
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 04 June 2021
Sec. Lipids in Cardiovascular Disease
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.696413
This article is part of the Research TopicHighlights in Lipids in Cardiovascular Disease: 2021View all 6 articles
 Joseph Adu-Amankwaah1
Joseph Adu-Amankwaah1 Gabriel Komla Adzika1
Gabriel Komla Adzika1 Adebayo Oluwafemi Adekunle1
Adebayo Oluwafemi Adekunle1 Marie Louise Ndzie Noah1
Marie Louise Ndzie Noah1 Richard Mprah1
Richard Mprah1 Aisha Bushi2
Aisha Bushi2 Nazma Akhter1
Nazma Akhter1 Yaxin Xu1
Yaxin Xu1 Fei Huang1
Fei Huang1 Benard Chatambarara2
Benard Chatambarara2 Hong Sun1*
Hong Sun1*Due to its reversible nature, Takotsubo cardiomyopathy (TTC) is considered an intriguing and fascinating cardiovascular disease characterized by a transient wall motion abnormality of the left ventricle, affecting more than one coronary artery territory, often in a circumferential apical distribution. Takotsubo cardiomyopathy was discovered by a Japanese cardiovascular expert and classified as acquired primary cardiomyopathy by the American Heart Association (AHA) in 1990 and 2006, respectively. Regardless of the extensive research efforts, its pathophysiology is still unclear; therefore, there are no well-established guidelines specifically for treating and managing TTC patients. Increasing evidence suggests that sympatho-adrenergic stimulation is strongly associated with the pathogenesis of this disease. Under acute stressful conditions, the hyperstimulation of beta-adrenergic receptors (β-ARs) resulting from excessive release of catecholamines induces intracellular kinases capable of phosphorylating and activating “A Disintegrin and Metalloprotease 17” (ADAM17), a type-I transmembrane protease that plays a central role in acute myocardial inflammation and metabolic lipids dysregulation which are the main hallmarks of TTC. However, our understanding of this is limited; hence this concise review provides a comprehensive insight into the key role of ADAM17 in acute myocardial inflammation and metabolic lipids dysregulation during acute stress. Also, how the synergy of ADAM17-induced acute inflammation and lipids dysregulation causes TTC is explained. Finally, potential therapeutic targets for TTC are also discussed.
Takotsubo cardiomyopathy (TTC) is an acute, stress-induced cardiac syndrome characterized by a transient wall motion abnormality of the left ventricle, affecting more than one coronary artery territory, often in a circumferential apical distribution (1). This condition is also known as; stress cardiomyopathy, ampulla cardiomyopathy, stress-induced cardiomyopathy, apical ballooning cardiomyopathy, transient left ventricular dysfunction, Gebrochenes-Herz-syndrome, and broken heart syndrome (2). Takotsubo cardiomyopathy was first described in 1990 by a Japanese cardiovascular expert (3). In 2006, it was classified as acquired primary cardiomyopathy by the American Heart Association (AHA) (4). It is initiated by extreme physical or emotional stress and can occur in females and males at any age; however, postmenopausal females are commonly affected. Takotsubo cardiomyopathy can also be caused by infections, such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (5, 6). The diagnosis of this condition is generally based on clinical criteria combined with a multi-modality imaging approach which includes coronary angiography (with left ventriculography), electrocardiography (ECG), cardiac magnetic resonance imaging (CMR), and transthoracic echocardiography (TTE) (7). Takotsubo cardiomyopathy is usually reversible within a few weeks; hence, its prognosis was initially considered favorable (8). Nevertheless, research suggests that the long-term prognosis of TTC is poorer than anticipated (8) since it accounts for substantial morbidity and mortality worldwide (9). Regardless of the extensive research efforts, its pathophysiology is still not clear (8). Due to this, there are no well-established guidelines specifically for treating and managing TTC patients. It is, therefore, vital to understand the pathomechanisms that enhance the development of TTC.
Sympatho-adrenergic stimulation is strongly associated with the pathogenesis of TTC (10, 11). Indisputably, the histological alterations in the myocardium during TTC are comparable to those found in catecholamine-induced cardiotoxicity in humans and animals (12). Current studies have also revealed that acute myocardial inflammation and metabolic lipids dysregulation are implicated in the pathogenesis of TTC (9, 13). However, how sympatho-adrenergic stimulation leads to TTC characterized by the latter is still unclear.
Intriguingly, “A Disintegrin and Metalloprotease 17” (ADAM17) forming part of the 560 proteases encoded in the human genome has recently emerged as a key regulator of inflammation and lipids dysregulation following stressful conditions. “A Disintegrin and Metalloprotease 17” is ubiquitously expressed in human tissues, including the brain, heart, kidney, and skeletal muscle (14, 15). Although ADAM17's expression is downregulated in physiological states, several studies have demonstrated its upregulation along with its substrates [tumor necrosis factor-alpha (TNFα) and soluble interleukin-6 receptor (sIL-6R)] in dilated cardiomyopathies (16–19). Hence, this indicates ADAM17's essential role in the etiology of TTC. Besides ADAM17 inducing cardiomyopathies via exacerbating inflammatory responses and lipids dysregulation, its variants and mutants have been associated with mild cardiomyopathies and congenital heart defects, including Tetralogy of Fallot (20, 21).
In acute stress state, the hyperstimulation of beta-adrenergic receptors (β-ARs) due to excessive release of catecholamines from sympathetic responses exert negative inotropic and chronotropic effects on the heart (11), thereby increasing the activities of intracellular kinases such as receptor-stimulated p38 mitogen-activated-protein-kinases (p38 MAPKs) and extracellular signal-regulated kinases (ERKs) (22, 23). These kinases are well-known activators of ADAM17 via their intracellular phosphorylation (24). Hyperactive ADAM17 plays a crucial role in acute cardiac inflammation (17) and myocardial lipids dysregulation (9), which successfully results in left ventricular abnormalities characterizing TTC. However, our understanding of this is limited; hence this concise review provides a comprehensive insight into the key role of ADAM17 in acute myocardial inflammation and metabolic lipids dysregulation during acute stress. Also, how the synergy of ADAM17-induced acute inflammation and lipids dysregulation causes TTC is explained. Finally, potential therapeutic targets for TTC are discussed.
