 Fahmida Jahan
Fahmida Jahan Rushita A. Bagchi
Rushita A. Bagchi- 1Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada
- 2Division of Cardiology, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
Introduction
Nicotinamide adenine dinucleotide (NAD) is not only the master regulator of energy metabolism pathways but also an essential substrate for several key enzymes of health and disease states (1, 2). NAD+/NADH redox coupling is vital for fueling glycolysis, tricarboxylic acid (TCA) cycle and electron transport chain (ETC) for producing ATP. In addition to that, NAD+ is a substrate for NAD-dependent deacetylases called sirtuins (SIRTs), poly-ADP-ribose polymerases (PARPs), monoADP-ribosyltransferases (ARTDs), ADP ribose synthetases (CD38/CD157), and sterile alpha and toll/interleukin-1 receptor motif-containing protein 1 (SARM1), etc. (1). NAD kinase also utilizes NAD+ to generate nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) which is consumed by reactive oxygen species producing enzymes, NADPH oxidases. For maintaining cellular homeostasis, NAD+ levels are tightly regulated through its salvage pathway where the by-product of enzymatic reactions, nicotinamide adenine dinucleotide (NAM), is converted back to NAD+ by the rate limiting enzyme NAM phosphoribosyl transferase (NAMPT) (1, 2). Other biosynthesis pathways include de novo synthesis from tryptophan (Trp), and from the dietary uptake of its precursors- nicotinic acid (NA) or nicotinamide riboside (NR) (1, 2).
NAD+ Depletion in Aging and Disease States
Maintaining the levels of NAD+ is highly critical for normal cellular function and this is indicated by the embryonic lethality of homozygous knockout of the NAD+ salvage enzyme Nampt in mice (3). Various tissue-specific conditional knockout Nampt mice also demonstrated the importance of maintaining NAD+ levels within an organ (Table 1). During metabolic stress, chronic inflammatory conditions, and aging NAD+ content has been shown to decline (1, 10, 11). In healthy humans, plasma NAD+ levels and the levels of its related metabolites such as NADP+ and nicotinic acid adenine dinucleotide (NAAD) have been reported to decline with age (12). Several studies have shown that with aging, the major NAD+ consuming enzymes such as PARP1 and CD38 are hyperactivated due to age-related increase in DNA damage, inflammation and oxidative stress leading to impaired NAD+ signaling (1, 11, 13, 14). In mouse models of muscular dystrophy, increasing levels of NAD+ was shown to improve muscle function, decrease muscle stem cell senescence, and increase lifespan (15, 16). In aging- related neurodegenerative diseases replenishment of NAD+ improved health span by reducing neuroinflammation and DNA damage in mouse brain (17–19). In an animal model of high-fat high-sucrose (HFHS) diet, NAD+ supplementation prevented hepatic steatosis by activating sirtuin mediated mitochondrial unfolded protein response (UPRmt) (20). NAD+ boosting was also shown to be beneficial for type 2 diabetes and diabetic neuropathy in mice (21). A clinical trial showed that NR can improve physical activity acutely (22) and chronic NR supplementation has been shown to decrease levels of inflammatory cytokines in sera of aged humans. NR also enhanced the levels of NAD+ and its related metabolites in muscle (10). In contrast, some clinical trials did not observe beneficial effects of NAD+ boosting in certain disease conditions. For instance, in obese men, either non-diabetic or insulin resistant, NR supplementation for a period of 12 weeks did not improve insulin sensitivity or glucose tolerance (23, 24). Moreover, the research group did not find any changes in skeletal muscle mitochondrial function or content in these subjects (25). This suggests that the beneficial effect of NAD+ supplementation may be tissue-specific or may require a long-term and dosage-optimized treatment regime, or may be in certain conditions it could be only effective as a preventative measure rather than a therapeutic (26).

Table 1. Organ specific knockout models of NAD+ salvage enzyme Nampt and their phenotypes.
