 Yi Lu
Yi Lu Tan Yuan
Tan Yuan Xinjia Min1
Xinjia Min1 Zhen Yuan
Zhen Yuan Zhejun Cai
Zhejun Cai- 1Department of Cardiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Jiaxing Key Laboratory of Cardiac Rehabilitation, Jiaxing, China
Vascular calcification (VC) is an urgent worldwide health issue with no available medical treatment. It is an active cell-driven process by osteogenic differentiation of vascular cells with complex mechanisms. The AMP-activated protein kinase (AMPK) serves as the master sensor of cellular energy status. Accumulating evidence reveals the vital role of AMPK in VC progression. AMPK is involved in VC in various ways, including inhibiting runt-related transcription factor 2 signaling pathways, triggering autophagy, attenuating endoplasmic reticulum stress and dynamic-related protein 1-mediated mitochondrial fission, and activating endothelial nitric oxide synthase. AMPK activators, like metformin, are associated with reduced calcification deposits in certain groups of patients, indicating that AMPK is a potential therapeutic target for VC.
Introduction
Vascular calcification (VC) is characterized by accumulating calcium deposits in the tunica intima and tunica media of the vessel wall. The mineral deposition results in stiffness of conduit arteries and impaired elasticity of vessels that preserve distal perfusion during the cardiac cycle (1). The burden of VC, especially coronary artery calcification, is an independent risk factor for cardiovascular events and long-term all-cause mortality (2), which constitutes a critical medical problem with aggregating economic burden (3). However, due to the complexity of the underlying mechanism of VC (4), invasive transcatheter procedures and surgeries are the only available options for severe calcific vascular diseases (5), and there is no clinically approved medical therapy for VC, so far.
VC was once deemed as a passive, unregulated, degenerative process in the past (6). However, accumulating evidence suggests that VC is an active cell-driven process (6, 7). Vascular smooth muscle cells (VSMCs) are the essential constituents of the vascular wall. Those contraction-related proteins secreted by contractile type of VSMCs are critical for regulating blood pressure and maintaining the extracellular matrix (ECM) of vessels (8). However, the contractile phenotype is predisposed to osteoblastic phenotypic transition under certain local stimuli like inflammation (9). This phenotypic transdifferentiation is a hallmark in the pathogenesis of VC, which is characterized by the loss of contraction-related proteins and the accumulation of osteoblastic-involved proteins, including runt-related transcription factor 2 (Runx2), alkaline phosphatase (ALP), and osteopontin (9–11).
The AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine-protein kinase. It is a heterotrimeric complex consisting of a catalytic α subunit and two regulatory β and γ subunits (12). The α subunit has two isoforms (α1 and α2), which are differentially distributed in different cells (13). AMPK serves as a critical cellular energy sensor that is expressed ubiquitously in eukaryotic cells (14). Knowledge of AMPK upstream inducers and downstream targets is expanding (15, 16). AMPK can be activated via canonical and non-canonical mechanisms (16). The increase of AMP: ATP and ADP: ATP ratio during cellular energy stress triggers AMPK through canonical mechanisms (16). 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), which can be converted into AMP analogs that bind the γ subunit of AMPK, is wildly used in laboratory research as a non-canonical AMPK activator. Intracellular Ca2+ can activate AMPK by phosphorylating Thr172 by calmodulin-dependent protein kinase CaMKKβ (17, 18). Both canonical and non-canonical mechanisms are involved in the Ca2+/CaMKKβ pathway. What is more, multiple commercialized drugs, like metformin, simvastatin, and resveratrol, can activate AMPK indirectly by inhibiting ATP synthesis (15).
Besides the classical role of AMPK in metabolic regulation, increasing evidence indicates that AMPK is a crucial player in the pathogenesis of VC. Pharmacological activation of AMPK can significantly inhibit VSMC calcification (19, 20). Treatment of AMPK activator metformin was associated with lower coronary and extremity artery calcification burden in diabetic patients (21, 22). In this review, we will focus on the recent advances concerning the role of AMPK in VC and interpret its potential therapeutic utility.
The Protective Role of AMPK Against VC
AMPK-Runx2 Signaling Pathways
Runx2, also named Core-binding factor alpha 3 subunit (CBFA1), is a well-accepted activator of osteoblast differentiation-related genes (23). The elevated expression of Runx2, together with other osteoclastic-associated proteins, in calcified human vascular tissues (11) and mice VC models (24, 25), cast light on its role in vascular calcification.
The relationship between AMPK and Runx2 was first reported in osteogenesis (26, 27). Jang et al. found that metformin increased the expression of Runx2 via AMPK, which resulted in the stimulation of osteoblast differentiation (27). However, AMPK-Runx2 signaling pathway seemingly exerts an opposite role in VSMCs differentiation. Previous studies had demonstrated that Runx2 was expressed in VSMCs rather than macrophages in the calcified lesions (19, 28), and VSMC autonomous Runx2 was essential for vascular osteogenesis (28). Vascular calcification was markedly inhibited in VSMC-specific Runx2-deficiency mice fed with high-fat diet, which was accompanied by decreased macrophage infiltration and osteogenic differentiation (29). Cao et al. investigated AMPK-Runx2 in VSMCs and reported that AMPK activation downregulated the Runx2 expression in VSMCs (30). Our previous work further investigated the underlying mechanisms. We found that the activation of AMPKα1 could phosphorylate PIAS1, the SUMO E3-ligase of Runx2, to enhance the instability of Runx2. Moreover, deficiency of AMPKα1 in VSMC resulted in the upregulated expression of Runx2 and promoted osteoblastic differentiation of VSMCs. On the other hand, chronic metformin treatment could prevent the VC process and down-regulate Runx2 level in Apoe−/− mice through activating AMPKα1 (19). One possible explanation for the opposite roles of AMPK-Runx2 in osteoblasts and VSMC differentiation might be the different responses of Smurf1 (the ubiquitin E3-ligase of Runx2) upon AMPK phosphorylation (19, 31).