Acute stress has been recognized as a modifiable risk factor for several cardiomyopathies (25). The impact of acute stress on physiological and psychological processes is determined by the stress stimulus's characteristics, either emotional or physical. However, in both, the autonomic nervous system is one of the central neural pathways activated (26). Thus, in an acute stress state, the sympathetic system's hyperstimulation results in elevated catecholamines (epinephrine and norepinephrine). Physiologically, epinephrine and norepinephrine serve as neurotransmitters and hormones necessary for homeostasis maintenance; however, excessive increase in their levels leads to the hyperactivation of β-ARs, which are the main receptors mediating the inotropic and chronotropic function of the heart (9, 23). Beta-adrenergic receptors are 7-transmembrane, G-protein coupled receptors (GPCRs) which exist in three subtypes, namely; β1-AR, β2-AR, β3-AR (27, 28). All are widely expressed in the heart, with β1-AR having the highest expression and β3-AR with the least expression (23, 27). β1-AR can only signal through Gαs when activated while β2-AR and β3-AR can signal through Gαs or Gαi upon activation depending on the condition (physiological or pathological) (23, 29). Physiologically, upon activation, β2-AR signals through Gαs (23) while β3-AR signals through Gαi (29).
Hyperstimulation of β-ARs under acute stressful conditions, characterized by an excessive increase in catecholamines, causes desensitization of β1-AR and hyperactivation of β2-AR coupling to Gαi. This occurs because, among the three β-AR subtypes, β2-AR and β3-AR are rarely depleted during acute stress (30, 31). To prevent cardiac injury caused by acute stress, Gαi signaling via the non-canonical pathway increases the activities of intracellular kinases such as p38 MAPKs and ERKs (22, 23). Interestingly, studies have revealed that p38 MAPKs and ERKs can positively influence the activation of ADAM17 via their intracellular phosphorylation, either directly or indirectly, through the activation of iRhoms (32, 33). Physiologically, ADAM17's expression can be regulated by transcriptional factors such as nuclear factor kappa B (NF-kB) and ETS Like-1 (Elk-1) (34). However, post-transcriptional mechanisms such as chromatin remodeling protein BRG1 affect its expression. Also, subcellular localization in the perinuclear region has been shown to regulate the activities of ADAM17 (35). Nonetheless, phosphorylation of threonine 735 in ADAM17 by active kinases can rapidly activate its cleavage activity, possibly by triggering and facilitating its translocation from the Golgi network to the cell surface where its proteolytic activity is reported (36, 37). Structurally, ADAM17 comprises a prodomain, a catalytic domain, a disintegrin-like domain, a membrane-proximal domain (MPD), and a short stalk region, which together form the extracellular part of the protease and are connected to an intracellular region by a transmembrane part (38). The stalk region of this protease contains the CANDIS motif (Conserved ADAM 17 Dynamic Interaction Sequence), which is found closer to the MPD around the plasma membrane and is vital for substrate recognition. (39). Intracellular phosphorylation of ADAM17 induces the phosphatidylserine exposure at the outer leaflet of the cell membrane, causing it to bind to the membrane through its MPD and CANDIS, thereby initiating its activation and cleaving process (40). Upon activation, ADAM17 cleaves and stimulates pro-inflammatory cytokines and their cognate receptors (14, 15, 41, 42), resulting in acute inflammation and metabolic lipids dysregulation in the heart (Figure 1).
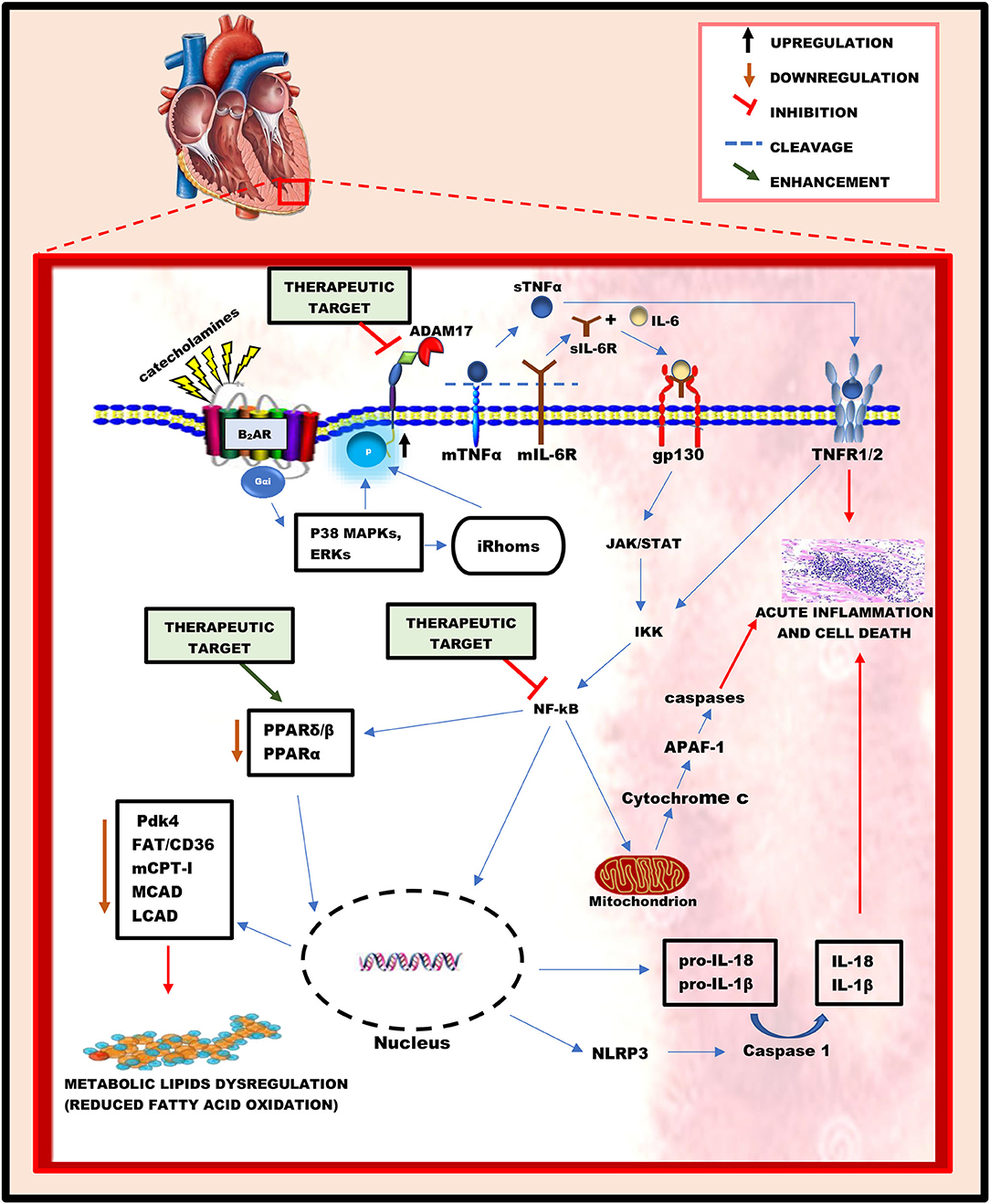
Figure 1. A schematic diagram illustrating the key roles played by ADAM17 and its substrates in inducing acute myocardial inflammation and metabolic lipids dysregulation during acute stressful conditions. In an acute stress state, the hyperactivation of beta-adrenergic receptors resulting from the excessive release of catecholamines influences the activation of ADAM17 via intracellular phosphorylation by ERKs and p38 MAPKs. Hyperactive ADAM17 cleaves and activates TNFα and IL-6R to trigger cell signaling cascades, leading to NF-kB activation. Active NF-kB successfully causes acute myocardial inflammation via the gene elevation of pro-inflammatory cytokines (IL-18, IL-1β) and inflammasomes (NLRP3) in the nucleus, as well as the release of cytochrome c from the mitochondria. Additionally, the activation of NF-kB causes a reduction in the activity of PPARβ/δ and PPARα, thereby leading to a decreased mRNA and protein levels of key regulatory enzymes of fatty acid oxidation, which characterizes metabolic lipids dysregulation. Hence, the proposed therapeutic targets for the attenuation of acute myocardial inflammation and metabolic lipids dysregulation may include; the inhibition of ADAM17 and NF-kB's activation and enhancing the activities of PPARβ/δ and PPARα.
