–5. doi: 10.1161/01.CIR.0000147610.41984.E8
86. Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, et al. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. (2006) 20:926–34. doi: 10.1096/fj.05-5148com
87. Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L873–90. doi: 10.1152/ajplung.00006.2015
88. Pinzone JJ, Stevenson H, Strobl JS, Berg PE. Molecular and cellular determinants of estrogen receptor alpha expression. Mol Cell Biol. (2004) 24:4605–12. doi: 10.1128/MCB.24.11.4605-4612.2004
89. Ihionkhan CE, Chambliss KL, Gibson LL, Hahner LD, Mendelsohn ME, Shaul PW. Estrogen causes dynamic alterations in endothelial estrogen receptor expression. Circul Res. (2002) 91:814–20. doi: 10.1161/01.RES.0000038304.62046.4C
90. Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, et al. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. (2012) 185:965–80. doi: 10.1164/rccm.201107-1293OC
91. Lindner V, Kim SK, Karas RH, Kuiper GG, Gustafsson JA, Mendelsohn ME. Increased expression of estrogen receptor-beta mRNA in male blood vessels after vascular injury. Circul Res. (1998) 83:224–9. doi: 10.1161/01.RES.83.2.224
92. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology. (1997) 138:4613–21. doi: 10.1210/endo.138.11.5496
93. Post WS, Goldschmidt-Clermont PJ, Wilhide CC, Heldman AW, Sussman MS, Ouyang P, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. (1999) 43:985–91. doi: 10.1016/S0008-6363(99)00153-4
94. Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguère V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol. (2010) 24:1349–58. doi: 10.1210/me.2009-0441
95. Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. (2006) 20:1479–93. doi: 10.1210/me.2005-0531
96. Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J Mol Endocrinol. (2008) 40:173–84. doi: 10.1677/JME-07-0165
97. Garbán HJ, Márquez-Garbán DC, Pietras RJ, Ignarro LJ. Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc Natl Acad Sci USA. (2005) 102:2632–6. doi: 10.1073/pnas.0409854102
98. Lindberg MK, Movérare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, et al. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol. (2003) 17:203–8. doi: 10.1210/me.2002-0206
99. Tsutsumi S, Zhang X, Takata K, Takahashi K, Karas RH, Kurachi H, et al. Differential regulation of the inducible nitric oxide synthase gene by estrogen receptors 1 and 2. J Endocrinol. (2008) 199:267–73. doi: 10.1677/JOE-07-0292
100. Nikolic I, Liu D, Bell JA, Collins J, Steenbergen C, Murphy E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J Mol Cell Cardiol. (2007) 42:769–80. doi: 10.1016/j.yjmcc.2007.01.014
101. Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, et al. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol Endocrinol. (2003) 17:2070–83. doi: 10.1210/me.2003-0146
102. Schnoes KK, Jaffe IZ, Iyer L, Dabreo A, Aronovitz M, Newfell B, et al. Rapid recruitment of temporally distinct vascular gene sets by estrogen. Mol Endocrinol. (2008) 22:2544–56. doi: 10.1210/me.2008-0044
103. Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER. Estrogen receptor-beta prevents cardiac fibrosis. Mol Endocrinol. (2010) 24:2152–65. doi: 10.1210/me.2010-0154
104. Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology. (2008) 149:3361–9. doi: 10.1210/en.2008-0133
105. Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol. (2001) 280:L88–97. doi: 10.1152/ajplung.2001.280.1.L88
106. Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, et al. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circul Res. (2002) 90:1087–92. doi: 10.1161/01.RES.0000021114.92282.FA
107. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. (2001) 103:423–8. doi: 10.1161/01.CIR.103.3.423
108. Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res. (2012) 94:96–104. doi: 10.1093/cvr/cvs090
109. Weil BR, Manukyan MC, Herrmann JL, Wang Y, Abarbanell AM, Poynter JA, et al. Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery. (2010) 148:436–43. doi: 10.1016/j.surg.2010.03.011
110. Lösel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. (2003) 4:46–56. doi: 10.1038/nrm1009
111. Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J Biol Chem. (2001) 276:13615–21. doi: 10.1074/jbc.M008384200
112. O'Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol Endocrinol. (2004) 18:1859–75. doi: 10.1210/me.2003-0044