Besides the direct effect of AMPK-Runx2 pathway on VSMC transdifferentiation, a recent study indicated that AMPK could also inhibit VC by regulating receptor activator of nuclear factor kappa-B ligand (RANKL) (32). RANKL serves as a chemoattractant that induces the infiltration of macrophage and the transformation of macrophage into bone-resorbing osteoclast-like cells, which further accelerate the process of VC (29, 32, 33). Since RANKL is a known downstream factor of Runx2 (34), it is reasonable to hypothesize that Runx2 mediates the AMPK-RANKL pathway.
To sum up, the activation of AMPK could down-regulate the expression and activity of Runx2 both in the translational and post-translational levels, which results in the inhibition of osteoblastic differentiation of VSMCs. Besides, AMPK-Runx2 signaling pathway may be involved in the infiltration and transformation of macrophages by down-regulating the level of RANKL.
AMPK and Autophagy Pathways
Autophagy is indispensable for human health by degrading cellular components like dysfunctional proteins or organelles in lysosomes. This catabolic process is up-regulated under specific stimuli like nutrient deprivation (35), resulting in cellular death and metabolic stress (15). Recent evidence suggests that autophagy was also implicated in VC development (36–38). Autophagy is enhanced in VC models, and the activation of autophagy ameliorates the pathology of VC both in calcified VSMCs (39) and rat VC models (40). One possible explanation is autophagy could inhibit the apoptosis and osteoblastic transformation of VSMCs (41).
AMPK is an integral part of autophagy with complex mechanisms. By phosphorylating tuberous sclerosis complex 2 (42), subunit raptor of the mechanistic target of rapamycin (mTOR) (43) and Unc-51-like kinase 1 (ULK1) (44), AMPK initiates autophagy directly (15). Meanwhile, by regulating the expression of relevant downstream transcription factors like FOXO3, AMPK can also initiate autophagy indirectly (45). Recent studies had investigated the protective role of autophagy in VC through AMPK activation. By activating AMPK with melatonin (41) or ghrelin (36), the process of autophagy was enhanced, which resulted in reduced VSMC osteoblastic differentiation both in cell culture (41) and rat models of VC (36). On the other hand, treatment of aldosterone or advanced glycation end products (AGEs) facilitated VC by inhibiting AMPK-dependent autophagy (46). Pretreatment of AMPK activator AICAR could upregulate the autophagy level and reverse the effect of AGEs on osteoblastic differentiation of VSMCs (41, 47). AMPK/mTOR signaling pathway was the possible involving mechanism (41, 47, 48).
Taken together, AMPK activation was associated with the enhancement of autophagy and, subsequently, inhibited VSMC calcification. However, most of the studies mentioned above were conducted in cultured VSMCs. More studies with direct evidence are needed to verify these findings in vivo.
AMPK and Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is an essential intracellular organelle that acts as a protein synthesis factory. ER has pivotal roles in coordinating energetic disturbance via regulating metabolism and cell fate decisions (49). The disruption of ER homeostasis is defined as ER stress (ERS), resulting in the activation of PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6), which causes an adaptive signaling pathway named unfolded protein response (UPR) (50, 51). Prolonged ER stress ultimately leads to the modulation of multiple cellular pathways, including apoptosis, necroptosis, autophagy, and UPR-associated morphological changes (49).
Increasing evidence reveals the tight connection between AMPK activation and ERS in different disease models (52–54). AMPK antagonists abolished deficiency-mediated inhibition of ERS in VSMCs incubated with calcifying media (55). Biomarkers of ERS were increased significantly in calcification lesions (56, 57) and were associated with VSMC apoptosis (56). Compared with Apoe−/− littermates, the ERS and prevalence of atherosclerosis was significantly increased in Apoe−/−Prkaa2−/− mice (58, 59). However, as the last stage of atherosclerosis, the role of AMPK in ERS-mediated vascular calcification has not been fully elucidated.
Among all the effector molecules in response to ERS, transcription factor 4 (ATF4) is proved to be of enormous significance in VC. ATF4 was up-regulated in calcified aortas and VSMCs, while inhibition of ERS alleviated calcification (55). Previous studies found that PERK-eukaryotic initiation factor 2α(eIF2α)-ATF4 signaling pathway was involved in ERS-induced VSMCs apoptosis and osteoblast differentiation during the process of VC (60, 61). Li et al. found that death-associated protein kinase 3 (DAPK3) inactivated AMPK signaling and promote the expression of ERS-related protein (including ATF4), thus leading to osteogenic differentiation of VSMCs and VC (55).
In summary, the activation of AMPK inhibits ERS to ameliorate VC. ERS downstream protein like ATF4 mediates phenotypic transformation and apoptosis of VSMC that promotes the VC process. More specific inhibitors like ATF4 inhibitors may be further validated in clinical application in VC.
