
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 12 April 2021
Sec. Cardiovascular Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.649512
This article is part of the Research Topic Oxidative Stress in Cardiovascular Diseases and Pulmonary Hypertension View all 8 articles
 Srikanth Karnati1*†
Srikanth Karnati1*† Michael Seimetz2†Florian Kleefeldt1
Michael Seimetz2†Florian Kleefeldt1 Avinash Sonawane3Thati Madhusudhan4Akash Bachhuka5
Avinash Sonawane3Thati Madhusudhan4Akash Bachhuka5 Djuro Kosanovic2,6
Djuro Kosanovic2,6 Norbert Weissmann2
Norbert Weissmann2 Karsten Krüger7
Karsten Krüger7 Süleyman Ergün1
Süleyman Ergün1Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality worldwide and encompasses chronic bronchitis and emphysema. It has been shown that vascular wall remodeling and pulmonary hypertension (PH) can occur not only in patients with COPD but also in smokers with normal lung function, suggesting a causal role for vascular alterations in the development of emphysema. Mechanistically, abnormalities in the vasculature, such as inflammation, endothelial dysfunction, imbalances in cellular apoptosis/proliferation, and increased oxidative/nitrosative stress promote development of PH, cor pulmonale, and most probably pulmonary emphysema. Hypoxemia in the pulmonary chamber modulates the activation of key transcription factors and signaling cascades, which propagates inflammation and infiltration of neutrophils, resulting in vascular remodeling. Endothelial progenitor cells have angiogenesis capabilities, resulting in transdifferentiation of the smooth muscle cells via aberrant activation of several cytokines, growth factors, and chemokines. The vascular endothelium influences the balance between vaso-constriction and -dilation in the heart. Targeting key players affecting the vasculature might help in the development of new treatment strategies for both PH and COPD. The present review aims to summarize current knowledge about vascular alterations and production of reactive oxygen species in COPD. The present review emphasizes on the importance of the vasculature for the usually parenchyma-focused view of the pathobiology of COPD.
Respiratory diseases are a major cause of morbidity and mortality worldwide. COPD is caused by a persistent obstruction of the airflow in the lungs, which has profound effects on cardiac function and gas exchange, with systemic consequences. The condition arises either because of emphysema, where the pulmonary air sacs are damaged, or because of chronic bronchitis, which is characterized by continuous airway inflammation (1). According to the 2017 World Health Organization (WHO) Global Burden of Disease Study, 3.17 million people died because of COPD in the year 2015, and 251 million individuals were reported to have COPD in 2016. Alarmingly, COPD will be the third-leading cause of mortality worldwide by 2030 (2). COPD is most prevalent in low- and middle-income countries. In developing countries, exposure to biomass smoke, especially during cooking, exposure to harmful smokes during work and underlying diseased conditions (such as tuberculosis) acts as trigger during COPD infections (3). Individuals who have high levels of exposure to tobacco, dust, harmful chemicals, and fumes from burning fuel, as well as individuals with alpha-1-antitrypsin deficiency, are more prone to developing COPD (4). In Western countries, long-term tobacco smoking is the main reason for the development of COPD.
The systemic consequences of COPD can initiate various comorbid diseases, such as ischemic heart disease, heart failure, osteoporosis, normocytic anemia, lung cancer, depression, and diabetes (3). On a cellular and molecular level, these changes are initiated by important upstream events, encompassing the influx of leukocytes, an imbalance of proteases/antiproteases, and increased production of reactive oxygen species (ROS) (4–6). COPD is a multifactorial disease; however, the most studied fundamental mediators are oxidative stress, inflammation, and a lack of physical activity. An important COPD-associated pathophysiology is the spillover of pulmonary inflammation into the systemic circulation (7, 8). Inflammation gives rise to neutrophil extravasation (markers include elastase and calprotectin) and production of inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β), interferon α/γ (IFN-α/γ), interleukin 6 (IL-6), interleukin 8 (IL-8), reactive proteins, and leukotrienes. The constant recruitment of inflammatory immune cells encourages neutrophil infiltration into the lungs, thereby activating the release of proteases and free radicals and resulting in decreased lung elasticity (9). Thus, COPD in the lungs is accompanied by destruction of the elastic architecture of the lung parenchyma, leading to the enlargement of distal airspaces (10). In addition to being an airway and systemic inflammatory disease, COPD also appears to be a vascular disease. It is assumed that cigarette smoke (CS) is vasoactive and directly affects the pulmonary vasculature. Consequently, dysfunction of the blood vessels promotes vascular remodeling, pulmonary hypertension (PH), and finally cor pulmonale (11–13).
In patients with COPD who smoke, oxidative stress is elevated due to chronic exposure to CS and other toxic air pollutants (9). Epidemiological studies that have explored tobacco smoke exposure in patients with PH showed that ~49% of these patients were smokers, of whom 71% were male. In females, PH was often caused by second-hand (passive) exposure to tobacco smoke (14). Lungs are a common site for oxidative stress due to their oxygen-rich microenvironment and frequent exposure to environmental toxins and pathogens. Chronic cigarette smoking results in the progression of COPD due to excessive endogenous ROS production, both from dysfunction of mitochondrial complexes I and III and persistent activation of inflammatory cytokines. In addition, ROS-generating enzymes, such as NADPH oxidases, xanthine oxidases, and heme peroxidases, promote the infiltration of inflammatory cells inside the airways (15). Another major pathophysiological characteristic of COPD is an imbalance in protease/antiprotease levels. There are three classes of proteases linked with COPD pathology: serine proteases, matrix-metalloproteinases (MMPs), and cysteine proteases. Serine proteases are mucus stimulators, which exacerbate airflow obstruction. MMPs degrade protein components of the extracellular matrix (ECM), leading to tissue damage and increased macrophage infiltration, while cysteine proteases, which include caspases, stimulate apoptosis in alveolar epithelial cells (16). Thus, the imbalance is due to excessive neutrophil accumulation, which triggers pulmonary dysfunction.
The pathophysiological interlink between vascular disease and COPD embraces conditions such as PH, hypoxia, systemic inflammation, and oxidative stress (17). The severity of disease in PH-associated COPD significantly increases in individuals who also have pulmonary fibrosis or emphysema, with the survival rate decreasing by up to 50% (18, 19). PH in COPD is characterized by a mean pulmonary artery pressure (mPAP) between 21 and 24 mmHg in the presence of pulmonary vascular resistance (PVR≥3 Wood Units) or an elevation of mPAP 25–34 mmHg, with nearly normal cardiac output (20, 21). An increase in mPAP≥ 35 mmHg or a mPAP≥ 25 mmHg with a low cardiac index (<2.0 L·min−1·m−2) is considered severe PH in COPD. An mPAP of more than 40 mmHg is frequently observed in patients with severe COPD. Since the presence of PH clearly increases mortality, the occurrence of PH in patients with COPD is of important prognostic relevance (18, 19, 22). The exact prevalence of PH in patients with mild or moderate COPD has not been accurately determined. However, the mortality rate is ~30% in cases of heart disease-related COPD (23). Some other published reports have suggested that the occurrence of PH with mild, moderate, and severe cases of COPD is 16–44, 43–56, and 59–84%, respectively (24–26). The incidence of severe PH in patients with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage IV was reported to be 3–5% (mPAP>35 to 40 mmHg) (21).
PH is suggested to be the result of hypoxia associated with COPD (27). It has been shown repeatedly that vascular alterations often appear before alveolar destruction is detectable (28–31). A decrease in alveolar oxygen tension in COPD results in constriction of the pulmonary arteries, leading to hypoxia in the body. Elevated alveolar hypoxia is not restricted to smokers with COPD, however, there have been few reports demonstrated that smokers who had not diagnosed COPD also exhibited conditions of cor pulmonale. A study performed using the C57BL/6 mouse model showed that tobacco smoke induced emphysema, promoted remodeling of pulmonary vasculature, increased airspaces (which included changes in parameters such as the surface area and volume of the alveolar walls/septa), and decreased the number of alveoli; this was accompanied by alterations in lung compliance, tidal volume, and airway resistance (29). Drugs such as tadalafil (a phosphodiesterase type 5 inhibitor) and piclamilast (a phosphodiesterase type 4 inhibitor) have been reported to prevent CS-induced emphysema in a mouse model by improving pulmonary performance, lung tidal volume, pulmonary vascular remodeling, systolic pressure, and hypertrophy of the right ventricle (27). The present review summarizes the state of current knowledge about vascular alterations that occur in COPD.
PH in COPD is slowly progressive, and mPAP can often remain stable over a period of 3–12 years (32–34). It has been shown that the average change in mPAP can be just+0.5 mmHg/year, independent of the presence of initial PH (defined by mPAP>20 mmHg) (34). Another study that investigated the pathobiology of PH in COPD over time (initial mPAP <20 mmHg) demonstrated that only 33/121 patients developed PH after 6.8 ± 2.9 years (35). However, ~30% of patients with severe COPD exhibited a remarkable worsening of mPAP during follow-up. According to WHO, COPD patients with PH are categorized as Group 3 PH. Group 3 PH patients have a significantly lower survival rate in comparison with PH patients without COPD (Group 1), followed by Group 4 (patients with chronic thromboembolic PH) and Group 5 (patients with hematologic disorders, systemic disorders, and metabolic disorders) (36). A population-based study reported that a higher proportion of older males (aged more than 70 years) are prone to PH with COPD. This population also suffered from co-morbidities such as diabetes, hypertension, coronary artery disease, and atrial fibrillation. In addition, patients with Group 3 PH showed increased left ventricular mass and end-diastolic diameter. The prevalence of PH in patients with COPD depends on the definition of PH, the severity of COPD, and the mPAP, which ranges from 20 to 91% (37). Most patients (90%) with PH have mPAP>20 mmHg, with the majority varying between 20 and 35 mmHg. These patients were characterized by a progressive worsening of partial oxygen/carbon dioxide pressure (PaO2/PaCO2) over time. In addition, there was an association between alterations in PaO2 and mPAP (33, 34). Pulmonary anatomic changes can result in respiratory failure, both type I, where PaCO2 <45 mmHg (6 kPa) i.e., normal or low and the partial pressure of oxygen, PaO2, is low (hypoxemia); and type II, where PaCO2>45 mmHg (6kPa) and PaO2 <60 mmHg (8kPa) (38, 39). Within 5 years of diagnosis, 7% of patients with COPD will experience hypoxemia. The pathology of inpatients with right heart failure (RHF) is commonly preceded by PH. The severity of PH and the development of RHF are closely associated. PH increases the workload of the right ventricle, leading to hypertrophy, dilatation, and ventricular dysfunction. RHF is frequently accompanied by peripheral edema and can be observed in patients with advanced COPD (40, 41). Peripheral edema is considered to reflect RHF, but the possible occurrence of RHF is sometimes assumed to simply indicate the presence of secondary hyperaldosteronism induced by functional renal insufficiency (42).
The effect of pressure overload in the development of RHF has been intensively discussed, probably due to additional causes independent of PH. In patients with stable COPD, right ventricular contractility, measured by the end-systolic pressure–volume relationship, is not abnormal in COPD patients suffering from PH. Notably, many patients with advanced COPD never develop RHF. The level of mPAP is suggested to be a valuable prognostic indicator for patients with COPD (39, 43). Accordingly, life expectancy is less in patients suffering with PH compared with patients that do not have PH (18, 44, 45). The 5-year survival rate of COPD patients with PH (mPAP>20 mmHg) is about 50%. In PH, there is an increased mean pulmonary arterial blood pressure (46) that causes an increased afterload for the right ventricle (RV) of the heart leading to right heart hypertrophy. This adaptive hypertrophy helps the heart to deal with the high pulmonary vascular resistance (PVR). However, this beneficial adaptive hypertrophy can result in maladaptation, RV dilatation and finally failure. The term “cor pulmonale” was used to define a right ventricular dilation due to COPD. However, present studies describing patients with mild-to-moderate COPD demonstrated reduced RV volumes compared with healthy controls (47). This discrepancy can be explained by the fact that majority of the patients with severe COPD primarily suffer from increased intrathoracic pressures due to hyperinflation and airway obstruction, but not from right heart failure. Indeed, increased intrathoracic pressures reduce deoxygenated blood returning into the thorax, thereby reducing the cardiac chambers volumes. To support this notion, the recent CLAIM study by Hohlfeld et al. showed that the reduced volume can be reversed by means of combined long-acting bronchodilators, causing deflation of the lung and increased end-diastolic filling of both the right and left ventricle and a significant increase in stroke volume (48). Moreover, up to 30% of COPD patients suffer from systolic or diastolic heart failure thereby enhancing in both pulmonary arterial wedge pressure and mPAP due to lung hyperinflation (49). Further, COPD patients do not show an increased hypertrophy of the left ventricle, but dysfunction. There is evidence that the systemic inflammation occurring in these patients might have a causal role in the pathogenesis of atherosclerosis (50). The high prevalence of left ventricular systolic dysfunction in individuals with COPD can be explained by the acceleration of the progression of coronary atherosclerosis by systemic inflammation, which leads to the development of ischemic heart disease. The high incidence of motor disorders of the left ventricle wall observed in patients with COPD and left ventricular dysfunction could also justify the relationship between both chronic processes.
Further, another explanation may be the existence of predominant COPD subphenotypes. Interestingly, Burrows et al. found that COPD patients with emphysema were less likely to demonstrate RV hypertrophy than other COPD patients, under the same pulmonary vascular resistance (51). Moreover, patients dying with emphysema did not exhibit RV hypertrophy, which was more common in COPD patients with chronic bronchitis (52). Further, Kawut et al. reported that cardiac complications are linked to more prominent airways disease and less parenchymal destruction, supporting a stronger link between the “chronic bronchitis” subphenotype than the “emphysema” subphenotype (53). The mechanism of reduced RV filling in emphysematous COPD may relate to several factors. As Watz et al. suggested that pulmonary hyperinflation reduces right atrial and RV filling in moderate-severe COPD (54), and lung volume reduction surgery for very severe COPD (which decreases hyperinflation) is associated with increased oxygen pulse (55). Long-term oxygen therapy (LTOT) can significantly improve the survival of hypoxemic COPD patients who also suffer from PH. Accordingly, the prognosis for PH will improve with LTOT therapy. In fact, LTOT is the only recommended therapeutic intervention to increase the survival rate of COPD patients with chronic hypoxemia. Approximately 20% of patients with COPD are prescribed LTOT (56). LTOT (13 h/per day) was prescribed for patients with COPD during the very early phase of the condition. This therapy was observed to be successful in patients with resting hypoxemia, however in nocturnal or exercise-induced hypoxemia LTOT exhibited no significant relief (56–58). Cor pulmonale also contributes to the mortality associated with COPD. Treatment in such cases involves LTOT administration for more than 16 h per day or the use of vasodilator drugs (59).
Vascular alterations have been shown to play an important role in the development of emphysema in both animal models and in patients with COPD. In patients with end-stage COPD, such remodeling is characterized by thickened walls or vascular occlusion, reducing the vascular lumen resulting in increased resistance and intravascular pressure. In PH-associated COPD, pulmonary ventricles and arteries undergo structural modifications (60). The most prominent feature of the vascular remodeling of blood vessels is the varying degree of thickening of the intimal and/or medial layer of muscular vessel layers in distal pre-capillary arterioles (distal muscularization) (61). Although smooth muscle cells (SMCs) are not resident intimal cells, studies in animal models have shown that SMCs can migrate from the media and proliferate in the intima following endothelial injury (61). It has become evident that intimal hyperplasia can be detected during early-stage COPD, resulting from the proliferation and migration of SMCs and associated with elastic and collagen fiber deposition (62). Immunohistochemistry analysis of ECM proteins from lung specimens of patients with COPD has shown that abundant elastin can be detected during the early stages of COPD and that the abundance of collagen is correlated with the degree of intimal thickness, suggesting that collagen deposition has important consequences for pulmonary vascular remodeling associated with COPD (60, 63–65). Sekhon et al. showed that, in rats, CS triggered proliferation of polymorphonuclear leucocytes (66). The effect of muscularization is prominent in small arteries (diameter <500 μm) (67–69). Immunohistochemistry, using SMC markers, of lung specimens from patients with PH-associated COPD showed positive expression of vimentin (indicating the expression of mesenchymal cells) and negative staining for desmin (an intermediate filament protein characteristic of cells of myogenic origin), indicating that less-differentiated SMCs contribute to an ongoing process of vascular remodeling (13, 67). Although the detailed molecular processes have not been identified, the occurrence of SMCs might be explained by the infiltration and differentiation of circulating bone marrow-derived progenitor cells, differentiation from resident precursor cells, the dedifferentiation of mature SMCs from the media that migrate to the intima (70), or transdifferentiation of endothelial cells to SMCs by endothelial-to-mesenchymal-transition. In this regard, bone marrow-derived progenitor cells are suggested to contribute on the one hand to vascular repair via differentiation into endothelial cells and on the other to vessel remodeling through differentiation into SMCs (71–73). Chronic hypoxia at high altitudes can also cause PH, but this condition is reversible upon returning to sea level. Together, these findings provide evidence of the primary reason for medial hypertrophy. By contrast, the remodeling of all vessel layers cannot be reversed by supplemental oxygen, either in cases of acute (74) or chronic COPD (75).
The muscularization of vessels in the pulmonary region is a response to oxidative stress and endothelial cell (EC) injury. The integrity of ECs is lost due to apoptosis, following the accumulation of fluids and immune cells in the perivascular region. As a result of the immune response, bone marrow-derived precursor cells are recruited to the site, leading to the transition from ECs to mesenchymal cells (76, 77). Internal hypoxia has been proposed to be the primary mechanism underlying PH-associated COPD. Hypoxemia promotes vascular constriction via recruitment of immune cells and results in muscularization of arterioles. This remodeling affects the intima, media, and adventitia of vessels in the lung. Various studies have shown that pulmonary vascular remodeling and endothelial dysfunction occurs in animal models of lung emphysema (78), in patients with mild COPD not suffering from hypoxemia, and in smokers with normal lung function (72, 79). Presumably, oxygen therapy is unable to reverse PH in many patients with COPD. Nevertheless, many studies have shown that hypoxia plays a role in COPD, at least in severe forms of the disease.
PH occurs because of increased pulmonary vascular resistance (PVR) during chronic respiratory diseases. Multiple factors contribute to the increase in PVR (41, 80), but hypoxia during COPD is thought to be the major cause (41, 81, 82). Acute hypoxia causes pulmonary vasoconstriction, while chronic hypoxia induces structural vascular changes (remodeling) over time. During acute hypoxia, increases in PVR and mPAP are features of hypoxic pulmonary vasoconstriction. Chronic alveolar hypoxia causes morphological changes in the pulmonary vascular bed (remodeling) that are comparable to those seen in COPD patients with PH (including muscularization of pulmonary arterioles and thickening of the intima in muscular pulmonary arteries and arterioles).
Generalized hypoxia due to partial pressure of oxygen has both systemic and organ-specific effects (83–85). This type of hypoxia induces pulmonary vasoconstriction, peripheral vasodilation, and activation of a sympathetic-adrenergic stress response to increase cardiac output (86), while erythropoietin-stimulated red cell production is activated in the bone marrow. Hypoxic pulmonary vasoconstriction here represents an adaptive response in local blood perfusion to the alveolar ventilation situation, to prevent hypoxemia.
