 Ayiguli Abudukeremu1†Canxia Huang2†Hongwei Li1Runlu Sun1,3
Ayiguli Abudukeremu1†Canxia Huang2†Hongwei Li1Runlu Sun1,3 Xiao Liu1,3Xiaoying Wu1,3
Xiao Liu1,3Xiaoying Wu1,3 Xiangkun Xie1,3Jingjing Huang1,3Jie Zhang1,3Jinlan Bao4
Xiangkun Xie1,3Jingjing Huang1,3Jie Zhang1,3Jinlan Bao4 Yuling Zhang1,3*
Yuling Zhang1,3*- 1Department of Cardiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
- 2Critical Care Medicine Department, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
- 3Guangdong Province Key Laboratory of Arrhythmia and Electrophysiology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
- 4Comprehensive Department, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
Background: Although elevation of HDL-C levels by pharmaceutical drugs have no benefit of cardiovascular endpoint, the effect of high-density lipoprotein/apolipoprotein A1 (HDL/apoA-1) replacement therapy on atherosclerosis is controversial. The current meta-analysis analyzed the effects of HDL/apoA-1 replacement therapies on atherosclerotic lesions both in humans and mice.
Methods: The PubMed, Cochrane Library, Web of Science, and EMBASE databases were searched through June 6, 2020. The methodological quality of the human studies was assessed using Review Manager (RevMan, version 5.3.). The methodological quality of the mouse studies was assessed using a stair list. STATA (version 14.0) was used to perform all statistical analyses.
Results: Fifteen randomized controlled human trials and 17 animal studies were included. The pooled results showed that HDL/apoA-1 replacement therapy use did not significantly decrease the percent atheroma volume (p = 0.766) or total atheroma volume (p = 0.510) in acute coronary syndrome (ACS) patients (N = 754). However, HDL/apoA-1 replacement therapies were significantly associated with the final percent lesion area, final lesion area, and changes in lesion area (SMD, −1.75; 95% CI: −2.21~-1.29, p = 0.000; SMD, −0.78; 95% CI: −1.18~-0.38, p = 0.000; SMD: −2.06; 95% CI, −3.92~-0.2, p = 0.03, respectively) in mice.
Conclusions: HDL/apoA-1 replacement therapies are safe but do not significantly improve arterial atheroma volume in humans. The results in animals suggest that HDL/apoA-1 replacement therapies decrease the lesion area. Additional studies are needed to investigate and explain the differences in HDL/apoA-1 replacement therapy efficacies between humans and animals.
Trial registration number: Human pooled analysis: PROSPERO, CRD42020210772. prospectively registered.
Key Points
1 HDL/apoA-1 replacement therapies significantly benefit plaque inhibition in atherosclerosis mouse models but do not significantly benefit human atheroma volume.
2 Trials with mice preliminarily suggest that HDL/apoA-1 replacement therapies more effectively inhibit atherosclerosis development in earlier stages than in mature lesions.
Introduction
Cardiovascular disease (CVD) claimed an estimated 17.9 million lives in 2016, and it remains the leading cause of death worldwide, representing 31% of all global deaths (1). Effective strategies for preventing and treating CVD is a major public health challenge worldwide. Lipid modification is the major method for cardiovascular risk reduction, especially low-density lipoprotein cholesterol (LDL-C)-lowering therapy, but patients with controlled LDL-C in the normal range still have increased residual cardiovascular risk (2). High-density lipoprotein (HDL) plays a crucial role in cholesterol reverse transportation and metabolism. Since Miller discovered that a reduction in plasma HDL concentrations may accelerate the development of atherosclerosis, HDL-C has become a new focus in the investigation of preventive therapy for CVD (3). However, studies over the past several decades that increased the self-production of HDL-C showed different results regarding CVD prevention (4–8). A great number of studies have been performed to explain these differences. (1) HDL is a conglomerate of protein, triglycerides, phospholipids, cholesterol esters, and cholesterol that performs various functions, including cholesterol efflux and reverse cholesterol transportation, anti-inflammatory effects, antioxidant effects, nitric oxide-promoting and endothelial function-enhancing effects, antithrombotic effects, and antiapoptotic effects (9). (2) HDL particles that fail to perform the biological functions mentioned above are termed “dysfunctional.” (3) Changes in HDL composition may cause dysfunction. Serum amyloid A is a significant predictor of cardiovascular disease risk, and it replaces apoA-1 in HDL during acute phase induction and prevents access of HDL to the plasma membrane (10, 11). Reductions in the antioxidative and anti-inflammatory properties of HDL-associated enzymes, such as paraoxonase 1 activity, lecithin cholesterol acyltransferase deficiency were accompanied by HDL oxidation and the promotion of atherosclerotic lesion formation (12, 13). Changes in lipid components, including triglyceride(s) and cholesteryl ester, also caused HDL dysfunction (14). In addition to losing its cardioprotective role, dysfunctional HDL may be harmful to patients via conversion into a proinflammatory and pro-oxidant component that promotes LDL oxidation (15, 16). Therefore, the use of normal-functioning HDL/apoA-1 or mimetics may ultimately elucidate the role of HDL/apoA-1 in CVD prevention.
Thus, in the present meta-analysis, we aimed to investigate the efficiency of HDL/apoA-1 replacement therapies in mouse models and patients with atherosclerosis.
Methods
The meta-analysis was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement (Supplementary Table 1). The methodological quality of the human studies was assessed using Review Manager (RevMan) software (version 5.3). The methodological quality of the mouse studies was assessed by using a stair list.
Participants included in the efficacy analysis were acute coronary syndrome (ACS) patients. The intervention was HDL/apoA-1 replacement therapy. The comparison was atheroma volume in the coronary artery measured via intravascular ultrasonography. The outcomes were changes in percent atheroma volume (PAV) and total atheroma volume (TAV), and the study type was randomized controlled trials. The mice included in the efficacy analysis were apolipoprotein E-deficient or low-density lipoprotein receptor-deficient mice fed with regular rodent chow or a high-fat diet. The intervention was HDL/apo-A1 replacement therapy. Comparisons included the lesion areas in the aortic sinus, aortic root, aorta, innominate artery, or carotid arteries. The outcomes were the final percent lesion area, final lesion area, and changes in lesion area. The study type was limited to animal-controlled studies.
