 Weiming Wang
Weiming Wang Tingting Zhao
Tingting Zhao Kang Geng1
Kang Geng1 Gang Yuan
Gang Yuan Yue Chen
Yue Chen Youhua Xu
Youhua Xu- 1The State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Macau, China
- 2Department of General Surgery (Vascular Surgery), The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 3Key Laboratory of Medical Electrophysiology, Ministry of Education and Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
- 4Department of Nuclear Medicine, The Affiliated Hospital of Southwest Medical University, Luzhou, China
Smoking is one of the most important preventable factors causing peripheral artery disease (PAD). The purpose of this review is to comprehensively analyze and summarize the pathogenesis and clinical characteristics of smoking in PAD based on existing clinical, in vivo, and in vitro studies. Extensive searches and literature reviews have shown that a large amount of data exists on the pathological process underlying the effects of cigarette smoke and its components on PAD through various mechanisms. Cigarette smoke extracts (CSE) induce endothelial cell dysfunction, smooth muscle cell remodeling and macrophage phenotypic transformation through multiple molecular mechanisms. These pathological changes are the molecular basis for the occurrence and development of peripheral vascular diseases. With few discussions on the topic, we will summarize recent insights into the effect of smoking on regulating PAD through multiple pathways and its possible pathogenic mechanism.
Introduction
With changes in global population epidemiology, the incidence of PAD may increase in the future (1). In a broad sense, PAD includes arterial stenosis and occlusion caused by atherosclerotic plaques and thrombosis, arterial inflammation, arterial dilatation, peripheral artery dysfunction, external pressure lesions, etc., and its most strict definition refers to atherosclerotic occlusion disease (2, 3). The occurrence of peripheral vascular disease is closely related to traditional cardiovascular factors (smoking, hypertension, diabetes, and dyslipidemia) and the aging of the population (4). However, for some low- and middle-income countries, environmental factors such as poverty, industrialization, and infection may also affect the occurrence and development of peripheral vascular diseases. Smoking is a particularly strong risk factor for peripheral vascular disease, and with further research, existing studies have indicated that smoking independently correlates with peripheral vascular disease (1, 3).
Clinical Evidence of Smoking-Associated PAD
Numerous studies have confirmed the link between active smoking and PAD. A systematic review showed that half of PAD cases are due to smoking (5). Compared with non-smokers, people who have ever smoked are still at higher risk of developing PAD (6, 7). Meanwhile, passive smoking also increases the risk of vascular damage or the diagnosis of PAD (8).
The harm caused by smoking cannot be underestimated, but researchers have not clearly determined whether quitting smoking effectively reduces the incidence of cardiovascular disease. Although quitting smoking reduces the risk of cardiovascular morbidity and mortality in patients with a history of smoking, the continuing effects of smoking are still relatively high. Therefore, smoking cessation is never too late to reduce the cardiovascular risk (9). At the same time, David Conen also confirmed that quitting smoking significantly reduces the risk of PAD, but the incidence of PAD is still increasing, even for those who quit smoking (10).
In terms of the effect of smoking on PAD after surgery, studies have shown that smoking intensity exerts an unusually large effect on the prognosis of patients with symptomatic lower extremity revascularization. Compared with patients who continue to smoke, the mortality rate of patients who quit smoking is lower and these patients have a higher survival rate without amputation (11, 12).
Smoking and Atherosclerosis
Clinical Evidence
Smoking has been widely regarded as the leading risk factor for clinical cardiovascular disease that directly affects atherosclerosis. Active smoking and passive smoking directly correlate with the occurrence of atherosclerosis (13, 14). Strict smoking bans have reduced the number of active smokers worldwide, but non-smokers may still be exposed to environmental smoke in their homes or workplaces through passive smoke inhalation. Therefore, passive smoking must be prevented and controlled (8). At the same time, in terms of passive smoking, children's long-term cigarette exposure is significantly correlated with the risk of developing severe carotid atherosclerotic plaques in early adulthood, which is ~2 times higher than those without exposure (15). A dose-response relationship has been observed between the time and amount of smoking and the occurrence of atherosclerosis. The only effective measure to reduce the risk of smoking-related atherosclerosis is to quit smoking. However, after quitting smoking, the continuous effects caused by smoking are not completely eliminated (16, 17).
With the transformation of tobacco marketing methods, in addition to traditional smoking and its methods, some new types of tobacco substitutes (such as electronic cigarettes, hookahs, etc.) have gradually become new consumption methods and are more commonly used by young people (18, 19). Hundreds of different chemical components are present in various tobacco substitutes (20–22). Although qualitative and quantitative comparisons of the compositions of e-cigarettes, hookah aerosols and cigarette smoke have been performed, the risks of e-cigarettes and hookah aerosols are considered lower than those of cigarette use (22–24). Therefore, the current misconceptions about this new form of tobacco use and its potentially harmful health effects are gradually contributing to the risk of cardiovascular disease in this group of people. In summary, e-cigarettes, hookahs or cigarettes, as well as active or passive smoking, exert serious effects on cardiovascular disease in the population (Figure 1).
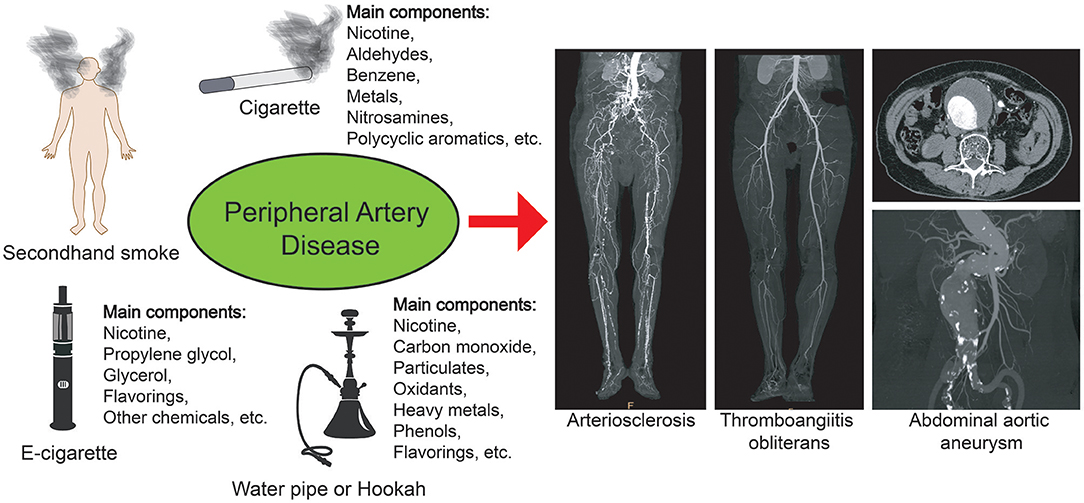
Figure 1. Smoking and peripheral artery disease. Different types of smoking may cause peripheral vascular diseases, such as arteriosclerosis, thromboangiitis obliterans, and abdominal aortic aneurysm. The ingredients in different cigarettes and substitutes are different, but they may all pose a risk of cardiovascular disease.
Smoking-Related Molecular Mechanisms
Atherosclerosis is the primary pathological basis of PAD. Its main feature is the formation of atherosclerotic plaques, which are characterized by the accumulation of lipids and immune cells in the blood vessel wall covered by a collagen fiber cap. The abnormal metabolism and distribution of lipids play an important role in the occurrence and development of atherosclerosis (25, 26). Studies have shown that smoking affect the composition and distribution of blood lipids in ApoE-/- and LDLr-/- mice, accelerates the formation of aortic plaques and increases the total cholesterol content, thereby inducing the occurrence of atherosclerosis. Smoking cessation slows plaque progression and leads to decreased levels of many lipid types in the plasma and aorta (27–29). However, Kunimoto et al. reported that cigarette smoke had no effect on serum cholesterol and triglyceride levels in ApoE-/- mice (30). The author proposes that this difference may be due to a certain difference in the technology and composition of smoke exposure, which ultimately led to the ambiguity of the results of the study. Therefore, some researchers adopted the model of atherosclerosis induced by nicotine, which is the major addictive component of all components of cigarette smoke, to avoid the confounding effects of other components (31). A large body of evidence also supports the role of nicotine in promoting atherosclerosis (23, 32). However, due to the particular characteristics of nicotine, its acquisition requires strict approval procedures, and thus it is not widely used in basic research.
In addition to abnormal lipid metabolism, various cells are also involved in the occurrence of atherosclerosis (33). At present, various studies have shown that for long-term smokers, whether they are active smokers or passive smokers, harmful substances may induce various phenotypic changes and dysfunction of macrophages, endothelial cells and smooth muscle cells through various mechanisms, thus promoting the occurrence and development of vascular diseases (34, 35) (Figure 2).
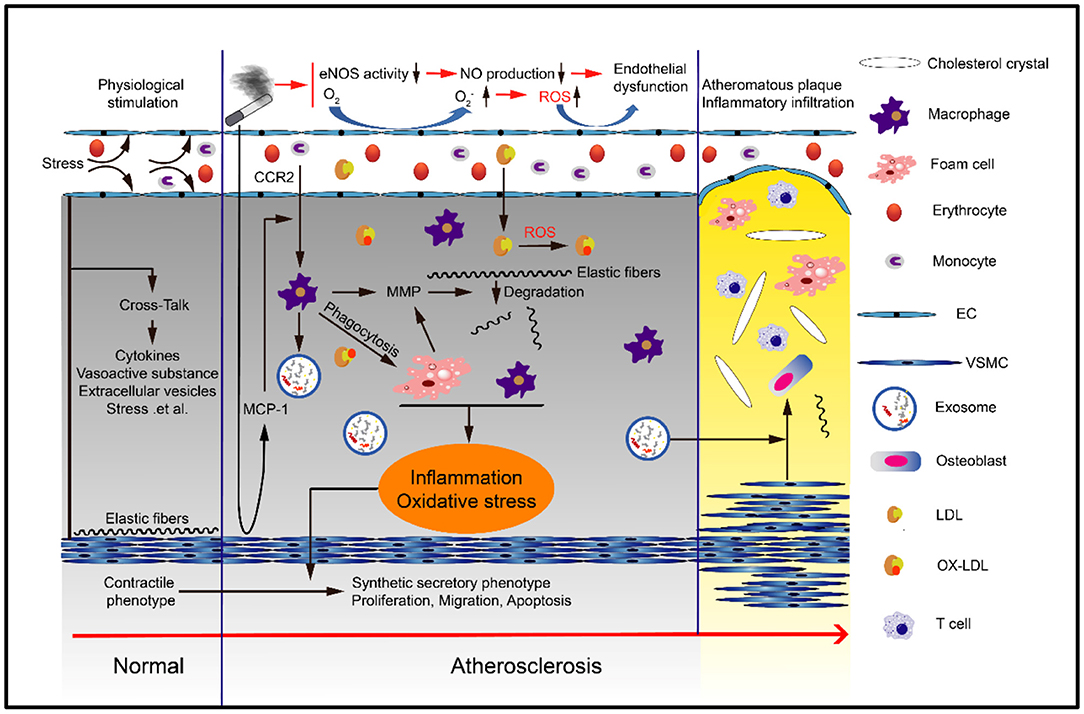
Figure 2. Smoking can cause atherosclerosis. Smoking can cause endothelial cell dysfunction, monocyte differentiation into macrophages and eventually foam cells, smooth muscle cell proliferation, migration and phenotypic transformation, and extracellular matrix degradation and remodeling. These processes are typically mediated by oxidative stress and inflammation.
Smoking and Endothelial Cells
Endothelial dysfunction is one of the earliest abnormalities that is observed during the development of atherosclerosis, and endothelial nitric oxide synthase (eNOS) plays a crucial role in regulating endothelial dysfunction (36, 37) (Figure 3). The monomer eNOS has no activity. However, when monomeric eNOS forms a stable dimer through heme and BH4, it has biological activity. GTP cyclohydrolase (GTPCH) is an important rate-limiting enzyme in the synthesis of BH4. CSE reduces the synthesis of GTPCH by inhibiting the transport of HuR from the nucleus to the cytoplasm, thereby affecting the synthesis of BH4. Finally, it affects the activity of eNOS and reduces the production of nitric oxide (NO) (38–40). At the same time, CSE can also inhibit the synthesis of BH4 by reducing the expression of dihydrofolate reductase (DHFR), and thus affect the activity of eNOS (41). CSE increases the expression of thiocyanate (SCN-), which is used by myeloperoxidase (MPO) to form hypothiocyanic acid (HOSCN). HOSCN reduces the level of active dimerized eNOS and increases the release of inactive monomers and Zn2+ (42, 43). HOSCN-modified low-density lipoprotein also impairs eNOS activity, thereby reducing NO production (44). CSE-derived reactive oxygen/nitrogen species interact with VEGFR2, resulting in post-translational modifications of VEGFR2 that decrease the activity of the downstream Akt/eNOS/NO signaling pathway, leading to endothelial dysfunction (45).