NAD+ Boosting in Cardioprotection
Ischemia, ischemia reperfusion (IR) injury, or myocardial infarction has been shown to result in reduced NAD+ levels in the heart (27). Friedreich's ataxia cardiomyopathy mouse model has been shown to be associated with increased mitochondrial protein acetylation along with a decrease in mitochondrial Sirt3 deacetylase transcript levels as well as altered expression of NAD+ biosynthesis enzymes. The authors demonstrated that NAD+ boosting by NR normalized cardiac efficiency by activating mitochondrial Sirt3 function and thereby reducing mitochondrial protein acetylation (28). Moreover, a mouse cardiac hypertrophy model showed that NAD+ treatment can inhibit pro-hypertrophic pathway and activate anti-hypertrophic LKB1-AMPK pathway by Sirt3-dependent deacetylation of LKB1 (29). Mouse models of dilated cardiomyopathy and cardiac hypertrophy showed 30% decline in NAD+ levels in the heart. Expression of NAD+ salvage enzyme NAMPT was decreased and, an increase in NMRK2 enzyme that converts nicotinamide riboside (NR) to nicotinamide mononucleotide (NMN) was noted. This was consistent with results obtained from failing human hearts. NR administration prevented heart failure in mice with dilated cardiomyopathy and partially improved cardiac function in transverse aortic constriction (TAC) mouse model by enhancing NAD+ metabolome and cardiac citrate metabolism (30). A genetic model of mitochondrial complex I dysfunction (Ndufs4; cKO) was shown to be more susceptible to heart failure which was accompanied with increased NADH/NAD+ ratio and protein hyperacetylation (31). Administration of NMN or overexpressing NAMPT normalized NADH/NAD+ ratio and reduced protein acetylation; and thus prevented heart failure in the Ndufs4 cKO and chronic pressure overload models (31).
Clinical trials with various NAD+ precursors such as Trp, NA, or NR showed that increasing NAD+ levels is associated with reduced risk of developing CVD, lowers blood pressure and improves hypocholesteremia, enhances cardiac mitochondrial function and decreases aortic stiffness (32) (Figure 1). For example, in a long term safety and efficacy study, niacin treatment showed lower cholesterol levels and decreased occurrence of non-fatal myocardial re-infarction which led to 11% decrease in mortality compared to placebo (33). Another study showed that elevated plasma tryptophan level is associated with decreased CVD incidence (34). A systematic review reported that niacin treatment increased levels of serum high-density lipoprotein (HDL) levels (35). Moreover, a randomized controlled trial showed that NR administration for 6 weeks led to reduction in blood pressure and aortic stiffness in middle-aged and older adults (36). A recent study reported that diastolic heart failure in humans is associated with depleted NAD+ content (37). Using three murine models of heart failure, (i) aging, (ii) hypertension, and (iii) ZSF1 obese rats, the study showed that NAM treatment prevented diastolic dysfunction by improving cardiometabolic function and bioenergetics (Figure 1). Moreover, they performed a long-term population-based analysis which suggested that supplementation of NA or its equivalent is capable of reducing blood pressure and, thereby it was associated with lower risks of cardiac mortality (37).
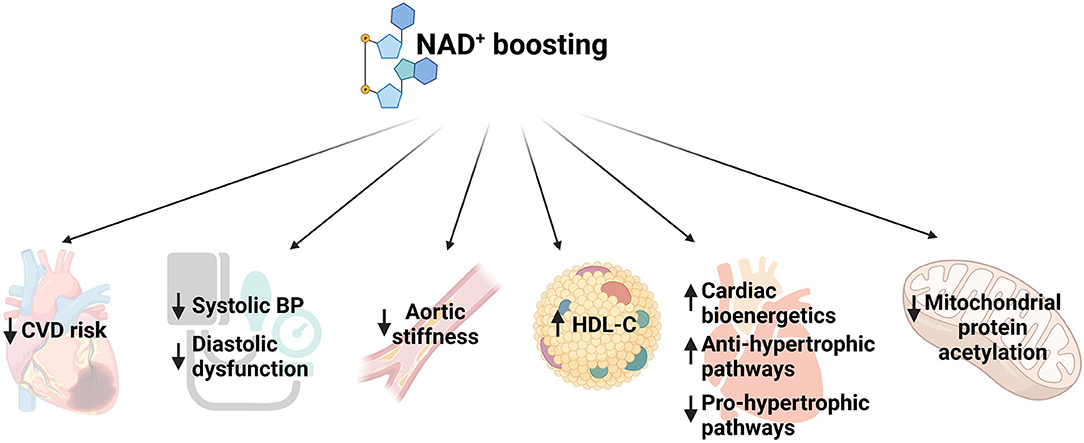
Figure 1. Schematic representing function of NAD+ boosting in multiple aspects of cardiovascular diseases. CVD, cardiovascular disease; BP, blood pressure; HDL, high density lipoprotein.