Search Strategy
Two authors (AA and CH) searched the PubMed, Cochrane Library, Web of Science, and EMBASE databases up to June 6, 2020, for eligible studies using wide search terms and included all publications that met the inclusion criteria (Supplementary Table 2). We used the following terms: “Lipoproteins, HDL” or “Apolipoprotein A-I” and “mimetic” or “recombinant HDL” and “coronary artery disease” or “Cardiovascular Diseases” for the literature search. The reference lists from relevant articles were reviewed to identify additional studies.
Inclusion Criteria and Study Selection
Studies meeting the following criteria were included: (1) studies reporting the effect or safety of HDL/apoA-1 replacement therapies on atherosclerosis in mouse models and patients with atherosclerosis, (2) human studies designed as randomized controlled trials (RCTs), and (3) studies written in English. Studies with insufficient data or gray literature were excluded.
Data Extraction
Two independent reviewers (AA and CH) screened the titles and abstracts for relevance. Disagreements between reviewers were discussed until a consensus was reached. The manuscripts of selected titles/abstracts were assessed for inclusion. Using the selection criteria listed above, the two reviewers independently extracted baseline information using predefined extraction flow sheets. We resolved any disagreements between the authors via discussion. A third review author (HL) arbitrated when differences in opinions emerged. If a study compared different HDL/apoA-1 replacement therapies with a replacement therapy-naïve cohort, the data of all replacement therapies were separately compared with the control group.
Data Analysis
Statistical analyses were performed using Stata (version 14.0). A random-effects model with I-V heterogeneity (continuous data) or the Mantel–Haenszel (binary data) method was used to calculate pooled standardized mean differences (SMDs), or odds ratios (ORs) and 95% confidence intervals (CIs). We assessed several outcomes in human trials: PAV, TAV in the coronary artery, and other outcomes representing safety. We assessed three outcomes in animal trials: final percent lesion area, final lesion area, and changes in lesion area in arteries. Heterogeneity between studies was assessed using the Q-test and I2 statistic. Heterogeneity was considered significant when the p-value was < 0.05. Publication bias was assessed using visual inspection of funnel plots and Egger's regression test. The influence of individual studies was examined by the removal of one study at a time.
Results
Human Trials
Study Outlines and Characteristics
We identified 15 RCTs, of which 6 trials, including 754 ACS patients (replacement therapies = 414, placebo = 340), were used for efficacy analysis, and all 15 trials were used for safety analysis (Table 1) (17–31). A flowchart of the selection of eligible trails is shown in Figure 1. The duration of replacement therapy administration in the included studies ranged from a single administration to 10 weeks (weekly administration). The risk of bias of the included studies is shown in the Supplementary Figure 1.
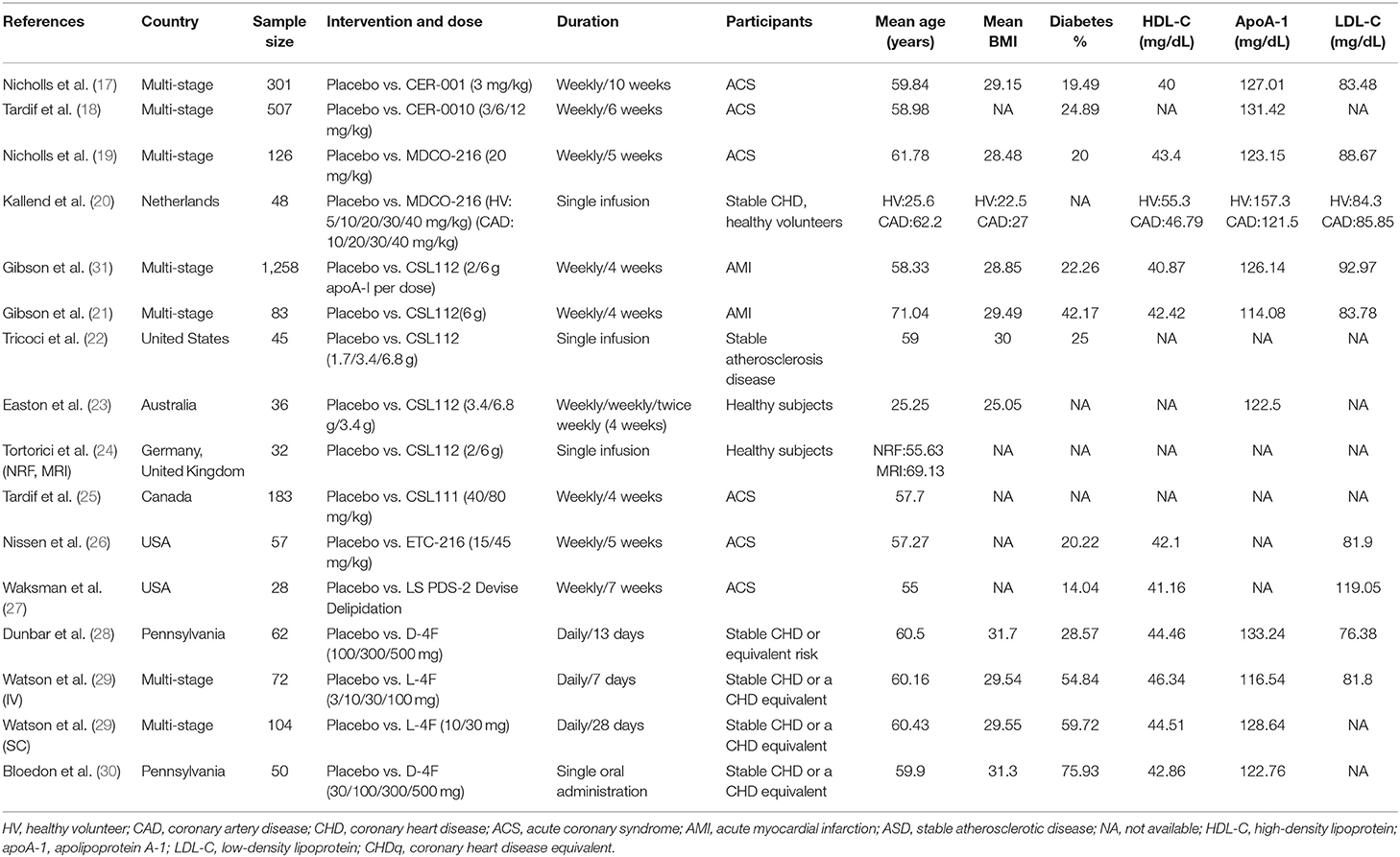
Table 1. Characteristics of the studies included in the human meta-analysis.