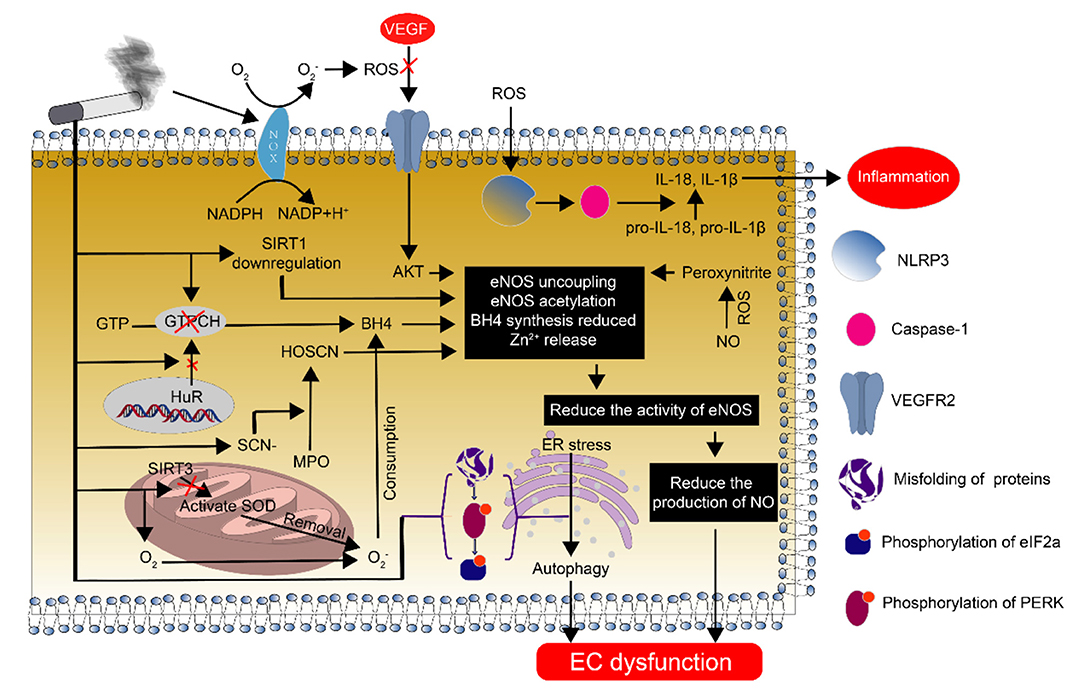
Figure 3. Endothelial cell dysfunction caused by smoking and its mechanism. Smoking causes endothelial cell dysfunction through various pathways. The most important pathway is the decrease in NO synthesis due to eNOS deficiency and the reduction in eNOS function, which plays an important regulatory role in endothelial dysfunction.
Oxidative stress is the interaction between the body's response to oxidation caused by various pathological factors and antioxidants. Normally, it is in a balanced state. However, when the effect of oxidation is greater than the antioxidant effect, the infiltration of inflammatory cells occurs, the secretion of various proteases and cytokines increases, and a large amount of oxidative intermediate products are generated, which in turn cause a series of physiological and pathological changes (46, 47). Studies have confirmed that oxidative stress is also involved in CSE-induced endothelial cell dysfunction (48–50). Mitochondria are an important source of O free radicals, and excessive O accumulation induces endothelial cell dysfunction through multiple pathways. Under normal physiological conditions, the removal of O mainly occurs through the antioxidant SOD, and SIRT3 is indispensable for the regulation of SOD activation and mitochondrial metabolism in mitochondria (51). CSE reduces the expression of the mitochondrial deacetylase SIRT3, thereby increasing SOD hyperacetylation, subsequently hindering O clearance and resulting in its excessive accumulation in the body (52). Meanwhile, thiol-stabilized compounds in CSE activate NADPH oxidase and increase the production of endothelial O (53). Increased O production consumes the cofactor tetrahydrobiopterin necessary for eNOS activity (39). CSE-mediated oxidative stress downregulates SIRT1 (it activates eNOS by reducing the acetylation of eNOS), causing increased eNOS acetylation and thereby reducing NO-mediated signaling and endothelial dysfunction (54).
The production of a large amount of ROS leads to the rapid inactivation of NO and the formation of peroxynitrite, thereby reducing the biological activity of NO (23). Peroxynitrite also destabilizes the eNOS dimer by detaching BH4 from the active site of the zinc finger complex (55–57). At the same time, peroxynitrite is also a strong biological oxidant that promotes post-translational modification of proteins (including the eNOS protein), alters metabolic pathways, and ultimately leads to increased superoxide production and reduced NO production (58, 59). CSE results in reduced eNOS protein quality, decreased mRNA expression, and inhibited calpain activity, resulting in impaired irreversible inhibition of the activity of eNOS and a reduced content in endothelial cells, which ultimately impairs endothelium-dependent vasodilation and decreases NO synthesis (60, 61). In addition, CSE activates the NLRP3 inflammasome through ROS, causing the release of downstream IL-1β and IL-18 and other inflammatory factors, leading to functional changes such as autophagy, pyrolysis, and apoptosis of endothelial cells (56, 62, 63).
Studies have shown that CSE can cause vascular wall contraction by activating Rho kinase (64, 65). On the one hand, activated Rho kinase directly phosphorylates eNOS (Thr113), thereby inhibiting NO production in endothelial cells (66, 67). On the other hand, it can promote the contraction of smooth muscle cells by regulating the sensitivity of calcium ions (68, 69). At the same time, CSE can also cause the activation of Rho kinase in macrophages, and then participate in the occurrence and development of atherosclerosis (70, 71).
According to clinical studies, plasma levels of asymmetric dimethylarginine (ADMA) are significantly higher in smoking patients than in non-smokers (72). ADMA, a metabolite of L-arginine, is an endogenous inhibitor of eNOS (73). Studies have further confirmed that cigarette extract not only inhibits L-arginine uptake but also reduces the expression of L-arginine transporter CAT1, thereby reducing NO production (74). However, L-arginine supplementation improves arterial diastolic function in healthy smokers (75).
In vitro studies have shown that CSE reduces protein synthesis by inducing protein misfolding in the endoplasmic reticulum. At the same time, a large number of unfolded proteins interact with each other and lead to the phosphorylation of PERK and eIF2a, which will prolong endoplasmic reticulum stress and activate autophagy-related programs, resulting in endothelial cell dysfunction (76). In other organelles, CSE induces mitochondrial damage and changes in the membrane potential by activating the NF-KB and NLRP3 signaling pathways. However, this process is reversed by MitoQ (77). In addition, cigarette extracts also upregulate the expression of galectin-3, thereby increasing the levels of p-AMPK and decreasing the levels of p-mTOR, resulting in increased autophagy in endothelial cells. At the same time, cigarette extract increases ROS production and downregulates eNOS expression, thereby affecting the tube formation and migration ability of endothelial cells (78). At present, there are many studies on smoking and endothelial cells, but most of them focus on endothelial dysfunction (Figures 3, 4).
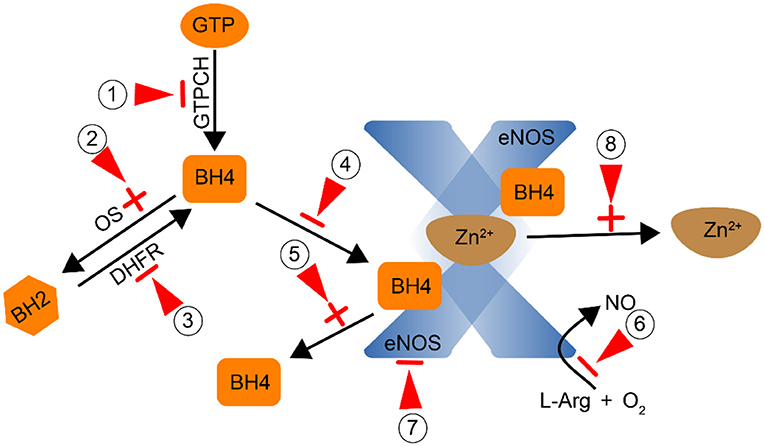
Figure 4. Smoking affects the activity of eNOS by direct or indirect action (Red arrow: point of action).① Inhibit GTPCH and reduce the synthesis of BH4;② Promote the conversion of BH4 to BH2 through oxidative stress;③ Inhibit DHFR and reduce the conversion of BH2 to BH4;, ④,⑤ Prevent the combination of BH4 and eNOS to form a stable dimer; ⑥ Inhibit the conversion of L-Arg to NO; ⑦ Inhibit the activity of eNOS; ⑧ Promote the dissociation of Zn2+ from the dimer.
Smoking and Monocytes-Macrophages
Circulating monocytes are derived from hematopoietic progenitor cells in the bone marrow. According to the expression levels of CD14 and FCγRIII immunoglobulin receptor CD16, which are components of the lipopolysaccharide receptor complex, three subgroups of monocytes have been identified in humans, including CD14++ CD16–, CD14+ CD16+, and CD14+ CD16++ monocytes. The activation of monocytes participates in the formation of PAD (79, 80). Cigarettes induce the production of various chemokines and proinflammatory cytokines. These factors promote the adhesion of monocytes to endothelial cells, drive monocytes to enter the endothelium, and undergo phenotypic changes. The most important regulatory factor is CC chemokine receptor 2 (CCR2) and its ligands monocyte chemoattractant protein 1 (MCP-1) and monocyte chemoattractant protein 3 (MCP-3) (81–83). Second, the particulate components of CSE activate protein kinase C (PKC), resulting in increased expression of the peripheral blood monocyte adhesion ligand CD11b and endothelial cell counter receptors VCAM1, ICAM-1 and ELAM-1. The expression of these ligands and counter receptors leads to enhanced adhesion of monocytes to endothelial cells, which is a factor initiating the pathogenesis of CSE and causing inflammation in blood vessel walls (84, 85).
Aryl hydrocarbon receptor (AHR) is a member of the transcriptional regulator bHLH (basic helix-loop-helix)-PAS (Per-ARNT-Sim) family, which mainly regulates various developmental and physiological functions (86, 87). Normally, AHR forms a dormant complex with HSP90, XAP2, and p23 in the cytoplasm (88). When exposed to smoke for a long time, the polycyclic aromatic hydrocarbon (PAH) ligand in cigarette smoke binds to AHR and causes a conformational change (89). The continuously activated AHR may promote the occurrence of atherosclerosis (90–92). At the same time, AHR can induce the production of AHRR (AHR repressor) and then regulate the activation of AHR through negative feedback. However, cigarette smoke continuously activates the AhR signaling pathway, which potentially drives changes in the methylation of the AHRR gene, accompanied by histone modification and enhancer activation. Methylated AhRR will undergo regional DNA conformation changes that affect the interaction of protein factors and DNA, subsequently altering gene transcription and expression and participating in the regulatory process of atherosclerosis (93–95). After a conformational change in AHR, HSP90 is released from the complex. Studies have shown that HSP90 interacts with Akt to mediate the phosphorylation of eNOS (96–98).
Monocytes and macrophages that enter the endothelium release large amounts of MPO that forms hypochlorous acid (HOCl) and hypothiocyanic acid (HOSCN). Macrophages exposed to HOSCN exhibit increased release of inflammatory cytokines and chemokines, including MCP-1, TNF-α, and IL6, and the subsequent of amplification of the inflammatory cascade. However, HOSCN-induced cytokine/chemokine expression and cell death are reduced by inhibiting the NF-kB signaling pathway (99). Further research showed the differential expression of miRNAs in smokers and non-smokers. This differential expression also participates in the regulation of monocyte phenotypic changes. The expression of miR-124-3p in smokers is significantly higher than that in non-smokers, and changes in surface markers are accompanied by changes in the monocyte phenotype. When miR-124-3p is overexpressed, the expression of CD206 on the surface of monocytes is significantly upregulated (100).
