 Helena Kerp1
Helena Kerp1 Georg Sebastian Hönes1
Georg Sebastian Hönes1 Elen Tolstik2Judith Hönes-Wendland1
Elen Tolstik2Judith Hönes-Wendland1 Janina Gassen1Lars Christian Moeller1Kristina Lorenz2,3*†Dagmar Führer1*†
Janina Gassen1Lars Christian Moeller1Kristina Lorenz2,3*†Dagmar Führer1*†- 1Department of Endocrinology, Diabetes and Metabolism, University Hospital Essen, University of Duisburg-Essen, Essen, Germany
- 2Leibniz-Institut für Analytische Wissenschaften-ISAS-e.V., Dortmund, Germany
- 3Institute of Pharmacology and Toxicology, University of Würzburg, Würzburg, Germany
Purpose: Thyroid hormones (TH) play a central role for cardiac function. TH influence heart rate and cardiac contractility, and altered thyroid function is associated with increased cardiovascular morbidity and mortality. The precise role of TH in onset and progression of heart failure still requires clarification.
Methods: Chronic left ventricular pressure overload was induced in mouse hearts by transverse aortic constriction (TAC). One week after TAC, alteration of TH status was induced and the impact on cardiac disease progression was studied longitudinally over 4 weeks in mice with hypo- or hyperthyroidism and was compared to euthyroid TAC controls. Serial assessment was performed for heart function (2D M-mode echocardiography), heart morphology (weight, fibrosis, and cardiomyocyte cross-sectional area), and molecular changes in heart tissues (TH target gene expression, apoptosis, and mTOR activation) at 2 and 4 weeks.
Results: In diseased heart, subsequent TH restriction stopped progression of maladaptive cardiac hypertrophy and improved cardiac function. In contrast and compared to euthyroid TAC controls, increased TH availability after TAC propelled maladaptive cardiac growth and development of heart failure. This was accompanied by a rise in cardiomyocyte apoptosis and mTOR pathway activation.
Conclusion: This study shows, for the first time, a protective effect of TH deprivation against progression of pathological cardiac hypertrophy and development of congestive heart failure in mice with left ventricular pressure overload. Whether this also applies to the human situation needs to be determined in clinical studies and would infer a critical re-thinking of management of TH status in patients with hypertensive heart disease.
Introduction
Despite great therapeutic advances, cardiovascular diseases remain the most common cause of death, globally (1). Many clinical and animal studies have described a close association of thyroid dysfunction with cardiovascular diseases (2–5). Both systemic hyperthyroidism and hypothyroidism have been identified as risk factors for heart failure. However, the precise role of TH in development and progression of heart failure is still not fully understood.
TH exert positive inotropic and chronotropic effects in the heart via direct and indirect mechanisms (3, 6–8). For example, TH stimulate cardiac protein synthesis, promote physiological cardiac growth and angiogenesis, and reduce cardiac afterload by decreasing arterial vascular smooth muscle tone (2, 9). In the heart, these physiological TH effects involve sarcomeric proteins, such as myosin heavy chain alpha (MYH6) and beta (MYH7), regulators of calcium homeostasis such as sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and phospholamban (PLN) as well as beta1-adrenoreceptors (10, 11). For a limited duration, treatment with TH has been shown to improve cardiac output, reduce afterload, and lead to a compensated, so-called physiological or “athletic,” cardiac hypertrophy (3, 12–18). These effects could even be shown in relation to cardiac diseases in a small patient cohort with advanced heart failure (19) and in patients with dilated cardiomyopathy (20). While this supports a beneficial role of TH in heart failure, large cohort studies and comprehensive experimental characterization of heart disease under low and high TH availability are lacking. Thus, the aim of our study was to investigate longitudinally whether modulation of TH status at early stages in an already diseased heart could affect cardiac outcome. To this end, we induced chronic left ventricular pressure overload in mice by transverse aortic constriction (TAC) and studied changes in heart function and structure under TH deprivation and high TH availability over a period of 4 weeks. Modulation of thyroid status commenced 1 week after TAC, when first signs of cardiac hypertrophy and impaired cardiac function have evolved (21). In this heart disease model, we show that reducing TH availability effectively slows down pathological cardiac growth and improves cardiac function even under continued left ventricular pressure overload, while opposite effects occur with increased TH availability.
Materials and Methods
Animals
Male wild-type C57BL/6JRj mice (Janvier Labs, France) aged 2 months (n = 6–9 animals per treatment) were studied. All mice were housed in groups of three to five animals in temperature (23 ± 1°C)- and light (inverse 12:12-h light–dark cycle)-controlled conditions. Food and water were provided ad libitum. All animal experiments were performed in accordance with the German regulations for Laboratory Animal Science (GVSOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The protocols for animal studies were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany (LANUV-NRW, AZ 84-02.04.2016.A261).
Transverse Aortic Constriction
Mice were subjected to TAC to induce chronic left ventricular pressure overload using a 27-gauge needle for 4 weeks as described previously (22). Before TAC, as well as 2 and 4 weeks after TAC, echocardiography was performed. Hearts were isolated for biochemical, histological, and weight analyses. The surgeon was blinded to treatment groups. Banded mice with an aortic pressure gradient below 60 mmHg were excluded from this study.
Echocardiography
Transthoracic echocardiograms of male mice were performed in a blinded manner using the Vevo2100 high-resolution imaging systems (VisualSonics) and a 30-MHz probe. Values for end-diastolic and -systolic intraventricular septal (IVSd/IVSs) and left ventricular posterior wall thicknesses (LVPWd/LVPWs) as well as end-diastolic and -systolic left ventricular internal diameters (LVIDd/LVIDs) were obtained from 2D M-mode images in the short-axis view at the proximal level of the papillary muscles. Peak blood flow velocities at the site of TAC [Vmax (mm/s)] were derived from pulsed-wave Doppler measurements. Fractional shortening (FS) as a measure for cardiac contractility (23), ejection fraction (EF), and aortic pressure gradients (mmHg) were calculated by VisualSonics Cardiac Measurements software. The data shown represent averages of at least six cardiac cycles per animal. The investigator was blinded to experimental settings during measurements and data analysis. Mice with heart rates below 450 bpm were excluded from the study.
Treatment
Mice were subjected to three treatment protocols: (1) For increased TH availability (TH high) mice were fed normal diet (MD.1572, Envigo, USA) and received 1 μg/ml T4 (Sigma-Aldrich (T2376), USA; stock solution: 100 μg/ml T4 solved in 40 mM NaOH and 2 g/L bovine serum albumin) in drinking water. (2) For TH deprivation (TH low), animals were fed a low-iodine diet (LoI; MD.1571, Envigo, USA) and received drinking water supplemented with anti-thyroid drugs 0.04% methimazole [MMI, Sigma-Aldrich (301507), USA], 0.5% sodium perchlorate [, Sigma-Aldrich (310514), USA]), and 0.3% saccharine as sweetener [Sigma-Aldrich (240931), USA] (LoI/MMI/). (3) In the control group (ctrl), animals were fed a normal diet (MD.1572, Envigo, USA) and received drinking water without supplements. Other than the low iodine content in the TH deprivation group, caloric and nutritional composition of chow was comparable in all three mouse groups. The treatment protocols started 1 week after TAC surgery and were continued for 2 or 4 weeks as indicated (Figure 1).
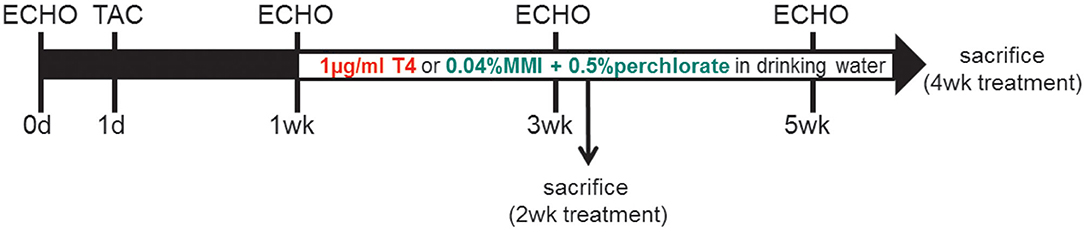
Figure 1. Study design: Two-month-old male C57Bl/6 mice were subjected to TAC at experimental day 1. One week after banding, when cardiac hypertrophy had developed, animals were exposed to either TH treatment or TH deprivation by 1 μg/ml T4 or LoI/MMI/ in drinking water, respectively. Control mice were maintained on normal chow and drinking water without supplements. Serial echocardiography (ECHO) was conducted at day 0 and at 1, 3, and 5 weeks (wk) after TAC surgery. Mice were sacrificed at 2 and 4 weeks after start of treatment protocol (3 and 5 weeks after TAC).
Male mice under chronic high or low TH availability without TAC surgery have been extensively studied by us earlier (24–26) and TAC sham operation has previously been shown not to affect parameters of cardiac function or morphology (22, 27, 28). For these reasons and in line with the “3R concept” in animal research, the present study was done without including an additional sham-operated control group.