AMPK-eNOS-NO Signaling Pathway
Endothelium-derived nitric oxide (NO) is a messenger molecule that is crucial in the maintenance of vascular function (62, 63). Endogenous NO functions as a modulator of VSMC proliferation and migration (64, 65), which can inhibit VC by interfering with transforming growth factor-beta (TGF-β) signaling (66). Endothelial nitric oxide synthase (eNOS) is the primary enzyme for NO production in endothelial cells. Genetic lack of eNOS was associated with raised atherosclerotic lesions and valvular calcification in mice models (67, 68). A recent study revealed that eNOS deficiency was also associated with the exacerbation of aortic calcification (69). On the other hand, exercise training prevented eNOS down-regulation and resulted in fewer calcification deposits in rat VC models (70). However, in contrast to previous findings that eNOS mainly play a protective role in VC, Tziakas et al. found that erythrocyte-origin eNOS might be harmful in the development of VC (71).
AMPK is a well-defined regulator of eNOS. By phosphorylating eNOS at Ser1177/1179, AMPK enhances the activity of eNOS in a post-translation manner (72). Kanazawa et al. found that metformin could induce the differentiation and mineralization of osteoblasts via activating AMPK (73). The elevated AMPK expression protects human coronary artery endothelial cells from diabetic lipoapoptosis via increasing eNOS synthesis (74). Daily injections of AMPK activator AICAR attenuated high-fat diet-induced arterial stiffening in Klotho-deficient mice, together with increased level of phosphorylated eNOS (75). Besides endothelial cells, VSMCs are also known origins of eNOS (66). Cao et al. explored the underlying mechanism in rat aortic VSMCs with a β-glycerophosphate-induced VC model and found that metformin-mediated calcification protection was AMPK-eNOS-NO-dependent (30). AMPK activation by metformin treatment was accompanied by increased eNOS level and NO overproduction (30, 76). Inhibition of either AMPK or eNOS abolished metformin-mediated VC prevention, indicating an essential protective role of the AMPK-eNOS-NO pathway in VC development (30). Due to the limitation of in vitro study, further study is needed to verify the protective role of AMPK-eNOS-NO in VC in animal models.
These findings suggest the activation of AMPK-eNOS-NO signaling pathway is associated with the amelioration of VC. Since endothelial cells rather than VSMCs are the primary source of eNOS, it is more reasonable that AMPK acts on endothelial cells' eNOS signaling to prevent VC.
AMPK and Mitochondrial Dynamics
Mitochondria are continually undergoing fission and fusion, termed as mitochondrial dynamics, under the control of specific fission and fusion machinery (77, 78). A proper balance in mitochondrial dynamics is critical for mitochondrial morphology, biogenesis, degradation, and cellular apoptosis (79, 80). Recruitment of dynamic-related protein 1 (DRP1) from the cytosol is required in mitochondrial fission, which causes constriction and eventual division of the mitochondria (78, 81). Not until recently did scientists uncovered that DRP1 promoted cardiovascular calcification via regulating osteogenic differentiation (82). The inhibition of expression and phosphorylation of DRP1 ameliorated the apoptosis of VSMC and attenuated VC in rodent VC models (83, 84).
AMPK is genetically required for cells to process rapid mitochondrial fission. By direct phosphorylating mitochondrial fission factor (MFF, the dominant receptor of DRP1), AMPK can acutely trigger mitochondrial fission (85, 86). Also, Drp1 is a known downstream factor of AMPK. In addition to regulating the expression of Drp1, AMPK can phosphorylate DRP1 at Ser-637, resulting in the inhibition of Drp1 activity and its translocation to mitochondria (87, 88). Previous studies had shown that AMPK activation could suppress atherosclerosis and endothelial dysfunction by reducing DRP1-mediated mitochondrial fission (88, 89). Activation of AMPK by metformin reduced DRP1 expression, mitochondrial fragmentation, and plaque formation in diabetic mice models, while AMPKα2 deficiency abolished the effect s of metformin on atherosclerosis in Apoe−/−Prkaa2−/− mice. Another recently published work described the protective role of the AMPK-DRP1 pathway in the calcification of VSMCs (90). By activating AMPK expression with melatonin, the expression of Drp1 was decreased and subsequently inhibited mitochondrial fission, which resulted in reduced apoptosis, Runx2 expression, and calcium deposition (90).
In general, AMPK-mediated mitochondrial fission attenuates VC by inhibiting the expression of DRP1. However, the role of mitochondrial dynamics in VC has not been fully elucidated. More studies are needed to prove the function of AMPK-dependent DRP1-mediated mitochondrial fission in VC. DRP1 activators may also be applied as a target for VC treatment.
Endogenous AMPK Activator and VC
As mentioned above, the expression and activity of AMPK are under strict and delicate regulation, which is tightly associated with ATP metabolism. ATP is metabolized by ENPP1 into AMP and pyrophosphate, which was then further hydrolyzed by CD73 to build adenosine and phosphate (91). Phosphate, calcium, pyrophosphate (PPi), and adenosine are important inducers that activate AMPK canonically. These endogenous AMPK activators can also influence the process of VC. High phosphate level is considered the main determinants of VC in patients with chronic kidney disease (92). Hyperphosphatemic triggers diverse signaling pathways (including Runx2) that enhance the sensitivity of VSMCs to calcification (93), and reduces levels of calcification inhibitors (94). On the other hand, PPi is a well-accept endogenous inhibitor of biomineralization (95). The presence of PPi inhibits the calcification of rat aortas in vitro (96). Humans lacking ectonucleotide pyrophosphorylase1 (extracellular PPi synthesizer) develop severe VC at an early age (97). What is more, recent studies demonstrate that adenosine might serve as an endogenous inhibitor of VC through regulating the expression of tissue non-specific alkaline phosphatase (TNAP) (98, 99).