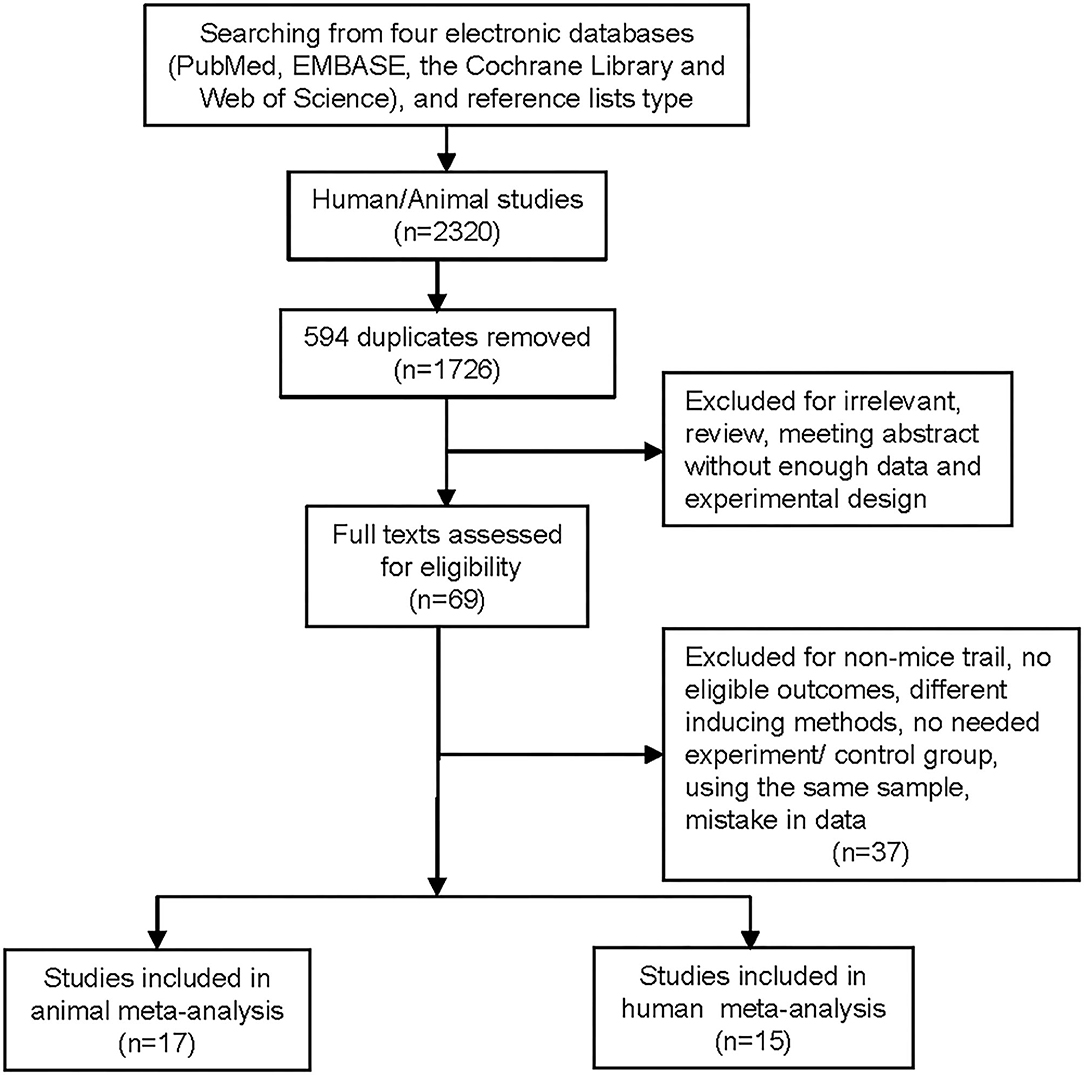
Figure 1. Flowchart of the literature search and study selection process.
Percent Atheroma Volume and Total Atheroma Volume
Pooled analysis revealed that HDL/apoA-1 replacement therapies did not significantly decrease PAV (SMD:0.03; 95% CI −0.17~0.23, p = 0.766, I2 = 39.7%) or TAV (SMD: −0.06; 95% CI −0.23~0.11, p = 0.510, I2 = 20.0%) in patients with atherosclerosis (Figure 2) (17–19, 25–27). The results did not significantly change in the sensitivity analysis (Supplementary Figures 2A,B).