Smoking and Vascular Smooth Muscle Cells
Under normal physiological conditions, the relaxation and contraction of smooth muscle cells in the arterial wall is necessary to maintain relatively stable blood flow and blood pressure. However, when cells are stimulated with pathological factors, smooth muscle cell proliferation, migration, apoptosis and phenotypic changes are crucial in the pathological process of atherosclerosis (101). Nicotinic acetylcholine receptors (nAChRs) are members of the superfamily of cys-loop ligand-gated ion channels (cysLGICs). They are widely distributed transmembrane proteins in vascular smooth muscle cells and are involved in regulating ion homeostasis and maintaining functional changes in blood vessels (102–104). Nicotine in cigarette smoke is an important ligand for the α1 nicotinic acetylcholine receptor, which directly induces the transformation of VSMCs from a contractile phenotype to a synthetic phenotype through the nicotinic acetylcholine receptor and G protein-coupled receptor. It also promotes the migration of VSMCs from the media to atheromatous plaques in the vascular intima (105). At the same time, the gated ion channel is open, resulting in the influx of a large amount of Ca2+. When the concentration of Ca2+ in the cytoplasm is further increased, it will cause a conformational change in calpain-1 that triggers its proteolytic activity. Therefore, calpain promotes remodeling of the actin cytoskeleton in VSMCs to form podosomes by activating PKC, accompanied by the degradation of extracellular matrix (ECM) (106). On the other hand, the extracellular matrix is degraded by activated MMP-2/MMP-9 (107, 108). Second, an increased influx of Ca2+ activates the ERK1/2-Egr-1 signaling pathway and form a cascade amplification effect, thereby participating in nicotine-induced smooth muscle cell proliferation (109).
Nicotine also participates in the functional and phenotypic changes of smooth muscle cells through nAChR-induced regulation of multiple related downstream signaling pathways. Knockdown of nAChRα1 significantly reduces the CSE-induced phosphorylation of STAT3, Akt, and mTOR, while overexpression of nAChRα1 exerts the opposite effect. STAT3 directly interacts with the nicotine receptor nAChRα1 to regulate the nuclear translocation of STAT3 and its binding to the Akt promoter region, thereby promoting the proliferation and migration of VSMCs and inflammation mediated by macrophages, which in turn causes atherosclerosis. STAT3 inhibition significantly reduces nicotine-induced atherosclerosis (110). Nicotine also modulates the release of vascular endothelial growth factor (VEGF) in VSMCs in rats, and this process is blocked by selective inhibitors of epidermal growth factor receptor (EGFR) kinase and MEK, leading to reduced ERK phosphorylation and decreased VEGF release. Therefore, nicotine promotes VEGF release from VSMCs by activating the EGFR-ERK pathway and increases the risk of cardiovascular disease in smokers (111). In addition, nicotine causes oxidative stress to increase the release of inflammatory bodies through nAChRs and promote smooth muscle cell autophagy through the NF-κB signaling pathway, which in turn causes smooth muscle cell phenotypic transformation (112, 113).
Endothelin plays an important role in maintaining circulatory homeostasis (114). Studies have shown that cigarette smoke or nicotine can affect the expression of ETA (mainly distributed in the smooth muscle cells of the blood vessel wall) and ETB (mainly distributed in the endothelial cells of the blood vessel wall) receptor expression, thereby regulating the vasomotor reactivity (115–117). Cigarette smoke exposure can promote the upregulation of ETA receptor expression through MEK1/2 (118), ERK1/2 (119), MAPK (120), and NF-kB pathways (121), and thus induce the contraction of smooth muscle cells dependent on Ca2+ signaling channels (122, 123). At the same time, cigarette smoke can also increase the expression of ETB receptor in endothelial cells, and cause endothelial dysfunction and contraction of VSMCs through the effect of ET-1 and the reduction of NO synthesis (66, 121, 124).
CSE or nicotine promotes bFGF production in smooth muscle cells, which subsequently promotes the mitosis of cells, accompanied by the phosphorylation of ERK and c-Jun and increased expression of cyclinD1. When ERK phosphorylation is inhibited, the levels of p-c-Jun and cyclinD1 decrease correspondingly, along with a decrease in cell viability. When c-Jun is overexpressed or knocked down, the expression of cyclinD1 is also affected accordingly. CSE promotes smooth muscle cell proliferation through the p-ERK-p-c-Jun-cyclin D1 pathway (125, 126).
In addition, smoking reduces acetylcholine (ACh)-induced vasodilation. This mechanism may be caused by the hyperpolarization of VSMCs induced by stimulating endothelial cells to reduce the production of NO and PGI2 (127). Based on recent studies, the functional and phenotypic changes in smooth muscle cells play a vital role in the onset and progression of atherosclerosis (Figure 5), but the specific mechanism by which smoking regulates smooth muscle cells still requires further research.
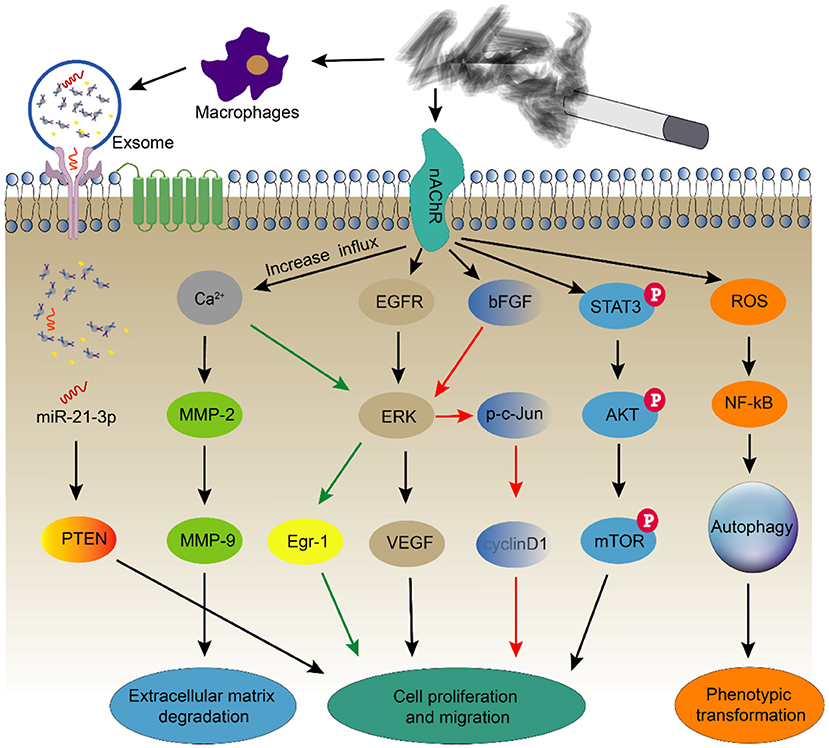
Figure 5. Smoking alters the function and phenotype of smooth muscle cells. Smoking induces smooth muscle cell autophagy through oxidative stress and causes smooth muscle cell proliferation and migration through multiple signaling pathways. At the same time, it also regulates changes in the functions of smooth muscle cells by stimulating macrophages to secrete exosomes.
Smoking and Animal Models of Atherosclerosis
Smoking can regulate ECs, VSMCs and mononuclear macrophages through a variety of mechanisms to participate in the occurrence and development of atherosclerosis (Table 1). However, in vivo experiments, many studies have also confirmed the relationship between smoking and atherosclerosis in animals (138, 139). Currently, the animal models used to study atherosclerosis mainly include ApoE-/- mice, LDLR-/- mice, and ApoE-/- and LDLR-/- mice. Models have also been derived from different species, such as mice, rabbits, pigs and non-human primates (140). However, different animal models (focus on comparing rabbits and mice) have different advantages and limitations (140–148) (Table 2).
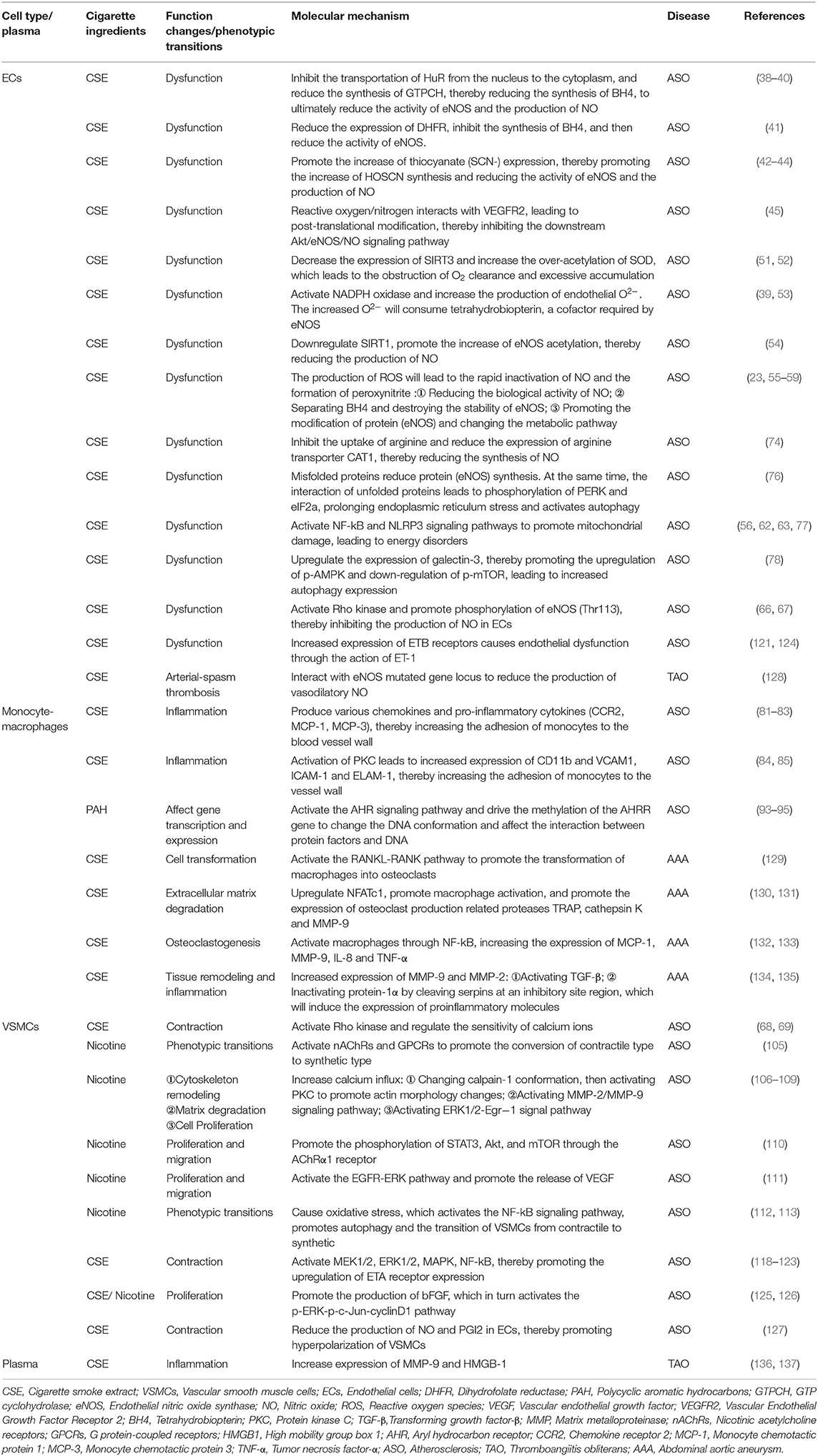
Table 1. Smoking and various types of cell function changes, phenotypic transitions and their molecular mechanisms.