113. Vanacker JM, Pettersson K, Gustafsson JA, Laudet V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. EMBO J. (1999) 18:4270–9. doi: 10.1093/emboj/18.15.4270
114. Carascossa S, Dudek P, Cenni B, Briand PA, Picard D. CARM1 mediates the ligand-independent tamoxifen-resistant activation of the estrogen receptor alpha by cAMP. Genes Dev. (2010) 24:708–19. doi: 10.1101/gad.568410
115. Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. (2000) 407:538–41. doi: 10.1038/35035131
116. Hsieh YC, Yu HP, Frink M, Suzuki T, Choudhry MA, Schwacha MG, et al. G protein-coupled receptor 30-dependent protein kinase A pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma-hemorrhage. Am J Pathol. (2007) 170:1210–8. doi: 10.2353/ajpath.2007.060883
117. Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME, Pfister SL, et al. Estrogen acutely activates prostacyclin synthesis in ovine fetal pulmonary artery endothelium. Am J Respir Cell Mol Biol. (2002) 26:610–6. doi: 10.1165/ajrcmb.26.5.4528
118. Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Investig. (2010) 120:2319–30. doi: 10.1172/JCI38291
119. Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. (2011) 286:14737–43. doi: 10.1074/jbc.R110.191791
120. Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y, et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. (2001) 276:3459–67. doi: 10.1074/jbc.M005036200
121. Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Investig. (1999) 103:401–6. doi: 10.1172/JCI5347
122. Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol. (2002) 16:938–46. doi: 10.1210/mend.16.5.0827
123. Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Tan J, et al. Selective estrogen receptor-alpha and estrogen receptor-beta agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integrative Compar Physiol. (2008) 295:R1486–93. doi: 10.1152/ajpregu.90667.2008
124. Ba ZF, Lu A, Shimizu T, Szalay L, Schwacha MG, Rue LW, et al. 17beta-Estradiol modulates vasoconstriction induced by endothelin-1 following trauma-hemorrhage. Am J Physiol Heart Circul Physiol. (2007) 292:H245-50. doi: 10.1152/ajpheart.00809.2006
125. Alencar AK, Montes GC, Montagnoli T, Silva AM, Martinez ST, Fraga AG, et al. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Euro J Pharm Sci. (2017) 97:208–17. doi: 10.1016/j.ejps.2016.11.009
126. Courand PY, Pina Jomir G, Khouatra C, Scheiber C, Turquier S, Glérant JC, et al. Prognostic value of right ventricular ejection fraction in pulmonary arterial hypertension. Euro Respir J. (2015) 45:139–49. doi: 10.1183/09031936.00158014
127. Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, et al. Sex hormones are associated with right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med. (2011) 183:659–67. doi: 10.1164/rccm.201007-1027OC
128. Nadadur RD, Umar S, Wong G, Eghbali M, Iorga A, Matori H, et al. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J Appl Physiol. (2012) 113:149–58. doi: 10.1152/japplphysiol.01349.2011
129. Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Kararigas G, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integrative Compar Physiol. (2010) 298:R1597–606. doi: 10.1152/ajpregu.00825.2009
130. Pedram A, Razandi M, Aitkenhead M, Levin ER. Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1. J Biol Chem. (2005) 280:26339–48. doi: 10.1074/jbc.M414409200
131. Babiker FA, De Windt LJ, van Eickels M, Thijssen V, Bronsaer RJ, Grohé C, et al. 17beta-estradiol antagonizes cardiomyocyte hypertrophy by autocrine/paracrine stimulation of a guanylyl cyclase A receptor-cyclic guanosine monophosphate-dependent protein kinase pathway. Circulation. (2004) 109:269–76. doi: 10.1161/01.CIR.0000105682.85732.BD
132. Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation. (2009) 120:245–54. doi: 10.1161/CIRCULATIONAHA.109.868729
133. Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circul Res. (2006) 98:403–11. doi: 10.1161/01.RES.0000202707.79018.0a
134. Liu A, Schreier D, Tian L, Eickhoff JC, Wang Z, Hacker TA, et al. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am J Physiol Heart Circul Physiol. (2014) 307:H273–83. doi: 10.1152/ajpheart.00758.2013
135. Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, et al. Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integrative Compar Physiol. (2009) 296:R972–8. doi: 10.1152/ajpregu.00045.2009
136. Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, et al. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circul Physiol. (2005) 288:H469–76. doi: 10.1152/ajpheart.00723.2004
137. Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circul Physiol. (2006) 290:H2204–9. doi: 10.1152/ajpheart.01219.2005
138. Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circul Physiol. (2010) 298:H16–23. doi: 10.1152/ajpheart.00588.2009
139. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. (2004) 43:13s−24s. doi: 10.1016/j.jacc.2004.02.029
140. Morecroft I, Pang L, Baranowska M, Nilsen M, Loughlin L, Dempsie Y, et al. In vivo effects of a combined 5-HT1B receptor/SERT antagonist in experimental pulmonary hypertension. Cardiovasc Res. (2010) 85:593–603. doi: 10.1093/cvr/cvp306
141. Lawrie A, Spiekerkoetter E, Martinez EC, Ambartsumian N, Sheward WJ, MacLean MR, et al. Interdependent serotonin transporter and receptor pathways regulate S100A4/Mts1, a gene associated with pulmonary vascular disease. Circul Res. (2005) 97:227–35. doi: 10.1161/01.RES.0000176025.57706.1e
142. Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biology of sex differences. (2012) 3:6. doi: 10.1186/2042-6410-3-6
143. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. (2001) 104:790–5. doi: 10.1161/hc3201.094152
144. Yu PB, Deng DY, Beppu H, Hong CC, Lai C, Hoyng SA, et al. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J Biol Chem. (2008) 283:3877–88. doi: 10.1074/jbc.M706797200
145. Mair KM, Yang XD, Long L, White K, Wallace E, Ewart MA, et al. Sex affects bone morphogenetic protein type II receptor signaling in pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. (2015) 191:693–703. doi: 10.1164/rccm.201410-1802OC
146. Ichimori H, Kogaki S, Takahashi K, Ishida H, Narita J, Nawa N, et al. Drastic shift from positive to negative estrogen effect on bone morphogenetic protein signaling in pulmonary arterial endothelial cells under hypoxia. Circul J. (2013) 77:2118–26. doi: 10.1253/circj.CJ-12-0997
147. Zacharia LC, Gogos JA, Karayiorgou M, Jackson EK, Gillespie DG, Barchiesi F, et al. Methoxyestradiols mediate the antimitogenic effects of 17beta-estradiol: direct evidence from catechol-O-methyltransferase-knockout mice. Circulation. (2003) 108:2974–8. doi: 10.1161/01.CIR.0000106900.66354.30
148. Barchiesi F, Jackson EK, Fingerle J, Gillespie DG, Odermatt B, Dubey RK. 2-Methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circul Res. (2006) 99:266–74. doi: 10.1161/01.RES.0000233318.85181.2e
149. Escuin D, Kline ER, Giannakakou P. Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1alpha accumulation and activity by disrupting microtubule function. Cancer Res. (2005) 65:9021–8. doi: 10.1158/0008-5472.CAN-04-4095
150. Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. (2003) 3:363–75 doi: 10.1016/s1535-6108(03)00077-1
151. Muyan M, Roser JF, Dybdal N, Baldwin DM. Modulation of gonadotropin-releasing hormone-stimulated luteinizing hormone release in cultured male equine anterior pituitary cells by gonadal steroids. Biology of reproduction. (1993) 49:340–5. doi: 10.1095/biolreprod49.2.340
152. Ogola B, Zhang Y, Iyer L, Thekkumkara T. 2-Methoxyestradiol causes matrix metalloproteinase 9-mediated transactivation of epidermal growth factor receptor and angiotensin type 1 receptor downregulation in rat aortic smooth muscle cells. Am J Physiol Cell Physiol. (2018) 314:C554–68. doi: 10.1152/ajpcell.00152.2017
153. Koganti S, Snyder R, Gumaste U, Karamyan VT, Thekkumkara T. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Euro J Pharmacol. (2014) 723:131–40. doi: 10.1016/j.ejphar.2013.10.064
154. de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:780–9. doi: 10.1164/rccm.201203-0411OC
155. Spyridopoulos I, Sullivan AB, Kearney M, Isner JM, Losordo DW. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis. Estradiol as a survival factor. Circulation. (1997) 95:1505–14. doi: 10.1161/01.CIR.95.6.1505
156. Tofovic SP, Zhang X, Jackson EK, Dacic S, Petrusevska G. 2-Methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vasc Pharmacol. (2006) 45:358–67. doi: 10.1016/j.vph.2006.05.007
157. Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. (2004) 64:423–8. doi: 10.1158/0008-5472.CAN-03-2446
158. Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. (1998) 19:1–27.