“A Disintegrin and Metalloprotease 17” has emerged as a chief regulatory hub in inflammation due to cleavage and activation of several pro-inflammatory cytokines and their cognate receptors. The most noticeable examples include; TNFα, tumor necrosis receptor 1 and 2 (TNFR1 and 2), and IL-6R (14, 15, 41, 42). These cytokines are widely expressed in the heart as membrane-bound proteins (43). Excessive increase in their soluble forms following the cleavage process of ADAM17 can trigger cascades resulting in acute cardiac inflammation, myocardial lipotoxicity, and low energy production.
Soluble TNFα (sTNFα), when released under acute stressful conditions, can signal via TNFR1 or TNFR2 (44, 45), which are predominantly expressed on myocytes (44). Interestingly, soluble TNFRs (sTNFR1/2) resulting from ADAM17's cleavage process can also bind to membrane TNFα (mTNFα) and act as antagonistic decoy receptors known as “reverse signaling,” which is seen chiefly among other TNF family members (46, 47). Interleukin-6 (IL-6) is a pleiotropic cytokine with distinct pro-and anti-inflammatory properties when released under stressful conditions. It can signal via two different ways, namely; (1) Signaling through its membrane-bound receptor, IL-6R, which is known as “classic signaling” and can only occur on cell types expressing surface IL-6R, including hepatocytes and certain leukocytes' subpopulations (48). (2) Signaling via the soluble form of its receptor, sIL-6R, which is termed as “IL-6 trans-signaling” and can occur on all body cells, including cardiomyocytes, since the IL-6/sIL-6R complex can directly bind to and activate the ubiquitously expressed glycoprotein-130 (gp130) without the need of a membrane-bound IL-6R (43, 48) (Figure 1).
The binding of sTNFα and IL-6/sIL-6R complex to TNFR1/2 and gp130, respectively, can trigger intracellular signaling cascades leading to acute myocardial inflammation (49, 50) and metabolic lipids dysregulation (9, 51, 52). The intracellular region of TNFR1 contains a death domain that can directly induce apoptosis and inflammation when activated (43). However, this domain is absent in the intracellular region of TNFR2. The activation of TNFR2 by sTNFα mediates the phosphorylation of the p65 subunit of NF-Kb at ser536 via interaction with the IkB kinase (IKK), subsequently resulting in the activation of NF-kB dimer (52, 53). Similarly, the binding of the IL-6/sIL-6R complex to gp130 is capable of inducing phosphorylation and activation of NF-kB dimer via gp130/JAK/STAT pathway (49). Following activation, NF-kB migrates into the mitochondria (54, 55) and nucleus (56) to induce signaling cascades and up/down-regulate genes, respectively. According to Liu et al., NF-kB can stimulate the intrinsic apoptotic pathway in the mitochondria via the release of cytochrome c (50). In the cytoplasm, cytochrome c binds to apoptotic protease activating factor 1 (APAF-1), causing it to undergo conformational changes and oligomerization into a heptameric wheel-like structure known as apoptosome, which recruits and activates the initiator caspase-9 (57). Active caspase-9 cleaves and activates the executioner caspases-3 and −7, resulting in rapid apoptosis and inflammation in cardiac cells (50, 57). Additionally, in the nucleus, active NF-kB upregulates genes of pro-inflammatory cytokines (pro-IL-18 and pro-IL-1β) and NLR family pyrin domain containing 3 (NLRP3), hence elevating their protein levels. NLRP3 is an intracellular sensor that can be triggered under acute stressful conditions, resulting in the formation and activation of NLRP3 inflammasome (58, 59). Active NLRP3 inflammasome is known to activate caspase 1, which in turn cleaves pro-IL-1β and pro- IL-18 to release their soluble forms, successively inducing necrosis and inflammation in cardiac cells (60) (Figure 1).
Interestingly, studies have also revealed that the activation of NF-kB downregulates the activity of peroxisome proliferator-activated receptor (PPAR)β/δ, α (51, 52) and genes regulating fatty acid (FA) oxidation in the heart (52). Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptors that serve as transcription factors regulating the expression of metabolic genes within cells (61). PPARs comprises three subtypes, namely; PPARα, PPARγ, and PPARβ/δ (61). Fatty acids are well-known endogenous ligands of the PPAR family (62). PPARβ/δ and PPARα are widely expressed on cardiac cells, which serve as transcriptional regulators of myocardial energy and lipid homeostasis (51). According to Planavila et al., activation of NF-kB caused a reduction in the expression of pyruvate dehydrogenase kinase 4 (Pdk4), a target gene of PPARβ/δ, which plays a vital role in fatty acid utilization and palmitate oxidation. The reduction in the activity of PPARβ/δ was proposed to be caused by the physical interaction between the p65 subunit of NF-kB and PPAR β/δ during the phosphorylation and activation of NF-kB (51). Also, a study carried out by Pellieux et al., revealed that the activation of NF-kB is associated with the downregulation of PPARα's activity (52). Physiologically, the activation of PPARα upregulates the mRNA and proteins levels of key regulatory enzymes of fatty acid oxidation, hence reduction in its activity due to the activation of NF-kB decreases the levels of these key enzymes, which includes; fatty acid translocase/cluster of differentiation 36 (FAT/CD36), carnitine palmitoyltransferase I (mCPT-I), medium-chain acyl-CoA dehydrogenase (MCAD), and long-chain-acyl-CoA dehydrogenase (LCAD) (52, 63, 64). Fatty acid enters the cell via FA transporters on the cell membrane, including FAT/CD36 (65). Within the cell, the conversion of long-chain fatty acyl CoA to an acylcarnitine required for entry into the mitochondria is carried out by mCPT-I (62). In the mitochondria, MCAD and LCAD play vital roles in converting FA into energy during β-oxidation (62, 66). Thus, these enzymes play crucial roles in the uptake and utilization of FA. In short, activation of NF-kB subsequently causes a reduction in FA oxidation via downregulation of FA oxidation pathways. The decrease of FA oxidation in the heart is accompanied by intramyocardial lipid accumulation and reduced myocardial energy, characterizing myocardial lipids dysregulation (9) (Figure 1).