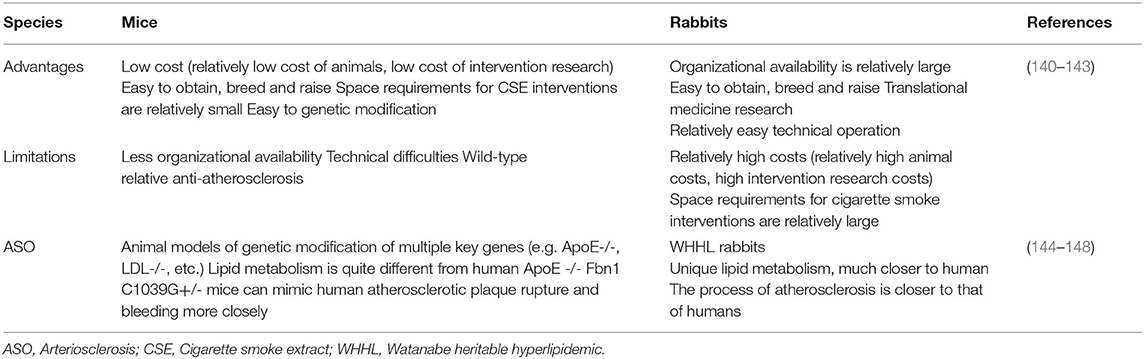
Table 2. Advantages and limitations of mice and rabbits in animal models of ASO.
Smoking and Thromboangiitis Obliterans
Clinical Evidence
Thromboangiitis obliterans (TAO), or Buerger's disease, is a group of unexplained non-atherosclerotic inflammatory diseases mainly caused by small and medium-sized arteriovenous vascular lesions. It is characterized by the involvement of vascular thrombosis (with or without recanalization), infiltration of inflammatory cells (in the tissue surrounding the thrombus or the adventitia), fibrosis of the intima and media, and changes in the internal elastic layer. Among people with a low socioeconomic status, smoking history is an important risk factor for this condition, and a strong correlation has been observed between heavy tobacco use and thromboembolic vasculitis (149, 150). At present, a definite diagnostic standard for diagnosing the disease is unavailable, but it mainly depends on the medical history, physical examination, laboratory examination, imaging examination and exclusion of other diagnoses. The simplest and most effective method to prevent further aggravation of this diseases is to stop smoking completely or use tobacco in any form (151, 152). After the diagnosis of TAO, patients who stop smoking have a lower risk of amputation than those who continue to smoke (153).
Smoking-Related Molecular Mechanisms
Currently, although a large number of studies have confirmed that smoking is closely related to the occurrence and development of TAO, its specific mechanism is not yet clear. Under physiological or pathological conditions, the role of NO in maintaining vascular wall tension is mainly regulated by eNOS (154). However, the T allele of the eNOS 894 G-T polymorphism is associated with the prevention of TAO (155). Charles J. Glueck et al. also considered that mutations in the eNOS gene locus interact with cigarettes to reduce the production of vasodilatory NO and promote arterial spasm thrombosis. However, TAO has potential therapeutic value by promoting NO production and improving homocysteine metabolism (128). A pathological examination of the diseased vascular intima of patients with TAO revealed an immune response (156). Anticardiolipin antibody (ACA) is an autoantibody that uses negatively charged cardiolipin on the platelet and endothelial cell membrane as the target antigen and is commonly detected in patients with systemic lupus erythematosus and other autoimmune diseases (157). Clinical studies have confirmed that ACA may be related to Buerger's disease, which may exacerbate thrombotic events in patients with the disease (158). An analysis of clinical cases found that thromboembolic vasculitis was associated with an increased prevalence of anticardiolipin antibodies and that the presence of high antibody titers in these patients was associated with increased morbidity, including large limb amputation (156, 159). Previous reports have documented the correlation between smoking and anti-cardiolipin antibodies. As in-depth clinical research is conducted, researchers have proposed that smoking may cause physiological changes in some patients, resulting in the formation of aPL. Of course, this finding has also been confirmed in studies of many healthy individuals who later developed vascular disease (160, 161).
In addition, studies have shown that long-term cigarette exposure will lead to damage to the antioxidant defense system, which in turn increases the expression of MMP-9 and high mobility group box 1 (HMGB-1); high expression of MMP-9 and HMGB-1 also plays a role in TAO-related vascular lesions (136, 137). In summary, a large body of literature has established a link between smoking and TAO. However, the occurrence of TAO is not caused by a single factor. It is affected by a number of factors, including tobacco sensitization, cold, autoimmunity and changes in hormone levels. Therefore, clinical and molecular studies on TAO still must overcome numerous challenges.
Smoking and Animal Models of TAO
The existing research on TAO mainly focuses on in vitro and clinical research. Although animal models of TAO (induced by sodium laurate) have been generated, the establishment and confirmation of these models are only confirmed by pathological examinations (162, 163). However, it still has its advantages and limitations in vivo animal models (162, 164) (Table 3). Due to our limited understanding of the pathogenesis of TAO, it is difficult to simulate the true pathogenesis of TAO in vivo (169, 170).
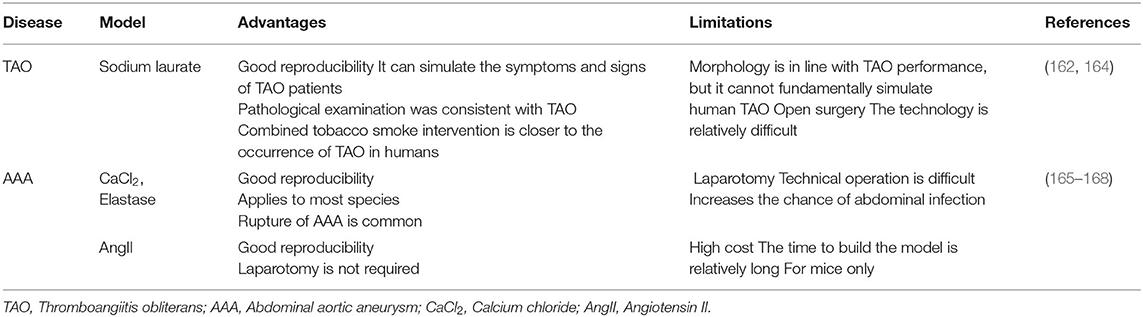
Table 3. Advantages and limitations of animal models of TAO and AAA.
Smoking and Aneurysm
Clinical Evidence
Abdominal aortic aneurysm (AAA) is an abnormal enlargement of the infrarenal aorta. The pathophysiology of AAA causes the aortic wall to become increasingly thinner, which may rupture and cause life-threatening bleeding. Various factors may cause AAA. According to the classification of risk factors for AAA, smoking is the most important risk factor leading to aneurysm-related hospitalization and death, followed by the male sex and hypertension (171–173). A large number of retrospective studies, systematic reviews and other studies have shown that both active smoking and passive smoking may lead to the occurrence of aortic aneurysms and increase the risk of death. Women are often more sensitive than men. However, the occurrence of abdominal aortic aneurysms after quitting smoking is also strongly linked to a previous smoking history, but the risk of developing abdominal aortic aneurysms decreases faster in women than in men after quitting smoking (174–176). A study of the effect on smoking on small abdominal aortic aneurysms showed that continuous smoking is directly related to the prognosis of patients with small AAA, and patients with higher plasma cotinine concentrations (smokers) have an increased risk of rupture. The long-term survival rate is reduced (177).
In addition, smoking may exert a direct effect on perioperative and long-term complications after AAA. In terms of the effect of smoking on post-operative AAA, quitting smoking more than 8 weeks before opening and repairing AAA significantly reduces the incidence of post-operative pulmonary complications. However, elective open AAA repair after smoking cessation for <8 weeks has no correlation with the perioperative prognosis (178, 179). Although smoking cessation may reduce the risk of rapid progression of AAA, it still exerts a lasting effect on the development and operation of AAA, and the condition is more likely to occur in smokers than non-smokers. Therefore, the prohibition of smoking may effectively control the occurrence of AAA and the occurrence of serious complications (129).
Smoking-Related Molecular Mechanisms
At present, the pathogenesis of AAA is mainly attributed to inflammatory cell infiltration, the destruction of elastin and collagen in the media and adventitia, biological changes in smooth muscle cells and angiogenesis (180, 181). Studies have confirmed that cigarette extracts induce the conversion of macrophages to osteoclasts through the classic RANKL-RANK pathway, upregulate the expression of NFATc1, promote macrophage activation, and induce the expression of the osteoclastogenesis-related proteases TRAP, cathepsin K and MMP-9 (129–131). Second, CSE also enhances the inflammatory response through the NF-kB pathway, which leads to the activation of macrophages and increased expression of MCP-1, MMP-9, IL-8, and TNF-α. These proinflammatory mediators and proteases are closely related to osteoclastogenesis and play a vital role in the formation of aneurysms (132, 133). Upon stimulation with cigarette smoke extracts, MMP-9 secreted by macrophages and MMP-2 secreted by mesenchymal cells cooperate to produce AAA. On the one hand, the mechanism is to activate TGF-β by cleaving its latent form, and TGF-β activation plays an important role in controlling tissue remodeling under physiological and pathological conditions. On the other hand, both enzymes inactivate protein-1α by cleaving serpins at an inhibitory site, and cleaved serpins induce the expression of proinflammatory molecules in monocytes (134, 135). Keisuke Hashimoto et al. added deoxyribonucleic acid (DNA) to the diet of mice to understand the effect of dietary DNA on blood vessel walls. An examination of the aorta revealed that dietary DNA may attenuate nicotine-induced elastic fiber degradation by inhibiting the MMP-2-dependent pathway, thereby preventing the weakened elasticity of the aortic wall (182). The role of MMPs in the formation and progression of AAA is indisputable (183, 184). The dialogue between cells (ECs, VSMCs, and macrophages) plays an important role in CSE-induced MMPs production in the aortic wall (135, 185), but its specific molecular mechanisms need to be further in-depth explored.
Smoking and Animal Models of AAA
How does smoking affect animal models of AAA? At present, the three most commonly used animal models of AAA are calcium chloride-induced AAA, subrenal abdominal aortic perfusion with elastase, and subcutaneous injection of angiotensin II (AngII) (186, 187). Smoking not only affects the mechanical properties of the aorta and substantially changes the structure of the arterial wall but also changes gene expression through epigenetic mechanisms, thereby promoting the formation of AAA (188, 189). In a model of AAA induced by AngII, smoking further increased the expression of a matrix metalloproteinase (MMP) gene in the abdominal aorta, thereby enhancing the proteolytic activity of MMP (190). At the same time, 3,4-benzo pyrene in cigarette smoke also promotes the formation of AngII-induced AAA in mice through the same mechanism (191). However, in the model of AAA induced by elastase perfusion, the effect of smoking on the development of AAA does not depend on the activity of elastase, and smoking does not alter the expression of MMP-9 or MMP-12. However, smoking increases elastin degradation and the aneurysm size (192, 193). Of course, in order to simulate the aneurysm model more vividly, some special surgical treatments (such as aortic coarctation) on the basis of drug induction are often more effective in increasing the diameter of the aneurysm (194). However, AAA are often associated with arteriosclerosis factors. Therefore, establishing an animal model of AAA on the basis of arteriosclerosis (such as a high-fat diet) will be closer to the occurrence of human AAA (195). Although the existing AAA models do not fully replicate the pathological changes in AAA, the development of animal models has substantially improved our knowledge and understanding of the disease (165–168) (Table 3).
Summary
With aging of the population, the incidence of PAD is increasing annually. Many factors promote the development of this disease, and smoking is an independent risk factor for PAD. The effect of tobacco exposure on PAD is continuous. Quitting smoking can partially reverse or delay disease progression, but it is not completely eliminated. As for the pathogenesis of smoking and PAD, in vitro studies, cigarettes and their substitutes can regulate the occurrence and development of PAD through a variety of pathways. In vivo studies, numerous studies confirm the association between smoking and cardiovascular disease. However, due to the advantages and limitations of different animal models, as well as the differences in the components of cigarette smoke and intervention techniques, the mechanism of smoking on cardiovascular diseases cannot completely mimic the pathogenesis of humans. Therefore, in the study of animal models of smoking and peripheral arterial disease, we should select appropriate animal models for related research based on the purpose of the research and combining the pathophysiological mechanisms of human diseases. At the same time, with the development of gene editing technology, we also look forward to the development of genetically modified animal models related to PAD, which can better simulate the occurrence and development of human PAD.
Author Contributions
WW and TZ designed the study and searched literature. WW, KG, and GY contributed to drafting of the manuscript. All authors have read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy and integrity of any part of the work are appropriately investigated and resolved.
