 Olina Dagher1,2,3*
Olina Dagher1,2,3* Pauline Mury3
Pauline Mury3 Nathalie Thorin-Trescases3Pierre Emmanuel Noly2,3
Nathalie Thorin-Trescases3Pierre Emmanuel Noly2,3 Eric Thorin2,3Michel Carrier2,3
Eric Thorin2,3Michel Carrier2,3- 1Department of Cardiac Sciences, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 2Department of Surgery, Faculty of Medicine, Université de Montréal, Montreal, QC, Canada
- 3Center for Research, Montreal Heart Institute, Montreal, QC, Canada
The vascular endothelium occupies a catalog of functions that contribute to the homeostasis of the cardiovascular system. It is a physically active barrier between circulating blood and tissue, a regulator of the vascular tone, a biochemical processor and a modulator of coagulation, inflammation, and immunity. Given these essential roles, it comes to no surprise that endothelial dysfunction is prodromal to chronic age-related diseases of the heart and arteries, globally termed cardiovascular diseases (CVD). An example would be ischemic heart disease (IHD), which is the main cause of death from CVD. We have made phenomenal advances in treating CVD, but the aging endothelium, as it senesces, always seems to out-run the benefits of medical and surgical therapies. Remarkably, many epidemiological studies have detected a correlation between a flavonoid-rich diet and a lower incidence of mortality from CVD. Quercetin, a member of the flavonoid class, is a natural compound ubiquitously found in various food sources such as fruits, vegetables, seeds, nuts, and wine. It has been reported to have a wide range of health promoting effects and has gained significant attention over the years. A growing body of evidence suggests quercetin could lower the risk of IHD by mitigating endothelial dysfunction and its risk factors, such as hypertension, atherosclerosis, accumulation of senescent endothelial cells, and endothelial-mesenchymal transition (EndoMT). In this review, we will explore these pathophysiological cascades and their interrelation with endothelial dysfunction. We will then present the scientific evidence to quercetin's anti-atherosclerotic, anti-hypertensive, senolytic, and anti-EndoMT effects. Finally, we will discuss the prospect for its clinical use in alleviating myocardial ischemic injuries in IHD.
Introduction
It is impressive to think that one single layer of cells tightly regulates homeostasis of the cardiovascular system. With its enormous surface area and its key location at the interface between circulating blood and tissue, the vascular endothelium has multiple physiological functions, such as modulation of the vascular tone and local regulation of coagulative, immune and inflammatory stimuli, in addition to providing a semipermeable barrier (1). A normally functioning endothelium appropriately arbitrates between opposing states of vasodilatation and constriction, permeability and non-permeability, adhesion and non-adhesion, as well as anti-thrombotic and pro-thrombotic conditions (1). Therefore, it is intuitive to imagine that a distortion in this equilibrium can result in adverse effects (2). Indeed, many cardiovascular diseases (CVD) are either a direct or indirect result of a dysfunction of the endothelium that fails to maintain moment-to-moment homeostasis, ultimately creating maladaptation in meeting organ metabolic demand and chronic damages (3). An example would be ischemic heart disease (IHD), which is the main cause of death from CVD (1). IHD itself represents an umbrella term for a group of clinical syndromes characterized by myocardial ischemia such as stable angina and acute coronary syndromes. Risk factors for endothelial dysfunction, and, by extension, IHD, include smoking, obesity, insulin resistance, diabetes, hypercholesterolemia, and physical inactivity (4). Phenomenal advances in pharmacology have enabled us to therapeutically target many of these risk factors, resulting in a significant decline in cardiovascular mortality over the last four decades (5). However, the use of drugs remains hampered by their toxicity, patients' tolerance and the limits of their clinical efficacy. In addition, endothelial dysfunction inevitably occurs with normal aging, fuelled by a process of irreversible cell cycle arrest termed senescence (6). For these reasons, there has been a burgeoning interest in introducing complementary therapies, such as dietary components, in the prevention of CVD (7).
Among promising nutraceuticals, a group of naturally occurring compounds found in plants, called flavonoids, have become increasingly popular. As early as in the 1990s, data from epidemiological studies have established a connection between a higher intake of flavonoid rich diets and a lower incidence of CVD (8). Quercetin has been singled out among flavonoids mainly because of its ubiquitous presence in our diets. It was also the first flavonoid to be discovered, precisely in the context of a vascular pathology. Indeed, in 1936, Albert Szent-Gyorgyi and his collaborators published the case of a patient who recovered from a bleeding disorder after receiving an infusion of a substance extracted from a Hungarian red pepper, which they called vitamin P, for “permeability” (9). Quercetin has since gained significant attention for its wide range of biological activities, some of which can mediate cardioprotective effects (10). In this review, we will examine quercetin's potential to alleviate CVD by protecting endothelial function. We will focus on three core pathophysiological mechanisms: atherosclerosis, hypertension and endothelial senescence. We will also cover quercetin's effects against endothelial-mesenchymal transition (EndoMT), as an additional, yet poorly explored, therapeutic avenue. Finally, we will discuss its potential use in secondary and tertiary prevention of endothelial dysfunction by taking the example of myocardial ischemic injury in IHD.
Endothelial Dysfunction as a Target for Preventing Cardiovascular Diseases
Conceptually, the core feature of endothelial dysfunction is a disrupted nitric oxide (NO) bioavailability as a consequence of a reduced production by endothelial NO synthase (eNOS) from L-arginine and in favor of free-radicals generation (11). Different causal paths have been implicated, including shear stress, dyslipidemia, hyperglycemia, insulin resistance, hyperhomocysteinemia and, more recently, senescence and EndoMT. The mechanisms by which they can lead to endothelial dysfunction and CVD pathogenesis are broad and complex. Most often, many of these factors accumulate in one person where they cross talk and synergistically enhance dysfunction of the arterial wall. Treatment of these cardiovascular risk factors was shown to reverse endothelial dysfunction and simultaneously improve the incidence of cardiac events (12). Here, we will focus our attention on the mechanistic connections between hypertension, atherosclerosis, senescence, and endothelial dysfunction.
Endothelial Dysfunction in Hypertension and Atherosclerosis
Endothelial dysfunction is seen as an early step in the development of hypertension and atherosclerosis (13, 14). Indeed, the functional characteristics of endothelial dysfunction include an impairment of endothelium-dependent vasodilation and endothelial activation marked by pro-inflammatory, proliferative, and procoagulatory states (14).
Upon activation, endothelial cells switch from a predominant NO signaling to an oxidative stress signaling mediated by reactive oxygen species (ROS) (15). While NO promotes inhibition of pro-inflammatory cytokine secretion, thrombosis, smooth muscle cell proliferation and immune cell extravasation, ROS induce nuclear transcription factor kappa B (NFκB) signaling, the main regulator of inflammation (15). In addition, the diseased endothelium acquires a pro-inflammatory state and becomes more permeable, allowing the avid accumulation of oxidized low-density lipoproteins (ox-LDLs) and macrophages in the subintimal layer, culminating in foam cell formation and fatty streaks which are hallmarks of atherosclerosis development (15). On the other hand, a defective L-arginine/NO pathway, impaired responsiveness to exogenous NO and reduced generation of platelet NO result in a state of predominant vasoconstriction and higher resting blood pressure (14). Furthermore, atherosclerotic lesions develop preferentially at arterial bifurcations, branching points and vessel curvatures, where the blood flow is disturbed (16). This suggests the importance of hemodynamic forces and mechanical stress, hence of hypertension, in the initiation of atherosclerosis. When considering the role of atherosclerosis in hypertension, a number of studies reported that atherosclerotic segments were accompanied by an altered function of eNOS in which it produces superoxide instead of NO (17). NADPH oxidase (NOX), which is induced by ox-LDLs, was shown to lie upstream to this eNOS alteration (17). Referred to as “eNOS uncoupling,” this oxidative pathway is also present in aged microvessels (18). It goes without saying that oxidative stress plays a critical role in endothelial dysfunction, and, as we will next, in stress-induced senescence.
This interconnection between endothelial dysfunction, atherosclerosis and hypertension has been confirmed clinically: using arterial dilatation as a non-invasive measure for assessing endothelial function, endothelial dysfunction has been documented in both hypertensive and atherosclerotic patients (12, 19–22). Using acetylcholine to induce endothelium-dependent dilation, a reduction in arterial dilation was observed in the forearm and coronary beds of patients with essential hypertension (12). Furthermore, the response to acetylcholine and adenosine was significantly decreased in patients with hypertension and left ventricular hypertrophy, indicating an impairment in both endothelium-dependent and endothelium-independent vasodilation (19). Ludmer et al. provided the first evidence of compromised endothelium-dependent vasodilation in the presence of atherosclerosis in humans (20). Using the acetylcholine test, they reported a paradoxical constriction in the coronary arteries of patients with both mild and advanced coronary artery disease (20). Endothelial dysfunction was also present in the vasculature of patients with coronary risk factors but no angiographic or ultrasound evidence of structural coronary artery disease (21). These studies suggest that endothelial dysfunction is detectable from the early stages of atherosclerosis and that it might even be a trigger mechanism (22).
Now endothelial dysfunction can be extended beyond the concept of a damaged conduit vessel to that of a defective vascular wall composed of layers of cells that are prone to aging. If endothelial dysfunction is the primum movens of hypertension and atherosclerosis, an upstream connection between the three could be linked to senescence.
Senescence: The Natural Fate of Aging Cells
Successive replication (23) and harmful stimuli such as DNA damage, oxidative stress, and induction of mitochondrial dysfunction eventually impose a state of permanent proliferative arrest on cells (24, 25). This phenomenon, termed “senescence,” is well-recognized as one of the nine hallmarks of aging (26).
Despite being in cell cycle arrest, senescent cells (SCs) undergo profound phenotypic changes and remain metabolically active. In response to stress, they secrete a set of proteins collectively termed the senescence-associated secretory phenotype (SASP) (27, 28). These include pro-inflammatory cytokines (interleukin (IL)-6, IL-8, membrane cofactor proteins (MCPs) and macrophage inflammatory proteins) and chemokines, immune modulators, growth factors [hepatocyte growth factor, fibroblast growth factors, granulocyte-macrophage colony-stimulating factor, or transforming growth factor beta (TGF-β)] and hundreds of signaling molecules such as damage-associated molecular patterns, proteases, extracellular matrix (ECM) components [matrix metalloproteinases (MMPs)], serine/cysteine proteinase inhibitors (SERPINs), tissue inhibitor of metalloproteinases and cathepsins), proteases, bradykinins, and hemostatic factors (27–29). Without a doubt, the SASP plays an essential role in normal tissue development (30), wound healing (31), and cardiac repair (32). Transient expression of SASP during the acute phase of a tissue injury assists with repair and remodeling by recruiting the immune system to clear damaged cells and by stimulating progenitor cells to repopulate the damaged tissue (33). However, senescence becomes a double-edged phenomenon when ineffective clearance of SCs prolongs their residency. In the concept of “inflamm-aging,” aberrant focal accumulation of SCs creates a pro-inflammatory environment favorable for the onset of various pathological conditions, including endothelial dysfunction (34). Indeed, a growing body of evidence shows that SCs are prominent in diseased vascular walls (35), including in intact arteries from IHD patients (36, 37). Furthermore, although vascular cells have a finite replicative capacity, a combination of both damage-dependent replicative senescence and stress-induced senescence might be especially relevant to premature vascular aging and endothelial dysfunction (3, 38).
The Vicious Circle of Endothelial Dysfunction
Senescence of a vascular wall leads to two immediate consequences: induction of a pro-inflammatory environment by the SASP and a reduction in the turnover of vascular cells (Figure 1). In atherogenesis, plaque initiation could be driven by senescent endothelial cells through their increased secretion of chemoattractant factors and adhesion molecules, which allow for the initial invasion of circulating monocytes into the vessel wall (39). Conversely, clearance of senescent vascular cells lowered the pathogenesis of atherosclerosis in a mouse model of severe dyslipidemia (36). In addition, a senescent endothelium presents an altered cellular lining, causing a break in selective permeability (40). This can facilitate migration of ox-LDLs to the subendothelial layers. The SASP can also stimulate vascular smooth muscle cells to secrete elastase and MMPs, which can digest components of the extracellular matrix (41). An amplified degradation of the extracellular matrix could create a rupture-prone vulnerable plaque. Thus, senescence of vascular cells leads to vascular inflammation and plaque progression. This vascular inflammation also raises the possibility of a multistep role of senescence in hypertension, although the link between the two is less clearly established. The SASP could play a role in the dysregulation of the vascular tone: as an example, it was found to activate the renin–angiotensin–aldosterone system (35).
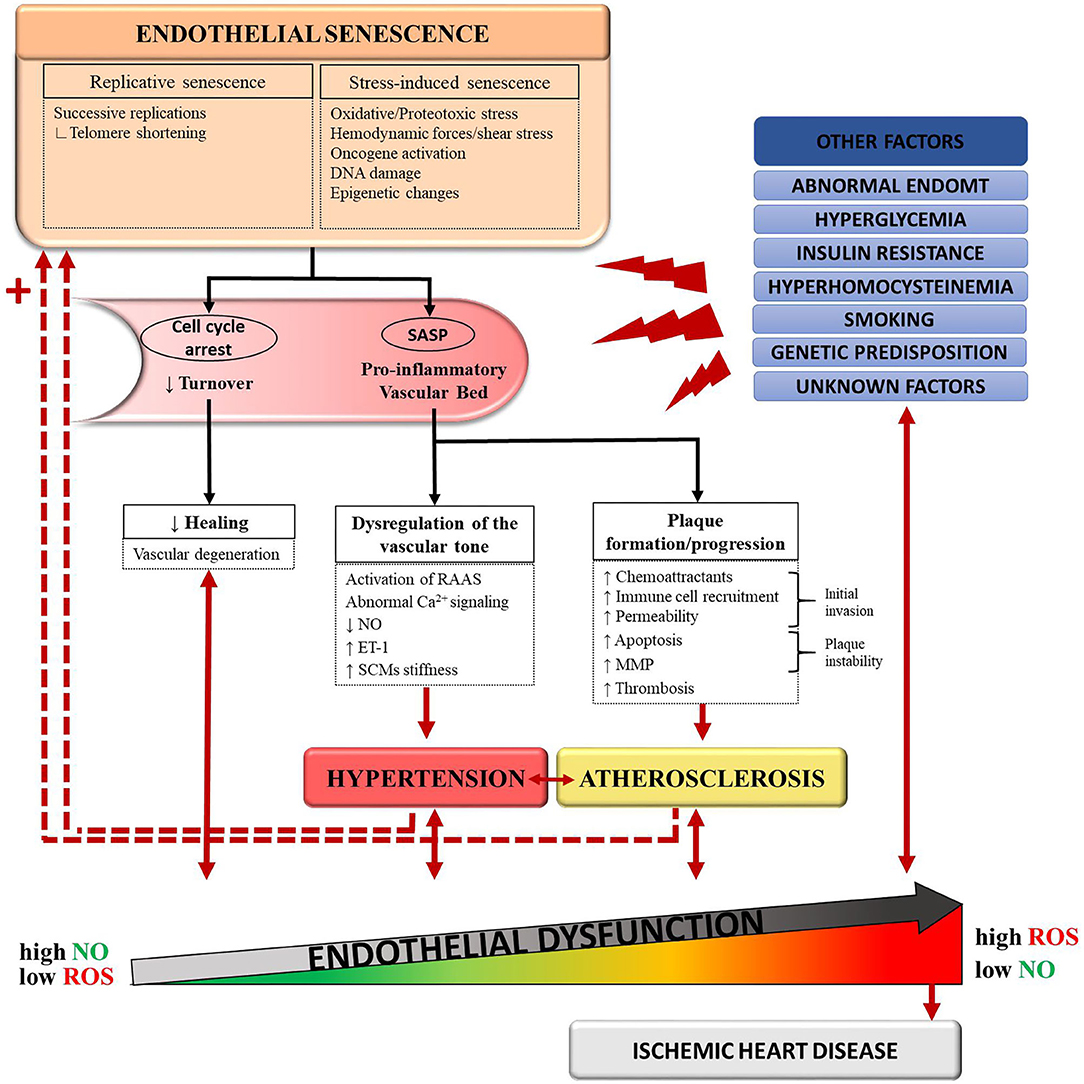
Figure 1. Schematic representation of the proposed connections between senescence, hypertension, atherosclerosis, and endothelial dysfunction. Normal aging and deleterious stimuli induce senescence in endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and foam cells. Accumulation of these senescent cells favors a pro-inflammatory state of the vascular bed through the senescence-associated secretory pathway (SASP). In turn, the SASP promotes pathological changes leading to the development of hypertension and atherosclerosis. In a feedback manner, hypertension and atherosclerosis induce more stressors to an already dysfunctional and senescent vessel wall. This vicious circle translates into endothelial dysfunction and, eventually, ischemic heart disease. Other causal pathways of endothelial dysfunction include hyperglycemia, insulin resistance, abnormal endothelial-to-mesenchymal transition (EndoMT), genetic predisposition and detrimental lifestyle habits such as smoking. ET-1, endothelin-1; MMP, matrix metalloproteases; NO, nitric-oxide; RAAS, renin–angiotensin–aldosterone system; ROS, reactive oxygen species.
With aging, the clearance of SCs by the immune system is decreased, contributing to the accumulation of SCs (33, 42). Senescence therefore begets senescence (Figure 1), a phenomenon that has been validated in mice (43). As senescence induces more senescence, the processes involved in the pathogenesis of atherosclerosis and hypertension are further amplified. In parallel, important changes in the extra-cellular matrix (ECM) protein composition occur with aging and promote arterial stiffening (44). A rigid arterial wall causes systolic hypertension, which in turn, contributes to atherosclerosis through shear stress (3). As mentioned before, other cardiovascular risk factors often coexist, such as metabolic disturbances, obesity, smoking or genetic predisposition, accelerating this deleterious process (12). The vascular wall eventually gets caught into a vicious circle where it must face more stressors with less protective capacities (3). Therefore, it is possible to acknowledge a cyclical, rather than sequential, relationship between senescence, hypertension and atherosclerosis, all contributing to endothelial dysfunction. In the next sections, we will explore quercetin's potential to target this triad of endothelial dysfunction.
