
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med., 07 May 2021
Sec. Pediatric Cardiology
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.668231
This article is part of the Research TopicCardiovascular Genetics – Focus on Paediatric CardiomyopathyView all 15 articles
 Tatiana Kovalchuk1*
Tatiana Kovalchuk1* Elena Yakovleva1
Elena Yakovleva1 Svetlana Fetisova1Tatiana Vershinina1
Svetlana Fetisova1Tatiana Vershinina1 Viktoriya Lebedeva2
Viktoriya Lebedeva2 Tamara Lyubimtseva2Dmitriy Lebedev2Lubov Mitrofanova3Anton Ryzhkov4Polina Sokolnikova5Yuliya Fomicheva5Alexandra Kozyreva5Sergey Zhuk5Natalia Smolina5Anna Zlotina5Tatiana Pervunina6Anna Kostareva5,7
Tamara Lyubimtseva2Dmitriy Lebedev2Lubov Mitrofanova3Anton Ryzhkov4Polina Sokolnikova5Yuliya Fomicheva5Alexandra Kozyreva5Sergey Zhuk5Natalia Smolina5Anna Zlotina5Tatiana Pervunina6Anna Kostareva5,7 Elena Vasichkina1
Elena Vasichkina1Emery-Dreifuss muscular dystrophy (EDMD) is inherited muscle dystrophy often accompanied by cardiac abnormalities in the form of supraventricular arrhythmias, conduction defects and sinus node dysfunction. Cardiac phenotype typically arises years after skeletal muscle presentation, though, could be severe and life-threatening. The defined clinical manifestation with joint contractures, progressive muscle weakness and atrophy, as well as cardiac symptoms are observed by the third decade of life. Still, clinical course and sequence of muscle and cardiac signs may be variable and depends on the genotype. Cardiac abnormalities in patients with EDMD in pediatric age are not commonly seen. Here we describe five patients with different forms of EDMD (X-linked and autosomal-dominant) caused by the mutations in EMD and LMNA genes, presented with early onset of cardiac abnormalities and no prominent skeletal muscle phenotype. The predominant forms of cardiac pathology were atrial arrhythmias and conduction disturbances that progress over time. The presented cases discussed in the light of therapeutic strategy, including radiofrequency ablation and antiarrhythmic devices implantation, and the importance of thorough neurological and genetic screening in pediatric patients presenting with complex heart rhythm disorders.
Emery-Dreifuss muscular dystrophy (EDMD) is a group of inherited muscle-joint-cardio syndromes (1). The cardiac involvement in EDMD can be severe and life-threatening (2). Various genetic backgrounds contribute in different ways to the broad spectrum of cardiac manifestations, mainly in the form of conduction disorders, supraventricular arrhythmias and more rarely functional and structural abnormalities of the heart (3).
EDMD is a rare disease. The general prevalence of EDMD is 0.39–1 per 100,000, with significant heterogeneity of estimates; in the pediatric population −0.22 per 100,000 (4–6). The classic clinical triad includes early joint contractures (elbows, neck, ankles, and spine), slowly progressive muscle weakness/atrophy and cardiac abnormalities, but the clinical course and sequence of symptoms depend on the genotype. The severity of cardiac involvement does not correspond to the progression of muscular weakness (3). In the case of EDMD1, associated with mutations in the EMD gene, the contractures frequently emerge in the first decade of life, become more significant during the growth spurt followed by muscle atrophy and weakness in the second decade of life and usually precede cardiac phenotype. In EDMD2, caused by mutations in the LMNA gene, symptoms varied widely from a mild phenotype with a later onset and slow progression to a severe one with life-threatening complications. Lack of information for the reported cases EDMD3, EDMD4, and EDMD5 linked to the genes encoding proteins of nuclear envelope does not allow to define a unified clinical picture of each of those subtypes (7, 8).
Cardiac involvement in EDMD usually follows muscle phenotypes and predominantly presents after the second decade of life (9, 10). After the diagnosis is established due to typical muscular manifestation, the cardiac abnormalities are screened in the frame of expected disease phenotype (11). Several cases of early cardiac debut preceding muscle dystrophy have been reported, and even isolated cardiac involvement has been described; still, these cases remain scarce and atypical (10, 12–15). The cardiac disease commonly manifests from atrial arrhythmias and conduction disturbances. Syncope and sudden cardiac death (SCD), caused by complete heart block or ventricular tachyarrhythmia, can also occur, and single cases of left ventricular non-compaction have been described (16, 17). Systolic dysfunction and dilated cardiomyopathy found in a minority of patients and are mainly associated with autosomal-dominant disease (AD-EDMD) due to LMNA-mutations (18–22). Creatine kinase (CK) level can range from normal to 15 times the upper limit, without direct correlation with muscular and cardiac involvement, so in patients with severe cardiac phenotype CK levels could remain normal (23).
The genetic spectrum of EDMD includes mutations in EMD, LMNA, SYNE1, SYNE2, FHL1, TMEM43, SUN1, SUN2, and TTN genes (5, 24–28). These genes mainly encode for the nuclear envelope proteins, which give rise to the term “nuclear envelopathies” (5, 23, 29). An exception is FHL1 protein which localizes to the sarcomere and the sarcolemma but may also shuttle between cytosolic and nucleoplasmic fraction, thus becoming a part of the nuclear envelope (30). Mutations in LMNA and EMD are the most common causes of EDMD, and together they account for about 36% of the cases (5). Thus, for EDMD, there are still a number of undetected causative genes (30). An AD-EDMD mainly arises from mutations in LMNA, which contribute to ~28% of the cases. In LMNA-associated EDMD, males and females are equally affected and left ventricular dysfunction is more commonly observed as well as ventricular arrhythmias and SCD (9, 19, 20). Several cases of LMNA-associated autosomal-recessive form of EDMD presented in pediatric patients have also been described (31, 32). The X-linked form is usually caused by mutations in the EMD gene (8%) and, rarely, in the FHL1 gene (2%) and predominantly affects males with rare cases of disease manifestation in female carriers (18). Mutations in the EMD gene occur sporadically and rarely, but mutations in the LMNA gene are increasingly identified (23).
Due to the typical presentation during the second-third decades of life and a debut from muscle symptoms, there are only several reports on EDMD presented in children with the isolated cardiac phenotype (13, 14, 33–35). Here we describe five cases of EDMD1 and EDMD2 with a cardiac manifestation in childhood and discuss the need for target screening of neuromuscular phenotypes in children with unexplained atrial dysfunction, conduction abnormalities, and ventricular arrhythmias.
