 Alexander Guldmann Clausen
Alexander Guldmann Clausen Oliver Bundgaard Vad
Oliver Bundgaard Vad Julie Husted Andersen1
Julie Husted Andersen1 Morten Salling Olesen
Morten Salling Olesen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 09 March 2021
Sec. Cardiovascular Genetics and Systems Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.650667
This article is part of the Research Topic Genomic Architecture of Cardiovascular Diseases View all 15 articles
Multiple genome-wide association studies (GWAS) have identified numerous loci associated with atrial fibrillation (AF). However, the genes driving these associations and how they contribute to the AF pathogenesis remains poorly understood. To identify genes likely to be driving the observed association, we searched the FinnGen study consisting of 12,859 AF cases and 73,341 controls for rare genetic variants predicted to cause loss-of-function. A specific splice site variant was found in the SYNPO2L gene, located in an AF associated locus on chromosome 10. This variant was associated with an increased risk of AF with a relatively high odds ratio of 3.5 (p = 9.9 × 10−8). SYNPO2L is an important gene involved in the structural development and function of the cardiac myocyte and our findings thus support the recent suggestions that AF can present as atrial cardiomyopathy.
Atrial Fibrillation (AF) is the most common cardiac arrhythmia with more than 33 million people affected worldwide (1). The disease has been associated with an increased risk of stroke, heart failure as well as increased all-cause mortality (2). While AF is a complex disease associated with multiple risk factors including age, sex, lifestyle and existing heart disease, a significant genetic component of the disease has also been established (3). In 2018, a meta-analysis of genome wide association studies (GWAS) of AF identified a total of 134 loci associated with AF (4). Many of these loci have been linked to genes related to ion channels, structural proteins of the myocardium and transcription factors (5). However, a large number of the identified loci are located in non-coding regions and in the vicinity of multiple genes. More work is needed to identify the causal genes mediating the observed associations. The purpose of this paper is to investigate potentially causal genes at known GWAS loci in order to further elucidate the mechanisms behind AF pathogenesis. This was done by searching genes in a publicly available dataset containing phenotypes linked with imputed genotypes for AF-associated variants in loci previously associated with AF in GWAS.
To identify genes likely to be driving the association between the observed GWAS loci and AF, we searched the FinnGen database (6) for variants significantly enriched in AF cases. Only variants located in one of the 134 GWAS loci previously identified in our GWAS meta-analysis (4) were selected for further investigation. To increase the likelihood of the identified variants contributing to the pathogenesis of AF development, we confined our analysis of the FinnGen data to only include predicted loss-of-functions variants. These were defined as variants leading to a pre-mature stop codon, deletions or insertions leading to a frameshift or variants involving splice sites. To further evaluate the identified candidate genes, we analyzed mRNA expression in right atrial appendage tissue integrated with genotype data obtained through the Genotype-Tissue Expression (GTEx) project. This was done by searching the publicly available GTEx release v8 database (https://gtexportal.org/home/gene/SYNPO2L). A brief description of the used data from the FinnGen database and the GTEx project is given below.
The publicly available FinnGen database contains genotype data from Finnish biobanks linked with corresponding national health registry data from 12,859 AF cases and 73,341 controls. Data from the FinnGen database was retrieved from freeze number 3. FinnGen participants were genotyped by the FinnGen investigators using various Illumina and Affymetrix GWAS arrays. This was followed by a genotype imputation by the Beagle v.4.1 software using 3,775 high coverage whole genome sequences of Finnish origin as a population specific reference panel (6). We used the FinnGen disease endpoint “Atrial Fibrillation and Flutter.” The SAIGE software was used for the genetic association tests of the FinnGen data. SAIGE is a mixed model logistic regression R/C++ package that is able to adjust for relatedness of the analyzed samples while also controlling for unbalanced case-control ratios that can otherwise lead to inflated type 1 error rates (7). SAIGE first generates relatedness estimates and a null model. Next, the null model is fitted with the respective SNP, and the genetic kinship matrix and the model itself is then adjusted for the following covariates: sex, age, 10 principal components and genotyping batch. For more information on the included biobanks, disease endpoint definitions and genotype data in the FinnGen database, please visit https://finngen.gitbook.io/documentation/v/r3/.
The GTEx Project database contains analyses of mRNA levels in 47 different tissues, including atrial appendage tissue obtained from 429 deceased donors with available genotype data. The data was obtained by massive parallel sequencing of RNA strands using Illumina TrueSeq RNA sequencing followed by qPCR (8).