Quercetin
Classification and Structure
Quercetin is part of a larger family of molecular compounds, named flavonoids, which share a common hydroxylated 3-ringed skeleton with attached hydroxyl groups (45) (Figure 2). Combined with the pyrocatechol, a benzene ring, this chemical structure allows them to act as radical scavengers, explaining, in part, flavonoids' antioxidant property (45). Flavonoids are themselves part of a large class of plant-derived substances named polyphenols (46). Flavonoids include several subclasses such as flavonols, flavones, flavanols, flavanones, isoflavones, and anthocyanins (46). They exist in most of the plants and play a variety of biological activities involved in vegetative growth (46). Being phytochemicals, flavonoids cannot be synthesized by humans or animals, but they are ubiquitously present in our diet (46). They are found in virtually all fruits and vegetables, as well as in seeds, nuts, tea and red wine (46). The mean daily intake of flavonoids in Australian, European and US adult populations has been estimated at 435 mg/day (47).
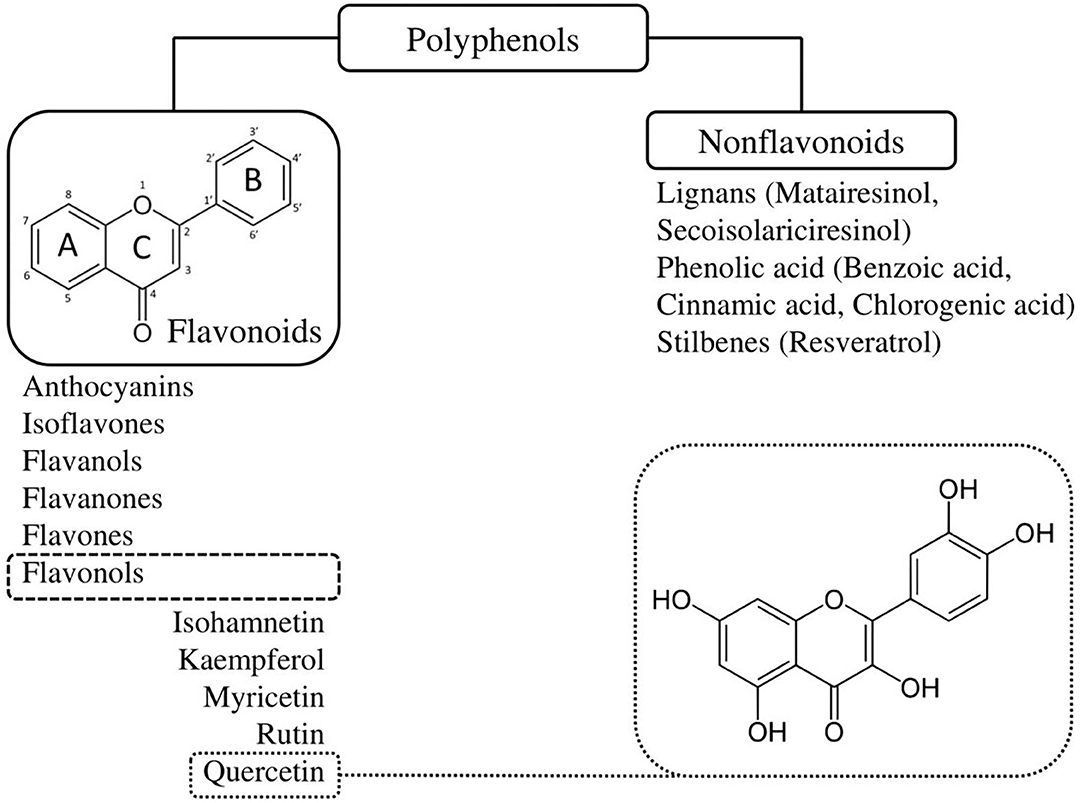
Figure 2. Classification and chemical structure of quercetin, a family member of flavonoids. Quercetin is a pentahydroxyflavone, having five hydroxyl groups placed at the 3-, 3'-, 4'-, 5-, and 7-positions. Combined with the pyrocatechol, a benzene ring, this chemical structure allows them to act as radical scavengers, explaining, in part, quercetin's strong antioxidant properties.
Structurally, quercetin is not only found in its free (aglycone) form, but also in various conjugated forms with glycosides or methyl ethers attached to the hydroxyl groups. Glycosylation preferentially occurs at the 3-hydroxyl position, such as quercetin 3-O-β-D-glucoside (isoquercetrin) or quercetin 3-O-galactoside (hyperoside), whereas methylation usually occurs at the 3', 4', or 7-hydroxyl positions, such as 3-methylquercetin (isorhamnetin) (48). Some quercetin derivatives even contain both glycosyl and ethyl groups. For example, tamarixetin has a glucose residue at the 3' position and a methyl group at the 4' position (48). Extensive studies of the biological activities of quercetin have shown that the various derivatives have different levels of efficacy. For example, free quercetin was found to have the strongest antioxidant activity, confirming the important contribution of unbound hydroxyl groups (49). Among its metabolites, free quercetin was also the most effective recombinant human angiotensin-converting enzyme (ACE) 2 inhibitor (50). Tamarixetin and isorhamnetin demonstrated a stronger inhibition of lipid peroxidation compared to quercetin (49, 51). Tamarixetin also exhibited the highest anti-inflammatory activity, suggesting that unlike the antioxidant activity, anti-inflammatory activity is not correlated with the number of free hydroxyl groups (49). These disparities in biological activities prompt the synthesis of particular metabolites that present the highest efficacy of a desired effect. This can be achieved by inducing glycosylation or methylation using purified biocatalysts in vitro and native or metabolically engineered microorganisms (48).
Bioavailability and Pharmacology
The chemical structure of aglycone quercetin makes it hydrophobic (52). Its solubility in water is 2.1 mg/L at 25°C, while it is up to 2 g/L in ethanol (52). This physical property limits its absorption and practical use in preparation forms as a dietary supplement. Initial investigations on the pharmacokinetics of quercetin in humans suggested very poor oral bioavailability after a single oral dose (~2%) (53). The absorption was found to increase to 3–17% when quercetin was consumed in a glycosidic bond compared to its aglycone form (53). Different delivery systems using nanotechnology have since been developed to further improve its water solubility and bioavailability, for example, by binding it to solid lipid carriers or nanosized polymeric micelles (54). A pharmacokinetic study in beagle dogs showed that quercetin encapsulated in polymeric micelles induces a 2.19-fold longer half-life and a relative oral bioavailability increased by 286% as compared to free quercetin (55).
Since dietary quercetin is usually present in its glycosylated form, it can be rapidly hydrolyzed by β-glucosidases in the digestive tract, which makes it easier for absorption by the colonic mucosa (56). It is then transferred to the liver through the portal circulation where it undergoes first-pass metabolism and is almost completely metabolized by glucuronidation, methylation, or sulfonylation (56, 57). Peak plasma concentration following an oral quercetin dose is reached anywhere from 0.6 to 4 h (58–60). Quercetin glucuronides are the main circulating metabolites and are rapidly eliminated in the urines (57, 60). This short elimination half-life is another limit to quercetin's medical use. Furthermore, quercetin's metabolism seems to be dependent on individual characteristics. A correlation between β-glucuronidase activity and the apolipoprotein (apo) E phenotype may explain the efficacy of quercetin in patients with apoE3 phenotype as opposed to those expressing apoE4 (61, 62). On the other hand, an increased expression of β-glucuronidase was correlated with inflammation, raising the hypothesis that quercetin may be more effective under inflammatory conditions (63). This is especially favorable as endothelial dysfunction is often associated with a pro-inflammatory state.
When it comes to pharmacokinetic interactions, conclusions are still open to debate. Some studies investigated the effects of quercetin on the cytochrome P450 system and have noted a potential inhibitory effect of quercetin on the activity of selected enzymes (64). Studies in pigs have shown that quercetin can decrease bioavailability of cyclosporine and increase that of digoxin, verapamil and various chemotherapeutic agents (65–67). However, conflicting results between in vitro and in vivo studies have been found (67). Quercetin was also reported to bind to DNA gyrase enzyme in bacteria, which could competitively inhibit fluoroquinolone antibiotics' activity (68). One case report of a clinically relevant warfarin interaction resulting in supratherapeutic international normalized ratio values has been documented in an elderly patient who ate large quantities of scuppernongs, a quercetin-containing muscadine grape (69).
Safety Profile
In the 1970s, in vitro mutagenicity of quercetin in the Ames test was reported, leading to concerns about its safety (70). Later, in vivo studies contradicted these findings and showed that quercetin may be protective against carcinogens (70). Since 1999, it is classified as a group 3 agent (“not classifiable as to its carcinogenicity” to humans) by the International Agency for Research on Cancer (70). In 2010, QU995, a highly pure form of quercetin, was granted a “generally recognized as safe” (GRAS) status by the U.S. Food and Drug Administration (59). Many other quercetin formulas have since been developed and made widely available over the counter as oral dietary supplements or added ingredient to numerous multivitamin preparations.
Quercetin is generally well-tolerated. Some minor side-effects such as mild headache, nausea, and tingling of the extremities were observed in long-term supplementation at 1,000 mg/day (67). In Canada, the recommended maximum daily dose is 1,200 mg (67). A therapy as long as 12 weeks showed no evidence of toxicity, but data on long-term safety are lacking (67). Nephrotoxicity has been reported with high intravenous doses in cancer patients (67). Quercetin was not found to cause critical adverse effects on fetal growth in rats, but human studies are not available (67). Therefore, dosages above those found in foods should be avoided by pregnant women and nursing mothers (67).
A Recent Resurgence in Interest
While it was previously known as “vitamin P,” the National Nutrition Institute withdrew its status in 1950 when it was found to be a non-essential nutrient (71). Added to a mislabeling of genotoxicity, altogether this contributed to a loss of interest in the molecule. In 1993, however, the Zutphen Elderly Study first reported a 50% reduction of mortality from IHD in Dutch men who consumed >29 mg flavonoids/day compared with those who consumed <19 mg (72). Around the same time, the concept of the French paradox emerged from the contradictory observation of a low IHD-related mortality despite high intakes of dietary saturated fat among the French population (73). Most debates have focused on high consumption of red wine, which contains a variety of polyphenols, including flavonoids (73). Other epidemiological studies soon followed and showed a positive correlation between dietary intake of flavonoids and a reduced incidence of stroke, myocardial infarction and mortality from IHD (74).
Over the years, quercetin was found to have a diverse array of biological properties, such as anti-inflammatory, anti-oxidative, anti-platelet, anti-diabetic, anti-histaminic, anti-carcinogenic, anti-bacterial, immunomodulating, and neuroprotective (75). These prominent effects have sparked attention and hope among the scientific community. As of the end of 2020, there are more than 20,000 published articles on quercetin, and this number exceeds 120,000 when including all flavonoids (Figure 3). Despite quercetin being discovered for its role in treating capillary wall dysfunction, it has gained more popularity in oncology and sports medicine, each counting 50% more publications than the field of cardiovascular research. However, its promising benefits for the endothelium cannot be ignored (Table 1).
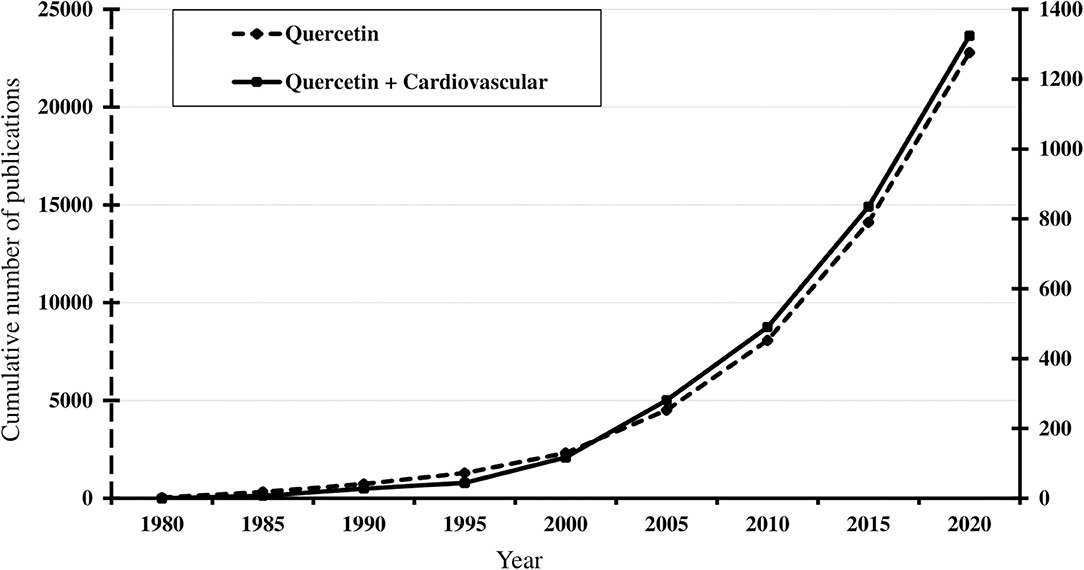
Figure 3. Timeline of the cumulative number of published results, from 1980 to 2020, of an online PubMed literature search using “Quercetin” (dotted line) and “Quercetin [and] Cardiovascular” (solid line) as the search term. Note the progressive increase from the mid-1990s, coinciding with publication of observational studies associating flavonoid consumption with lower cardiovascular risks. Search performed January 10, 2021 (www.ncbi.nlm.nih.gov/pubmed).
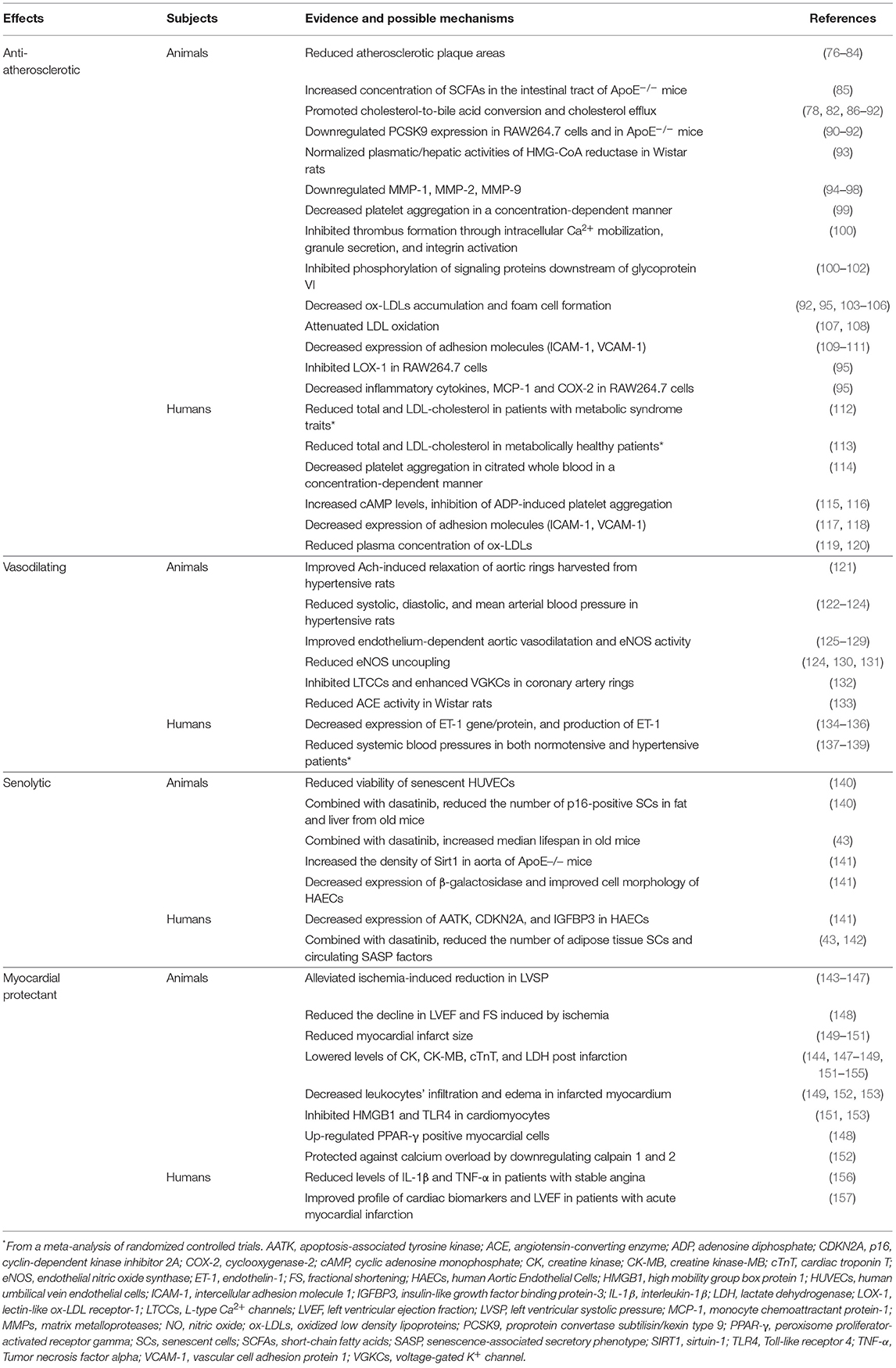
Table 1. Summary of the main in vitro and in vivo cardiovascular effects of quercetin.
Cardiovascular Protective Properties of Quercetin
Anti-Atherosclerotic Effects of Quercetin
With the increasing epidemic of the metabolic syndrome, the burden of atherosclerosis-related disorders persists despite the current pharmacologic treatment of dyslipidemia (64). Therefore, finding additional anti-atherogenic drugs remains a topic of interest (158). Considerable evidence from experimental data indicates that quercetin may protect against atherosclerosis by interfering with multiple pathways involved in disease progression (Table 1). Several high-fat animal models exhibited reduced atherosclerotic plaque areas when exposed to quercetin (76–84). This observation was associated with a prevention of atherosclerosis-related acute aortic syndromes: in a mouse model with an exaggerated degeneration of the elastic lamina: administration of quercetin 2 weeks before inducing aortic diseases was found to reduce the incidence of aneurysms, dissections and aortic ruptures (109).