Studies have shown that acute myocardial inflammation (13) and lipotoxicity (9) are implicated in the pathogenesis of TTC. It is well-known that cytokines released during inflammation exert detrimental effects on the heart. Thus, cytokines like TNF-α, IL-6, and IL-1ß cause cardiac necrosis and apoptosis (60) and downregulate the expression of calcium (Ca2+)-regulating genes, including sarcoplasmic reticulum Ca2+ ATPase (67) and Ca2+-release channel (68), resulting in a direct negative inotropic effect on the heart (69, 70). It is well-established that Ca2+ ions are responsible for the electrical activation and mechanical contraction of the the myocardia (71), hence reduction in intracellular Ca2+ coupling with cell death following acute inflammation can lead to abnormalities in the contraction of the left ventricle. Also, ADAM17-induced lipid dysregulation under acute stress state is accompanied by intramyocardial lipid accumulation and reduced myocardial production of adenosine triphosphate (ATP) (9). Excessive accumulation of lipids in the myocardia increases levels of toxic intermediates leading to lipotoxicity (72). Low ATP production and lipotoxicity may affect left ventricular function via abnormal cardiac contraction development (72). In a nutshell, acute myocardial inflammation coupling with lipids dysregulation triggered by active ADAM17 under acute stressful events may be the main inducers of the transient wall motion abnormalities of the left ventricle characterizing TTC, which is usually reversible after few days or may progress to heart failure (Figure 2).
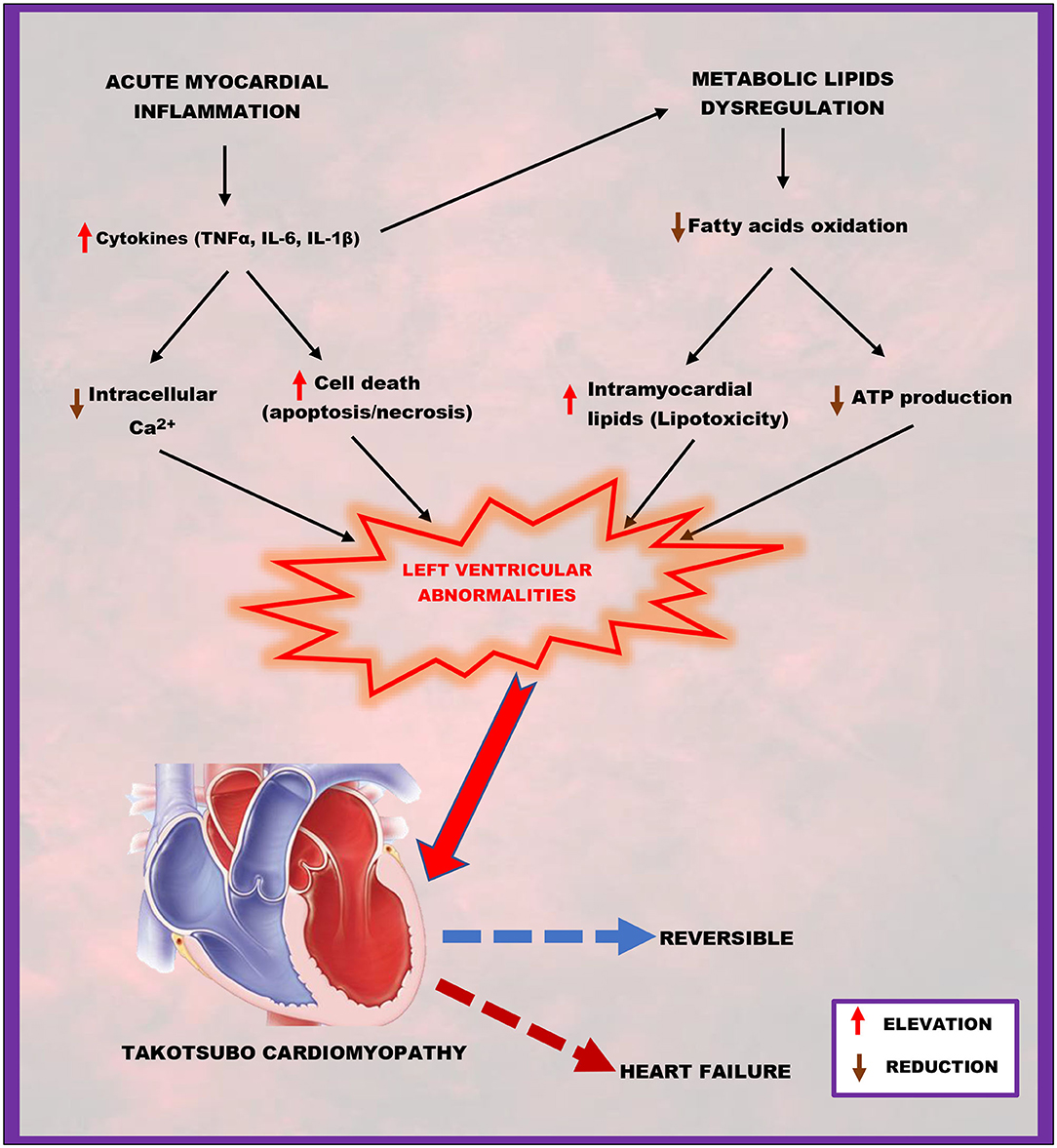
Figure 2. A schematic representation of how acute myocardial inflammation, in combination with metabolic lipid dysregulation, contributes to takotsubo cardiomyopathy. Elevated cytokines (TNFα, IL-6, IL-1β) and reduced fatty acid oxidation resulting from ADAM17-induced acute myocardial inflammation and metabolic lipids dysregulation successfully lead to left ventricular abnormalities characterized by reduced intracellular calcium and adenosine triphosphate (ATP) production; increased cell death (apoptosis/necrosis) and intramyocardial lipids accumulation (lipotoxicity), which are the main hallmarks of takotsubo cardiomyopathy.