159. Gómez A, Bialostozky D, Zajarias A, Santos E, Palomar A, Martínez ML, et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. (2001) 38:1137–42. doi: 10.1016/S0735-1097(01)01496-6
160. Frump AL, Bonnet S, de Jesus Perez VA, Lahm T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L443–60. doi: 10.1152/ajplung.00374.2017
161. Morales DE, McGowan KA, Grant DS, Maheshwari S, Bhartiya D, Cid MC, et al. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation. (1995) 91:755–63. doi: 10.1161/01.CIR.91.3.755
162. Sanchez AM, Flamini MI, Zullino S, Gopal S, Genazzani AR, Simoncini T. Estrogen receptor-{alpha} promotes endothelial cell motility through focal adhesion kinase. Mol Human Reprod. (2011) 17:219–26. doi: 10.1093/molehr/gaq097
163. Krasinski K, Spyridopoulos I, Asahara T, van der Zee R, Isner JM, Losordo DW. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation. (1997) 95:1768–72. doi: 10.1161/01.CIR.95.7.1768
164. Oviedo PJ, Sobrino A, Laguna-Fernandez A, Novella S, Tarín JJ, García-Pérez MA, et al. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol Cell Endocrinol. (2011) 335:96–103. doi: 10.1016/j.mce.2010.06.020
165. Mueller MD, Vigne JL, Minchenko A, Lebovic DI, Leitman DC, Taylor RN. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors alpha and beta. Proc Natl Acad Sci USA. (2000) 97:10972–7. doi: 10.1073/pnas.200377097
166. Iwakura A, Shastry S, Luedemann C, Hamada H, Kawamoto A, Kishore R, et al. Estradiol enhances recovery after myocardial infarction by augmenting incorporation of bone marrow-derived endothelial progenitor cells into sites of ischemia-induced neovascularization via endothelial nitric oxide synthase-mediated activation of matrix metalloproteinase-9. Circulation. (2006) 113:1605–14. doi: 10.1161/CIRCULATIONAHA.105.553925
167. Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Weil B, et al. Exogenous estrogen rapidly attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction. Shock. (2008) 30:660–7. doi: 10.1097/SHK.0b013e31816f239f
168. Lahm T, Patel KM, Crisostomo PR, Markel TA, Wang M, Herring C, et al. Endogenous estrogen attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction: the effects of sex and menstrual cycle. Am J Physiol Endocrinol Metab. (2007) 293:E865–71. doi: 10.1152/ajpendo.00201.2007
169. Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. (2004) 18:1746–8. doi: 10.1096/fj.04-2317fje
170. Caulin-Glaser T, García-Cardeña G, Sarrel P, Sessa WC, Bender JR. 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circul Res. (1997) 81:885–92. doi: 10.1161/01.RES.81.5.885
171. Gonzales RJ, Walker BR, Kanagy NL. 17beta-estradiol increases nitric oxide-dependent dilation in rat pulmonary arteries and thoracic aorta. Am J Physiol Lung Cell Mol Physiol. (2001) 280:L555–64. doi: 10.1152/ajplung.2001.280.3.L555
172. Parker TA, Kinsella JP, Galan HL, Le Cras TD, Richter GT, Markham NE, et al. Prolonged infusions of estradiol dilate the ovine fetal pulmonary circulation. Pediatric research. (2000) 47:89–96. doi: 10.1203/00006450-200001000-00017
173. Liu A, Tian L, Golob M, Eickhoff JC, Boston M, Chesler NC. 17β-estradiol attenuates conduit pulmonary artery mechanical property changes with pulmonary arterial hypertension. Hypertension. (2015) 66:1082–8. doi: 10.1161/HYPERTENSIONAHA.115.05843
174. Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, et al. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. (2003) 278:2118–23. doi: 10.1074/jbc.M210828200