In order to further explore the genes responsible for the identified genome-wide associated risk locus, we performed a colocalization analysis. Using the LocusFocus tool (9), we tested whether the GWAS signals are colocalized with eQTL signals, and therefore are likely to share a causal variant. The LocusFocus tool utilizes the Simple Sum colocalization method based on a frequentist framework. To perform the colocalization analysis, we used the publicly available FinnGen AF GWAS summary statistics and integrated them with cis-eQTL data from atrial appendage tissue from the GTEx project v8. For an identified locus, we used GWAS summary statistics from all SNPs within a 100 kb window centered around the identified lead SNP. The performance of the Simple Sum colocalization method compared with other colocalization methods has been described by Gong et al. (10).
We performed an in-sillico pathway analysis of interactions between the protein product of SYNPO2L and other proteins in AF, using the tool String (v11) (11). String is a large database of protein-protein interactions, derived from multiple sources, including both physical and functional associations. We conducted a search of proteins thought to be important in structural function of cardiomyocytes, as defined in a recent review of AF genetics by Andersen et al. (12). Afterwards a search of all proteins was done, limited to the 10 interactions predicted with highest confidence. Evaluation of confidence in interactions have been described in detail by Szklarczyk et al. (11).
Variants in candidate genes were evaluated using the in silico tool Combined Annotation Dependent Depletion Score (CADD), in order to predict the effects and deleteriousness of the concerned variants (13). This method uses a machine learning model to incorporate multiple different forms of variant annotations into one integrated score, in order to better identify causal variants. Variants with a CADD-score >20 are predicted to be among the 1% most deleterious variants in the human genome. Those with a CADD-score >30 are predicted to be among the 0.1% most deleterious.
A total of 119 variants were associated with AF in the FinnGen study. Of these, 13 variants were located in genes in loci previously identified in our GWAS meta-analysis. The 13 variants are presented in Table 1. Of the 13 variants, only one variant, rs766868752, was predicted to be a loss-of-function variant. This variant was located in the SYNPO2L gene. The variant was imputed with an INFO-score of 0.972, indicating a very high confidence imputation.
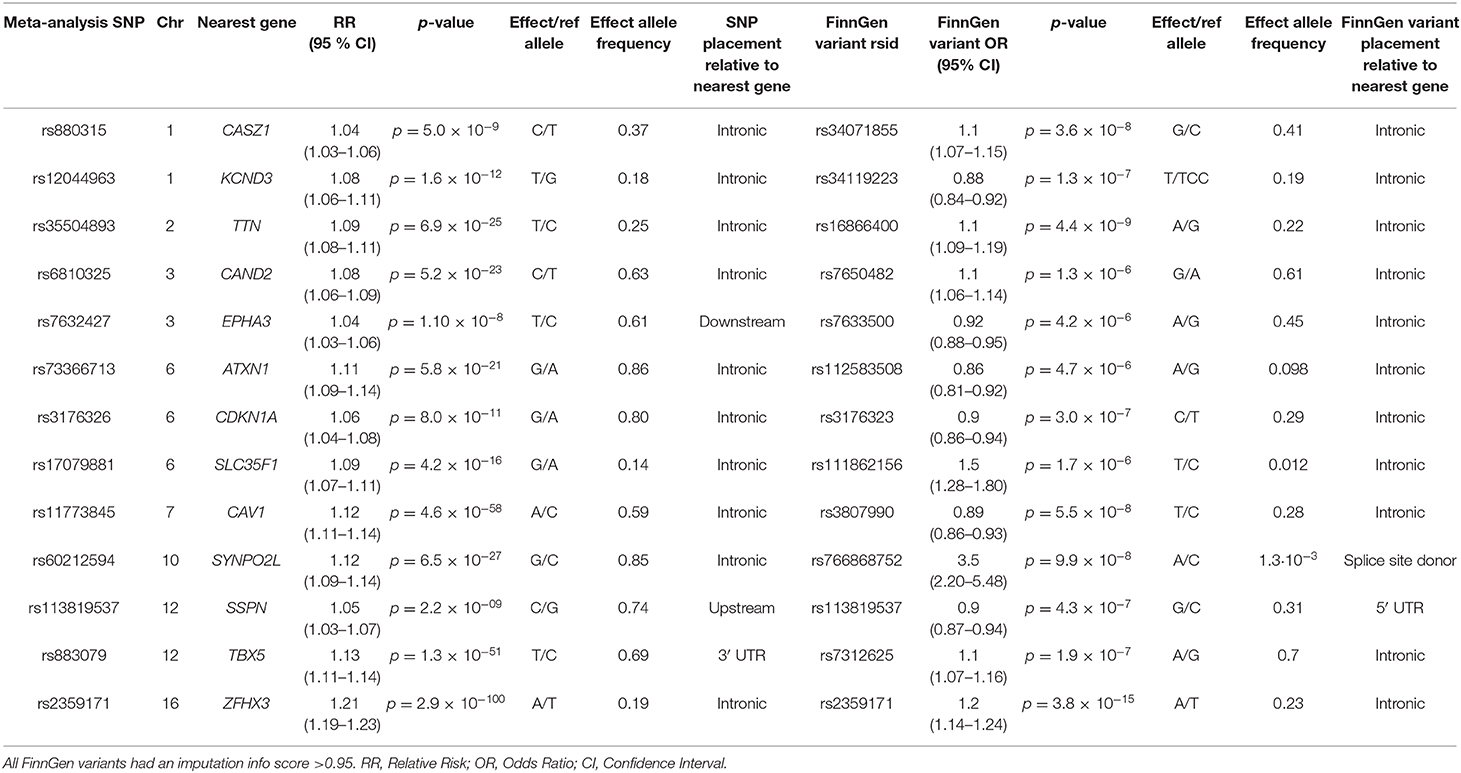
Table 1. Variants associated with AF in the GWAS meta-analysis by Roselli et al. that were located near genes also associated with AF in the FinnGen study.