Funding
This study was funded by Education Department of Sichuan Province Science and Technology Plan Project (No. 18ZA0521).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circ Res. (2015) 116:1509–26. doi: 10.1161/CIRCRESAHA.116.303849
2. Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, et al. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation. (2006) 113:e463–654. doi: 10.1161/CIRCULATIONAHA.106.174526
3. Creager MA. Decade in review–peripheral vascular disease: 10 years of breakthroughs in peripheral vascular disease. Nat Rev Cardiol. (2014) 11:635–6. doi: 10.1038/nrcardio.2014.153
4. Fowkes FG, Aboyans V, Fowkes FJ, McDermott MM, Sampson UK, Criqui MH. Peripheral artery disease: epidemiology and global perspectives. Nat Rev Cardiol. (2017) 14:156–70. doi: 10.1038/nrcardio.2016.179
5. Morley RL, Sharma A, Horsch AD, Hinchliffe RJ. Peripheral artery disease. BMJ. (2018) 360:j5842. doi: 10.1136/bmj.j5842
6. Willigendael EM, Teijink JA, Bartelink ML, Kuiken BW, Boiten J, Moll FL, et al. Influence of smoking on incidence and prevalence of peripheral arterial disease. J Vasc Surg. (2004) 40:1158–65. doi: 10.1016/j.jvs.2004.08.049
7. Aday AW, Matsushita K. Epidemiology of peripheral artery disease and polyvascular disease. Circ Res. (2021) 128:1818–32. doi: 10.1161/CIRCRESAHA.121.318535
8. Ngu NL, McEvoy M. Environmental tobacco smoke and peripheral arterial disease: a review. Atherosclerosis. (2017) 266:113–20. doi: 10.1016/j.atherosclerosis.2017.09.024
9. Joosten MM, Pai JK, Bertoia ML, Rimm EB, Spiegelman D, Mittleman MA, et al. Associations between conventional cardiovascular risk factors and risk of peripheral artery disease in men. JAMA. (2012) 308:1660–7. doi: 10.1001/jama.2012.13415
10. Conen D, Everett BM, Kurth T, Creager MA, Buring JE, Ridker PM, et al. Smoking, smoking cessation, [corrected] and risk for symptomatic peripheral artery disease in women: a cohort study. Ann Intern Med. (2011) 154:719–26. doi: 10.7326/0003-4819-154-11-201106070-00003
11. Young JC, Paul NJ, Karatas TB, Kondrasov SA, McGinigle KL, Crowner JR, et al. Cigarette smoking intensity informs outcomes after open revascularization for peripheral artery disease. J Vasc Surg. (2019) 70:1973–83.e5. doi: 10.1016/j.jvs.2019.02.066
12. Armstrong EJ, Wu J, Singh GD, Dawson DL, Pevec WC, Amsterdam EA, et al. Smoking cessation is associated with decreased mortality and improved amputation-free survival among patients with symptomatic peripheral artery disease. J Vasc Surg. (2014) 60:1565–71. doi: 10.1016/j.jvs.2014.08.064
13. Howard G, Wagenknecht LE, Burke GL, Diez-Roux A, Evans GW, McGovern P, et al. Cigarette smoking and progression of atherosclerosis: the atherosclerosis risk in communities (ARIC) study. JAMA. (1998) 279:119–24. doi: 10.1001/jama.279.2.119
14. Jones MR, Magid HS, Al-Rifai M, McEvoy JW, Kaufman JD, Hinckley Stukovsky KD, et al. Secondhand smoke exposure and subclinical cardiovascular disease: the multi-ethnic study of atherosclerosis. J Am Heart Assoc. (2016) 5:e002965. doi: 10.1161/JAHA.115.002965
15. Mendelson MM, de Ferranti SD. Childhood environmental tobacco smoke exposure: a smoking gun for atherosclerosis in adulthood. Circulation. (2015) 131:1231–3. doi: 10.1161/CIRCULATIONAHA.115.015705
16. Jiang CQ, Lao XQ, Yin P, Thomas GN, Zhang WS, Liu B, et al. Smoking, smoking cessation and aortic arch calcification in older Chinese: the Guangzhou Biobank Cohort Study. Atherosclerosis. (2009) 202:529–34. doi: 10.1016/j.atherosclerosis.2008.03.004
17. Kim JA, Chun EJ, Lee MS, Kim KJ, Choi SI. Relationship between amount of cigarette smoking and coronary atherosclerosis on coronary CTA in asymptomatic individuals. Int J Cardiovasc Imaging. (2013) 29(Suppl. 1):21–8. doi: 10.1007/s10554-013-0224-8
18. Bhatnagar A, Maziak W, Eissenberg T, Ward KD, Thurston G, King BA, et al. Water pipe (Hookah) smoking and cardiovascular disease risk: a scientific statement from the American Heart Association. Circulation. (2019) 139:e917–36. doi: 10.1161/CIR.0000000000000671
19. Osei AD, Mirbolouk M, Orimoloye OA, Dzaye O, Uddin SMI, Benjamin EJ, et al. Association between E-cigarette use and cardiovascular disease among never and current combustible-cigarette smokers. Am J Med. (2019) 132:949–4.e2. doi: 10.1016/j.amjmed.2019.02.016
20. Rustemeier K, Stabbert R, Haussmann HJ, Roemer E, Carmines EL. Evaluation of the potential effects of ingredients added to cigarettes. Part 2: chemical composition of mainstream smoke. Food Chem Toxicol. (2002) 40:93–104. doi: 10.1016/s0278-6915(01)00085-0
21. Margham J, McAdam K, Forster M, Liu C, Wright C, Mariner D, et al. Chemical composition of aerosol from an e-cigarette: a quantitative comparison with cigarette smoke. Chem Res Toxicol. (2016) 29:1662–78. doi: 10.1021/acs.chemrestox.6b00188
22. Rezk-Hanna M, Benowitz NL. Cardiovascular effects of hookah smoking: potential implications for cardiovascular risk. Nicotine Tob Res. (2019) 21:1151–61. doi: 10.1093/ntr/nty065
23. Benowitz NL, Fraiman JB. Cardiovascular effects of electronic cigarettes. Nat Rev Cardiol. (2017) 14:447–56. doi: 10.1038/nrcardio.2017.36
24. Glantz SA, Bareham DW. E-Cigarettes: use, effects on smoking, risks, and policy implications. Annu Rev Public Health. (2018) 39:215–35. doi: 10.1146/annurev-publhealth-040617-013757
25. Takami R, Takeda N, Hayashi M, Sasaki A, Kawachi S, Yoshino K, et al. Body fatness and fat distribution as predictors of metabolic abnormalities and early carotid atherosclerosis. Diabetes Care. (2001) 24:1248–52. doi: 10.2337/diacare.24.7.1248
26. Ong KL, Ding J, McClelland RL, Cheung BM, Criqui MH, Barter PJ, et al. Relationship of pericardial fat with lipoprotein distribution: the multi-ethnic study of atherosclerosis. Atherosclerosis. (2015) 241:664–70. doi: 10.1016/j.atherosclerosis.2015.06.027
27. Han SG, Howatt DA, Daugherty A, Gairola CG. Atherogenic and pulmonary responses of ApoE- and LDL receptor-deficient mice to sidestream cigarette smoke. Toxicology. (2012) 299:133–8. doi: 10.1016/j.tox.2012.05.015
28. Lietz M, Berges A, Lebrun S, Meurrens K, Steffen Y, Stolle K, et al. Cigarette-smoke-induced atherogenic lipid profiles in plasma and vascular tissue of apolipoprotein E-deficient mice are attenuated by smoking cessation. Atherosclerosis. (2013) 229:86–93. doi: 10.1016/j.atherosclerosis.2013.03.036
29. Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Oxidants in cigarette smoke extract modify low-density lipoprotein in the plasma and facilitate atherogenesis in the aorta of Watanabe heritable hyperlipidemic rabbits. Atherosclerosis. (2001) 156:109–17. doi: 10.1016/s0021-9150(00)00637-7
30. Kunitomo M, Yamaguchi Y, Kagota S, Yoshikawa N, Nakamura K, Shinozuka K. Biochemical evidence of atherosclerosis progression mediated by increased oxidative stress in apolipoprotein E-deficient spontaneously hyperlipidemic mice exposed to chronic cigarette smoke. J Pharmacol Sci. (2009) 110:354–61. doi: 10.1254/jphs.09100fp
31. Lee J, Cooke JP. The role of nicotine in the pathogenesis of atherosclerosis. Atherosclerosis. (2011) 215:281–3. doi: 10.1016/j.atherosclerosis.2011.01.003
32. Centner AM, Bhide PG, Salazar G. Nicotine in senescence and atherosclerosis. Cells. (2020) 9:1035. doi: 10.3390/cells9041035
33. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
34. Siasos G, Tsigkou V, Kokkou E, Oikonomou E, Vavuranakis M, Vlachopoulos C, et al. Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Curr Med Chem. (2014) 21:3936–48. doi: 10.2174/092986732134141015161539
35. Armstrong AW, Armstrong EJ, Fuller EN, Sockolov ME, Voyles SV. Smoking and pathogenesis of psoriasis: a review of oxidative, inflammatory and genetic mechanisms. Br J Dermatol. (2011) 165:1162–8. doi: 10.1111/j.1365-2133.2011.10526.x
36. Gimbrone Jr MA, García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
37. Donato AJ, Morgan RG, Walker AE, Lesniewski LA. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol. (2015) 89:122–35. doi: 10.1016/j.yjmcc.2015.01.021
38. Zhao Q, Zheng K, Ma C, Li J, Zhuo L, Huang W, et al. PTPS facilitates compartmentalized LTBP1 S-nitrosylation and promotes tumor growth under hypoxia. Mol Cell. (2020) 77:95–107.e5. doi: 10.1016/j.molcel.2019.09.018
39. Abdelghany TM, Ismail RS, Mansoor FA, Zweier JR, Lowe F, Zweier JL. Cigarette smoke constituents cause endothelial nitric oxide synthase dysfunction and uncoupling due to depletion of tetrahydrobiopterin with degradation of GTP cyclohydrolase. Nitric Oxide. (2018) 76:113–21. doi: 10.1016/j.niox.2018.02.009
40. Li J, Liu S, Cao G, Sun Y, Chen W, Dong F, et al. Nicotine induces endothelial dysfunction and promotes atherosclerosis via GTPCH1. J Cell Mol Med. (2018) 22:5406–17. doi: 10.1111/jcmm.13812
41. Chen LM, Nergard JC, Ni L, Rosser CJ, Chai KX. Long-term exposure to cigarette smoke extract induces hypomethylation at the RUNX3 and IGF2-H19 loci in immortalized human urothelial cells. PLoS ONE. (2013) 8:e65513. doi: 10.1371/journal.pone.0065513
42. Talib J, Kwan J, Suryo Rahmanto A, Witting PK, Davies MJ. The smoking-associated oxidant hypothiocyanous acid induces endothelial nitric oxide synthase dysfunction. Biochem J. (2014) 457:89–97. doi: 10.1042/BJ20131135