175. Selemidis S, Dusting GJ, Peshavariya H, Kemp-Harper BK, Drummond GR. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc Res. (2007) 75:349–58. doi: 10.1016/j.cardiores.2007.03.030
176. Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, et al. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci. (2007) 120:492–501. doi: 10.1242/jcs.03361
177. Chakrabarti S, Lekontseva O, Peters A, Davidge ST. 17beta-Estradiol induces protein S-nitrosylation in the endothelium. Cardiovasc Res. (2010) 85:796–805. doi: 10.1093/cvr/cvp368
178. Nuedling S, Kahlert S, Loebbert K, Doevendans PA, Meyer R, Vetter H, et al. 17 Beta-estradiol stimulates expression of endothelial and inducible NO synthase in rat myocardium in-vitro and in-vivo. Cardiovasc Res. (1999) 43:666–74. doi: 10.1016/S0008-6363(99)00093-0
179. Tan E, Gurjar MV, Sharma RV, Bhalla RC. Estrogen receptor-alpha gene transfer into bovine aortic endothelial cells induces eNOS gene expression and inhibits cell migration. Cardiovasc Res. (1999) 43:788–97. doi: 10.1016/S0008-6363(99)00159-5
180. Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Euro Respir J. (2003) 22:358–63. doi: 10.1183/09031936.03.00038903
181. Nicolls MR, Voelkel NF. The roles of immunity in the prevention and evolution of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2017) 195:1292–9. doi: 10.1164/rccm.201608-1630PP
182. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circul Res. (2014) 115:165–75. doi: 10.1161/CIRCRESAHA.113.301141
183. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. (1995) 151:1628–31. doi: 10.1164/ajrccm.151.5.7735624
184. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:261–72. doi: 10.1164/rccm.201201-0164OC
185. Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. (2006) 168:659–69. doi: 10.2353/ajpath.2006.050599
186. Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, et al. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. (2011) 123:1986–95. doi: 10.1161/CIRCULATIONAHA.110.978627
187. Reed MJ, Coldham NG, Patel SR, Ghilchik MW, James VH. Interleukin-1 and interleukin-6 in breast cyst fluid: their role in regulating aromatase activity in breast cancer cells. J Endocrinol. (1992) 132:R5–8. doi: 10.1677/joe.0.132R005
188. Seriolo B, Accardo S, Garnero A, Fasciolo D, Cutolo M. Association between anticardiolipin antibody positivity and increased 17-beta-estradiol levels in premenopausal women with rheumatoid arthritis. Ann N Y Acad Sci. (1999) 876:159–63. doi: 10.1111/j.1749-6632.1999.tb07635.x
189. Speyer CL, Rancilio NJ, McClintock SD, Crawford JD, Gao H, Sarma JV, et al. Regulatory effects of estrogen on acute lung inflammation in mice. Am J Physiol Cell Physiol. (2005) 288:C881–90. doi: 10.1152/ajpcell.00467.2004
190. Leventhal L, Brandt MR, Cummons TA, Piesla MJ, Rogers KE, Harris HA. An estrogen receptor-beta agonist is active in models of inflammatory and chemical-induced pain. Euro J Pharmacol. (2006) 553:146–8. doi: 10.1016/j.ejphar.2006.09.033
191. Edvardsson K, Ström A, Jonsson P, Gustafsson J, Williams C. Estrogen receptor β induces antiinflammatory and antitumorigenic networks in colon cancer cells. Mol Endocrinol. (2011) 25:969–79. doi: 10.1210/me.2010-0452
192. Shand FH, Langenbach SY, Keenan CR, Ma SP, Wheaton BJ, Schuliga MJ, et al. In vitro and in vivo evidence for anti-inflammatory properties of 2-methoxyestradiol. J Pharmacol Exp Ther. (2011) 336:962–72. doi: 10.1124/jpet.110.174854
193. Tofovic SP, Dubey RK, Jackson EK. 2-Hydroxyestradiol attenuates the development of obesity, the metabolic syndrome, and vascular and renal dysfunction in obese ZSF1 rats. J Pharmacol Exp Ther. (2001) 299:973–7.