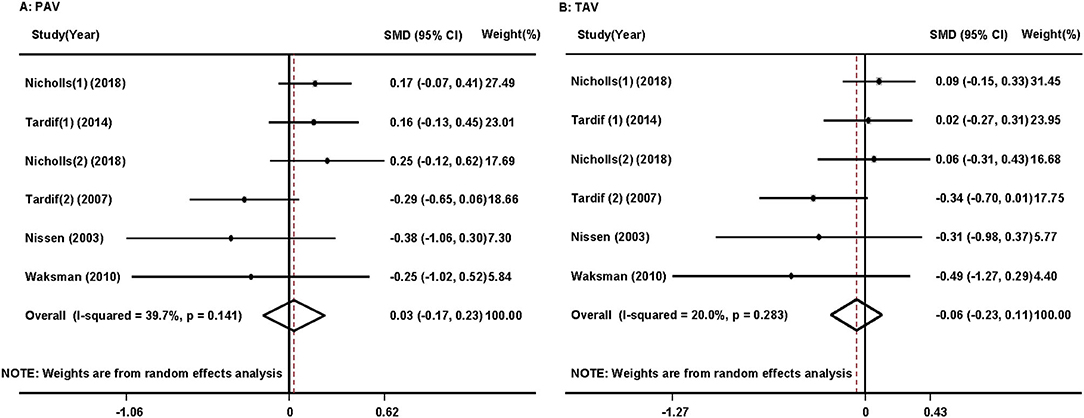
Figure 2. Forest plots of the meta-analysis of the associations between percent atheroma volume (PAV) (A) and total atheroma volume (TAV) (B) in the coronary artery and high-density lipoprotein/apolipoprotein A1 (HDL/apoA-1) replacement therapy administration in acute coronary syndrome patients using a random-effects model. SMD, standardized mean difference; CI, confidence interval. This result shows that HDL/apoA-1 replacement therapies did not significantly decrease PAV or TAV in patients with acute coronary syndrome.
Safety Assessment of High-Density Lipoprotein/Apolipoprotein A1 Replacement Therapies
Several studies separately reported the adverse effects of HDL/apoA-1 replacement therapies, including headache (n = 8), renal impairment (n = 6), hepatic impairment (n = 11), and nausea, vomiting, and abdominal pain (n = 7). Pooled analysis revealed that HDL/apoA-1 replacement therapies were safe, with no significantly increased risk of adverse events (headache: OR 1.58, 95% CI 0.84–2.97, I2 = 0.0%; renal impairment: OR 0.76, 95% CI 0.29–1.99, I2 = 8.7%; hepatic impairment: OR 1.37, 95% CI 0.51–3.64, I2 = 34.9%; nausea, vomiting, or abdominal pain: OR 0.81; 95% CI 0.47–1.38, I2 = 0.0%) (Supplementary Figure 3). The results did not significantly change in the sensitivity analysis (Supplementary Figure 4).
Mouse Trials
Study Outlines and Characteristics
We identified 17 controlled trials that included 479 atherosclerotic mice (HDL/apoA-1 replacement therapies = 285, placebo = 194) (Supplementary Table 3) (32–45). A flowchart of the selection of eligible trails is shown in Figure 1. The therapy administration duration ranged from 30 days to 16 weeks. The scientific inquiry of the animal trials was assessed using a stair list that included 12 “yes,” 95 “no,” and 5 “unclear” (Supplementary Table 5). Funnel plots were provided in Supplementary Figures 6A,B.
Final Percent Lesion Area
Eleven studies reported the effects of HDL/apoA-1 replacement therapies on the percent lesion area in mice (32–37, 40, 41, 46–48). A reduced final percent lesion area was observed in the HDL/apoA-1 replacement therapy-treated groups (SMD, −1.75; 95% CI: −2.21~-1.29, I2 = 64.6%) (Figure 3A). The results did not significantly change in the sensitivity analysis (Supplementary Figure 6C).
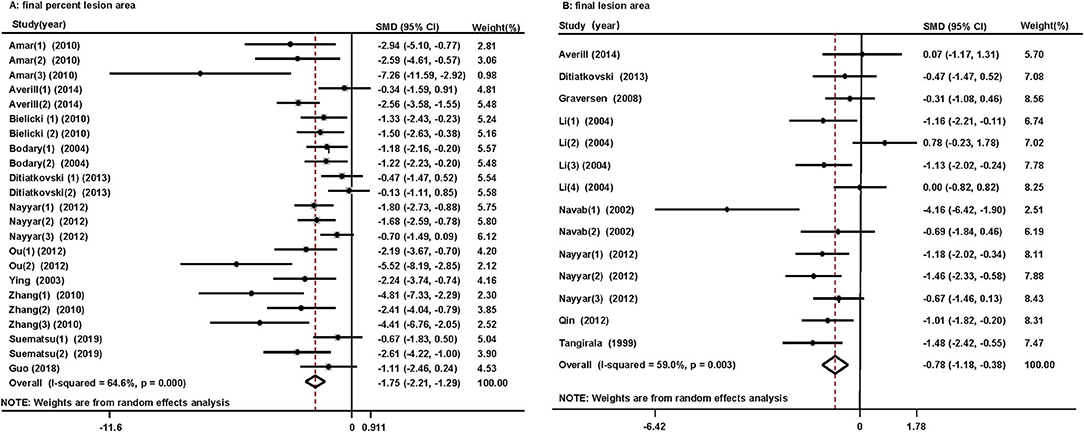
Figure 3. Forest plots of the meta-analysis of the associations between final percent lesion area (A), final lesion area (B) in arteries, and HDL/apoA-1 replacement therapy administration in mice using a random-effects model. SMD, standardized mean difference; CI, confidence interval. This result shows that HDL/apoA-1 replacement therapies significantly decreased the final percent lesion area and final lesion area in mice with coronary atherosclerosis.
Final Lesion Area
Eight studies reported the effect of HDL/apoA-1 replacement therapies on the final lesion area in mice (33, 36, 38–40, 42, 43, 45). A reduced final lesion area was observed in the HDL/apoA-1 replacement therapy group (SMD, −0.78; 95% CI: −1.18~-0.38, I2 = 59%) (Figure 3B). The results did not significantly change in the sensitivity analysis (Supplementary Figure 6D).
Changes in Lesion Area
Three studies reported the effects of HDL/apoA-1 replacement therapies on changes in lesion area in mice (37, 43, 44). More changes in lesion area (reduction) were observed in the HDL/apoA-1 group than the control group (SMD: −2.06; 95% CI, −3.92~-0.2, I2 = 83.9%) (Supplementary Figure 5).
Subgroup Analysis
A reduced final percent lesion area was observed in HDL/apoA-1 subgroups based on LDL−/− mice, apoE−/− mice, males, and females. A reduced final lesion area was observed in HDL/apoA-1 subgroups based on females and apoE−/− mice (Supplementary Table 4).