Organ Isolation and Serum Measurements
Mice were deeply anesthetized at 3 or 5 weeks after TAC surgery, i.e., 2 or 4 weeks after start of treatment protocols, by ip injection of 200 μl of Ketamine/Xylazine mixture [150 μl of 100 mg/ml Ketamine (Bela-pharm, Germany) and 50 μl of 20 mg/ml Xylazine (Ceva, Germany)], and final blood was obtained by right ventricular heart puncture. For tissue collection, mice were perfused with heparinized saline through a needle placed in the left heart ventricle. Hearts were shock frozen in liquid nitrogen, and stored at −80°C until further processing or fixed in 4% buffered formalin (Formafix, Germany). Final blood samples were stored 30 min on ice and centrifuged, and free triiodothyronine (fT3), free thyroxine (fT4), and total T4 (TT4) concentrations in serum of mice were measured using commercial ELISA kits according to the manufacturer's instructions (DRG Instruments GmbH, Marburg, Germany). Detection limits were 0.5 μg/dl, 0.05 ng/dl, and 0.05 pg/ml for TT4, fT4, and fT3, respectively. Values below were calculated from the standard curve.
RNA Isolation and qRT-PCR
Total RNA from hearts was isolated and reverse transcribed into cDNA as previously described (29). In compliance with the MIQE guidelines for RT-PCR, we used a set of three reference genes to assure accurate normalization and calculation, Gapdh (glyceraldehyde-3-phosphate dehydrogenase), Rn18s (18S ribosomal RNA), and Polr2a (polymerase RNA II). Primer sequences of all quantified genes are listed in Supplementary Table 1. Analysis and calculation of the fold change in gene expression were done on Ct ≤ 35 using the efficiency corrected method.
TUNEL Assay
After dewaxing and rehydration, tissue sections were permeabilized with proteinase K. For TUNEL staining, an in situ cell death detection kit (Sigma-Aldrich, USA) was used according to the manufacturer's protocol. Pretreatment with DNase I served as a positive control and TUNEL reaction mixture lacking terminal transferase (TdT) served as a negative control. Cell nuclei and membranes were counterstained with DAPI (D1306, Thermo Fisher Scientific, USA) and wheat germ agglutinin (W11261, Thermo Fisher Scientific, USA). For quantification, blind analysis of sections pictured with the Olympus BX51 upright microscope (Olympus, Germany) was conducted.
Histological Staining and Quantification of Cardiomyocyte Sizes
Formalin-fixed hearts were embedded in paraffin and 5-μm-thick sections were used for staining. Hematoxylin and eosin (H&E) staining was performed for determination of cardiomyocyte sizes and Sirius Red for fibrosis as described previously (27). For quantification, left ventricular sections were recorded using Olympus BX51. Cross-sectional areas of cardiomyocytes in H&E- or wheat germ agglutinin-stained vertical sections were determined (n = 37–107 cells per animal) using ImageJ. Only cells with visible central nucleus were included. Fibrosis was calculated as the ratio of red stained/myocardial area via Adobe Photoshop. Analysis and quantification were done in a blinded manner.
Immunoblot
Extraction of whole protein lysates, protein quantification, and immunoblotting were performed as described previously (30). The following antibodies were used: anti-AKT (9,272, Cell Signaling, 1:1,000, rabbit), anti-p-AKT (S473) (9271S, Cell Signaling, 1:1,000, rabbit), anti-ERK 1/2 (4695S, Cell Signaling, 1:1,000, rabbit), anti-p-ERK 1/2 (T202/Y204) (4370T, Cell Signaling, rabbit), anti-mTOR (2972S, Cell Signaling, 1:1,000, rabbit), anti-p-mTOR (Ser2448) (5536S, Cell Signaling, 1:1,000, rabbit), anti-BAX (2,772, Cell Signaling, 1:1,000, rabbit), anti-GAPDH (ACR001P, Acris Antibodies, 1:6,000, mouse), anti-mouse IgG (7,076, Cell Signaling, 1:2,000, goat), and anti-rabbit IgG (7,074, Cell Signaling, 1:2,000, goat). Visualization was done by luminescence using the Immun-Star™ WesternC™ Kit (BioRad, USA) and VersaDoc System (BioRad, USA). Differences in protein expression levels were quantified by densitometry using the Image Lab™ Software (BioRad, USA).
Two-Photon and Polarized Light Microscopy
Cardiac fibrotic tissue was visualized by two-photon microscopy using Leica TCS SP8 microscope and Leica Application Suite (LAS X) software, equipped with a Chameleon Vision II Titan-Sapphire laser and a 25 × HCX IRAPO L water-immersion objective with a NA of 0.95. Autofluorescence of the heart and second harmonic signal (SHG) were detected using an excitation wavelength of 900 nm. A hybrid detector (HyD), bandpass emission filter 525/50 was used for autofluorescence detection, and SHG was detected using a hybrid detector (HyD), bandpass emission filter 460/50. Z-stacks of 12 μm with a spacing of 1 μm were acquired and pictures were projected with ImageJ. Cardiac fibrotic tissues from all six groups were additionally examined with a polarization microscope to assess the birefringence pattern of collagen fibers. Polarization imaging was performed using a Leica TCS SP8 DMi8 CS Bino DLS microscope with a magnification of 20 ×. Single random xy mappings were acquired in 512 × 512 image format, where x and y described the length and width of the mapping in pixels. Each image was acquired in polarized and bright-field light. Images were captured and exported using the Leica Application Suite (LAS X) integrated software.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 6 Software. Two-way ANOVA followed by Tukey's post-hoc analysis or unpaired Student's t-test was applied as indicated. Values of *p < 0.05, **p < 0.01, ***p < 0.001, and #p < 0.0001 were considered statistically significant. Outliers were identified using GraphPad Outlier test and were excluded if significantly (p < 0.05) different from the group.
Results
To investigate how TH status impacts progression of pathological cardiac hypertrophy and development of heart failure, we used a heart disease mouse model with chronic left ventricular pressure overload induced by ligation of the transverse aorta. Well-characterized features of this model are pathological growth of the heart, i.e., maladaptive cardiac hypertrophy associated with interstitial fibrosis and apoptosis that subsequently leads to decreased FS and congestive heart failure (31). In this model and at an early stage of evolving cardiac disease at 1 week after TAC surgery, we changed TH status in mice by either oral T4 supplementation or TH deprivation through anti-thyroidal drug treatment (LoI/MMI/) of mice. As depicted in the study design (Figure 1), these treatments were conducted over 4 weeks.
Notably, TAC surgery per se had no impact on serum TH status in mice, since TT4, fT4, and fT3 serum concentrations in TAC operated control mice were comparable to TH levels in 2-month-old TAC naïve mice (24). In contrast, T4 treatment or TH deprivation led to the expected alterations in TH serum concentrations with a hyperthyroid and hypothyroid serum status in TAC mice at 2 and 4 weeks [Figure 2; (32, 33)]. Additionally, cardiac TH receptor expression α (TRα) and β (TRβ) assessed by qRT-PCR showed decreased TRα availability upon hyperthyroidism and increased during TH deprivation. TRβ expression was not altered by TH status (Figure 2D).
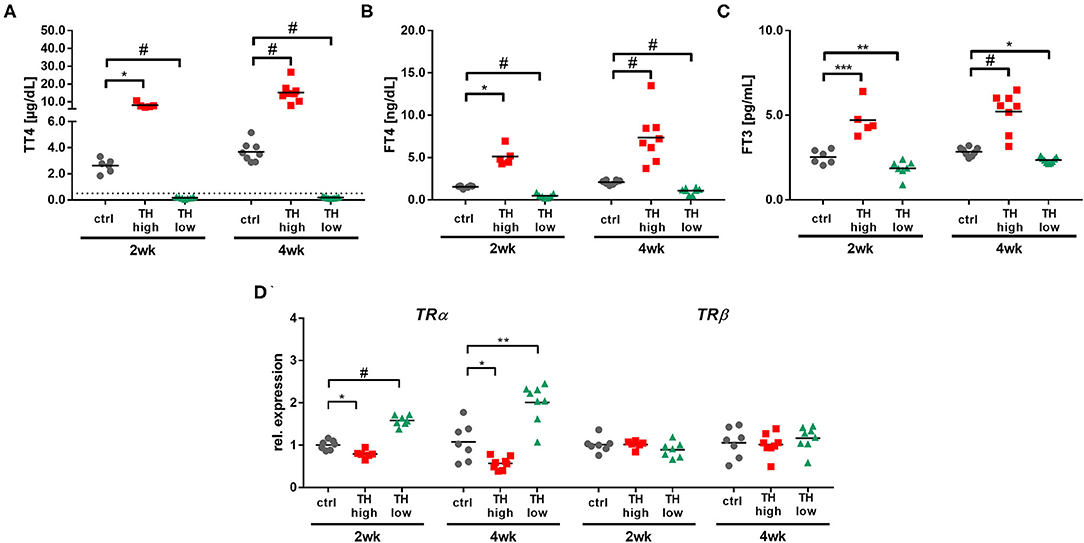
Figure 2. Confirmation of increased (TH high) or decreased (TH low) TH serum concentrations in T4- or LoI/MMI/-treated TAC mice (n = 5–9 per group) compared to euthyroid control TAC mice (ctrl). Dotted line represents detection limit of assay and values below were calculated from the standard curve. Serum TH concentrations (TT4, fT4, and fT3; A–C) and cardiac TRα and TRβ expression (D) were measured after 2 weeks and 4 weeks of treatment. Scatter dot plot and mean in all panels, *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.0001 by two-way ANOVA and Tukey's post-hoc analysis for TH serum measurement and by unpaired t-test for qRT-PCR, ctrl, control; wk, weeks.