First, quercetin could positively regulate the metabolism of lipids. A recent systematic and meta-analysis of 16 randomized controlled trials (RCTs) published between 2007 and 2017 looked at the effects of quercetin on lipid profiles of patients with metabolic syndrome traits (112). A pooled analysis revealed that quercetin leads to a significant reduction in total and LDL cholesterol, without affecting triglyceride levels (112). The daily doses and treatment durations used in the trials varied greatly, from 3.12 to 3,000 mg/ day and from 3 to 12 weeks (112). Another meta-analysis of 9 RCTs done in overweight and obese subjects found that quercetin supplementation significantly reduces LDL cholesterol levels at doses of ≥250 mg/day and for a total dose ≥14,000 mg (159). Similar findings were observed in metabolically healthy non-obese adults after an 8-week regimen, with comparable effects among men and women (113). Recent studies have highlighted the influence of the gut microbiota on host metabolic health through its metabolites, especially short-chain fatty acids, which have been linked with improved lipid metabolism (160, 161). Quercetin was shown to increase the concentration of short-chain fatty acids in the intestinal tract of ApoE knockout (ApoE−/−) mice (85). Experiments on both in vivo rodent models and murine cultured macrophages (RAW264.7 cells) have suggested that quercetin promotes cholesterol-to-bile acid conversion and cholesterol efflux by upregulating activity of hepatic CYP7A1, liver X receptor α, ABCG1, ABCA1, and LDLR (78, 82, 86–92). Quercetin also downregulated PCSK9 expression in RAW264.7 cells and in ApoE−/− mice (90–92). HMG-CoA reductase plays a major role in the regulation of cholesterol metabolism as a rate limiting enzyme in the pathway of cholesterol biosynthesis (162). Results relating the effects of quercetin on HMG-CoA reductase activity have been inconsistent (86, 93). Wistar rats fed with a diet containing 0.4% quercetin for 5 weeks did not express a change in the enzyme's activity (86). However, in a model of isoproterenol (ISO)-induced myocardial infarction in Wistar rats, a 2 week oral quercetin pre-treatment at a dose of 10 mg/kg normalized plasmatic and hepatic activities of HMG-CoA reductase (93). Another protective mechanism of quercetin involving enhancement of autophagy by aortic macrophages was highlighted in ApoE−/− mice (84).
Second, quercetin has been suggested to downregulate the expression of MMP-1, MMP-2, and MMP-9 in studies using molecular modeling techniques, cultured endothelial cells, murine macrophage cells and in hypertensive rats, an effect that translates in the prevention of plaque instability (94–98).
Third, platelet aggregation at the site of an unstable plaque also contributes to acute complications of atherosclerosis. Quercetin was found to have an antiaggregatory effect on rat platelet-rich plasma in a concentration-dependent manner (99). This was also observed in human citrated whole blood: using samples from 100 healthy volunteers, the minimal antiaggregatory concentration of quercetin was estimated at 15.26 μM (114). A synergistic enhancement of antiplatelet effect was noted when quercetin was added to aspirin (100). The half maximal inhibitory concentrations (IC50) values for the inhibition of platelet aggregation decreased from 10.83 μM when using aspirin alone, to 3.32, 2.99, and 1.11 μM upon co-administration of 2.5, 5, and 10 μM quercetin, respectively (100). In addition, isorhamnetin and tamarixetin, two methylated metabolites of quercetin, were shown to inhibit platelet aggregation and thrombus formation in vitro through effects on activation processes such as intracellular Ca2+ mobilization, granule secretion, and integrin activation (100). Their antithrombotic effect was confirmed with laser-induced thrombi in mouse cremaster arterioles (100). In human platelets, quercetin significantly increases cyclic AMP levels and inhibits arachidonic acid and adenosine diphosphate (ADP)-induced platelet aggregation (115, 116). Antiplatelet effects of quercetin and its metabolites have also been associated to inhibition of the phosphorylation of signaling proteins downstream of glycoprotein VI, namely the Src family tyrosine kinases Fyn and Syk, the phospholipase Cγ2 and the linker for activation of T cells (100–102).
Fourth, once oxidized in the intima, LDLs transform into an antigenic factor, ox-LDLs, which attract monocyte-derived macrophages to the vascular wall, thereby initiating a phagocytic process leading to foam cell formation (163). Accumulation of foam cells is an early step in the pathogenesis of atherosclerosis (163). In their study, Kawai et al. used mAb14A2, a novel monoclonal antibody binding quercetin, to stain aortic samples in Japanese subjects (164). Their results revealed that quercetin metabolites accumulate in atherosclerotic lesions, but not in normal-appearing aorta (164). In addition, intense staining was primarily localized with foam cells, suggesting a potential cellular target of quercetin (164). Several studies done on cultured cells showed that quercetin can attenuate ox-LDLs accumulation, foam cell formation, as well as ox-LDLs induced cytotoxicity and calcification (92, 95, 103–106). Interestingly, quercetin significantly reduced plasma concentrations of ox-LDLs in two RCTs (119, 120). A retrospective comparison of the participants' apoE genotypes revealed no significant inter-group difference in the reduction of ox-LDLs between the apoE3 and apoE4 subgroups (62). This lowering effect on ox-LDLs might be achieved through direct attenuation of LDL oxidation: the lag time of LDL oxidation was increased by 3- to 4-fold after administration of quercetin in vitro and in rats (107, 108). The authors proposed two mechanisms contributing to this attenuation of LDL oxidation: inhibition of copper-induced LDL oxidation, as well as up-regulation of Paraoxonase 1 (PON1) and its protective capacity against LDL oxidation (107, 108). Lectin-like ox-LDL receptor-1 (LOX-1) is a scavenger receptor that mediates uptake of ox-LDLs by macrophages (165). Administration of anti-LOX-1 antibodies was shown to inhibit atherosclerosis by decreasing these cellular events (165). Quercetin was shown to block LOX-1 in RAW264.7 cells (95). Moreover, ox-LDLs activate endothelial cells by inducing cell adhesion molecules, especially vascular cell adhesion molecule-1 (VCAM-1) and intracellular cell adhesion molecule-1 (ICAM-1) (166). Quercetin was found to downregulate ICAM-1 expression in diabetic rats and human endothelial cells (110, 117). Quercetin and isoquercetin were shown to attenuate VCAM-1 expression in mice, HUVECs and rat intestinal microvascular endothelial cells by suppressing multiple pathways including caveolin-1 (CAV-1), Toll-like receptor 4 (TLR4) and NFκB (109, 111, 118). As previously mentioned, ox-LDLs also stimulate eNOS uncoupling and ROS overproduction by macrophages and endothelial cells via activation of NOX (167, 168). In ApoE−/− mice, quercetin partially reversed NOX expression and inhibited ox-LDL induced ROS formation in macrophages (83).
Finally, atherosclerosis is also a chronic inflammatory disease mediated by a network of pro-inflammatory cytokines. Quercetin's administration was associated with a decrease in multiple inflammatory cytokines, such as IL-1α, IL-1β, IL-2, IL-10, TNF-α, macrophage chemoattractant protein-1 and cyclooxygenase-2 (95). The impacts of quercetin on such a wide range of inflammatory markers are in favor of a multi-target effect of signal transduction.
Vasodilating Effects of Quercetin
Several ex-vivo reactivity studies have shown a vasodilating ability of quercetin in rat aorta, portal vein, mesenteric arteries and coronary arteries (169–171) (Table 1). In addition, Choi et al. reported that quercetin acutely improved acetylcholine-induced relaxation of aortic rings harvested from two-kidney, one-clip (2K1C) hypertensive rats (121).
Quercetin's BP lowering effects were first documented in vivo in spontaneously hypertensive rats (122). Rats exposed to quercetin had a significant lower systolic (−18%), diastolic (−23%), and mean (−21%) arterial BP (122). In normotensive rats, endothelial dysfunction induced by a high-fat high-sucrose diet was prevented by the supplementation with quercetin for 28 days: both endothelium-dependent aortic vasodilatation and eNOS activity were improved by quercetin (125). The vasomotor protective effects of quercetin were also demonstrated in mice exposed to lipopolysaccharide-induced endotoxemia. Whether given before or after lipopolysaccharide injection, quercetin dose-dependently restored eNOS expression while abolishing inducible NO synthase (iNOS) (126).
Several hypotheses have been formulated regarding the up regulation of eNOS activity induced by quercetin. Some authors have suggested that quercetin phosphorylates eNOS by an AMP-activated protein kinase-dependent mechanism (127). Li et al. observed, in bovine aortic endothelial cells, that quercetin induced phosphorylation of eNOS at serine 1179 in a concentration, time-dependent manner; this effect was abolished by H-89, an inhibitor of protein kinase A (128). Using the same primary cell cultures, Khoo et al. proposed that quercetin stimulates eNOS phosphorylation at serine 1179 by causing a rapid increase in intracellular Ca2+ (129).
Other calcium-mediated vasoactive effects of quercetin have been proposed. L-type Ca2+ channels (LTCCs) and voltage-gated K+ channels (VGKCs) play a tonic role in the regulation of arterial vasomotricity and are commonly expressed in vascular smooth muscle cells (172, 173). LTCCs are involved in excitation-contraction coupling while VGKCs are critical for restoring the resting membrane potential (172, 173). Large (big)-conductance Ca2+-sensitive potassium channels (BK) and VGKCs are closely associated with coronary arterial smooth muscle vasodilatation (174). Of note, aging is associated with a reduced expression of BK channels in coronary arteries, which is consistent with a higher frequency of spontaneous vasospasmic activity in elderly people (174). Hou et al. showed that quercetin can inhibit LTCCs and enhance VGKCs in rat coronary artery rings, resulting in a decrease of the vasocontractions induced by high-K+ depolarizing solution (132). Moreover, coronary vasodilation induced by quercetin was not lost after denuding the arterial rings of their endothelium, suggesting that quercetin can also promote its vasodilatory effect through VSMC-mediated mechanisms (132). Cogolludo et al. noted that quercetin could activate BK channels in coronary artery myocytes while generating hydrogen peroxide (H2O2) (175). Although H2O2 is considered as a relaxing endothelium-derived hyperpolarizing factor (176) and can also activate the soluble guanylate cyclase as does NO (177, 178), the data of Cogolludo et al. may nonetheless represent an instance where quercetin behaves as a pro-oxidant rather than a vasodilator.
Endothelin (ET) is one of the most potent vasoconstrictors and is mainly produced by the vascular endothelium (179). ET-1 plays a major role in the homeostasis of the cardiovascular system. ET-1 has been associated with increased oxidative stress and endothelial dysfunction in humans (179). It was shown to stimulate eNOS uncoupling, therefore superoxide production, and promote vasoconstriction via activation of NOX (130, 180). ET-1 can further reduce NO bioavailability by interfering with eNOS expression through protein kinase C (PKC)-mediated activation of STAT3 (181). These data indicate that diminished ET-1 concentrations may be accompanied by elevated NO bioavailability (181). Lodi et al. showed that quercetin significantly decreased expression of ET-1 in human umbilical artery smooth muscle cells and human vein endothelial cells (HUVECs) co-culture model exposed to TNF-α-induced change in vasomodulatory molecules (134). Zhao et al. also showed that quercetin decreases ET-1 production in thrombin-stimulated HUVECs in a concentration-dependent manner, with an IC50 of 1.54 μmol/L (135). In rat aortic rings, quercetin prevented ET-1-induced PKC activation, with a subsequent decrease in superoxide production (130). Moreover, chronic treatment with quercetin reduced blood pressure and improved endothelial function in deoxycorticosterone acetate (DOCA)-salt rats, a low renin model of hypertension in which ET-1 is overexpressed (123, 124). These effects of quercetin were associated with a reduction in both vascular and systemic oxidative stress (124). Quercetin protective effects against eNOS uncoupling were even maintained under glucotoxic conditions (131).
A RCT studied the acute effects of administering 200 mg of quercetin in 12 healthy men (136). Blood and urine samples taken, respectively, 2 and 5 h after oral ingestion of quercetin revealed a significant acute reduction in both the plasma and urinary concentrations of ET-1, translated into a reduced ET-1 production (136). This effect is substantial given the relatively small dose used and the low bioavailability of quercetin. Interestingly, it was reported that NO inhibits ET-1 production through the suppression of NFκB (182). A second mechanism involves the renin–angiotensin system: ACE inhibitors neutralize ACE by binding a zinc atom at the active site of the enzyme, which slows conversion of angiotensin I to angiotensin II, a powerful vasoconstrictor (183), including in human coronary arteries (184). Quercetin can chelate metal ions, including zinc (185), and it is tempting to presume it can act as an ACE inhibitor. However, available results have been discordant. In vitro, quercetin inhibited ACE activity in a concentration-dependent manner, with an IC50 of 310 μM (186). This value was significantly higher than that of captopril (0.02 μM) (186). In Wistar rats receiving an angiotensin-1 infusion, Hackl et al. showed that an attenuation of the BP was obtained with both oral and intravenous administration of quercetin (133). They also reported a 31% reduction in ACE activity in the quercetin group compared to the control group (133). In contrast, one double-blind placebo-controlled RCT did not find ACE activity inhibition after a single-dose of quercetin (187). In this study, five normotensive men and twelve hypertensive men ingested a total of 1,095 mg quercetin and 10 h later, the mean BP was reduced among the hypertensive patients by 5 mm Hg compared to the placebo group (187). Plasma ACE activity, ET-1, and brachial artery flow-mediated dilation were unaffected by quercetin, suggesting that the reduction in BP in hypertensive men was independent of the changes in ACE and ET-1 activity, or NO bioavailability (187).
Serban et al. conducted a systematic review of 7 RCTs published between 1998 and 2014, looking at the effects of quercetin on BP (137). Their meta-analysis revealed a significant reduction in systemic BPs associated with oral supplementation of quercetin (137). The weighed mean differences for the systolic and diastolic BPs were 3.04 mm Hg (p = 0.028) and 2.63 mm Hg (p < 0.001), respectively (137). These values are appreciable considering that the cohorts were largely made up of normotensive subjects. The doses of quercetin ranged from 100 to 1,000 mg/day. Interestingly, when using a meta-regression analysis, the systolic BP-lowering effect was only associated with the duration of supplementation, and not the administered dose, contrarily to the diastolic BP-lowering effects (137). Furthermore, when the RCTs were stratified according to the duration of supplementation, quercetin had no significant benefit in the subsets of studies lasting <8 weeks. Likewise, the BP values did not differ significantly between the two treatment arms in the subset of trials administering doses <500 mg/day. Altogether, these results indicate a significant anti-hypertensive effect of quercetin supplementation only when doses ≥500 mg/day are taken for ≥8weeks (137). Another meta-analysis which included 896 participants across 17 RCTS mirrored the results obtained by Serban et al., which indeed is the meta-analysis done by Huang et al. (138). More recently, a meta-analysis of 8 RCTs conducted among patients with metabolic syndrome traits showed that quercetin supplementation significantly reduced systolic BP, yet did not affect diastolic BP (139). Clearly, trials directly comparing different doses and regimen durations are needed.
Senolytic Properties of Quercetin
The accumulation of SCs in the aging and diseased vessel wall raises the possibility that reducing senescence might delay deterioration of vascular structure and function. In an elegant and seminal experiment, Baker et al. demonstrated that the health span in progeroid mice can be enhanced by killing SCs using a transgenic suicide gene (188). Elimination of SCs also delayed progression of multiple age-related phenotypes, such as cancer, cataract, sarcopenia, lordokyphosis, loss of adipose tissue and skeletal muscle fibers, as well as improved exercise capacity (188). Translating that same effect into a druggable compound sparked research interest and led to the recent concept of “senolytic therapy” (140). Formed by the words “senescence” and “lytic” (destroying), a senolytic represents a molecule that could specifically induce cell death in SCs (189). Based on the knowledge that SCs survive despite their harsh internal state, the hypothesis was that this would be achieved by targeting their survival pathways and anti-apoptotic mechanisms (189). An alternative strategy to interfere with senescence would be to reduce the burden of SASP. The advent of antibody-based techniques such as sandwich enzyme-linked immunosorbent assay, and large-scale molecular biology techniques such as mRNA profiling, antibody arrays, proteomics or multiplex assays have made the detection and measurement of several SASP factors possible (190). These powerful tools therefore serve to test pharmaceutical efficacy of drugs that target SASP (190). In the following sections, we will see evidence suggesting that quercetin eliminates SCs and reduces the SASP.
In 2008, quercetin was reported to increase longevity of worms (191), but it was not until 2015 that its potential as a senolytic was highlighted in Kirkland's laboratory (140) (Table 1). First, the investigators identified a series of senolytic transcripts on pre-adipocytes. These included components of the ephrin regulating system, ephrin ligands B (EFNB), as well as the plasminogen-activated inhibitor-1 (PAI-1) and a member of the phosphatidylinositol-4,5-bisphosphate 3 kinase (PI3K) family, involved in regulating multiple cellular functions including survival (140). Then, they tested whether drugs that target any of these gene products would effectively induce apoptosis in radiation-induced senescent human pre-adipocytes and HUVECs. Of the 46 agents tested, quercetin and dasatinib, a non-specific tyrosine kinase inhibitor used for cancer therapy, were noticeably promising (140). Dasatinib is known to block EFNB-dependent suppression of apoptosis, while quercetin inhibits PI3K, other kinases and PAI-1, from the SERPIN family member (140, 192). In contrast to dasatinib, which was more effective on pre-adipocytes, quercetin preferentially reduced viability of senescent HUVECs (140). Parallel cultures of non-senescent HUVECs proliferated 2- to 3-fold in the presence of quercetin over the same period of 3 days, indicating that quercetin's induction of apoptosis is selective to SCs (140). In addition, the combination of dasatinib and quercetin achieved a synergistic effect by selectively killing both senescent pre-adipocytes and HUVECs, whom viability was, respectively, reduced by ~70% and ~50% (140). This suggests that using a mix of senolytics to target a broader range of anti-apoptotic networks may be a strategy to follow in developing future senolytic therapies (189). Used in vivo, the senolytic cocktail also reduced the number of p16-positive SCs in fat and liver from old mice (140). After a single 5 day treatment course of dasatinib+quercetin, the rodents exhibited an improved left ventricular ejection fraction (LVEF) and fractional shortening with no alteration of cardiac mass, as well as increased smooth muscle vascular reactivity to nitroprusside (140). Similar results were obtained by Xu et al.: 20 month-old mice who were fed dasatinib+quercetin intermittently for 4 months performed better at physical endurance tests compared to the control group (43). Next, they administered biweekly oral doses of dasatinib+quercetin to 24 to 27 month-old mice, equivalent to a human age of 75–90 years; compared to the controls, these mice had a 36% higher median post-treatment lifespan and a 65% lower mortality hazard (43). This was neither associated with an increased physical morbidity nor an increased age-related disease burden (43). In addition, in ApoE−/− mice fed with a high-fat diet, dasatinib+quercetin given once monthly for 3 months was shown to decrease aortic calcifications and increase vascular reactivity (193). When used alone, quercetin increased the density of sirtuin 1 (Sirt1) in aorta of ApoE−/− mice (141). Sirt1 functions as a nicotinamide adenosine dinucleotide (NAD+)-dependent deacetylase and is involved in genomic stability, basal level autophagy and cell survival (194). Sirt1 was found to delay both replicative and stress-induced senescence (194).