As yet, there are no well-established therapeutic guidelines specifically for treating TTC patients (73); however, based on the broad knowledge gained from this concise review, potential therapeutic targets for the treatment and management of TTC might lie in the direct inhibition of ADAM17 and NF-kB or indirectly antagonizing their activation and activities. The most promising therapeutic inhibitors of ADAM17 includes tissue inhibitor of metalloproteinase 3 (TIMP3), protein disulfide isomerases (PDIs), and integrins (43). Remarkably, direct injection of TIMP3 in the heart has been shown to downregulate ADAM17's activity by binding to its catalytic domain, thereby inactivating it (74, 75). Also, PDIs can directly interact with the MPD of ADAM17, where it catalyzes the isomerization of two disulfide bridges, thus downregulating ADAM17's activity (76, 77). A study conducted by Bax et al., revealed that the binding of intergrin α5β1 to ADAM17 via its disintegrin domain, downregulated its activity by affecting its mediated cell adhesion and migration (78). Aside from its natural inhibitors, a variety of miRNAs, including miR-145 (79), miR-124 (80), miR-152 (81), and miR-326 (82), have been proven to downregulate ADAM17's expression and reduce its substrate release by directly binding to the ADAM17 3′-UTR. Regarding NF-kB, there are several approaches to inhibiting its transduction pathway, including receptor inhibition, adaptor inhibition, IKK inhibition, IkB stabilization, cytoplasmic retention, and transcription factor inhibition (83). NF-kB inhibitors have been grouped into three categories: biomolecular inhibitors, synthetic chemicals, and natural products (and their derivatives) (83). Biomolecular inhibitors include siRNAs, decoy oligonucleotides (containing the kB site), ribozymes, the IkB super-repressor, interfering peptides, and dominant-negative molecules (83). Most synthetic chemicals are molecules engineered against components of the IKK complex, which plays a central role in NF-kB activation (84, 85). Natural products include a wide range of marine-, plant-, and microbe-derived compounds that target different steps in the NF-kB pathway (86–88). These natural products are categorized into three main groups; IKK inhibitors, antioxidants, and thiol-reactive compounds that can target several stages of the NF-kB signaling pathway. Among 19 drugs reported in previous studies, digitoxin, ectinascidin 743, ouabain, chromomycin A3, and bortezomib were the most potent NF-kB inhibitors (89). The inhibition of ADAM17 and NF-kB is key in attenuating acute myocardial inflammation and lipids dysregulation, which are the hallmarks of TTC (Figure 1).
Additionally, the usage of pharmacologic agents capable of enhancing the activities of PPARβ/δ and PPARα might be a promising target for the treatment and management of TTC by preventing the reduction of key regulatory proteins of FA oxidation via two different mechanisms (52). The first mechanism involves direct activation of the transcription of target genes by binding to peroxisome proliferator responsive elements in the promoter region (52). The second mechanism is a direct interaction with transcription factors involved in the hypertrophic response, including NF-kB and activator protein 1 (AP-1), which may, directly or indirectly, contribute to reducing the proteins of FA oxidation (90). Studies suggest that the activity of PPARα can be enhanced by fibrate hypolipidemic drugs (91, 92). However, there are currently no commercially available drugs capable of enhancing the function of PPARβ/δ (92). The maintenance of optimal levels of key regulatory proteins of FA oxidation is vital in ameliorating cardiac function via the attenuation of lipotoxicity and increasing myocardial ATP production (Figure 1).
Although TTC is considered a unique and interesting cardiomyopathy due to its reversible nature, it is still underappreciated among clinicians and researchers. Several promising pathophysiologic theories have been suggested, but this condition's exact underlying mechanistic processes remain unclear. This concise review has provided some general insights into this disease's pathogenesis; however, considering the prevalence in postmenopausal women, TTC could be linked to the modulation of acute myocardial inflammation and metabolic lipids dysregulation by sex hormones and the endocrine system at large. Thus, much remains to be discovered about TTC. Hence many areas require further exploration to understand this multifaceted cardiomyopathy perfectly.
The review idea was conceived by JA-A. JA-A drafted and wrote the manuscript with the supervision of HS. JA-A, GA, AA, MN, RM, AB, NA, FH, YX, and BC revised and proofread the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grant nos. 81461138036 and 81370329), The Natural Science Foundation of the Jiangsu Higher Education Institutes of China (grant no. 17KJB180016) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the help of Prof. Festus Adzaku, Dr. Innocent Afeke, and Miss Mary Nyarko for proofreading the entire manuscript.
1. Akashi YJ, Nef HM, Lyon AR. Epidemiology and pathophysiology of Takotsubo syndrome. Nat Rev Cardiol. (2015) 12:387–97. doi: 10.1038/nrcardio.2015.39
2. Rathish D, Karalliyadda M. Takotsubo syndrome in patients with myasthenia gravis: a systematic review of previously reported cases. BMC Neurol. (2019) 19:281. doi: 10.1186/s12883-019-1523-z
3. Sato TH, Uchida T, Dote K, Ishihara M. Tako-tsubo-like left ventricular dysfunction due to multivessel coronary spasm, In: Kodama K, Haze K, Hori M, editors. Clinical Aspect of Myocardial Injury: From Ischemia to Heart Failure. Tokyo: Kagakuhyoronsha Publishing Co. (1990). p. 56–64.