Hypoxia-dependent vasoconstriction is mediated by hypoxia-inducible transcription factor 1 alpha (HIF-1α), which promotes the activation of innate immune responses and inflammation in arterioles. Other transcription factors, such as forkhead box O (FoxO), CBF1/RBP-Jκ (recombination signal-binding protein for immunoglobulin kappa J region), peroxisome proliferator-activated receptor gamma (PPAR-γ), Krüppel-like factor 4 (KLF4), transcriptional coactivator pyruvate kinase isozyme PKM2, the corepressor CtBP1 [a member of the C-terminal binding protein (CtBP) family], and the Twist family bHLH transcription factor 1 (TWIST1) (87), were found to play a crucial role in PH and dysfunction of the right ventricle. However, of these transcription factors, HIF is most strongly implicated in PH pathogenesis, as it was observed that conditional deletions of HIF isoforms in mice improved vascular remodeling and augmented pulmonary arterial pressure post-chronic hypoxia (88). In alveolar macrophages, hypoxemia induces the expression of FIZZ1, also known as hypoxia-induced mitogenic factor (HIMF), thereby promoting smooth muscle contraction in pulmonary arterioles via interleukin-linked kinase 4 signaling mechanisms (76, 89). Patients with COPD who were regular smokers exhibited higher expression of HIF-1α, VEGF (a potent regulator of vascular permeability), and VEGF receptors (90). This in turn can activate the proinflammatory transcription factor nuclear factor-kappaB (NF-κB) (91, 92).
NF-κB modulates the expression of cytokine expression and thereby manipulates proliferative homeostasis in immune cells. Mechanistic insights into the hypoxia-induced expression of HIF and NF-κB suggest that both factors are activated in an IKK–transforming growth factor β-activated kinase 1 (TAK1)-dependent manner (93, 94). The hypoxic microenvironment inside the lungs also releases chemotactic factors, such as leukotriene B4 (LTB4), VEGF, and FIZZ1, which tend to increase hypoxia in the bone marrow as there is active mobilization of bone marrow-derived cells (such as mast cells, mesenchymal precursor cells megakaryocytes, and dendritic cells) to the lungs. Hypoxia-dependent transcription of NF-κB leads to transcription of phospholipase A2 (PLA2). Transcriptional activation of PLA2 results in activation of 5-lipoxygenase and chemotactic factor LTB4, eventually leading to the deposition of bone marrow-derived precursor cells (85).
IL-6 has received considerable interest as a mediator of COPD progression. IL-6 levels in serum from patients with COPD were found to be significantly elevated in comparison with healthy individuals (95). During hypoxia, IL-6-deficient mice showed less inflammation and a marginal reduction in pulmonary hypertension (96). IL-6 contributes to the increased migration of pulmonary artery smooth muscle cells (PASMCs) in chronic hypoxia-exposed vessels of the lung that are non-muscularized (96). IL-6 is upregulated following chronic hypoxia in mouse lungs, but it does not seem to be essential for the development of chronic hypoxia-induced PH. Chronic hypoxia with excessive IL-6 seems to change the mode of vascular remodeling toward angioproliferation (85). It has been reported that overexpression of IL-6 results in a significant decrease in the expression of lung protective protein bone morphogenetic protein receptor type 2 (BMRP2), through a signal transducer and activator of transcription 3 (STAT3)-microRNA cluster 17/92 pathway. The decrease in BMRP2 is driven by the modulation of the STAT3 pathway. BMPR2 is a member of the transforming growth factor-β (TGF-β) superfamily of growth factor receptors, which is involved in signaling pathways including protein kinase B/phosphatidylinosital 3-kinase (Akt/PI3K), phosphorylation of extracellular-signal regulated kinase (pERK), phosphorylation of c-Jun N-terminal kinase (JNK), phosphorylation of Smad1, and phosphorylation of phospho-mitogen activated protein kinase (p-p38MAPK) (97–99). Conversely, the effects of IL-6 overexpression have been found to be insufficient to cause pulmonary occlusions and aid the mobilization of bone marrow cells to vessels in the lung (85). It has been shown that mutations in the BMPR2 gene are linked to the development of PH (100) and that BMPR2 expression is reduced in the pulmonary vasculature in patients with PH (101). On the other hand, it is assumed that IL-6 affects the balance between apoptosis and proliferation of PASMCs and pulmonary artery endothelial cells (PAECs), leading to vascular remodeling (102). Therefore, overexpression of IL-6 induces the angioproliferative growth factor VEGF and intracellular ERK, resulting in increased proliferation. In parallel, IL-6 expression is followed by a downregulation of TGF-β and proapoptotic MAP kinases (JNK1, p38MAPK) (103) and upregulation of B-cell lymphoma 2 (Bcl2), an inhibitor of apoptosis. Accordingly, IL-6 can trigger vascular remodeling by inducing signaling pathways that lead to increased proliferation and decreased apoptosis of PASMCs and PAECs (Figure 1). There are some reports about the role played by BMPR1αin pulmonary arterial hypertension (PAH). In a mouse model, deletion of BMPR1α did not significantly modify the dynamics of blood flow in the distal vasculature of the lung as a response to hypoxia, however the hemodynamics of proximal pulmonary arteries were changed; a deficiency in BMPR1α further decreased excessive dilation, as a result of collagen accumulation (104). Arterial stiffness in turn altered the function of the right ventricle.
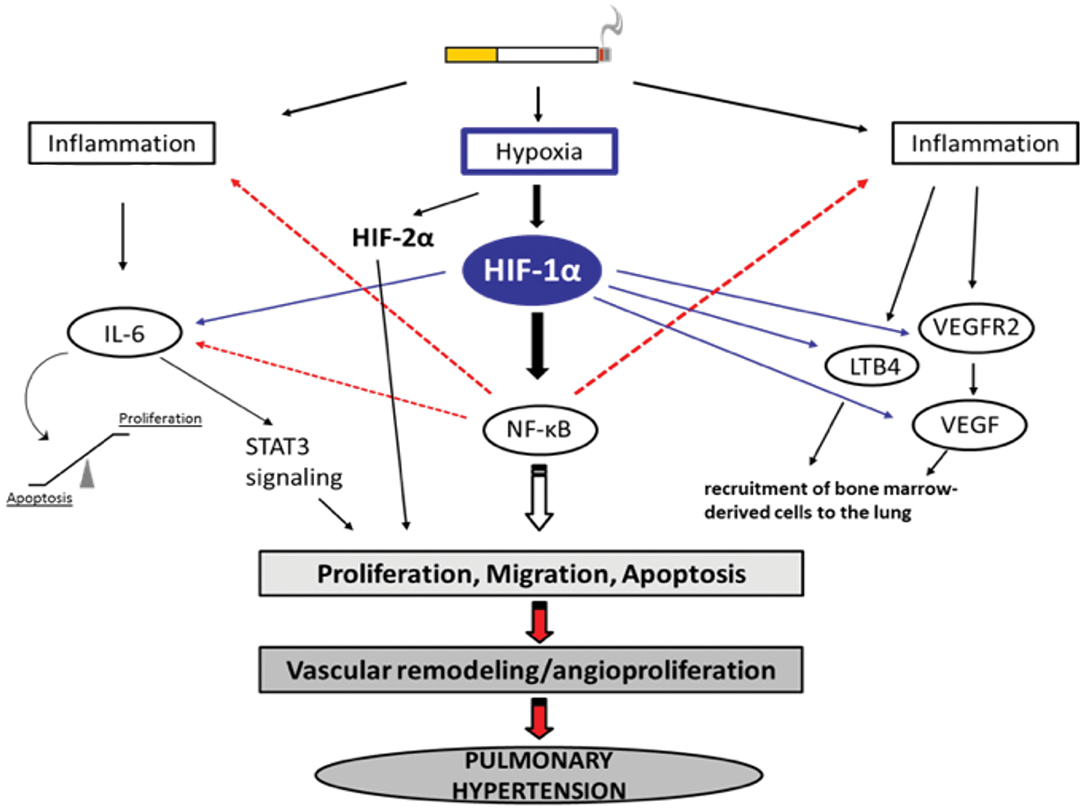
Figure 1. Selection of possible hypoxia-dependent mechanisms contributing to pulmonary vascular remodeling in COPD-PH. Cigarette smoke-induced airway obstruction and emphysema can result in hypoxia. Hypoxia, amongst others, activates HIF-1α which can trigger pathways associated with inflammation, the recruitment of bone marrow-derived cells, and alterations in proliferation/apoptosis balance of vascular endothelial and smooth muscle cells (SMC). Increased proliferation of SMCs causes narrowing of the vessels resulting in pulmonary hypertension. All important acronyms of the molecules are explained in the text.
In addition to the modulation of signaling cascades, IL-6 also modulates the homeostasis between pro- and anti-apoptotic proteins, thereby encouraging vascular remodeling. IL-6 overexpression upregulates Bcl2 and survivin, inhibitors of apoptosis, leading to the inhibition of apoptosis. IL-6 promotes cell proliferation via activation of the VEGF and MAPK pathways (85). This cytokine also participates in cell differentiation and proliferation of the ECM, thereby causing hyperplasia in airway goblet cells and squamous metaplasia in small airway cells. The proliferation of ECM activates the ERK and p38 signaling pathways by increasing the expression of type I/III collagens, laminin, and fibronectin and decreasing the expression of proteoglycans and elastin (94).
Hypoxemia indirectly modulates the expression of angiotensin II, serotonin, platelet-derived growth factor, and metalloproteinases present on the walls of arteries in the lungs, leading to alterations in vascular cross-sectional areas and pulmonary hypertension. The pathways may be interrelated with intracellular ion concentrations. It has been previously reported that potassium channels play a crucial role in inducing excitation in smooth muscle. Inhibition or activation of vascular SMCs can therefore cause changes in their membrane potential, leading to augmentation of calcium ion concentrations inside the cells and propagating vasoconstriction (Figure 2). It has been shown that, under chronic hypoxic conditions, voltage-gated K+ channel (Kv) currents are decreased (105–107), which is most likely mediated by ROS derived from mitochondria (108–112) and/or NADPH oxidases, such as NOX4 (113). Furthermore, influx of calcium ions via transient receptor potential ion channels (TRP) has also been associated SMC proliferation and hypoxia induced vasoconstrictions in the lungs (114). Under hypoxia, the membrane potential is depolarized by between 15 and 20 mV and arteries are constricted to about 300 μM diameter. The generation of ROS in mitochondria via NADPH oxidase has a considerable effect on potassium ion conductance and membrane potential (115, 116). The voltage-independent, two-pore domain K+ channel, TWIK-related acid-sensitive K+channel, (TASK)-1, was shown to be inhibited by hypoxia, leading to membrane depolarization and calcium ion entry through L-type channels. Mutations in the potassium channel subfamily K member 3 (KCNK3) gene, which encodes for TASK-1, have been reported in patients with PH. To date, six different mutations in KCNK3 have been studied in patients diagnosed with PH (117–119). Another protein, the 30-kDa four and a half LIM domain protein 1 (FHL1), has been shown to participate in the induction of hypoxia-induced migration as well as in proliferation of PASMCs thus indicating its importance in vascular remodeling during PH-associated COPD and in patients with idiopathic PAH. The increase in FHL-1 causes migration of PASMCs and their elevated proliferation, contributing to vascular remodeling (120). However, the precise molecular mechanisms underlying this remain unclear (120–122).
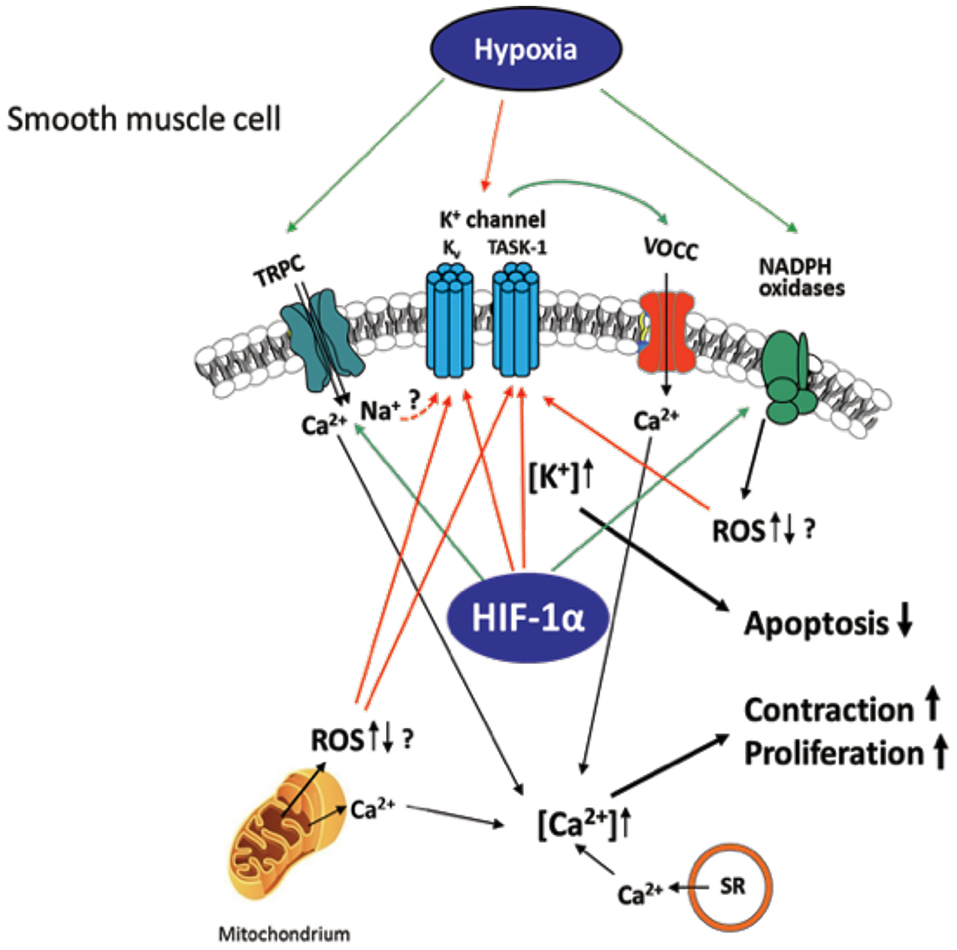
Figure 2. Hypoxia-induced ion channel-mediated increase in proliferation, contraction, and decrease of apoptosis of SMCs contributing to pulmonary vascular remodeling. Kv and TASK-1 channels are downregulated and less active after chronic hypoxia leading to accumulation of K+ within the cell (mediating apoptosis-resistance) and membrane potential depolarization of the SMCs. This depolarization causes opening of voltage-operated Ca2+ channels (VOCC), especially L-type channels, which mediate Ca2+ entry. Hypoxia-dependent ROS regulation derived from NADPH oxidases and/or mitochondria is suggested to inhibit the K+ channels, although it is unclear whether an increase or decrease of ROS occurs in hypoxia. Transient receptor potential channels (TRPC)-mediated Ca2+ or Na+ influx (speculatively by gating K+ channels) was also shown to be essential for the intracellular Ca2+ increase in at least acute hypoxia. Ca2+ release from mitochondria and sarcoplasmic reticulum (SR) was shown to additionally increase Ca2+ within the cell. Thus, mediated contraction and proliferation of the SMCs can contribute to vascular remodeling. Colored arrows depict either activation (green) or inhibition (red).
It has long been suggested that hypoxia is the primary driving force behind the development of PH in COPD. This was supported by studies that showed a close relationship between mPAP and/or pulmonary resistance and alveolar hypoxia (123, 124). However, there is also evidence for other causal factors, independent of hypoxia. First, it has been demonstrated that oxygen therapy is unable to completely reverse PH in COPD (75, 125). Second, analysis of pulmonary vessels from COPD patients with PH demonstrated prominent intimal thickening, medial hypertrophy, and muscularization of small arterioles (126). In contrast, hypoxia-induced vascular remodeling has been shown to be restricted to the media. In addition, these pulmonary vascular alterations occurred in non-hypoxic patients with mild airflow obstruction and smokers without any parenchymal disorder, suggesting that vascular remodeling may be driven by mechanisms independent of hypoxia/hypoxemia (79). Consistent with these findings in humans, studies of mice exposed to tobacco smoke demonstrated that pulmonary vascular remodeling and PH preceded the development of emphysema and was independent of hypoxia (27, 29). Furthermore, these studies showed that gene expression patterns linked with pathways associated with PH and COPD, such as apoptosis, proliferation, oxidative stress, ECM, and inflammation, were completely different compared with gene expression patterns during chronic hypoxia-induced vascular remodeling. Interestingly, the combination of CS and hypoxia act synergistically to affect the vasculature. Experiments with guinea pigs exposed to CS and to hypoxia showed increased mPAP and more pronounced remodeling in small vessels compared with guinea pigs exposed to a single stimulus (28).
Animal studies have shown that CS has a direct effect on the parenchyma and the vasculature. CS has also been shown to increase the expression of genes that encode vasoactive mediators in pulmonary arteries (127, 128). Guinea pigs exposed to chronic CS developed emphysema that was associated with reduced lung capillary density (129). Additionally, cigarette smoke extract (CSE) can induce endothelin-1 (ET-1) in PAECs (130) and reduce prostacyclin synthase expression (131). Furthermore, CSE induces the production of superoxide in ECs, promoting peroxynitrite formation (132). This strong oxidant radical has been shown to suppress VEGFR2 expression, followed by a reduction in EC maintenance and growth. It has been suggested that the cGMP pathway is downregulated following exposure to CS. Congruently, CSE-induced EC apoptosis via p53 (133) can be prevented by the PDE5 inhibitor sildenafil (134), followed by an increase in cGMP levels.
The inability of LTOT to completely reverse the vasoconstriction and remodeling seen in patients with PH-associated COPD indicates that hypoxia-independent factors are also involved in the development of PH in COPD patients. This is supported by finding that pulmonary vascular alterations were also observed in non-hypoxic patients with mild airflow obstruction and smokers with no parenchymal disorders. CS-exposed C57BL/6 mice developed emphysema. In addition, the incursion of large numbers of neutrophils and macrophages and activation of NF-κB and inflammatory cytokines were observed. Further studies have reported that smoke-induced PH and emphysema in mice can be reduced by inhibiting the expression of inducible nitric oxide synthase (iNOS) and activating the expression of soluble guanylate cyclase (sGC) (29). Furthermore, these authors showed that the inhibition of iNOS regulates genes that support lung regeneration through the formation of new alveoli (neoalveolarization) (29, 135). Studies with L-NIL (N6-(1-iminoethyl)-L-lysine dihydrochloride), an iNOS inhibitor, showed repression of matrix metallopeptidase 9 (MMP9) via amplified transcription of expression of metalloproteinase inhibitor 3, TIMP3, resulting in decreased parenchymal destruction. It was also reported that L-NIL treatment supported the formation of elastic fibers in the lungs, thereby inducing active pulmonary repair. In addition, the repression of pro-proliferative (Fgf10 and Ccna1) and apoptosis-inducing factors and reduced proliferation of granulocytes, macrophages, and activated T cells post iNOS inhibition was found to have a positive effect on vessels and neoalveolarization, in support of vascular regeneration (29). CS increases EC permeability via the activation of Ras homolog family member A (RhoA) and myosin light chain (MLC) kinase. Further, CS promotes the release of vasoconstrictors and pro-mitogenic markers, such as ET-1 and thromboxane A2, which eventually results in the remodeling of vessels and pulmonary cell dysfunction (27, 130, 134, 136). Prostacyclin, a well-known vasodilator present in ECs and SMCs, is known to be inhibited in smoke-induced PH. A loss of prostacyclin in the lungs was found to result in increased platelet adhesion and endothelial dysfunction in PH-associated COPD patients (137).