194. Purohit A, Singh A, Ghilchik MW, Reed MJ. Inhibition of tumor necrosis factor alpha-stimulated aromatase activity by microtubule-stabilizing agents, paclitaxel and 2-methoxyestradiol. Biochem Biophys Res Commun. (1999) 261:214–7. doi: 10.1006/bbrc.1999.1010
195. Vegeto E, Cuzzocrea S, Crisafulli C, Mazzon E, Sala A, Krust A, et al. Estrogen receptor-alpha as a drug target candidate for preventing lung inflammation. Endocrinology. (2010) 151:174–84. doi: 10.1210/en.2009-0876
196. Rowell KO, Hall J, Pugh PJ, Jones TH, Channer KS, Jones RD. Testosterone acts as an efficacious vasodilator in isolated human pulmonary arteries and veins: evidence for a biphasic effect at physiological and supra-physiological concentrations. J Endocrinol Investig. (2009) 32:718–23. doi: 10.1007/BF03346526
197. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Euro Heart J. (2016) 37:67–119. doi: 10.1183/13993003.01032-2015
198. Carranza-Lira S, Hernández F, Sánchez M, Murrieta S, Hernández A, Sandoval C. Prolactin secretion in molar and normal pregnancy. Int J Gynaecol Obstetr. (1998) 60:137–41. doi: 10.1016/S0020-7292(97)00251-8
199. O'Leary P, Boyne P, Flett P, Beilby J, James I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin Chem. (1991) 37:667–72. doi: 10.1093/clinchem/37.5.667
200. Larrea F, Méndez I, Parra A, Espinosa de los Monteros A. Serum pattern of different molecular forms of prolactin during normal human pregnancy. Human Reprod. (1993) 8:1617–22. doi: 10.1093/oxfordjournals.humrep.a137901
201. Montelongo A, Lasunción MA, Pallardo LF, Herrera E. Longitudinal study of plasma lipoproteins and hormones during pregnancy in normal and diabetic women. Diabetes. (1992) 41:1651–9. doi: 10.2337/diab.41.12.1651
202. English KM, Jones RD, Jones TH, Morice AH, Channer KS. Gender differences in the vasomotor effects of different steroid hormones in rat pulmonary and coronary arteries. Hormone Metabolic Res. (2001) 33:645–52. doi: 10.1055/s-2001-18689
203. Hatakeyama H, Nishizawa M, Nakagawa A, Nakano S, Kigoshi T, Uchida K. Testosterone inhibits tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in human aortic endothelial cells. FEBS Lett. (2002) 530:129–32. doi: 10.1016/S0014-5793(02)03440-3
204. Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. (2010) 56:696–708. doi: 10.1097/FJC.0b013e3181f9ea8d
205. Pritchard JA. Changes in the blood volume during pregnancy and delivery. Anesthesiology. (1965) 26:393–9.
206. Bernstein IM, Ziegler W, Badger GJ. Plasma volume expansion in early pregnancy. Obstetr Gynecol. (2001) 97:669–72. doi: 10.1016/s0029-7844(00)01222-9
207. Gardner JD, Brower GL, Voloshenyuk TG, Janicki JS. Cardioprotection in female rats subjected to chronic volume overload: synergistic interaction of estrogen and phytoestrogens. Am J Physiol Heart Circul Physiol. (2008) 294:H198–204. doi: 10.1152/ajpheart.00281.2007
208. Bédard E, Dimopoulos K, Gatzoulis MA. Has there been any progress made on pregnancy outcomes among women with pulmonary arterial hypertension? Euro Heart J. (2009) 30:256–65. doi: 10.1093/eurheartj/ehn597
209. Bonnin M, Mercier FJ, Sitbon O, Roger-Christoph S, Jaïs X, Humbert M, et al. Severe pulmonary hypertension during pregnancy: mode of delivery and anesthetic management of 15 consecutive cases. Anesthesiology. (2005) 102:1133–7; discussion: 5A−6A. doi: 10.1097/00000542-200506000-00012
210. Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemostasis. (2003) 29:125–30. doi: 10.1055/s-2003-38897
Keywords: pulmonary arterial hypertension, estrogen paradox, estrogen metabolism, estrogen receptors, pulmonary hypertension
Citation: Sun Y, Sangam S, Guo Q, Wang J, Tang H, Black SM and Desai AA (2021) Sex Differences, Estrogen Metabolism and Signaling in the Development of Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 8:719058. doi: 10.3389/fcvm.2021.719058
Received: 01 June 2021; Accepted: 11 August 2021;
Published: 10 September 2021.
Edited by:
Emma Louise Robinson, University of Colorado, United StatesReviewed by:
Shokoufeh Mahmoodzadeh, Max Delbrück Center for Molecular Medicine, GermanyCopyright © 2021 Sun, Sangam, Guo, Wang, Tang, Black and Desai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiyang Tang, dGFuZ2h5MjAwOEB5YWhvby5jb20=; Stephen M. Black, c3RibGFja0BmaXUuZWR1; Ankit A. Desai, YW5rZGVzYWlAaXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.