Discussion
To our knowledge, this study is the first meta-analysis of the effects of HDL/apoA-1 replacement therapies on atherosclerotic lesions. The pooled estimate in animal studies showed that HDL/apoA-1 replacement therapies were associated with a significant reduction in the final percent lesion area and final lesion area and a substantial decrease in the lesion area. However, we failed to find a significant association between HDL/apoA-1 replacement therapies and PAV or TAV in the pooled human results.
HDL plays several crucial roles in atherosclerotic prevention, including cholesterol efflux capacity, antioxidant activity, anti-inflammatory activity, cytoprotective activity, and vasodilatory activity (49). However, some abnormal conditions, including inflammatory conditions and metabolic diseases, cause HDL dysfunction by affecting its components, as described in the Introduction section (50–52). This change in HDL may underlie the failure to prevent atherosclerosis by increasing HDL production without considering its dysfunction. For example, increasing HDL by inhibiting cholesteryl ester transfer protein (CETP) did not reduce the rate of cardiovascular events despite inducing a 133.2% increase in HDL-C levels (53). One case reported a 52-year-old white female with an extremely high HDL level of 218 mg/dl who suffered from coronary artery disease (54). Therefore, improving HDL dysfunction may be more beneficial for atherosclerosis prevention than increasing its level.
Previously published animal studies suggest that HDL/apoA-1 replacement therapies (considered equal to normal-functioning HDL/apoA-1) have a significant beneficial effect on atherosclerosis prevention. Our pooled animal trials also revealed that HDL/apoA-1 replacement therapies had a significant preventive effect on the lesion area, which is consistent with our hypothesis that providing normal-functioning HDL/apoA-1 would be effective for atherosclerosis prevention. This strategy should also be effective in atherosclerosis prevention in humans. However, the cause of the failure of HDL/apoA-1 replacement therapies to provide beneficial effects on atherosclerosis in humans is not known. We summarized the possible reasons below.
1. HDL/apoA-1 replacement therapies may be more effective in preventing early-stage atherosclerotic lesions than altering mature lesions, which was demonstrated in animal trials (38, 44) because atherosclerosis is a chronic inflammatory process. Mohanta et al. proposed a new concept that the disruption of the balance between pro- and antiatherogenic immune cell subsets due to the exhaustion of anti-inflammatory and immune-suppressing cells during sustained inflammatory processes may trigger clinically overt atherosclerosis (55). Reconstituted discoidal HDL and apoA-1 inhibit inflammation in endothelial cells (56, 57). Lipid-free apoA-1 also inhibits macrophage activation via three main pathways (58–60). These studies suggest that earlier administration of HDL/apoA-1 replacement therapy would prevent the sustained inflammatory process that triggers a clinically overt atherosclerosis process, which is consistent with the discovery of Li et al. that D4F decreased the intraplaque lipid and macrophage content in evolving atherosclerosis (38). However, the balance between pro- and antiatherogenic immune cell subsets is already damaged in mature atherosclerotic lesions, which weakens the atheroprotective role of HDL/apoA-1 replacement therapy.
2. Does the administered HDL/apoA-1 replacement therapy maintain normal functioning in participants with a dysfunctional internal environment or after conversion into pro-inflammatory and pro-oxidant environments? The apoA-1 mimetic peptide D4F caused pre-β HDL formation, improved HDL-mediated cholesterol efflux, and reversed cholesterol transportation from macrophages in mice and rendered HDL less inflammatory in high-risk coronary heart disease patients (28, 61). However, two distinct clinical studies that included patients with coronary heart disease tested the impact of the apoA-1 mimetic peptide L-4F and failed to demonstrate its role in the HDL-inflammatory index and paraoxonase activity improvement, but it increased the high-sensitivity C-reactive protein levels, which was paradoxical to an ex vivo study (29). Therefore, the functional transformation of HDL/apoA-1 replacement therapy after administration to the body should be further analyzed to guide the prevention of atherosclerosis by increasing the “good” HDL.
3. Differences in lipid metabolism-relevant gene expression between humans and mice, including cholesteryl ester transfer protein, which is absent from the mouse genome but provides an avenue for the delivery of cholesterol from HDL to LDL in humans, may also be responsible for the different outcomes both in vivo and in vitro (mentioned above) after HDL/apoA-1 replacement therapy administration (62). Therefore, further human-specific analyses must be performed.
4. The duration and frequency of HDL/apoA-1 replacement therapy administration may also explain the different results in human and mouse trials (Table 1 and Supplementary Table 3). The human lifespan is much longer than that of mice, and the process of lesion development in humans is likely much more complicated. However, the duration and frequency of replacement therapy administration in some human studies were shorter than those in mouse studies. Therefore, the frequency and doses at the beginning of the replacement therapy administration period should be further investigated.
Further RCTs should investigate the preventive role of HDL/apoA-1 replacement therapies on atherosclerosis development. The participants would be healthy volunteers with atherosclerotic lesions in the early stage, and the duration and dose of the replacement therapy would be long and adequate.
However, before comparing the role of HDL/apoA-1 replacement therapy on atherosclerotic plaques between mice and humans, we must consider atherosclerosis progression in apoE−/− and LDLR−/− mice accompanied by expansive remodeling, which causes the absence of luminal narrowing that is different from humans, especially in ACS patients (63). However, mouse models have shown that apo A-I overexpression consistently reduces atherosclerosis progression independent of the genetic and metabolic context by increasing HDL cholesterol, which suggests their partial comparability (64). Therefore, animal trials may be a “lighthouse” for clinical trials, but we should not overestimate their guiding function.
Limitation
Our pooled analysis has several limitations. The methods used to evaluate vascular atherosclerosis were different in humans and mice (human: atheroma volume, mice: lesion area) because expansive remodeling of the vasculature accompanying atherosclerosis progression in mice caused the absence of luminal narrowing (63). The small number of studies in the human pooled analyses of efficacy is a limitation, and further investigations with earlier and longer administration times and increased doses should be performed. Comparisons of functional tests, including anti-inflammatory, paraoxonase activity, and RCTs, between humans and mice are absent because of data restrictions.