Applications of AMPK in VC Clinical Settings
Accumulating evidence shows that VC is an active cell-driven process (6, 7), which poses a potential for therapeutic targeting (41). Considering the protective effects of AMPK in VC, it is reasonable to speculate that AMPK activators can prevent VC. Multiple communalized drugs can activate AMPK indirectly, either by inhibiting ATP synthesis (like metformin, statins) (16, 100), or by inhibiting tetrahydrofolate-utilizing enzymes that catalyze ZMP to purine nucleotides (like pemetrexed, methotrexate) (16, 101). Since pemetrexed and methotrexate function as immune suppressors, we mainly discuss drugs, including metformin and statins in current review.
Metformin, the first-line oral anti-diabetic drug, can activate AMPK in a dose- and time-dependent manner both in vivo and in vitro (73, 102, 103), which might be implicated in VC treatment and diabetic complications prevention (104). Metformin prescription resulted in a significant reduction of circulating osteoprotegerin, a biomarker of VC, in diabetic patients (105). The VC progression in the coronary artery and peripheral artery was inhibited by metformin usage (22, 106). Our group had reaffirmed that metformin prescription was associated with lower coronary artery calcification levels among patients with type 2 diabetic mellitus (T2DM) (21). The association was independent of age, gender, duration of T2DM and renal function (21). Another group investigated metformin usage in prediabetic patients, which demonstrated its consistent VC protective effect in male prediabetic subjects (107). We believe that metformin's protective effect on VC, which was seemingly independent of serum glucose, is at least partly achieved through AMPK activation (21).
Discussion and Perspectives
VC is an urgent worldwide health issue with no available medical treatment. Accumulating evidence shows that AMPK plays a vital and protective role in developing VC via distinct signaling pathways, including the Runx2, autophagy, ERS, eNOS activation, and DRP1 (Figure 1).
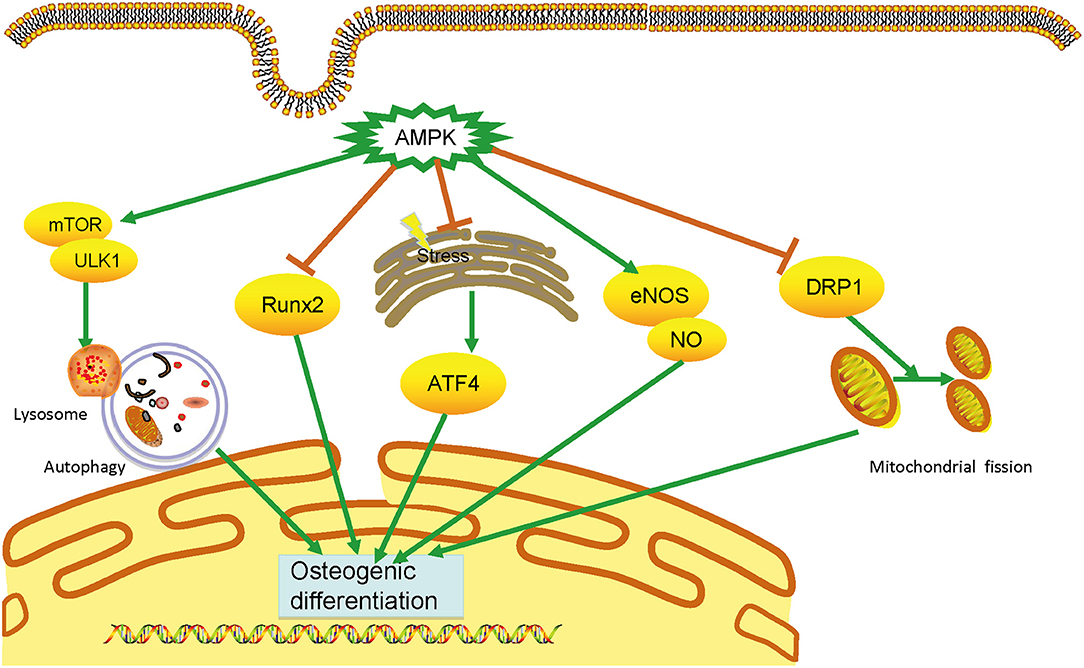
Figure 1. Mechanisms of AMPK in vascular calcification. AMPK plays an essential role in calcification through multiple mechanisms, including inhibiting runt-related transcription factor 2 (Runx2) signaling pathways, triggering autophagy, attenuating endoplasmic reticulum stress, and dynamic-related protein 1(DRP1)-mediated mitochondrial fission, and activating endothelial nitric oxide synthase. By preventing the osteogenic differentiation of vascular smooth muscle cells, AMPK can prevent vascular calcification development. AMPK, AMP-activated protein kinase; DRP1, dynamic-related protein 1; eNOS, endothelial nitric oxide synthase; ERS, endoplasmic reticulum stress; VSMC, vascular smooth muscle cell.