The rs766868752 variant in SYNPO2L is characterized as a splice site donor variant and is located on position 73,655,817 on the first base of intron 1. In carriers of the variant, the C base of the non-template strand is replaced with an A base. Thus, the normal splice donor site of 5′ GT 3′ in the template strand is changed to a 5′ TT 3 ′. In the 12,859 AF cases in the FinnGen study, the variant was found with a minor allele frequency (MAF) of 0.002585. In the 73,341 control participants, the variant was found with a MAF of 0.001022. We found an increased Odds Ratio (OR) of developing AF of 3.5 in persons carrying the c.105+1G>T variant in SYNPO2L (p = 9.9 × 10−8). According to the gnomAD browser, a database containing more than 76,000 exome sequences, the specific variant was almost exclusively found in people of Finnish descent (14). Of the 41 carriers of the mutation in the gnomAD database, 40 was of Finnish descent and only one carrier was of non-Finnish European descent. Only one other splice site donor variant has been identified in the gnomAD v3 database, suggesting that splice site mutations in SYNPO2L are rare and likely deleterious.
Two isoforms of the SYNPO2L genes are expressed in-vivo. A long isoform with the ID ENST00000394810.2 as well as a short isoform with the ID ENST00000372873.8 (14). The splice site mutation found in the FinnGen study is located in the long transcript as seen in Figure 1. As the only gene positioned in a locus identified in our GWAS meta-analysis that also was found to have predicted loss-of-function variants enriched in the FinnGen study population, the SYNPO2L gene was selected for further investigation.
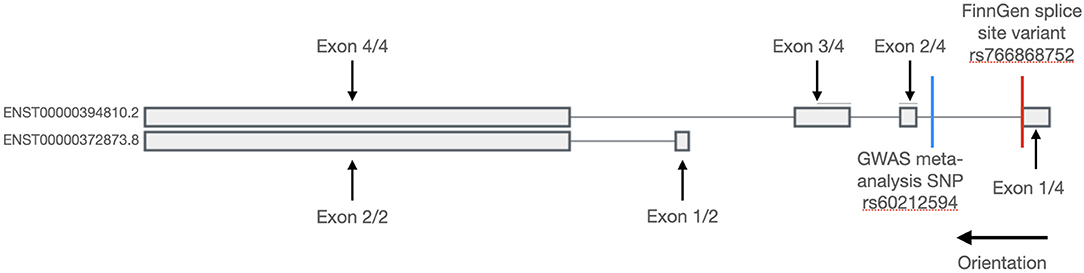
Figure 1. Transcripts of the SYNPO2L gene. Exons are shown with thick bars and introns are shown with thin lines. The SYNPO2L gene is expressed in a short and a long isoform. The splice site variant found in the FinnGen database is shown with a red line and is located in the first intron of the long isoform. The position of the common SNP, rs60212594, identified in the GWAS meta-analysis is shown with a blue line.
To determine whether the GWAS signals at the SYNPO2L locus are likely to be driven by changes in SYNPO2L expression in atrial appendage tissue, we performed a colocalization analysis using the LocusFocus tool. Our analysis supports a strong colocalization of GWAS signals with eQTLs of SYNPO2L in atrial appendage tissue with a Simple Sum p-value of 2.1 × 10−6. The colocalization analysis has been visualized in Figure 2.