Conclusion
Evidence from a pooled analysis suggests that HDL/apoA-1 replacement therapies reduce the atherosclerotic lesion area in mice but do not show a beneficial effect on human atheroma volume despite being considered safe. Additional studies are needed to further investigate and explain the different effects of HDL/apoA1 replacement therapies in humans and mice.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
AA and CH: searched for articles, filtered the included articles, performed statistical analyses, and drafted the manuscript. HL: helped establish the inclusion and exclusion criteria, settled conflicts in literature inclusion, and evaluated the included studies. RS: performed statistical analyses and created the tables and figures. XL and XW: evaluated the included studies and performed the statistical analyses. XX and JH: searched for articles and selected included articles. JZ and JB: helped generate the figures and tables. YZ: designed the topic of the review, established the inclusion and exclusion criteria, and revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was funded by grants from the National Natural Science Foundation of China (Grant number: 81970388), the Natural Science Foundation of Guangdong Province (Grant number: 2019A1515011682), and the Yat-sen Scientific Research Launching Project (Grant number: YXQH201804).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.700233/full#supplementary-material
References
1. WHO Newsroom. Online Referencing. (2016). Available online at:https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed March 13, 2021).
2. Barter P, Gotto AM, LaRosa JC, Maroni J, Szarek M, Grundy SM, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. (2007) 357:1301–10. doi: 10.1056/NEJMoa064278
3. Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. (1975) 1:16–9. doi: 10.1016/s0140-6736(75)92376-4
4. Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, et al. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. (1977) 55:767–72. doi: 10.1161/01.cir.55.5.767
5. Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease_ the Framingham Study. Am J Med. (1977) 62:707–14. doi: 10.1016/0002-9343(77)90874-9
6. Agerholm-Larsen B, Nordestgaard BG, Steffensen R, Jensen G, Tybjaerg-Hansen A. Elevated HDL cholesterol is a risk factor for ischemic heart disease in white women when caused by a common mutation in the cholesteryl ester transfer protein gene. Circulation. (2000) 101:1907–12. doi: 10.1161/01.cir.101.16.1907
7. Andersen RV, Wittrup HH, Tybjærg-Hansen A, Steffensen R, Schnohr P, Nordestgaard BG. Hepatic lipase mutations, elevated high-density lipoprotein cholesterol, and increased risk of ischemic heart disease. J Am Coll Cardiol. (2003) 41:1972–82. doi: 10.1016/s0735-1097(03)00407-8
8. Madsen CM, Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J. (2017) 38:2478–86. doi: 10.1093/eurheartj/ehx163
9. Eren E, Yilmaz N, Aydin O. High density lipoprotein and it's dysfunction. Open Biochem J. (2012) 6:78–93. doi: 10.2174/1874091X01206010078
10. Parks JS, Rudel LL. Alteration of high density lipoprotein subfraction distribution with induction of serum amyloid A protein (SAA) in the nonhuman primate. J Lipid Res. (1985) 26:82–91.
11. Han CY, Tang C, Guevara ME, Wei H, Wietecha T, Shao B, et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J Clin Invest. (2016) 126:266–81. doi: 10.1172/JCI83475
12. Jaouad L, Milochevitch C, Khalil A. PON1 paraoxonase activity is reduced during HDL oxidation and is an indicator of HDL antioxidant capacity. Free Radic Res. (2003) 37:77–83. doi: 10.1080/1071576021000036614
13. Dong Z, Shi H, Zhao M, Zhang X, Huang W, Wang Y, et al. Loss of LCAT activity in the golden Syrian hamster elicits pro-atherogenic dyslipidemia and enhanced atherosclerosis. Metabolism. (2018) 83:245–55. doi: 10.1016/j.metabol.2018.03.003
14. Kontush A, Chapman MJ. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev. (2006) 58:342–74. doi: 10.1124/pr.58.3.1
15. Rosenson RS, Brewer HB Jr., Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. (2016) 13:48–60. doi: 10.1038/nrcardio.2015.124
16. Bindhu H, Rao VS, Kakkar VV. Friend turns foe: transformation of anti-inflammatory HDL to proinflammatory HDL during acute-phase response. Cholesterol. (2011) 2011:274629. doi: 10.1155/2011/274629
17. Nicholls S, Andrews J, Kastelein J, Merkely B, Nissen S, Ray K, et al. Effect of serial infusions of CER-001, a pre-β high-density lipoprotein mimetic, on coronary atherosclerosis in patients following acute coronary syndromes in the CER-001 atherosclerosis regression acute coronary syndrome trial: a randomized clinical trial. JAMA Cardiol. (2018) 3:815–22 doi: 10.1001/jamacardio.2018.2121
18. Tardif J, Ballantyne C, Barter P, Dasseux J, Fayad Z, Guertin M, et al. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J. (2014) 7:3277–86. doi: 10.1093/eurheartj/ehu171
19. Nicholls S, Puri R, Ballantyne C, Jukema J, Kastelein J, Koenig W, et al. Effect of infusion of high-density lipoprotein mimetic containing recombinant apolipoprotein A-I Milano on coronary disease in patients with an acute coronary syndrome in the MILANO-PILOT trial: a randomized clinical trial. JAMA Cardiol. (2018) 3:806–14. doi: 10.1001/jamacardio.2018.2112