TH Deprivation Stops Progression of Maladaptive Cardiac Hypertrophy and Prevents Development of Heart Failure
The progression of pathological cardiac hypertrophy and cardiac dysfunction in response to TAC was monitored by serial echocardiograpy. One week after surgery—before manipulation of TH status was started—wall thickness of left ventricles was already significantly increased in TAC mice as depicted by the diameter of posterior and septum walls compared to corresponding baseline values prior to surgery (Figures 3A,B). Additionally, all TAC-treated mice showed significantly reduced FS (Figure 3C); however, no dilatation of the left ventricle was yet evident (Figure 3D).
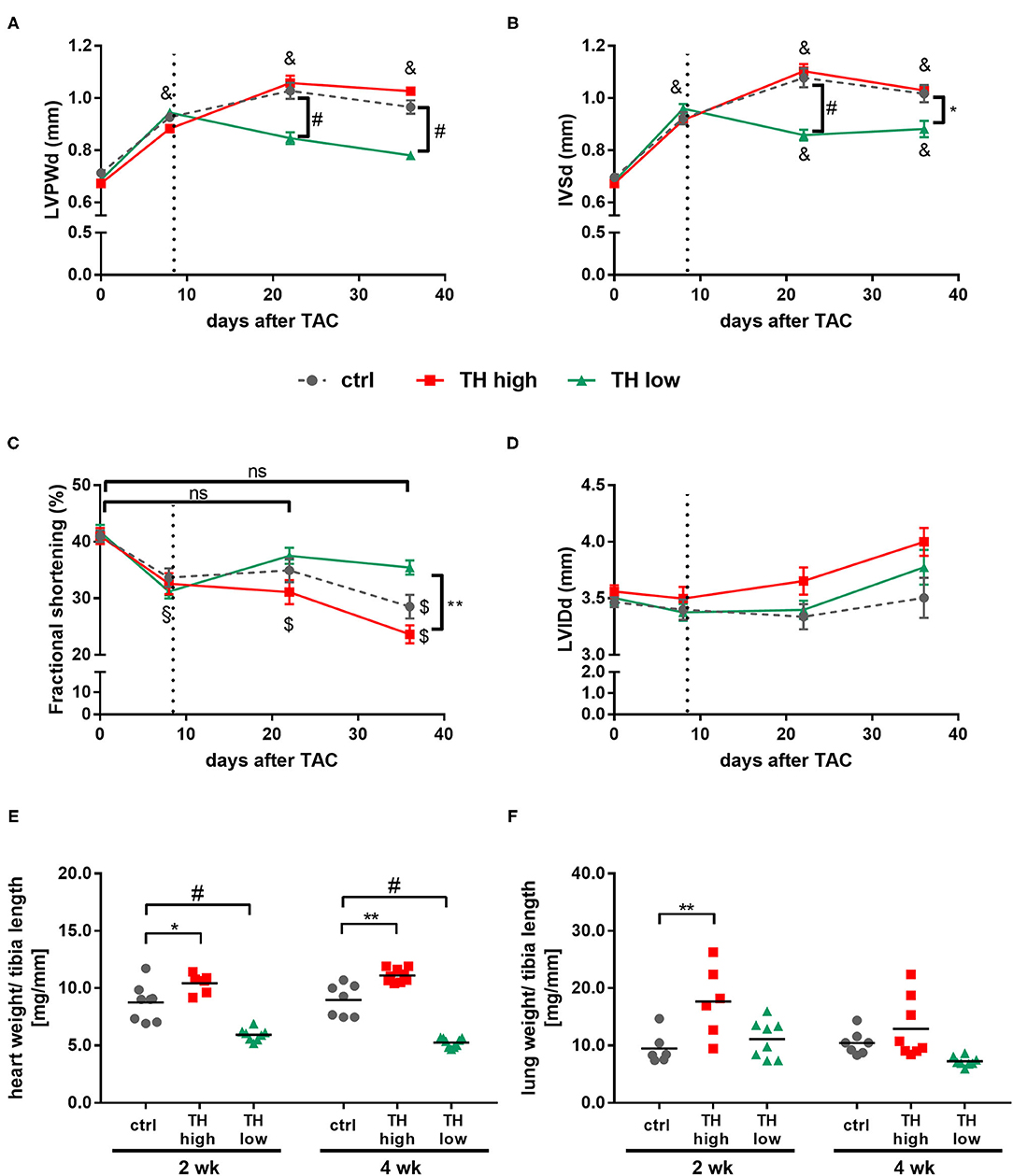
Figure 3. TH deprivation stops progression of maladaptive cardiac hypertrophy, improves fractional shortening, and effectively prevents development of heart failure. Echocardiography was conducted prior to TAC and 1, 3, and 5 weeks after surgery. Diastolic left ventricular posterior wall (LVPWd; A), intraventricular septum (IVSd; B) thickness, fractional shortening (C), and diastolic left ventricular inner diameter (LVIDd; D) were determined in all mice. Values are indicated as mean ± SEM, for n, see Supplementary Table 2. *p < 0.05 TH high vs. TH low, **p < 0.01 TH high vs. TH low, #p < 0.0001 to TH low at indicated time, §p < 0.05 to baseline, $p < 0.001 to baseline, &p < 0.0001 to baseline by two-way ANOVA and Tukey's post-hoc analysis, ctrl, control; ns, not significant; wk, weeks. Dotted line represents start of T4 or LoI/MMI/ treatment. Analysis of heart weight-to-tibia length ratio (E, n = 6–9) and lung weight-to-tibia length ratio (F, n = 6–8) was determined at 2 and 4 weeks after start of treatment protocol. Scatter dot plot and mean, *p < 0.05, **p < 0.01, #p < 0.0001 by two-way ANOVA and Tukey's post-hoc analysis.
In the following 4 weeks, the increase in wall thickness continued in control mice and was accompanied by a further significant decrease in FS over time. Interestingly, no differences in FS were observed between control and T4-treated mice (Figures 3A–C). In contrast, TH deprivation stopped progression of pathological cardiac hypertrophy and improved cardiac performance with a marked increase in FS after 2 and 4 weeks of treatment compared to control and T4-treated mice [Figure 3C and Supplementary Figure 1]. These differences in the progression of TAC-induced cardiac hypertrophy and changes in cardiac function under increased or decreased TH availability were also illustrated by changes in heart weight. Thus, after 2 and 4 weeks, heart weight-to-tibia length ratios in TH-deprived mice were significantly lower, while those for T4-treated mice were significantly higher compared to control animals (Figure 3E). Cardiomyocyte cross-sectional areas correlated with heart weight and were significantly smaller in TH-deprived mice compared to controls after 4 weeks of treatment [Supplementary Figure 2]. Furthermore, significantly increased lung weight, a functional indicator of congestive heart failure, was noted in T4-treated TAC mice, an effect that was significant after 2 weeks of treatment compared to TAC controls (Figure 3F). In line with these findings—even though not of statistic relevance−2 of 17 T4-treated mice died, while all TH-deprived (n = 17) and all control (n = 16) mice survived the 4-week treatment period after TAC.
T4 Treatment in Chronic Pressure Overload Increases Cardiomyocyte Death Rate and Expression of Heart Failure Markers
Interstitial fibrosis due to proliferation of fibroblasts, collagen accumulation, and cardiomyocyte death causes cardiac muscle stiffness (34). To evaluate whether tissue remodeling contributes to the functional outcome, mouse hearts were histologically analyzed for interstitial fibrosis, apoptotic cell death by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL), and pro-apoptotic signaling by protein expression of pro-apoptotic Bcl2-associated protein X (BAX).
Although a trend toward a transient decrease after 2 weeks of TH deprivation was noted, no significant alterations in the degree of fibrosis were identified by quantification of Sirius Red staining (Figures 4A,B) and visualization by two-photon and polarized light microscopy (Figures 4C,D). Moreover, apoptosis rate (Figures 5A,B) differed not between the three treatment groups after 2 weeks. However, after 4 weeks, TUNEL staining revealed an increase in apoptotic cell death and a significant increase in BAX protein expression was found in hearts of T4-treated TAC mice (Figures 5A–D), while fibrosis was not affected. Moreover, quantification of heart failure markers revealed an increase in cardiac atrial natriuretic peptide (Anp) and brain natriuretic peptide (Bnp) expression under T4 treatment compared to controls or TH-deprived animals (Figure 5E). Of note, hypothyroid hearts displayed the lowest Anp levels of all three treatments groups at the end of experiments, 5 weeks after TAC surgery (p < 0.001, Figure 5E). These changes in T4-treated TAC mice, however, were not accompanied by increased fibrosis, suggesting that T4 treatment might have diverged impact on cardiomyocyte apoptosis and cardiomyocyte function in heart.