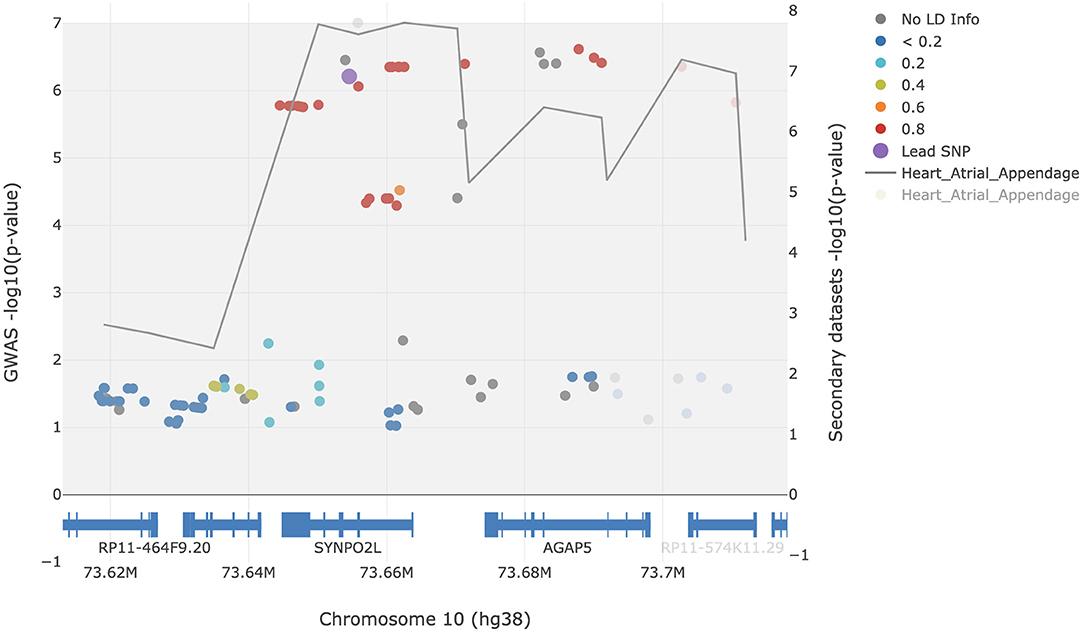
Figure 2. Colocalization analysis of AF GWAS signals with eQTL signals of SYNPO2L in atrial appendage tissue. Filled circles represent AF GWAS -log10(p-values) (left y-axis). The gray line represents the eQTL signals and traces the lowest p-value [right y-axis, showing -log10(p-values)] per window of 22.5 bp. The rs60212594 SNP was defined as the lead SNP and is presented in purple. The European 1000 Genomes Project was used for the LD calculations in reference to the lead SNP. The figure was generated using the LocusFocus tool.
To further assess the involvement of the SYNPO2L gene in AF, we examined whether the expression of the SYNPO2L gene was affected in carriers of the SNP rs60212594. This common SNP is located at the SYNPO2L locus and was associated with an increased risk of AF in our GWAS meta-analysis (4). This was done by a quantitative trait locus (cis-eQTL) analysis using data from the GTEx project. The rs60212594 SNP identified in the GWAS was shown to be associated with a minor but significant change in SYNPO2L expression (15). Carriers of the C risk allele had a 0.08-fold decrease in SYNPO2L expression with a p-value of 2.9 × 10−8, which is illustrated in Figure 3.
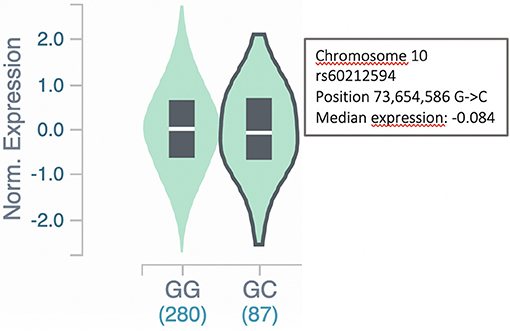
Figure 3. SYNPO2L expression in carriers of the rs60212594 SNP. Expression of the SYNPO2L gene is decreased (p = 2.9 × 10−8) in heterozygous carriers of the C allele of the rs60212594 SNP associated with AF. The data and figure were obtained from the GTEx project eQTL analysis (https://gtexportal.org/home/snp/rs60212594).
Using the tool String, we examined interactions between the protein product of SYNPO2L and those of other structural genes previously associated with AF. The protein product of SYNPOL2 as well as TTN was found to be co-expressed in numerous species as illustrated in Figures 4A–E, indicating a functional association between the protein products of these genes. When searching for interactions between SYNPO2L and all genes, we found the SYNPO2L to interact with and be co-expressed with the protein products of several other genes. These genes are associated with the structure of sarcomeres, calcium signaling and cardiomyopathies. The proteins interacting with the SYNPO2L product are summarized in Supplementary Table 1.