Activation of AMPK by medicines is a potential therapeutic approach for vascular calcification. However, there are still many unanswered questions in the field. Agents that precisely targeting AMPK or the subunits of AMPK are yet to be developed. While metformin is mainly prescribed for diabetes patients, it will be interesting to determine metformin's effect on VC in otherwise non-diabetic subjects. Whether AMPK activation could prevent or reverse the pathological process of VC needs to be explored. For conditions like chronic kidney disease prone to develop severe VC, whether AMPK activation is protective against VC is unknown. More clinical studies, especially prospective randomized clinical trials, are required to confirm the therapeutic target of AMPK for VC.
Author Contributions
ZC and YL designed the study. YL and TY drafted the manuscript. XM and ZY contributed data. All authors were involved in critically revising the manuscript.
Funding
This work was supported by funding from the National Natural Science Foundation of China (No. 81970396 to ZC and No. 81900416 to YL), and the Zhejiang Provincial Natural Science Foundation for Distinguished Young Scholars (No. LR20H020002 to ZC), and the Zhejiang Provincial Natural Science Foundation (No. LQ19H020006 to YL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Soldatos G, Jandeleit-Dahm K, Thomson H, Formosa M, D'Orsa K, Calkin AC, et al. Large artery biomechanics and diastolic dysfunctionin patients with Type 2 diabetes. Diabet Med. (2011) 28:54–60. doi: 10.1111/j.1464-5491.2010.03146.x
2. Grandhi GR, Mirbolouk M, Dardari ZA, Al-Mallah MH, Rumberger JA, Shaw LJ, et al. Interplay of coronary artery calcium and risk factors for predicting CVD/CHD mortality: the CAC consortium. JACC Cardiovasc Imaging. (2020) 13:1175–86. doi: 10.1016/j.jcmg.2019.08.024
3. Garrison LP Jr, Lewin J, Young CH, Genereux P, Crittendon J, Mann MR, et al. The clinical and cost burden of coronary calcification in a Medicare cohort: an economic model to address under-reporting and misclassification. Cardiovasc Revasc Med. (2015) 16:406–12. doi: 10.1016/j.carrev.2015.08.002
4. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol. (2019) 39:1307–16. doi: 10.1161/ATVBAHA.118.311576
5. Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol. (2019) 16:261–74. doi: 10.1038/s41569-018-0123-8
6. Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. (2004) 24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42
7. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. (2001) 89:1147–54. doi: 10.1161/hh2401.101070
8. Lu CL, Liao MT, Hou YC, Fang YW, Zheng CM, Liu WC, et al. Sirtuin-1 and its relevance in vascular calcification. Int J Mol Sci. (2020) 21:1593. doi: 10.3390/ijms21051593
9. Alves RD, Eijken M J. van de Peppel J, van Leeuwen JP. Calcifying vascular smooth muscle cells and osteoblasts: independent cell types exhibiting extracellular matrix and biomineralization-related mimicries. BMC Genomics. (2014) 15:965. doi: 10.1186/1471-2164-15-965
10. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
11. Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol. (2003) 23:489–94. doi: 10.1161/01.ATV.0000059406.92165.31
12. Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J. (2016) 283:2987–3001. doi: 10.1111/febs.13698
13. Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. (1996) 271:611–4. doi: 10.1074/jbc.271.2.611
14. Gonzalez A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. (2020) 31:472–92. doi: 10.1016/j.cmet.2020.01.015
15. Xiang H, Zhang J, Lin C, Zhang L, Liu B, Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. (2020) 10:569–81. doi: 10.1016/j.apsb.2019.10.003
16. Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. (2016) 26:190–201. doi: 10.1016/j.tcb.2015.10.013
17. Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. (2005) 2:21–33. doi: 10.1016/j.cmet.2005.06.005
18. Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, Hardie DG. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. (2010) 426:109–18. doi: 10.1042/BJ20091372
19. Cai Z, Ding Y, Zhang M, Lu Q, Wu S, Zhu H, et al. Ablation of adenosine monophosphate-activated protein kinase alpha1 in vascular smooth muscle cells promotes diet-induced atherosclerotic calcification in vivo. Circ Res. (2016) 119:422–33. doi: 10.1161/CIRCRESAHA.116.308301
20. Son BK, Akishita M, Iijima K, Kozaki K, Maemura K, Eto M, et al. Adiponectin antagonizes stimulatory effect of tumor necrosis factor-alpha on vascular smooth muscle cell calcification: regulation of growth arrest-specific gene 6-mediated survival pathway by adenosine 5'-monophosphate-activated protein kinase. Endocrinology. (2008) 149:1646–53. doi: 10.1210/en.2007-1021
21. Lu Y, Wang Y, Weng T, Chen Z, Sun X, Wei J, et al. Association between metformin use and coronary artery calcification in type 2 diabetic patients. J Diabetes Res. (2019) 2019:9484717. doi: 10.1155/2019/9484717
22. Mary A, Hartemann A, Liabeuf S, Aubert CE, Kemel S, Salem JE, et al. Association between metformin use and below-the-knee arterial calcification score in type 2 diabetic patients. Cardiovasc Diabetol. (2017) 16:24. doi: 10.1186/s12933-017-0509-7