43. Love DT, Barrett TJ, White MY, Cordwell SJ, Davies MJ, Hawkins CL. Cellular targets of the myeloperoxidase-derived oxidant hypothiocyanous acid (HOSCN) and its role in the inhibition of glycolysis in macrophages. Free Radic Biol Med. (2016) 94:88–98. doi: 10.1016/j.freeradbiomed.2016.02.016
44. Abdo AI, Rayner BS, van Reyk DM, Hawkins CL. Low-density lipoprotein modified by myeloperoxidase oxidants induces endothelial dysfunction. Redox Biol. (2017) 13:623–32. doi: 10.1016/j.redox.2017.08.004
45. Edirisinghe I, Rahman I. Cigarette smoke-mediated oxidative stress, shear stress, and endothelial dysfunction: role of VEGFR2. Ann N Y Acad Sci. (2010) 1203:66–72. doi: 10.1111/j.1749-6632.2010.05601.x
46. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. (2015) 4:180–3. doi: 10.1016/j.redox.2015.01.002
47. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. (2006) 8:1865–79. doi: 10.1089/ars.2006.8.1865
48. García N, Zazueta C, Aguilera-Aguirre L. Oxidative stress and inflammation in cardiovascular disease. Oxid Med Cell Longev. (2017) 2017:5853238. doi: 10.1155/2017/5853238
49. Banerjee J, Khanna S, Bhattacharya A. MicroRNA regulation of oxidative stress. Oxid Med Cell Longev. (2017) 2017:2872156. doi: 10.1155/2017/2872156
50. Lee WH, Ong SG, Zhou Y, Tian L, Bae HR, Baker N, et al. Modeling cardiovascular risks of E-cigarettes with human-induced pluripotent stem cell-derived endothelial cells. J Am Coll Cardiol. (2019) 73:2722–37. doi: 10.1016/j.jacc.2019.03.476
51. Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. (2016) 23:128–42. doi: 10.1016/j.cmet.2015.10.013
52. Dikalov S, Itani H, Richmond B, Vergeade A, Rahman SMJ, Boutaud O, et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol. (2019) 316:H639–46. doi: 10.1152/ajpheart.00595.2018
53. Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. (2004) 24:1031–6. doi: 10.1161/01.ATV.0000127083.88549.58
54. Arunachalam G, Yao H, Sundar IK, Caito S, Rahman I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: role of resveratrol. Biochem Biophys Res Commun. (2010) 393:66–72. doi: 10.1016/j.bbrc.2010.01.080
55. Raij L, DeMaster EG, Jaimes EA. Cigarette smoke-induced endothelium dysfunction: role of superoxide anion. J Hypertens. (2001) 19:891–7. doi: 10.1097/00004872-200105000-00009
56. Ismaeel A, Brumberg RS, Kirk JS, Papoutsi E, Farmer PJ, Bohannon WT, et al. Oxidative stress and arterial dysfunction in peripheral artery disease. Antioxidants (Basel). (2018) 7:145. doi: 10.3390/antiox7100145
57. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. (2012) 33:829–37, 837a–d. doi: 10.1093/eurheartj/ehr304
58. Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. (2002) 109:817–26. doi: 10.1172/JCI14442
59. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. (2017) 87:315–424. doi: 10.1152/physrev.00029.2006
60. Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. (1998) 19:819–25. doi: 10.1165/ajrcmb.19.5.3091
61. Cui Z, Han Z, Li Z, Hu H, Patel JM, Antony V, et al. Involvement of calpain-calpastatin in cigarette smoke-induced inhibition of lung endothelial nitric oxide synthase. Am J Respir Cell Mol Biol. (2005) 33:513–20. doi: 10.1165/rcmb.2005-0046OC
62. Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. (2018) 9:171. doi: 10.1038/s41419-017-0257-3
63. Wang X, Bian Y, Zhang R, Liu X, Ni L, Ma B, et al. Melatonin alleviates cigarette smoke-induced endothelial cell pyroptosis through inhibiting ROS/NLRP3 axis. Biochem Biophys Res Commun. (2019) 519:402–8. doi: 10.1016/j.bbrc.2019.09.005
64. Duong-Quy S, Dao P, Hua-Huy T, Guilluy C, Pacaud P, Dinh-Xuan AT. Increased Rho-kinase expression and activity and pulmonary endothelial dysfunction in smokers with normal lung function. Eur Respir J. (2011) 37:349–55. doi: 10.1183/09031936.00056610
65. Hidaka T, Hata T, Soga J, Fujii Y, Idei N, Fujimura N, et al. Increased leukocyte rho kinase (ROCK) activity and endothelial dysfunction in cigarette smokers. Hypertens Res. (2010) 33:354–9. doi: 10.1038/hr.2010.3
66. Sugimoto M, Nakayama M, Goto TM, Amano M, Komori K, Kaibuchi K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochem Biophys Res Commun. (2007) 361:462–7. doi: 10.1016/j.bbrc.2007.07.030
67. Noma K, Goto C, Nishioka K, Hara K, Kimura M, Umemura T, et al. Smoking, endothelial function, and Rho-kinase in humans. Arterioscler Thromb Vasc Biol. (2005) 25:2630–5. doi: 10.1161/01.ATV.0000189304.32725.bd
68. Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. (2004) 24:1842–7. doi: 10.1161/01.ATV.0000142813.33538.82
69. Richens TR, Linderman DJ, Horstmann SA, Lambert C, Xiao YQ, Keith RL, et al. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am J Respir Crit Care Med. (2009) 179:1011–21. doi: 10.1164/rccm.200807-1148OC
70. Wei Y, Zhu M, Corbalán-Campos J, Heyll K, Weber C, Schober A. Regulation of Csf1r and Bcl6 in macrophages mediates the stage-specific effects of microRNA-155 on atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:796–803. doi: 10.1161/ATVBAHA.114.304723
71. Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. (2014) 16:431. doi: 10.1007/s11906-014-0431-2
72. Sobczak A, Prokopowicz A, Radek M, Szula M, Zaciera M, Kurek J, et al. Tobacco smoking decreases plasma concentration of the emerging cardiovascular risk marker, L-homoarginine. Circ J. (2014) 78:1254–8. doi: 10.1253/circj.cj-13-1334
73. Gawrys J, Gajecki D, Szahidewicz-Krupska E, Doroszko A. Intraplatelet L-arginine-nitric oxide metabolic pathway: from discovery to clinical implications in prevention and treatment of cardiovascular disorders. Oxid Med Cell Longev. (2020) 2020:1015908. doi: 10.1155/2020/1015908
74. Zhang WZ, Venardos K, Chin-Dusting J, Kaye DM. Adverse effects of cigarette smoke on NO bioavailability: role of arginine metabolism and oxidative stress. Hypertension. (2006) 48:278–85. doi: 10.1161/01.HYP.0000231509.27406.42
75. Siasos G, Tousoulis D, Vlachopoulos C, Antoniades C, Stefanadi E, Ioakeimidis N, et al. Short-term treatment with L-arginine prevents the smoking-induced impairment of endothelial function and vascular elastic properties in young individuals. Int J Cardiol. (2008) 126:394–9. doi: 10.1016/j.ijcard.2007.04.057
76. Csordas A, Kreutmayer S, Ploner C, Braun PR, Karlas A, Backovic A, et al. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc Res. (2011) 92:141–8. doi: 10.1093/cvr/cvr165
77. Chen S, Wang Y, Zhang H, Chen R, Lv F, Li Z, et al. The antioxidant MitoQ protects against CSE-induced endothelial barrier injury and inflammation by inhibiting ROS and autophagy in human umbilical vein endothelial cells. Int J Biol Sci. (2019) 15:1440–51. doi: 10.7150/ijbs.30193
78. Pei C, Wang X, Lin Y, Fang L, Meng S. Inhibition of galectin-3 alleviates cigarette smoke extract-induced autophagy and dysfunction in endothelial progenitor cells. Oxid Med Cell Longev. (2019) 2019:7252943. doi: 10.1155/2019/7252943
79. Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol. (2017) 37:35–42. doi: 10.1161/ATVBAHA.116.308198
80. Frodermann V, Nahrendorf M. Macrophages and cardiovascular health. Physiol Rev. (2018) 98:2523–69. doi: 10.1152/physrev.00068.2017
81. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. (2011) 145:341–55. doi: 10.1016/j.cell.2011.04.005
82. Giebe S, Cockcroft N, Hewitt K, Brux M, Hofmann A, Morawietz H, et al. Cigarette smoke extract counteracts atheroprotective effects of high laminar flow on endothelial function. Redox Biol. (2017) 12:776–86. doi: 10.1016/j.redox.2017.04.008
83. Komiyama M, Takanabe R, Ono K, Shimada S, Wada H, Yamakage H, et al. Association between monocyte chemoattractant protein-1 and blood pressure in smokers. J Int Med Res. (2018) 46:965–74. doi: 10.1177/0300060517723415
84. Kalra VK, Ying Y, Deemer K, Natarajan R, Nadler JL, Coates TD. Mechanism of cigarette smoke condensate induced adhesion of human monocytes to cultured endothelial cells. J Cell Physiol. (1994) 160:154–62. doi: 10.1002/jcp.1041600118
85. Giunzioni I, Bonomo A, Bishop E, Castiglioni S, Corsini A, Bellosta S. Cigarette smoke condensate affects monocyte interaction with endothelium. Atherosclerosis. (2014) 234:383–90. doi: 10.1016/j.atherosclerosis.2014.03.029
86. Uemura S, Nakajima Y, Yoshida Y, Furuya M, Matsutani S, Kawate S, et al. Biochemical properties of human full-length aryl hydrocarbon receptor (AhR). J Biochem. (2020) 168:285–94. doi: 10.1093/jb/mvaa047
87. Hao N, Whitelaw ML. The emerging roles of AhR in physiology and immunity. Biochem Pharmacol. (2013) 86:561–70. doi: 10.1016/j.bcp.2013.07.004
88. Wang GZ, Zhang L, Zhao XC, Gao SH, Qu LW, Yu H, et al. The aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat Commun. (2019) 10:1125. doi: 10.1038/s41467-019-08887-7
89. Cole JW, Xu H. Aryl hydrocarbon receptor repressor methylation: a link between smoking and atherosclerosis. Circ Cardiovasc Genet. (2015) 8:640–2. doi: 10.1161/CIRCGENETICS.115.001243
90. Wang C, Petriello MC, Zhu B, Hennig B. PCB 126 induces monocyte/macrophage polarization and inflammation through AhR and NF-κB pathways. Toxicol Appl Pharmacol. (2019) 367:71–81. doi: 10.1016/j.taap.2019.02.006
91. Kim JB, Zhao Q, Nguyen T, Pjanic M, Cheng P, Wirka R, et al. Environment-sensing aryl hydrocarbon receptor inhibits the chondrogenic fate of modulated smooth muscle cells in atherosclerotic lesions. Circulation. (2020) 142:575–90. doi: 10.1161/CIRCULATIONAHA.120.045981