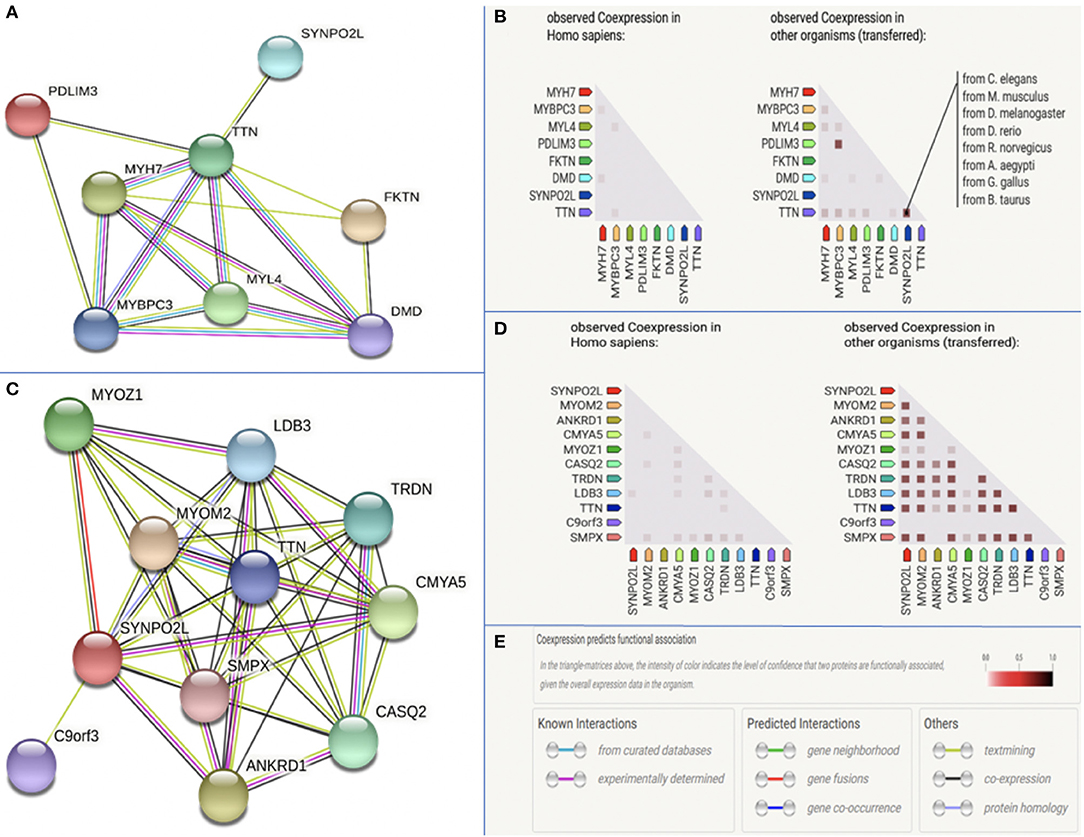
Figure 4. Protein-protein interaction analysis. The figure is modified from data provided by the tool String v11. (A) Protein interactions between the product of SYNPO2L and seven other structural genes previously associated with AF. (B) Co-expression data across Homo Sapiens and other species for the product of SYNPO2L and seven other structural genes previously associated with AF. Species in which co-expression has been observed has been highlighted for SYNPO2L and TTN. (C) Protein interactions between the product of SYNPO2L and the products of the 10 genes with the highest confidence in interaction prediction. (D) Co-expression data across Homo Sapiens and other species for the product of SYNPO2L and the products of the 10 genes with the highest confidence in interaction prediction. (E) Legend of color codes used in this figures (A–D).
Using in silico analyses we investigated the potential deleteriousness of the rs766868752 variant in the SYNPO2L gene. The variant was found to have a CADD score of 35, indicating that it is among the 0.1% most deleterious variants in the human genome.
In a previously performed meta-analysis of GWAS, 134 loci associated with AF were identified in the human genome (4). However, these are all common variants with small effect sizes. In this study we report on the first loss-of-function variant in the FinnGen study significantly associated with atrial fibrillation. This rare variant has a substantial effect size and is found in the SYNPO2L gene located on chromosome 10q22. This region has previously been associated with AF as the intronic SNP rs60212594 in SYNPO2L has been identified in our GWAS meta-analysis. This common variant was found with an associated OR of 1.12 (p = 6,48 × 10−27). Interestingly, a study by Brugada et al. (16) from 1997 identified a family of 26 with AF being inherited in an autosomal dominant pattern. The responsible genetic locus was localized to chromosome 10q22-10q24. Combined with GWAS, this strongly implicate genes in chromosome 10q22 as candidate genes with a potential large significant effect on the risk of AF.
Different studies have suggested both MYOZ1 (17, 18) and SYNPO2L (19, 20) both located on chromosome 10q22, as the likely causal genes driving the association. Studies suggesting MYOZ1 as the causal gene, have done so primarily based on eQTL studies showing an altered expression of the MYOZ1 gene in carriers of AF risk alleles identified through GWAS studies. However, expression of the MYOZ1 gene has been shown to be primarily restricted to skeletal muscle, with very low MYOZ1 mRNA levels in cardiac muscle tissue (21, 22). Additionally, no variants in the MYOZ1 gene were found to be significantly enriched in the FinnGen study. These findings suggest MYOZ1 as an unlikely candidate gene in the locus. Conversely, the findings of this study showing a predicted loss-of-function variant in the SYNPO2L gene with an associated OR of 3.5, strongly suggests SYNPO2L as the likely causal gene in the locus. This is supported by the colocalization of the AF GWAS signals with the eQTLs of SYNPO2L as seen in Figure 2.