Patients were examined between 2009 and 2020 in tertiary pediatric cardiac care center – Almazov National Medical Research Centre, St. Petersburg, Russia. All data, including the clinical history, case notes, reports of instrumental methods and surgical protocols were extracted from paper and electronic databases. Clinical examination included physical, 12-lead electrocardiography (ECG), Holter monitoring (HM), transthoracic echocardiography, neurological examination and laboratory tests. Additionally, we performed CMR, electrophysiological study (EPS) and electroneuromyography (ENMG). One patient underwent an endomyocardial biopsy to exclude inflammatory heart disease.
Target sequencing was performed using a panel of 108 or 172 genes, as previously described (36). For Patients 1 and 2, a targeted panel of 108 cardiomyopathy-associated genes has been initially studied using Haloplex Target Enrichment System (Agilent, Waldbronn, Germany) with an Illumina MiSeq instrument (for gene list, see Supplementary Table 1). For Patients 3–5, a targeted panel of 172 cardiomyopathy-associated genes was studied using the SureSelect Target Enrichment System (Agilent; Waldbronn, Germany) (see Supplementary Table 2). Data processing and variant calling were performed according to GATK BestPractice recommendations (Broad Institute, Cambridge, MA, USA) using hg19 and hg38 human genome references. For variant validation, bidirectional Sanger sequencing was performed using ABI 3500 machine (Applied Biosystems). All novel and previously reported variants of interest with a frequency below 0.01% were classified according to the recommendations of American College of Medical Genetics (37). The described variants were submitted to Gene bank repository under submission numbers SCV001548550 - SCV001548554.
The study was performed according to the Declaration of Helsinki, and approval was obtained from the local ethical committee of Almazov National Medical Research Centre. Written informed consents were obtained from the parents of the minor prior to investigation.
We observed five patients with EDMD and cardiac symptoms in childhood: three with EDMD1 and two with EDMD2. All patients were males. The mean age of cardiac manifestation was 13.2 ± 3.11 (from 9 to 16 y.o.). The mean follow-up period was 7.4 ± 2.6 years. All patients had sinus node dysfunction and four out of five - atrioventricular block (AVB). The leading arrhythmic phenotypes included various types of supraventricular arrhythmias: multifocal atrial tachycardia (mAT) (n = 4), premature atrial captures (PACs) (n = 4), atrial flutter (AF) (n = 3), atrial fibrillation (AFib) (n = 3) and AV nodal recurrent tachycardia (AVRNT). Arrythmias were the first manifestation in four patients. Patients predominantly complained of palpitation (n = 4), dizziness (n = 1), fatigue, and reduced physical tolerance (n = 2) (Table 1).
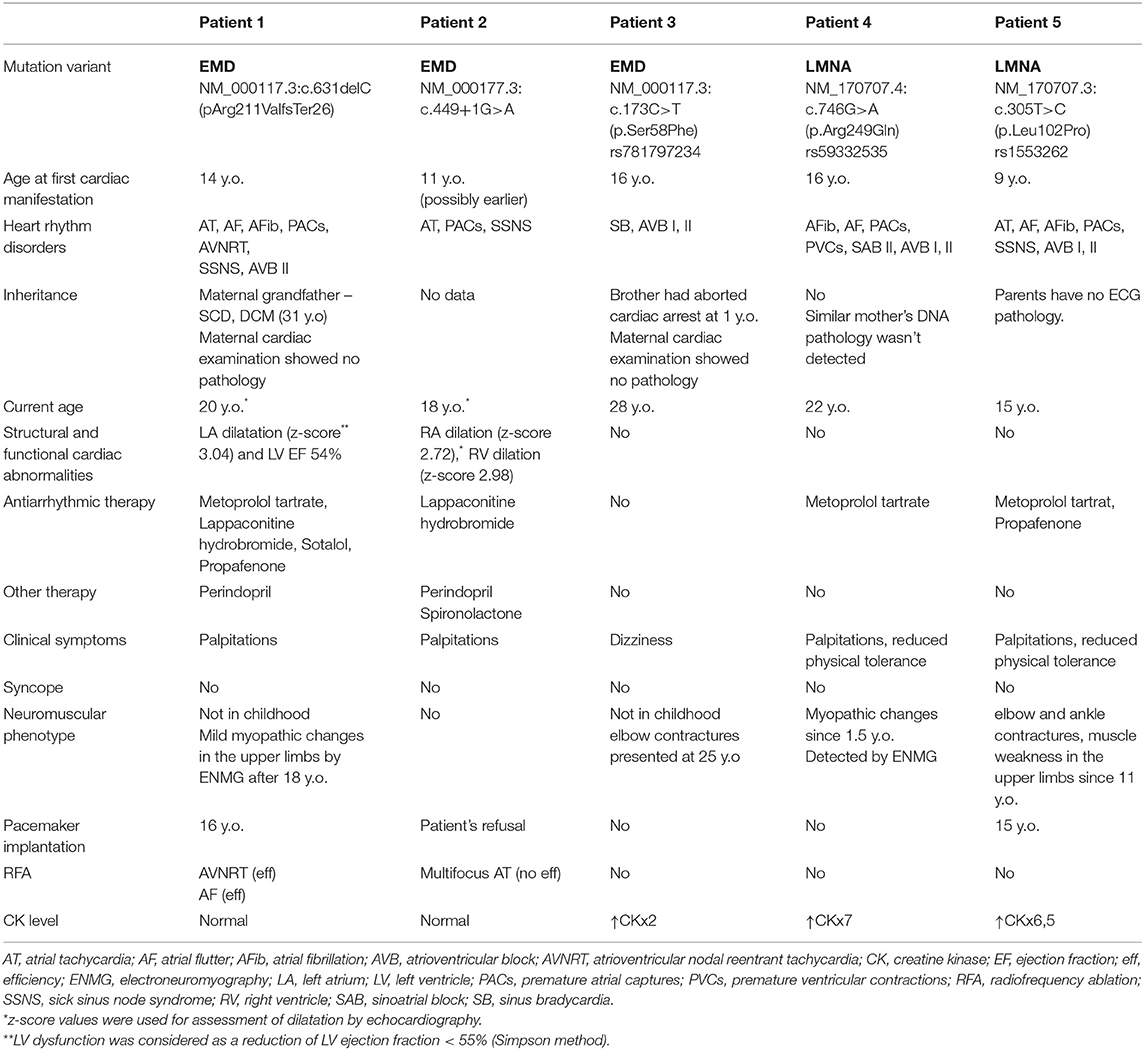
Table 1. Clinical and genetic characteristics of patients affected by EDMD and presented with heart rhythm disorders.