Hwang et al. conducted in vitro experiments with adult human coronary artery endothelial cells (HCAEC) from three deceased female donors using replicative senescence as a relevant model for human arterial aging (195). Contrary to the previous results reported in HUVECs (140), their findings showed that quercetin induces death in both early (non-senescent) and late-passage (senescent) HCAECs, without any selectivity for the latter (195). Quercetin's cytotoxicity was evident in all three donors at a concentration of 10 μM, which was half the amount used with HUVECs (140). Late-passage cells were more sensitive to quercetin's toxic effects as their relative cell abundance was already significantly decreased at a concentration of 6 μM (195). Their study also investigated hyperoside, also known as quercetin 3-D-galactoside, as an alternative to quercetin (195). Hyperoside is a natural derivative of quercetin produced by St. John's Wort and structurally identical except for a galactoside group attached in position 3 (Figure 2) (195). In contrast to quercetin, hyperoside had no significant cytotoxicity to either proliferating or late-passage HCAECs but was unable to display any senolytic activity (195). A second in vitro model of an adult human vasculature model was investigated by Jiang et al. using human aortic endothelial cells (HAECs) (141). In their study, senescence was induced by ox-LDLs. Their results revealed that quercetin decreased the expression of senescence-associated β-galactosidase and improved cell morphology of HAECs (141). The senolytic effect was dose dependent, as 0.3, 1, and 3 μmol/L of quercetin improved cells viability by 10.8, 40.9, and 48.9%, respectively (141). Quercetin simultaneously decreased ROS generation, also in a concentration-dependent manner. In addition, transcriptome microarray assays were performed and identified differentially expressed genes in the mRNAs profile of senescent HAECs treated with quercetin (141). Among them, several were involved in p53 and mammalian target of rapamycin (mTOR) signaling pathways, NO metabolism, maintenance of the cytoskeleton, extracellular matrix-receptor interaction, as well as complement and coagulation cascades, suggesting the potential mechanisms by which quercetin was effective against ox-LDLs (141). Quercetin also decreased the genetic expression of AATK, CDKN2A, and IGFBP3 (141). AATK is induced during apoptosis, while CDKN2A (p16) is one of the most important senescence markers (141). Interestingly, a high circulating concentration of IGFBP3 was found to be a predictor of IHD (196). One can therefore wonder if quercetin could alleviate the risks of IHD in patients by decreasing IGFBP3.
Clinical trials studying the senolytic effects of quercetin remain scarce (Table 1). In the first clinical trial of senolytics, an intermittent regimen of dasatinib+quercetin (dasatinib: 100 mg/day, quercetin: 1,250 mg/day, 3 days/week over 3 weeks) improved physical tolerance, but not pulmonary function, in patients with idiopathic pulmonary fibrosis, a fatal senescence-associated disease (197). Another open label pilot study was conducted by Hickson et al. in 9 adults aged 50–80 years with diabetic kidney disease (142). The patients received a 3 day oral treatment regimen with dasatinib 100 mg daily and quercetin 500 mg bid. Eleven days after treatment completion, there was a significant reduction in the number of adipose tissue SCs and circulating SASP factors, including IL-1α, IL-6, MMP-9, and MMP-12, accompanied by an increase of adipocyte progenitors, suggesting a selective cytotoxic effect for SCs (142). These results are in agreement with a previous in vitro study performed on human omental tissue resected during gastric bypass surgery (43). The surgical explants treated with a dasatinib+quercetin medium for 48 h had significantly less SCs and a lower secretion of SASP components compared to the explants treated with a vehicle (43). To the best of our knowledge, no clinical trial has yet examined the senolytic effects of quercetin on endothelial dysfunction in humans, in the context of CVD.
Myocardial Protective Effects of Quercetin
The beneficial effects of quercetin on dyslipidemia, hypertension, senescence and other risk factors can be seen as a primary prevention measure against endothelial dysfunction. Once the endothalial dysfunction has resulted in an adverse cardiac event, secondary and tertiary prevention strategies become crucial in order to reduce the progression of the disease and its impacts on patients' quality of life. One of the most striking examples is myocardial ischemia. In the latter, dysfunctional endothelial cells of the coronary arteries induce a local disturbance in other cell lines, including cardiomyocytes and fibroblasts (198). They trigger a host response which includes increased oxidative stress, calcium imbalance, as well as cytokine, platelets, and leukocytes activation (198). This endothelial dysfunction is also a critical mediator of myocardial dysfunction after reperfusion (198). In response, many pathological adaptations occur, such as increased extracellular matrix deposition leading to myocardial interstitial fibrosis, changes in the myocardial cell morphology, and eventually, ventricular dilatation (199). The latter, called “ventricular remodeling” is detrimental to ventricular compliance and contractility (199). Clinically, it translates into debilitating conditions, ranging from stable angina to myocardial infarction and heart failure. In addition, experimental data showed that endothelial dysfunction correlates with the degree of myocardial injury, both from the ischemic and reperfusion insults (198). These observations suggest that quercetin's ability to minimize myocardial injury following an ischemic event may be, at least partly, mediated by its effects on the endothelium.
Many in vivo and ex vivo murine studies have shown both functional and structural benefits of exposing myocardium to quercetin in an acute ischemic setting (143–153) (Table 1). These studies used rodent models in which transient myocardial injury was induced by ISO injections, surgical occlusion of the left coronary artery (LAD) or interruption of Langendorff perfusion (123, 124, 130, 131, 134–136, 178–181). Quercetin was either given as an oral gavage, an intravenous or intraperitoneal infusion (123, 124, 130, 131, 134–136, 178–181). Measured functional hemodynamic parameters included left ventricular end-diastolic pressure (LVEDP), left ventricular systolic pressure (LVSP) and maximal ratio of pressure change during isovolumetric contraction (peak dP/dt). While myocardial ischemia systematically decreased LVSP and peak dP/dt, and increased LVEDP, this effect was counteracted by quercetin (143–147). Liu et al. used echocardiography in mice to estimate left ventricular function (148). They showed that quercetin significantly slowed the decline in LVEF and fractional shortening compared with the control group (148). On macroscopic examination, treatment with quercetin induced a significant reduction of myocardial infarct size on triphenyl tetrazolium chloride (TTC) staining (149–151). This was further supported by lower levels of serum creatine kinase (CK), CK-MB, cardiac troponin T and lactate dehydrogenase, all enzymatic markers of myocardial insult (144, 147–149, 151–155). Histopathological examinations also revealed lower infiltration of leukocytes to the site of infarction, less edema and overall maintained tissue architecture (149, 152, 153). All these findings are in favor of a preservation of cardiomyocytes' membrane integrity and global improvement in myocardial function after exposure with quercetin. Interestingly, these cardioprotective effects were observed whether quercetin was administered before induction of ischemia or during reperfusion. This suggests that quercetin may have both ischemic preconditioning and postconditioning capacities.
Although timely reperfusion is essential for myocardial salvage, it is accompanied by a stress reaction known as “myocardial ischemia-reperfusion injury” (MIRI), which paradoxically increases the degree of myocardial damage (200). As restoration of the circulation allows blood to reach cells that were previously subjected to ischemia, sudden availibility of oxygen leads to a burst in the generation of ROS, mainly deriving from the Fenton reaction, NOX, and xanthine oxidase (XO) (200). These redox reactions lead to formation of oxygen radicals, lipid peroxidation, calcium overload, activation of inflammatory cascades, and apoptosis, which propagate and cause myocardial damage even distant to the original site of insult (200). This has important clinical implications as it limits the benefits of current revascularization therapies such as thrombolysis, angioplasty or coronary artery bypass surgery (200). A number of studies based on the rodent models of transient myocardial infarction have suggested that quercetin attenuates MIRI by interfering with several of these pathways (Figure 4).
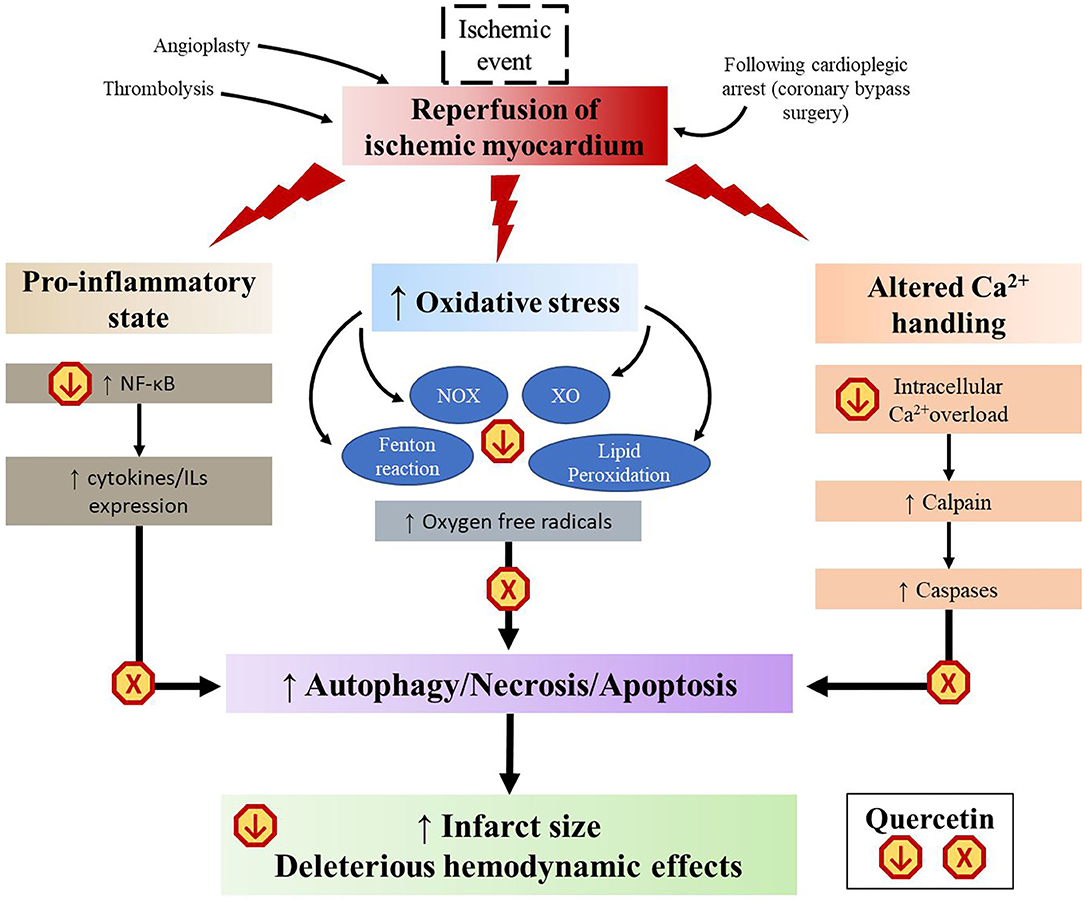
Figure 4. Schematic representation of the multistep mechanisms of quercetin to mitigate myocardial ischemic reperfusion injury. XO, xanthine oxidase; NOX, NADPH oxidase.
First, quercetin has well-documented antioxidant properties. Thanks to its chemical structure (Figure 2), it is able to directly scavenge free radicals such as superoxide, hydrogen peroxide, peroxyl, and hydroxyl radicals (45). Quercetin can also reduce the formation of ROS by inhibiting NOX and XO, decreasing the activity of cyclooxygenase and LOX, as well as regulating the activity of intracellular signaling cascades involved in inflammatory reactions (53). Chemical studies revealed that quercetin can reversibly inhibit XO-catalyzed uric acid and superoxide radicals formation in a double-displacement reaction (201, 202). However, results of in vivo studies remain controversial (203). In a hyperuricemic mouse model, quercetin given orally at 100 mg/kg for 1 to 7 consecutive days reduced serum urate levels and XO hepatic activity in a concentration-dependent manner (204). In another study using the same model, a 7 day treatment of 400 mg/kg orally administered quercetin failed to reduce both serum urate levels and XO expression (205). In a rabbit model of surgically-induced MIRI, an intravenous injection of quercetin given 5 min before ligation attenuated the enzymatic activity of NOX2 expressed in endothelial cells (206). On the other hand, quercetin acts as a chelating agent. It can inhibit the Fenton reaction by interfering with ferrous iron (207). It can also bind to zinc and facilitate zinc trafficking into cells (208), which in turn functions as an antioxidant (209). Lipid peroxidation is the process by which unsaturated fatty acids are converted to lipid peroxyl radicals by hydrogen oxidation, which, in turn, extract hydrogen from other fatty acid molecules to create more free radicals (210). Some studies reported that quercetin offers a protection against lipid peroxidation chain reaction by neutralizing peroxyl radicals and by binding to transition metal ions, catalyzers of lipid peroxidation (154, 211, 212). Finally, quercetin pretreatment was shown to decrease the content of malondialdehyde (MDA), a mutagenic product of lipid peroxidation chain reaction, and to potentiate the activity of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px), two most important antioxidases in cardiomyocytes (144, 147, 148, 152, 213, 214). All these properties allow quercetin to slow down the domino effects of free radical injury in MIRI.
With myocardial ischemia and MIRI, there is a shift toward a pro-inflammatory and pro-apoptotic phenotype caused by an increased secretion of cytokines (200). As seen previously, quercetin was shown to significantly repress this inflammatory cascade, both in vivo and in vitro (95). The pro-inflammatory response is further exacerbated by activation of NFκB, which is a pivot transcription factor in promoting cytokine expression. Enhanced NFκB signaling induces a positive feedback, which further prompts inflammasome assembly (35). NFκB can be activated through the interaction of high mobility group box-1 (HMGB1) with toll-like receptors (TLRs) located in cardiomyocytes (215). HMGB1 has been found to be released by necrotic cardiomyocytes under ischemic conditions and may serve as an early mediator of inflammation following MIRI (215). Western blot analyses revealed a strong activation of the HMGB1/TLR/NFκB pathway in heart tissues after ischemic/reperfusion stimulation in LAD ligated rats (151). Treatment with quercetin significantly inhibited expression of HMGB1 and TLR4 (151, 153). In addition, up-regulation by quercetin of peroxisome proliferation-activated receptor gamma (PPAR-γ) further supports the targetting of NFκB activation (148). Several reports revealed that PPAR-γ, a ligand-activated nuclear transcription factor, could suppress the signal transduction of NFκB pathway in vascular diseases (216). A study demonstrated that mice with transient LAD ligation that received a 10 day pre-treatment of quercetin had a significantly higher number of PPAR-γ positive myocardial cells (148). The authors also found that quercetin partially reversed the effects of a PPAR-γ inhibitor, GW9662, compared to non-quercetin-treated mice, with an associated improvement in LVEF, fractional shortening and cardiac biomarkers (148). Lastly, quercetin was shown to protect against calcium overload. Elevated intracellular Ca2+ is involved in the deleterious biochemical and functional changes accompanying MIRI (217). In vitro, quercetin decreased Ca2+-dependent cell death when added to H9C2 cardiomyocyte 30 min before application of H2O2-induced oxidative stress (218). Furthermore, the downstream Ca2+ activated calpain pathways may lead to contractile dysfunction and cytoskeleton damage (219). Increased calpain activity has been reported as an aggravating factor in myocardial infarction (219). Oral quercetin (50 mg/kg) pre-treatment of Wistar rats exposed to an ISO-induced myocardial infarction downregulated the genetic expression of calpain 1 and 2, protecting the myocardium from their overactivity (152). This cardioprotective effect was also supported by a reduction of CK-MB and cardiac troponin T in quercetin-treated rats compared to the control group (152). Another in vivo study found that quercetin prevented inhibition of the sodium-potassium and the calcium pumps caused by myocardial infarction (149).
Despite extensive experimental data suggesting that quercetin can attenuate MIRI, very few trials have explored the use of quercetin for the treatment of myocardial ischemia in humans. In the study done by Chekalina et al., 30 out of 85 patients with stable angina on optimal medical therapy were given quercetin at a daily dose of 120 mg for 2 months (156). The quercetin patients had lower levels of IL-1β and TNF-α compared to the control group (156). An open-label clinical trial conducted in Ukraine studied the administration of intravenous quercetin (Corvitin) over 10 days in patient admitted with an acute myocardial infarction and heart failure symptoms. After 3 days, there was a significant improvement in their profile of cardiac biomarkers and LVEF (157). Altogether, these clinical data suggest that quercetin has potential cardioprotective effects, and form a solid foundation for a potential application of quercetin in the prevention of IHD and its complications. This hypothesis remains to be tested in large clinical trials.