Since PARP1 hyperactivation has been implicated as a cause of NAD+ depletion in various pathologies, pharmacological inhibitors of PARP1 have been tested in pre-clinical models. An in vitro study showed that PARP1 inhibition protects tachypacing-induced and gamma-irradiation-induced cardiomyocyte dysfunction in HL1 and rat atrial cardiomyocytes (38). In a mouse TAC model of heart failure, PARP1 inhibition improved cardiac contractile function (39). Moreover, in murine models of chronic heart failure and ischemia/reperfusion (I/R) injury, PARP inhibition has been found to be protective (40, 41). Using a genetic model of mouse preeclampsia, a study reported that deletion of NAD+ consuming enzyme CD38 gene leads to reduced blood pressure in this model. CD38 inhibitors are also available which could be tested in CVD models (42). However, such inhibitors may not be highly selective; and as these enzymes are involved in various pathways and tissue functions, their global inhibition may pose unwanted side effects for human application.
NAD+ Precursors and Concerns Associated With Their High Dose Administration
Although several studies have successfully demonstrated the beneficial effects of NAD+ boosting, the dosage and precursors used need to be carefully evaluated for human application. Studies conducted have tested Trp, NA, NAM, NAM-mono nucleotide (NMN) and NR as NAD+ precursors. As Trp is also involved in other biosynthetic pathways, it seems to be less efficient in elevating NAD+ levels in disease conditions (1). NA has lipid lowering effects in blood but at doses higher than 50 mg/day it can cause flushing (43, 44). On the other hand, high doses of NAM have been reported to inhibit SIRT1 (45). NA and NAM have also been shown to affect the cellular methyl pool which is critical for maintaining DNA and protein methylation states. NAM accepts methyl groups from S-adenosylmethionine (SAMe) by the action of nicotinamide N-methyltransferase (NNMT) and generates methylated NAM (MeNAM) and homocysteine. Under physiologic conditions, methylation of NAM is regulated as the Km value of NNMT for NAM is much higher compared to plasma levels of NAM. Thus, high doses of NAM treatment will lead to NNMT activation which may result in depletion of the methyl pool. Consistent with this, NAM has been reported to cause liver toxicity at high doses (46, 47). For example, one study reported that male rats treated with 1 or 4 grams of NAM for 8 weeks had altered hepatic DNA methylation and homocysteine metabolism; along with increased oxidative damage and insulin resistance compared to untreated rats (48). Another research study showed that healthy rats treated with nicotinamide (500 mg/kg) had an increase in the parameters of hepatic and renal toxicity (46). Moreover, NAM treatment in pregnant rats led to decreased placental and fetal DNA methylation which suggests that high doses of NAM treatment may modify epigenetic profile in offspring (49).
Sterile alpha and TIR motif-containing protein 1 (SARM1), an NADase enzyme, when activated causes axonal degeneration. Accumulation of NMN due to loss of NMNAT2, an enzyme that converts NMN to NAD+, or an increase in NMN/NAD+ ratio, has been shown to activate SARM1 which leads to axonal death. This raises safety concerns for pharmacological use of NMN in humans, although pharmacokinetics study of NMN administration at 100, 250, and 500 mg in humans did not show any deleterious effects after 5 h of intervention (50–52). NMN treatment in mice at a high dose (2,860 mg/kg, twice daily) for 7 days led to elevated alanine aminotransferase indicating liver toxicity. Moreover, NMN treatment at 1,340 mg/day twice daily in beagle dogs showed an increase in body weight; and elevated serum creatinine and uric acid levels indicating compromised kidney function compared to controls (53).
Pharmacokinetics studies and several clinical trials have reported that NR is effective in enhancing NAD+ metabolome and that it is orally available and well-tolerated in humans (10, 36, 54, 55). In a randomized double-blinded placebo-controlled trial, unlike NAM treatment, long term NR administration (56 days) at 10, 300 and 1,000 mg/day doses did not affect levels of plasma homocysteine (55). A rodent study showed that rats treated with 300 mg/kg NR for 21 days displayed a non-significant decrease in incremental swimming performance test compared to non-treated rats (56). However, in aged humans, a double-blinded crossover study showed that NR treatment can acutely enhance physical performance (22). Another study showed that NR supplementation at 1 g/day dosage for 21 days seemed to maintain mitochondrial bioenergetics and elevate NAD+ metabolome in muscle despite downregulation of mitochondrial pathways. NR supplementation also led to a decrease in age-associated inflammation in these subjects (10). Here, exercise performance or physical activity was not evaluated which would be essential information to determine the outcome of the study accurately.