The patient first presented with atrial rhythm and rare PACs on the ECG at the age of 14 years. He had no complaints, was a member of a football team and was undergoing ECG annually. No pathology was noticed on echocardiography. At 14.5 y.o. he complained on palpitations. An EPS revealed three types of arrhythmias: non-sustained mAT with heart rate (HR) 114 bpm, non-sustained AF and slow-slow AVRNT with HR 130 bpm. The radiofrequency ablation (RFA) of AVRNT was performed. Metoprolol tartrate was prescribed but canceled due to progressive AVB with pauses up to 3,261 ms. Six months later, AF with irregular AV conduction (2:1–4:1) was registered and followed by to RFA of the inferior vena cava-tricuspid isthmus. During the procedure, extensive low-amplitude areas corresponding to fibrosis fields were observed in the right atrium (Figure 1). CMR showed mild-dilated right atrium, atrial fibrosis and no acute inflammatory (Figure 1). The progression of the sinus and AV nodes dysfunction with bradycardia to 34–43 bpm and pauses to 7,104 ms (Figure 2), increasing atrial ectopy led to dual-chamber pacemaker (PM) implantation and Propafenone therapy. Increasing of pacing percentage, no intrinsic rhythm, non-sustained AFib during PM programming registered over time. Target genetic screening using 108-gene panel identified novel genetic variant in EMD gene (NM_000117.3):c.631delC, p.Arg211ValfsTer26 (chrX:153609422) classified as pathogenic according to ACMG criteria (PVS1, PM2, PP3). Parental genetic testing hasn't been performed. CK level was not elevated. Neurological examination showed no pathology, but mild myogenic changes in the upper limbs were detected by ENMG later, at the age of 18 years. Maternal grandfather died suddenly at the age of 31 with dilated cardiomyopathy according to the autopsy. Maternal cardiac examination revealed no pathology.
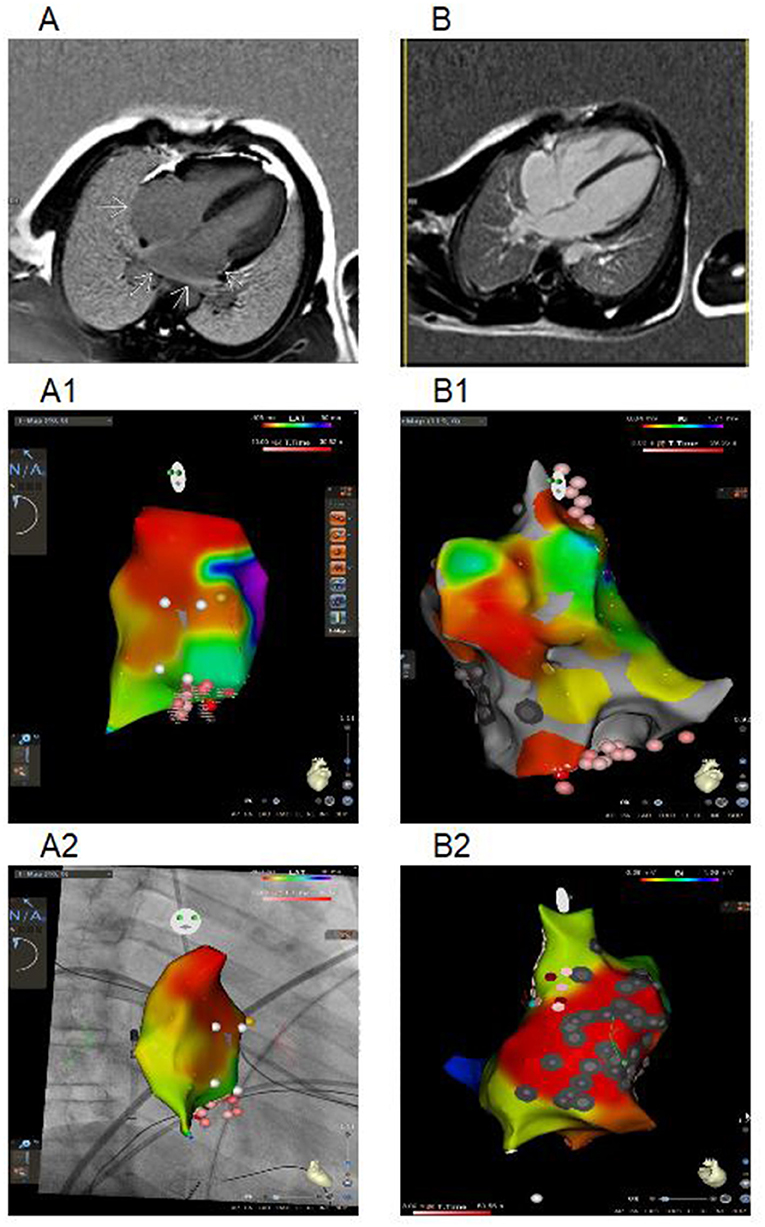
Figure 1. Cardiac maps using three-dimensional mapping system and CMR (imaging in the 4-chamber plane) of Patient 1 and 2. (A) CMR from a patient 1: delayed-enhancement CMR shows areas of myocardial scarring in both atria (arrows). (A1) Right atrial activation map (CARTO 3 system), LAO position. White dots – location points, yellow dots – part of conduction system, red and pink dots – RF ablation points (the degree of color intensity depends on the RF exposure time). (A2) Right atrial activation map (CARTO 3 system with CARTOUNIVU module), RAO position. White dots – location points; yellow dots – part of conduction system, red and pink dots – RF ablation points (the degree of color intensity depends on the RF exposure time). (B) CMR from a patient 2 with RA dilatation. (B1) Bipolar map of right atrium (CARTO 3 system), LAO position. (B2) Bipolar map of right atrium (CARTO 3 system), non-standard position with focus on the scar and ablation sites. Gray dots – scar points, white dots – location points, blue dots – double potential, red and pink dots – RF ablation points (the degree of color intensity depends on the RF exposure time). Bipolar color scale: red fill color has the lowest voltage.