The pathogenesis of PH in COPD is thought to be driven by an endothelium-derived vasoconstrictor/dilator imbalance caused by ED. Thus, ED in the walls of arteries in the lungs contributes to the development of PH in patients with COPD. This dysfunction has been measured by analyzing the nitric oxide-dependent relaxation of arterial rings in the lungs in response to dose-dependent increases in exogenous acetylcholine and adenosine diphosphate (ADP) (78, 138, 139). In emphysematous lungs, the expression of VEGF and VEGFR was significantly reduced. The extent of pulmonary injury was further increased following exposure of the lungs to CS. The growth and proliferation of inadequately differentiated SMCs and deposition of ECM proteins in arterial walls in the lungs contributed to the dysfunction and progression of the disease. The process of ED is also associated with decreased expression or uncoupling of endothelial nitric oxide synthase (eNOS), which might contribute to the development of PH (Figure 3). In contrast, the expression of VEGF and serotonin transporters seems to be increased (140, 141). ECs are known to be important for the regulation of vascular homeostasis. They control the vascular tone and affect pulmonary vessel adaptations to changes in blood flow and in response to hypoxia (142–145). Dysfunction of the endothelium has been reported in patients with end-stage COPD after lung transplantation (140), as well as in patients with mild-to-moderate COPD (146). In general, endothelial function is affected by the expression of various vasoreactive substances that control vasoconstriction (and are also pro-proliferative for SMCs) or vasodilation (and are also anti-proliferative for SMCs). In patients with primary or secondary PH and COPD, the protein ET-1 was shown to increase (147). Vasodilative mediators, such as eNOS (147, 148) and prostacyclin synthase (PGI2-S), were downregulated in pulmonary arteries (131). CSE can decrease the expression of PGI2-S in human PAECs (131), suggesting that its downregulation is a direct effect from the ingredients in CSE. After exposure to CS for 8 months, the downregulation of eNOS in lungs and vessels was observed, concomitant with the development of emphysema and PH (29). In addition, eNOS-deficient mice developed emphysema and PH following exposure to CS, whereas iNOS-deficient mice did not (29). Other changes in the presence of CS include increased injuries to ECs, increased infiltration of neutrophils, increased lipid peroxidation, excessive oxidative stress, and imbalances between the expression of vasoconstrictors and modulators (78) (Figure 3).
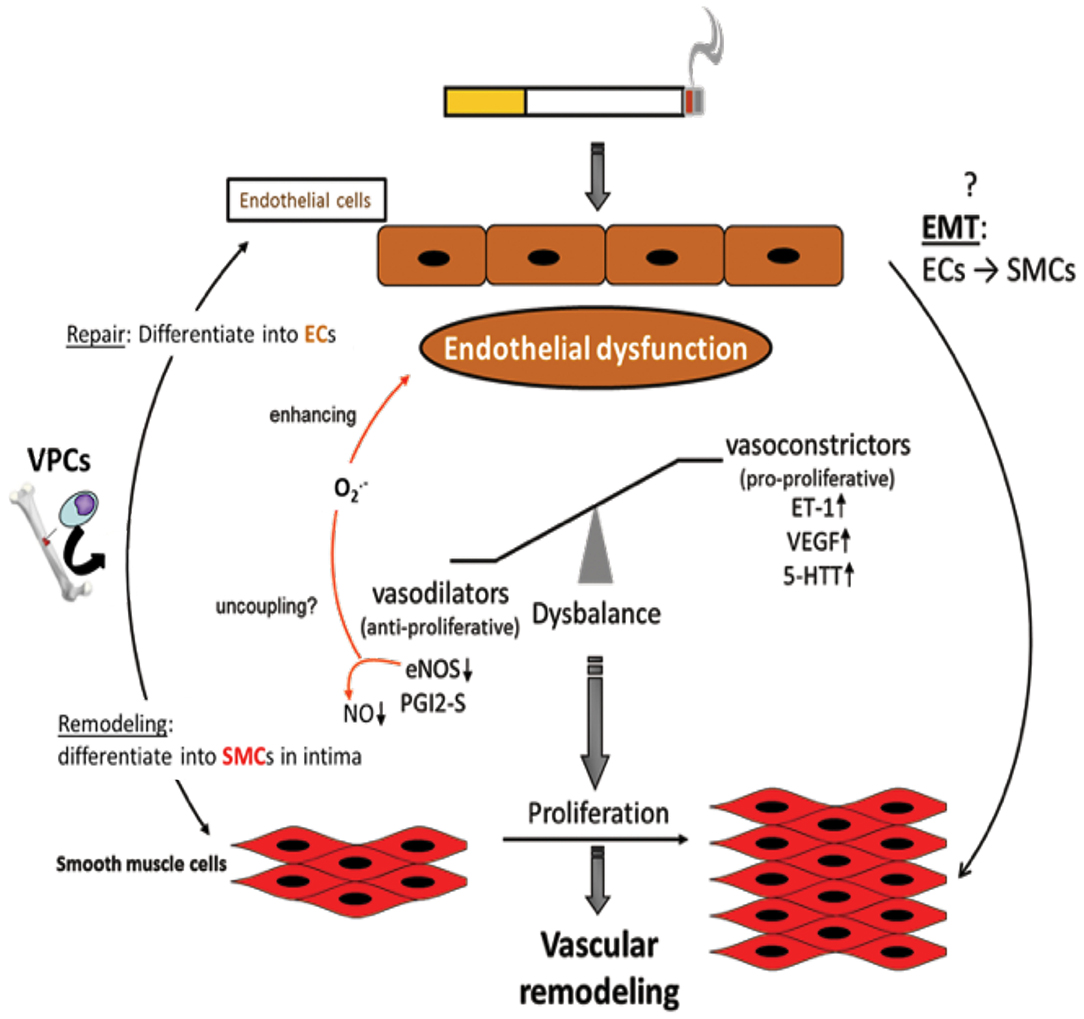
Figure 3. Endothelial dysfunction as causing factor for development of vascular remodeling. Cigarette smoke and inflammatory mediators can cause endothelial dysfunction which is triggered by a disbalance of vasodilative and vasoconstrictive molecules towards an excess of vasoconstrictors and damage/dysregulation of EC signaling. Additionally, vascular progenitor cells (VPCs) are attracted to the damaged endothelium. Such VPCs can either contribute to repair by differentiation into ECs or to remodeling by differentiation into SMCs. Furthermore, an endothelial-to-mesenchymal-transition (EMT) may occur resulting in an SMC phenotype. Vasoconstriction and altered endothelial cell signaling are stimuli for SMCs to proliferate resulting in vascular remodeling, increased pulmonary artery pressure, and finally in pulmonary hypertension.
Acute exacerbation due to increased inflammation in the lungs is the most important characteristic identified and studied in patients with COPD. During progression of the disease, excessive mucus, containing inflammatory cells, accumulates in the blood vessels thereby increasing the tissue volume in the bronchial wall. The increase in tissue volume can be measured by the infiltration of both innate and adaptive inflammatory markers, such as neutrophils, macrophages, and CD4 and CD8 lymphocytes. Neutrophils contribute significantly to the production of ROS, cytokines, and chemokines during lung inflammation. The inflammation is triggered via the activation of Toll-like receptors, followed by transcriptional activation of NF-κB and activation of STAT pathways. During pulmonary inflammation, patients with COPD exhibit higher expression of inflammatory cytokines, such as IFN-γ and TNF-α, along with IL-1, IL-6, and IL-8. Reports have also indicated that IL-17 plays a role in COPD progression and inflammation (149–152). In patients with COPD, an increased number of inflammatory cells invade the adventitia of pulmonary muscular arteries. These cells are predominantly activated CD8+ T-cells (140, 153). Numbers of these lymphocytes are also increased in the arterial adventitia of smokers with normal lung function. The ratio of CD4+/CD8+T-cells is reduced compared with the ratio in non-smokers and is comparable to the situation in patients with mild-to-moderate COPD (153). In addition, an association between IL-6 expression and elevations in mPAP supports a role for inflammation in the pathogenesis of PH (in COPD) (154). This is particularly relevant, because the vascular adventitia has been shown to harbor inflammatory cell progenitors, e.g., CD45+ macrophage progenitors, independent from bone marrow-derived monocytes (155–157). Upon activation, these progenitors deliver F4/80+ macrophages that serve as a local source for high levels of VEGF production (157). The potential contribution pulmonary vessel adventitia-derived inflammatory cells, e.g., macrophages, make to PH and the proliferation of vascular adventitia-resident SMC progenitors has yet to be studied.
Increased production of ROS in the endothelium is an important characteristic of pulmonary endothelium dysfunction in PH. During oxidative stress in patients with PH, the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) fails to activate the expression of antioxidant enzymes, such as superoxide dismutase (SOD) and catalase, thereby increasing the level of ROS in the body. Also, increased production of superoxide radicals reduces the availability of NO, as superoxide ions merge with NO to form peroxynitrite, which in turn increases the expression of inflammatory markers and leukocyte infiltration via the expression of adhesion molecules in the endothelium. The formation of peroxynitrite further contributes in depolarizing potassium ion channels and increasing calcium ion concentration inside cells, eventually leading to tissue injury due to increased vascular permeability. Superoxide ions can generate downstream toxic products, including hydrogen peroxide (H2O2), which in turn activates the phosphorylation of NF-kB and leads to the activation of inflammatory responses. Activation of inflammasomes due to oxidative stress further perpetuates inflammation in the body (151, 158). The elevated production of superoxide and H2O2, together with reduced NO bioavailability, make a fundamental contribution to vascular remodeling and the development of emphysema. Elevated concentrations of H2O2 and 8-isoprostane, both of which are oxidative stress markers, are found in the exhaled breath condensate of smokers and ex-smokers, as well as during exacerbations (159, 160). ROS have been shown to negatively affect the function of antiproteases, such as α1-antitrypsin and secretory leukocyte proteinase inhibitor (SLPI) (16, 161). Consequently, a protease/anti-protease imbalance accelerates the degradation of elastin in the lung parenchyma, resulting in emphysema. Angiotensin II is involved in NADPH oxidase-generated superoxide production, mediated by angiotensin type I receptor, which is converted to H2O2, with SOD acting as a second messenger. This pathway induces hypertrophy or hyperplasia in vascular SMCs (162, 163). This angiotensin II-induced process has been shown to be inhibited by the flavoprotein inhibitor DPI (162), catalase, and knockdown of p22phox, which supports the involvement of NADPH oxidases in the vasculature (164–166).
ROS promote vascular remodeling by increasing the deposition of ECM proteins. In particular, collagen and elastic fibers are degraded by proteinases, specifically MMPs. MMPs are secreted, in an inactive form, by macrophages and vascular SMCs (167). ROS, such as peroxynitrite, have been shown to activate MMP-2 and -9 in human SMCs, followed by degradation of the basement membrane and elastin (168, 169). A hypertension model involving aldosterone-induced systemic oxidative stress revealed that endothelin-1-associated processes are the main contributors to vascular remodeling (170, 171). Similarly, redox-sensitive inflammatory processes are known to induce vascular remodeling. In particular, increased expression of the inflammation-related intracellular adhesion molecule-1 (ICAM-1) has been shown in the aorta of aldosterone-treated rats (170, 171). Furthermore, angiotensin II-induced oxidative stress results in tissue hypertrophy associated with an increase in ICAM-1 expression. Macroscopic and microscopic examinations of COPD emphysematous lungs using hematoxylin and eosin staining showed that the alveolar septa were extremely thin and avascular (172). This indicates that pulmonary endothelial dysfunction might be the key element in COPD pathogenesis.
The endothelium forms a continuous monolayer and thereby a regulated barrier that separates the intravascular blood compartment from surrounding tissues (173, 174). ED is classically defined as impaired NO-mediated vascular relaxation (Figure 4). In a broader sense, ED encompasses a state in which ECs are activated, additionally characterized by endothelial barrier impairment and reduced anti-adhesive and antithrombotic properties (175).
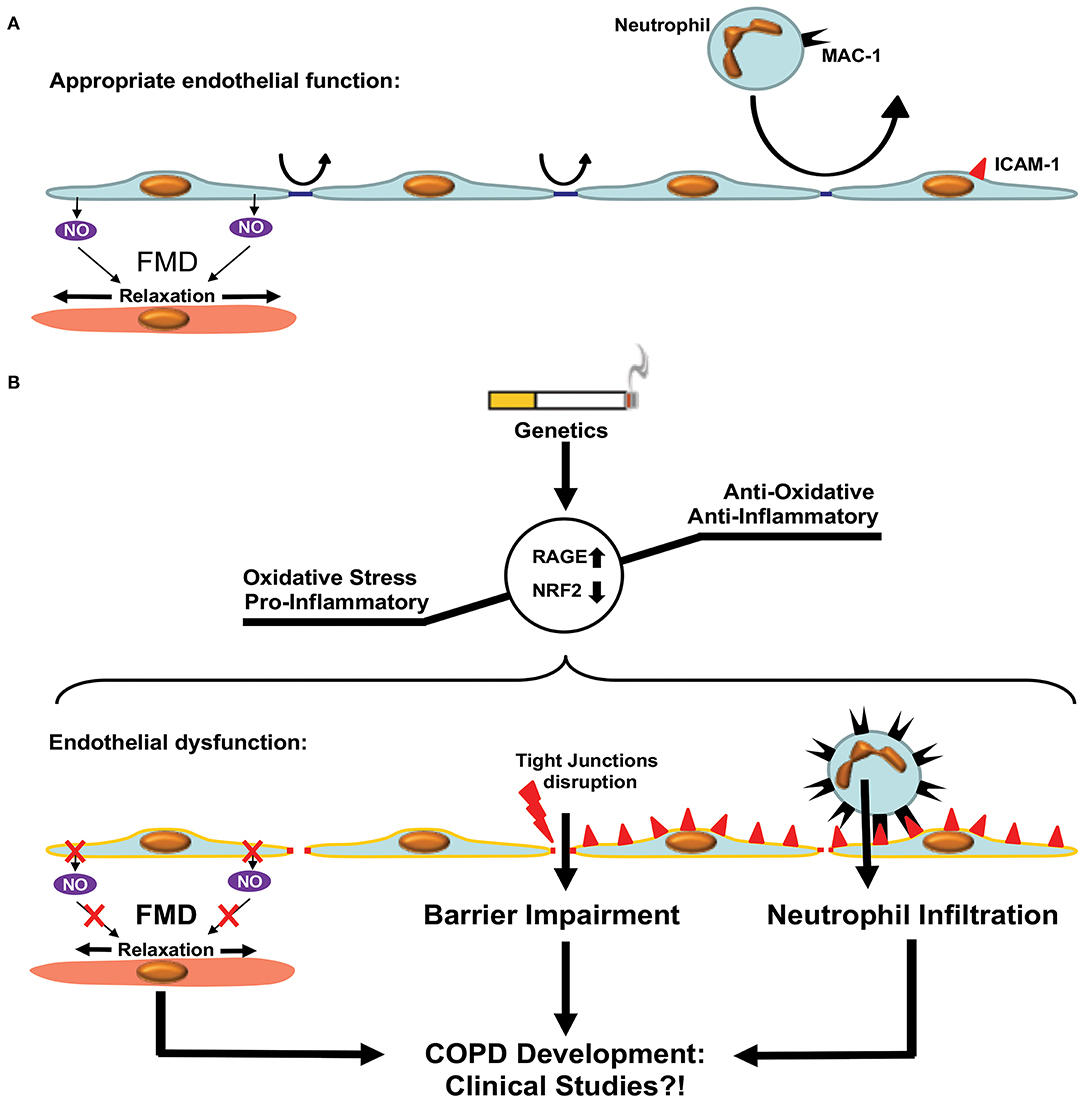
Figure 4. Endothelial dysfunction in COPD. (A) Appropriate endothelial function under physiological conditions. The endothelium shows adequate flow mediated dilation (FMD) via relaxation of smooth muscle cells within the vascular media. Paracellular permeability is limited by intact tight junctions. Low adhesion molecule expression limits neutrophil extravasation in pulmonary tissue. (B) Development of COPD is accompanied by enhanced oxidative stress and a pro-inflammatory state resulting in a dysfunctional endothelium. Dysfunction is characterized by reduced FMD, impaired endothelial barrier function due to disruption of tight junctions and enhanced expression of adhesion molecules facilitating neutrophil extravasation. These processes promote COPD development via increased inflammation. Clinical studies are required in order to test the beneficial effects of therapies targeting ED in COPD patients.
Recent studies have shown that the microvascular barrier is impaired in patients with COPD and that the level of impairment is correlated with the severity of airway obstruction (176, 177). This could be attributed to the disruption of endothelial tight junctions observed in patients with COPD, even in the absence of CS (178, 179). Immune cells, especially neutrophils, are critically involved in the pathogenesis of COPD. This has been reviewed in details elsewhere (180, 181). Neutrophil accumulation was observed in the lungs of patients with COPD, in clinical settings, using radiolabeled neutrophils and single-photon emission computerized tomography (SPECT/CT) imaging (182). Neutrophil accumulation within the pulmonary tissue requires extravasation through the endothelial barrier, which is an active process involving adherence to ECs and migration either between (paracellular diapedesis) or through (transcellular diapedesis) ECs. Neutrophil MAC-1 (αmβ2, CD11b/CD18, and complement receptor type 3) interactions with endothelial ICAM-1 promote endothelial transmigration of neutrophils. Neutrophil adhesion and migration across the endothelium were shown to be upregulated in patients with COPD, presumably via upregulation of MAC-1 expression in neutrophils (183). Accordingly, levels of ICAM-1 were shown to be elevated in patients with COPD (184). Furthermore, ICAM-1, as well as P-selectin, another endothelial adhesion molecule involved in neutrophil transmigration, was inversely associated with first forced expiratory volume (FEV1). Furthermore, enhanced soluble ICAM-1 levels are independently associated with emphysema progression in the general population (185).
In addition to the presence of ED in animal studies of COPD, clinical studies suggest a dysfunction of the endothelium even prior to the onset of COPD. This has been ascribed to excessive tobacco consumption, since normal lungs of smokers showed intimal thickening of small pulmonary arteries, similar to that seen in the lungs of COPD patients, when compared with lung tissues of non-smokers (146).
Flow-mediated dilation of the brachial artery analyzed by ultrasound, the reference method used to determine ED in humans, is reduced in early COPD and associated with FEV1 reduction and a higher percentage of emphysema in CT scans of former smokers (186–188). In addition to ED, direct injury of ECs seems to play a critical role in COPD, as pulmonary septa appear almost avascular (172) and CT scans of patients with COPD show vascular pruning of small arteries that can predict the clinical severity of disease and mortality (189, 190). This has been linked to impaired VEGF signaling, as VEGF and VEGFR2 expression are reduced in areas of the lungs with emphysema in patients with COPD (191), and VEGFR inhibition resulted in an emphysema phenotype in animal studies (192–194). In contrast, enhanced expression of HIF-1α, VEGF, and VEGFR was observed in human patients with COPD and reflected disease severity. This led to the assumption that VEGF signaling is increased in non-emphysema tissue in COPD patients, while it is decreased in emphysematous COPD parenchyma (90). It is important to mention that current knowledge on the vascular involvement in COPD/emphysema is based upon the pioneering work of Averill A. Liebow (172).
Patients with COPD have an increased risk of suffering with CVDs (50). This is of particular relevance, as ED is a common feature in both COPD and the development of atherosclerosis, which in turn can cause CVDs such as myocardial infarction and stroke (50, 175, 195). Interestingly, ED in patients with COPD exhibits an intermediate state between healthy patients and patients suffering from coronary artery disease (196). In a murine model of atherosclerosis (Apo.E−/−), increased oxidative stress was suggested to link ED with COPD pathogenesis, especially with regard to the development of emphysema (197). Several studies have shown increased vascular oxidative stress levels in COPD patients, which is associated with a reduction in FEV1 (198–200). The receptor for advanced glycation end products (RAGE) seems to play a major role in this process (Figure 4). Genetic deletion or pharmacologic inhibition of RAGE protects against the development of CS-induced emphysema (201). Furthermore, genome-wide studies of single nucleotide polymorphisms linked RAGE to the development of emphysema in COPD (202, 203). Therefore, targeting vascular oxidative stress-mediated ED seems to be a promising treatment for COPD. However, studies investigating anti-inflammatory therapy in patients with COPD were mainly conducted without determining the effect on vascular dysfunction (204–208). So far, only animal studies have provided evidence that anti-oxidative treatment of ED shows a beneficial effect on COPD, i.e., by the activation of the transcription factor Nrf2 (94, 207, 209). Therefore, investigating the targeting of ED in COPD, e.g., with anti-oxidative pharmaceuticals, with a concomitant analysis of effects on flow-mediated dilation, remains a task for the future.