Conclusion
Overall, the findings discussed in this article suggest that NAD+ boosting is a promising therapeutic strategy for cardiovascular diseases. However, the choice of NAD+ precursor, their appropriate dosing and long-term effects in humans need to be critically evaluated and investigated. Long-term effect of these precursors could be tested in animal models at various doses to determine impact on molecular parameters such as DNA methylation, homocysteine metabolism and SARM1 activation. Exercise performance or physical activity should also be evaluated. In non-medicated, metabolically healthy obese individuals, NAD+ boosting for 12 weeks did not improve glucose tolerance even though in pre-clinical models of Type 2 diabetes, NAD+ precursor supplementation has been shown to be effective. This may suggest that a therapeutic benefit may only be achieved in conditions with metabolic dysfunction and a preventative benefit may require even longer or repeated treatment periods. Thus, such aspects need to be carefully considered for designing clinical trials.
Author Contributions
FJ and RB designed the concept. FJ drafted and RB edited and finalized the manuscript.
Funding
FJ holds a Frederick Banting and Charles Best Canada Graduate Scholarship from Canadian Institutes of Health Research (FRN-167027). RB was a recipient of a CIHR Postdoctoral Fellowship (FRN-216927).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Mr. Mohammed Ariff Iqbal, University of Ottawa, for critical input on the manuscript. Figure 1 was created with Biorender.com.
References
1. Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. (2015) 22:31–53. doi: 10.1016/j.cmet.2015.05.023
2. Xu W, Li L, Zhang L. NAD(+) metabolism as an emerging therapeutic target for cardiovascular diseases associated with sudden cardiac death. Front Physiol. (2020) 11:901. doi: 10.3389/fphys.2020.00901
3. Zhang LQ, Van Haandel L, Xiong M, Huang P, Heruth DP, Bi C, et al. Metabolic and molecular insights into an essential role of nicotinamide phosphoribosyltransferase. Cell Death Dis. (2017) 8:e2705. doi: 10.1038/cddis.2017.132
4. Nielsen KN, Peics J, Ma T, Karavaeva I, Dall M, Chubanava S, et al. NAMPT-mediated NAD(+) biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol Metab. (2018) 11:178–88. doi: 10.1016/j.molmet.2018.02.014
5. Dall M, Trammell SAJ, Asping M, Hassing AS, Agerholm M, Vienberg SG, et al. Mitochondrial function in liver cells is resistant to perturbations in NAD(+) salvage capacity. J Biol Chem. (2019) 294:13304–26. doi: 10.1074/jbc.RA118.006756
6. Muraoka H, Hasegawa K, Sakamaki Y, Minakuchi H, Kawaguchi T, Yasuda I, et al. Role of nampt-sirt6 axis in renal proximal tubules in extracellular matrix deposition in diabetic nephropathy. Cell Rep. (2019) 27:199–212 e5. doi: 10.1016/j.celrep.2019.03.024
7. Wang X, Zhang Q, Bao R, Zhang N, Wang Y, Polo-Parada L, et al. Deletion of nampt in projection neurons of adult mice leads to motor dysfunction, neurodegeneration, and death. Cell Rep. (2017) 20:2184–200. doi: 10.1016/j.celrep.2017.08.022
8. de Guia RM, Hassing AS, Ma T, Plucinska K, Holst B, Gerhart-Hines Z, et al. Ablation of Nampt in AgRP neurons leads to neurodegeneration and impairs fasting- and ghrelin-mediated food intake. FASEB J. (2021) 35:e21450. doi: 10.1096/fj.202002740R
9. Basse AL, Agerholm M, Farup J, Dalbram E, Nielsen J, Ortenblad N, et al. Nampt controls skeletal muscle development by maintaining Ca(2+) homeostasis and mitochondrial integrity. Mol Metab. (2021) 53:101271. doi: 10.1016/j.molmet.2021.101271
10. Elhassan YS, Kluckova K, Fletcher RS, Schmidt MS, Garten A, Doig CL, et al. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. (2019) 28:1717–28.e6. doi: 10.1016/j.celrep.2019.07.043
11. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 dictates age-related NAD Decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
12. Clement J, Wong M, Poljak A, Sachdev P, Braidy N. The plasma NAD(+) metabolome is dysregulated in “normal” aging. Rejuvenation Res. (2019) 22:121–30. doi: 10.1089/rej.2018.2077
13. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE. (2011) 6:e19194. doi: 10.1371/journal.pone.0019194
14. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. (2013) 154:430–41. doi: 10.1016/j.cell.2013.06.016
15. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. (2016) 352:1436–43. doi: 10.1126/science.aaf2693
16. Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L, et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med. (2016) 8:361ra139. doi: 10.1126/scitranslmed.aaf5504