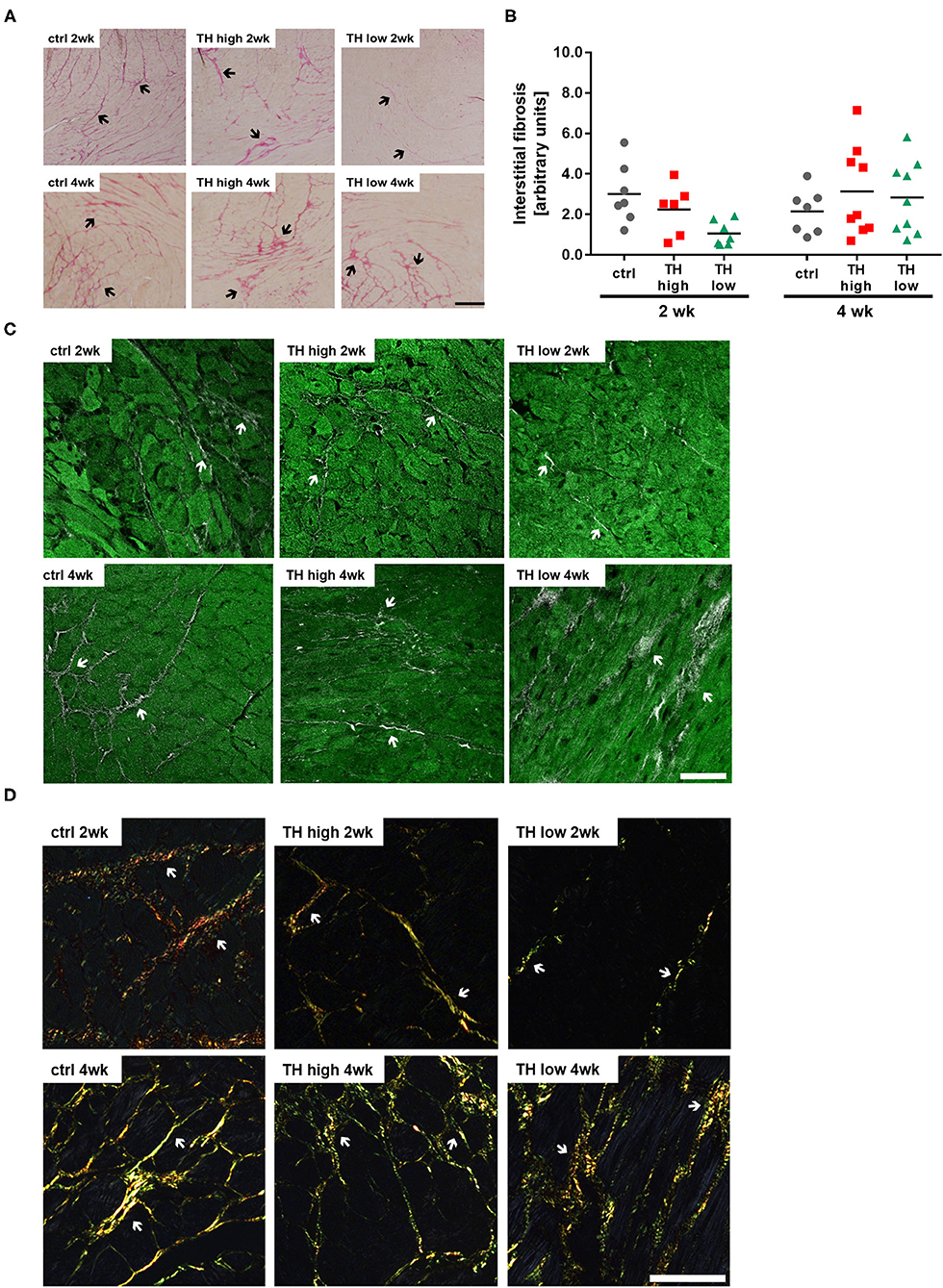
Figure 4. T4 status has no effect on fibrosis in TAC-operated hearts. Representative pictures of Sirius Red-stained sections (A, scale bar = 200 μm, black arrows indicate fibrotic fibers) and relative quantification of interstitial fibrosis (B, n = 6–9). Overlaid second harmonic generation images (gray) and autofluorescence signals (green) (C, scale bar = 50 μm, white arrows indicate fibrotic fibers) of heart slices were compared to polarized light imaging (D, scale bar = 50 μm, white arrows indicate fibrotic fibers).
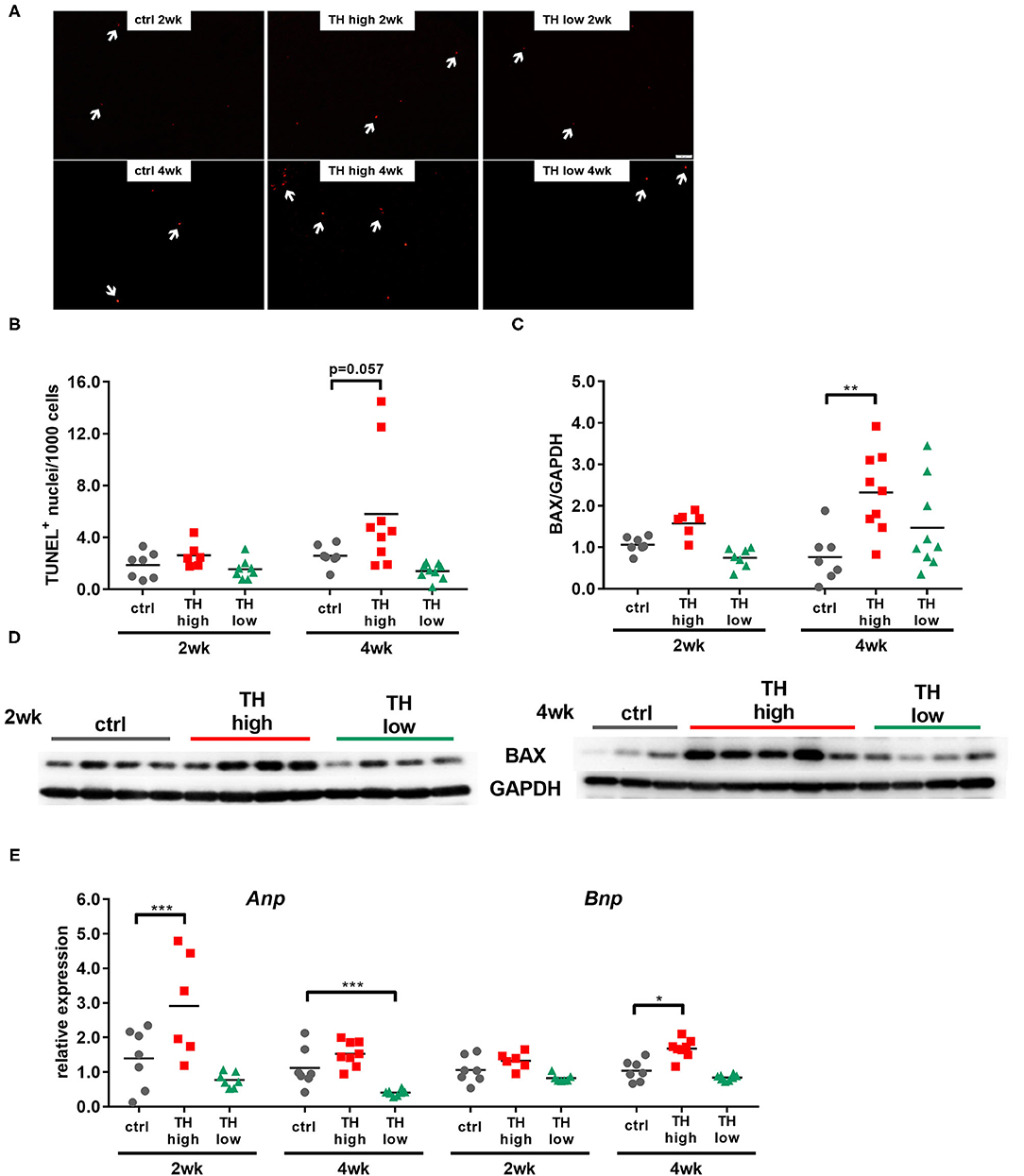
Figure 5. Cardiomyocyte apoptosis and expression of heart failure markers are increased in T4-treated TAC-operated hearts. TUNEL-positive nuclei (A, scale bar = 50 μm, white arrows indicate TUNEL-positive nuclei) were quantified in heart sections (B, n = 6–9), and cardiac protein expression of Bax was assessed by Western blotting after 2 and 4 weeks of treatment (C, D n = 6–9). Expression of Anp and Bnp was determined in mouse hearts by qRT-PCR (E, n = 6–9). Scatter dot plot and mean, **p < 0.01, by two-way ANOVA and Tukey's post-hoc analysis for TUNEL and BAX quantification; mean and SEM, *p < 0.05, ***p < 0.001 by unpaired t-test for qRT-PCR; ctrl, control; wk, weeks.
mTOR Signaling in Hearts Subjected to TAC Depends on TH Availability
Several molecular pathways have been implicated in maladaptive cardiac hypertrophy (5, 35–37). One signaling pathway that is a key mediator of pathological growth and has recently gained increasing attention in the heart is the mTOR pathway (9, 38, 39). In fact, increased cardiac mTOR signaling has previously been demonstrated after TAC in mice (38, 40, 41). In our study, 4 weeks of T4 treatment resulted in significantly increased mTOR expression in TAC mouse hearts (Figure 6). Conversely, TH deprivation resulted in strongly reduced mTOR expression after 2 and 4 weeks in TAC mouse hearts (Figures 6A,B) compared to controls and T4-treated mice.