Endothelial-to-Mesenchymal Transition: a Less Explored Player in Endothelial Dysfunction
In the late 1990's, endothelial cells were found to undergo a highly dynamic process of dedifferentiation known as endothelial-to-mesenchymal transition (EndoMT) (220). During EndoMT, endothelial cells progressively acquire a wide spectrum of phenotypes characteristic of multipotent cells (221). This phenotype switch involves a reduced expression of distinctive endothelial cells markers such as von Willebrand factor, vascular endothelial-cadherin or CD31/PECAM-1 and an increased expression of mesenchymal cells markers such as alpha-smooth muscle actin, vimentin and N-cadherin (220, 222). Similar to senescence, EndoMT is a double-edged sword. EndoMT-derived cells exhibit hallmarks of invasive cells through cytoskeletal reorganization, increased ECM production, loss of cellular adhesion and resultant enhanced migratory potential (223, 224). This process is critical in the developing embryo where it was shown to generate vasculogenesis and the cardiac cushions for valve development (225). EndoMT was also shown to contribute to wound healing (226). However, when triggered under certain pathological conditions such as inflammation or shear-stress injuries, EndoMT can give rise to cancer progression (224), fibrodysplasia ossificans progressive (221), pulmonary arterial hypertension (227, 228) or cardiac and renal fibrosis (229, 230). The best-studied mediator of EndoMT is TGF-β (231). The latter can induce EndoMT either directly, through both Smad-dependent and Smad-independent pathways (232, 233), or indirectly, through ET-1 (234, 235), CAV-1 (236), or NFκB (151, 153, 237).
Strong lines of evidence support the cross-links between endothelial dysfunction, atherosclerosis, hypertension, senescence and EndoMT (228, 238–241). Targeting EndoMT opens therefore a new therapeutic avenue against CVD. However, contrarily to the other players of endothelial dysfunction, few studies specifically looked at the potential contribution of quercetin. In their study performed in vitro, Huang et al. showed that quercetin effectively inhibited TGF-β1-induced human pulmonary arterial endothelial cells proliferation and transdifferentiation (242). This suggests that quercetin may be a potential antagonist for a pathogenic model of pulmonary artery hypertension secondary to pulmonary arterial endothelial cells excessive growth. Moreover, as discussed in previous sections, experiments done outside the scope of EndoMT have demonstrated that quercetin can downregulate ET-1 (134, 135), CAV-1 (118), and NFκB, which are all mediators of EndoMT. Other studies explored the effects of quercetin on a similar process as EndoMT but involving epithelial cells, hence the name “epithelial-to-mesenchymal transition” (EMT). In human retinal pigment epithelial cells (ARPE-19) exposed to TGF-β1, quercetin suppressed proliferation, migration, and collagen I secretion (243). It also downregulated EMT-related markers such as alpha-smooth muscle actin and N-cadherin; conversely, it upregulated the expression of tight junction proteins and epithelial-cadherin (243). In addition, quercetin inhibited Smad2/3 phosphorylation and translocation of Smad4, suggesting that the progression of EMT in ARPE-19 cells was reversed via the Smad pathway (243). Incubation with quercetin also reduced EMT in mammary carcinoma and prostate cancer cell lines (244, 245). Together, these results support a role for quercetin against EndoMT and EMT. However, despite multiple similarities between the two processes, including canonical TGF-β signaling as their driving force (246), more research in EndoMT models is needed to further confirm the efficacy of quercetin in targeting its trigger mechanisms.
Discussion
The magnitude of the role of endothelial dysfunction in CVD is well-established. Many pathophysiological processes are involved, and they contribute to each other in a feedback manner, as seen with the triad of vascular senescence, hypertension and atherosclerosis. This also means that each pathway is a potential target for alleviating endothelial dysfunction. Numerous drugs are already available to effectively treat dyslipidemia and hypertension. In comparison, anti-senescence therapy is only a nascent yet promising research field. Development of senolytic drugs would bring a conceptual change that an aging vessel is not an immutable process. On the other hand, an important clinical consequence of endothelial dysfunction is manifested in IHD. The burden of myocardial ischemia has been improved with more timely and effective reperfusion strategies such as angioplasty, bypass surgery, antiplatelet, and antithrombotic agents used to restore the patency of infarct-related coronary arteries. However, at present, there is still no effective therapy to prevent MIRI. In this review, we have described mechanistic, experimental and clinical evidence that suggests quercetin can manifest a wide range of cardioprotective biological activities (Table 1). Not only does it have anti-hypertensive and anti-atherosclerotic effects, but it also seems to mitigate senescence and MIRI, two Achilles' heels in the modern treatment of CVD. Moreover, although still scarce, encouraging data suggest that quercetin may also act against abnormal EndoMT, an important yet less explored player in endothelial dysfunction. These properties of quercetin form the basis for its potential benefits against aging-related endothelial dysfunction and CVD (Figure 5).
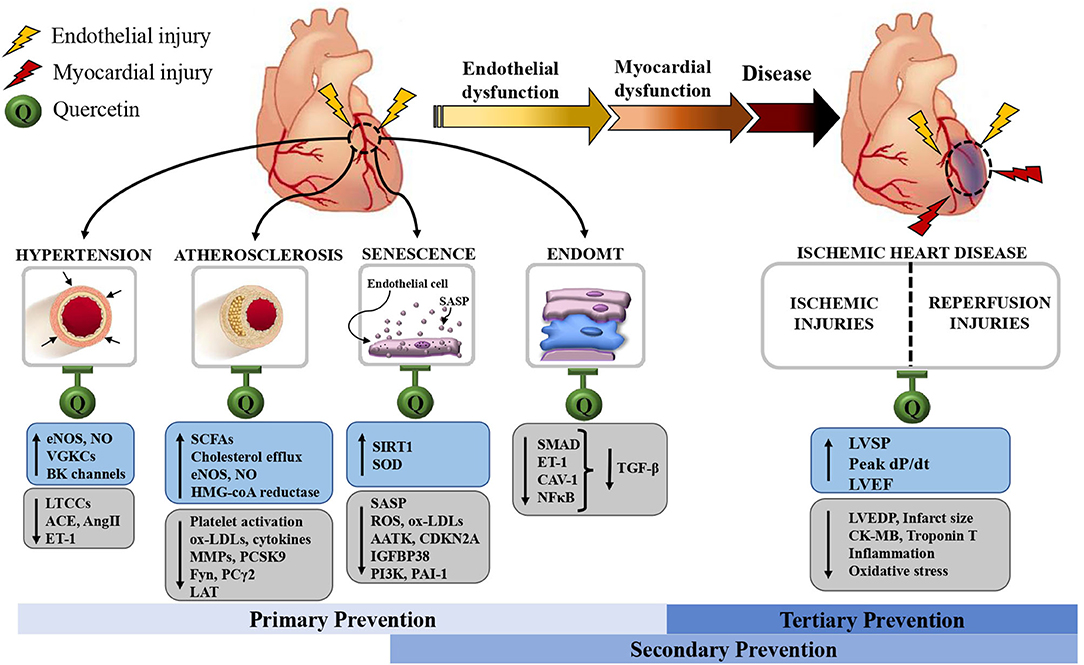
Figure 5. Schematic representation of the endothelial and, by extension, myocardial protective effects of quercetin. These allow quercetin to act as a primary, secondary and tertiary preventive measure against cardiovascular diseases. AATK: apoptosis-associated tyrosine kinase; ACE: angiotensin-converting enzyme; AngII, angiotensin II; BK, big K, large-conductance Ca2+-sensitive K+ channels; CAV-1, caveolin-1; CDKN2A, p16, cyclin-dependent kinase inhibitor 2A; CK-MB, creatinine kinase-MB; EndoMT, endothelial-to-mesenchymal transition; ET-1, endothelin-1; IGFBP3, insulin-like growth factor binding protein-3; eNOS, endothelial nitric oxide synthase; NFκB, nuclear factor-kappa B; NO, nitric oxide; ox-LDLs, oxidized low density lipoproteins; Fyn, Src family 59 kDa non-receptor protein tyrosine-kinase; LAT, linker for activation of T cells; LTCCs, L-type Ca2+ channels; LVEDP, left ventricular end-diastolic pressure; LVEF, left ventricular ejection fraction; LVSP, left ventricular systolic pressure; MMPs, matrix metalloproteases; PAI-1, plasminogen-activated inhibitor-1; PCSK9, proprotein convertase subtilisin/kexin type 9; PI3K, phosphatidylinositol-4,5-bisphosphate 3 kinase; PLCγ2, phospholipase Cγ2; SCFAs, short-chain fatty acids; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; SIRT1, sirtuin-1, nicotinamide adenine dinucleotide [NAD(+)]-dependent protein deacetylase; SOD, superoxide dismutase; TGF-β, transforming growth factor beta; VGKCs, voltage-gated K+ channels.
While this review has focused on conditions originating from a diseased endothelial layer, prevention of endothelial dysfunction can be achieved by intervening beyond the endothelium itself. Indeed, endothelial dysfunction is undeniably associated with the remaining entities of the metabolic syndrome: obesity and insulin resistance (247–251). Their role as independent risk factors for CVD merits as much attention as hypertension and dyslipidemia (252). In fact, the increasing incidence of obesity and corresponding rise in type-2 diabetes are further challenging the prevention and treatment of CVD (253). The metabolic effects of quercetin against these two conditions have been equally encouraging and were recently reviewed elsewhere (254–258). In addition, it should be emphasized that, for clarity reasons, the effects of quercetin on the various biochemical and biomechanical signaling cascades involved in endothelial dysfunction were discussed as separate entities. However, in reality, senescence, vasomotor dysfunction, atherosclerosis, EndoMT, inflammation, oxidative stress, and altered endothelial cellular metabolism all interact, cross talk and occur simultaneously. The resulting chain reactions create a vicious circle, which, once it is established in one person, can easily multiply their cardiovascular risks. The ability of quercetin to act as such a versatile multi-target agent against the domino effect of endothelial dysfunction becomes very appealing. It seems to be promising not only in primary prevention, but also in secondary and tertiary prevention against the dysfunctional coronary endothelium and myocardium exposed to ischemia-reperfusion injuries.
However, after being studied for two decades and showing encouraging in vitro and in vivo results, quercetin still occupies a modest title as a dietary supplement. This could be explained by a couple of factors. First, quercetin lacks molecular specificity. It does not radically block a metabolic pathway nor inhibit a receptor of interest. Instead, quercetin has a wide variety of biological activities, which makes it difficult to establish a clear association between its administration and the observed positive effects. As a matter of fact, quercetin's role as a direct senolytic agent is still open for discussion. Does it selectively target SCs, or does it improve their clearance by off-target mechanisms such as antioxidant activity? Second, clinical trials using more consistent protocols are needed to consolidate the medical findings attributed to quercetin. Published trials have been using various treatment durations, doses and routes of administration, certainly contributing to heterogenous findings. The considerable variation in bioavailability of quercetin among individuals might result in subtherapeutic plasma concentrations, especially when using lower doses. Furthermore, in vitro experiments most often administered a hit-and-run treatment with quercetin. We could hypothesize that replicating this regimen in humans would yield more substantial effects. Lastly, to quote Samuel Butler, “medicine is not practiced as an art of drawing sufficient conclusions from insufficient premises.” In the case of quercetin, the absence of its medical use despite its appealing properties might suggest that we should reconsider our expectations for its potential therapeutic implications. If a defect of the endothelium was accountable for a disease, and a drug could be given which would correct the defect, the disease would obviously be cured. However, drugs that were to act through less direct principles might still be useful. For example, if quercetin's well-established anti-inflammatory and antioxidant effects, albeit less specific, could make the endothelium less vulnerable to injury and senescence, it could potentially reinforce the efficacy of other cardiovascular agents. Ultimately, managing CVD has never been about carrying a hammer and treating everything “as if it were a nail.” Instead, it revolves around adding different tools to tackle different issues of a large-scale problem.
In conclusion, quercetin represents a promising natural compound that appears to satisfy all the requirements to develop a nutraceutical against endothelial dysfunction. There is a pressing need for well-designed clinical trials that explore its intriguing potential for senolytic therapy and myocardial protection.
Author Contributions
OD, NT-T, and ET designed the project and its main conceptual ideas. OD performed the research strategy, data collection, data analysis and interpretation, drafted the manuscript, and designed the figures, with input from all authors. All authors provided critical revisions to the article and approved the final version for publication.
Funding
This work was supported by the Canadian Institutes of Health Research [PJT 166110 and PJT 162446 to ET]; and the Foundation of the Montreal Heart Institute [ET and MC]. PM is a post-doctoral scholar from the Fonds de la recherche du Québec.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. (2017) 174:1591–619. doi: 10.1111/bph.13517
2. Thorin E. Life [ageing] is like riding a bicycle. To keep your [coronary and heart] balance you must keep moving. J Physiol. (2017) 595:3701–2. doi: 10.1113/JP274297
3. Thorin E, Thorin-Trescases N. Vascular endothelial ageing, heartbeat after heartbeat. Cardiovasc Res. (2009) 84:24–32. doi: 10.1093/cvr/cvp236
4. Matsuzawa Y, Lerman A. Endothelial dysfunction and coronary artery disease: assessment, prognosis, and treatment. Coron Artery Dis. (2014) 25:713–24. doi: 10.1097/MCA.0000000000000178
5. Mensah GA, Wei GS, Sorlie PD, Fine LJ, Rosenberg Y, Kaufmann PG, et al. Decline in cardiovascular mortality: possible causes and implications. Circ Res. (2017) 120:366–80. doi: 10.1161/CIRCRESAHA.116.309115
6. Alfaras I, Di Germanio C, Bernier M, Csiszar A, Ungvari Z, Lakatta EG, et al. Pharmacological strategies to retard cardiovascular aging. Circ Res. (2016) 118:1626–42. doi: 10.1161/CIRCRESAHA.116.307475
7. Zuchi C, Ambrosio G, Luscher TF, Landmesser U. Nutraceuticals in cardiovascular prevention: lessons from studies on endothelial function. Cardiovasc Ther. (2010) 28:187–201. doi: 10.1111/j.1755-5922.2010.00165.x
8. Mozaffarian D, Wu JHY. Flavonoids, dairy foods, and cardiovascular and metabolic health: a review of emerging biologic pathways. Circ Res. (2018) 122:369–84. doi: 10.1161/CIRCRESAHA.117.309008
9. RusznyÁK ST, Szent-GyÖRgyi A. Vitamin P: flavonols as vitamins. Nature. (1936) 138:27. doi: 10.1038/138027a0
10. Patel RV, Mistry BM, Shinde SK, Syed R, Singh V, Shin HS. Therapeutic potential of quercetin as a cardiovascular agent. Eur J Med Chem. (2018) 155:889–904. doi: 10.1016/j.ejmech.2018.06.053
11. Rosen GM, Tsai P, Pou S. Mechanism of free-radical generation by nitric oxide synthase. Chem Rev. (2002) 102:1191–200. doi: 10.1021/cr010187s
12. Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. (2005) 1:183–98.
13. Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. (2003) 23:168–75. doi: 10.1161/01.atv.0000051384.43104.fc
14. Konukoglu D, Uzun H. Endothelial dysfunction and hypertension. Adv Exp Med Biol. (2017) 956:511–40. doi: 10.1007/5584_2016_90
15. Gimbrone MA Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
16. Warboys CM, Amini N, de Luca A, Evans PC. The role of blood flow in determining the sites of atherosclerotic plaques. F1000 Med Rep. (2011) 3:5. doi: 10.3410/M3-5
17. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. (2006) 113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532
18. Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. (2009) 297:H1829–36. doi: 10.1152/ajpheart.00230.2009
19. Hamasaki S, Al Suwaidi J, Higano ST, Miyauchi K, Holmes DR Jr, et al. Attenuated coronary flow reserve and vascular remodeling in patients with hypertension and left ventricular hypertrophy. J Am Coll Cardiol. (2000) 35:1654–60. doi: 10.1016/s0735-1097(00)00594-5
20. Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. (1986) 315:1046–51. doi: 10.1056/NEJM198610233151702
21. Reddy KG, Nair RN, Sheehan HM, Hodgson JM. Evidence that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis. J Am Coll Cardiol. (1994) 23:833–43. doi: 10.1016/0735-1097(94)90627-0
22. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. (2004) 109(Suppl. 1):III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8
23. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
24. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
25. Paez-Ribes M, Gonzalez-Gualda E, Doherty GJ, Munoz-Espin D. Targeting senescent cells in translational medicine. EMBO Mol Med. (2019) 11:e10234. doi: 10.15252/emmm.201810234
26. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
27. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
28. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
29. Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. (2019) 117:15–22. doi: 10.1016/j.cyto.2019.01.013
30. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. (2013) 155:1119–30. doi: 10.1016/j.cell.2013.10.041
31. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. (2010) 12:676–85. doi: 10.1038/ncb2070
32. Feng T, Meng J, Kou S, Jiang Z, Huang X, Lu Z, et al. CCN1-induced cellular senescence promotes heart regeneration. Circulation. (2019) 139:2495–8. doi: 10.1161/CIRCULATIONAHA.119.039530
33. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. (2017) 31:172–83. doi: 10.1101/gad.290635.116
34. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
35. Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1802–9. doi: 10.1016/j.bbadis.2018.08.008
36. Caland L, Labbe P, Mamarbachi M, Villeneuve L, Ferbeyre G, Noly PE, et al. Knockdown of angiopoietin-like 2 induces clearance of vascular endothelial senescent cells by apoptosis, promotes endothelial repair and slows atherogenesis in mice. Aging. (2019) 11:3832–50. doi: 10.18632/aging.102020
37. Noly PE, Labbe P, Thorin-Trescases N, Fortier A, Nguyen A, Thorin E, et al. Reduction of plasma angiopoietin-like 2 after cardiac surgery is related to tissue inflammation and senescence status of patients. J Thorac Cardiovasc Surg. (2019) 158:792–802 e5. doi: 10.1016/j.jtcvs.2018.12.047
38. Voghel G, Thorin-Trescases N, Mamarbachi AM, Villeneuve L, Mallette FA, Ferbeyre G, et al. Endogenous oxidative stress prevents telomerase-dependent immortalization of human endothelial cells. Mech Ageing Dev. (2010) 131:354–63. doi: 10.1016/j.mad.2010.04.004
39. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. (2015) 21:1424–35. doi: 10.1038/nm.4000
40. Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell. (2012) 4:12. doi: 10.1186/2045-824X-4-12
41. Freitas-Rodriguez S, Folgueras AR, Lopez-Otin C. The role of matrix metalloproteinases in aging: tissue remodeling and beyond. Biochim Biophys Acta Mol Cell Res. (2017) 1864(Pt. A):2015–25. doi: 10.1016/j.bbamcr.2017.05.007
42. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. (2011) 479:547–51. doi: 10.1038/nature10599
43. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
44. Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet. (2015) 6:112. doi: 10.3389/fgene.2015.00112
45. Xu D, Hu MJ, Wang YQ, Cui YL. Antioxidant activities of quercetin and its complexes for medicinal application. Molecules. (2019) 24:1123. doi: 10.3390/molecules24061123
46. Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. (2016) 5:e47. doi: 10.1017/jns.2016.41
47. Peterson JJ, Dwyer JT, Jacques PF, McCullough ML. Improving the estimation of flavonoid intake for study of health outcomes. Nutr Rev. (2015) 73:553–76. doi: 10.1093/nutrit/nuv008
48. Magar RT, Sohng JK. A review on structure, modifications and structure-activity relation of quercetin and its derivatives. J Microbiol Biotechnol. (2020) 30:11–20. doi: 10.4014/jmb.1907.07003
49. Lesjak M, Beara I, Simin N, Pintać D, Majkić T, Bekvalac K, et al. Antioxidant and anti-inflammatory activities of quercetin and its derivatives. J Funct Foods. (2018) 40:68–75. doi: 10.1016/j.jff.2017.10.047
50. Liu X, Raghuvanshi R, Ceylan FD, Bolling BW. Quercetin and its metabolites inhibit recombinant human angiotensin-converting enzyme 2 (ACE2) activity. J Agric Food Chem. (2020) 68:13982–9. doi: 10.1021/acs.jafc.0c05064
51. Santos AC, Uyemura SA, Lopes JL, Bazon JN, Mingatto FE, Curti C. Effect of naturally occurring flavonoids on lipid peroxidation and membrane permeability transition in mitochondria. Free Radic Biol Med. (1998) 24:1455–61. doi: 10.1016/s0891-5849(98)00003-3
52. Srinivas K, King JW, Howard LR, Monrad JK. Solubility and solution thermodynamic properties of quercetin and quercetin dihydrate in subcritical water. J Food Eng. (2010) 100:208–18. doi: 10.1016/j.jfoodeng.2010.04.001
53. Li Y, Yao J, Han C, Yang J, Chaudhry MT, Wang S, et al. Quercetin, inflammation and immunity. Nutrients. (2016) 8:167. doi: 10.3390/nu8030167
54. Rahman HS, Othman HH, Hammadi NI, Yeap SK, Amin KM, Abdul Samad N, et al. Novel drug delivery systems for loading of natural plant extracts and their biomedical applications. Int J Nanomed. (2020) 15:2439–83. doi: 10.2147/IJN.S227805
55. Wu LD, Enjiang Y, Xiaona C, Xinguo W, Zhengzan Z, Lingzhen Q, et al. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res Lett. (2014) 9:1–11. doi: 10.1186/1556-276X-9-684
56. Zhang L, Dong M, Guangyong X, Yuan T, Tang H, Wang Y. Metabolomics reveals that dietary ferulic acid and quercetin modulate metabolic homeostasis in rats. J Agric Food Chem. (2018) 66:1723–31. doi: 10.1021/acs.jafc.8b00054
57. Tanaka S, Oyama M, Nishikawa M, Ikushiro S, Hara H. Simultaneous collection of the portal and superior vena cava blood in conscious rats defined that intestinal epithelium is the major site of glucuronidation, but not sulfation and methylation, of quercetin. Biosci Biotechnol Biochem. (2018) 82:2118–29. doi: 10.1080/09168451.2018.1515615
58. Moon YJ, Wang L, DiCenzo R, Morris ME. Quercetin pharmacokinetics in humans. Biopharm Drug Dispos. (2008) 29:205–17. doi: 10.1002/bdd.605
59. Kaushik D, O'Fallon K, Clarkson PM, Dunne CP, Conca KR, Michniak-Kohn B. Comparison of quercetin pharmacokinetics following oral supplementation in humans. J Food Sci. (2012) 77:H231–8. doi: 10.1111/j.1750-3841.2012.02934.x
60. Dabeek WM, Marra MV. Dietary quercetin and kaempferol: bioavailability and potential cardiovascular-related bioactivity in humans. Nutrients. (2019) 11:2288. doi: 10.3390/nu11102288
61. Boesch-Saadatmandi C, Niering J, Minihane AM, Wiswedel I, Gardeman A, Wolffram S, et al. Impact of apolipoprotein E genotype and dietary quercetin on paraoxonase 1 status in apoE3 and apoE4 transgenic mice. Atherosclerosis. (2010) 211:110–3. doi: 10.1016/j.atherosclerosis.2010.02.027
62. Egert S, Boesch-Saadatmandi C, Wolffram S, Rimbach G, Muller MJ. Serum lipid and blood pressure responses to quercetin vary in overweight patients by apolipoprotein E genotype. J Nutr. (2010) 140:278–84. doi: 10.3945/jn.109.117655
63. Shimoi K, Nakayama T. Glucuronidase deconjugation in inflammation. Methods Enzymol. (2005) 400:263–72. doi: 10.1016/S0076-6879(05)00015-7
64. Elbarbry F, Ung A, Abdelkawy K. Studying the inhibitory effect of quercetin and thymoquinone on human cytochrome P450 enzyme activities. Pharmacogn Mag. (2018) 13(Suppl. 4):S895–9. doi: 10.4103/0973-1296.224342
65. Choi JS, Choi BC, Choi KE. Effect of quercetin on the pharmacokinetics of oral cyclosporine. Am J Health Syst Pharm. (2004) 61:2406–9. doi: 10.1093/ajhp/61.22.2406
66. Wang YH, Chao PD, Hsiu SL, Wen KC, Hou YC. Lethal quercetin-digoxin interaction in pigs. Life Sci. (2004) 74:1191–7. doi: 10.1016/j.lfs.2003.06.044
67. Andres S, Pevny S, Ziegenhagen R, Bakhiya N, Schafer B, Hirsch-Ernst KI, et al. Safety aspects of the use of quercetin as a dietary supplement. Mol Nutr Food Res. (2018) 62:1700447. doi: 10.1002/mnfr.201700447
68. Hilliard JJ, Krause HM, Bernstein JI, Fernandez JA, Nguyen V, Ohemeng KA, et al. A comparison of active site binding of 4-quinolones and novel flavone gyrase inhibitors to DNA gyrase. Adv Exp Med Biol. (1995) 390:59–69. doi: 10.1007/978-1-4757-9203-4_5
69. Woodward CJ, Deyo ZM, Donahue KE, Deal AM, Hawes EM. Clinically relevant interaction between warfarin and scuppernongs, a quercetin containing muscadine grape: continued questions surrounding flavonoid-induced warfarin interactions. BMJ Case Rep. (2014) 2014:bcr2013009608. doi: 10.1136/bcr-2013-009608
70. Harwood M, Danielewska-Nikiel B, Borzelleca JF, Flamm GW, Williams GM, Lines TC. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem Toxicol. (2007) 45:2179–205. doi: 10.1016/j.fct.2007.05.015
71. Vickery HB, Nelson EM, Almquist HJ, Elvehjem CA. Term “vitamin P” recommended to be discontinued. Science. (1950) 112:628. doi: 10.1126/science.112.2917.628
72. Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet. (1993) 342:1007–11. doi: 10.1016/0140-6736(93)92876-u
73. Haseeb S, Alexander B, Baranchuk A. Wine and cardiovascular health: a comprehensive review. Circulation. (2017) 136:1434–48. doi: 10.1161/CIRCULATIONAHA.117.030387
74. Sanchez M, Romero M, Gomez-Guzman M, Tamargo J, Perez-Vizcaino F, Duarte J. Cardiovascular effects of flavonoids. Curr Med Chem. (2019) 26:6991–7034. doi: 10.2174/0929867326666181220094721
75. D'Andrea G. Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia. (2015) 106:256–71. doi: 10.1016/j.fitote.2015.09.018
76. Juzwiak S, Wojcicki J, Mokrzycki K, Marchlewicz M, Bialecka M, Wenda-Rozewicka L, et al. Effect of quercetin on experimental hyperlipidemia and atherosclerosis in rabbits. Pharmacol Rep. (2005) 57:604–9.
77. Hayek T, Fuhrman B, Vaya J, Rosenblat M, Belinky P, Coleman R, et al. Reduced progression of atherosclerosis in apolipoprotein E-deficient mice following consumption of red wine, or its polyphenols quercetin or catechin, is associated with reduced susceptibility of LDL to oxidation and aggregation. Arterioscler Thromb Vasc Biol. (1997) 17:2744–52. doi: 10.1161/01.atv.17.11.2744
78. Li SS, Cao H, Shen DZ, Chen C, Xing SL, Dou FF, et al. Effect of quercetin on atherosclerosis based on expressions of ABCA1, LXR-alpha and PCSK9 in ApoE(-/-) mice. Chin J Integr Med. (2020) 26:114–21. doi: 10.1007/s11655-019-2942-9
79. Garelnabi M, Mahini H, Wilson T. Quercetin intake with exercise modulates lipoprotein metabolism and reduces atherosclerosis plaque formation. J Int Soc Sports Nutr. (2014) 11:22. doi: 10.1186/1550-2783-11-22
80. Lu XL, Zhao CH, Yao XL, Zhang H. Quercetin attenuates high fructose feeding-induced atherosclerosis by suppressing inflammation and apoptosis via ROS-regulated PI3K/AKT signaling pathway. Biomed Pharmacother. (2017) 85:658–71. doi: 10.1016/j.biopha.2016.11.077
81. Nie J, Zhang L, Zhao G, Du X. Quercetin reduces atherosclerotic lesions by altering the gut microbiota and reducing atherogenic lipid metabolites. J Appl Microbiol. (2019) 127:1824–34. doi: 10.1111/jam.14441
82. Jia Q, Cao H, Shen D, Li S, Yan L, Chen C, et al. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARgamma, LXRalpha and ABCA1. Int J Mol Med. (2019) 44:893–902. doi: 10.3892/ijmm.2019.4263
83. Xiao L, Liu L, Guo X, Zhang S, Wang J, Zhou F, et al. Quercetin attenuates high fat diet-induced atherosclerosis in apolipoprotein E knockout mice: a critical role of NADPH oxidase. Food Chem Toxicol. (2017) 105:22–33. doi: 10.1016/j.fct.2017.03.048
84. Cao H, Jia Q, Shen D, Yan L, Chen C, Xing S, et al. Quercetin has a protective effect on atherosclerosis via enhancement of autophagy in ApoE-/- mice. Exp Ther Med. (2021) 18:2451–8. doi: 10.3892/etm.2019.7851
85. Wu DN, Guan L, Jiang YX, Ma SH, Sun YN, Lei HT, et al. Microbiome and metabonomics study of quercetin for the treatment of atherosclerosis. Cardiovasc Diagn Ther. (2019) 9:545–60. doi: 10.21037/cdt.2019.12.04
86. Zhang M, Xie Z, Gao W, Pu L, Wei J, Guo C. Quercetin regulates hepatic cholesterol metabolism by promoting cholesterol-to-bile acid conversion and cholesterol efflux in rats. Nutr Res. (2016) 36:271–9. doi: 10.1016/j.nutres.2015.11.019
87. Son HY, Lee MS, Chang E, Kim SY, Kang B, Ko H, et al. Formulation and characterization of quercetin-loaded oil in water nanoemulsion and evaluation of hypocholesterolemic activity in rats. Nutrients. (2019) 11:244. doi: 10.3390/nu11020244
88. Kang HJ, Pichiah PBT, Abinaya RV, Sohn HS, Cha YS. Hypocholesterolemic effect of quercetin-rich onion peel extract in C57BL/6J mice fed with high cholesterol diet. Food Sci Biotechnol. (2016) 25:855–60. doi: 10.1007/s10068-016-0141-4
89. Tian H, Liu Q, Qin S, Zong C, Zhang Y, Yao S, et al. Synthesis and cardiovascular protective effects of quercetin 7-O-sialic acid. J Cell Mol Med. (2017) 21:107–20. doi: 10.1111/jcmm.12943
90. Chang YC, Lee TS, Chiang AN. Quercetin enhances ABCA1 expression and cholesterol efflux through a p38-dependent pathway in macrophages. J Lipid Res. (2012) 53:1840–50. doi: 10.1194/jlr.M024471
91. Ren K, Jiang T, Zhao GJ. Quercetin induces the selective uptake of HDL-cholesterol via promoting SR-BI expression and the activation of the PPARgamma/LXRalpha pathway. Food Funct. (2018) 9:624–35. doi: 10.1039/c7fo01107e
92. Li S, Cao H, Shen D, Jia Q, Chen C, Xing SL. Quercetin protects against oxLDLinduced injury via regulation of ABCAl, LXRalpha and PCSK9 in RAW264.7 macrophages. Mol Med Rep. (2018) 18:799–806. doi: 10.3892/mmr.2018.9048
93. Punithavathi VR, Prince PS. Combined effects of quercetin and alpha-tocopherol on lipids and glycoprotein components in isoproterenol induced myocardial infarcted Wistar rats. Chem Biol Interact. (2009) 181:322–7. doi: 10.1016/j.cbi.2009.07.002
94. Song L, Xu M, Lopes-Virella MF, Huang Y. Quercetin inhibits matrix metalloproteinase-1 expression in human vascular endothelial cells through extracellular signal-regulated kinase. Arch Biochem Biophys. (2001) 391:72–8. doi: 10.1006/abbi.2001.2402
95. Xue F, Nie X, Shi J, Liu Q, Wang Z, Li X, et al. Quercetin inhibits LPS-induced inflammation and ox-LDL-induced lipid deposition. Front Pharmacol. (2017) 8:40. doi: 10.3389/fphar.2017.00040
96. Saragusti AC, Ortega MG, Cabrera JL, Estrin DA, Marti MA, Chiabrando GA. Inhibitory effect of quercetin on matrix metalloproteinase 9 activity molecular mechanism and structure-activity relationship of the flavonoid-enzyme interaction. Eur J Pharmacol. (2010) 644:138–45. doi: 10.1016/j.ejphar.2010.07.001
97. Scoditti E, Calabriso N, Massaro M, Pellegrino M, Storelli C, Martines G, et al. Mediterranean diet polyphenols reduce inflammatory angiogenesis through MMP-9 and COX-2 inhibition in human vascular endothelial cells: a potentially protective mechanism in atherosclerotic vascular disease and cancer. Arch Biochem Biophys. (2012) 527:81–9. doi: 10.1016/j.abb.2012.05.003
98. Wang L, Wang B, Li H, Lu H, Qiu F, Xiong L, et al. Quercetin, a flavonoid with anti-inflammatory activity, suppresses the development of abdominal aortic aneurysms in mice. Eur J Pharmacol. (2012) 690:133–41. doi: 10.1016/j.ejphar.2012.06.018
99. Ko EY, Nile SH, Jung YS, Keum YS. Antioxidant and antiplatelet potential of different methanol fractions and flavonols extracted from onion (Allium cepa L.). 3 Biotech. (2018) 8:155. doi: 10.1007/s13205-018-1184-4
100. Stainer AR, Sasikumar P, Bye AP, Unsworth AJ, Holbrook LM, Tindall M, et al. The metabolites of the dietary flavonoid quercetin possess potent antithrombotic activity, and interact with aspirin to enhance antiplatelet effects. TH Open. (2019) 3:e244–58. doi: 10.1055/s-0039-1694028
101. Hubbard GP, Wolffram S, Lovegrove JA, Gibbins JM. Ingestion of quercetin inhibits platelet aggregation and essential components of the collagen-stimulated platelet activation pathway in humans. J Thromb Haemost. (2004) 2:2138–45. doi: 10.1111/j.1538-7836.2004.01067.x