17. Hou Y, Lautrup S, Cordonnier S, Wang Y, Croteau DL, Zavala E, et al. NAD(+) supplementation normalizes key alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci USA. (2018) 115:E1876–E85. doi: 10.1073/pnas.1718819115
18. Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. (2016) 24:566–81. doi: 10.1016/j.cmet.2016.09.004
19. Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. (2014) 157:882–96. doi: 10.1016/j.cell.2014.03.026
20. Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. (2016) 63:1190–204. doi: 10.1002/hep.28245
21. Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. (2016) 6:26933. doi: 10.1038/srep26933
22. Dolopikou CF Kourtzidis IA Margaritelis NV Vrabas IS Koidou I Kyparos A. Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: a double-blind cross-over study. Eur J Nutr. (2020) 59:505–15. doi: 10.1007/s00394-019-01919-4
23. Dollerup OL, Christensen B, Svart M, Schmidt MS, Sulek K, Ringgaard S, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. (2018) 108:343–53. doi: 10.1093/ajcn/nqy132
24. Dollerup OL, Trammell SAJ, Hartmann B, Holst JJ, Christensen B, Moller N, et al. Effects of nicotinamide riboside on endocrine pancreatic function and incretin hormones in nondiabetic men with obesity. J Clin Endocrinol Metab. (2019) 104:5703–14. doi: 10.1210/jc.2019-01081
25. Dollerup OL, Chubanava S, Agerholm M, Sondergard SD, Altintas A, Moller AB, et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J Physiol. (2020) 598:731–54. doi: 10.1113/JP278752
26. Dall M, Hassing AS, Treebak JT. NAD(+) and NAFLD - caution, causality and careful optimism. J Physiol. (2021) 1–20. doi: 10.1113/JP280908
27. Mericskay M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis. (2016) 109:207–15. doi: 10.1016/j.acvd.2015.10.004
28. Martin AS, Abraham DM, Hershberger KA, Bhatt DP, Mao L, Cui H, et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a friedreich's ataxia cardiomyopathy model. JCI Insight. (2017) 2:e93885. doi: 10.1172/jci.insight.93885
29. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. (2010) 285:3133–44. doi: 10.1074/jbc.M109.077271
30. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. (2018) 137:2256–73. doi: 10.1161/CIRCULATIONAHA.116.026099
31. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. (2016) 134:883–94. doi: 10.1161/CIRCULATIONAHA.116.022495
32. Lin Q, Zuo W, Liu Y, Wu K, Liu Q. NAD(+) and cardiovascular diseases. Clin Chim Acta. (2021) 515:104–10. doi: 10.1016/j.cca.2021.01.012
33. Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, et al. Fifteen year mortality in coronary drug project patients: long-term benefit with niacin. J Am Coll Cardiol. (1986) 8:1245–55. doi: 10.1016/S0735-1097(86)80293-5
34. Yu E, Ruiz-Canela M, Guasch-Ferre M, Zheng Y, Toledo E, Clish CB, et al. Increases in plasma tryptophan are inversely associated with incident cardiovascular disease in the prevencion con dieta mediterranea (PREDIMED) study. J Nutr. (2017) 147:314–22. doi: 10.3945/jn.117.251801
35. Garg A, Sharma A, Krishnamoorthy P, Garg J, Virmani D, Sharma T, et al. Role of niacin in current clinical practice: a systematic review. Am J Med. (2017) 130:173–87. doi: 10.1016/j.amjmed.2016.07.038
36. Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun. (2018) 9:1286. doi: 10.1038/s41467-018-03421-7
37. Abdellatif M, Trummer-Herbst V, Koser F, Durand S, Adao R, Vasques-Novoa F, et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med. (2021) 13:eabd7064. doi: 10.1126/scitranslmed.abd7064
38. Zhang D, Hu X, Li J, Liu J, Baks-Te Bulte L, Wiersma M, et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD(+) depletion in experimental atrial fibrillation. Nat Commun. (2019) 10:1307. doi: 10.1038/s41467-019-09014-2
39. Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, et al. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther. (2005) 312:891–8. doi: 10.1124/jpet.104.077164
40. Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. (2002) 40:1006–16. doi: 10.1016/S0735-1097(02)02062-4
41. Song ZF, Chen DY, Du B, Ji XP. Poly (ADP-ribose) polymerase inhibitor reduces heart ischaemia/reperfusion injury via inflammation and Akt signalling in rats. Chin Med J. (2013) 126:1913–7. doi: 10.3760/cma.j.issn.0366-6999.20121234
42. Li F, Fushima T, Oyanagi G, Townley-Tilson HW, Sato E, Nakada H, et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc Natl Acad Sci USA. (2016) 113:13450–5. doi: 10.1073/pnas.1614947113
43. Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate Other B Vitamins and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Collection: Reports funded by National Institutes of Health (1998).