23. Vimalraj S, Arumugam B, Miranda PJ, Selvamurugan N. Runx2: structure, function, and phosphorylation in osteoblast differentiation. Int J Biol Macromol. (2015) 78:202–8. doi: 10.1016/j.ijbiomac.2015.04.008
24. Shen J, Zhang N, Lin YN, Xiang P, Liu XB, Shan PF, et al. Regulation of vascular calcification by growth hormone-releasing hormone and its agonists. Circ Res. (2018) 122:1395–408. doi: 10.1161/CIRCRESAHA.117.312418
25. Liu L, Zeng P, Yang X, Duan Y, Zhang W, Ma C, et al. Inhibition of vascular calcification. Arterioscler Thromb Vasc Biol. (2018) 38:2382–95. doi: 10.1161/ATVBAHA.118.311546
26. Kim EK, Lim S, Park JM, Seo JK, Kim JH, Kim KT, et al. Human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by AMP-activated protein kinase. J Cell Physiol. (2012) 227:1680–7. doi: 10.1002/jcp.22892
27. Jang WG, Kim EJ, Bae IH, Lee KN, Kim YD, Kim DK, et al. Metformin induces osteoblast differentiation via orphan nuclear receptor SHP-mediated transactivation of Runx2. Bone. (2011) 48:885–93. doi: 10.1016/j.bone.2010.12.003
28. Lin ME, Chen TM, Wallingford MC, Nguyen NB, Yamada S, Sawangmake C, et al. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc Res. (2016) 112:606–616. doi: 10.1093/cvr/cvw205
29. Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, et al. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. (2012) 111:543–52. doi: 10.1161/CIRCRESAHA.112.267237
30. Cao X, Li H, Tao H, Wu N, Yu L, Zhang D, et al. Metformin inhibits vascular calcification in female rat aortic smooth muscle cells via the AMPK-eNOS-NO pathway. Endocrinology. (2013) 154:3680–9. doi: 10.1210/en.2013-1002
31. Shimazu J, Wei J, Karsenty G. Smurf1 inhibits osteoblast differentiation, bone formation, and glucose homeostasis through serine 148. Cell Rep. (2016) 15:27–35. doi: 10.1016/j.celrep.2016.03.003
32. Chen Y. Zhao X, Wu H. Transcriptional programming in arteriosclerotic disease: a multifaceted function of the Runx2 (Runt-related transcription factor 2). Arterioscler Thromb Vasc Biol. (2020) 41:20–34. doi: 10.1161/ATVBAHA.120.313791
33. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, et al. Runx2-upregulated receptor activator of nuclear factor kappaB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. (2011) 31:1387–96. doi: 10.1161/ATVBAHA.110.222547
34. Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, et al. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arterioscler Thromb Vasc Biol. (2006) 26:2117–24. doi: 10.1161/01.ATV.0000236428.91125.e6
35. Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. (2011) 13:495–504. doi: 10.1016/j.cmet.2011.04.004
36. Xu M, Liu L, Song C, Chen W, Gui S. Ghrelin improves vascular autophagy in rats with vascular calcification. Life Sci. (2017) 179:23–9. doi: 10.1016/j.lfs.2016.11.025
37. Frauscher B, Kirsch AH, Schabhuttl C, Schweighofer K, Ketszeri M, Pollheimer M, et al. Autophagy protects from uremic vascular media calcification. Front Immunol. (2018) 9:1866. doi: 10.3389/fimmu.2018.01866
38. Zhang Z, Lin J, Tian N, Wu Y, Zhou Y, Wang C, et al. Melatonin protects vertebral endplate chondrocytes against apoptosis and calcification via the Sirt1-autophagy pathway. J Cell Mol Med. (2019) 23:177–93. doi: 10.1111/jcmm.13903
39. Liu D, Cui W, Liu B, Hu H, Liu J, Xie R, et al. Atorvastatin protects vascular smooth muscle cells from TGF-beta1-stimulated calcification by inducing autophagy via suppression of the beta-catenin pathway. Cell Physiol Biochem. (2014) 33:129–41. doi: 10.1159/000356656
40. Zhao Y, Zhao MM, Cai Y, Zheng MF, Sun WL, Zhang SY, et al. Mammalian target of rapamycin signaling inhibition ameliorates vascular calcification via Klotho upregulation. Kidney Int. (2015) 88:711–21. doi: 10.1038/ki.2015.160
41. Chen WR, Yang JQ, Liu F, Shen XQ, Zhou YJ. Melatonin attenuates vascular calcification by activating autophagy via an AMPK/mTOR/ULK1 signaling pathway. Exp Cell Res. (2020) 389:111883. doi: 10.1016/j.yexcr.2020.111883
42. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. (2003) 115:577–90. doi: 10.1016/S0092-8674(03)00929-2
43. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
44. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
45. Li Y, Chen Y. AMPK and autophagy. Adv Exp Med Biol. (2019) 1206:85–108. doi: 10.1007/978-981-15-0602-4_4
46. Gao JW, He WB, Xie CM, Gao M, Feng LY, Liu ZY, et al. Aldosterone enhances high phosphate-induced vascular calcification through inhibition of AMPK-mediated autophagy. J Cell Mol Med. (2020) 24:13648–59. doi: 10.1111/jcmm.15813
47. Liu Y, Li J, Han Y, Chen Y, Liu L, Lang J, et al. Advanced glycation end-products suppress autophagy by AMPK/mTOR signaling pathway to promote vascular calcification. Mol Cell Biochem. (2020) 471:91–100. doi: 10.1007/s11010-020-03769-9
48. Qiu X, Xu Q, Xu T, Wan P, Sheng Z, Han Y, et al. Metformin alleviates beta-glycerophosphate-induced calcification of vascular smooth muscle cells via AMPK/mTOR-activated autophagy. Exp Ther Med. (2021) 21:58. doi: 10.3892/etm.2020.9490
49. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. (2019) 286:241–78. doi: 10.1111/febs.14608
50. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
51. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
52. Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. (2005) 25:9554–75. doi: 10.1128/MCB.25.21.9554-9575.2005
53. Steinberg GR, Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol. (2014) 92:340–5. doi: 10.1038/icb.2014.11
54. Kuznetsov JN, Leclerc GJ, Leclerc GM, Barredo JC. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol Cancer Ther. (2011) 10:437–47. doi: 10.1158/1535-7163.MCT-10-0777
55. Li KX, Du Q, Wang HP, Sun HJ. Death-associated protein kinase 3 deficiency alleviates vascular calcification via AMPK-mediated inhibition of endoplasmic reticulum stress. Eur J Pharmacol. (2019) 852:90–8. doi: 10.1016/j.ejphar.2019.03.007
56. Duan X, Zhou Y, Teng X, Tang C, Qi Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun. (2009) 387:694–9. doi: 10.1016/j.bbrc.2009.07.085
57. Cai Z, Li F, Gong W, Liu W, Duan Q, Chen C, et al. Endoplasmic reticulum stress participates in aortic valve calcification in hypercholesterolemic animals. Arterioscler Thromb Vasc Biol. (2013) 33:2345–54. doi: 10.1161/ATVBAHA.112.300226
58. Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. (2010) 121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928
59. Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, et al. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes. (2010) 59:1386–96. doi: 10.2337/db09-1637
60. Duan XH, Chang JR, Zhang J, Zhang BH, Li YL, Teng X, et al. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis. (2013) 18:1132–44. doi: 10.1007/s10495-013-0861-3
61. Masuda M, Miyazaki-Anzai S, Levi M, Ting TC, Miyazaki M. PERK-eIF2alpha-ATF4-CHOP signaling contributes to TNFalpha-induced vascular calcification. J Am Heart Assoc. (2013) 2:e000238. doi: 10.1161/JAHA.113.000238
62. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. (2004) 109(23 Suppl. 1):III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8
63. Walford G, Loscalzo J. Nitric oxide in vascular biology. J Thromb Haemost. (2003) 1:2112–8. doi: 10.1046/j.1538-7836.2003.00345.x
64. Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. (1989) 83:1774–7. doi: 10.1172/JCI114081
65. Dubey RK, Jackson EK, Luscher TF. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J Clin Invest. (1995) 96:141–9. doi: 10.1172/JCI118014
66. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc Res. (2008) 77:221–30. doi: 10.1093/cvr/cvm049
67. Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J Clin Invest. (2000) 105:451–8. doi: 10.1172/JCI8376
68. El Accaoui RN, Gould ST, Hajj GP, Chu Y, Davis MK, Kraft DC, et al. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. (2014) 306:H1302–13. doi: 10.1152/ajpheart.00392.2013
69. Oe Y, Mitsui S, Sato E, Shibata N, Kisu K, Sekimoto A, et al. Lack of endothelial nitric oxide synthase accelerates ectopic calcification in uremic mice fed an adenine and high phosphorus diet. Am J Pathol. (2020) 191:283–93. doi: 10.1016/j.ajpath.2020.10.012
70. Park JH, Iemitsu M, Maeda S, Kitajima A, Nosaka T, Omi N. Voluntary running exercise attenuates the progression of endothelial dysfunction and arterial calcification in ovariectomized rats. Acta Physiol. (2008) 193:47–55. doi: 10.1111/j.1748-1716.2007.01799.x
71. Tziakas DN, Chalikias G, Pavlaki M, Kareli D, Gogiraju R, Hubert A, et al. Lysed erythrocyte membranes promote vascular calcification. Circulation. (2019) 139:2032–48. doi: 10.1161/CIRCULATIONAHA.118.037166
72. Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. (1999) 443:285–9. doi: 10.1016/S0014-5793(98)01705-0
73. Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Metformin enhances the differentiation and mineralization of osteoblastic MC3T3-E1 cells via AMP kinase activation as well as eNOS and BMP-2 expression. Biochem Biophys Res Commun. (2008) 375:414–9. doi: 10.1016/j.bbrc.2008.08.034
74. Eriksson L, Nystrom T. Activation of AMP-activated protein kinase by metformin protects human coronary artery endothelial cells against diabetic lipoapoptosis. Cardiovasc Diabetol. (2014) 13:152. doi: 10.1186/s12933-014-0152-5
75. Lin Y, Chen J, Sun Z. Antiaging gene klotho deficiency promoted high-fat diet-induced arterial stiffening via inactivation of AMP-activated protein kinase. Hypertension. (2016) 67:564–73. doi: 10.1161/HYPERTENSIONAHA.115.06825
76. Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, et al. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. (2006) 26:1281–7. doi: 10.1161/01.ATV.0000221230.08596.98
77. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. (2015) 163:560–9. doi: 10.1016/j.cell.2015.10.001
78. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. (2014) 15:634–46. doi: 10.1038/nrm3877
79. Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. (2007) 8:870–9. doi: 10.1038/nrm2275
80. Yin F, Cadenas E. Mitochondria: the cellular hub of the dynamic coordinated network. Antioxid Redox Signal. (2015) 22:961–4. doi: 10.1089/ars.2015.6313
81. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
82. Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, et al. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res. (2017) 121:220–33. doi: 10.1161/CIRCRESAHA.116.310293