102. Wright B, Moraes LA, Kemp CF, Mullen W, Crozier A, Lovegrove JA, et al. A structural basis for the inhibition of collagen-stimulated platelet function by quercetin and structurally related flavonoids. Br J Pharmacol. (2010) 159:1312–25. doi: 10.1111/j.1476-5381.2009.00632.x
103. Choi JS, Kang SW, Li J, Kim JL, Bae JY, Kim DS, et al. Blockade of oxidized LDL-triggered endothelial apoptosis by quercetin and rutin through differential signaling pathways involving JAK2. J Agric Food Chem. (2009) 57:2079–86. doi: 10.1021/jf803390m
104. Yang MY, Huang CN, Chan KC, Yang YS, Peng CH, Wang CJ. Mulberry leaf polyphenols possess antiatherogenesis effect via inhibiting LDL oxidation and foam cell formation. J Agric Food Chem. (2011) 59:1985–95. doi: 10.1021/jf103661v
105. Yao S, Sang H, Song G, Yang N, Liu Q, Zhang Y, et al. Quercetin protects macrophages from oxidized low-density lipoprotein-induced apoptosis by inhibiting the endoplasmic reticulum stress-C/EBP homologous protein pathway. Exp Biol Med. (2012) 237:822–31. doi: 10.1258/ebm.2012.012027
106. Liang Q, Chen Y, Li C, Lu L. [Quercetin attenuates Ox-LDL-induced calcification in vascular smooth muscle cells by regulating ROS-TLR4 signaling pathway]. Nan Fang Yi Ke Da Xue Xue Bao. (2018) 38:980–5. doi: 10.3969/j.issn.1673-4254.2018.08.13
107. Janisch KM, Williamson G, Needs P, Plumb GW. Properties of quercetin conjugates: modulation of LDL oxidation and binding to human serum albumin. Free Radic Res. (2004) 38:877–84. doi: 10.1080/10715760410001728415
108. Gong M, Garige M, Varatharajalu R, Marmillot P, Gottipatti C, Leckey LC, et al. Quercetin up-regulates paraoxonase 1 gene expression with concomitant protection against LDL oxidation. Biochem Biophys Res Commun. (2009) 379:1001–4. doi: 10.1016/j.bbrc.2009.01.015
109. Kondo M, Izawa-Ishizawa Y, Goda M, Hosooka M, Kagimoto Y, Saito N, et al. Preventive effects of quercetin against the onset of atherosclerosis-related acute aortic syndromes in mice. Int J Mol Sci. (2020) 21:7226. doi: 10.3390/ijms21197226
110. Tong F, Liu S, Yan B, Li X, Ruan S, Yang S. Quercetin nanoparticle complex attenuated diabetic nephropathy via regulating the expression level of ICAM-1 on endothelium. Int J Nanomed. (2017) 12:7799–813. doi: 10.2147/IJN.S146978
111. Bian Y, Liu P, Zhong J, Hu Y, Zhuang S, Fan K, et al. Quercetin attenuates adhesion molecule expression in intestinal microvascular endothelial cells by modulating multiple pathways. Dig Dis Sci. (2018) 63:3297–304. doi: 10.1007/s10620-018-5221-2
112. Tabrizi R, Tamtaji OR, Mirhosseini N, Lankarani KB, Akbari M, Heydari ST. The effects of quercetin supplementation on lipid profiles and inflammatory markers among patients with metabolic syndrome and related disorders: a systematic review and meta-analysis of randomized controlled trials. Crit Rev Food Sci Nutr. (2020) 60:1855–68. doi: 10.1080/10408398.2019.1604491
113. Burak C, Wolffram S, Zur B, Langguth P, Fimmers R, Alteheld B, et al. Effect of alpha-linolenic acid in combination with the flavonol quercetin on markers of cardiovascular disease risk in healthy, non-obese adults: a randomized, double-blinded placebo-controlled crossover trial. Nutrition. (2019) 58:47–56. doi: 10.1016/j.nut.2018.06.012
114. Bojic M, Debeljak Z, Tomicic M, Medic-Saric M, Tomic S. Evaluation of antiaggregatory activity of flavonoid aglycone series. Nutr J. (2011) 10:73. doi: 10.1186/1475-2891-10-73
115. Boyanov KO, Maneva AI. Influence of platelet aggregation modulators on cyclic amp production in human thrombocytes. Folia Med. (2018) 60:241–7. doi: 10.1515/folmed-2017-0091
116. Beretz A, Stierle A, Anton R, Cazenave JP. Role of cyclic AMP in the inhibition of human platelet aggregation by quercetin, a flavonoid that potentiates the effect of prostacyclin. Biochem Pharmacol. (1982) 31:3597–600. doi: 10.1016/0006-2952(82)90581-0
117. Kobuchi H, Roy S, Sen CK, Nguyen HG, Packer L. Quercetin inhibits inducible ICAM-1 expression in human endothelial cells through the JNK pathway. Am J Physiol. (1999) 277:C403–11. doi: 10.1152/ajpcell.1999.277.3.C403
118. Kamada C, Mukai R, Kondo A, Sato S, Terao J. Effect of quercetin and its metabolite on caveolin-1 expression induced by oxidized LDL and lysophosphatidylcholine in endothelial cells. J Clin Biochem Nutr. (2016) 58:193–201. doi: 10.3164/jcbn.16-2
119. Egert S, Bosy-Westphal A, Seiberl J, Kurbitz C, Settler U, Plachta-Danielzik S, et al. Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: a double-blinded, placebo-controlled cross-over study. Br J Nutr. (2009) 102:1065–74. doi: 10.1017/S0007114509359127
120. Chopra M, Fitzsimons PE, Strain JJ, Thurnham DI, Howard AN. Nonalcoholic red wine extract and quercetin inhibit LDL oxidation without affecting plasma antioxidant vitamin and carotenoid concentrations. Clin Chem. (2000) 46(Pt. 1):1162–70. doi: 10.1093/clinchem/46.8.1162
121. Choi S, Ryu KH, Park SH, Jun JY, Shin BC, Chung JH, et al. Direct vascular actions of quercetin in aorta from renal hypertensive rats. Kidney Res Clin Pract. (2016) 35:15–21. doi: 10.1016/j.krcp.2015.12.003
122. Duarte J, Perez-Palencia R, Vargas F, Ocete MA, Perez-Vizcaino F, Zarzuelo A, et al. Antihypertensive effects of the flavonoid quercetin in spontaneously hypertensive rats. Br J Pharmacol. (2001) 133:117–24. doi: 10.1038/sj.bjp.0704064
123. Galisteo M, Garcia-Saura MF, Jimenez R, Villar IC, Wangensteen R, Zarzuelo A, et al. Effects of quercetin treatment on vascular function in deoxycorticosterone acetate-salt hypertensive rats. Comparative study with verapamil. Planta Med. (2004) 70:334–41. doi: 10.1055/s-2004-818945
124. Galisteo M, Garcia-Saura MF, Jimenez R, Villar IC, Zarzuelo A, Vargas F, et al. Effects of chronic quercetin treatment on antioxidant defence system and oxidative status of deoxycorticosterone acetate-salt-hypertensive rats. Mol Cell Biochem. (2004) 259:91–9. doi: 10.1023/b:mcbi.0000021360.89867.64
125. Sanchez M, Galisteo M, Vera R, Villar IC, Zarzuelo A, Tamargo J, et al. Quercetin downregulates NADPH oxidase, increases eNOS activity and prevents endothelial dysfunction in spontaneously hypertensive rats. J Hypertens. (2006) 24:75–84. doi: 10.1097/01.hjh.0000198029.22472.d9
126. Kukongviriyapan U, Sompamit K, Pannangpetch P, Kukongviriyapan V, Donpunha W. Preventive and therapeutic effects of quercetin on lipopolysaccharide-induced oxidative stress and vascular dysfunction in mice. Can J Physiol Pharmacol. (2012) 90:1345–53. doi: 10.1139/y2012-101
127. Shen Y, Croft KD, Hodgson JM, Kyle R, Lee IL, Wang Y, et al. Quercetin and its metabolites improve vessel function by inducing eNOS activity via phosphorylation of AMPK. Biochem Pharmacol. (2012) 84:1036–44. doi: 10.1016/j.bcp.2012.07.016
128. Li PG, Sun L, Han X, Ling S, Gan WT, Xu JW. Quercetin induces rapid eNOS phosphorylation and vasodilation by an Akt-independent and PKA-dependent mechanism. Pharmacology. (2012) 89:220–8. doi: 10.1159/000337182
129. Khoo NK, White CR, Pozzo-Miller L, Zhou F, Constance C, Inoue T, et al. Dietary flavonoid quercetin stimulates vasorelaxation in aortic vessels. Free Radic Biol Med. (2010) 49:339–47. doi: 10.1016/j.freeradbiomed.2010.04.022
130. Romero M, Jimenez R, Sanchez M, Lopez-Sepulveda R, Zarzuelo MJ, O'Valle F, et al. Quercetin inhibits vascular superoxide production induced by endothelin-1: role of NADPH oxidase, uncoupled eNOS and PKC. Atherosclerosis. (2009) 202:58–67. doi: 10.1016/j.atherosclerosis.2008.03.007
131. Qian Y, Babu PVA, Symons JD, Jalili T. Metabolites of flavonoid compounds preserve indices of endothelial cell nitric oxide bioavailability under glucotoxic conditions. Nutr Diabetes. (2017) 7:e286. doi: 10.1038/nutd.2017.34
132. Hou X, Liu Y, Niu L, Cui L, Zhang M. Enhancement of voltage-gated K+ channels and depression of voltage-gated Ca2+ channels are involved in quercetin-induced vasorelaxation in rat coronary artery. Planta Med. (2014) 80:465–72. doi: 10.1055/s-0034-1368320
133. Hackl LP, Cuttle G, Dovichi SS, Lima-Landman MT, Nicolau M. Inhibition of angiotesin-converting enzyme by quercetin alters the vascular response to brandykinin and angiotensin I. Pharmacology. (2002) 65:182–6. doi: 10.1159/000064341
134. Lodi F, Winterbone MS, Tribolo S, Needs PW, Hughes DA, Kroon PA. Human quercetin conjugated metabolites attenuate TNF-alpha-induced changes in vasomodulatory molecules in an HUASMCs/HUVECs co-culture model. Planta Med. (2012) 78:1571–3. doi: 10.1055/s-0032-1315148
135. Zhao X, Gu Z, Attele AS, Yuan CS. Effects of quercetin on the release of endothelin, prostacyclin and tissue plasminogen activator from human endothelial cells in culture. J Ethnopharmacol. (1999) 67:279–85. doi: 10.1016/s0378-8741(99)00055-0
136. Loke WM, Hodgson JM, Proudfoot JM, McKinley AJ, Puddey IB, Croft KD. Pure dietary flavonoids quercetin and (-)-epicatechin augment nitric oxide products and reduce endothelin-1 acutely in healthy men. Am J Clin Nutr. (2008) 88:1018–25. doi: 10.1093/ajcn/88.4.1018
137. Serban MC, Sahebkar A, Zanchetti A, Mikhailidis DP, Howard G, Antal D, et al. Effects of quercetin on blood pressure: a systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc. (2016) 5:e002713. doi: 10.1161/JAHA.115.002713
138. Huang H, Liao D, Dong Y, Pu R. Effect of quercetin supplementation on plasma lipid profiles, blood pressure, and glucose levels: a systematic review and meta-analysis. Nutr Rev. (2020) 78:615–26. doi: 10.1093/nutrit/nuz071
139. Tamtaji OR, Milajerdi A, Dadgostar E, Kolahdooz F, Chamani M, Amirani E. The effects of quercetin supplementation on blood pressures and endothelial function among patients with metabolic syndrome and related disorders: a systematic review and meta-analysis of randomized controlled trials. Curr Pharm Des. (2019) 25:1372–84. doi: 10.2174/1381612825666190513095352
140. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. (2015) 14:644–58. doi: 10.1111/acel.12344
141. Jiang YH, Jiang LY, Wang YC, Ma DF, Li X. Quercetin attenuates atherosclerosis via modulating oxidized ldl-induced endothelial cellular senescence. Front Pharmacol. (2020) 11:512. doi: 10.3389/fphar.2020.00512
142. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
143. Jin HB, Yang YB, Song YL, Zhang YC, Li YR. Protective roles of quercetin in acute myocardial ischemia and reperfusion injury in rats. Mol Biol Rep. (2012) 39:11005–9. doi: 10.1007/s11033-012-2002-4
144. Liu H, Guo X, Chu Y, Lu S. Heart protective effects and mechanism of quercetin preconditioning on anti-myocardial ischemia reperfusion (IR) injuries in rats. Gene. (2014) 545:149–55. doi: 10.1016/j.gene.2014.04.043
145. Bartekova M, Carnicka S, Pancza D, Ondrejcakova M, Breier A, Ravingerova T. Acute treatment with polyphenol quercetin improves postischemic recovery of isolated perfused rat hearts after global ischemia. Can J Physiol Pharmacol. (2010) 88:465–71. doi: 10.1139/y10-025
146. Ferenczyova K, Kalocayova B, Kindernay L, Jelemensky M, Balis P, Berenyiova A, et al. Quercetin exerts age-dependent beneficial effects on blood pressure and vascular function, but is inefficient in preventing myocardial ischemia-reperfusion injury in zucker diabetic fatty rats. Molecules. (2020) 25:187. doi: 10.3390/molecules25010187
147. Shu Z, Yang Y, Yang L, Jiang H, Yu X, Wang Y. Cardioprotective effects of dihydroquercetin against ischemia reperfusion injury by inhibiting oxidative stress and endoplasmic reticulum stress-induced apoptosis via the PI3K/Akt pathway. Food Funct. (2019) 10:203–15. doi: 10.1039/c8fo01256c
148. Liu X, Yu Z, Huang X, Gao Y, Wang X, Gu J, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) mediates the protective effect of quercetin against myocardial ischemia-reperfusion injury via suppressing the NF-kappaB pathway. Am J Transl Res. (2016) 8:5169–86.
149. Liang Y, Zhang Y, Liu M, Han X, Zhang J, Zhang X, et al. Protective effect of quercetin against myocardial ischemia as a Ca(2+) channel inhibitor: involvement of inhibiting contractility and Ca(2+) influx via L-type Ca(2+) channels. Arch Pharm Res. (2020) 43:808–20. doi: 10.1007/s12272-020-01261-y
150. Annapurna A, Reddy CS, Akondi RB, Rao SR. Cardioprotective actions of two bioflavonoids, quercetin and rutin, in experimental myocardial infarction in both normal and streptozotocin-induced type I diabetic rats. J Pharm Pharmacol. (2009) 61:1365–74. doi: 10.1211/jpp/61.10.0014
151. Dong LY, Chen F, Xu M, Yao LP, Zhang YJ, Zhuang Y. Quercetin attenuates myocardial ischemia-reperfusion injury via downregulation of the HMGB1-TLR4-NF-kappaB signaling pathway. Am J Transl Res. (2018) 10:1273–83.
152. Kumar M, Kasala ER, Bodduluru LN, Kumar V, Lahkar M. Molecular and biochemical evidence on the protective effects of quercetin in isoproterenol-induced acute myocardial injury in rats. J Biochem Mol Toxicol. (2017) 31:1–8. doi: 10.1002/jbt.21832
153. Ma C, Jiang Y, Zhang X, Chen X, Liu Z, Tian X. Isoquercetin ameliorates myocardial infarction through anti-inflammation and anti-apoptosis factor and regulating TLR4-NF-kappaB signal pathway. Mol Med Rep. (2018) 17:6675–80. doi: 10.3892/mmr.2018.8709
154. Punithavathi VR, Prince PS. Pretreatment with a combination of quercetin and alpha-tocopherol ameliorates adenosine triphosphatases and lysosomal enzymes in myocardial infarcted rats. Life Sci. (2010) 86:178–84. doi: 10.1016/j.lfs.2009.11.021
155. Wang Y, Zhang ZZ, Wu Y, Ke JJ, He XH, Wang YL. Quercetin postconditioning attenuates myocardial ischemia/reperfusion injury in rats through the PI3K/Akt pathway. Braz J Med Biol Res. (2013) 46:861–7. doi: 10.1590/1414-431X20133036
156. Chekalina N, Burmak Y, Petrov Y, Borisova Z, Manusha Y, Kazakov Y, et al. Quercetin reduces the transcriptional activity of NF-kB in stable coronary artery disease. Indian Heart J. (2018) 70:593–7. doi: 10.1016/j.ihj.2018.04.006
157. Parkhomenko A, Kozhukhov S, Lutay Y. Multicenter randomized clinical trial of the efficacy and safety of intravenous quercetin in patients with ST-elevation acute myocardial infarction. Euro Heart J. (2018) 39(suppl_1):2152. doi: 10.1093/eurheartj/ehy565.2152
158. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. (2018) 20:12. doi: 10.1007/s11906-018-0812-z
159. Guo W, Gong X, Li M. Quercetin actions on lipid profiles in overweight and obese individuals: a systematic review and meta-analysis. Curr Pharm Des. (2019) 25:3087–95. doi: 10.2174/1381612825666190829153552
160. Schoeler M, Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. (2019) 20:461–72. doi: 10.1007/s11154-019-09512-0
161. Ohira H, Tsutsui W, Fujioka Y. Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? J Atheroscler Thromb. (2017) 24:660–72. doi: 10.5551/jat.RV17006
162. Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. (2001) 292:1160–4. doi: 10.1126/science.1059344
163. Faxon DP, Fuster V, Libby P, Beckman JA, Hiatt WR, Thompson RW, et al. Atherosclerotic Vascular Disease Conference: Writing Group III: pathophysiology. Circulation. (2004) 109:2617–25. doi: 10.1161/01.CIR.0000128520.37674.EF
164. Kawai Y, Nishikawa T, Shiba Y, Saito S, Murota K, Shibata N, et al. Macrophage as a target of quercetin glucuronides in human atherosclerotic arteries: implication in the anti-atherosclerotic mechanism of dietary flavonoids. J Biol Chem. (2008) 283:9424–34. doi: 10.1074/jbc.M706571200
165. Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. (2013) 70:2859–72. doi: 10.1007/s00018-012-1194-z
166. Frostegard J, Haegerstrand A, Gidlund M, Nilsson J. Biologically modified LDL increases the adhesive properties of endothelial cells. Atherosclerosis. (1991) 90:119–26. doi: 10.1016/0021-9150(91)90106-d
167. Heinloth A, Heermeier K, Raff U, Wanner C, Galle J. Stimulation of NADPH oxidase by oxidized low-density lipoprotein induces proliferation of human vascular endothelial cells. J Am Soc Nephrol. (2000) 11:1819–25. Available online at: https://jasn.asnjournals.org/content/11/10/1819
168. Katouah H, Chen A, Othman I, Gieseg SP. Oxidised low density lipoprotein causes human macrophage cell death through oxidant generation and inhibition of key catabolic enzymes. Int J Biochem Cell Biol. (2015) 67:34–42. doi: 10.1016/j.biocel.2015.08.001
169. Perez-Vizcaino F, Ibarra M, Cogolludo AL, Duarte J, Zaragoza-Arnaez F, Moreno L, et al. Endothelium-independent vasodilator effects of the flavonoid quercetin and its methylated metabolites in rat conductance and resistance arteries. J Pharmacol Exp Ther. (2002) 302:66–72. doi: 10.1124/jpet.302.1.66
170. Ibarra M, Perez-Vizcaino F, Cogolludo A, Duarte J, Zaragoza-Arnaez F, Lopez-Lopez JG, et al. Cardiovascular effects of isorhamnetin and quercetin in isolated rat and porcine vascular smooth muscle and isolated rat atria. Planta Med. (2002) 68:307–10. doi: 10.1055/s-2002-26752
171. Chiwororo WD, Ojewole JA. Dual effect of quercetin on rat isolated portal vein smooth muscle contractility. Cardiovasc J Afr. (2010) 21:132–6.
172. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The Physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. (2015) 67:821–70. doi: 10.1124/pr.114.009654
173. Jackson WF. KV channels and the regulation of vascular smooth muscle tone. Microcirculation. (2018) 25:e12421. doi: 10.1111/micc.12421
174. Carvalho-de-Souza JL, Varanda WA, Tostes RC, Chignalia AZ. BK channels in cardiovascular diseases and aging. Aging Dis. (2013) 4:38–49. Available online at: http://www.aginganddisease.org/EN/Y2013/V4/I1/38
175. Cogolludo A, Frazziano G, Briones AM, Cobeno L, Moreno L, Lodi F, et al. The dietary flavonoid quercetin activates BKCa currents in coronary arteries via production of H2O2. Role in vasodilatation. Cardiovasc Res. (2007) 73:424–31. doi: 10.1016/j.cardiores.2006.09.008
176. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. (2000) 106:1521–30. doi: 10.1172/JCI10506
177. Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. (2007) 73:73–81. doi: 10.1016/j.cardiores.2006.10.005
178. Drouin A, Thorin E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke. (2009) 40:1827–33. doi: 10.1161/STROKEAHA.108.536805
179. Thorin E, Webb DJ. Endothelium-derived endothelin-1. Pflugers Arch. (2010) 459:951–8. doi: 10.1007/s00424-009-0763-y
180. Cerrato R, Cunnington C, Crabtree MJ, Antoniades C, Pernow J, Channon KM, et al. Endothelin-1 increases superoxide production in human coronary artery bypass grafts. Life Sci. (2012) 91:723–8. doi: 10.1016/j.lfs.2012.03.024
181. Sud N, Black SM. Endothelin-1 impairs nitric oxide signaling in endothelial cells through a protein kinase Cdelta-dependent activation of STAT3 and decreased endothelial nitric oxide synthase expression. DNA Cell Biol. (2009) 28:543–53. doi: 10.1089/dna.2009.0865
182. Ohkita M, Takaoka M, Shiota Y, Nojiri R, Matsumura Y. Nitric oxide inhibits endothelin-1 production through the suppression of nuclear factor kappa B. Clin Sci. (2002) 103(Suppl. 48):68–71S. doi: 10.1042/CS103S068S
183. Carretero OA. Novel mechanism of action of ACE and its inhibitors. Am J Physiol Heart Circ Physiol. (2005) 289:H1796–7. doi: 10.1152/ajpheart.00781.2005
184. Thorin E. Different contribution of endothelial nitric oxide in the relaxation of human coronary arteries of ischemic and dilated cardiomyopathic hearts. J Cardiovasc Pharmacol. (2001) 37:227–32. doi: 10.1097/00005344-200102000-00010
185. Primikyri A, Mazzone G, Lekka C, Tzakos AG, Russo N, Gerothanassis IP. Understanding zinc(II) chelation with quercetin and luteolin: a combined NMR and theoretical study. J Phys Chem B. (2015) 119:83–95. doi: 10.1021/jp509752s
186. Loizzo MR, Said A, Tundis R, Rashed K, Statti GA, Hufner A, et al. Inhibition of angiotensin converting enzyme (ACE) by flavonoids isolated from Ailanthus excelsa (Roxb) (Simaroubaceae). Phytother Res. (2007) 21:32–6. doi: 10.1002/ptr.2008
187. Larson AJ, Bruno RS, Guo Y, Gale D, Tanner J, Jalili T, et al. Acute quercetin supplementation does not lower blood pressure or ace activity in normotensive males. J Am Diet Assoc. (2009) 109:A16. doi: 10.1016/j.jada.2009.06.388
188. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. (2011) 479:232–6. doi: 10.1038/nature10600
189. Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL. Discovery, development, and future application of senolytics: theories and predictions. FEBS J. (2020) 287:2418–27. doi: 10.1111/febs.15264
190. Rodier F. Detection of the senescence-associated secretory phenotype (SASP). Methods Mol Biol. (2013) 965:165–73. doi: 10.1007/978-1-62703-239-1_10
191. Kampkotter A, Timpel C, Zurawski RF, Ruhl S, Chovolou Y, Proksch P, et al. Increase of stress resistance and lifespan of Caenorhabditis elegans by quercetin. Comp Biochem Physiol B Biochem Mol Biol. (2008) 149:314–23. doi: 10.1016/j.cbpb.2007.10.004
192. Olave NC, Grenett MH, Cadeiras M, Grenett HE, Higgins PJ. Upstream stimulatory factor-2 mediates quercetin-induced suppression of PAI-1 gene expression in human endothelial cells. J Cell Biochem. (2010) 111:720–6. doi: 10.1002/jcb.22760
193. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. (2016) 15:973–7. doi: 10.1111/acel.12458
194. Lamichane S, Baek SH, Kim YJ, Park JH, Dahal Lamichane B, Jang WB, et al. MHY2233 attenuates replicative cellular senescence in human endothelial progenitor cells via SIRT1 signaling. Oxid Med Cell Longev. (2019) 2019:6492029. doi: 10.1155/2019/6492029
195. Hwang HV, Tran DT, Rebuffatti MN, Li CS, Knowlton AA. Investigation of quercetin and hyperoside as senolytics in adult human endothelial cells. PLoS ONE. (2018) 13:e0190374. doi: 10.1371/journal.pone.0190374
196. Juul A, Scheike T, Davidsen M, Gyllenborg J, Jorgensen T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: a population-based case-control study. Circulation. (2002) 106:939–44. doi: 10.1161/01.cir.0000027563.44593.cc
197. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
198. Singhal AK, Symons JD, Boudina S, Jaishy B, Shiu YT. Role of endothelial cells in myocardial ischemia-reperfusion injury. Vasc Dis Prev. (2010) 7:1–14. doi: 10.2174/1874120701007010001
199. Pagliaro BR, Cannata F, Stefanini GG, Bolognese L. Myocardial ischemia and coronary disease in heart failure. Heart Fail Rev. (2020) 25:53–65. doi: 10.1007/s10741-019-09831-z
200. Gonzalez-Montero J, Brito R, Gajardo AI, Rodrigo R. Myocardial reperfusion injury and oxidative stress: therapeutic opportunities. World J Cardiol. (2018) 10:74–86. doi: 10.4330/wjc.v10.i9.74
201. Van Hoorn DE, Nijveldt RJ, Van Leeuwen PA, Hofman Z, M'Rabet L, De Bont DB, et al. Accurate prediction of xanthine oxidase inhibition based on the structure of flavonoids. Eur J Pharmacol. (2002) 451:111–8. doi: 10.1016/s0014-2999(02)02192-1
202. Lin CM, Chen CS, Chen CT, Liang YC, Lin JK. Molecular modeling of flavonoids that inhibits xanthine oxidase. Biochem Biophys Res Commun. (2002) 294:167–72. doi: 10.1016/S0006-291X(02)00442-4
203. Mohos V, Panovics A, Fliszar-Nyul E, Schilli G, Hetenyi C, Mladenka P, et al. Inhibitory effects of quercetin and its human and microbial metabolites on xanthine oxidase enzyme. Int J Mol Sci. (2019) 20:2681. doi: 10.3390/ijms20112681
204. Huang J, Wang S, Zhu M, Chen J, Zhu X. Effects of genistein, apigenin, quercetin, rutin and astilbin on serum uric acid levels and xanthine oxidase activities in normal and hyperuricemic mice. Food Chem Toxicol. (2011) 49:1943–7. doi: 10.1016/j.fct.2011.04.029
205. Zhu JX, Wang Y, Kong LD, Yang C, Zhang X. Effects of Biota orientalis extract and its flavonoid constituents, quercetin and rutin on serum uric acid levels in oxonate-induced mice and xanthine dehydrogenase and xanthine oxidase activities in mouse liver. J Ethnopharmacol. (2004) 93:133–40. doi: 10.1016/j.jep.2004.03.037
206. Wan LL, Xia J, Ye D, Liu J, Chen J, Wang G. Effects of quercetin on gene and protein expression of NOX and NOS after myocardial ischemia and reperfusion in rabbit. Cardiovasc Ther. (2009) 27:28–33. doi: 10.1111/j.1755-5922.2009.00071.x
207. Guo M, Perez C, Wei Y, Rapoza E, Su G, Bou-Abdallah F, et al. Iron-binding properties of plant phenolics and Cranberry's bio-effects. Dalton Trans. (2007) 43:4951–61. doi: 10.1039/b705136k
208. Dabbagh-Bazarbachi H, Clergeaud G, Quesada IM, Ortiz M, O'Sullivan CK, Fernandez-Larrea JB. Zinc ionophore activity of quercetin and epigallocatechin-gallate: from Hepa 1-6 cells to a liposome model. J Agric Food Chem. (2014) 62:8085–93. doi: 10.1021/jf5014633
209. Bao B, Prasad AS, Beck FW, Fitzgerald JT, Snell D, Bao GW, et al. Zinc decreases C-reactive protein, lipid peroxidation, and inflammatory cytokines in elderly subjects: a potential implication of zinc as an atheroprotective agent. Am J Clin Nutr. (2010) 91:1634–41. doi: 10.3945/ajcn.2009.28836
210. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. (2011) 111:5944–72. doi: 10.1021/cr200084z
211. Dugas AJ Jr, Castaneda-Acosta J, Bonin GC, Price KL, Fischer NH, et al. Evaluation of the total peroxyl radical-scavenging capacity of flavonoids: structure-activity relationships. J Nat Prod. (2000) 63:327–31. doi: 10.1021/np990352n
212. Kumar P, Sharma S, Khanna M, Raj HG. Effect of Quercetin on lipid peroxidation and changes in lung morphology in experimental influenza virus infection. Int J Exp Pathol. (2003) 84:127–33. doi: 10.1046/j.1365-2613.2003.00344.x
213. Tang L, Peng Y, Xu T, Yi X, Liu Y, Luo Y, et al. The effects of quercetin protect cardiomyocytes from A/R injury is related to its capability to increasing expression and activity of PKCepsilon protein. Mol Cell Biochem. (2013) 382:145–52. doi: 10.1007/s11010-013-1729-0
214. Li B, Yang M, Liu JW, Yin GT. Protective mechanism of quercetin on acute myocardial infarction in rats. Genet Mol Res. (2016) 15:15017117. doi: 10.4238/gmr.15017117
215. Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. (2008) 117:3216–26. doi: 10.1161/CIRCULATIONAHA.108.769331
216. Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. (2008) 102:283–94. doi: 10.1161/CIRCRESAHA.107.164384
217. Pittas K, Vrachatis DA, Angelidis C, Tsoucala S, Giannopoulos G, Deftereos S. The role of calcium handling mechanisms in reperfusion injury. Curr Pharm Des. (2018) 24:4077–89. doi: 10.2174/1381612825666181120155953
218. Guo X, Chen M, Zeng H, Liu P, Zhu X, Zhou F, et al. Quercetin attenuates ethanol-induced iron uptake and myocardial injury by regulating the angiotensin II-L-type calcium channel. Mol Nutr Food Res. (2018) 62:1700772. doi: 10.1002/mnfr.201700772
219. Neuhof C, Neuhof H. Calpain system and its involvement in myocardial ischemia and reperfusion injury. World J Cardiol. (2014) 6:638–52. doi: 10.4330/wjc.v6.i7.638
220. Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. (2002) 90:1189–96. doi: 10.1161/01.res.0000021432.70309.28
221. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. (2010) 16:1400–6. doi: 10.1038/nm.2252
222. Sanchez-Duffhues G, Orlova V, Ten Dijke P. In brief: endothelial-to-mesenchymal transition. J Pathol. (2016) 238:378–80. doi: 10.1002/path.4653
223. Moonen JR, Krenning G, Brinker MG, Koerts JA, van Luyn MJ, Harmsen MC. Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res. (2010) 86:506–15. doi: 10.1093/cvr/cvq012
224. Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. (2008) 99:1375–9. doi: 10.1038/sj.bjc.6604662
225. Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. (2004) 95:459–70. doi: 10.1161/01.RES.0000141146.95728.da
226. Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. (2012) 125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352
227. Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. (2015) 131:1006–18. doi: 10.1161/CIRCULATIONAHA.114.008750
228. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, et al. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. (2015) 185:1850–8. doi: 10.1016/j.ajpath.2015.03.019
229. Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to mesenchymal transition (endomt) in the pathogenesis of human fibrotic diseases. J Clin Med. (2016) 5:45. doi: 10.3390/jcm5040045
230. Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. (2011) 121:468–74. doi: 10.1172/jci44595
231. Ma J, Sanchez-Duffhues G, Goumans MJ, Ten Dijke P. TGF-beta-induced endothelial to mesenchymal transition in disease and tissue engineering. Front Cell Dev Biol. (2020) 8:260. doi: 10.3389/fcell.2020.00260
232. van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res. (2012) 347:177–86. doi: 10.1007/s00441-011-1222-6
233. Medici D, Potenta S, Kalluri R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. (2011) 437:515–20. doi: 10.1042/BJ20101500
234. Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. (2010) 121:2407–18. doi: 10.1161/CIRCULATIONAHA.110.938217
235. Wermuth PJ, Li Z, Mendoza FA, Jimenez SA. Stimulation of transforming growth factor-beta1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (ET-1): a novel profibrotic effect of ET-1. PLoS ONE. (2016) 11:e0161988. doi: 10.1371/journal.pone.0161988
236. Li Z, Wermuth PJ, Benn BS, Lisanti MP, Jimenez SA. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am J Pathol. (2013) 182:325–31. doi: 10.1016/j.ajpath.2012.10.022
237. Maleszewska M, Moonen JR, Huijkman N, van de Sluis B, Krenning G, Harmsen MC. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology. (2013) 218:443–54. doi: 10.1016/j.imbio.2012.05.026
238. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res. (2018) 114:565–77. doi: 10.1093/cvr/cvx253
239. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. (2018) 9:294. doi: 10.3389/fimmu.2018.00294
240. Fleenor BS, Marshall KD, Rippe C, Seals DR. Replicative aging induces endothelial to mesenchymal transition in human aortic endothelial cells: potential role of inflammation. J Vasc Res. (2012) 49:59–64. doi: 10.1159/000329681
241. Lee ES. Biomechanical regulation of endothelial phenotype [Thesis fully internal (DIV)]. University of Groningen, Groningen, Netherlands (2015).
242. Huang S, Zhu X, Huang W, He Y, Pang L, Lan X, et al. Quercetin inhibits pulmonary arterial endothelial cell transdifferentiation possibly by Akt and Erk1/2 pathways. Biomed Res Int. (2017) 2017:6147294. doi: 10.1155/2017/6147294
243. Cai W, Yu D, Fan J, Liang X, Jin H, Liu C, et al. Quercetin inhibits transforming growth factor beta1-induced epithelial-mesenchymal transition in human retinal pigment epithelial cells via the Smad pathway. Drug Des Devel Ther. (2018) 12:4149–61. doi: 10.2147/DDDT.S185618
244. Balakrishnan S, Bhat FA, Raja Singh P, Mukherjee S, Elumalai P, Das S, et al. Gold nanoparticle-conjugated quercetin inhibits epithelial-mesenchymal transition, angiogenesis and invasiveness via EGFR/VEGFR-2-mediated pathway in breast cancer. Cell Prolif. (2016) 49:678–97. doi: 10.1111/cpr.12296
245. Lu X, Chen D, Yang F, Xing N. Quercetin inhibits epithelial-to-mesenchymal transition (emt) process and promotes apoptosis in prostate cancer via downregulating lncRNA MALAT1. Cancer Manag Res. (2020) 12:1741–50. doi: 10.2147/CMAR.S241093
246. Saito A. EMT and EndMT: regulated in similar ways? J Biochem. (2013) 153:493–5. doi: 10.1093/jb/mvt032
247. McVeigh GE, Cohn JN. Endothelial dysfunction and the metabolic syndrome. Curr Diab Rep. (2003) 3:87–92. doi: 10.1007/s11892-003-0059-0
248. Tziomalos K, Athyros VG, Karagiannis A, Mikhailidis DP. Endothelial dysfunction in metabolic syndrome: prevalence, pathogenesis and management. Nutr Metab Cardiovasc Dis. (2010) 20:140–6. doi: 10.1016/j.numecd.2009.08.006
249. Virdis A, Masi S, Colucci R, Chiriaco M, Uliana M, Puxeddu I, et al. Microvascular endothelial dysfunction in patients with obesity. Curr Hypertens Rep. (2019) 21:32. doi: 10.1007/s11906-019-0930-2
250. Engin A. Endothelial dysfunction in obesity. Adv Exp Med Biol. (2017) 960:345–79. doi: 10.1007/978-3-319-48382-5_15
251. Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes. (2017) 9:434–49. doi: 10.1111/1753-0407.12521
252. Bozkurt B, Aguilar D, Deswal A, Dunbar SB, Francis GS, Horwich T, et al. Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American Heart Association. Circulation. (2016) 134:e535–78. doi: 10.1161/CIR.0000000000000450
253. Green V. The domino effect: obesity, type 2 diabetes and cardiovascular disease. Br J Community Nurs. (2005) 10:358–61. doi: 10.12968/bjcn.2005.10.8.18573
254. Sato S, Mukai Y. Modulation of chronic inflammation by quercetin: the beneficial effects on obesity. J Inflamm Res. (2020) 13:421–31. doi: 10.2147/JIR.S228361
255. Williamson G, Sheedy K. Effects of polyphenols on insulin resistance. Nutrients. (2020) 12:3135. doi: 10.3390/nu12103135
256. Eid HM, Haddad PS. The antidiabetic potential of quercetin: underlying mechanisms. Curr Med Chem. (2017) 24:355–64. doi: 10.2174/0929867323666160909153707
257. Chen S, Jiang H, Wu X, Fang J. Therapeutic effects of quercetin on inflammation, obesity, and type 2 diabetes. Mediators Inflamm. (2016) 2016:9340637. doi: 10.1155/2016/9340637
Keywords: endothelial (dys)function, flavonoids, quercetin, hypertension, atherosclerosis, senescence, aging, ischemia-reperfusion
Citation: Dagher O, Mury P, Thorin-Trescases N, Noly PE, Thorin E and Carrier M (2021) Therapeutic Potential of Quercetin to Alleviate Endothelial Dysfunction in Age-Related Cardiovascular Diseases. Front. Cardiovasc. Med. 8:658400. doi: 10.3389/fcvm.2021.658400
Received: 25 January 2021; Accepted: 05 March 2021;
Published: 30 March 2021.
Edited by:
Lamiaa A. Ahmed, Cairo University, EgyptReviewed by:
Suowen Xu, University of Science and Technology of China, ChinaOwen Llewellyn Woodman, Monash University, Australia
Copyright © 2021 Dagher, Mury, Thorin-Trescases, Noly, Thorin and Carrier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olina Dagher, b2xpbmEuZGFnaGVyQGFocy5jYQ==