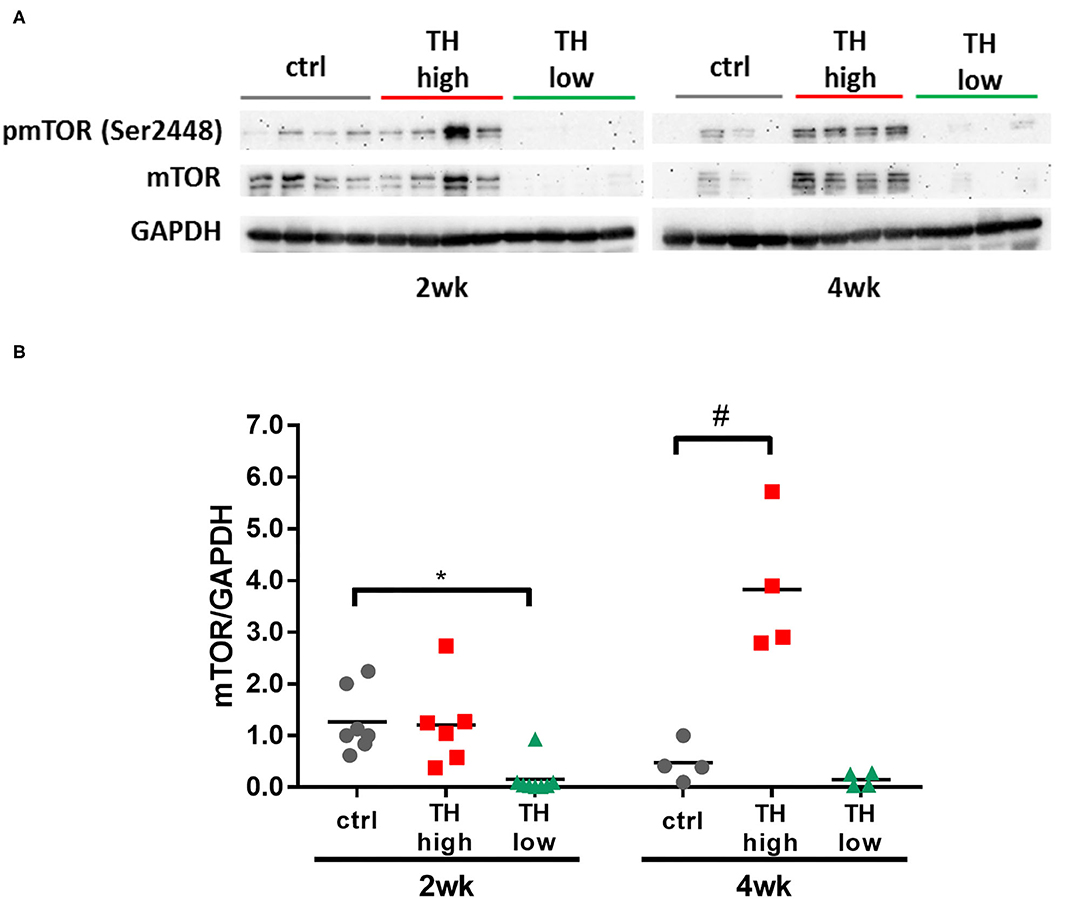
Figure 6. Cardiac expression of mTOR increased with T4 administration and decreased with TH deprivation. mTOR expression was significantly elevated after 4 weeks of T4 treatment, whereas TH deprivation resulted in drastic reduction of mTOR expression within 2 weeks (A,B, n = 4–8). Scatter dot plot and mean, *p < 0.05, #p < 0.0001 by two-way ANOVA and Tukey's post-hoc analysis; ctrl, control; wk, weeks.
Other pathways that are important signaling transducers are the serine/threonine-specific protein kinase AKT and extracellular signal-regulated kinase 1/2 (ERK 1/2). Transient activation of both kinases, expressed by increased ratio of pAKT/AKT and pERK/ERK, was noted in hearts of TH-deprived mice (Supplementary Figure 3). However, 4 weeks of treatment of low or high TH condition did not activate these pathways.
TH Availability Dominates the Regulation of Genes Involved in Cardiac Contractility
Cardiac contractility is determined by multiple factors in normal and failing hearts and several involved molecules are also described to be TH responsive (13, 42–44). To assess their regulation upon TAC-induced pressure overload in relation to TH serum status, transcript levels of cardiac contractility genes Myh6, Myh7, Pln, and Serca2a were quantified by qRT-PCR.
High TH availability repressed Myh7 expression at 2 and 4 weeks and Pln expression after 4 weeks of treatment (Figures 7A,B), while TH deprivation increased transcript levels of Myh7 and Pln and downregulated Myh6 and Serca2a. In addition, expression of a gene regulating differentiation and calcium-dependent gene expression in muscle cells [myocyte-specific enhancer factor (Mef2a)] and its downstream target Myomaxin was investigated (Figure 7C). T4 treatment lowered Mef2a expression after 2 weeks, but not after 4 weeks. In contrast, low TH status elevated Mef2a transcripts at both time points. No significant changes were found for Myomaxin expression. Thus, TH status had a more pronounced impact on regulation of cardiac contractility genes than TAC-induced chronic pressure overload.
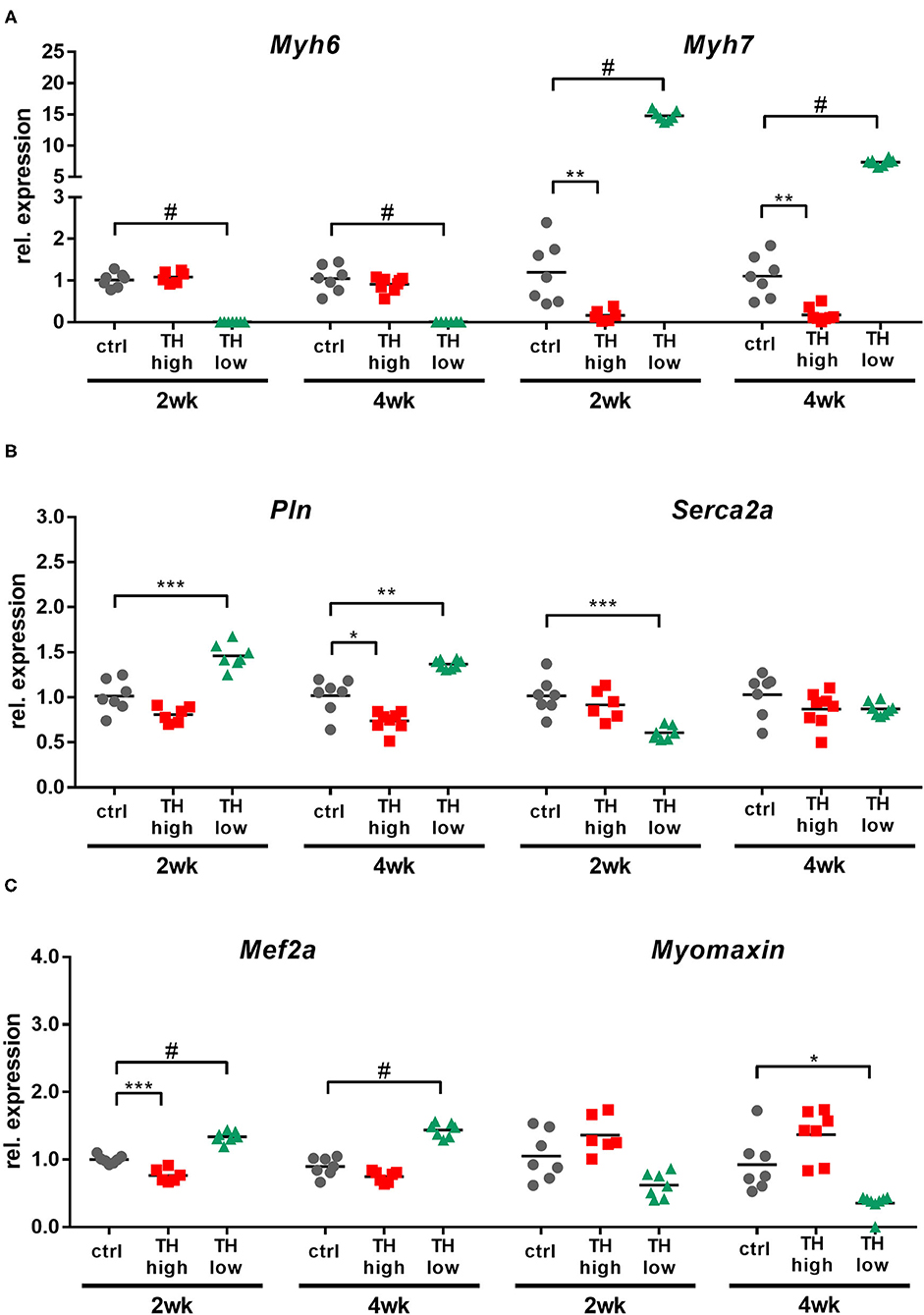
Figure 7. Cardiac expression of cardiac contractility genes and TH-regulated genes was primarily affected by high and low TH serum state. TH availability determined the transcript amounts of Myh6, Myh7, Pln, Serca2a, Mef2a, and Myomaxin in mouse hearts (A–C). Scatter dot plot and mean, *p < 0.05, **p < 0.01, ***p < 0.001, and #p < 0.0001 by unpaired t-test for qRT-PCR; ctrl, control; wk, weeks.
Discussion
Chronic pressure overload of the left ventricle is a pathological condition and predictor of adverse cardiovascular events. It can be caused by systemic hypertension or aortic stenosis due to increased peripheral resistance or obstruction, respectively (45). Here, we have used the so-called “TAC model” to induce chronic left ventricular pressure overload in order to simulate a central aspect of hypertensive heart disease. Interestingly, in these conditions—different to expectations from some preclinical and clinical studies (5, 20, 46, 47)—we found that high TH treatment was not beneficial for diseased heart while TH deprivation was highly protective.