Nitric oxide has been suggested to play an important role in CS-induced emphysema and PH in mice (29). It has been shown that CS induces upregulation of iNOS; this was predominantly observed in small pulmonary vessels and associated with increased NO generation. Interestingly, iNOS was downregulated during the early phase of disease, in both mice and human patients with COPD. The vasodialative effect of NO was most likely abolished because of the abundance of ROS from both external (CS) and internal sources. It was also suggested that the formation of peroxynitrite had pro-apoptotic and anti-proliferative effects on alveolar epithelial cells type II (AECII) and ECs, resulting in the development of emphysema, vessel loss, vascular remodeling, and an increase in the level of nitrotyrosine. It is assumed that the decrease in eNOS level was associated with the uncoupling of this enzyme, followed by a switch from NO to superoxide production. Interestingly, in iNOS-deficient but not eNOS-deficient mice, vascular remodeling, PH, and emphysema did not occur. Additionally, treatment with an iNOS inhibitor (L-NIL) prevented the development of disease and promoted lung regeneration in mice exposed to CS.
It has been demonstrated that iNOS-carrying bone marrow-derived cells mediate the development of PH, while emphysema is dependent on iNOS in non-bone marrow-derived cells (29) (Figure 5). These results clearly show that the pathophysiology of PH and emphysema is partly independent. Furthermore, this might also be a reason why not all patients with COPD suffer from PH. In animal studies, it was further demonstrated that the stimulation of sGC, an enzyme that uses (i) NOS-generated NO to produce cGMP from guanosine triphosphate (GTP), prevented the CS-induced development of vascular remodeling but also emphysema (210). cGMP acts as a second messenger mediating vasodilation. In addition to the vasodilatory effect, cGMP was shown to effect on proliferation, platelet aggregation and recruitment of inflammatory cells (211). These authors further showed that, riociguat (approved for PAH and chronic thromboembolic PH treatment) which promotes the NO-cGMP pathway, not only prevented tobacco smoke-driven PH development but also prevented airspace enlargement in smoke-exposed mice (210, 211). Similarly, in another recent study from Pichl et al. showed the riociguat treatment in the mouse model of smoke-induced PH and emphysema reversed fully established emphysema, muscularization of small pulmonary vessels, and RH hypertrophy and had beneficial effects on small cohort of COPD patients (212). Moreover, the same group also investigated another drug BAY 41-2272 which also stimulates sGC in tobacco smoke- exposed guinea pigs reduced vascular remodeling and prevented emphysema development (210). In, another study by Paul et al. also demonstrated by treating this drug BAY 41-2272 to guinea pigs that were chronically exposed to smoke exhibited similar effects with decreased extent of emphysema, RV hypertrophy, and improved pulmonary haemodynamics (213). These studies demonstrate the importance of sGC playing crucial role in the pathology of COPD. Therefore, other downstream molecules of this pathway were focused in preclinical studies. Namely, blocking of cGMP degradation by PDE5 inhibitors such as sildenafil (214) or tadalafil (27), prevented the development of PH in smoke-exposed guinea pigs and mice, respectively. Moreover, treatment with the PDE4 inhibitor demonstrated a significant protective effect on emphysema and PH development, suggesting that cAMP plays an important role in the pathology of COPD. These findings implicate an important role for the NO–sGC–cGMP axis in the physiology and pathophysiology of the pulmonary vasculature (29). The dysregulation of this system has previously been suggested to contribute to pulmonary diseases and PH (215–217). In line with previous findings (29), impairment in vascular remodeling was associated with the prevention of emphysema, although causality was not investigated in these studies.
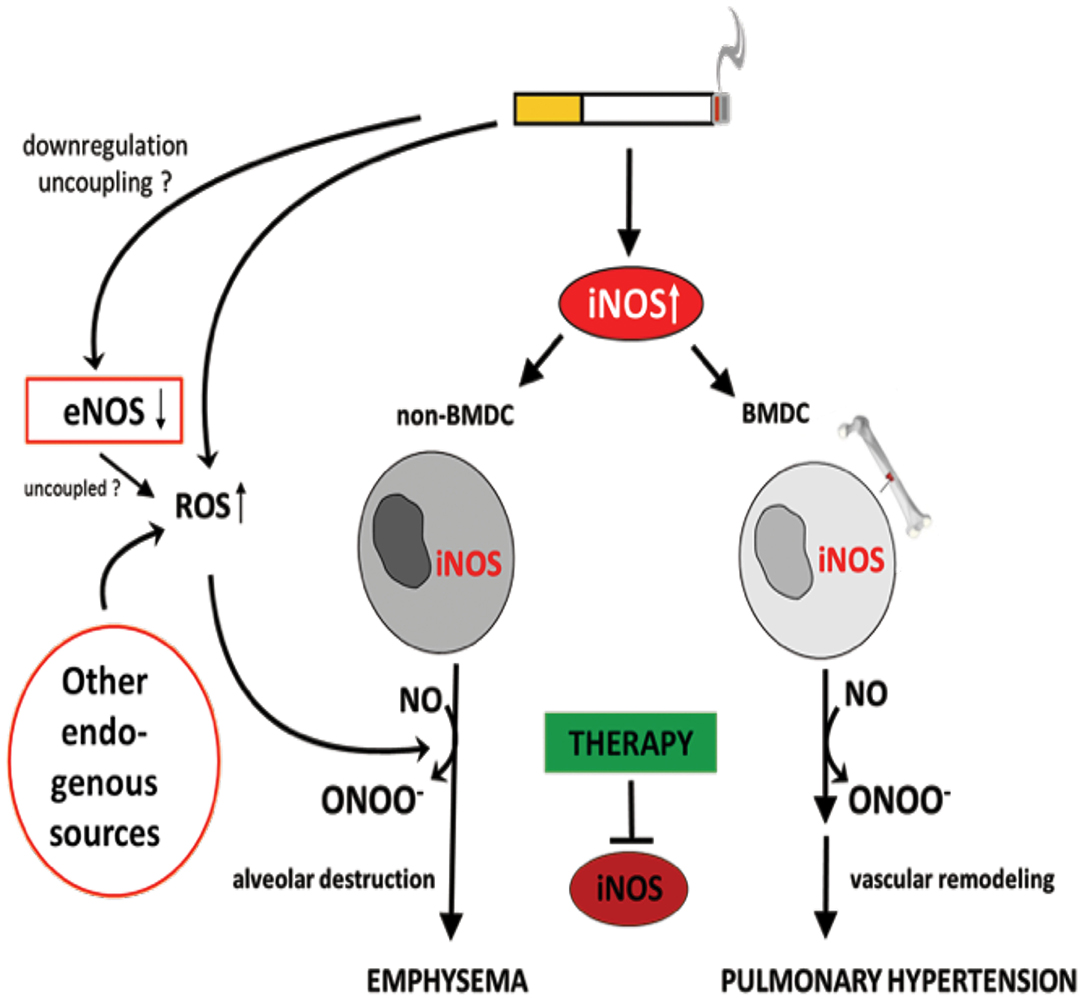
Figure 5. Identification of inducible NO synthase (iNOS) as an essential factor for the development of cigarette smoke-induced emphysema and pulmonary hypertension in mice. Cigarette smoke-mediated upregulation of iNOS leads to excessive NO production. The formation of peroxynitrite, resulting from the reaction of NO with superoxide, was suggested to mediate emphysema and PH development. Superoxide can derive from cigarette smoke itself and/or from uncoupled eNOS, NADPH oxidases, xanthine oxidases, cyclo- and lipooxidases, and mitochondria. Of interest, iNOS generated by non-bone marrow-derived cells (N-BMDC), possibly vascular cells, leading to lung destruction resulting in emphysema whereas elevated iNOS expression in bone marrow-derived cells (BMDC) causes vascular remodeling. Treatment with specific iNOS inhibitor L-NIL prevents or even reverses pathological alterations.
While it is known that CS leads to ROS-mediated oxidative stress, other sources of ROS that affect vascular remodeling have yet to be fully resolved. Various sources of ROS, such as mitochondria, NADPH oxidases, xanthine oxidase, cyclooxygenases, lipooxygenases, and uncoupled eNOS, must be considered (215, 218–221). Furthermore, especially in the lung, mitochondria have been implicated in vascular remodeling, and imbalances in mitochondrial ROS production seem to play an important a role in this process (222–226). Moreover, recent evidence suggests that mitochondrial ROS are causatively linked to the development of PH (227). However, the mechanisms of mitochondrial ROS production have not yet been fully elucidated. The vasculature in non-hypoxic PAH models has suggested a decrease in mitochondrial ROS, whereas a chronic hypoxia model of PH demonstrated increased mitochondrial ROS (222, 228, 229). According to some researchers, mechanistic discrepancies in mitochondrial ROS production might be based on “different experimental conditions, species differences, and perhaps complexities of how pure hypoxic stress may interface with other triggers of PH” (227). In addition to this phenomenon, significant alterations in mitochondrial metabolic pathways may drive a metabolic shift (“cancer theory of PH”) in PASMCs, triggering vascular remodeling and PH (227, 230, 231). In terms of COPD, recent data provide some evidence that mitochondrial ROS might play a role in lung remodeling/emphysema development. The overproduction of the alternative oxidase (AOX), which bypasses the cytochrome segment of the respiratory chain, attenuated CS-induced lung tissue destruction and loss of function in mice chronically exposed to CS for 9 months. This implicates mitochondrial respiratory inhibition as a key pathogenic mechanism of CS toxicity in the lung (232).
Regarding vascular remodeling, ROS are known to affect various intracellular signaling cascades, such as activation of ERK, MAPKs, protein tyrosine phosphatases, transcription factors such as NF-kB and AP-1, and receptor and non-receptor tyrosine kinases, which have been shown to be involved in cardiovascular remodeling and vascular damage. In addition, monocytes and lymphocytes are able to infiltrate cardiovascular tissues and pulmonary vessels, while inflammatory processes are often related to immune defense or CS. Previous studies have also shown that macrophages contribute to COPD development and that NADPH oxidase plays a crucial role in this regard (233, 234).
Neprilys in (neutral endopeptidase, NEP) may be an important factor for the regulation of susceptibility to pulmonary vascular remodeling in response to smoke inhalation and hypoxia (235) (Figure 6). NEP is a transmembrane zinc peptidase that is widely expressed in PASMCs, ECs, and fibroblasts (236). NEP expression and activity is decreased by CS (237), hypoxia (238, 239), and oxidative stress (240). The depletion of NEP in mice resulted in increased severity of PH, associated with greater proliferation of PASMCs. Therefore, it has been suggested NEP plays a protective role against PH, partly by suppressing the proliferation and migration of PASMCs (239).
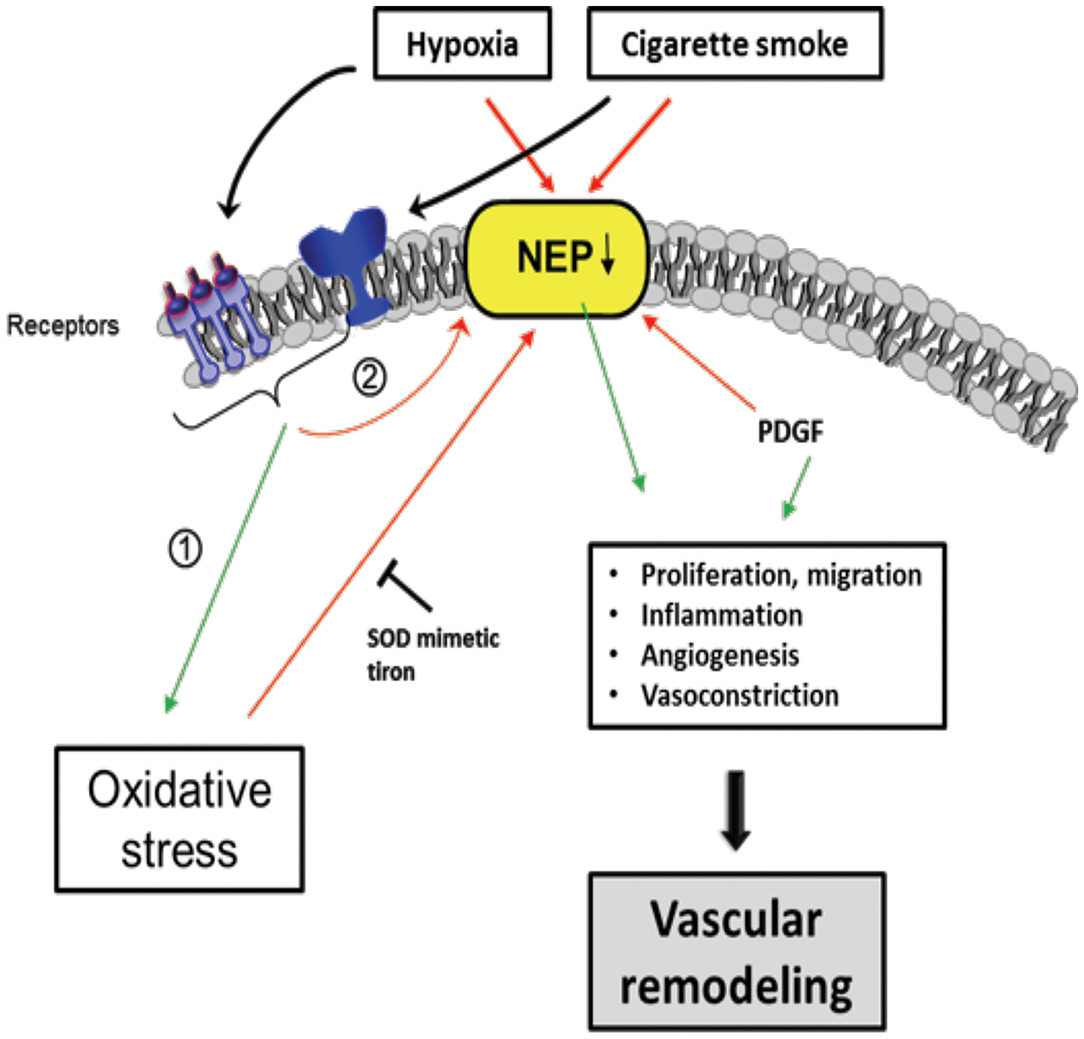
Figure 6. Scheme of proposed Neprilysin (NEP)-dependent mechanisms leading to vascular remodeling. Extracellular stimuli, such as hypoxia and cigarette smoke activate pathways in vascular cells causing downregulation of NEP expression and/or activity which was seen in patients with COPD associated with PH. Stimuli can have direct negative effects on NEP, but also indirectly by (1) increasing ROS, PDGF and (2) activation of other pathways, in part receptor-mediated. NEP downregulation leads to elevated proliferation, migration, inflammation, Angiogenesis, and vasoconstriction mediated amongst others by depicted molecules causing vascular remodeling. The ROS effect on NEP can be inhibited by the superoxide dismutase (SOD) mimetic tiron. Colored boxes indicate involvement in the respective pathway; red arrows, inhibition/downregulation; green arrows, activation/upregulation; PDGF, platelet-derived growth factor; ET-1, endothelin-1; FGF-2, fibroblast growth factor; AT-II, angiotensin-II; PASMC, pulmonary artery smooth muscle cells; EC, endothelial cells; FB, fibroblasts.
Wick et al. found that NEP expression decreased in the lungs of COPD with PH as well as in non-COPD PH patients (235). NEP is involved in many peptidase-dependent (e.g., degradation of vasoactive neuropeptides) and -independent (e.g., interaction of signaling molecules with the intracellular cytosolic domain of NEP) signaling pathways (241, 242), but its role affecting vascular remodeling remains to be elucidated. In this regard, Wick et al. suggested that the proliferation/migration of dedifferentiated SMCs or myofibroblasts promotes pulmonary vascular remodeling and PH if NEP is less active or downregulated. It is assumed that this process is mediated by platelet-derived growth factor (PDGF), the expression of which is inversely correlated with NEP. Karoor et al. supported this assumption by showing that PDGF receptor (PDGFR) signaling was constitutively active in NEP−/− cells and in lungs, an effect that is attenuated by the endothelin A (ETA) receptor antagonist ambrisentan (243). The decrease in NEP following CS and hypoxia may also enhance the angiogenic effect of fibroblast growth factor-2 (FGF-2) (242) and the pro-proliferative and vasoconstrictive responses of ET-1 (244) and bombesin-like peptides (245), which are substrates of NEP.
In PASMCs, both FGF-2 and ET-1 were shown to synergize with PDGF in increasing the phosphorylation of Src kinase and PDGFR (243). The phosphatase PTEN (phosphatase and tensin homolog) was also shown to play an important role in vascular biology, because a loss of PTEN results in PH (246). PTEN is inactivated by phosphorylation (mediated by Src and PDGFR) and downregulated in NEP-deficient PASMCs. This downregulation can be rescued by NEP overexpression in NEP null cells or by a reduction in Src or PDGFR by small interfering RNA (siRNA). Accordingly, it has been suggested that NEP-dependent mechanisms may protect against the inactivation of PTEN (243). In addition, NEP can be inactivated by ROS, as shown by its decreased activity in the presence of H2O2 and improved activity when an antioxidant, the SOD mimetic Tiron, was added (235).
Early studies suggested that the inhibition of NEP exerted beneficial effects in the treatment of PH (247, 248). This idea resulted from the fact that NEP can inactivate atrial/brain natriuretic peptides (ANP/BNP) that promote vasodilation by increasing cGMP, mediated by natriuretic peptide receptor-A (NPR-A) (248). cGMP-dependent protein kinase (PKG), cGMP binding phosphodiesterases (PDEs), and cyclic nucleotide-gated ion channels bind cGMP, with PKG seeming to be the main mediator of cGMP signals (249, 250). Binding of ANP/BNP-induced cGMP activates PKG, followed by the catalytic transfer of phosphate from ATP to target proteins. The phosphorylated proteins then translate the extracellular stimuli into specific biological outputs (251), such as vasodilation.
NEP antagonists alone and in combination with ACE (angiotensin converting enzyme) and ECE (endothelin converting enzyme) inhibitors were able to improve cardiac function, limit cardiac hypertrophy, and decrease systemic blood pressure (252–255). Nevertheless, side effects were observed if single NEP inhibitors or dual inhibitors (NEP/ACE or NEP/ECE) were used. Triple vasopeptidase inhibitors (NEP/ACE/ECE) showed promising preliminary results, with fewer side effects. In particular, an increase in ET-1 can be antagonized by simultaneous application of an ECE inhibitor (255). Combination therapies such as these, and the different mechanisms of NEP, ACE, and ECE inhibition, were reviewed by Daull et al. (255).
The existing literature supports the notion that NEP protects against PH (235, 239, 256). The discrepancy between beneficial and harmful pulmonary effects with NEP inhibition might be because pulmonary and systemic circulation usually respond to hypoxia (a major stimulus for PH) by divergent pathways: while pulmonary vessels contract to redirect blood flow to better oxygenated areas of the lung, systemic vessels dilate to increase the flow of oxygenated blood to areas of tissue hypoxia or ischemia (239). In conclusion, in terms of the lung, it is suggested that NEP be increased to treat PH, whereas cardiac NEP inhibition could be used for the treatment of hypertrophy and improvement of cardiac function. These studies provide insights into distinct regulation of NEP in cardiovascular disease and PH. Additional investigation are required whether therapeutic activation of NEP selectively in lung ameliorates PH.
The regeneration or replacement of structurally impaired lung tissue would represent a breakthrough in the treatment of pulmonary diseases, including COPD. Therefore, stem and progenitor cells may be a promising therapeutic approach for COPD. Various types of stem cells, including mesenchymal stem cells (MSCs), induced pluripotent stem cells (iPSCs), embryonic stem cells (ESCs), and stem cells derived from lung tissue, were tested in animal models and in a limited number of clinical studies as COPD treatment options (257–259).