20. Kallend DG, Reijers JA, Bellibas SE, Bobillier A, Kempen H, Burggraaf J, et al. A single infusion of MDCO-216 (ApoA-1 Milano/POPC) increases ABCA1-mediated cholesterol efflux and pre-beta 1 HDL in healthy volunteers and patients with stable coronary artery disease. Eur Heart J Cardiovasc Pharmacother. (2016) 2:23–9. doi: 10.1093/ehjcvp/pvv041
21. Gibson CM, Kerneis M, Yee MK, Daaboul Y, Korjian S, Mehr AP, et al. The CSL112-2001 trial: Safety and tolerability of multiple doses of CSL112 (apolipoprotein A-I [human]), an intravenous formulation of plasma-derived apolipoprotein A-I, among subjects with moderate renal impairment after acute myocardial infarction. Am Heart J. (2019) 208:81–90. doi: 10.1016/j.ahj.2018.11.008
22. Tricoci P, D'Andrea DM, Gurbel PA, Yao Z, Cuchel M, Winston B, et al. Infusion of reconstituted high-density lipoprotein, CSL112, in patients with atherosclerosis: safety and pharmacokinetic results from a phase 2a randomized clinical trial. J Am Heart Assoc. (2015) 4:e002171. doi: 10.1161/JAHA.115.002171
23. Easton R, Gille A, D'Andrea D, Davis R, Wright SD, Shear C. A multiple ascending dose study of CSL112, an infused formulation of ApoA-I. J Clin Pharmacol. (2014) 54:301–10. doi: 10.1002/jcph.194
24. Tortorici MA, Duffy D, Evans R, Feaster J, Gille A, Mant TGK, et al. Pharmacokinetics and safety of CSL112 (Apolipoprotein A-I [Human]) in adults with moderate renal impairment and normal renal function. Clin Pharmacol Drug Dev. (2019) 8:628–36. doi: 10.1002/cpdd.618
25. Tardif J-C, Grégoire J, L'Allier PL, Ibrahim R, Lespérance J, Heinonen TM, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis. A randomized controlled trial. JAMA. (2007) 297:1675–82. doi: 10.1001/jama.297.15.jpc70004
26. Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. (2003) 290:2292–300. doi: 10.1001/jama.290.17.2292
27. Waksman R, Torguson R, Kent KM, Pichard AD, Suddath WO, Satler LF, et al. A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. (2010) 55:2727–35. doi: 10.1016/j.jacc.2009.12.067
28. Dunbar RL, Movva R, Bloedon LT, Duffy D, Norris RB, Navab M, et al. Oral apolipoprotein A-I mimetic D-4F lowers HDL-inflammatory index in high-risk patients: a first-in-human multiple-dose, randomized controlled trial. Clin Transl Sci. (2017) 10:455–69. doi: 10.1111/cts.12487
29. Watson CE, Weissbach N, Kjems L, Ayalasomayajula S, Zhang Y, Chang I, et al. Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J Lipid Res. (2011) 52:361–73. doi: 10.1194/jlr.M011098
30. Bloedon LT, Dunbar R, Duffy D, Pinell-Salles P, Norris R, DeGroot BJ, et al. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res. (2008) 49:1344–52. doi: 10.1194/jlr.P800003-JLR200
31. Michael GC, Korjian S, Tricoci P, Daaboul Y, Yee M, Jain P, et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I, after acute myocardial infarction: the AEGIS-I trial (ApoA-I event reducing in ischemic syndromes I). Circulation. (2016) 134:1918–30. doi: 10.1161/CIRCULATIONAHA.116.025687
32. Amar MJA, D'Souza W, Turner S, Demosky S, Sviridov D, Stonik J, et al. 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J Pharmacol Exp Ther. (2010). 334:634–41. doi: 10.1124/jpet.110.167890
33. Averill MM, Kim EJ, Goodspeed L, Wang S, Subramanian S, Den Hartigh LJ, et al. The apolipoprotein-AI mimetic peptide L4F at a modest dose does not attenuate weight gain, inflammation, or atherosclerosis in LDLR-null mice. PLoS ONE. (2014) 9:e109252. doi: 10.1371/journal.pone.0109252
34. Bielicki JK, Zhang H, Cortez Y, Zheng Y, Narayanaswami V, Patel A, et al. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J Lipid Res. (2010) 51:1496–503. doi: 10.1194/jlr.M003665
35. Bodary PF, Shen YC, Westrick RJ, Lalwani ND, Drake SL, Dasseux JLH, et al. Gene transfer of an ApoA-I mimetic peptide reduces atherosclerosis in mice. J Am Coll Cardiol. (2000) 36:959–69. doi: 10.1016/s0735-1097(04)91969-9
36. Ditiatkovski M, D'Souza W, Kesani R, Chin-Dusting J, de Haan JB, Remaley A, et al. An apolipoprotein A-I mimetic peptide designed with a reductionist approach stimulates reverse cholesterol transport and reduces atherosclerosis in mice. PLoS ONE. (2013) 8:e68802. doi: 10.1371/journal.pone.0068802
37. Guo Y, Yuan W, Yu B, Kuai R, Hu W, Morin EE, et al. Synthetic high-density lipoprotein-mediated targeted delivery of liver X receptors agonist promotes atherosclerosis regression. EBioMedicine. (2018) 28:225–33. doi: 10.1016/j.ebiom.2017.12.021
38. Li X, Chyu KY, Neto JRF, Yano J, Nathwani N, Ferreira C, et al. Differential effects of apolipoprotein A-I-mimetic peptide on evolving and established atherosclerosis in apolipoprotein E-null mice. Circulation. (2004) 110:1701–5. doi: 10.1161/01.CIR.0000142857.79401.69
39. Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, et al. Oral administration of an apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. (2002) 105:290–2. doi: 10.1161/hc0302.103711
40. Nayyar G, Mishra VK, Handattu SP, Palgunachari MN, Shin R, McPherson DT, et al. Sidedness of interfacial arginine residues and anti-atherogenicity of apolipoprotein A-I mimetic peptides. J Lipid Res. (2012) 53:849–58. doi: 10.1194/jlr.M019844
41. Ou ZJ, Li L, Liao XL, Wang YM, Hu XX, Zhang QL, et al. Apolipoprotein A-I mimetic peptide inhibits atherosclerosis by altering plasma metabolites in hypercholesterolemia. Am J Physiol Endocrinol Metab. (2012) 303:E683–94. doi: 10.1152/ajpendo.00136.2012
42. Qin S, Kamanna VS, Lai JH, Liu T, Ganji SH, Zhang L, et al. Reverse D4F, an apolipoprotein-AI mimetic peptide, inhibits atherosclerosis in ApoE-null mice. J Cardiovasc Pharmacol Ther. (2012) 17:334–43. doi: 10.1177/1074248411434598
43. Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, Rader DJ. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. (1999) 100:1816–22. doi: 10.1161/01.Cir.100.17.1816
44. Wool GD, Cabana VG, Lukens J, Shaw PX, Binder CJ, Witztum JL, et al. 4F Peptide reduces nascent atherosclerosis and induces natural antibody production in apolipoprotein E-null mice. FASEB J. (2011). 25:290–300. doi: 10.1096/fj.10-165670
45. Graversen JH, Laurberg JM, Andersen MH, Falk E, Nieland J, Christensen J, et al. Trimerization of apolipoprotein A-I retards plasma clearance and preserves antiatherosclerotic properties. J Cardiovasc Pharmacol. (2008) 51:170–7. doi: 10.1097/FJC.0b013e31815ed0b9
46. Zhang X, Zhu X, Chen B. Inhibition of collar-induced carotid atherosclerosis by recombinant apoA-I cysteine mutants in apoE-deficient mice. J Lipid Res. (2010) 51:3434–42. doi: 10.1194/jlr.M008573
47. Ying R, Yuan Y, Qin YF, Tian D, Feng L, Guo ZG, et al. The combination of L-4F and simvastatin stimulate cholesterol efflux and related proteins expressions to reduce atherosclerotic lesions in apoE knockout mice. Lipids Health Dis. (2013) 12:180. doi: 10.1186/1476-511X-12-180
48. Suematsu Y, Kawachi E, Idemoto Y, Matsuo Y, Kuwano T, Kitajima K, et al. Anti-atherosclerotic effects of an improved apolipoprotein A-I mimetic peptide. Int J Cardiol. (2019) 297:111–7. doi: 10.1016/j.ijcard.2019.08.043
49. Camont L, Chapman MJ, Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol Med. (2011) 17:594–603. doi: 10.1016/j.molmed.2011.05.013
50. Jakob P, Luscher TF. Dysfunctional HDL and inflammation: a noxious liaison in adolescents with type 1 diabetes. Eur Heart J. (2019) 40:3567–70. doi: 10.1093/eurheartj/ehz502
51. Van Lenten BJWA, Nayak DP, Hama S, Navab M, Fogelman AM. High-density lipoprotein loses its anti-inflammatory properties during acute influenza A infection. Circulation. (2001) 103:2283–8. doi: 10.1161/01.cir.103.18.2283
52. de Souza JA, Vindis C, Hansel B, Negre-Salvayre A, Therond P, Serrano CV, et al. Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis. (2008) 197:84–94. doi: 10.1016/j.atherosclerosis.2007.08.009
53. Fernandez-Ruiz I. Dyslipidaemia: is CETP inhibition a viable therapeutic strategy? Nat Rev Cardiol. (2017) 14:382–3. doi: 10.1038/nrcardio.2017.87
54. Sawalha K, Kamoga GR. Extremely high high-density lipoprotein cholesterol with coronary artery disease: case report. Clin Case Rep. (2021) 9:e04092. doi: 10.1002/ccr3.4092
55. Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, et al. Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res. (2014) 114:1772–87. doi: 10.1161/CIRCRESAHA.114.301137
56. Pan B, Kong J, Jin J, Kong J, He Y, Dong S, et al. A novel anti-inflammatory mechanism of high density lipoprotein through up-regulating annexin A1 in vascular endothelial cells. Biochim Biophys Acta. (2016) 1861:501–12. doi: 10.1016/j.bbalip.2016.03.022
57. Theofilatos D, Fotakis P, Valanti E, Sanoudou D, Zannis V, Kardassis D. HDL-apoA-I induces the expression of angiopoietin like 4 (ANGPTL4) in endothelial cells via a PI3K/AKT/FOXO1 signaling pathway. Metabolism. (2018) 87:36–47. doi: 10.1016/j.metabol.2018.06.002
58. Tang C, Houston BA, Storey C, LeBoeuf RC. Both STAT3 activation and cholesterol efflux contribute to the anti-inflammatory effect of apoA-I/ABCA1 interaction in macrophages. J Lipid Res. (2016) 57:848–57. doi: 10.1194/jlr.M065797
59. Yvan-Charvet L, Matsuura F, Wang N, Bamberger MJ, Nguyen T, Rinninger F, et al. Inhibition of cholesteryl ester transfer protein by torcetrapib modestly increases macrophage cholesterol efflux to HDL. Arterioscler Thromb Vasc Biol. (2007) 27:1132–8. doi: 10.1161/ATVBAHA.106.138347
60. Zhang M, Zhao GJ, Yin K, Xia XD, Gong D, Zhao ZW, et al. Apolipoprotein A-1 binding protein inhibits inflammatory signaling pathways by binding to apolipoprotein A-1 in THP-1 macrophages. Circ J. (2018) 82:1396–404. doi: 10.1253/circj.CJ-17-0877
61. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, et al. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. (2004) 109:3215–20. doi: 10.1161/01.CIR.0000134275.90823.87
62. Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem. (2002) 277:3801–4. doi: 10.1074/jbc.R100057200
63. Van Craeyveld E, Gordts SC, Singh N, Jacobs F, De Geest B. A critical reassessment of murine and rabbit models of atherosclerosis: focus on lesion progression and remodelling. Acta Cardiol. (2012) 67:11–21. doi: 10.1080/ac.67.1.2146561
Keywords: lipoproteins, HDL, apolipoprotein A-I, mimetic, atherosclerosis
Citation: Abudukeremu A, Huang C, Li H, Sun R, Liu X, Wu X, Xie X, Huang J, Zhang J, Bao J and Zhang Y (2021) Efficacy and Safety of High-Density Lipoprotein/Apolipoprotein A1 Replacement Therapy in Humans and Mice With Atherosclerosis: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 8:700233. doi: 10.3389/fcvm.2021.700233
Received: 25 April 2021; Accepted: 28 June 2021;
Published: 02 August 2021.
Edited by:
Tetsuro Miyazaki, Juntendo University Urayasu Hospital, JapanReviewed by:
Mehmet Ozaydin, Akdeniz Sifa Hastanesi, TurkeyDan Rudic, Augusta University, United States
Copyright © 2021 Abudukeremu, Huang, Li, Sun, Liu, Wu, Xie, Huang, Zhang, Bao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuling Zhang, emh5dWxAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work