44. Guyton JR, Bays HE. Safety considerations with niacin therapy. Am J Cardiol. (2007) 99:22C−31C. doi: 10.1016/j.amjcard.2006.11.018
45. Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. (2002) 277:45099–107. doi: 10.1074/jbc.M205670200
46. Mahmoud YI, Mahmoud AA. Role of nicotinamide (vitamin B3) in acetaminophen-induced changes in rat liver: nicotinamide effect in acetaminophen-damged liver. Exp Toxicol Pathol. (2016) 68:345–54. doi: 10.1016/j.etp.2016.05.003
47. Winter SL, Boyer JL. Hepatic toxicity from large doses of vitamin B3 (nicotinamide). N Engl J Med. (1973) 289:1180–2. doi: 10.1056/NEJM197311292892208
48. Li D, Tian YJ, Guo J, Sun WP, Lun YZ, Guo M, et al. Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br J Nutr. (2013) 110:2156–64. doi: 10.1017/S0007114513001815
49. Tian YJ, Luo N, Chen NN, Lun YZ, Gu XY, Li Z, et al. Maternal nicotinamide supplementation causes global DNA hypomethylation, uracil hypo-incorporation and gene expression changes in fetal rats. Br J Nutr. (2014) 111:1594–601. doi: 10.1017/S0007114513004054
50. Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron. (2021) 109:1118–36.e11. doi: 10.1016/j.neuron.2021.02.009
51. Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, et al. A cell-permeant mimetic of NMN activates SARM1 to produce cyclic ADP-ribose and induce non-apoptotic cell death. iScience. (2019) 15:452–66. doi: 10.1016/j.isci.2019.05.001
52. Irie J, Inagaki E, Fujita M, Nakaya H, Mitsuishi M, Yamaguchi S, et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr J. (2020) 67:153–60. doi: 10.1507/endocrj.EJ19-0313
53. You Y, Gao Y, Wang H, Li J, Zhang X, Zhu Z, et al. Subacute toxicity study of nicotinamide mononucleotide via oral administration. Front Pharmacol. (2020) 11:604404. doi: 10.3389/fphar.2020.604404
54. Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. (2016) 7:12948. doi: 10.1038/ncomms12948
55. Conze D, Brenner C, Kruger CL. Safety and metabolism of long-term administration of NIAGEN (nicotinamide riboside chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci Rep. (2019) 9:9772. doi: 10.1038/s41598-019-46120-z
Keywords: nicotinamide adenine dinucleotide, metabolome, cardiovascular disease, aging, therapeutic
Citation: Jahan F and Bagchi RA (2021) Enhancing NAD+ Metabolome in Cardiovascular Diseases: Promises and Considerations. Front. Cardiovasc. Med. 8:716989. doi: 10.3389/fcvm.2021.716989
Received: 29 May 2021; Accepted: 09 August 2021;
Published: 27 August 2021.
Edited by:
Kristine DeLeon-Pennell, Medical University of South Carolina, United StatesReviewed by:
Dipsikha Biswas, University of Copenhagen, DenmarkGanesh V. Halade, University of South Florida, United States
Copyright © 2021 Jahan and Bagchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rushita A. Bagchi, cnVzaGl0YS5iYWdjaGlAY3VhbnNjaHV0ei5lZHU=