92. Assefa EG, Yan Q, Gezahegn SB, Salissou MTM, He S, Wu N, et al. Role of resveratrol on indoxyl sulfate-induced endothelial hyperpermeability via aryl hydrocarbon receptor (AHR)/src-dependent pathway. Oxid Med Cell Longev. (2019) 2019:5847040. doi: 10.1155/2019/5847040
93. Wan M, Bennett BD, Pittman GS, Campbell MR, Reynolds LM, Porter DK, et al. Identification of smoking-associated differentially methylated regions using reduced representation bisulfite sequencing and cell type-specific enhancer activation and gene expression. Environ Health Perspect. (2018) 126:047015. doi: 10.1289/EHP2395
94. Reynolds LM, Magid HS, Chi GC, Lohman K, Barr RG, Kaufman JD, et al. Secondhand tobacco smoke exposure associations with DNA methylation of the aryl hydrocarbon receptor repressor. Nicotine Tob Res. (2017) 19:442–51. doi: 10.1093/ntr/ntw219
95. Reynolds LM, Wan M, Ding J, Taylor JR, Lohman K, Su D, et al. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circ Cardiovasc Genet. (2015) 8:707–16. doi: 10.1161/CIRCGENETICS.115.001097
96. Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J Biol Chem. (2003) 278:30821–7. doi: 10.1074/jbc.M304471200
97. Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol. (2015) 74:38–48. doi: 10.1016/j.vph.2015.05.008
98. Brouet A, Sonveaux P, Dessy C, Moniotte S, Balligand JL, Feron O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res. (2001) 89:866–73. doi: 10.1161/hh2201.100319
99. Pan GJ, Rayner BS, Zhang Y, van Reyk DM, Hawkins CL. A pivotal role for NF-κB in the macrophage inflammatory response to the myeloperoxidase oxidant hypothiocyanous acid. Arch Biochem Biophys. (2018) 642:23–30. doi: 10.1016/j.abb.2018.01.016
100. de Ronde MWJ, Kok MGM, Moerland PD, Van den Bossche J, Neele AE, Halliani A, et al. High miR-124-3p expression identifies smoking individuals susceptible to atherosclerosis. Atherosclerosis. (2017) 263:377–84. doi: 10.1016/j.atherosclerosis.2017.03.045
101. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
102. Liu W, Li MD. Insights into nicotinic receptor signaling in nicotine addiction: implications for prevention and treatment. Curr Neuropharmacol. (2018) 16:350–70. doi: 10.2174/1570159X15666170801103009
103. Egleton RD, Brown KC, Dasgupta P. Angiogenic activity of nicotinic acetylcholine receptors: implications in tobacco-related vascular diseases. Pharmacol Ther. (2009) 121:205–23. doi: 10.1016/j.pharmthera.2008.10.007
104. Zou Q, Leung SW, Vanhoutte PM. Activation of nicotinic receptors can contribute to endothelium-dependent relaxations to acetylcholine in the rat aorta. J Pharmacol Exp Ther. (2012) 341:756–63. doi: 10.1124/jpet.112.192229
105. Yoshiyama S, Chen Z, Okagaki T, Kohama K, Nasu-Kawaharada R, Izumi T, et al. Nicotine exposure alters human vascular smooth muscle cell phenotype from a contractile to a synthetic type. Atherosclerosis. (2014) 237:464–70. doi: 10.1016/j.atherosclerosis.2014.10.019
106. Gu Z, Fonseca V, Hai CM. Nicotinic acetylcholine receptor mediates nicotine-induced actin cytoskeletal remodeling and extracellular matrix degradation by vascular smooth muscle cells. Vascul Pharmacol. (2013) 58:87–97. doi: 10.1016/j.vph.2012.08.003
107. Li ZZ, Guo ZZ, Zhang Z, Cao QA, Zhu YJ, Yao HL, et al. Nicotine-induced upregulation of VCAM-1, MMP-2, and MMP-9 through the α7-nAChR-JNK pathway in RAW264 7 and MOVAS cells. Mol Cell Biochem. (2015) 399:49–58. doi: 10.1007/s11010-014-2231-z
108. Ren A, Wu H, Liu L, Guo Z, Cao Q, Dai Q. Nicotine promotes atherosclerosis development in apolipoprotein E-deficient mice through α1-nAChR. J Cell Physiol. (2018) 108. doi: 10.1002/jcp.27728
109. Vazquez-Padron RI, Mateu D, Rodriguez-Menocal L, Wei Y, Webster KA, Pham SM. Novel role of Egr-1 in nicotine-related neointimal formation. Cardiovasc Res. (2010) 88:296–303. doi: 10.1093/cvr/cvq213
110. Xu S, Ni H, Chen H, Dai Q. The interaction between STAT3 and nAChRα1 interferes with nicotine-induced atherosclerosis via Akt/mTOR signaling cascade. Aging (Albany NY). (2019) 11:8120–38. doi: 10.18632/aging.102296
111. Kanda Y, Watanabe Y. Nicotine-induced vascular endothelial growth factor release via the EGFR-ERK pathway in rat vascular smooth muscle cells. Life Sci. (2007) 80:1409–14. doi: 10.1016/j.lfs.2006.12.033
112. Wang Z, Liu B, Zhu J, Wang D, Wang Y. Nicotine-mediated autophagy of vascular smooth muscle cell accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway. Atherosclerosis. (2019) 284:1–10. doi: 10.1016/j.atherosclerosis.2019.02.008
113. Yao Y, Mao J, Xu S, Zhao L, Long L, Chen L, et al. Rosmarinic acid inhibits nicotine-induced C-reactive protein generation by inhibiting NLRP3 inflammasome activation in smooth muscle cells. J Cell Physiol. (2019) 234:1758–67. doi: 10.1002/jcp.27046
114. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev. (2016) 68:357–418. doi: 10.1124/pr.115.011833
115. Tanus-Santos JE, Sampaio RC, Hyslop S, Franchini KG, Moreno Jr H. Endothelin ET(A) receptor antagonism attenuates the pressor effects of nicotine in rats. Eur J Pharmacol. (2000) 396:33–7. doi: 10.1016/s0014-2999(00)00194-1
116. Hashimoto T, Yoneda M, Shimada T, Kurosawa M, Terano A. Intraportal nicotine infusion in rats decreases hepatic blood flow through endothelin-1 and both endothelin A and endothelin B receptors. Toxicol Appl Pharmacol. (2004) 196:1–10. doi: 10.1016/j.taap.2003.09.004
117. Granström BW, Xu CB, Nilsson E, Vikman P, Edvinsson L. Smoking particles enhance endothelin A and endothelin B receptor-mediated contractions by enhancing translation in rat bronchi. BMC Pulm Med. (2006) 6:6. doi: 10.1186/1471-2466-6-6
118. Cao L, Ping NN, Cao YX, Li W, Cai Y, Warfvinge K, et al. The effects of MEK1/2 inhibition on cigarette smoke exposure-induced ET receptor upregulation in rat cerebral arteries. Toxicol Appl Pharmacol. (2016) 304:70–8. doi: 10.1016/j.taap.2016.05.013
119. Zhang Y, Zhang W, Edvinsson L, Xu CB. Lipid-soluble cigarette smoke particles induced vascular endothelin type a receptor up-regulation through activation of ERK1/2 signal pathways. Basic Clin Pharmacol Toxicol. (2017) 120:327–34. doi: 10.1111/bcpt.12688
120. Xu CB, Zheng JP, Zhang W, Zhang Y, Edvinsson L. Lipid-soluble smoke particles upregulate vascular smooth muscle ETB receptors via activation of mitogen-activating protein kinases and NF-kappaB pathways. Toxicol Sci. (2008) 106:546–55. doi: 10.1093/toxsci/kfn173
121. Xie YH, Wang SW, Zhang Y, Edvinsson L, Xu CB. Up-regulation of G-protein-coupled receptors for endothelin and thromboxane by lipid-soluble smoke particles in renal artery of rat. Basic Clin Pharmacol Toxicol. (2010) 107:803–12. doi: 10.1111/j.1742-7843.2010.00585.x
122. Sudjarwo SA, Hori M, Tanaka T, Matsuda Y, Karaki H. Coupling of the endothelin ETA and ETB receptors to Ca2+ mobilization and Ca2+ sensitization in vascular smooth muscle. Eur J Pharmacol. (1995) 289:197–204. doi: 10.1016/0922-4106(95)90095-0
123. Bailey MA, Haton C, Orea V, Sassard J, Bailly C, Unwin RJ, et al. ETA receptor-mediated Ca2+ signaling in thin descending limbs of Henle's loop: impairment in genetic hypertension. Kidney Int. (2003) 63:1276–84. doi: 10.1046/j.1523-1755.2003.00880.x
124. Milara J, Ortiz JL, Juan G, Guijarro R, Almudever P, Martorell M, et al. Cigarette smoke exposure up-regulates endothelin receptor B in human pulmonary artery endothelial cells: molecular and functional consequences. Br J Pharmacol. (2010) 161:1599–615. doi: 10.1111/j.1476-5381.2010.00979.x
125. Li T, Song T, Ni L, Yang G, Song X, Wu L, et al. The p-ERK-p-c-Jun-cyclinD1 pathway is involved in proliferation of smooth muscle cells after exposure to cigarette smoke extract. Biochem Biophys Res Commun. (2014) 453:316–20. doi: 10.1016/j.bbrc.2014.09.062
126. Carty CS, Soloway PD, Kayastha S, Bauer J, Marsan B, Ricotta JJ, et al. Nicotine and cotinine stimulate secretion of basic fibroblast growth factor and affect expression of matrix metalloproteinases in cultured human smooth muscle cells. J Vasc Surg. (1996) 24:927–34. doi: 10.1016/s0741-5214(96)70038-1
127. Fujii N, Reinke MC, Brunt VE, Minson CT. Impaired acetylcholine-induced cutaneous vasodilation in young smokers: roles of nitric oxide and prostanoids. Am J Physiol Heart Circ Physiol. (2013) 304:H667–73. doi: 10.1152/ajpheart.00731.2012
128. Glueck CJ, Haque M, Winarska M, Dharashivkar S, Fontaine RN, Zhu B, et al. Stromelysin-1 5A/6A and eNOS T-786C polymorphisms, MTHFR C677T and A1298C mutations, and cigarette-cannabis smoking: a pilot, hypothesis-generating study of gene-environment pathophysiological associations with Buerger's disease. Clin Appl Thromb Hemost. (2006) 12:427–39. doi: 10.1177/1076029606293429
129. Norman PE, Curci JA. Understanding the effects of tobacco smoke on the pathogenesis of aortic aneurysm. Arterioscler Thromb Vasc Biol. (2013) 33:1473–7. doi: 10.1161/ATVBAHA.112.300158
130. Igari K, Kelly MJ, Yamanouchi D. Cigarette smoke extract activates tartrate-resistant acid phosphatase-positive macrophage. J Vasc Res. (2019) 56:139–51. doi: 10.1159/000498893
131. Tanaka T, Kelly M, Takei Y, Yamanouchi D. RANKL-mediated osteoclastogenic differentiation of macrophages in the abdominal aorta of angiotensin II-infused apolipoprotein E knockout mice. J Vasc Surg. (2018) 68:48S−59S.e1. doi: 10.1016/j.jvs.2017.11.091
132. Yavropoulou MP, Yovos JG. Osteoclastogenesis–current knowledge and future perspectives. J Musculoskelet Neuronal Interact. (2008) 8:204–16.
133. Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L46–57. doi: 10.1152/ajplung.00241.2005
134. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. (2002) 110:625–32. doi: 10.1172/JCI15334
135. Ghosh A, Pechota LV, Upchurch Jr., GR, Eliason JL. Cross-talk between macrophages, smooth muscle cells, and endothelial cells in response to cigarette smoke: the effects on MMP2 and 9. Mol Cell Biochem. (2015) 410:75–84. doi: 10.1007/s11010-015-2539-3
136. Bezerra FS, Valença SS, Pires KM, Lanzetti M, Pimenta WA, Schmidt AC, et al. Long-term exposure to cigarette smoke impairs lung function and increases HMGB-1 expression in mice. Respir Physiol Neurobiol. (2011) 177:120–6. doi: 10.1016/j.resp.2011.03.023
137. De Caridi G, Bitto A, Massara M, Pallio G, Pizzino G, Serra R, et al. Increased Serum HMGB-1, ICAM-1 and Metalloproteinase-9 Levels in Buerger's Patients. Curr Vasc Pharmacol. (2016) 14:382–7. doi: 10.2174/1570161114666160303111355
138. Qin W, Zhang L, Li Z, Xiao D, Zhang Y, Zhang H, et al. Endothelial to mesenchymal transition contributes to nicotine-induced atherosclerosis. Theranostics. (2020) 10:5276–89. doi: 10.7150/thno.42470
139. Espinoza-Derout J, Hasan KM, Shao XM, Jordan MC, Sims C, Lee DL, et al. Chronic intermittent electronic cigarette exposure induces cardiac dysfunction and atherosclerosis in apolipoprotein-E knockout mice. Am J Physiol Heart Circ Physiol. (2019) 317:H445–59. doi: 10.1152/ajpheart.00738.2018
140. Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, et al. Animal models of atherosclerosis. Eur J Pharmacol. (2017) 816:3–13. doi: 10.1016/j.ejphar.2017.05.010
141. Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. (2012) 32:1104–15. doi: 10.1161/ATVBAHA.111.237693
142. Zhao Y, Qu H, Wang Y, Xiao W, Zhang Y, Shi D. Small rodent models of atherosclerosis. Biomed Pharmacother. (2020) 129:110426. doi: 10.1016/j.biopha.2020.110426
143. Fan J, Chen Y, Yan H, Niimi M, Wang Y, Liang J. Principles and applications of rabbit models for atherosclerosis research. J Atheroscler Thromb. (2018) 25:213–20. doi: 10.5551/jat.RV17018
144. von Scheidt M, Zhao Y, Kurt Z, Pan C, Zeng L, Yang X, et al. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab. (2017) 25:248–61. doi: 10.1016/j.cmet.2016.11.001
145. Poznyak AV, Silaeva YY, Orekhov AN, Deykin AV. Animal models of human atherosclerosis: current progress. Braz J Med Biol Res. (2020) 53:e9557. doi: 10.1590/1414-431x20209557
146. Van der Donckt C, Van Herck JL, Schrijvers DM, Vanhoutte G, Verhoye M, Blockx I, et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. (2015) 36:1049–58. doi: 10.1093/eurheartj/ehu041
147. Yamaguchi Y, Kagota S, Haginaka J, Kunitomo M. Evidence of modified LDL in the plasma of hypercholesterolemic WHHL rabbits injected with aqueous extracts of cigarette smoke. Environ Toxicol Pharmacol. (2000) 8:255–60. doi: 10.1016/s1382-6689(00)00050-8
148. Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Fluvastatin reduces modification of low-density lipoprotein in hyperlipidemic rabbit loaded with oxidative stress. Eur J Pharmacol. (2002) 436:97–105. doi: 10.1016/s0014-2999(01)01576-x
149. Olin JW. Thromboangiitis obliterans (Buerger's disease). N Engl J Med. (2000) 343:864–9. doi: 10.1056/NEJM200009213431207
150. Klein-Weigel PF, Richter JG. Thromboangiitis obliterans (Buerger's disease). Vasa. (2014) 43:337–46. doi: 10.1024/0301-1526/a000371
151. Olin JW, Shih A. Thromboangiitis obliterans (Buerger's disease). Curr Opin Rheumatol. (2006) 18:18–24. doi: 10.1097/01.bor.0000198000.58073.aa
152. Igari K, Inoue Y, Iwai T. The epidemiologic and clinical findings of patients with buerger disease. Ann Vasc Surg. (2016) 30:263–9. doi: 10.1016/j.avsg.2015.07.014
153. Le Joncour A, Soudet S, Dupont A, Espitia O, Koskas F, Cluzel P, et al. Long-term outcome and prognostic factors of complications in thromboangiitis obliterans (Buerger's disease): a multicenter study of 224 patients. J Am Heart Assoc. (2018) 7:e010677. doi: 10.1161/JAHA.118.010677
154. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. (1993) 329:2002–12. doi: 10.1056/NEJM199312303292706
155. Adigüzel Y, Yilmaz E, Akar N. Effect of eNOS and ET-1 polymorphisms in thromboangiitis obliterans. Clin Appl Thromb Hemost. (2010) 16:103–6. doi: 10.1177/1076029609336854
156. Chavoshan A, Sharebiani H, Taheri H, Fazeli B. Antiphospholipid antibodies in Buerger's disease. Thromb Res. (2019) 181:64–6. doi: 10.1016/j.thromres.2019.07.015
157. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. (2011) 7:330–9. doi: 10.1038/nrrheum.2011.52
158. Pereira de. Godoy JM, Braile DM. Buerger's disease and anticardiolipin antibodies. J Cardiovasc Med (Hagerstown). (2009) 10:792–4. doi: 10.2459/JCM.0b013e32832ce8d0
159. Maslowski L, McBane R, Alexewicz P, Wysokinski WE. Antiphospholipid antibodies in thromboangiitis obliterans. Vasc Med. (2002) 7:259–64. doi: 10.1191/1358863x02vm452oa
160. Binder SR, Litwin CM. Anti-phospholipid antibodies and smoking: an overview. Clin Rev Allergy Immunol. (2017) 53:1–13. doi: 10.1007/s12016-016-8565-4
161. Gustafsson JT, Gunnarsson I, Källberg H, Pettersson S, Zickert A, Vikerfors A, et al. Cigarette smoking, antiphospholipid antibodies and vascular events in Systemic Lupus Erythematosus. Ann Rheum Dis. (2015) 74:1537–43. doi: 10.1136/annrheumdis-2013-205159
162. Zhang Z, Ji J, Zhang D, Ma M, Sun L. Protective effects and potential mechanism of salvianolic acid B on sodium laurate-induced thromboangiitis obliterans in rats. Phytomedicine. (2020) 66:153110. doi: 10.1016/j.phymed.2019.153110
163. Kong X, Yuan H, Wu X, Zhang J, Zhou H, Wang M, et al. High-mobility-group box protein 1A box reduces development of sodium laurate-induced thromboangiitis obliterans in rats. J Vasc Surg. (2013) 57:194–204. doi: 10.1016/j.jvs.2012.06.083
164. Ashida S, Ishihara M, Ogawa H, Abiko Y. Protective effect of ticlopidine on experimentally induced peripheral arterial occlusive disease in rats. Thromb Res. (1980) 18:55–67. doi: 10.1016/0049-3848(80)90170-x
165. Patelis N, Moris D, Schizas D, Damaskos C, Perrea D, Bakoyiannis C, et al. Animal models in the research of abdominal aortic aneurysms development. Physiol Res. (2017) 66:899–915. doi: 10.33549/physiolres.933579
166. Lysgaard Poulsen J, Stubbe J, Lindholt JS. Animal models used to explore abdominal aortic aneurysms: a systematic review. Eur J Vasc Endovasc Surg. (2016) 52:487–99. doi: 10.1016/j.ejvs.2016.07.004
167. Paraskevas KI, Mikhailidis DP, Perrea D. Experimental models of abdominal aortic aneurysms: an overview. Curr Pharm Des. (2008) 14:325–37. doi: 10.2174/138161208783497697
168. Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2004) 24:429–34. doi: 10.1161/01.ATV.0000118013.72016.ea
169. Li MD, Wang YF, Yang MW, Hong FF, Yang SL. Risk factors, mechanisms and treatments of thromboangiitis obliterans: an overview of recent research. Curr Med Chem. (2020) 27:6057–72. doi: 10.2174/0929867326666190816233042
170. Małecki R, Zdrojowy K, Adamiec R. Thromboangiitis obliterans in the 21st century–a new face of disease. Atherosclerosis. (2009) 206:328–34. doi: 10.1016/j.atherosclerosis.2009.01.042
171. Sakalihasan N, Michel JB, Katsargyris A, Kuivaniemi H, Defraigne JO, Nchimi A, et al. Abdominal aortic aneurysms. Nat Rev Dis Primers. (2018) 4:34. doi: 10.1038/s41572-018-0030-7
172. Sode BF, Nordestgaard BG, Grønbæk M, Dahl M. Tobacco smoking and aortic aneurysm: two population-based studies. Int J Cardiol. (2013) 167:2271–7. doi: 10.1016/j.ijcard.2012.06.003
173. Lee AJ, Fowkes FG, Carson MN, Leng GC, Allan PL. Smoking, atherosclerosis and risk of abdominal aortic aneurysm. Eur Heart J. (1997) 18:671–6. doi: 10.1093/oxfordjournals.eurheartj.a015314
174. Kihara T, Yamagishi K, Iso H, Tamakoshi A. JACC Study Group. Passive smoking and mortality from aortic dissection or aneurysm. Atherosclerosis. (2017) 263:145–50. doi: 10.1016/j.atherosclerosis.2017.06.022
175. Aune D, Schlesinger S, Norat T, Riboli E. Tobacco smoking and the risk of abdominal aortic aneurysm: a systematic review and meta-analysis of prospective studies. Sci Rep. (2018) 8:14786. doi: 10.1038/s41598-018-32100-2
176. Stackelberg O, Björck M, Larsson SC, Orsini N, Wolk A. Sex differences in the association between smoking and abdominal aortic aneurysm. Br J Surg. (2014) 101:1230–7. doi: 10.1002/bjs.9526
177. Smoking lung function and the prognosis of abdominal aortic aneurysm. The UK small aneurysm trial participants. Eur J Vasc Endovasc Surg. (2000) 19:636–42. doi: 10.1053/ejvs.2000.1066
178. Arinze N, Farber A, Levin SR, Cheng TW, Jones DW, Siracuse CG, et al. The effect of the duration of preoperative smoking cessation timing on outcomes after elective open abdominal aortic aneurysm repair and lower extremity bypass. J Vasc Surg. (2019) 70:1851–61. doi: 10.1016/j.jvs.2019.02.028
179. Koole D, Moll FL, Buth J, Hobo R, Zandvoort H, Pasterkamp G, et al. The influence of smoking on endovascular abdominal aortic aneurysm repair. J Vasc Surg. (2012) 55:1581–6. doi: 10.1016/j.jvs.2011.12.018
180. Kent KC. Clinical practice. Abdominal aortic aneurysms. N Engl J Med. (2014) 371:2101–8. doi: 10.1056/NEJMcp1401430
181. Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. (2017) 14:457–71. doi: 10.1038/nrcardio.2017.52
182. Hashimoto K, Zaima N, Sekiguchi H, Kugo H, Miyamoto C, Hoshino K, et al. Dietary DNA attenuates the degradation of elastin fibers in the aortic wall in nicotine-administrated mice. J Nutr Sci Vitaminol (Tokyo). (2018) 64:271–6. doi: 10.3177/jnsv.64.271
183. Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. (2001) 104:304–9. doi: 10.1161/01.cir.104.3.304
184. Wilson WR, Anderton M, Schwalbe EC, Jones JL, Furness PN, Bell PR, et al. Matrix metalloproteinase-8 and−9 are increased at the site of abdominal aortic aneurysm rupture. Circulation. (2006) 113:438–45. doi: 10.1161/CIRCULATIONAHA.105.551572
185. Yamamoto Y, Osanai T, Nishizaki F, Sukekawa T, Izumiyama K, Sagara S, et al. Matrix metalloprotein-9 activation under cell-to-cell interaction between endothelial cells and monocytes: possible role of hypoxia and tumor necrosis factor-α. Heart Vessels. (2012) 27:624–33. doi: 10.1007/s00380-011-0214-5
186. Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. (2019) 16:225–42. doi: 10.1038/s41569-018-0114-9
187. Davis FM, Daugherty A, Lu HS. Updates of recent aortic aneurysm research. Arterioscler Thromb Vasc Biol. (2019) 39:e83–90. doi: 10.1161/ATVBAHA.119.312000
188. Jaldin RG, Castardelli É, Perobelli JE, Yoshida WB, de Castro Rodrigues A, Sequeira JL, et al. Morphologic and biomechanical changes of thoracic and abdominal aorta in a rat model of cigarette smoke exposure. Ann Vasc Surg. (2013) 27:791–800. doi: 10.1016/j.avsg.2013.03.002
189. Greenway J, Gilreath N, Patel S, Horimatsu T, Moses M, Kim D, et al. Profiling of histone modifications reveals epigenomic dynamics during abdominal aortic aneurysm formation in mouse models. Front Cardiovasc Med. (2020) 7:595011. doi: 10.3389/fcvm.2020.595011
190. Stolle K, Berges A, Lietz M, Lebrun S, Wallerath T. Cigarette smoke enhances abdominal aortic aneurysm formation in angiotensin II-treated apolipoprotein E-deficient mice. Toxicol Lett. (2010) 199:403–9. doi: 10.1016/j.toxlet.2010.10.005
191. Ji K, Zhang Y, Jiang F, Qian L, Guo H, Hu J, et al. Exploration of the mechanisms by which 3,4-benzopyrene promotes angiotensin II-induced abdominal aortic aneurysm formation in mice. J Vasc Surg. (2014) 59:492–9. doi: 10.1016/j.jvs.2013.03.022
192. Bergoeing MP, Arif B, Hackmann AE, Ennis TL, Thompson RW, Curci JA. Cigarette smoking increases aortic dilatation without affecting matrix metalloproteinase-9 and−12 expression in a modified mouse model of aneurysm formation. J Vasc Surg. (2007) 45:1217–27. doi: 10.1016/j.jvs.2007.01.058
193. Jin J, Arif B, Garcia-Fernandez F, Ennis TL, Davis EC, Thompson RW, et al. Novel mechanism of aortic aneurysm development in mice associated with smoking and leukocytes. Arterioscler Thromb Vasc Biol. (2012) 32:2901–9. doi: 10.1161/ATVBAHA.112.300208
194. Bi Y, Zhong H, Xu K, Qi X, Zhang Z, Wu G, et al. Novel experimental model of enlarging abdominal aortic aneurysm in rabbits. J Vasc Surg. (2015) 62:1054–63. doi: 10.1016/j.jvs.2014.02.062
Keywords: smoking, peripheral artery disease, clinical evidence, molecular mechanism, review
Citation: Wang W, Zhao T, Geng K, Yuan G, Chen Y and Xu Y (2021) Smoking and the Pathophysiology of Peripheral Artery Disease. Front. Cardiovasc. Med. 8:704106. doi: 10.3389/fcvm.2021.704106
Received: 01 May 2021; Accepted: 31 July 2021;
Published: 27 August 2021.
Edited by:
Lars Edvinsson, Lund University, SwedenReviewed by:
Xiaofeng Yang, Temple University, United StatesDan Rudic, Augusta University, United States
Copyright © 2021 Wang, Zhao, Geng, Yuan, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Chen, Y2hlbnl1ZTU1MjNAMTI2LmNvbQ==; Youhua Xu, eWh4dUBtdXN0LmVkdS5tbw==
†These authors have contributed equally to this work