For the efficient treatment of COPD, regeneration of alveolar epithelium as well as capillary formation is required. Multiple stem cells have been shown to differentiate into ECs capable of capillary formation, both in vitro and in vivo (257, 260–263). Transplantation of pre-differentiated ESCs and iPSCs reduced lung injury in a bleomycin mouse model, presumably by reducing inflammation (257, 264). MSC transplantation was shown to protect against EC apoptosis in animal emphysema models and promoted restoration of both alveolar and endothelial structures (265–267). Furthermore, MSC-conditioned medium restored endothelial barrier impairment caused by CS (268). Li et al. reported that MSCs derived from iPCSs were superior to bone marrow-derived MSCs regarding CS-induced pulmonary airspace enlargement (258). This indicates that stem cells of different origin vary in their pulmonary regenerative potential. In contrast with these beneficial effects, the engraftment of transplanted cells has been found to be quite low (<2%) in murine models of lung emphysema (269–271). This is in accordance with a study by Huh et al., who reported that not only bone marrow cells or MSCs (which largely disappeared in pulmonary tissue over time post-transplantation) but also cell-free conditioned media obtained from MSCs alleviated CS-induced emphysema (272). This indicates a paracrine effect rather than a direct effect of stem cell engraftment and could explain the positive effects observed in studies with low stem cell engraftment rates (Figure 7). Therefore, it remains questionable how long stem cells engrafted after transplantation remain viable in patients. Moreover, this raises the question whether efficient effects in patients might require recurring cell transplantations.
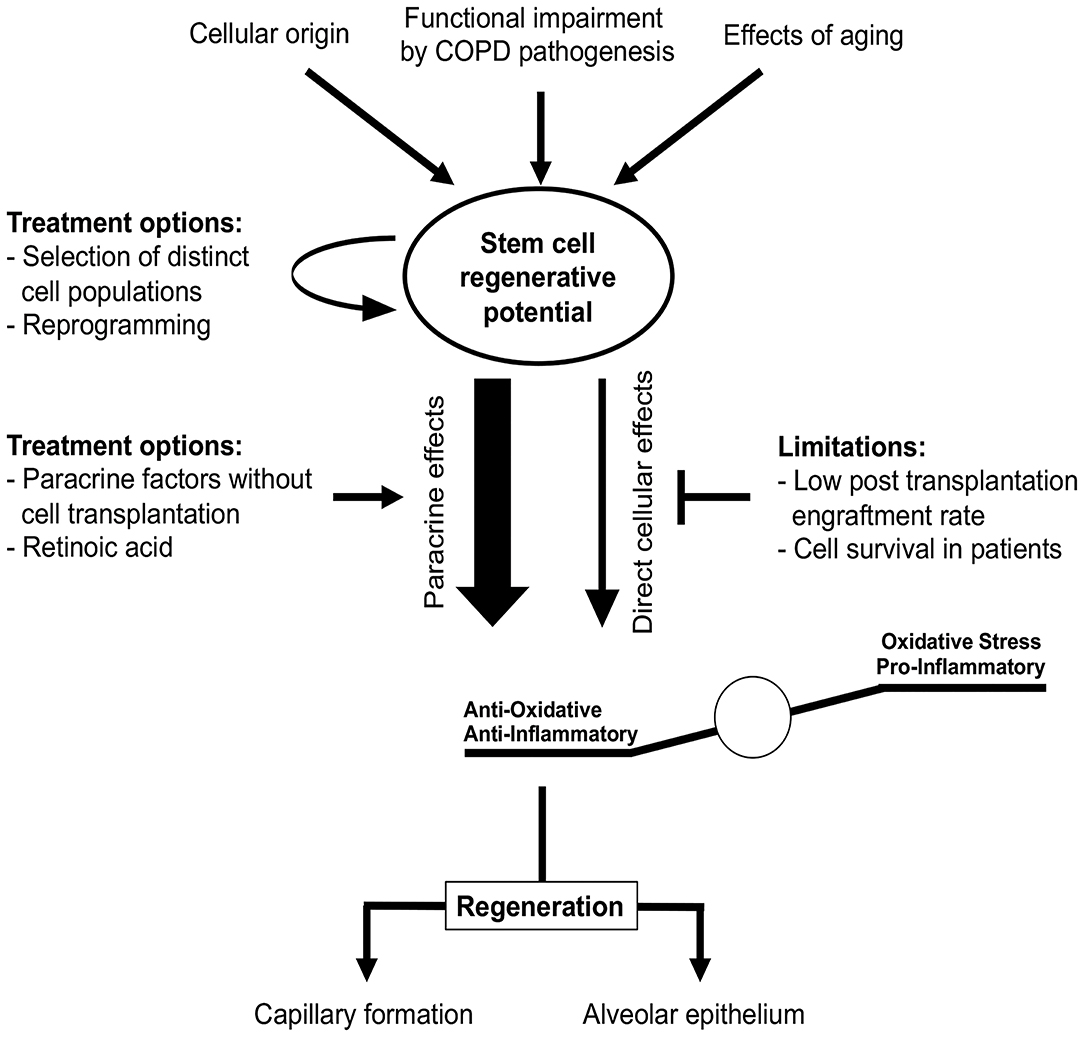
Figure 7. Stem cells in COPD treatment. Stem cell regenerative potential critically depends on cellular origin and can be impaired by different processes, e.g., due to aging or COPD itself. These impairments might be overcome by selection of distinct stem cell sub-populations or cellular reprogramming prior to transplantation. Regenerative potential of stem cells in COPD is rather mediated by paracrine effects reducing oxidative stress and pro-inflammatory stimuli than direct cellular effects, since cell engraftment and survival rates observed in vivo are low. Studies of COPD treatment have to focus not only on the functional regeneration of alveolar epithelium, but also on vascular aspects as both are required for effective therapy.
At present, identifying the beneficial underlying paracrine mechanisms of stem cell effects in pre-clinical COPD models might reveal new treatment options without the need for cellular transplantation (Figure 7). Based on current studies, the beneficial paracrine effects of stem cells in COPD appear to be mediated by reduced inflammation and oxidative stress (269, 273, 274). Using these paracrine effects for cell-free therapy is clinically relevant, since ESCs and iPSCs can induce teratoma formation post-transplantation (275–277). This currently limits the utility of these cells in clinical studies. Furthermore, recent studies indicate that dissemination of cells or pre-differentiation of iPSCs in lung progenitor cells might overcome this problem (278, 279). However, this must be confirmed prior to the first clinical implementation.
Low engraftment rates and survival of transplanted stem cells might be one of the reasons for the failure of the first studies of clinical stem cell transplantation in COPD. Cell transplantation had no significant impact on pulmonary functions or exacerbations. However, circulating c-reactive protein was reduced in some studies, although for a limited time (280, 281). In other clinical studies, the number of patients included were either too low for sufficient interpretation (282) or the improvements in FEV1 were attributable to lung volume reduction surgery (283). Nevertheless, these studies provide substantial evidence for the safety of MSC transplantation in humans and thereby the basis for future studies.
It has been suggested that patients with advanced stages of COPD who are included in clinical studies might display pulmonary damage too severe to be reversed or repaired by transplanted cells (280). Therefore, patient inclusion criteria will be important for successful study protocols in the future. In addition, aging bone marrow-derived stem cells from animals and humans show impaired proliferation, decreased differentiation potential, and secretion of paracrine factors (284, 285). However, these age-dependent changes seem not to affect the entire stem cell population, as some subpopulations retain a more “youthful” phenotype (286). Therefore, pre-selection and perhaps reprogramming of suitable cells also needs to be considered in future studies. In this context, reprogramming of epigenetic modifications in COPD lung fibroblasts in vitro through an iPSC intermediate state was shown to result in fibroblasts similar to fibroblasts from non-COPD patients, indicating a possible treatment option without cellular transfer (287). Furthermore, the vitamin retinoic acid is important for pulmonary development, e.g., for progenitor cell differentiation, and was shown to be beneficial in animal emphysema models (288–293). Although clinical evidence is lacking (294), the reprogramming or activation of intrinsic stem and progenitor cells suggests an additional therapeutic treatment option.
Recent studies provide evidence that EPCs play a critical role in COPD. Circulating endothelial progenitor cell reduction in COPD is correlated with disease severity and inversely correlated with the extent of emphysema, whereas hematopoietic progenitor cells are unaltered (295–297). Intratracheal application of bone marrow-derived EPCs attenuated the development of pulmonary emphysema in mice exposed to CS in a long term murine study, via the alleviation of inflammatory infiltration, decreased proteolytic enzyme activity, and improved antioxidant activity (298). In contrast, enhanced progenitor cell numbers in pulmonary arteries obtained from patients with COPD were associated with decreased endothelium-dependent dilation and inversely correlated with arterial lumen area (299). However, the characterization of these cells by progenitor marker expression was limited in this study. As the level of circulating EPCs is decreased but their migratory potential and adhesion is increased in COPD, it was suggested that circulating ECs are recruited to pulmonary tissue in COPD (296, 299, 300). This might not be beneficial in COPD patients, since circulating progenitor cells in these patients show impaired angiogenic ability, increased apoptosis, and impaired NO production compared with these features of EPCs obtained from healthy, non-smoking controls (301, 302). The beneficial effects of endothelial progenitor cell transplantation obtained by Shi et al. (298) might at least partially be attributed to the non-COPD origin of stem cells in this study. Therefore, modulation of EPCs in COPD in vivo or pre-transplantation to re-achieve physiological cell functions might be a promising approach in the treatment of COPD.
Taking the above evidence together, the mechanisms underlying COPD and PH are still not fully understood. However, experimental studies and clinical observations have mechanistically linked vascular dysfunction with the development of COPD. Vascular remodeling and PH can occur in cases of COPD, not only in severe cases but also in mild-to-moderate forms of the disease and even in smokers with no airflow limitations. COPD associated with PH and pulmonary vascular remodeling is a multifactorial disease which involves hypoxia-related, hypoxia-unrelated, inflammatory, and endothelial dysfunction-associated mechanisms. Recent investigations have changed the view of the pathophysiology of COPD. In the past, vascular remodeling was suggested to be a secondary event occurring following the destruction of the parenchyma, with the predominant causes being hypoxia and hypoxemia. This perception has changed, with recent observations demonstrating that vascular abnormalities can be early events in COPD, which precede airflow limitations and emphysema independent of hypoxia. PH and lung emphysema can occur independently, suggesting that vascular molecular alterations can be a trigger for the development of lung emphysema. In this regard, the NO–sGC–cGMP axis might play an important role in both pathology and regenerative potential. However, further investigations of the contribution of pulmonary vascular changes to the development of COPD are needed to identify new therapeutic targets for this disease. The endothelium is known to regulate homeostasis between vasoconstriction and vasodilation, providing adequate perfusion pressure. However, exposure to toxic radicals can lead to ED via leukocyte infiltration and adhesion, resulting in tissue injury. Thus, ED changes the permeability of tissues and leads to structural damage of arterial walls via the proliferation of SMCs, resulting in CVD (303, 304). Cigarette smoke-induced CVD reduces the regenerative capability of the cardiovascular system; however, EPCs modulate vasculogenesis. EPCs modify cardiovascularity via the release of cytokines, vascular growth factors, and chemokines, thereby promoting transdifferentiation of cardiac SMCs and reducing the risk of CVD (305–307). Stem cell-based therapeutic strategies should therefore be investigated for the treatment of ED in PH and CVD-associated COPD patients.
SK, MS, FK, AS, TM, AB, DK, and KK conceived the focus of the review, wrote, and drafted the paper. NW and SE contributed intellectual content and expertise. SK, MS, and FK prepared the figures. SK supervised the study. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Patel AR, Patel AR, Singh S, Singh S, Khawaja I. Global initiative for chronic obstructive lung disease: the changes made. Cureus. (2019) 11:e4985. doi: 10.7759/cureus.4985
2. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. (2006) 3:2011–30. doi: 10.1371/journal.pmed.0030442
3. Sertogullarindan B, Gumrukcuoglu HA, Sezgi C, Akil MA. Frequency of pulmonary hypertension in patients with COPD due to biomass smoke and tobacco smoke. Int J Med Sci. (2012) 9:406–12. doi: 10.7150/ijms.4715
4. Barnes PJ, Burney PGJ, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Prim. (2015) 1:15076. doi: 10.1038/nrdp.2015.76
5. Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L612–31. doi: 10.1152/ajplung.00390.2007
6. Sharafkhaneh A, Hanania NA, Kim V. Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc. (2008) 5:475–7. doi: 10.1513/pats.200708-126ET
7. Austin V, Crack PJ, Bozinovski S, Miller AA, Vlahos R. COPD and stroke: are systemic inflammation and oxidative stress the missing links? Clin Sci. (2016) 130:1039–50. doi: 10.1042/CS20160043
8. Chan SMH, Selemidis S, Bozinovski S, Vlahos R. Pathobiological mechanisms underlying metabolic syndrome (MetS) in chronic obstructive pulmonary disease (COPD): clinical significance and therapeutic strategies. Pharmacol Ther. (2019) 198:160–88. doi: 10.1016/j.pharmthera.2019.02.013
9. McGuinness A, Sapey E. Oxidative stress in COPD: sources, markers, and potential mechanisms. J Clin Med. (2017) 6:21. doi: 10.3390/jcm6020021
10. Black PN, Ching PST, Beaumont B, Ranasinghe S, Taylor G, Merrilees MJ. Changes in elastic fibres in the small airways and alveoli in COPD. Eur Respir J. (2008) 31:998–1004. doi: 10.1183/09031936.00017207
11. Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. (2003) 21:892–905. doi: 10.1183/09031936.03.00115402
12. Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax. (2005) 60:605–9. doi: 10.1136/thx.2005.042994
13. Peinado VI, Pizarro S, Barberà JA. Pulmonary vascular involvement in COPD. Chest. (2008) 134:808–14. doi: 10.1378/chest.08-0820
14. Keusch S, Hildenbrand FF, Bollmann T, Halank M, Held M, Kaiser R, et al. Tobacco smoke exposure in pulmonary arterial and thromboembolic pulmonary hypertension. Respiration. (2014) 88:38–45. doi: 10.1159/000359972
15. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. (2013) 144:266–73. doi: 10.1378/chest.12-2664
16. Pandey KC, De S, Mishra PK. Role of proteases in chronic obstructive pulmonary disease. Front Pharmacol. (2017) 8:512. doi: 10.3389/fphar.2017.00512
17. Rabe KF, Hurst JR, Suissa S. Cardiovascular disease and COPD: dangerous liaisons? Eur Respir Rev. (2018) 27:180057. doi: 10.1183/16000617.0057-2018
18. Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, et al. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest. (1995) 107:1193–8. doi: 10.1378/chest.107.5.1193
19. Cuttica MJ, Kalhan R, Shlobin OA, Ahmad S, Gladwin M, Machado RF, et al. Categorization and impact of pulmonary hypertension in patients with advanced COPD. Respir Med. (2010) 104:1877–82. doi: 10.1016/j.rmed.2010.05.009
20. Zangiabadi A, De Pasquale CG, Sajkov D. Pulmonary hypertension and right heart dysfunction in chronic lung disease. Biomed Res Int. (2014) 2014:739674. doi: 10.1155/2014/739674
21. Opitz I, Ulrich S. Pulmonary hypertension in chronic obstructive pulmonary disease and emphysema patients: prevalence, therapeutic options and pulmonary circulatory effects of lung volume reduction surgery. J Thorac Dis. (2018) 10:S2763–74. doi: 10.21037/jtd.2018.07.63
22. Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc. (2005) 2:20–2. doi: 10.1513/pats.200407-037MS
23. Leitao Filho FS, Sin DD. COPD and cardiovascular diseases: now is the time for action! Thorax. (2018) 73:799–800. doi: 10.1136/thoraxjnl-2018-211553
24. Arbex MA, Varella MdeCL, de Siqueira HR, de Mello FAF. Drogas antituberculose: Interações medicamentosas, efeitos adversos e utilização em situações especiais. parte 1: fármacos de primeira linha. J. Bras. Pneumol. (2010) 36:626–40. doi: 10.1590/S1806-37132010000500016
25. Blanco I, Gimeno E, Munoz PA, Pizarro S, Gistau C, Rodriguez-Roisin R, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. (2010) 181:270–8. doi: 10.1164/rccm.200907-0988OC
26. Pynnaert C, Lamotte M, Naeije R. Aerobic exercise capacity in COPD patients with and without pulmonary hypertension. Respir Med. (2010) 104:121–6. doi: 10.1016/j.rmed.2009.06.006
27. Seimetz M, Parajuli N, Pichl A, Bednorz M, Ghofrani HA, Schermuly RT, et al. Cigarette smoke-induced emphysema and pulmonary hypertension can be prevented by phosphodiesterase 4 and 5 inhibition in mice. PLoS ONE. (2015) 10:e0129327. doi: 10.1371/journal.pone.0129327
28. Ferrer E, Peinado VI, Castañeda J, Prieto-Lloret J, Olea E, González-Martín MC, et al. Effects of cigarette smoke and hypoxia on pulmonary circulation in the guinea pig. Eur Respir J. (2011) 38:617–27. doi: 10.1183/09031936.00105110
29. Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. (2011) 147:293–305. doi: 10.1016/j.cell.2011.08.035
30. Baraldo S, Turato G, Saetta M. Pathophysiology of the small airways in chronic obstructive pulmonary disease. Respiration. (2012) 84:89–97. doi: 10.1159/000341382
31. Berg K, Wright JL. The pathology of chronic obstructive pulmonary disease progress in the 20th and 21st centuries. Arch Pathol Lab Med. (2016) 140:1423–8. doi: 10.5858/arpa.2015-0455-RS
32. Schrijen F, Uffholtz H, Polu JM, Poincelot F. Pulmonary and systemic hemodynamic evolution in chronic bronchitis. Am Rev Respir Dis. (1978) 117:25–31.
33. Weitzenblum E, Loiseau A, Hirth C, Mirhom R, Rasaholinjanahary J. Course of pulmonary hemodynamics in patients with chronic obstructive pulmonary disease. Chest. (1979) 75:656–62. doi: 10.1378/chest.75.6.656
34. Weitzenblum E, Sautegeau A, Ehrhart M, Mammosser M, Hirth C, Roegel E. Long-term course of pulmonary arterial pressure in chronic obstructive pulmonary disease. Am Rev Respir Dis. (1984) 130:993–8.
35. Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducoloné A, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. (2001) 164:219–24. doi: 10.1164/ajrccm.164.2.2006129
36. Chebib N, Mornex JF, Traclet J, Philit F, Khouatra C, Zeghmar S, et al. Pulmonary hypertension in chronic lung diseases: comparison to other pulmonary hypertension groups. Pulm Circ. (2018) 8:2045894018775056. doi: 10.1177/2045894018775056
37. Shujaat A, Bajwa AA, Cury JD. Pulmonary hypertension secondary to COPD. Pulm Med. (2012) 2012:203952. doi: 10.1155/2012/203952
38. Cukic V. The changes of arterial blood gases in COPD during four-year period. Med Arh. (2014) 68:14–8. doi: 10.5455/medarh.2014.68.14-18
39. Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. (2019) 53:1801914. doi: 10.1183/13993003.01914-2018
40. Richens JM, Howard P. Oedema in cor pulmonale. Clin Sci. (1982) 62:255–9. doi: 10.1042/cs0620255
41. MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (1994) 150:833–52. doi: 10.1164/ajrccm.150.3.8087359
42. Farber MO, Weinberger MH, Robertson GL, Fineberg NS, Manfredi F. Hormonal abnormalities affecting sodium and water balance in acute respiratory failure due to chronic obstructive lung disease. Chest. (1984) 85:49–54. doi: 10.1378/chest.85.1.49
43. Weitzenblum E, Apprill M, Oswald M, Chaouat A, Imbs JL. Pulmonary hemodynamics in patients with chronic obstructive pulmonary disease before and during an episode of peripheral edema. Chest. (1994) 105:1377–82. doi: 10.1378/chest.105.5.1377
44. Chaouat A, Bugnet A-S, Kadaoui N, Schott R, Enache I, Ducoloné A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2005) 172:189–94. doi: 10.1164/rccm.200401-006OC
45. Elwing J, Panos RJ. Pulmonary hypertension associated with COPD. Int J COPD. (2008) 3:55–70. doi: 10.2147/COPD.S1170
46. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. (2019) 53:2018. doi: 10.1183/13993003.01913-2018
47. Kawut SM, Poor HD, Parikh MA, Hueper K, Smith BM, Bluemke DA, et al. Cor pulmonale parvus in chronic obstructive pulmonary disease and emphysema: the MESA COPD study. J Am Coll Cardiol. (2014) 64:2000–9. doi: 10.1016/j.jacc.2014.07.991
48. Hohlfeld JM, Vogel-Claussen J, Biller H, Berliner D, Berschneider K, Tillmann HC, et al. Effect of lung deflation with indacaterol plus glycopyrronium on ventricular filling in patients with hyperinflation and COPD (CLAIM): a double-blind, randomised, crossover, placebo-controlled, single-centre trial. Lancet Respir Med. (2018) 6:368–78. doi: 10.1016/S2213-2600(18)30054-7
49. Funk GC, Lang I, Schenk P, Valipour A, Hartl S, Burghuber OC. Left ventricular diastolic dysfunction in patients with COPD in the presence and absence of elevated pulmonary arterial pressure. Chest. (2008) 133:1354–9. doi: 10.1378/chest.07-2685
50. Sin DD, Paul Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. (2003) 107:1514–9. doi: 10.1161/01.CIR.0000056767.69054.B3
51. Burrows B, Kettel LJ, Niden AH, Rabinowitz M, Diener CF. Patterns of cardiovascular dysfunction in chronic obstructive lung disease. N Engl J Med. (1972) 286:912–8. doi: 10.1056/NEJM197204272861703
52. Jamal K, Fleetham JA, Thurlbeck WM. Cor pulmonale: correlation with central airway lesions, peripheral airway lesions, emphysema, and control of breathing. Am Rev Respir Dis. (1990) 141:1172–7. doi: 10.1164/ajrccm/141.5_Pt_1.1172
53. Kawut SM. COPD: cardiopulmonary disease? Eur Respir J. (2013) 41:1241–3. doi: 10.1183/09031936.00009413
54. Watz H, Waschki B, Meyer T, Kretschmar G, Kirsten A, Claussen M, et al. Decreasing cardiac chamber sizes and associated heart dysfunction in COPD: role of hyperinflation. Chest. (2010) 138:32–8. doi: 10.1378/chest.09-2810
55. Lammi MR, Ciccolella D, Marchetti N, Kohler M, Criner GJ. Increased oxygen pulse after lung volume reduction surgery is associated with reduced dynamic hyperinflation. Eur Respir J. (2012) 40:837–43. doi: 10.1183/09031936.00169311
56. Ergan B, Nava S. Long-term oxygen therapy in COPD patients who do not meet the actual recommendations. COPD J Chronic Obstr Pulm Dis. (2017) 14:351–66. doi: 10.1080/15412555.2017.1319918
57. Katsenos S, Constantopoulos SH. Long-term oxygen therapy in COPD: factors affecting and ways of improving patient compliance. Pulm Med. (2011) 2011:325362. doi: 10.1155/2011/325362
58. Koczulla AR, Schneeberger T, Jarosch I, Kenn K, Gloeckl R. Long-term oxygen therapy current evidence and practical, day to day consideration. Dtsch Arztebl Int. (2018) 115:871–7. doi: 10.3238/arztebl.2018.0871
59. Weitzenblum E, Chaouat A, Kessler R. Pulmonary hypertension in chronic obstructive pulmonary disease. Pneumonol Alergol Pol. (2013) 81:390–8.
60. Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. (2017) 367:643–9. doi: 10.1007/s00441-016-2539-y
61. Jain M, Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Cornelissen A, et al. Smooth muscle cell-specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J Clin Invest. (2020) 130:295–314. doi: 10.1172/JCI124708
62. Gabbiani G, Rungger-Brändle E, de Chastonay C, Franke WW. Vimentin-containing smooth muscle cells in aortic intimal thickening after endothelial injury. Lab Invest. (1982) 47:265–9.
63. Rodríguez-Roisin R, Drakulovic M, Rodríguez DA, Roca J, Barberà JA, Wagner PD. Ventilation-perfusion imbalance and chronic obstructive pulmonary disease staging severity. J Appl Physiol. (2009) 106:1902–8. doi: 10.1152/japplphysiol.00085.2009
64. Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med. (2013) 91:297–309. doi: 10.1007/s00109-013-0998-0
65. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. (2017) 50:1601805. doi: 10.1183/13993003.01805-2016
66. Sekhon HS, Wright JL, Churg A. Cigarette smoke causes rapid cell proliferation in small airways and associated pulmonary arteries. Am J Physiol Lung Cell Mol Physiol. (1994) 267:L557–63. doi: 10.1152/ajplung.1994.267.5.L557
67. Barberà JA, Riverola A, Roca J, Ramirez J, Wagner PD, Ros D, et al. Pulmonary vascular abnormalities and ventilation-perfusion relationships in mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (1994) 149:423–9. doi: 10.1164/ajrccm.149.2.8306040
68. Xu J, Shi GP. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta Mol Basis Dis. (2014) 1842:2106–19. doi: 10.1016/j.bbadis.2014.07.008
69. Stenmark KR, Frid MG, Graham BB, Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res. (2018) 114:551–64. doi: 10.1093/cvr/cvy004
70. Rabinovitch M. Elastase and the pathobiology of unexplained pulmonary hypertension. Chest. (1998) 114:213S−24S. doi: 10.1378/chest.114.3_Supplement.213S
71. Wilkinson M, Langhorne CA, Heath D, Barer GR, Howard P. A pathophysiological study of 10 cases of hypoxic cor pulmonale. Q J Med. (1988) 66:65–85.
72. Barberà JA. Mechanisms of development of chronic obstructive pulmonary disease-associated pulmonary hypertension. Pulm Circ. (2013) 3:160–4. doi: 10.4103/2045-8932.109949
73. Naeije R, Barberà JA. Pulmonary hypertension associated with COPD. Crit Care. (2001) 5:286–9. doi: 10.1186/cc1049
74. Lejeune P, Mols P, Naeije R, Hallemans R, Melot C. Acute hemodynamic effects of controlled oxygen therapy in decompensated chronic obstructive pulmonary disease. Crit Care Med. (1984) 12:1032–5. doi: 10.1097/00003246-198412000-00006
75. Timms RM, Khaja FU, Williams GW. Hemodynamic response to oxygen therapy in chronic obstructive pulmonary disease. Ann Intern Med. (1985) 102:29–36. doi: 10.7326/0003-4819-102-1-29
76. Voelkel NF, Gomez-Arroyo J, Mizuno S. COPD/emphysema: the vascular story. Pulm Circ. (2011) 1:320–6. doi: 10.4103/2045-8932.87295
77. Coppolino I, Ruggeri P, Nucera F, Cannavò MF, Adcock I, Girbino G, et al. Role of stem cells in the pathogenesis of chronic obstructive pulmonary disease and pulmonary emphysema. COPD J Chronic Obstr Pulm Dis. (2018) 15:536–56. doi: 10.1080/15412555.2018.1536116
78. Polverino F, Celli BR, Owen CA. COPD as an endothelial disorder: endothelial injury linking lesions in the lungs and other organs? (2017 Grover Conference Series). In: Pulmonary Circulation. SAGE Publications Ltd. (2018). p. 2045894018758528. doi: 10.1177/2045894018758528
79. Santos S, Peinado VI, Ramírez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. (2002) 19:632–8. doi: 10.1183/09031936.02.00245902
80. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. (2008) 32:1371–85. doi: 10.1183/09031936.00015608
81. Fishman AP. Hypoxia on the pulmonary circulation. How and where it acts. Circ Res. (1976) 38:221–31. doi: 10.1161/01.RES.38.4.221
82. Fishman AP. State of the art: chronic cor pulmonale. Am Rev Respir Dis. (1976) 114:775–94. doi: 10.1164/arrd.1976.114.4.775
83. Chao J, Wood JG, Blanco VG, Gonzalez NC. The systemic inflammation of alveolar hypoxia is initiated by alveolar macrophage-borne mediator(s). Am J Respir Cell Mol Biol. (2009) 41:573–82. doi: 10.1165/rcmb.2008-0417OC
84. Chao J, Viets Z, Donham P, Wood JG, Gonzalez NC. Dexamethasone blocks the systemic inflammation of alveolar hypoxia at several sites in the inflammatory cascade. Am J Physiol Heart Circ Physiol. (2012) 303:H168–77. doi: 10.1152/ajpheart.00106.2012
85. Voelkel NF, Mizuno S, Bogaard HJ. The role of hypoxia in pulmonary vascular diseases: a perspective. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L457–65. doi: 10.1152/ajplung.00335.2012
86. Prabhakar NR, Kumar GK, Peng Y-J. Sympatho-adrenal activation by chronic intermittent hypoxia. J Appl Physiol. (2012) 113:1304–10. doi: 10.1152/japplphysiol.00444.2012
87. Pullamsetti SS, Perros F, Chelladurai P, Yuan J, Stenmark K. Transcription factors, transcriptional coregulators, and epigenetic modulation in the control of pulmonary vascular cell phenotype: therapeutic implications for pulmonary hypertension (2015 grover conference series). Pulm Circ. (2016) 6:448–64. doi: 10.1086/688908
88. Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med. (2014) 189:314–24. doi: 10.1164/rccm.201302-0302OC
89. Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L229–52. doi: 10.1152/ajplung.00238.2014
90. Fu X, Zhang F. Role of the hif-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med. (2018) 16:4553–61. doi: 10.3892/etm.2018.6785
91. Jiang H, Zhu Y, Xu H, Sun Y, Li Q. Activation of hypoxia-inducible factor-1 via nuclear factor-B in rats with chronic obstructive pulmonary disease. Acta Biochim Biophys Sin. (2010) 42:483–8. doi: 10.1093/abbs/gmq041
92. van Uden P, Kenneth NS, Webster R, Müller HA, Mudie S, Rocha S. Evolutionary conserved regulation of HIF-1β by nf-κb. PLoS Genet. (2011) 7:e1001285. doi: 10.1371/journal.pgen.1001285
93. D'Ignazio L, Bandarra D, Rocha S. NF-κB and HIF crosstalk in immune responses. FEBS J. (2016) 283:413–24. doi: 10.1111/febs.13578
94. Su YC, Jalalvand F, Thegerström J, Riesbeck K. The interplay between immune response and bacterial infection in COPD: focus upon non-typeable Haemophilus influenzae. Front Immunol. (2018) 9:2530. doi: 10.3389/fimmu.2018.02530
95. Wei J, Xiong XF, Lin YH, Zheng BX, Cheng DY. Association between serum interleukin-6 concentrations and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PeerJ. (2015) 2015:e1199. doi: 10.7717/peerj.1199
96. Maston LD, Jones DT, Giermakowska W, Resta TC, Ramiro-Diaz J, Howard TA, et al. Interleukin-6 trans-signaling contributes to chronic hypoxia-induced pulmonary hypertension. Pulm Circ. (2018) 8:2045894018780734. doi: 10.1177/2045894018780734
97. Kristensen SG, Andersen K, Clement CA, Franks S, Hardy K, Andersen CY. Expression of TGF-beta superfamily growth factors, their receptors, the associated SMADs and antagonists in five isolated size-matched populations of pre-antral follicles from normal human ovaries. Mol Hum Reprod. (2014) 20:293–308. doi: 10.1093/molehr/gat089
98. Rol N, Kurakula KB, Happé C, Bogaard HJ, Goumans MJ. TGF-β and BMPR2 signaling in PAH: two black sheep in one family. Int J Mol Sci. (2018) 19:2585. doi: 10.3390/ijms19092585
99. Yuan SM. Pulmonary artery hypertension in childhood: the transforming growth factor-β superfamily-related genes. Pediatr Neonatol. (2018) 59:112–9. doi: 10.1016/j.pedneo.2016.12.008
100. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. (2012) 122:4306–13. doi: 10.1172/JCI60658
101. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. (2002) 105:1672–8. doi: 10.1161/01.CIR.0000012754.72951.3D
102. Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, et al. Krüppel-like Factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res. (2011) 12:128. doi: 10.1186/1465-9921-12-128
103. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. (2009) 104:236–44. doi: 10.1161/CIRCRESAHA.108.182014
104. Vanderpool RR, El-Bizri N, Rabinovitch M, Chesler NC. Patchy deletion of Bmpr1a potentiates proximal pulmonary artery remodeling in mice exposed to chronic hypoxia. Biomech Model Mechanobiol. (2013) 12:33–42. doi: 10.1007/s10237-012-0379-6
105. Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol Circ Physiol. (1994) 266:H365–70. doi: 10.1152/ajpheart.1994.266.1.H365
106. Remillard CV, Yuan JX-J. High altitude pulmonary hypertension: role of K+ and Ca2+ channels. High Alt Med Biol. (2005) 6:133–46. doi: 10.1089/ham.2005.6.133
107. Moudgil R, Michelakis ED, Archer SL. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation. (2006) 13:615–32. doi: 10.1080/10739680600930222
108. Grimminger F, Weissmann N, Spriestersbach R, Becker E, Rosseau S, Seeger W. Effects of NADPH oxidase inhibitors on hypoxic vasoconstriction in buffer-perfused rabbit lungs. Am J Physiol Cell Mol Physiol. (1995) 268:L747–52. doi: 10.1152/ajplung.1995.268.5.L747
109. Weissmann N, Tadic A, Hänze J, Rose F, Winterhalder S, Nollen M, et al. Hypoxic vasoconstriction in intact lungs: a role for NADPH oxidase-derived H(2)O(2)? Am J Physiol Lung Cell Mol Physiol. (2000) 279:L683–90. doi: 10.1152/ajplung.2000.279.4.L683
110. Weissmann N, Zeller S, Schäfer RU, Turowski C, Ay M, Quanz K, et al. Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. (2006) 34:505–13. doi: 10.1165/rcmb.2005-0337OC
111. Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, et al. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. (2002) 90:1307–15. doi: 10.1161/01.RES.0000024689.07590.C2
112. Waypa GB, Schumacker PT. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol. (2005) 98:404–14. doi: 10.1152/japplphysiol.00722.2004
113. Mittal M, Gu XQ, Pak O, Pamenter ME, Haag D, Fuchs DB, et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med. (2012) 52:1033–42. doi: 10.1016/j.freeradbiomed.2011.12.004
114. Landsberg JW, Yuan JXJ. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci. (2004) 19:44–50. doi: 10.1152/nips.01457.2003
115. Lai N, Lu W, Wang J. Ca(2+) and ion channels in hypoxia-mediated pulmonary hypertension. Int J Clin Exp Pathol. (2015) 8:1081–92.
116. Weise-Cross L, Resta TC, Jernigan NL. Redox regulation of ion channels and receptors in pulmonary hypertension. Antioxidants Redox Signal. (2019) 31:898–915. doi: 10.1089/ars.2018.7699
117. Buckler KJ. TASK channels in arterial chemoreceptors and their role in oxygen and acid sensing. Pflugers Arch Eur J Physiol. (2015) 467:1013–25. doi: 10.1007/s00424-015-1689-1
118. Lambert M, Capuano V, Olschewski A, Sabourin J, Nagaraj C, Girerd B, et al. Ion channels in pulmonary hypertension: a therapeutic interest? Int J Mol Sci. (2018) 19:3162. doi: 10.3390/ijms19103162
119. Cheng J, Wen J, Wang N, Wang C, Xu Q, Yang Y. Ion channels and vascular diseases. Arterioscler Thromb Vasc Biol. (2019) 39:e146–56. doi: 10.1161/ATVBAHA.119.312004
120. Kwapiszewska G, Wygrecka M, Marsh LM, Schmitt S, Trösser R, Wilhelm J, et al. Fhl-1, a new key protein in pulmonary hypertension. Circulation. (2008) 118:1183–94. doi: 10.1161/CIRCULATIONAHA.107.761916
121. Li Y, Pu G, Chen C, Yang L. Inhibition of FHL1 inhibits cigarette smoke extract-induced proliferation in pulmonary arterial smooth muscle cells. Mol Med Rep. (2015) 12:3801–8. doi: 10.3892/mmr.2015.3787
122. Veith C, Schermuly RT, Brandes RP, Weissmann N. Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J Physiol. (2016) 594:1167–77. doi: 10.1113/JP270689
123. Thabut G, Dauriat G, Stern JB, Logeart D, Lévy A, Marrash-Chahla R, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. (2005) 127:1531–6. doi: 10.1378/chest.127.5.1531
124. Sims MW, Margolis DJ, Localio AR, Panettieri RA, Kawut SM, Christie JD. Impact of pulmonary artery pressure on exercise function in severe COPD. Chest. (2009) 136:412–9. doi: 10.1378/chest.08-2739
125. Weitzenblum E, Sautegeau A, Ehrhart M, Mammosser M, Pelletier A. Long-term oxygen therapy can reverse the progression of pulmonary hypertension in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. (1985) 131:493–8. doi: 10.1164/arrd.1985.131.4.493
126. Wright JL, Petty T, Thurlbeck WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trial. Lung. (1992) 170:109–24. doi: 10.1007/BF00175982
127. Wright JL, Tai H, Dai J, Churg A. Cigarette smoke induces rapid changes in gene expression in pulmonary arteries. Lab Investig. (2002) 82:1391–8. doi: 10.1097/01.LAB.0000032806.45023.08
128. Wright JL, Tai H, Churg A. Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries. Am J Respir Cell Mol Biol. (2004) 31:501–9. doi: 10.1165/rcmb.2004-0051OC
129. Yamato H, Sun JP, Churg A, Wright JL. Cigarette smoke-induced emphysema in guinea pigs is associated with diffusely decreased capillary density and capillary narrowing. Lab Invest. (1996) 75:211–9.