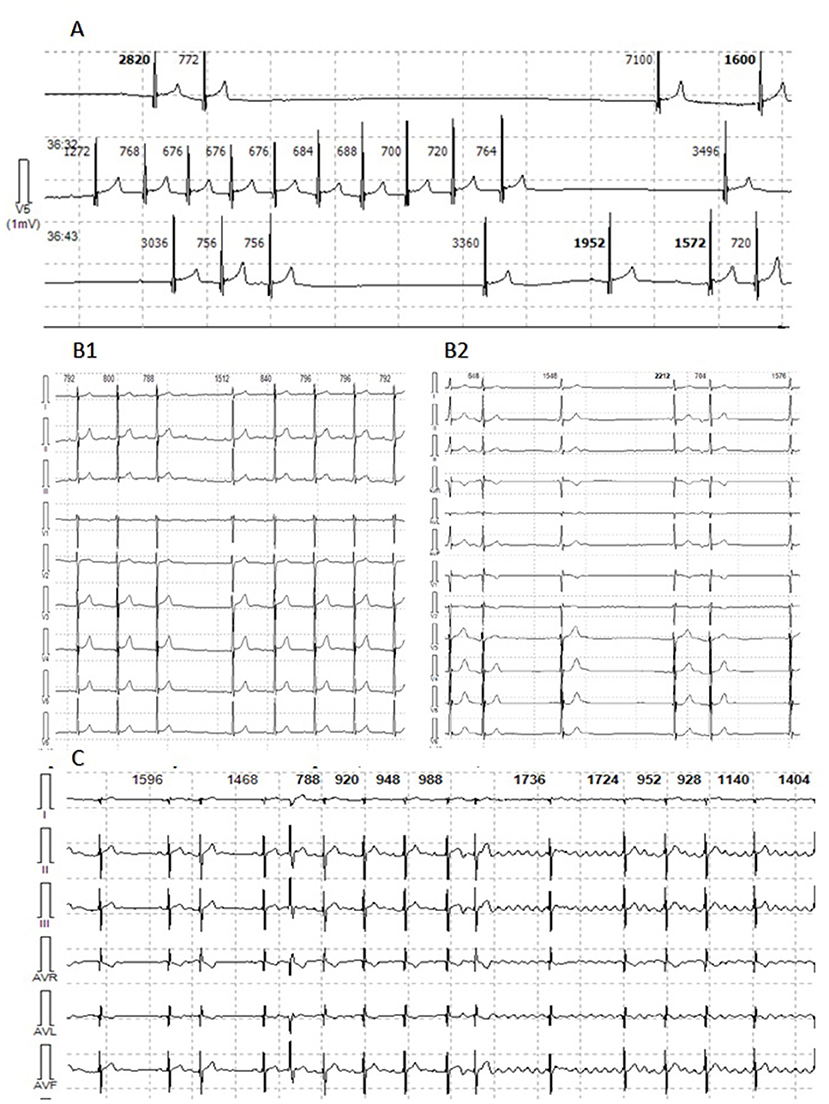
Figure 2. Holter monitor fragments of Patient 1, 2, and 5. (A) Patient 1. Sinus arrest with pauses from 3,036 to 7,104 ms. (B1) Patient 2. Episode of sustained AT with variable AV conduction. (B2) Patient 2. Multifocal PACs. (C) Patient 5. Episodes of multifocal PACs, AT and AFib with variable AV conduction.
Patient 2 was diagnosed with atrial rhythm with 93 bpm during ECG before being brought to the orphanage at the age of 11 years. His family history is unknown. He has been abusing nicotine and alcohol since the age of 15. On ECG, at the age of 14, atrial and junctional rhythm with PACs and non-sustained mAT were registered. Six months later, he was hospitalized in the emergency unit with palpitations. AT with HR to 200 bpm were registered and aborted by overdriving stimulation. Echocardiography and CMR confirmed right chambers dilatation without myocardial dysfunction. No signs of acute myocardial damage or fibrosis have been found (Figure 1). HM identified sinus bradycardia to 35 bpm, mainly atrial and junctional rhythm, mAT with variable AV conduction (2:1–7:1) and pauses to 4,404 ms (Figure 2). The RFA of mAT was performed (Figure 1) but wasn't effective. Endomyocardial biopsy showed no myocarditis signs. Target genetic screening using 172-gene panel identified EMD genetic variant (NM_000117.3):c.449+1G>A genetic variant, classified as pathogenic according to ACMG criteria (PVS1, PM2, PP3) previously reported in association with EDMD (38). He had not myopathy signs or contractures. ENMG was not performed due to the patient's unwillingness. CK level was normal. PM implantation was recommended, but the patient refused. Attempts to prescribe antiarrhythmic therapy (AAT) were unsuccessful.
Patient 3 first consulted a cardiologist at the age of 16 because of dizziness and fatigues. No structural abnormalities were detected by echocardiography. Rare symptomatic episodes of resting bradycardia to 30–34 bpm and pauses due to AVB II up to 2,500 ms were observed. Neurological examination didn't reveal any pathology. Later, at the age of 25, mild elbow contractures were noted. Two times higher CK level was documented at the age of 28. Genetic screening identified a variant in EMD (NM_000117.3):c.173C>T (p.Ser58Phe), classified as a variant of unknown significance according to ACMG criteria. Brother survived the aborted cardiac arrest at the age of 1 year, but his genotyping is currently infeasible. Maternal cardiac examination showed no pathology.
The patient was first referred to a neurologist at 1.5 years of age due to toe-walking. He had two times elevated CK level and myopathic changes detected by ENMG. Multifocal PACs were registered at the age of 16 years when the fatigue and palpitations appeared. A HM showed paroxysms of non-sustained AFib, multiple PACs, and transient AVB II and SA block. No pathology was detected on echocardiography and CMR. The CK level has been increased seven-fold. He had ankles contractures, muscle weakness in the shoulders and lower legs. Metoprolol tartrate was initiated with good response and without conduction worsening. Three months later, atrial arrhythmias were increased and rare premature ventricular contractions (PVCs) were registered. Metoprolol tartrate dose was doubled with a positive effect. The genetic investigation identified LMNA (NM_170707.4):c.746G>A (p.Arg249Gln) variant previously described in EDMD patients (rs59332535) and classified as pathogenic according to ACMG criteria. Mother doesn't have this variant, father died at 49 y.o. during a planned surgery due to cardiac arrest, no DNA was available for the genotyping. This patient has not indications for PM or cardioverter-defibrillator implantation (ICD).
The patient was first examined by a cardiologist at nine y.o. due to palpitations, but ECG revealed no pathology, HM and echocardiography were not performed. At the age of 11 episodes of mAT, PACs, and transient AVB I were detected by HM. Concurrently, elbow contractures and muscle weakness were noted. ENMG identified a moderate myopathic pattern in the lower limbs. Increase in CK by 6.5 times was detected. Within the next several years, arrhythmias progressed with the sustained episodes of mAT, AFib, deterioration of AV conduction and decrease of HR. Specific changes in the P-wave were detected (Figure 3). No structural abnormalities were detected by echocardiography. Metoprolol tartrate was up-titrated with a good but temporary effect. The genetic study identified an earlier reported variant in the LMNA gene (NM_170707.3):c.305T>C, (p.Leu102Pro). Family history was unremarkable. PM was implanted along with AAT (Propafenone) at the age of 15 years.