4. Maron BJ. The 2006 American Heart Association classification of cardiomyopathies is the gold standard. Circ Heart Fail. (2008) 1:72–5; discussion 76. doi: 10.1161/CIRCHEARTFAILURE.108.770826
5. Dabbagh MF, Aurora L, D'souza P, Weinmann AJ, Bhargava P, Basir MB. Cardiac tamponade secondary to COVID-19. JACC Case Rep. (2020). 2:1326–30. doi: 10.1016/j.jaccas.2020.04.009
6. Adu-Amankwaah J, Mprah R, Adekunle AO, Ndzie Noah ML, Adzika GK, Machuki JO, et al. The cardiovascular aspect of COVID-19. Ann Med. (2021) 53:227–36. doi: 10.1080/07853890.2020.1861644
7. Möller C, Stiermaier T, Brabant G, Graf T, Thiele H, Eitel I. Comprehensive assessment of sex hormones in Takotsubo syndrome. Int J Cardiol. (2018) 250:11–5. doi: 10.1016/j.ijcard.2017.10.047
8. Uribarri A, Núñez-Gil IJ, Conty DA, Vedia O, Almendro-Delia M, Duran Cambra A, et al. Short- and long-term prognosis of patients with takotsubo syndrome based on different triggers: importance of the physical nature. J Am Heart Assoc. (2019) 8:e013701. doi: 10.1161/JAHA.119.013701
9. Shao Y, Redfors B, Ståhlman M, Täng MS, Miljanovic A, Möllmann H, et al. A mouse model reveals an important role for catecholamine-induced lipotoxicity in the pathogenesis of stress-induced cardiomyopathy. Eur J Heart Fail. (2013) 15:9–22. doi: 10.1093/eurjhf/hfs161
10. Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. (2005) 352:539–48. doi: 10.1056/NEJMoa043046
11. Nef HM, Möllmann H, Hilpert P, Masseli F, Kostin S, Troidl C, et al. Sympathoadrenergic overstimulation in Tako-Tsubo cardiomyopathy triggered by physical and emotional stress. Int J Cardiol. (2008) 130:266–8. doi: 10.1016/j.ijcard.2007.05.119
12. Akashi YJ, Goldstein DS, Barbaro G, Ueyama T. Takotsubo cardiomyopathy: a new form of acute, reversible heart failure. Circulation. (2008) 118:2754–62. doi: 10.1161/CIRCULATIONAHA.108.767012
13. Scally C, Abbas H, Ahearn T, Srinivasan J, Mezincescu A, Rudd A, et al. Myocardial and systemic inflammation in acute stress-induced (Takotsubo) cardiomyopathy. Circulation. (2019) 139:1581–92. doi: 10.1161/CIRCULATIONAHA.118.037975
14. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. (1997) 385:729–33. doi: 10.1038/385729a0
15. Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. (1997) 385:733–6. doi: 10.1038/385733a0
16. Satoh M, Nakamura M, Saitoh H, Satoh H, Maesawa C, Segawa I, et al. Tumor necrosis factor-alpha-converting enzyme and tumor necrosis factor-alpha in human dilated cardiomyopathy. Circulation. (1999) 99:3260–5. doi: 10.1161/01.CIR.99.25.3260
17. Satoh M, Nakamura M, Satoh H, Saitoh H, Segawa I, Hiramori K. Expression of tumor necrosis factor-alpha–converting enzyme and tumor necrosis factor-alpha in human myocarditis. J Am Coll Cardiol. (2000) 36:1288–94. doi: 10.1016/S0735-1097(00)00827-5
18. Satoh M, Iwasaka J, Nakamura M, Akatsu T, Shimoda Y, Hiramori K. Increased expression of tumor necrosis factor-alpha converting enzyme and tumor necrosis factor-alpha in peripheral blood mononuclear cells in patients with advanced congestive heart failure. Eur J Heart Fail. (2004) 6:869–75. doi: 10.1016/j.ejheart.2004.02.007
19. Anderson DR, Poterucha JT, Mikuls TR, Duryee MJ, Garvin RP, Klassen LW, et al. IL-6 and its receptors in coronary artery disease and acute myocardial infarction. Cytokine. (2013) 62:395–400. doi: 10.1016/j.cyto.2013.03.020
20. Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. (2011) 365:1502–8. doi: 10.1056/NEJMoa1100721
21. Xie Y, Ma A, Wang B, Peng R, Jing Y, Wang D, et al. Rare mutations of ADAM17 from TOFs induce hypertrophy in human embryonic stem cell-derived cardiomyocytes via HB-EGF signaling. Clin Sci (Lond). (2019) 133:225–38. doi: 10.1042/CS20180842
22. Hou H, Zhao Z, Machuki JO, Zhang L, Zhang Y, Fu L, et al. Estrogen deficiency compromised the β(2)AR-Gs/Gi coupling: implications for arrhythmia and cardiac injury. Pflugers Arch. (2018) 470:559–70. doi: 10.1007/s00424-017-2098-4
23. Machuki JO, Zhang HY, Geng J, Fu L, Adzika GK, Wu L, et al. Estrogen regulation of cardiac cAMP-L-type Ca(2+) channel pathway modulates sex differences in basal contraction and responses to β(2)AR-mediated stress in left ventricular apical myocytes. Cell Commun Signal. (2019) 17:34. doi: 10.1186/s12964-019-0346-2
24. Xu P, Liu J, Sakaki-Yumoto M, Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci Signal. (2012) 5:ra34. doi: 10.1126/scisignal.2002689
25. Crestani CC. Emotional stress and cardiovascular complications in animal models: a review of the influence of stress type. Front Physiol. (2016) 7:251. doi: 10.3389/fphys.2016.00251
26. Won E, Kim YK. Stress, the autonomic nervous system, and the immune-kynurenine pathway in the etiology of depression. Curr Neuropharmacol. (2016) 14:665–73. doi: 10.2174/1570159X14666151208113006
27. Ahlquist RP. A study of the adrenotropic receptors. Am J Physiol. (1948) 153:586–600. doi: 10.1152/ajplegacy.1948.153.3.586
28. Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, et al. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. (1994) 46:121–36.