130. Lee SD, Lee DS, Chun YG, Shim TS, Lim CM, Koh Y, et al. Cigarette smoke extract induces endothelin-1 via protein kinase C in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L403–L411. doi: 10.1152/ajplung.2001.281.2.L403
131. Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med. (2007) 175:676–85. doi: 10.1164/rccm.200605-724OC
132. Peluffo G, Calcerrada P, Piacenza L, Pizzano N, Radi R. Superoxide-mediated inactivation of nitric oxide and peroxynitrite formation by tobacco smoke in vascular endothelium: studies in cultured cells and smokers. Am J Physiol Heart Circ Physiol. (2009) 296:H1781–H1792. doi: 10.1152/ajpheart.00930.2008
133. Damico R, Simms T, Kim BS, Tekeste Z, Amankwan H, Damarla M, et al. p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: inhibitory effects of macrophage migration inhibitor factor. Am J Respir Cell Mol Biol. (2011) 44:323–32. doi: 10.1165/rcmb.2009-0379OC
134. Milara J, Ortiz JL, Juan G, Guijarro R, Almudever P, Martorell M, et al. Cigarette smoke exposure up-regulates endothelin receptor B in human pulmonary artery endothelial cells: molecular and functional consequences. Br J Pharmacol. (2010) 161:1599–615. doi: 10.1111/j.1476-5381.2010.00979.x
135. Santos Valenca S, Castro P, Alves Pimenta W, Lanzetti M, Vargas Silva S, Barja-Fidalgo C, et al. Light cigarette smoke-induced emphysema and NFκB activation in mouse lung. Int J Exp Pathol. (2006) 87:373–81. doi: 10.1111/j.1365-2613.2006.00492.x
136. Xing AP, Du YC, Hu XY, Xu JY, Zhang HP, Li Y, et al. Cigarette smoke extract stimulates rat pulmonary artery smooth muscle cell proliferation via PKC-PDGFB signaling. J Biomed Biotechnol. (2012) 2012:534384. doi: 10.1155/2012/534384
137. Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. (1999) 159:1925–32. doi: 10.1164/ajrccm.159.6.9804054
138. Green CE, Turner AM. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir Res. (2017) 18:20. doi: 10.1186/s12931-017-0505-1
139. Weissmann N. Chronic obstructive pulmonary disease and pulmonary vascular disease a comorbidity? Ann Am Thorac Soc. (2018) 15:S278–81. doi: 10.1513/AnnalsATS.201808-532MG
140. Dinh-Xuan AT, Higenbottam TW, Clelland CA, Pepke-Zaba J, Cremona G, Butt AY, et al. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung disease. N Engl J Med. (1991) 324:1539–47. doi: 10.1056/NEJM199105303242203
141. Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J, et al. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2003) 167:1250–6. doi: 10.1164/rccm.200210-1233OC
142. Cremona G, Wood AM, Hall LW, Bower EA, Higenbottam T. Effect of inhibitors of nitric oxide release and action on vascular tone in isolated lungs of pig, sheep, dog and man. J Physiol. (1994) 481:185–95. doi: 10.1113/jphysiol.1994.sp020429
143. Stamler JS, Loh E, Roddy MA, Currie KE, Creager MA. Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation. (1994) 89:2035–40. doi: 10.1161/01.CIR.89.5.2035
144. Barberà JA, Roger N, Roca J, Rovira I, Higenbottam TW, Rodriguez-Roisin R. Worsening of pulmonary gas exchange with nitric oxide inhalation in chronic obstructive pulmonary disease. Lancet. (1996) 347:436–40. doi: 10.1016/S0140-6736(96)90011-2
145. Karamsetty VS, MacLean MR, McCulloch KM, Kane KA, Wadsworth RM. Hypoxic constrictor response in the isolated pulmonary artery from chronically hypoxic rats. Respir Physiol. (1996) 105:85–93. doi: 10.1016/0034-5687(96)00047-3
146. Peinado VI, Barbera JA, Ramirez J, Gomez FP, Roca J, Jover L, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. (1998) 274:L908–13. doi: 10.1152/ajplung.1998.274.6.L908
147. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. (1993) 328:1732–9. doi: 10.1056/NEJM199306173282402
148. Barberà JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med. (2001) 164:709–13. doi: 10.1164/ajrccm.164.4.2101023
149. O'Donnell R, Breen D, Wilson S, Djukanovic R. Inflammatory cells in the airways in COPD. Thorax. (2006) 61:448–54. doi: 10.1136/thx.2004.024463
150. King PT. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin Transl Med. (2015) 4:68. doi: 10.1186/s40169-015-0068-z
151. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2016) 138:16–27. doi: 10.1016/j.jaci.2016.05.011
152. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J COPD. (2018) 13:3341–8. doi: 10.2147/COPD.S176122
153. Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (1999) 160:711–7. doi: 10.1164/ajrccm.160.2.9812020
154. Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, et al. Role for interleukin-6 in COPD-related pulmonary hypertension. Chest. (2009) 136:678–87. doi: 10.1378/chest.08-2420
155. Zengin E, Chalajour F, Gehling UM, Ito WD, Treede H, Lauke H, et al. Vascular wall resident progenitor cells: a source for postnatal vasculogenesis. Development. (2006) 133:1543–51. doi: 10.1242/dev.02315
156. Psaltis PJ, Puranik AS, Spoon DB, Chue CD, Hoffman SJ, Witt TA, et al. Characterization of a resident population of adventitial macrophage progenitor cells in postnatal vasculature. Circ Res. (2014) 115:364–75. doi: 10.1161/CIRCRESAHA.115.303299
157. Mekala SR, Wörsdörfer P, Bauer J, Stoll O, Wagner N, Reeh L, et al. Generation of cardiomyocytes from vascular adventitia-resident stem cells. Circ Res. (2018) 123:686–99. doi: 10.1161/CIRCRESAHA.117.312526
158. Kar S, Kavdia M. Local oxidative and nitrosative stress increases in the microcirculation during leukocytes-endothelial cell interactions. PLoS ONE. (2012) 7:e38912. doi: 10.1371/journal.pone.0038912
159. Borrill ZL, Roy K, Singh D. Exhaled breath condensate biomarkers in COPD. Eur Respir J. (2008) 32:472–86. doi: 10.1183/09031936.00116107
160. Corhay JL, Moermans C, Henket M, Dang DN, Duysinx B, Louis R. Increased of exhaled breath condensate neutrophil chemotaxis in acute exacerbation of COPD. Respir Res. (2014) 15:115. doi: 10.1186/s12931-014-0115-0
161. Twigg MS, Brockbank S, Lowry P, Fitzgerald SP, Taggart C, Weldon S. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators Inflamm. (2015) 2015:293053. doi: 10.1155/2015/293053
162. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. (1994) 74:1141–8. doi: 10.1161/01.RES.74.6.1141
163. Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol. (2007) 18:2439–46. doi: 10.1681/ASN.2007020149
164. Ushio-Fukai M, Maziar Zafari A, Fukui T, Ishizaka N, Griendling KK. p22(phox) is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. (1996) 271:23317–21. doi: 10.1074/jbc.271.38.23317
165. Luo Z, Teerlink T, Griendling K, Aslam S, Welch WJ, Wilcox CS. Angiotensin II and NADPH oxidase increase ADMA in vascular smooth muscle cells. Hypertens. (2010) 56:498–504. doi: 10.1161/HYPERTENSIONAHA.110.152959
166. Manogue MR, Bennett JR, Holland DS, Choi CS, Drake DA, Taylor MS, et al. Smooth muscle specific overexpression of p22phox potentiates carotid artery wall thickening in response to injury. Oxid Med Cell Longev. (2015) 2015:305686. doi: 10.1155/2015/305686
167. Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. (2002) 90:251–62. doi: 10.1161/res.90.3.251
168. Ilumets H, Rytilä P, Demedts I, Brusselle GG, Sovijärvi A, Myllärniemi M, et al. Matrix metalloproteinases-8,-9 and-12 in smokers and patients with stage 0 COPD. Int J Chron Obstruct Pulmon Dis. (2007) 2:369–79.
169. Gilowska I, Kasper Ł, Bogacz K, Szczegielniak J, Szymasek T, Kasper M, et al. Impact of matrix metalloproteinase 9 on COPD development in polish patients: genetic polymorphism, protein level, and their relationship with lung function. Biomed Res Int. (2018) 2018:6417415. doi: 10.1155/2018/6417415
170. Pu Q, Neves MF, Virdis A, Touyz RM, Schiffrin EL. Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodeling. Hypertension. (2003) 42:49–55. doi: 10.1161/01.HYP.0000078357.92682.EC
171. Pollock DM. Endothelin, angiotensin, and oxidative stress in hypertension. Hypertension. (2005) 45:477–80. doi: 10.1161/01.HYP.0000158262.11935.d0
172. Liebow AA. Pulmonary emphysema with special reference to vascular changes. Am Rev Respir Dis. (1959) 80:67–93.
173. Cahill PA, Redmond EM. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis. (2016) 248:97–109. doi: 10.1016/j.atherosclerosis.2016.03.007
174. Ghavampour S, Kleefeldt F, Bömmel H, Volland J, Paus A, Horst A, et al. Endothelial barrier function is differentially regulated by CEACAM1-mediated signaling. FASEB J. (2018) 32:5612–25. doi: 10.1096/fj.201800331R
175. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
176. Harris B, Klein R, Jerosch-Herold M, Hoffman EA, Ahmed FS, Jacobs DR, et al. The association of systemic microvascular changes with lung function and lung density: a cross-sectional study. PLoS ONE. (2012) 7:e50224. doi: 10.1371/journal.pone.0050224
177. Kyomoto Y, Kanazawa H, Tochino Y, Watanabe T, Asai K, Kawaguchi T. Possible role of airway microvascular permeability on airway obstruction in patients with chronic obstructive pulmonary disease. Respir Med. (2019) 146:137–41. doi: 10.1016/j.rmed.2018.12.007
178. Heijink IH, Noordhoek JA, Timens W, Van Oosterhout AJM, Postma DS. Abnormalities in airway epithelial junction formation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2014) 189:1439–42. doi: 10.1164/rccm.201311-1982LE
179. Kim BG, Lee PH, Lee SH, Baek AR, Park JS, Lee J, et al. Impact of the endothelial tight junction protein claudin-5 on clinical profiles of patients with COPD. Allergy, Asthma Immunol Res. (2018) 10:533–42. doi: 10.4168/aair.2018.10.5.533
180. Butler A, Walton GM, Sapey E. Neutrophilic inflammation in the pathogenesis of chronic obstructive pulmonary disease. COPD J Chronic Obstr Pulm Dis. (2018) 15:392–404. doi: 10.1080/15412555.2018.1476475
181. Jasper AE, McIver WJ, Sapey E, Walton GM. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research. (2019) 8:F1000. doi: 10.12688/f1000research.18411.1
182. Tregay N, Begg M, Cahn A, Farahi N, Povey K, Madhavan S, et al. Use of autologous 99m Technetium-labelled neutrophils to quantify lung neutrophil clearance in COPD. Thorax. (2019) 74:659–66. doi: 10.1136/thoraxjnl-2018-212509
183. Woolhouse IS, Bayley DL, Lalor P, Adams DH, Stockley RA. Endothelial interactions of neutrophils under flow in chronic obstructive pulmonary disease. Eur Respir J. (2005) 25:612–7. doi: 10.1183/09031936.05.00086304
184. Blidberg K, Palmberg L, James A, Billing B, Henriksson E, Lantz AS, et al. Adhesion molecules in subjects with COPD and healthy non-smokers: a cross sectional parallel group study. Respir Res. (2013) 14:47. doi: 10.1186/1465-9921-14-47
185. Aaron CP, Schwartz JE, Bielinski SJ, Hoffman EA, Austin JHM, Oelsner EC, et al. Intercellular adhesion molecule 1 and progression of percent emphysema: the MESA Lung Study. Respir Med. (2015) 109:255–64. doi: 10.1016/j.rmed.2014.10.004
186. Barr RG, Mesia-Vela S, Austin JHM, Basner RC, Keller BM, Reeves AP, et al. Impaired flow-mediated dilation is associated with low pulmonary function and emphysema in ex-smokers: the Emphysema and Cancer Action Project (EMCAP) study. Am J Respir Crit Care Med. (2007) 176:1200–7. doi: 10.1164/rccm.200707-980OC
187. Clarenbach CF, Sievi NA, Kohler M. Annual progression of endothelial dysfunction in patients with COPD. Respir Med. (2017) 132:15–20. doi: 10.1016/j.rmed.2017.09.005
188. Vaes AW, Spruit MA, Theunis J, Goswami N, Vanfleteren LE, Franssen FME, et al. Endothelial function in patients with chronic obstructive pulmonary disease: a systematic review of studies using flow mediated dilatation. Expert Rev Respir Med. (2017) 11:1021–31. doi: 10.1080/17476348.2017.1389277
189. Estépar RSJ, Kinney GL, Black-Shinn JL, Bowler RP, Kindlmann GL, Ross JC, et al. Computed tomographic measures of pulmonary vascular morphology in smokers and their clinical implications. Am J Respir Crit Care Med. (2013) 188:231–9. doi: 10.1164/rccm.201301-0162OC
190. Washko GR, Nardelli P, Ash SY, Sanchez-Ferrero GV, Rahaghi FN, Come CE, et al. Arterial vascular pruning, right ventricular size, and clinical outcomes in chronic obstructive pulmonary disease a longitudinal observational study. Am J Respir Crit Care Med. (2019) 200:454–61. doi: 10.1164/rccm.201811-2063OC
191. Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. (2001) 163:737–44. doi: 10.1164/ajrccm.163.3.2002117
192. Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman SH, Hirth P, et al. Inhibition of vascular endothelial growth factor receptors causes lung cell apoptosis and emphysema. J Clin Invest. (2000) 106:1311–9. doi: 10.1172/JCI10259
193. Tang K, Rossiter HB, Wagner PD, Breen EC. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol. (2004) 97:1559–66. doi: 10.1152/japplphysiol.00221.2004
194. Wang L, Xu Z, Chen B, He W, Hu J, Zhang L, et al. The role of vascular endothelial growth factor in small-airway remodelling in a rat model of chronic obstructive pulmonary disease. Sci Rep. (2017) 7:41202. doi: 10.1038/srep41202
195. Bömmel H, Kleefeldt F, Zernecke A, Ghavampour S, Wagner N, Kuerten S, et al. Visualization of endothelial barrier damage prior to formation of atherosclerotic plaques. Histochem Cell Biol. (2017) 148:117–27. doi: 10.1007/s00418-017-1562-8
196. Bernardi E, Merlo C, Cogo A. Endothelial function in COPD is in an intermediate position between healthy subjects and coronary artery disease patients and is related to physical activity. Lung. (2018) 196:669–72. doi: 10.1007/s00408-018-0168-9
197. Arunachalam G, Sundar IK, Hwang JW, Yao H, Rahman I. Emphysema is associated with increased inflammation in lungs of atherosclerosis-prone mice by cigarette smoke: implications in comorbidities of COPD. J Inflamm. (2010) 7:34. doi: 10.1186/1476-9255-7-34
198. Rahman I, Van Schadewijk AAM, Crowther AJL, Hiemstra PS, Stolk J, MacNee W, et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2002) 166:490–5. doi: 10.1164/rccm.2110101
199. Montaño M, Cisneros J, Ramírez-Venegas A, Pedraza-Chaverri J, Mercado D, Ramos C, et al. Malondialdehyde and superoxide dismutase correlate with FEV(1) in patients with COPD associated with wood smoke exposure and tobacco smoking. Inhal Toxicol. (2010) 22:868–74. doi: 10.3109/08958378.2010.491840
200. Pasini AMF, Stranieri C, Peserico D, Garbin U, Cominacini L. Oxidative stress and NRF2 gene expression in peripheral blood mononuclear cells derived from COPD patients: a longitudinal study. Chest. (2019) 155:A363. doi: 10.1016/j.chest.2019.04.062
201. Sanders KA, Delker DA, Huecksteadt T, Beck E, Wuren T, Chen Y, et al. RAGE is a critical mediator of pulmonary oxidative stress, alveolar macrophage activation and emphysema in response to cigarette smoke. Sci Rep. (2019) 9:231. doi: 10.1038/s41598-018-36163-z
202. Cheng DT, Kim DK, Cockayne DA, Belousov A, Bitter H, Cho MH, et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2013) 188:948–57. doi: 10.1164/rccm.201302-0247OC
203. Cho MH, Castaldi PJ, Hersh CP, Hobbs BD, Barr RG, Tal-Singer R, et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am J Respir Crit Care Med. (2015) 192:559–69. doi: 10.1164/rccm.201501-0148OC
204. Goven D, Boutten A, Leçon-Malas V, Marchal-Sommé J, Amara N, Crestani B, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. (2008) 63:916–24. doi: 10.1136/thx.2007.091181
205. Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. (2008) 178:592–604. doi: 10.1164/rccm.200803-380OC
206. Blake DJ, Singh A, Kombairaju P, Malhotra D, Mariani TJ, Tuder RM, et al. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am J Respir Cell Mol Biol. (2010) 42:524–36. doi: 10.1165/rcmb.2009-0054OC
207. Cui W, Zhang Z, Zhang P, Qu J, Zheng C, Mo X, et al. Nrf2 attenuates inflammatory response in COPD/emphysema: crosstalk with Wnt3a/β-catenin and AMPK pathways. J Cell Mol Med. (2018) 22:3514–25. doi: 10.1111/jcmm.13628
208. Prange R, Thiedmann M, Bhandari A, Mishra N, Sinha A, Häsler R, et al. A Drosophila model of cigarette smoke induced COPD identifies Nrf2 signaling as an expedient target for intervention. Aging. (2018) 10:2122–35. doi: 10.18632/aging.101536
209. Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke–induced emphysema in mice. J Clin Invest. (2004) 114:1248–59. doi: 10.1172/JCI200421146
210. Weissmann N, Lobo B, Pichl A, Parajuli N, Seimetz M, Puig-Pey R, et al. Stimulation of soluble guanylate cyclase prevents cigarette smoke-induced pulmonary hypertension and emphysema. Am J Respir Crit Care Med. (2014) 189:1359–73. doi: 10.1164/rccm.201311-2037OC
211. Ghofrani HA, Humbert M, Langleben D, Schermuly R, Stasch JP, Wilkins MR, et al. Riociguat: mode of action and clinical development in pulmonary hypertension. Chest. (2017) 151:468–80. doi: 10.1016/j.chest.2016.05.024
212. Pichl A, Sommer N, Bednorz M, Seimetz M, Hadzic S, Kuhnert S, et al. Riociguat for treatment of pulmonary hypertension in COPD: a translational study. Eur Respir J. (2019) 53:1802445. doi: 10.1183/13993003.02445-2018
213. Paul T, Blanco I, Aguilar D, Tura-Ceide O, Bonjoch C, Smolders VF, et al. Therapeutic effects of soluble guanylate cyclase stimulation on pulmonary hemodynamics and emphysema development in guinea pigs chronically exposed to cigarette smoke. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L222–34. doi: 10.1152/ajplung.00399.2018
214. Domínguez-Fandos D, Valdés C, Ferrer E, Puig-Pey R, Blanco I, Tura-Ceide O, et al. Sildenafil in a cigarette smoke-induced model of COPD in the Guinea-pig. Eur Respir J. (2015) 46:346–54. doi: 10.1183/09031936.00139914
215. Li H, Förstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. (2000) 190:244–54. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8
216. Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. (2006) 113:286–95. doi: 10.1161/CIRCULATIONAHA.105.581405
217. Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HHHW, Stasch J-P. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. (2006) 5:755–68. doi: 10.1038/nrd2038
218. Kharitonov SA, Barnes PJ. Nitric oxide, nitrotyrosine, and nitric oxide modulators in asthma and chronic obstructive pulmonary disease. Curr Allergy Asthma Rep. (2003) 3:121–9. doi: 10.1007/s11882-003-0024-7
219. Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. (2004) 364:709–21. doi: 10.1016/S0140-6736(04)16900-6
220. Arif E, Ahsan A, Vibhuti A, Rajput C, Deepak D, Athar M, et al. Endothelial nitric oxide synthase gene variants contribute to oxidative stress in COPD. Biochem Biophys Res Commun. (2007) 361:182–8. doi: 10.1016/j.bbrc.2007.07.008
221. Förstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. (2008) 5:338–49. doi: 10.1038/ncpcardio1211
222. Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. (2006) 113:2630–41. doi: 10.1161/CIRCULATIONAHA.105.609008
223. Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. (2008) 294:H570–8. doi: 10.1152/ajpheart.01324.2007
224. Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, Dokic D, et al. Superoxide generated at mitochondrial complex iii triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med. (2013) 187:424–32. doi: 10.1164/rccm.201207-1294OC
225. Sommer N, Hüttemann M, Pak O, Scheibe S, Knoepp F, Sinkler C, et al. Mitochondrial complex IV subunit 4 isoform 2 is essential for acute pulmonary oxygen sensing. Circ Res. (2017) 121:424–38. doi: 10.1161/CIRCRESAHA.116.310482
226. Pak O, Scheibe S, Esfandiary A, Gierhardt M, Sydykov A, Logan A, et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur Respir J. (2018) 51:1701024. doi: 10.1183/13993003.01024-2017
227. Culley MK, Chan SY. Mitochondrial metabolism in pulmonary hypertension: beyond mountains there are mountains. J Clin Invest. (2018) 128:3704–15. doi: 10.1172/JCI120847
228. Chi AY, Waypa GB, Mungai PT, Schumacker PT. Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid Redox Signal. (2010) 12:603–10. doi: 10.1089/ars.2009.2861
229. Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, et al. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res. (2010) 106:526–35. doi: 10.1161/CIRCRESAHA.109.206334
230. Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. (2014) 115:148–64. doi: 10.1161/CIRCRESAHA.115.301130
231. Boucherat O, Vitry G, Trinh I, Paulin R, Provencher S, Bonnet S. The cancer theory of pulmonary arterial hypertension. Pulm Circ. (2017) 7:285–99. doi: 10.1177/2045893217701438
232. Giordano L, Farnham A, Dhandapani PK, Salminen L, Bhaskaran J, Voswinckel R, et al. Alternative oxidase attenuates cigarette smoke-induced lung dysfunction and tissue damage. Am J Respir Cell Mol Biol. (2019) 60:515–22. doi: 10.1165/rcmb.2018-0261OC
233. Hiemstra PS. Altered macrophage function in chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2013) 10(Suppl.):S180–5. doi: 10.1513/AnnalsATS.201305-123AW
234. Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. (2014) 5:435. doi: 10.3389/fimmu.2014.00435
235. Wick MJ, Buesing EJ, Wehling CA, Loomis ZL, Cool CD, Zamora MR, et al. Decreased neprilysin and pulmonary vascular remodeling in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 183:330–40. doi: 10.1164/rccm.201002-0154OC
236. Sunday ME, Hua J, Torday JS, Reyes B, Shipp MA. CD10/neutral endopeptidase 24.11 in developing human fetal lung. Patterns of expression and modulation of peptide-mediated proliferation. J Clin Invest. (1992) 90:2517–25. doi: 10.1172/JCI116145
237. Dusser DJ, Djokic TD, Borson DB, Nadel JA. Cigarette smoke induces bronchoconstrictor hyperresponsiveness to substance P and inactivates airway neutral endopeptidase in the guinea pig. Possible role of free radicals. J Clin Invest. (1989) 84:900–6. doi: 10.1172/JCI114251
238. Carpenter TC, Stenmark KR. Hypoxia decreases lung neprilysin expression and increases pulmonary vascular leak. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L941–8. doi: 10.1152/ajplung.2001.281.4.L941
239. Dempsey EC, Wick MJ, Karoor V, Barr EJ, Tallman DW, Wehling CA, et al. Neprilysin null mice develop exaggerated pulmonary vascular remodeling in response to chronic hypoxia. Am J Pathol. (2009) 174:782–96. doi: 10.2353/ajpath.2009.080345
240. Shinall H, Song ES, Hersh LB. Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral. Biochemistry. (2005) 44:15345–50. doi: 10.1021/bi050650l
241. Sumitomo M, Shen R, Nanus DM. Involvement of neutral endopeptidase in neoplastic progression. Biochim Biophys Acta Proteins Proteomics. (2005) 1751:52–9. doi: 10.1016/j.bbapap.2004.11.001
242. Papandreou CN, Nanus DM. Is methylation the key to CD10 loss? J Pediatr Hematol Oncol. (2010) 32:2–3. doi: 10.1097/MPH.0b013e3181c74aca
243. Karoor V, Oka M, Walchak SJ, Hersh LB, Miller YE, Dempsey EC. Neprilysin regulates pulmonary artery smooth muscle cell phenotype through a platelet-derived growth factor receptor-dependent mechanism. Hypertens. (2013) 61:921–30. doi: 10.1161/HYPERTENSIONAHA.111.199588
244. Chester AH, Yacoub MH. The role of endothelin-1 in pulmonary arterial hypertension. Glob Cardiol Sci Pract. (2014) 2014:29. doi: 10.5339/gcsp.2014.29
245. Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LXVIII Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol Rev. (2008) 60:1–42. doi: 10.1124/pr.107.07108
246. Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, et al. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1α. Circ Res. (2008) 102:1036–45. doi: 10.1161/CIRCRESAHA.107.169896
247. Thompson JS, Morice AH. Neutral endopeptidase inhibitors and the pulmonary circulation. Gen Pharmacol. (1996) 27:581–5. doi: 10.1016/0306-3623(95)02051-9
248. Casserly B, Klinger JR. Brain natriuretic peptide in pulmonary arterial hypertension: biomarker and potential therapeutic agent. Drug Des Devel Ther. (2009) 3:269–87. doi: 10.2147/DDDT.S4805
249. Silberbach M, Roberts CT. Natriuretic peptide signalling: molecular and cellular pathways to growth regulation. Cell Signal. (2001) 13:221–31. doi: 10.1016/S0898-6568(01)00139-5
250. Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. (2003) 93:907–16. doi: 10.1161/01.RES.0000100390.68771.CC
251. Willenbrock RC, Tremblay J, Garcia P, Hamet P. Dissociation of natriuresis and diuresis and heterogeneity of the effector system of atrial natriuretic factor in rats. J Clin Invest. (1989) 83:482–9. doi: 10.1172/JCI113907
252. Corti R, Burnett JC, Rouleau JL, Ruschitzka F, Lüscher TF. Vasopeptidase inhibitors: a new therapeutic concept in cardiovascular disease? Circulation. (2001) 104:1856–62. doi: 10.1161/hc4001.097191
253. Maki T, Nasa Y, Yamaguchi F, Yoshida H, Mori M, Takada T, et al. Long-term treatment with neutral endopeptidase inhibitor improves cardiac function and reduces natriuretic peptides in rats with chronic heart failure. Cardiovasc Res. (2001) 51:608–17. doi: 10.1016/S0008-6363(01)00258-9
254. Xu J, Carretero OA, Liu Y-H, Yang F, Shesely EG, Oja-Tebbe N, et al. Dual inhibition of ACE and NEP provides greater cardioprotection in mice with heart failure. J Card Fail. (2004) 10:83–9. doi: 10.1016/j.cardfail.2003.08.008
255. Daull P, Jeng AY, Battistini B. Towards triple vasopeptidase inhibitors for the treatment of cardiovascular diseases. J Cardiovasc Pharmacol. (2007) 50:247–56. doi: 10.1097/FJC.0b013e31813c6ca5
256. Sumitomo M, Iwase A, Zheng R, Navarro D, Kaminetzky D, Shen R, et al. Synergy in tumor suppression by direct interaction of neutral endopeptidase with PTEN. Cancer Cell. (2004) 5:67–78. doi: 10.1016/S1535-6108(03)00331-3
257. Wang D, Morales JE, Calame DG, Alcorn JL, Wetsel RA. Transplantation of human embryonic stem cell-derived alveolar epithelial type II cells abrogates acute lung injury in mice. Mol Ther. (2010) 18:625–34. doi: 10.1038/mt.2009.317
258. Li X, Zhang Y, Liang Y, Cui Y, Yeung SC, Ip MSM, et al. iPSC-derived mesenchymal stem cells exert SCF-dependent recovery of cigarette smoke-induced apoptosis/proliferation imbalance in airway cells. J Cell Mol Med. (2017) 21:265–77. doi: 10.1111/jcmm.12962
259. Lan YW, Yang JC, Yen CC, Huang TT, Chen YC, Chen HL, et al. Predifferentiated amniotic fluid mesenchymal stem cells enhance lung alveolar epithelium regeneration and reverse elastase-induced pulmonary emphysema. Stem Cell Res Ther. (2019) 10:163. doi: 10.1186/s13287-019-1282-1
260. Tsang KM, Hyun JS, Cheng KT, Vargas M, Mehta D, Ushio-Fukai M, et al. Embryonic stem cell differentiation to functional arterial endothelial cells through sequential activation of ETV2 and NOTCH1 signaling by HIF1α. Stem Cell Rep. (2017) 9:796–806. doi: 10.1016/j.stemcr.2017.07.001
261. Clayton ZE, Tan RP, Miravet MM, Lennartsson K, Cooke JP, Bursill CA, et al. Induced pluripotent stem cell-derived endothelial cells promote angiogenesis and accelerate wound closure in a murine excisional wound healing model. Biosci Rep. (2018) 38:BSR20180563. doi: 10.1042/BSR20180563
262. Olmer R, Engels L, Usman A, Menke S, Malik MNH, Pessler F, et al. Differentiation of human pluripotent stem cells into functional endothelial cells in scalable suspension culture. Stem Cell Rep. (2018) 10:1657–72. doi: 10.1016/j.stemcr.2018.03.017
263. Rosa S, Praça C, Pitrez PR, Gouveia PJ, Aranguren XL, Ricotti L, et al. Functional characterization of iPSC-derived arterial- and venous-like endothelial cells. Sci Rep. (2019) 9:3826. doi: 10.1038/s41598-019-40417-9
264. Zhou Q, Ye X, Sun R, Matsumoto Y, Moriyama M, Asano Y, et al. Differentiation of mouse induced pluripotent stem cells into alveolar epithelial cells in vitro for use in vivo. Stem Cells Transl Med. (2014) 3:675–85. doi: 10.5966/sctm.2013-0142
265. Shigemura N, Okumura M, Mizuno S, Imanishi Y, Nakamura T, Sawa Y. Autologous transplantation of adipose tissue-derived stromal cells ameliorates pulmonary emphysema. Am J Transplant. (2006) 6:2592–600. doi: 10.1111/j.1600-6143.2006.01522.x
266. Guan XJ, Song L, Han FF, Cui ZL, Chen X, Guo XJ, et al. Mesenchymal stem cells protect cigarette smoke-damaged lung and pulmonary function partly via VEGF-VEGF receptors. J Cell Biochem. (2013) 114:323–35. doi: 10.1002/jcb.24377
267. Antunes MA, Abreu SC, Cruz FF, Teixeira AC, Lopes-Pacheco M, Bandeira E, et al. Effects of different mesenchymal stromal cell sources and delivery routes in experimental emphysema. Respir Res. (2014) 15:118. doi: 10.1186/s12931-014-0118-x
268. Schweitzer KS, Johnstone BH, Garrison J, Rush NI, Cooper S, Traktuev DO, et al. Adipose stem cell treatment in mice attenuates lung and systemic injury induced by cigarette smoking. Am J Respir Crit Care Med. (2011) 183:215–25. doi: 10.1164/rccm.201001-0126OC
269. Katsha AM, Ohkouchi S, Xin H, Kanehira M, Sun R, Nukiwa T, et al. Paracrine factors of multipotent stromal cells ameliorate lung injury in an elastase-induced emphysema model. Mol Ther. (2011) 19:196–203. doi: 10.1038/mt.2010.192
270. Furuya N, Takenaga M, Ohta Y, Tokura Y, Hamaguchi A, Sakamaki A, et al. Cell therapy with adipose tissue-derived stem/stromal cells for elastase-induced pulmonary emphysema in rats. Regen Med. (2012) 7:503–12. doi: 10.2217/rme.12.25
271. Kim YS, Kim JY, Shin DM, Huh JW, Lee SW, Oh YM. Tracking intravenous adipose-derived mesenchymal stem cells in a model of elastase-induced emphysema. Tuberc Respir Dis. (2014) 77:116–23. doi: 10.4046/trd.2014.77.3.116
272. Huh JW, Kim SY, Lee JH, Lee JS, van Ta Q, Kim M, et al. Bone marrow cells repair cigarette smoke-induced emphysema in rats. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L255–66. doi: 10.1152/ajplung.00253.2010
273. Gu W, Song L, Li XM, Wang D, Guo XJ, Xu WG. Mesenchymal stem cells alleviate airway inflammation and emphysema in COPD through down-regulation of cyclooxygenase-2 via p38 and ERK MAPK pathways. Sci Rep. (2015) 5:8733. doi: 10.1038/srep08733
274. Zhang Y, Jiang X, Ren L. Optimization of the adipose-derived mesenchymal stem cell delivery time for radiation-induced lung fibrosis treatment in rats. Sci Rep. (2019) 9:5589. doi: 10.1038/s41598-019-41576-5
275. Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. (2009) 2:198–210. doi: 10.1016/j.scr.2009.02.002
276. Gutierrez-Aranda I, Ramos-Mejia V, Bueno C, Munoz-Lopez M, Real PJ, Mácia A, et al. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells. (2010) 28:1568–70. doi: 10.1002/stem.471
277. Gorecka J, Kostiuk V, Fereydooni A, Gonzalez L, Luo J, Dash B, et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res Ther. (2019) 10:87. doi: 10.1186/s13287-019-1185-1
278. Mitchell A, Wanczyk H, Jensen T, Finck C. Assessment of iPSC teratogenicity throughout directed differentiation toward an alveolar-like phenotype. Differentiation. (2019) 105:45–53. doi: 10.1016/j.diff.2019.01.003
279. Xiang M, Lu M, Quan J, Xu M, Meng D, Cui A, et al. Direct in vivo application of induced pluripotent stem cells is feasible and can be safe. Theranostics. (2019) 9:290–310. doi: 10.7150/thno.28671
280. Weiss DJ, Casaburi R, Flannery R, LeRoux-Williams M, Tashkin DP. A placebo-controlled, randomized trial of mesenchymal stem cells in COPD. Chest. (2013) 143:1590–8. doi: 10.1378/chest.12-2094
281. De Oliveira HG, Cruz FF, Antunes MA, De Macedo Neto AV, Oliveira GA, Svartman FM, et al. Combined bone marrow-derived mesenchymal stromal cell therapy and one-way endobronchial valve placement in patients with pulmonary emphysema: a phase i clinical trial. Stem Cells Transl Med. (2017) 6:962–9. doi: 10.1002/sctm.16-0315
282. Ribeiro-Paes JT, Bilaqui A, Greco OT, Ruiz MA, Marcelino MY, Stessuk T, et al. Unicentric study of cell therapy in chronic obstructive pulmonary disease/pulmonary emphysema. Int J COPD. (2011) 6:63–71. doi: 10.2147/COPD.S15292
283. Stolk J, Broekman W, Mauad T, Zwaginga JJ, Roelofs H, Fibbe WE, et al. A phase I study for intravenous autologous mesenchymal stromal cell administration to patients with severe emphysema. QJM. (2016) 109:331–6. doi: 10.1093/qjmed/hcw001
284. Cheng H, Qiu L, Ma J, Zhang H, Cheng M, Li W, et al. Replicative senescence of human bone marrow and umbilical cord derived mesenchymal stem cells and their differentiation to adipocytes and osteoblasts. Mol Biol Rep. (2011) 38:5161–8. doi: 10.1007/s11033-010-0665-2
285. Beane OS, Fonseca VC, Cooper LL, Koren G, Darling EM. Impact of aging on the regenerative properties of bone marrow-, muscle-, and adipose-derived mesenchymal stem/stromal cells. PLoS ONE. (2014) 9:e115963. doi: 10.1371/journal.pone.0115963
286. Block TJ, Marinkovic M, Tran ON, Gonzalez AO, Marshall A, Dean DD, et al. Restoring the quantity and quality of elderly human mesenchymal stem cells for autologous cell-based therapies. Stem Cell Res Ther. (2017) 8:239. doi: 10.1186/s13287-017-0688-x
287. Basma H, Gunji Y, Iwasawa S, Nelson A, Farid M, Ikari J, et al. Reprogramming of COPD lung fibroblasts through formation of induced pluripotent stem cells. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L552–65. doi: 10.1152/ajplung.00255.2013
288. Massaro GD, Massaro D. Postnatal treatment with retinoic acid increases the number of pulmonary alveoli in rats. Am J Physiol Cell Mol Physiol. (1996) 270:L305–10. doi: 10.1152/ajplung.1996.270.2.L305
289. Ishizawa K, Kubo H, Yamada M, Kobayashi S, Numasaki M, Ueda S, et al. Bone marrow-derived cells contribute to lung regeneration after elastase-induced pulmonary emphysema. FEBS Lett. (2004) 556:249–52. doi: 10.1016/S0014-5793(03)01399-1
290. Marquez HA, Cardoso WV. Vitamin A-retinoid signaling in pulmonary development and disease. Mol Cell Pediatr. (2016) 3:28. doi: 10.1186/s40348-016-0054-6
291. Ng-Blichfeldt JP, Schrik A, Kortekaas RK, Noordhoek JA, Heijink IH, Hiemstra PS, et al. Retinoic acid signaling balances adult distal lung epithelial progenitor cell growth and differentiation. EBioMed. (2018) 36:461–74. doi: 10.1016/j.ebiom.2018.09.002
292. Uniyal S, Dhasmana A, Tyagi A, Muyal JP. ATRA reduces inflammation and improves alveolar epithelium regeneration in emphysematous rat lung. Biomed Pharmacother. (2018) 108:1435–50. doi: 10.1016/j.biopha.2018.09.166
293. Morichika D, Miyahara N, Fujii U, Taniguchi A, Oda N, Senoo S, et al. A retinoid X receptor partial agonist attenuates pulmonary emphysema and airway inflammation. Respir Res. (2019) 20:2. doi: 10.1186/s12931-018-0963-0
294. Stolk J, Stockley RA, Stoel BC, Cooper BG, Piitulainen E, Seersholm N, et al. Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor. Eur Respir J. (2012) 40:306–12. doi: 10.1183/09031936.00161911
295. Palange P, Testa U, Huertas A, Calabrò L, Antonucci R, Petrucci E, et al. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Respir J. (2006) 27:529–41. doi: 10.1183/09031936.06.00120604
296. Salter BM, Manzoor F, Beaudin S, Kjarsgaard M, Nair P, Gauvreau GM, et al. Dysregulation of vascular endothelial progenitor cells lung-homing in subjects with COPD. Can Respir J. (2016) 2016:1472823. doi: 10.1155/2016/1472823
297. Doyle MF, Tracy RP, Parikh MA, Hoffman EA, Shimbo D, Austin JHM, et al. Endothelial progenitor cells in chronic obstructive pulmonary disease and emphysema. PLoS ONE. (2017) 12:e0173446. doi: 10.1371/journal.pone.0173446
298. Shi Z, Chen Y, Cao J, Zeng H, Yang Y, Chen P, et al. Intratracheal transplantation of endothelial progenitor cells attenuates smoking-induced COPD in mice. Int J COPD. (2017) 12:947–60. doi: 10.2147/COPD.S110781
299. Tura-Ceide O, Pizarro S, García-Lucio J, Ramírez J, Molins L, Blanco I, et al. Progenitor cell mobilisation and recruitment in pulmonary arteries in chronic obstructive pulmonary disease. Respir Res. (2019) 20:74. doi: 10.1186/s12931-019-1024-z
300. Peinado VI, Ramírez J, Roca J, Rodriguez-Roisin R, Barberà JA. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2006) 34:257–63. doi: 10.1165/rcmb.2005-0255OC
301. Paschalaki KE, Starke RD, Hu Y, Mercado N, Margariti A, Gorgoulis VG, et al. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased dna damage and senescence. Stem Cells. (2013) 31:2813–26. doi: 10.1002/stem.1488
302. Yang Y, Gan Y, Cao J, Chen Y, He Z-H, Luo H, et al. Decreased and dysfunctional circulating endothelial progenitor cells in patients with chronic obstructive pulmonary disease. Chin Med J. (2013) 126:3222–7.
303. Jay Widmer R, Lerman A. Endothelial dysfunction and cardiovascular disease. Glob Cardiol Sci Pract. (2014) 2014:43. doi: 10.5339/gcsp.2014.43
304. Barthelmes J, Nägele MP, Ludovici V, Ruschitzka F, Sudano I, Flammer AJ. Endothelial dysfunction in cardiovascular disease and Flammer syndrome-similarities and differences. EPMA J. (2017) 8:99–109. doi: 10.1007/s13167-017-0099-1
305. Lee PSS. Endothelial progenitor cells in cardiovascular diseases. World J Stem Cells. (2014) 6:355. doi: 10.4252/wjsc.v6.i3.355
306. Hou Y, Li C. Stem/Progenitor cells and their therapeutic application in cardiovascular disease. Front Cell Dev Biol. (2018) 6:139. doi: 10.3389/fcell.2018.00139
Keywords: COPD, emphysema, pulmonary hypertension, hypoxia, oxidative stress
Citation: Karnati S, Seimetz M, Kleefeldt F, Sonawane A, Madhusudhan T, Bachhuka A, Kosanovic D, Weissmann N, Krüger K and Ergün S (2021) Chronic Obstructive Pulmonary Disease and the Cardiovascular System: Vascular Repair and Regeneration as a Therapeutic Target. Front. Cardiovasc. Med. 8:649512. doi: 10.3389/fcvm.2021.649512
Received: 04 January 2021; Accepted: 08 March 2021;
Published: 12 April 2021.
Edited by:
Ippei Shimizu, Niigata University, JapanReviewed by:
Junichi Omura, Janssen Pharmaceutical K.K., JapanCopyright © 2021 Karnati, Seimetz, Kleefeldt, Sonawane, Madhusudhan, Bachhuka, Kosanovic, Weissmann, Krüger and Ergün. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Srikanth Karnati, c3Jpa2FudGgua2FybmF0aUB1bmktd3VlcnpidXJnLmRl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.