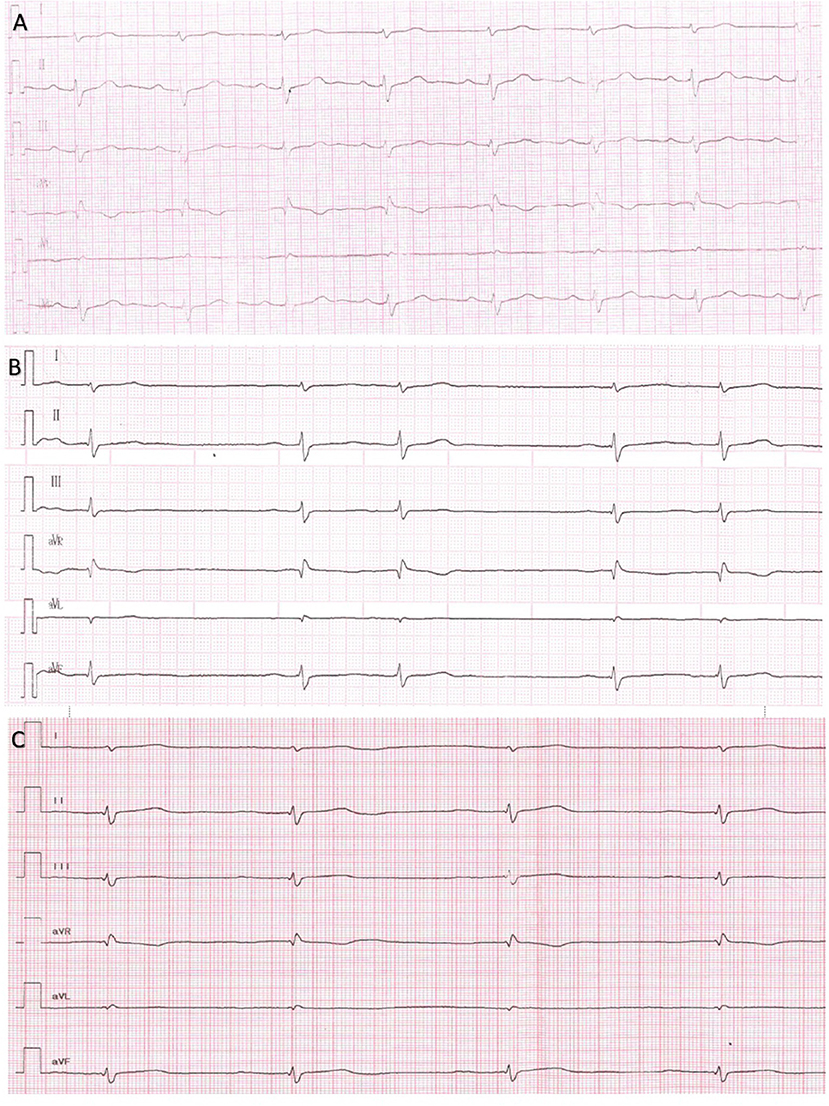
Figure 3. Series of Electrocardiograms from a patient 5 showing reduced amplitude of P wave and AV conduction in dynamics. (A) 13 y.o. P wave amplitude 2.4 mm, P wave duration 70 ms, PQ 160 ms. (B) 14 y.o. P wave amplitude 1.5 mm, P wave duration 100 ms, PQ 200 ms. (C) 15 y.o. P wave amplitude 1 mm, P wave duration 110 ms, PQ 250 ms.
Supraventricular tachycardia and conduction disorders with sinus and AV node dysfunction are the most common conditions in pediatric cardiology practice. The thorough diagnostic workup and precise nosology definition are important for the optimal therapeutic strategy. The presented cases as well as several earlier reports (Table 2) provide the evidence that EDMD has to be considered in children presenting with conduction disorders or atrial arrhythmias even without overt signs of muscular dystrophy and joint contractions. The natural cause of EDMD leads to the requirement of PM in 56% of the patients, AF/flutter developed in 61%, atria standstill in 45%, and embolic stroke in 36% of the patients (18). Therefore, identification of EDMD-underlying genetic background is important for following genetic cascade screening first-degree relatives of patients, that is recommended and includes ECG, HM, echocardiography, and CMR (43).
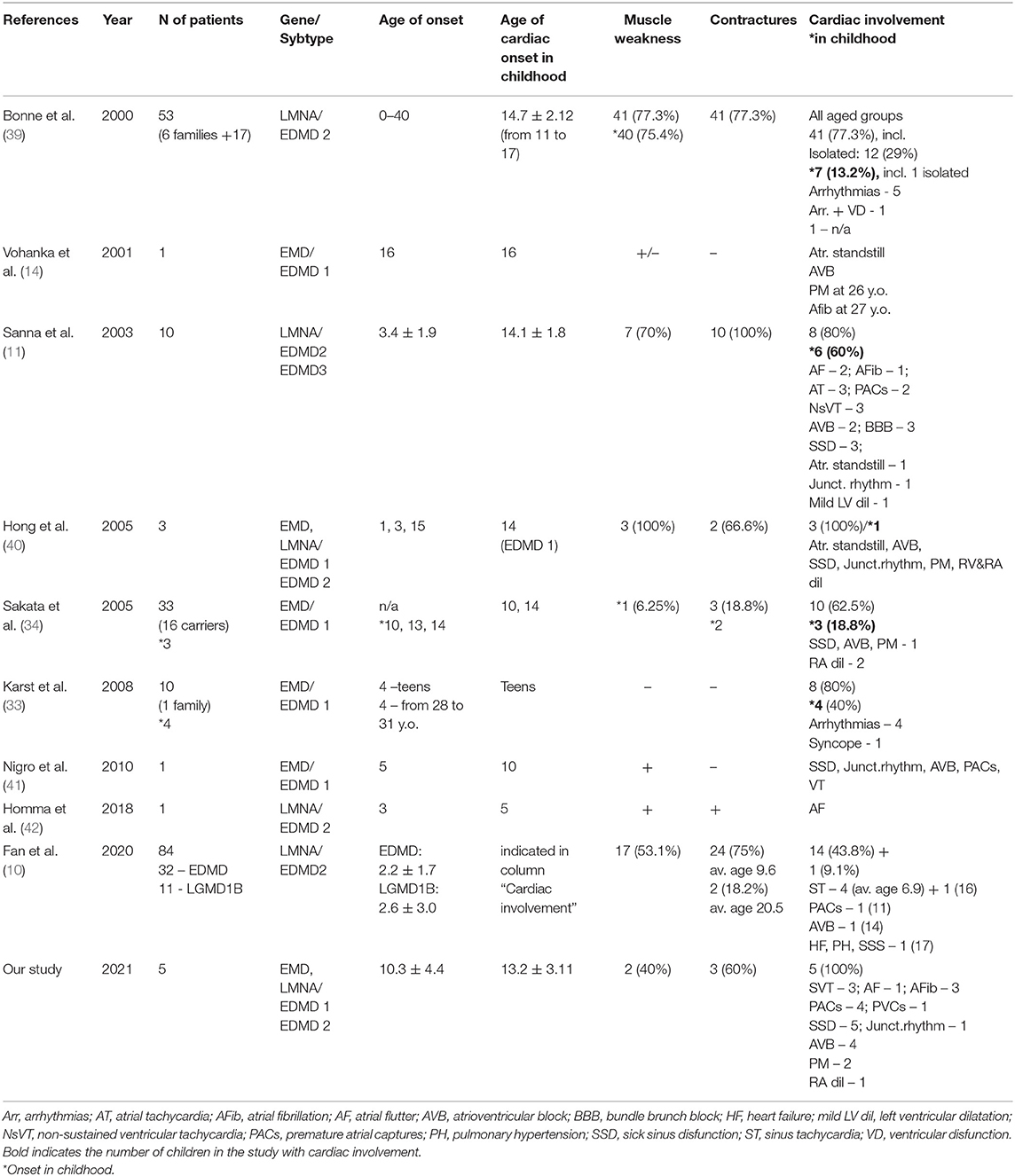
Table 2. Literature review on heart rhythm disturbances in EDMD in childhood.