The functions of the SYNPO2L gene product were first studied by Beqqali et al. (23), naming the protein product Cytoskeletal Heart-enriched Actin-associated Protein (CHAP). Beqqali et al. identified two different isoforms both expressed in cardiac tissue. The long isoform was named CHAPa (=978 aa) coded by the ENST00000394810.2 transcript, and the short isoform was named CHAPb (=749 aa) coded by the ENST00000372873.4 transcript. The authors furthermore found that the long isoform CHAPa was predominately expressed in adult cardiac tissue, whereas the short isoform, CHAPb, was primarily expressed in embryonic cardiac tissue. Expression of SYNPO2L in various tissues can be seen in Supplementary Figure 1 (24). The splice-site variant identified in the FinnGen database is only present in the long isoform coding the CHAPa. Using in situ hybrdization in mice, Beqqali et al. determined the subcellular localization of the proteins to the Z-disc of both skeletal and cardiac muscle tissue by co-localization with a-actinin (23). Both the CHAPa and CHAPb isoforms contain a nuclear localization signal (NLS) which was found to be highly conserved in both the mouse and zebrafish orthologs. Accordingly, CHAPb, the embryonic form, was also localized to the nucleus in embryonic mice cardiac tissue.
To further investigate the function of the SYNPO2L product, Beqqali et al. knocked down the CHAPa and CHAPb isoforms of the SYNPO2L gene product in a zebrafish model using antisense morpholino oligonucleotides (23). The zebrafish kncokdown-models showed a phenotype with highly disorganized sarcomeres and Z-disks, as well as decreased cardiac contractility. α-actinin-2, which was shown to interact with the CHAP proteins, was also displaced resulting in disorganized myofibrils. This suggests that the SYNPO2L gene product is highly involved in the structural development and function of the sarcomeres. Due to its nuclear and Z-disc localization, this role could be carried out by the protein acting as both a structural component and also being involved in cellular signaling processes. This is in line with previous findings suggesting other Z-disc proteins to be associated with a-actinin. One example is MLP, which can translocate from the cytoplasm to the nucleus and interact with varius transcription factors (25).
The consequences of splice site mutations on the protein product depend on multiple different factors. These are complex to predict, but can include exon skipping, intron inclusion or activation of different cryptic splice sites (26). If the splice site mutation in intron 1 of SYNPO2L leads to a change in the remaining reading frame, the mutation is likely to lead to a pre-mature stop codon followed by non-sense mediated decay (NMD) (27). Therefore, the splice site mutation could dispose to AF either by a lack of protein product or by an altered dysfunctional protein product. The pathogenicity of the splice variant identified in this study is underlined by in sillico analysis, predicting the variant to be among the 0.1% variants in the genome most likely to be deleterious. Future analysis of mRNA extracted from cell lines or patients with the splice site variant could help determine how the mutation affects splicing and protein product. Carriers of the common AF risk variant rs60212594 show a decreased expression of SYNPO2L, as shown in the eQTL analysis (Figure 3). Expression of SYNPO2L in carriers of this common variant was examined as the variant is associated with an increased risk of AF in our GWAS meta-analysis (4). The decreased expression is consistent with the present finding that the rs766868752 splice site variant, possibly leading to mRNA NMD, was strongly associated with AF. Both findings suggest that a decreased amount of SYNPO2L gene product pre-disposes to AF.
Genetic variants in other structural cardiac proteins e.g., titin, have previously been shown to be strongly associated with AF (28). TTN encodes a giant sarcome protein (titin) expressed in all chambers of the human heart. Ahlberg et al. (28) found sarcomere defects in mutant zebrafish carrying a truncated variant in ttn, the homologs TTN gene in zebrafish. This was also the case in the SYNPO2L knockdown zebrafish model studied by Beqqali et al. (23). Furthermore, a prolonged PR interval as well as an increased amount of fibrosis was shown in zebra fish models carrying the truncated titin variant (28). Another interesting gene, MYL4, is responsible for contractile, electrical and structural integrity of the atrium. Peng et al. found a loss-of function variant to cause early atrial fibrosis leading to atrial arrhytmia and atrial cardiomyopathy (29). In line with this, a mouse model with an overexpression of the SYNPO2L gene studied by Van Eldik et al. (30), also displayed both increased interstitial fibrosis and electrical abnormalities. Cardiac fibrosis has been shown to be increased in AF patients (31). The findings of increased cardiac fibrosis and atrial cardiomyopathy, resulting from abnormal sarcomere organization, propose a possible mechanism of how variants in the SYNPO2L gene and other structural proteins of the cardiomyocyte pre-dispose to AF.