It was previously shown that hypercorrection of a hypothyroid-like state, which occurs in heart failure, e.g., by decreased TH receptor expression or increased deiodinase type 3 activity in heart, may restore cardiac function (20, 48–51). Similarly, systemic hypothyroidism may confer cardiovascular morbidity and mortality and hence is widely regarded as a TH substitution indication (52–54). However, recent studies challenge this “one-for-all” concept (55–57) and have advocated a more risk-based TH treatment strategy in case of hypothyroidism (58, 59). While most therapeutic goals are directed at reducing cardiovascular risk, treatment studies addressing changes in TH status and their impact on progression of disease in different cardiovascular disease entities are still missing (60, 61). In this study, in the presence of chronic pressure overload, 4 weeks of TH deprivation resulted in stop of pathological cardiac growth with decreased wall thicknesses and lowered heart weight-to-tibia length ratio and cardiomyocyte size compared to TAC controls. This was associated with an impressively improved cardiac function as illustrated by a 25% increased FS compared to controls and a 50% increased FS compared to T4 treated TAC mice. Furthermore, several parameters suggest a detrimental impact of TH treatment in our model of congestive heart failure. For example, lung weight-to-tibia length ratio as a measure of congestive heart failure increased after 2 weeks of T4 treatment, two death events occurred, and on the molecular level, this was reflected in increased cardiomyocyte apoptosis, mTOR activation, and increased expression of heart failure markers Anp and Bnp in hearts of T4-treated TAC mice. Thus, other than expected (20, 47, 50), T4 treatment did not prevent cardiac remodeling under chronic pressure overload but worsened the outcome.
One explanation for the discrepancy between the expected and observed impact of TH status on cardiac disease progression might be the heart failure model used in our study, besides the high T4 dosage, which was used to counteract the low TH heart state observed in subacute phase of mouse heart disease (62). Thus, vasodilation and subsequent reduction in peripheral resistance is one major cardiovascular effect of TH and improved cardiac output (63); however, in the TAC model, peripheral resistance is preset and stably induced by aortic ligation. Similarly, a vascular impact of hypothyroidism, which normally increases peripheral resistance (64), can be neglected in our study. In fact, in the clinical setting, there are patients who suffer from chronic left ventricular pressure overload, e.g., due to aortic stenosis or hypertrophic obstructive cardiomyopathy, similarly to the situation reflected in the TAC mouse model. In this condition, increased TH availability that mainly targets the heart within the cardiovascular system seems to induce cardiac cell death and deterioration of functional parameters.
Increased TH availability may also confer harmful effects on the heart by an increase in cardiac sympathetic stress (65–67). Thus, an altered adrenergic responsiveness at heart level could also contribute to the observed harmful effects of T4 treatment or conversely the cardioprotective effects of TH deficiency in hearts under chronic pressure overload.
While TH status-dependent changes in cardiac function in our TAC model were in line with increased or decreased cardiomyocyte apoptosis and up- or downregulation of mTOR signaling in the T4-treated and TH-deprived TAC mouse groups, respectively, future studies are needed to examine the role of mTOR signaling in the TH-dependent recovery from pressure overload using specific inhibitors.
TH-dependent cardiac hypertrophy was described as physiological hypertrophy mediated by the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway (2, 9, 68). Over time, a compensated hypertrophy may result in left ventricular dilatation and develop pathological type of hypertrophy, which involves activation of additional intracellular signaling pathways such as ERK1/2, p38 MAPK, and JNK 1/2/3 (69) or the calcineurin system (70). Thus, previous studies described the activation of AKT and ERK upon TH treatment after myocardial infarction to be dose-dependent (71). Others found a TH-dependent ERK inhibition in pressure overload-induced mouse hearts (47). In our study, a transient activation of AKT and ERK signaling was noted after 2 weeks of TH deprivation and no change upon TH supply. However, due to the complex linkage and connection of intracellular signaling pathways during development and progression of cardiac hypertrophy, a dose- and time-dependent effect of TH is most likely.
Furthermore, no difference was found in the degree of fibrosis in the mouse hearts. The limited observation time of 4 weeks or anti-fibrotic effects of T4 reported by some groups (18, 72) may be reasons for the observation that the TH-dependent changes in TAC hearts did not (yet) translate into increased collagen deposition, a common histological finding in congestive heart failure.
Intrinsic effects on cardiac contractility such as changes in the expression of Myh6, Myh7, Serca2a, and Pln may determine function in normal and diseased hearts. Moreover, a switch involving decreased Myh6 and Serca2a and increased Myh7 and Pln transcripts was reported in conditions of hypertrophy and heart failure (42–44, 73, 74). Interestingly, this expression pattern was also found in the absence of TAC in hearts of hypothyroid mice while the opposite was observed in hyperthyroid mice in our previous experiments (13), suggesting that systemic TH availability may dominate the influence on cardiac gene expression rather than effects of pressure overload. In addition, TAC hearts under hyperthyroid conditions showed downregulation of TRα, while receptor expression was increased in TH-deprived TAC hearts, suggesting a local adaption of TH action in response to maladaptive hypertrophy and altered TH availability. In line, we found Mef2a to be similarly regulated as TRα most likely due to its function as a transcriptional co-factor of TRα (75). Decreased expression of Mef2a downstream-target Myomaxin was measured after 4 weeks in hypothyroid mice. This might be a protective state, as less myocardial damages when exposed to Angiotensin II were observed in mouse hearts with 20% of residual Myomaxin expression (76).
Limitations
For our study, we have chosen relatively strong hyper- and hypothyroid states to evaluate the effects of dysregulated hormone action. Such dosages are usually used in other experimental mice studies (24, 29, 32) and are well described. However, more moderate doses or short-term derailment may have different impact on development and progression of pressure overload. In addition, hypothyroidism may have only a transient beneficial effect and could change into detrimental outcome in the long-term. Also, the impact of different TH conditions on signaling pathways under pressure overload need to be further investigated to better understand the underlying molecular events.
Future Directions
Future animal studies should include more increments of TH states and treatment windows to fine-tune an optimal dosage as well as time frame. In the light of the outcome of our study, our data suggest that future clinical studies are warranted to define optimal thyroid status and reconsider differential TH therapy schemes in patients with cardiovascular disease conditions such as patients with aortic stenosis.
Conclusion
In summary, we found that TH deprivation is beneficial for diseased heart under chronic pressure overload with regard to slowing down hypertrophic remodeling and improving cardiac contractile performance. This represents an early protective effect of hypothyroidism in the progression of maladaptive hypertrophy and prevents development of congestive heart failure. In contrast, our data underline that high-dose TH administration accelerates cardiac dysfunction under condition of maladaptive hypertrophy. The latter findings clearly underline the detrimental role of increased if not excessive TH availability in the diseased heart as has been stated in several recent guidelines on management of subclinical and overt thyroid dysfunction (56, 77) and illustrated in a recent heart failure population study (78).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by LANUV AZ 84-02.04.2016.A261.