The precise molecular mechanisms underlying predominantly atrial and conduction pathology in EDMD are largely unknown. Most of the genes and genetic loci identified by GWAS linked to AFib represent transcriptional factors and signaling molecules responsible for cardiac development and cardiomyocyte differentiation (44). In this context, the ability of nuclear envelope proteins to modify chromatin organization and transcriptional activity could explain the effect of EMD deficiency on myocardial cell fate (45, 46). A deficiency or defects of this proteins could lead to nuclear instability in tissues undergoing mechanical stress, including cardiac and skeletal muscle. Of note, another nuclear envelope protein – nesprin 2 encoded by the SYNE2 gene and reported as a rare cause of EDMD was also identified among GWAS loci in association with AF (47, 48). In addition, Shimojima et al. reported that mutant forms of emerin caused abnormalities in nuclear Ca++ transients, which may further modulate nuclear transcriptional pathways in response to mechanical stress (49). As a result, alterations in cardiomyocyte differentiation gradually led to replacing the normal myocardial cells with fibrotic tissue leading to sinus node dysfunction, ectopic loci, and conduction defects. The question remains why these processes mainly start in the atria, often involve the atrioventricular node and only eventually affect the ventricles. This question is further difficult to address because of the unavailability of atrial tissue for morphological examination and limitation of CMR for atrial imaging because of difficulties in achieving adequate image resolution in thin-walled atria (50).
Cardiac structural abnormalities, ventricular arrhythmias and dysfunction are more common in AD-EDMD and are not typical of X-linked EDMD. Of note, in the presented case series, the only patient who demonstrated rare PVCs was the Patient 4 with LMNA-associated EDMD. Together with other reports on LMNA-associated SCD in patients with cardiac and muscle pathologies, this further draws attention for the more thorough follow-up of this group in terms of different arrhythmias (51).
It is commonly accepted that cardiac symptoms of EDMD follows the neuromuscular phenotypes and becomes evident in the second-third decade of life (7, 11, 23). Cases of isolated cardiac manifestations of EDMD remain rarely reported, especially in pediatric patients (7, 33, 40). However, such cases, together with our study, emphasize the importance of genetic testing and target search for EDMD in cases of progressive refractory arrhythmias with or without specific neurological and laboratory findings. Three patients with EDMD1 we observed did not have a typical clinical course with early contractures or muscle weakness, but two patients had severe cardiac abnormalities requiring medication and intervention treatment. Patients 4 and 5 with LMNA-genetic variants demonstrated the clinical variability of EDMD ranging from mild disease course and later cardiac debut to early manifestation and fast-progression that led to PM and AAT. Thus, the results of genetic testing helped us draw up a plan of management and further follow-up, including annual cardiological and neurological examinations.
The main therapeutic problem we faced was coexistence of symptomatic high-frequency atrial arrhythmias and conduction disorders, which limited the prescribing of AAT. The use of RFA in EDMD is a debatable issue (52). The literature search resulted in four cases of RFA in EDMD described in details: three with poor outcomes [1 – RFA of AF with recurrence of arrhythmia, leading to systolic dysfunction and heart transplantation (53); 2 – recurrent SVT with PM after AV nodal ablation, accession of ventricular tachycardia and death from irrecoverable asystole (54); 3 – repeated unsuccessful RFA of AFib leading to embolic stroke (42)] and one successful: RFA of AF by employing a three-dimensional mapping (52). In our study, RFA was performed in two patients: successful treatment of AVNRT and AF in Patient 1 and unsuccessful RFA of mAT in Patient 2.
Since sudden death in X-linked EDMD is primarily caused by a complete heart block, it can be reliably averted by PM implantation (3). The ICD, cardiac resynchronization therapy, mechanical circulatory support, and heart transplantation remain a rare but potentially applicable strategies in EDMD patients (21, 43, 55). In the long-term longitudinal study performed by Boriani et al., heart failure requiring transplantation occurred in 6% and asymptomatic LV dysfunction in 17% of patients (18). ICD is more often required for LMNA mutation carriers and should be considered with sustained or non-sustained ventricular tachyarrhythmias, especially in those with LVEF <45% or in patients with indications for PM implantations (41, 56). In line with that, three of five described patients had I class indications for PM, and none had indications for ICD implantation.
In conclusion, while being rare cases, heart rhythm disorders can represent the first and for a long time, the only clinical symptom of EDMD even in the pediatric group of patients. Therefore, thorough laboratory and neurological screening along with genetic studies are of importance in each pediatric patient presenting with complex arrhythmias of primary supraventricular origin to exclude EDMD or other neuromuscular disorders. Consideration of EDMD in diagnostic workup can facilitate the optimal strategy and personalized follow-up of this group of patients.
The data presented in the study are deposited in the Gene bank repository, accession numbers SCV001548550–SCV001548554 and publicly available.
The studies involving human participants were reviewed and approved by Almazov National Medical Research Centre Institute Ethical Review Boards. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
EY, SF, and TL contributed to the conception and design of the study, analysis, and interpretation of the data and drafting of the manuscript. TK contributed to the study concept and research design and wrote the manuscript. AKos, TP, and EV made contributions to the conception and design of the study and revision of the manuscript critically. VL, TV, DL, and LM took part in the analysis and interpretation of the data and have been involved in revising the manuscript critically. AR, PS, YF, AKoz, SZ, NS, and AZ conducted the experiments and performed the analysis and interpretation of the data. All authors have read and agreed to the published version of the manuscript.