83. Cui L, Li Z, Chang X, Cong G, Hao L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vascul Pharmacol. (2017) 88:21–9. doi: 10.1016/j.vph.2016.11.006
84. Zhu Y, Han XQ, Sun XJ, Yang R, Ma WQ, Liu NF. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. (2020) 25:321–40. doi: 10.1007/s10495-020-01592-7
85. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Loson OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. (2016) 351:275–81. doi: 10.1126/science.aab4138
86. Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, et al. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. (2014) 25:145–59. doi: 10.1091/mbc.e13-09-0525
87. Li A, Zhang S, Li J, Liu K, Huang F, Liu B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol Cell Endocrinol. (2016) 434:36–47. doi: 10.1016/j.mce.2016.06.008
88. Li J, Wang Y, Wang Y, Wen X, Ma XN, Chen W, et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol. (2015) 86:62–74. doi: 10.1016/j.yjmcc.2015.07.010
89. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. (2017) 66:193–205. doi: 10.2337/db16-0915
90. Chen WR, Zhou YJ, Sha Y, Wu XP, Yang JQ, Liu F. Melatonin attenuates vascular calcification by inhibiting mitochondria fission via an AMPK/Drp1 signalling pathway. J Cell Mol Med. (2020) 24:6043–54. doi: 10.1111/jcmm.15157
91. Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res. (2012) 95:269–80. doi: 10.1093/cvr/cvs187
92. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. (2011) 109:697–711. doi: 10.1161/CIRCRESAHA.110.234914
93. Chavkin NW, Chia JJ, Crouthamel MH, Giachelli CM. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp Cell Res. (2015) 333:39–48. doi: 10.1016/j.yexcr.2015.02.002
94. Ketteler M, Brandenburg V, Jahnen-Dechent W, Westenfeld R, Floege J. Do not be misguided by guidelines: the calcium x phosphate product can be a Trojan horse. Nephrol Dial Transplant. (2005) 20:673–7. doi: 10.1093/ndt/gfh734
95. Fleisch H, Bisaz S. Mechanism of calcification: inhibitory role of pyrophosphate. Nature. (1962) 195:911. doi: 10.1038/195911a0
96. Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI, O'Neill WC. Phosphate-induced vascular calcification: role of pyrophosphate and osteopontin. J Am Soc Nephrol. (2004) 15:1392–401. doi: 10.1097/01.ASN.0000128955.83129.9C
97. Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. (2001) 158:543–54. doi: 10.1016/S0002-9440(10)63996-X
98. Moorhead WJ 3rd, Chu CC, Cuevas RA, Callahan J 4th, Wong R, Regan C, et al. Dysregulation of FOXO1 (Forkhead box O1 protein) drives calcification in arterial calcification due to deficiency of CD73 and is present in peripheral artery disease. Arterioscler Thromb Vasc Biol. (2020) 40:1680–1694. doi: 10.1161/ATVBAHA.119.313765
99. Fujimoto K, Shioi A, Miki Y, Kakutani Y, Morioka T, Shoji T, et al. Adenosine attenuates aortic smooth muscle cell calcification through A3 adenosine receptor. Tohoku J Exp Med. (2019) 249:275–83. doi: 10.1620/tjem.249.275
100. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. (2010) 11:554–65. doi: 10.1016/j.cmet.2010.04.001
101. Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. (2009) 69:5467–74. doi: 10.1158/0008-5472.CAN-08-4979
102. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
103. Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. (2002) 51:2074–81. doi: 10.2337/diabetes.51.7.2074
104. Zhang X, Xiao J, Li R, Qin X, Wang F, Mao Y, et al. Metformin alleviates vascular calcification induced by vitamin D3 plus nicotine in rats via the AMPK pathway. Vascul Pharmacol. (2016) 81:83–90. doi: 10.1016/j.vph.2016.01.002
105. Schinzari F, Tesauro M, Bertoli A, Valentini A, Veneziani A, Campia U, et al. Calcification biomarkers and vascular dysfunction in obesity and type 2 diabetes: influence of oral hypoglycemic agents. Am J Physiol Endocrinol Metab. (2019) 317:E658–E666. doi: 10.1152/ajpendo.00204.2019
106. Fitch K, Abbara S, Lee H, Stavrou E, Sacks R, Michel T, et al. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. AIDS. (2012) 26:587–97. doi: 10.1097/QAD.0b013e32834f33cc
107. Goldberg RB, Aroda VR, Bluemke DA, Barrett-Connor E, Budoff M, Crandall JP, et al. Diabetes prevention program research: effect of long-term metformin and lifestyle in the diabetes prevention program and its outcome study on coronary artery calcium. Circulation. (2017) 136:52–64. doi: 10.1161/CIRCULATIONAHA.116.025483
Keywords: AMP-activated protein kinase, autophagy, endoplasmic reticulum stress, runt-related transcription factor 2, vascular calcification
Citation: Lu Y, Yuan T, Min X, Yuan Z and Cai Z (2021) AMPK: Potential Therapeutic Target for Vascular Calcification. Front. Cardiovasc. Med. 8:670222. doi: 10.3389/fcvm.2021.670222
Received: 20 February 2021; Accepted: 08 April 2021;
Published: 11 May 2021.
Edited by:
Esther Lutgens, Academic Medical Center, NetherlandsReviewed by:
Chieko Mineo, University of Texas Southwestern Medical Center, United StatesClaudia Goettsch, RWTH Aachen University, Germany
Copyright © 2021 Lu, Yuan, Min, Yuan and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhejun Cai, Y2FpemhlanVuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work