29. Schena G, Caplan MJ. Everything you always wanted to know about β(3)-AR * (*But were afraid to ask). Cells. (2019) 8:357. doi: 10.3390/cells8040357
30. Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O'gara P, et al. High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation. (2012) 126:697–706. doi: 10.1161/CIRCULATIONAHA.112.111591
31. Adzika GK, Machuki JO, Shang W, Hou H, Ma T, Wu L, et al. Pathological cardiac hypertrophy: the synergy of adenylyl cyclases inhibition in cardiac and immune cells during chronic catecholamine stress. J Mol Med (Berl). (2019) 97:897–907. doi: 10.1007/s00109-019-01790-0
32. Adrain C, Freeman M. New lives for old: evolution of pseudoenzyme function illustrated by iRhoms. Nat Rev Mol Cell Biol. (2012) 13:489–98. doi: 10.1038/nrm3392
33. Mcilwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science. (2012) 335:229–32. doi: 10.1126/science.1214448
34. Wawro K, Wawro M, Strzelecka M, Czarnek M, Bereta J. The role of NF-κB and Elk-1 in the regulation of mouse ADAM17 expression. Biol Open. (2019) 8:bio039420. doi: 10.1242/bio.039420
35. Chemaly M, McGilligan V, Gibson M, Clauss M, Watterson S, Alexander HD, et al. Role of tumour necrosis factor alpha converting enzyme (TACE/ADAM17) and associated proteins in coronary artery disease and cardiac events. Arch Cardiovasc Dis. (2017) 110:700–11. doi: 10.1016/j.acvd.2017.08.002
36. Soond SM, Everson B, Riches DW, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci. (2005) 118:2371–80. doi: 10.1242/jcs.02357
37. Schwarz J, Broder C, Helmstetter A, Schmidt S, Yan I, Müller M, et al. Short-term TNFα shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim Biophys Acta. (2013) 1833:3355–67. doi: 10.1016/j.bbamcr.2013.10.005
38. Grötzinger J, Lorenzen I, Düsterhöft S. Molecular insights into the multilayered regulation of ADAM17: the role of the extracellular region. Biochim Biophys Acta Mol Cell Res. (2017) 1864:2088–95. doi: 10.1016/j.bbamcr.2017.05.024
39. Düsterhöft S, Höbel K, Oldefest M, Lokau J, Waetzig GH, Chalaris A, et al. A disintegrin and metalloprotease 17 dynamic interaction sequence, the sweet tooth for the human interleukin 6 receptor. J Biol Chem. (2014) 289:16336–48. doi: 10.1074/jbc.M114.557322
40. Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. (2010) 45:146–69. doi: 10.3109/10409231003628015
41. Riethmueller S, Somasundaram P, Ehlers JC, Hung CW, Flynn CM, Lokau J, et al. Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol. (2017) 15:e2000080. doi: 10.1371/journal.pbio.2000080
42. Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. (2018) 17:395–412. doi: 10.1038/nrd.2018.45
43. Düsterhöft S, Lokau J, Garbers C. The metalloprotease ADAM17 in inflammation and cancer. Pathol Res Pract. (2019) 215:152410. doi: 10.1016/j.prp.2019.04.002
44. Defer N, Azroyan A, Pecker F, Pavoine C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J Biol Chem. (2007) 282:35564–73. doi: 10.1074/jbc.M704003200
45. Salmeri FM, Laganà AS, Sofo V, Triolo O, Sturlese E, Retto G, et al. Behavior of tumor necrosis factor-α and tumor necrosis factor receptor 1/tumor necrosis factor receptor 2 system in mononuclear cells recovered from peritoneal fluid of women with endometriosis at different stages. Reprod Sci. (2015) 22:165–72. doi: 10.1177/1933719114536472
46. Juhász K, Buzás K, Duda E. Importance of reverse signaling of the TNF superfamily in immune regulation. Expert Rev Clin Immunol. (2013) 9:335–48. doi: 10.1586/eci.13.14
47. Rego SL, Swamydas M, Kidiyoor A, Helms R, De Piante A, Lance AL, et al. Soluble tumor necrosis factor receptors shed by breast tumor cells inhibit macrophage chemotaxis. J Interferon Cytokine Res. (2013) 33:672–81. doi: 10.1089/jir.2013.0009
48. Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. (2014) 70:11–20. doi: 10.1016/j.cyto.2014.05.024
49. Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, et al. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. (2003) 107:798–802. doi: 10.1161/01.CIR.0000057545.82749.FF
50. Liu H, Ma Y, Pagliari LJ, Perlman H, Yu C, Lin A, et al. TNF-alpha-induced apoptosis of macrophages following inhibition of NF-kappa B: a central role for disruption of mitochondria. J Immunol. (2004) 172:1907–15. doi: 10.4049/jimmunol.172.3.1907
51. Planavila A, Laguna JC, Vázquez-Carrera M. Nuclear factor-kappaB activation leads to down-regulation of fatty acid oxidation during cardiac hypertrophy. J Biol Chem. (2005) 280:17464–71. doi: 10.1074/jbc.M414220200
52. Pellieux C, Montessuit C, Papageorgiou I, Lerch R. Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-alpha. Cardiovasc Res. (2009) 82:341–50. doi: 10.1093/cvr/cvp004
53. Li N, Karin M. Signaling pathways leading to nuclear factor-kappa B activation. Methods Enzymol. (2000) 319:273–9. doi: 10.1016/S0076-6879(00)19027-5
54. Bottero V, Busuttil V, Loubat A, Magné N, Fischel JL, Milano G, et al. Activation of nuclear factor kappaB through the IKK complex by the topoisomerase poisons SN38 and doxorubicin: a brake to apoptosis in HeLa human carcinoma cells. Cancer Res. (2001) 61:7785–91.
55. Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, et al. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem. (2003) 278:2963–8. doi: 10.1074/jbc.M209995200
56. Sen R, Smale ST. Selectivity of the NF-κB response. Cold Spring Harb Perspect Biol. (2010) 2:a000257. doi: 10.1101/cshperspect.a000257
57. Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci. (2010) 123:3209–14. doi: 10.1242/jcs.073643
58. Xing Y, Yao X, Li H, Xue G, Guo Q, Yang G, et al. Cutting edge: TRAF6 mediates TLR/IL-1R signaling-induced nontranscriptional priming of the NLRP3 inflammasome. J Immunol. (2017) 199:1561–6. doi: 10.4049/jimmunol.1700175
59. Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, et al. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflammation. (2019) 16:81. doi: 10.1186/s12974-019-1471-y
60. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
61. Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. (2011) 2:236–40. doi: 10.4103/2231-4040.90879
62. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. (2014) 171:2080–90. doi: 10.1111/bph.12475
63. Pellieux C, Aasum E, Larsen TS, Montessuit C, Papageorgiou I, Pedrazzini T, et al. Overexpression of angiotensinogen in the myocardium induces downregulation of the fatty acid oxidation pathway. J Mol Cell Cardiol. (2006) 41:459–66. doi: 10.1016/j.yjmcc.2006.06.004