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2020-901).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.668231/full#supplementary-material
1. Pillers DA, Von Bergen NH. Emery-Dreifuss muscular dystrophy: a test case for precision medicine. Appl Clin Genet. (2016) 9:27–32. doi: 10.2147/TACG.S75028
2. Holaska JM. Emerin and the nuclear lamina in muscle and cardiac disease. Circ Res. (2008) 103:16–23. doi: 10.1161/CIRCRESAHA.108.172197
3. Wang S, Peng D. Cardiac involvement in emery-dreifuss muscular dystrophy and related management strategies. Int Heart J. (2019) 60:12–8. doi: 10.1536/ihj.17-604
4. Mah JK, Korngut L, Fiest KM, Dykeman J, Day LJ, Pringsheim T, et al. A systematic review and meta-analysis on the epidemiology of the muscular dystrophies. Can J Neurol Sci. (2016) 43:163–77. doi: 10.1017/cjn.2015.311
5. Bonne G, Quijano-Roy S. Emery-dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol. (2013) 113:1367–6. doi: 10.1016/B978-0-444-59565-2.00007-1
6. Chung B, Wong V, Ip P. Prevalence of neuromuscular diseases in Chinese children: a study in southern China. J Child Neurol. (2003) 18:217–9. doi: 10.1177/08830738030180030201
7. Heller SA, Shih R, Kalra R, Kang PB. Emery-Dreifuss muscular dystrophy. Muscle Nerve. (2020) 61:436–48. doi: 10.1002/mus.26782
8. Madej-Pilarczyk A. Clinical aspects of Emery-Dreifuss muscular dystrophy. Nucleus. (2018) 9:268–74. doi: 10.1080/19491034.2018.1462635
9. van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. (2005) 83:79–83. doi: 10.1007/s00109-004-0589-1
10. Fan Y, Tan D, Song D, Zhang X, Chang X, Wang Z, et al. Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J Med Genet. (2020) 0:1–8. doi: 10.1136/jmedgenet-2019-106671
11. Sanna T, Dello Russo A, Toniolo D, Vytopil M, Pelargonio G, De Martino G, et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur Heart J. (2003) 24:2227–36. doi: 10.1016/j.ehj.2003.09.020
12. Zhang M, Chen J, Si D, Zheng Y, Jiao H, Feng Z, et al. Whole exome sequencing identifies a novel EMD mutation in a Chinese family with dilated cardiomyopathy. BMC Med Genet. (2014) 15:77. doi: 10.1186/1471-2350-15-77
13. Vytopil M, Vohanka S, Vlasinova J, Toman J, Novak M, Toniolo D, et al. The screening for X-linked Emery-Dreifuss muscular dystrophy amongst young patients with idiopathic heart conduction system disease treated by a pacemaker implant. Eur J Neurol. (2004) 11:531–4. doi: 10.1111/j.1468-1331.2004.00825.x
14. Vohanka S, Vytopil M, Bednarik J, Lukas Z, Kadanka Z, Schildberger J, et al. A mutation in the X-linked Emery-Dreifuss muscular dystrophy gene in a patient affected with conduction cardiomyopathy. Neuromuscul Disord. (2001) 11:411–3. doi: 10.1016/S0960-8966(00)00206-6
15. Kong D, Zhan Y, Liu C, Hu Y, Zhou Y, Luo J, et al. A novel mutation of the EMD gene in a family with cardiac conduction abnormalities and a high incidence of sudden cardiac death. Pharmgenomics Pers Med. (2019) 12:319–27. doi: 10.2147/PGPM.S221444
16. Wang C, Hata Y, Hirono K, Takasaki A, Ozawa SW, Nakaoka H, et al. A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc. (2017) 6:e006210. doi: 10.1161/JAHA.117.006210
17. Becane HM, Bonne G, Varnous S, Muchir A, Ortega V, Hammouda EH, et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin Electrophysiol. (2000) 23:1661–6. doi: 10.1046/j.1460-9592.2000.01661.x
18. Boriani G, Gallina M, Merlini L, Bonne G, Toniolo D, Amati S, et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: a long-term longitudinal study. Stroke. (2003) 34:901–8. doi: 10.1161/01.STR.0000064322.47667.49
19. Talkop UA, Talvik I, Sonajalg M, Sibul H, Kolk A, Piirsoo A, et al. Early onset of cardiomyopathy in two brothers with X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. (2002) 12:878–81. doi: 10.1016/S0960-8966(02)00134-7
20. Kichuk Chrisant MR, Drummond-Webb J, Hallowell S, Friedman NR. Cardiac transplantation in twins with autosomal dominant Emery-Dreifuss muscular dystrophy. J Heart Lung Transplant. (2004) 23:496–8. doi: 10.1016/S1053-2498(03)00204-3
21. Dell'Amore A, Botta L, Martin Suarez S, Lo Forte A, Mikus E, Camurri N, et al. Heart transplantation in patients with Emery-Dreifuss muscular dystrophy: case reports. Transplant Proc. (2007) 39:3538–40. doi: 10.1016/j.transproceed.2007.06.076
22. Higuchi Y, Hongou M, Ozawa K, Kokawa H, Masaki M. A family of Emery-Dreifuss muscular dystrophy with extreme difference in severity. Pediatr Neurol. (2005) 32:358–60. doi: 10.1016/j.pediatrneurol.2004.12.011
23. Astejada MN, Goto K, Nagano A, Ura S, Noguchi S, Nonaka I, et al. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol. (2007) 26:159–64.
24. Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. (2011) 69:1005–13. doi: 10.1002/ana.22338
25. Gueneau L, Bertrand AT, Jais JP, Salih MA, Stojkovic T, Wehnert M, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. (2009) 85:338–53. doi: 10.1016/j.ajhg.2009.07.015
26. Starr DA, Fridolfsson HN. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu Rev Cell Dev Biol. (2010) 26:421–44. doi: 10.1146/annurev-cellbio-100109-104037
27. Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and−2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. (2007) 16:2816–33. doi: 10.1093/hmg/ddm238
28. De Cid R, Ben Yaou R, Roudaut C, Charton K, Baulande S, Leturcq F, et al. A new titinopathy: childhood-juvenile onset Emery-Dreifuss-like phenotype without cardiomyopathy. Neurology. (2015) 85:2126–35. doi: 10.1212/WNL.0000000000002200
29. Meinke P, Nguyen TD, Wehnert MS. The LINC complex and human disease. Biochem Soc Trans. (2011) 39:1693–7. doi: 10.1042/BST20110658
30. Ziat E, Mamchaoui K, Beuvin M, Nelson I, Azibani F, Spuler S, et al. FHL1B interacts with lamin A/C and emerin at the nuclear lamina and is misregulated in Emery-Dreifuss muscular dystrophy. J Neuromuscul Dis. (2016) 3:497–510. doi: 10.3233/JND-160169
31. Wiltshire KM, Hegele RA, Innes AM, Brownell AK. Homozygous lamin A/C familial lipodystrophy R482Q mutation in autosomal recessive Emery Dreifuss muscular dystrophy. Neuromuscul Disord. (2013) 23:265–8. doi: 10.1016/j.nmd.2012.11.011
32. Jimenez-Escrig A, Gobernado I, Garcia-Villanueva M, Sanchez-Herranz A. Autosomal recessive Emery-Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve. (2012) 45:605–10. doi: 10.1002/mus.22324
33. Karst ML, Herron KJ, Olson TM. X-linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by emerin mutation. J Cardiovasc Electrophysiol. (2008) 19:510–5. doi: 10.1111/j.1540-8167.2007.01081.x
34. Sakata K, Shimizu M, Ino H, Yamaguchi M, Terai H, Fujino N, et al. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation. (2005) 111:3352–8. doi: 10.1161/CIRCULATIONAHA.104.527184
35. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. (2012) 59:493–500. doi: 10.1016/j.jacc.2011.08.078
36. Jorholt J, Formicheva Y, Vershinina T, Kiselev A, Muravyev A, Demchenko E, et al. Two new cases of hypertrophic cardiomyopathy and skeletal muscle features associated with ALPK3 homozygous and compound heterozygous variants. Genes. (2020) 11:1201. doi: 10.3390/genes11101201
37. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
38. Nevo Y, Ahituv S, Yaron Y, Kedmi M, Shomrat R, Legum C, et al. Novel mutations in the emerin gene in Israeli families. Hum Mutat. (2001) 17:522. doi: 10.1002/humu.1139
39. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. (2000) 48:170–80. doi: 10.1002/1531-8249(200008)48:2<170::AID-ANA6>3.0.CO;2-J
40. Hong JS, Ki CS, Kim JW, Suh YL, Kim JS, Baek KK, et al. Cardiac dysrhythmias,cardiomyopathy and muscular dystrophy in patients with Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy type 1B. J Korean Med Sci. (2005) 20:283–90. doi: 10.3346/jkms.2005.20.2.283
41. Nigro G, Russo V, Ventriglia VM, Della Cioppa N, Palladino A, Nigro V, et al. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. (2010) 20:174–7. doi: 10.1016/j.nmd.2009.12.004
42. Homma K, Nagata E, Hanano H, Uesugi T, Ohnuki Y, Matsuda S, et al. A young patient with emery-dreifuss muscular dystrophy treated with endovascular therapy for cardioembolic stroke: a case report. Tokai J Exp Clin Med. (2018) 43:103–5.
43. Feingold B, Mahle WT, Auerbach S, Clemens P, Domenighetti AA, Jefferies JL, et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation. (2017) 136:e200–31. doi: 10.1161/CIR.0000000000000526
44. Fatkin D, Santiago CF, Huttner IG, Lubitz SA, Ellinor PT. Genetics of atrial fibrillation: state of the art in 2017. Heart Lung Circ. (2017) 26:894–901. doi: 10.1016/j.hlc.2017.04.008
45. Bianchi A, Manti PG, Lucini F, Lanzuolo C. Mechanotransduction, nuclear architecture and epigenetics in Emery Dreifuss muscular dystrophy: tous pour un, un pour tous. Nucleus. (2018) 9:276–90. doi: 10.1080/19491034.2018.1460044
46. Janin A, Bauer D, Ratti F, Millat G, Mejat A. Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet J Rare Dis. (2017) 12:147. doi: 10.1186/s13023-017-0698-x
47. Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. (2012) 44:670–5. doi: 10.1038/ng.2261
48. Hall AW, Chaffin M, Roselli C, Lin H, Lubitz SA, Bianchi V, et al. Epigenetic analyses of human left atrial tissue identifies gene networks underlying atrial fibrillation. Circ Genom Precis Med. (2020) 13:e003085. doi: 10.1161/CIRCGEN.120.003085
49. Shimojima M, Yuasa S, Motoda C, Yozu G, Nagai T, Ito S, et al. Emerin plays a crucial role in nuclear invagination and in the nuclear calcium transient. Sci Rep. (2017) 7:44312. doi: 10.1038/srep44312
50. Harrison JL, Sohns C, Linton NW, Karim R, Williams SE, Rhode KS, et al. Repeat left atrial catheter ablation: cardiac magnetic resonance prediction of endocardial voltage and gaps in ablation lesion sets. Circ Arrhythm Electrophysiol. (2015) 8:270–8. doi: 10.1161/CIRCEP.114.002066
51. Nishiuchi S, Makiyama T, Aiba T, Nakajima K, Hirose S, Kohjitani H, et al. Gene-based risk stratification for cardiac disorders in LMNA mutation carriers. Circ Cardiovasc Genet. (2017) 10:e001603. doi: 10.1161/CIRCGENETICS.116.001603
52. Butt K, Ambati S. Atrial arrhythmias in Emery-Dreifuss muscular dystrophy: approach to successful ablation. HeartRhythm Case Rep. (2020) 6:318–21. doi: 10.1016/j.hrcr.2020.02.009
53. Blagova O, Nedostup A, Shumakov D, Poptsov V, Shestak A, Zaklyasminskaya E. Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy: from ablation to heart transplantation. J Atr Fibrillation. (2016) 9:1468. doi: 10.4022/jafib.1468
54. Carvalho AA, Levy JA, Gutierrez PS, Marie SK, Sosa EA, Scanavaca M. Emery-Dreifuss muscular dystrophy: anatomical-clinical correlation (case report). Arq Neuropsiquiatr. (2000) 58:1123–7. doi: 10.1590/S0004-282X2000000600023
55. Palladino A, D'Ambrosio P, Papa AA, Petillo R, Orsini C, Scutifero M, et al. Management of cardiac involvement in muscular dystrophies: paediatric versus adult forms. Acta Myol. (2016) 35:128–34.
Keywords: Emery-Dreifuss muscular dystrophy, children, atrial tachycardia, atrial fibrillation, cardiomyopathy, LMNA, EMD (emerin), pacemaker implantation
Citation: Kovalchuk T, Yakovleva E, Fetisova S, Vershinina T, Lebedeva V, Lyubimtseva T, Lebedev D, Mitrofanova L, Ryzhkov A, Sokolnikova P, Fomicheva Y, Kozyreva A, Zhuk S, Smolina N, Zlotina A, Pervunina T, Kostareva A and Vasichkina E (2021) Case Reports: Emery-Dreifuss Muscular Dystrophy Presenting as a Heart Rhythm Disorders in Children. Front. Cardiovasc. Med. 8:668231. doi: 10.3389/fcvm.2021.668231
Received: 15 February 2021; Accepted: 07 April 2021;
Published: 07 May 2021.
Edited by:
Juan Pablo Kaski, University College London, United KingdomReviewed by:
Gabrielle Norrish, Great Ormond Street Hospital Children's Charity, United KingdomCopyright © 2021 Kovalchuk, Yakovleva, Fetisova, Vershinina, Lebedeva, Lyubimtseva, Lebedev, Mitrofanova, Ryzhkov, Sokolnikova, Fomicheva, Kozyreva, Zhuk, Smolina, Zlotina, Pervunina, Kostareva and Vasichkina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatiana Kovalchuk, dHNrb3ZhbGNodWtAeWFuZGV4LnJ1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.