However, this study also has several limitations. While the association seems biologically plausible as the genetic locus is associated with AF and the variant has been predicted to be deleterious, the splice variant, rs766868752, is found with a very low allele frequency and is almost exclusively found in persons of Finnish heritage. Furthermore, the allele frequency of the variant has been imputed. Although the variant was imputed with a very high confidence in imputation, one must consider the possible risk of inaccuracies in imputation. Finally, the common variant analyzed in this study, rs60212594, while significantly associated with AF, has a quite modest effect on disease risk, and may not be of major clinical significance for individuals carrying this variant. We therefore urge our readers to interpret our results with caution.
For many years, AF has been considered an electrical disease, but our understanding of the pathophysiology of AF has improved and the field continues to grow as we explore new associations in AF genetics. Our findings suggest that mutations in the SYNPO2L gene encoding the structural protein CHAP pre-dispose to AF. This is primarily based on the loss-of-function splice site variant in the SYNPO2L gene that was associated with a significantly increased risk of AF in a Finnish population (p = 9.9 × 10−8). The loss-of-function variant in the SYNPO2L gene is too rare to be of any major clinical significance alone. However, the findings that mutations in another gene involved in the structural arrangement of the sarcomeres pre-dispose to AF, supports the theory that AF is not only an electrophysiological disease. Future studies are needed to investigate if SYNPO2L loss-of-function variants in general pre-dispose for AF with a similar effect size. The discovery that atrial fibrosis plays a significant role in AF might explain why existing treatment options of AF are sometimes inadequate and may help identify new directions of AF treatments and risk stratifications in the future. It also underlines the complexity of the disease and suggests that different genes in different geographic regions and populations may pre-dispose for the disease through diverse pathophysiological mechanisms.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.finngen.fi/en/access_results.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
AC and MO: conceived the idea. AC, MO, and OV: performed the analyses. AC: prepared the original draft. AC, MO, OV, and JA: helped write, edit and review the manuscript for key intellectual content. All authors have read and agreed to the published version of the manuscript.
This work was supported by The John and Birthe Meyer Foundation, The Research Foundation at Rigshospitalet, Villadsen Family Foundation, The Arvid Nilsson Foundation, and The Hallas-Møller Emerging Investigator Novo Nordisk (NNF17OC0031204). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We want to acknowledge the participants and investigators of FinnGen study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.650667/full#supplementary-material
1. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, et al. Worldwide epidemiology of atrial fibrillation. Circulation. (2014) 129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119
2. Ruddox V, Sandven I, Munkhaugen J, Skattebu J, Edvardsen T, Otterstad JE. Atrial fibrillation and the risk for myocardial infarction, all-cause mortality and heart failure: a systematic review and meta-analysis. Eur J Prev Cardiol. (2017) 24:1555–66. doi: 10.1177/2047487317715769
3. Fox CS, Parise H, D'Agostino Ralph BS, Lloyd-Jones DM, Vasan RS, Wang TJ, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. (2004) 291:2851–5. doi: 10.1001/jama.291.23.2851
4. Roselli C, Chaffin MD, Weng L-C, Aeschbacher S, Ahlberg G, Albert CM, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. (2018) 50:1225–33. doi: 10.1038/s41588-018-0133-9
5. Gutierrez A, Chung MK. Genomics of atrial fibrillation. Curr Cardiol Rep. (2016) 18:6. doi: 10.1007/s11886-016-0735-8
6. FinnGen. FinnGen Documentation of R3 Release. (2020). Available online at: https://finngen.gitbook.io/documentation/ (accessed November 22, 2020).
7. Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. (2018) 50:1335–41. doi: 10.1038/s41588-018-0184-y
8. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype-tissue expression (GTEx) project. Nat Genet. (2013) 45:580–5. doi: 10.1038/ng.2653
9. Panjwani N, Wang F, Mastromatteo S, Bao A, Wang C, He G, et al. LocusFocus: web-based colocalization for the annotation and functional follow-up of GWAS. PLoS Comput Biol. (2020) 16:e1008336. doi: 10.1371/journal.pcbi.1008336
10. Gong J, Wang F, Xiao B, Panjwani N, Lin F, Keenan K, et al. Genetic association and transcriptome integration identify contributing genes and tissues at cystic fibrosis modifier loci. PLoS Genet. (2019) 15:e1008007. doi: 10.1371/journal.pgen.1008007
11. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. (2019) 47:D607–13. doi: 10.1093/nar/gky1131
12. Andersen JH, Andreasen L, Olesen MS. Atrial fibrillation—a complex polygenetic disease. Eur J Hum Genet. (2020). doi: 10.1038/s41431-020-00784-8. [Epub ahead of print].
13. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–94. doi: 10.1093/nar/gky1016
14. GnomAD. GnomAD browser v3.1. Available online at: https://gnomad.broadinstitute.org/gene/ENSG00000166317?dataset=gnomad_r2_1 (accessed November 7, 2020).
15. GTEx Project Single Tissue eQTL Browser. Available online at: https://gtexportal.org/home/snp/rs60212594 (accessed November 27, 2020).
16. Brugada R, Tapscott T. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. (1997) 336:905–11.
17. Lin H, Dolmatova EV, Morley MP, Lunetta KL, McManus DD, Magnani JW, et al. Gene expression and genetic variation in human atria. Hear Rhythm. (2014) 11:266–71. doi: 10.1016/j.hrthm.2013.10.051
18. Martin RIR, Babaei MS, Choy MK, Owens WA, Chico TJA, Keenan D, et al. Genetic variants associated with risk of atrial fibrillation regulate expression of PITX2, CAV1, MYOZ1, C9orf3 and FANCC. J Mol Cell Cardiol. (2015) 85:207–14. doi: 10.1016/j.yjmcc.2015.06.005
19. Nielsen JB, Fritsche LG, Zhou W, Teslovich TM, Holmen OL, Gustafsson S, et al. Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am J Hum Genet. (2018) 102:103–15. doi: 10.1016/j.ajhg.2017.12.003
20. Lubitz SA, Brody JA, Bihlmeyer NA, Roselli C, Weng LC, Christophersen IE, et al. Whole exome sequencing in atrial fibrillation. PLoS Genet. (2016) 12:1–12. doi: 10.1371/journal.pgen.1006284
21. Gontier Y, Taivainen A, Fontao L, Sonnenberg A, van der Flier A, Carpen O, et al. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J Cell Sci. (2005) 118:3739–49. doi: 10.1242/jcs.02484
22. Posch MG, Perrot A, Dietz R, Ozcelik C, Pankuweit S, Ruppert V, et al. Mutations in MYOZ1 as well as MYOZ2 encoding the calsarcins are not associated with idiopathic and familial dilated cardiomyopathy. Mol Genet Metab. (2007) 91:207–8. doi: 10.1016/j.ymgme.2007.02.014
23. Beqqali A, Monshouwer-Kloots J, Monteiro R, Welling M, Bakkers J, Ehler E, et al. CHAP is a newly identified Z-disc protein essential for heart and skeletal muscle function. J Cell Sci. (2010) 123:1141–50. doi: 10.1242/jcs.063859
24. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. (2015) 347:1260419. doi: 10.1126/science.1260419
25. Ecarnot-Laubriet A, De Luca K, Vandroux D, Moisant M, Bernard C, Assem M, et al. Downregulation and nuclear relocation of MLP during the progression of right ventricular hypertrophy induced by chronic pressure overload. J Mol Cell Cardiol. (2000) 32:2385–95. doi: 10.1006/jmcc.2000.1269
26. Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. (2018) 59:253–68. doi: 10.1007/s13353-018-0444-7
27. Baker KE, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. (2004) 16:293–9. doi: 10.1016/j.ceb.2004.03.003
28. Ahlberg G, Refsgaard L, Lundegaard PR, Andreasen L, Ranthe MF, Linscheid N, et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun. (2018) 9:4316. doi: 10.1038/s41467-018-06618-y
29. Peng W, Li M, Li H, Tang K, Zhuang J, Zhang J, et al. Dysfunction of myosin light-chain 4 (MYL4) leads to heritable atrial cardiomyopathy with electrical, contractile, and structural components: evidence from genetically-engineered rats. J Am Heart Assoc. (2017) 6:e007030. doi: 10.1161/JAHA.117.007030
30. van Eldik W, den Adel B, Monshouwer-Kloots J, Salvatori D, Maas S, van der Made I, et al. Z-disc protein CHAPb induces cardiomyopathy and contractile dysfunction in the postnatal heart. PLoS ONE. (2017) 12:e0189139. doi: 10.1371/journal.pone.0189139
Keywords: atrial fibrillation, genetics, arrhythmia, splice site variant, cardiomyopathy, cardiology
Citation: Clausen AG, Vad OB, Andersen JH and Olesen MS (2021) Loss-of-Function Variants in the SYNPO2L Gene Are Associated With Atrial Fibrillation. Front. Cardiovasc. Med. 8:650667. doi: 10.3389/fcvm.2021.650667
Received: 07 January 2021; Accepted: 18 February 2021;
Published: 09 March 2021.
Edited by:
Seitaro Nomura, The University of Tokyo, JapanReviewed by:
Tom Robert Webb, University of Leicester, United KingdomCopyright © 2021 Clausen, Vad, Andersen and Olesen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Morten Salling Olesen, bW9ydGVuLnNhbGxpbmcub2xlc2VuQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.