64. Sekiguchi K, Tian Q, Ishiyama M, Burchfield J, Gao F, Mann DL, et al. Inhibition of PPAR-alpha activity in mice with cardiac-restricted expression of tumor necrosis factor: potential role of TGF-beta/Smad3. Am J Physiol Heart Circ Physiol. (2007) 292:H1443–51. doi: 10.1152/ajpheart.01056.2006
65. Nassir F, Wilson B, Han X, Gross RW, Abumrad NA. CD36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J Biol Chem. (2007) 282:19493–501. doi: 10.1074/jbc.M703330200
66. Tucci S, Herebian D, Sturm M, Seibt A, Spiekerkoetter U. Tissue-specific strategies of the very-long chain acyl-CoA dehydrogenase-deficient (VLCAD−/−) mouse to compensate a defective fatty acid β-oxidation. PLoS ONE. (2012) 7:e45429. doi: 10.1371/journal.pone.0045429
67. Wu CK, Lee JK, Chiang FT, Yang CH, Huang SW, Hwang JJ, et al. Plasma levels of tumor necrosis factor-α and interleukin-6 are associated with diastolic heart failure through downregulation of sarcoplasmic reticulum Ca2+ ATPase. Crit Care Med. (2011) 39:984–92. doi: 10.1097/CCM.0b013e31820a91b9
68. Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest. (1995) 96:1093–9. doi: 10.1172/JCI118095
69. Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. (1993) 92:2303–12. doi: 10.1172/JCI116834
70. Van Linthout S, Tschöpe C. Inflammation - cause or consequence of heart failure or both? Curr Heart Fail Rep. (2017) 14:251–65. doi: 10.1007/s11897-017-0337-9
71. Hobai IA, O'rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. (2001) 103:1577–84. doi: 10.1161/01.CIR.103.11.1577
72. Schulze PC. Myocardial lipid accumulation and lipotoxicity in heart failure. J Lipid Res. (2009) 50:2137–8. doi: 10.1194/jlr.R001115
73. Santoro F, Mallardi A, Leopizzi A, Vitale E, Rawish E, Stiermaier T, et al. Current knowledge and future challenges in Takotsubo syndrome: part 2-treatment and prognosis. J Clin Med. (2021) 10:468. doi: 10.3390/jcm10030468
74. Martz L. Taking TIMP3 to heart. Science-Business eXchange. (2014) 7:246. doi: 10.1038/scibx.2014.246
75. Chintalgattu V, Greenberg J, Singh S, Chiueh V, Gilbert A, O'neill JW, et al. Utility of Glycosylated TIMP3 molecules: inhibition of MMPs and TACE to improve cardiac function in rat myocardial infarct model. Pharmacol Res Perspect. (2018) 6:e00442. doi: 10.1002/prp2.442
76. Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, et al. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem J. (2010) 428:439–50. doi: 10.1042/BJ20100179
77. Calligaris M, Cuffaro D, Bonelli S, Spanò DP, Rossello A, Nuti E, et al. Strategies to target ADAM17 in disease: from its discovery to the iRhom revolution. Molecules. (2021) 26:944. doi: 10.3390/molecules26040944
78. Bax DV, Messent AJ, Tart J, Van Hoang M, Kott J, Maciewicz RA, et al. Integrin alpha5beta1 and ADAM-17 interact in vitro and co-localize in migrating HeLa cells. J Biol Chem. (2004) 279:22377–86. doi: 10.1074/jbc.M400180200
79. Doberstein K, Steinmeyer N, Hartmetz AK, Eberhardt W, Mittelbronn M, Harter PN, et al. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia. (2013) 15:218–30. doi: 10.1593/neo.121222
80. Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF, et al. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. (2013) 23:1270–83. doi: 10.1038/cr.2013.116
81. Su Y, Wang Y, Zhou H, Lei L, Xu L. MicroRNA-152 targets ADAM17 to suppress NSCLC progression. FEBS Lett. (2014) 588:1983–8. doi: 10.1016/j.febslet.2014.04.022
82. Cai M, Wang Z, Zhang J, Zhou H, Jin L, Bai R, et al. Adam17, a target of Mir-326, promotes EMT-induced cells invasion in lung adenocarcinoma. Cell Physiol Biochem. (2015) 36:1175–85. doi: 10.1159/000430288
83. Gilmore TD, Garbati MR. Inhibition of NF-κB signaling as a strategy in disease therapy. Curr Top Microbiol Immunol. (2011) 349:245–63. doi: 10.1007/82_2010_105
84. Lee DF, Hung MC. Advances in targeting IKK and IKK-related kinases for cancer therapy. Clin Cancer Res. (2008) 14:5656–62. doi: 10.1158/1078-0432.CCR-08-0123
85. Edwards MR, Bartlett NW, Clarke D, Birrell M, Belvisi M, Johnston SL. Targeting the NF-kappaB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol Ther. (2009) 121:1–13. doi: 10.1016/j.pharmthera.2008.09.003
86. Khanna D, Sethi G, Ahn KS, Pandey MK, Kunnumakkara AB, Sung B, et al. Natural products as a gold mine for arthritis treatment. Curr Opin Pharmacol. (2007) 7:344–51. doi: 10.1016/j.coph.2007.03.002
87. Folmer F, Jaspars M, Dicato M, Diederich M. Marine natural products as targeted modulators of the transcription factor NF-kappaB. Biochem Pharmacol. (2008) 75:603–17. doi: 10.1016/j.bcp.2007.07.044
88. Ríos JL, Recio MC, Escandell JM, Andújar I. Inhibition of transcription factors by plant-derived compounds and their implications in inflammation and cancer. Curr Pharm Des. (2009) 15:1212–37. doi: 10.2174/138161209787846874
89. Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene-Ramos MS, Shinn P, et al. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem Pharmacol. (2010) 79:1272–80. doi: 10.1016/j.bcp.2009.12.021
90. Planavila A, Rodríguez-Calvo R, Jové M, Michalik L, Wahli W, Laguna JC, et al. Peroxisome proliferator-activated receptor beta/delta activation inhibits hypertrophy in neonatal rat cardiomyocytes. Cardiovasc Res. (2005) 65:832–41. doi: 10.1016/j.cardiores.2004.11.011
91. Ginsberg HN, Elam MB, Lovato LC, Crouse JR III, Leiter LA, Linz P, Friedewald WT, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. (2010) 362:1563–74. doi: 10.1056/NEJMoa1001282
Keywords: takotsubo cardiomyopathy, pathophysiology, ADAM17, acute myocardial inflammation, metabolic lipids dysregulation, acute stress, therapeutic targets
Citation: Adu-Amankwaah J, Adzika GK, Adekunle AO, Ndzie Noah ML, Mprah R, Bushi A, Akhter N, Xu Y, Huang F, Chatambarara B and Sun H (2021) The Synergy of ADAM17-Induced Myocardial Inflammation and Metabolic Lipids Dysregulation During Acute Stress: New Pathophysiologic Insights Into Takotsubo Cardiomyopathy. Front. Cardiovasc. Med. 8:696413. doi: 10.3389/fcvm.2021.696413
Received: 16 April 2021; Accepted: 11 May 2021;
Published: 04 June 2021.
Edited by:
Xuewei Zhu, Wake Forest School of Medicine, United StatesReviewed by:
Zhao Wang, University of Texas Southwestern Medical Center, United StatesCopyright © 2021 Adu-Amankwaah, Adzika, Adekunle, Ndzie Noah, Mprah, Bushi, Akhter, Xu, Huang, Chatambarara and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Sun, c3VuaEB4emhtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.