Author Contributions
HK, ET, KL, and DF: material preparation, data collection, and analysis were performed. GH, JH-W, JG, and LM contributed to analysis and interpretation of data. The first draft of the manuscript was written by HK, KL, and DF. All authors commented on previous versions of the manuscript and contributed to the study conception, design, read, and approved the final manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft in the framework of SFB/TR 296 LOCOTACT (DF, LM, and KL) and SFB1116 (KL), the German Ministry of Research and Education (BMBF; ERK Casting), the Ministry for Innovation, Science and Research of the Federal State of North Rhine Westphalia, and the Drug Discovery Hub Dortmund (DDHD to KL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Imaging Core Facility Essen (IMCES) for support with the Olympus microscope and Dr. Anja Hasenberg for imaging heart sections with two-photon microscopy. We are also grateful to K. Schättel, S. Rehn, and J. Göbel for their dedicated technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.683522/full#supplementary-material
Abbreviations
ANP, atrial natriuretic peptide; BAX, Bcl2 associated protein X; BNP, brain natriuretic peptide; ECHO, echocardiography; FT3, free triiodothyronine; FT4, free thyroxine; FS, fractional shortening; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H&E, hematoxylin and eosin; IVSd, diastolic intraventricular septum thickness; LoI/MMI/, low iodine diet/methimazole/perchlorate; LVIDd, diastolic left ventricular inner diameter; LVPWd, diastolic left ventricular posterior wall thickness; mTOR, mechanistic target of rapamycin; MYH6, myosin heavy chain alpha; MYH7, myosin heavy chain beta; PLN, phospholamban; pmTOR, phosphorylated mechanistic target of rapamycin; T4, thyroxine; TAC, transverse aortic constriction; TH, thyroid hormones; TRα, thyroid hormone receptor alpha; TRβ, thyroid hormone receptor beta; TT4, total thyroxine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
References
1. Mendis S, Thygesen K, Kuulasmaa K, Giampaoli S, Mahonen M, Blackett KN, et al. World Health Organization definition of myocardial infarction: 2008-09 revision. Int J Epidemiol. (2011) 40:139–46. doi: 10.1093/ije/dyq165
2. Dillmann W. Cardiac hypertrophy and thyroid hormone signaling. Heart Fail Rev. (2010) 15:125–32. doi: 10.1007/s10741-008-9125-7
3. Pantos C, Mourouzis I, Cokkinos DV. New insights into the role of thyroid hormone in cardiac remodeling:time to reconsider? Heart Fail Rev. (2011) 16:79–96. doi: 10.1007/s10741-010-9185-3
4. Jabbar A, Pingitore A, Pearce SH, Zaman A, Iervasi G, Razvi S. Thyroid hormones and cardiovascular disease. Nat Rev Cardiol. (2017) 14:39–55. doi: 10.1038/nrcardio.2016.174
5. Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, Klein I, et al. Thyroid hormones and cardiovascular function and diseases. J Am Coll Cardiol. (2018) 71:1781–96. doi: 10.1016/j.jacc.2018.02.045
6. Li M, Iismaa SE, Naqvi N, Nicks A, Husain A, Graham RM. Thyroid hormone action in postnatal heart development. Stem Cell Res. (2014) 13:582–91. doi: 10.1016/j.scr.2014.07.001
7. Dillmann WH. Biochemical basis of thyroid hormone action in the heart. Am J Med. (1990) 88:626–30. doi: 10.1016/0002-9343(90)90530-Q
8. Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. (2005) 26:704–28. doi: 10.1210/er.2003-0033
9. Kenessey A, Ojamaa K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem. (2006) 281:20666–72. doi: 10.1074/jbc.M512671200
10. Holt E, Sjaastad I, Lunde PK, Christensen G, Sejersted OM. Thyroid hormone control of contraction and the Ca2+-ATPase/phospholamban complex in adult rat ventricular myocytes. J Mol Cell Cardiol. (1999) 31:645–56. doi: 10.1006/jmcc.1998.0900
11. Kiss E, Jakab G, Kranias EG, Edes I. Thyroid hormone-induced alterations in phospholamban protein expression. Regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. Circ Res. (1994) 75:245–51. doi: 10.1161/01.RES.75.2.245
12. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. (2016) 97:245–62. doi: 10.1016/j.yjmcc.2016.06.001
13. Klein I, Danzi S. Thyroid disease and the heart. Circulation. (2007) 116:1725–35. doi: 10.1161/CIRCULATIONAHA.106.678326
14. Thomas TA, Kuzman JA, Anderson BE, Andersen SM, Schlenker EH, Holder MS, et al. Thyroid hormones induce unique and potentially beneficial changes in cardiac myocyte shape in hypertensive rats near heart failure. Am J Physiol Heart Circ Physiol. (2005) 288:H2118–22. doi: 10.1152/ajpheart.01000.2004
15. Gerdes AM, Iervasi G. Thyroid replacement therapy and heart failure. Circulation. (2010) 122:385–93. doi: 10.1161/CIRCULATIONAHA.109.917922
16. Gerdes AM, Moore JA, Hines JM. Regional changes in myocyte size and number in propranolol-treated hyperthyroid rats. Lab Invest. (1987) 57:708–13.
17. Ito K, Kagaya Y, Shimokawa H. Thyroid hormone and chronically unloaded hearts. Vascul Pharmacol. (2010) 52:138–41. doi: 10.1016/j.vph.2009.10.004
18. Yao J, Eghbali M. Decreased collagen gene expression and absence of fibrosis in thyroid hormone-induced myocardial hypertrophy. Response of cardiac fibroblasts to thyroid hormone in vitro. Circ Res. (1992) 71:831–9. doi: 10.1161/01.RES.71.4.831
19. Hamilton MA, Stevenson LW, Fonarow GC, Steimle A, Goldhaber JI, Child JS, et al. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure. Am J Cardiol. (1998) 81:443–7. doi: 10.1016/S0002-9149(97)00950-8
20. Pingitore A, Galli E, Barison A, Iervasi A, Scarlattini M, Nucci D, et al. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome:a randomized, placebo-controlled study. J Clin Endocrinol Metab. (2008) 93:1351–8. doi: 10.1210/jc.2007-2210
21. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. (2010) 38:1729. doi: 10.3791/1729
22. Schmid E, Neef S, Berlin C, Tomasovic A, Kahlert K, Nordbeck P, et al. Cardiac RKIP induces a beneficial beta-adrenoceptor-dependent positive inotropy. Nat Med. (2015) 21:1298–306. doi: 10.1038/nm.3972
23. Lindsey ML, Kassiri Z, Virag JAI, de Castro Bras LE, Scherrer-Crosbie M. Guidelines for measuring cardiac physiology in mice. Am J Physiol Heart Circ Physiol. (2018) 314:H733–52. doi: 10.1152/ajpheart.00339.2017
24. Engels K, Rakov H, Hones GS, Brix K, Kohrle J, Zwanziger D, et al. Aging alters phenotypic traits of thyroid dysfunction in male mice with divergent effects on complex systems but preserved thyroid hormone action in target organs. J Gerontol A Biol Sci Med Sci. (2019) 74:1162–9. doi: 10.1093/gerona/glz040
25. Weiss RE, Korcarz C, Chassande O, Cua K, Sadow PM, Koo E, et al. Thyroid hormone and cardiac function in mice deficient in thyroid hormone receptor-alpha or -beta:an echocardiograph study. Am J Physiol Endocrinol Metab. (2002) 283:E428–35. doi: 10.1152/ajpendo.00019.2002
26. Kerp H, Engels K, Kramer F, Doycheva D, Hönes GS, Zwanziger D, et al. Age effect on thyroid hormone brain response in male mice. Endocrine. (2019) 66:596–606. doi: 10.1007/s12020-019-02078-6
27. Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. (2009) 15:75–83. doi: 10.1038/nm.1893
28. Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, et al. Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad Sci USA. (2013) 110:7440–5. doi: 10.1073/pnas.1221999110
29. Engels K, Rakov H, Zwanziger D, Hones GS, Rehders M, Brix K, et al. Efficacy of protocols for induction of chronic hyperthyroidism in male and female mice. Endocrine. (2016) 54:47–54. doi: 10.1007/s12020-016-1020-8
30. Zwanziger D, Rakov H, Engels K, Moeller LC, Fuhrer D. Sex-Dependent claudin-1 expression in the liver of euthyroid and hypothyroid mice. Eur Thyroid J. (2015) 4:67–73. doi: 10.1159/000431316
31. Richards DA, Aronovitz MJ, Calamaras TD, Tam K, Martin GL, Liu P, et al. Distinct phenotypes induced by three degrees of transverse aortic constriction in mice. Sci Rep. (2019) 9:5844. doi: 10.1038/s41598-019-42209-7
32. Rakov H, Engels K, Hones GS, Strucksberg KH, Moeller LC, Koehrle J, et al. Sex-specific phenotypes of hyperthyroidism and hypothyroidism in mice. Biol Sex Differ. (2016) 7:36. doi: 10.1186/s13293-016-0089-3
33. Johann K, Cremer AL, Fischer AW, Heine M, Pensado ER, Resch J, et al. Thyroid-Hormone-Induced browning of white adipose tissue does not contribute to thermogenesis and glucose consumption. Cell Rep. (2019) 27:3385–400. doi: 10.1016/j.celrep.2019.05.054
34. Leask A. Getting to the heart of the matter:new insights into cardiac fibrosis. Circ Res. (2015) 116:1269–76. doi: 10.1161/CIRCRESAHA.116.305381
35. Cokkinos DV, Chryssanthopoulos S. Thyroid hormones and cardiac remodeling. Heart Fail Rev. (2016) 21:365–72. doi: 10.1007/s10741-016-9554-7
36. Nicolini G, Pitto L, Kusmic C, Balzan S, Sabatino L, Iervasi G, et al. New insights into mechanisms of cardioprotection mediated by thyroid hormones. J Thyroid Res. (2013) 2013:264387. doi: 10.1155/2013/264387
37. Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure:signaling pathways and novel therapeutic targets. Arch Toxicol. (2015) 89:1401–38. doi: 10.1007/s00204-015-1477-x
38. Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. (2018) 122:489–505. doi: 10.1161/CIRCRESAHA.117.311147
39. Kuzman JA, O'Connell TD, Gerdes AM. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. (2007) 148:3477–84. doi: 10.1210/en.2007-0099
40. McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. (2004) 109:3050–5. doi: 10.1161/01.CIR.0000130641.08705.45
41. Morales CR, Li DL, Pedrozo Z, May HI, Jiang N, Kyrychenko V, et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci Signal. (2016) 9:ra34. doi: 10.1126/scisignal.aad5736
42. Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. (1997) 100:2362–70. doi: 10.1172/JCI119776
43. Park WJ, Oh JG. SERCA2a:a prime target for modulation of cardiac contractility during heart failure. BMB Rep. (2013) 46:237–43. doi: 10.5483/BMBRep.2013.46.5.077
44. Schwartz K, Boheler KR, de la Bastie D, Lompre AM, Mercadier JJ. Switches in cardiac muscle gene expression as a result of pressure and volume overload. Am J Physiol. (1992) 262:R364–9. doi: 10.1152/ajpregu.1992.262.3.R364
45. Antikainen RL, Peters R, Beckett NS, Fagard RH, Wang JG, Rajkumar C, et al. Left ventricular hypertrophy is a predictor of cardiovascular events in elderly hypertensive patients:Hypertension in the very elderly trial. J Hypertens. (2016) 34:2280–6. doi: 10.1097/HJH.0000000000001073
46. Amin A, Chitsazan M, Taghavi S, Ardeshiri M. Effects of triiodothyronine replacement therapy in patients with chronic stable heart failure and low-triiodothyronine syndrome:a randomized, double-blind, placebo-controlled study. ESC Heart Fail. (2015) 2:5–11. doi: 10.1002/ehf2.12025
47. Suarez J, Scott BT, Suarez-Ramirez JA, Chavira CV, Dillmann WH. Thyroid hormone inhibits ERK phosphorylation in pressure overload-induced hypertrophied mouse hearts through a receptor-mediated mechanism. Am J Physiol Cell Physiol. (2010) 299:C1524–9. doi: 10.1152/ajpcell.00168.2010
48. Belke DD, Gloss B, Swanson EA, Dillmann WH. Adeno-associated virus-mediated expression of thyroid hormone receptor isoforms-alpha1 and -beta1 improves contractile function in pressure overload-induced cardiac hypertrophy. Endocrinology. (2007) 148:2870–7. doi: 10.1210/en.2007-0009
49. Olivares EL, Marassi MP, Fortunato RS, da Silva AC, Costa-e-Sousa RH, Araujo IG, et al. Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats a time course study. Endocrinology. (2007) 148:4786–92. doi: 10.1210/en.2007-0043
50. Khalife WI, Tang YD, Kuzman JA, Thomas TA, Anderson BE, Said S, et al. Treatment of subclinical hypothyroidism reverses ischemia and prevents myocyte loss and progressive LV dysfunction in hamsters with dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2005) 289:H2409–15. doi: 10.1152/ajpheart.00483.2005
51. Paolino BS, Pomerantzeff PM, Dallan LAO, Gaiotto FA, Preite NZ, Latronico AC, et al. Myocardial Inactivation of Thyroid Hormones in Patients with Aortic Stenosis. Thyroid. (2017) 27:738–45. doi: 10.1089/thy.2016.0514
52. Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, Celi FS, et al. Guidelines for the treatment of hypothyroidism:prepared by the american thyroid association task force on thyroid hormone replacement. Thyroid. (2014) 24:1670–751. doi: 10.1089/thy.2014.0028
53. Rodondi N, den Elzen WP, Bauer DC, Cappola AR, Razvi S, Walsh JP, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. (2010) 304:1365–74. doi: 10.1001/jama.2010.1361
54. Cetin EH, Cetin MS, Konte HC, Ocak K, Yaman NM, Hepsen S, et al. Lower levels of triiodothyronine are associated with poor hemodynamic profile and all-cause mortality in heart failure. Biomark Med. (2021) 15:273–83. doi: 10.2217/bmm-2020-0425
55. Stott DJ, Rodondi N, Kearney PM, Ford I, Westendorp RGJ, Mooijaart SP, et al. Thyroid hormone therapy for older adults with subclinical hypothyroidism. N Engl J Med. (2017) 376:2534–44. doi: 10.1056/NEJMoa1603825
56. Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, et al. 2016 American thyroid association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. (2016) 26:1343–421. doi: 10.1089/thy.2016.0229
57. Corona G, Croce L, Sparano C, Petrone L, Sforza A, Maggi M, et al. Thyroid and heart, a clinically relevant relationship. J Endocrinol Invest. (2021). doi: 10.1007/s40618-021-01590-9. [Epub ahead of print].
58. Selmer C, Olesen JB, Hansen ML, von Kappelgaard LM, Madsen JC, Hansen PR, et al. Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events:a large population study. J Clin Endocrinol Metab. (2014) 99:2372–82. doi: 10.1210/jc.2013-4184
59. Cappola AR, Fried LP, Arnold AM, Danese MD, Kuller LH, Burke GL, et al. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA. (2006) 295:1033–41. doi: 10.1001/jama.295.9.1033
60. Cappola AR, Desai AS, Medici M, Cooper LS, Egan D, Sopko G, et al. Thyroid and cardiovascular disease:research agenda for enhancing knowledge, prevention, and treatment. Thyroid. (2019) 29:760–77. doi: 10.1089/thy.2018.0416
61. Panday P, Arcia Franchini AP, Iskander B, Anwer F, Oliveri F, Kakargias F, et al. Subclinical hypothyroidism in geriatric population and its association with heart failure. Cureus. (2021) 13:e14296. doi: 10.7759/cureus.14296
62. Pol CJ, Muller A, Zuidwijk MJ, van Deel ED, Kaptein E, Saba A, et al. Left-ventricular remodeling after myocardial infarction is associated with a cardiomyocyte-specific hypothyroid condition. Endocrinology. (2011) 152:669–79. doi: 10.1210/en.2010-0431
63. Moruzzi P, Doria E, Agostoni PG, Capacchione V, Sganzerla P. Usefulness of L-thyroxine to improve cardiac and exercise performance in idiopathic dilated cardiomyopathy. Am J Cardiol. (1994) 73:374–8. doi: 10.1016/0002-9149(94)90011-6
64. Stabouli S, Papakatsika S, Kotsis V. Hypothyroidism and hypertension. Expert Rev Cardiovasc Ther. (2010) 8:1559–65. doi: 10.1586/erc.10.141
65. Strauer BE, Schulze W. Experimental hypothyroidism:depression of myocardial contractile function and hemodynamics and their reversibility by substitution with thyroid hormones. Basic Res Cardiol. (1976) 71:624–44. doi: 10.1007/BF01906408
66. Ladenson PW. Recognition and management of cardiovascular disease related to thyroid dysfunction. Am J Med. (1990) 88:638–41. doi: 10.1016/0002-9343(90)90532-I
67. Zwaveling J, Batink HD, de Jong J, Winkler Prins EA, Pfaffendorf M, van Zwieten PA. Thyroid hormone modulates inotropic responses, alpha-adrenoceptor density and catecholamine concentrations in the rat heart. Naunyn Schmiedebergs Arch Pharmacol. (1996) 354:755–64. doi: 10.1007/BF00166902
68. Barreto-Chaves ML, Senger N, Fevereiro M, Parletta AC, Takano A. Impact of hyperthyroidism on cardiac hypertrophy. Endocr Connect. (2020) 9: R59–R69. doi: 10.1530/EC-19-0543
69. Wakatsuki T, Schlessinger J, Elson EL. The biochemical response of the heart to hypertension and exercise. Trends Biochem Sci. (2004) 29:609–17. doi: 10.1016/j.tibs.2004.09.002
70. Houser SR, Molkentin JD. Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci Signal. (2008) 1:pe31. doi: 10.1126/scisignal.125pe31
71. Mourouzis I, Mantzouratou P, Galanopoulos G, Kostakou E, Roukounakis N, Kokkinos AD, et al. Dose-dependent effects of thyroid hormone on post-ischemic cardiac performance:potential involvement of Akt and ERK signalings. Mol Cell Biochem. (2012) 363:235–43. doi: 10.1007/s11010-011-1175-9
72. Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. (2018) 24:39–49. doi: 10.1038/nm.4447
73. Schwartz K, Carrier L, Chassagne C, Wisnewsky C, Boheler KR. Regulation of myosin heavy chain and actin isogenes during cardiac growth and hypertrophy. Symp Soc Exp Biol. (1992) 46:265–72.
74. Schwartz K, Carrier L, Lompre AM, Mercadier JJ, Boheler KR. Contractile proteins and sarcoplasmic reticulum calcium-ATPase gene expression in the hypertrophied and failing heart. Basic Res Cardiol. (1992) 87(Suppl. 1):285–90. doi: 10.1007/978-3-642-72474-9_24
75. Lee Y, Nadal-Ginard B, Mahdavi V, Izumo S. Myocyte-specific enhancer factor 2 and thyroid hormone receptor associate and synergistically activate the alpha-cardiac myosin heavy-chain gene. Mol Cell Biol. (1997) 17:2745–55. doi: 10.1128/MCB.17.5.2745
76. McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sazo K, et al. Modulation of angiotensin II-mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res. (2010) 106:952–60. doi: 10.1161/CIRCRESAHA.109.209007
77. Biondi B, Bartalena L, Cooper DS, Hegedus L, Laurberg P, Kahaly GJ. The 2015 European thyroid association guidelines on diagnosis and treatment of endogenous subclinical hyperthyroidism. Eur Thyroid J. (2015) 4:149–63. doi: 10.1159/000438750
Keywords: thyroid hormones, maladaptive cardiac hypertrophy, pressure-overload, heart failure, mice
Citation: Kerp H, Hönes GS, Tolstik E, Hönes-Wendland J, Gassen J, Moeller LC, Lorenz K and Führer D (2021) Protective Effects of Thyroid Hormone Deprivation on Progression of Maladaptive Cardiac Hypertrophy and Heart Failure. Front. Cardiovasc. Med. 8:683522. doi: 10.3389/fcvm.2021.683522
Received: 22 March 2021; Accepted: 07 June 2021;
Published: 30 July 2021.
Edited by:
Leonardo Roever, Federal University of Uberlandia, BrazilReviewed by:
Hui-Hua Li, Capital Medical University, ChinaRushita Bagchi, University of Colorado Anschutz Medical Campus, United States
Copyright © 2021 Kerp, Hönes, Tolstik, Hönes-Wendland, Gassen, Moeller, Lorenz and Führer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristina Lorenz, bG9yZW56JiN4MDAwNDA7dG94aS51bmktd3VlcnpidXJnLmRl; Dagmar Führer, ZGFnbWFyLmZ1ZWhyZXImI3gwMDA0MDt1ay1lc3Nlbi5kZQ==
†These authors have contributed equally to this work and share last authorship