
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 20 May 2021
Sec. Cardiovascular Genetics and Systems Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.635141
This article is part of the Research Topic Genetic Architecture of Quantitative Cardiovascular Imaging and Electrocardiographic Traits View all 8 articles
 Maria Pia Leone1
Maria Pia Leone1 Pietro Palumbo1Johan Saenen2Sandra Mastroianno3Stefano Castellana4Cesare Amico3
Pietro Palumbo1Johan Saenen2Sandra Mastroianno3Stefano Castellana4Cesare Amico3 Tommaso Mazza4Domenico Rosario Potenza3
Tommaso Mazza4Domenico Rosario Potenza3 Antonio Petracca1Marco Castori1Massimo Carella1
Antonio Petracca1Marco Castori1Massimo Carella1 Giuseppe Di Stolfo3*
Giuseppe Di Stolfo3*Background: Arrhythmogenic cardiomyopathy (ACM) is a genetic disorder with an estimated prevalence between 1:2,000 and 1:5,000 and is characterized by the fibrofatty replacement of cardiomyocytes that predisposes to malignant arrhythmias, heart failure, and sudden cardiac death. The diagnosis is based on the 2010 Task Force Criteria including family history, electrocardiographic traits and arrhythmogenic pattern, specific gene mutations, and structural and/or histological abnormalities. Most ACMs display an autosomal dominant mode of inheritance often with incomplete penetrance and variable expressivity. Genetic screening of patients with ACM identifies pathogenic or likely pathogenic variants, prevalently in genes encoding the cardiac desmosome (PKP2, DSP, DSC2, DSG2, and JUP) or less frequently in non-desmosomal genes (CTNNA3, PLN, TMEM43, RYR2, SCN5A, CDH2, and DES).
Methods: In the present study, we performed molecular autopsy in a boy who died suddenly during physical exertion. In addition to post-mortem examination, a DNA sample was analyzed with next-generation sequencing (NGS).
Results: The genetic analysis revealed the presence of pathogenic heterozygous c.314del (p.Pro105Leufs*7) frameshift variant in the PKP2 gene. Cascade screening of family members allowed us to identify 12 mutation carriers and to intervene on subjects at risk, many of whom were athletes.
Conclusions: Molecular autopsy can establish cardiogenetic diagnosis and allow appropriate preventative measures in high-risk relatives.
Arrhythmogenic cardiomyopathy (ACM) is a genetic disorder with an estimated prevalence between 1:2,000 and 1:5,000 and is characterized by the fibrofatty replacement of cardiomyocytes (1). The progressive fibrotic replacement causes electrical instability with an increased risk of syncope and sudden cardiac death (SCD), or alteration of cardiac function with right- or bi-ventricular heart failure resembling dilated cardiomyopathy (2). The diagnosis is challenging and based on the conjugate of 2010 Task Force criteria including family history, peculiar electrocardiographic, arrhythmic, and structural and/or histological abnormalities (3). Inheritance of ACM is classically considered autosomal dominant; however, incomplete penetrance and highly variable and age-dependent disease expression are observed (4). Rarely, autosomal recessive inheritance is found in ACM both with or without cutaneous involvement.
Genetic screening of patients with ACM identifies pathogenic or likely pathogenic variants, predominantly in genes encoding the cardiac desmosome (PKP2, DSP, DSC2, DSG2, and JUP) or less frequently in non-desmosomal genes (CTNNA3, PLN, TMEM43, RYR2, SCN5A, CDH2, and DES) (5).
The PKP2 gene encodes a member of the Armadillo (ARM) repeat proteins. Plakophilin proteins contain numerous ARM repeats, localize to cell desmosomes and nuclei, and participate in linking cadherins to intermediate filaments in the cytoskeleton (6). Plakophilin-2 is important for the assembly of junctional proteins and represents an essential morphogenic factor and architectural component of the heart (7).
Mutations in the PKP2 gene, whose gene–phenotype relationships are available in Online Mendelian Inheritance in Man as OMIM*602861, encoding the main cardiac plakophilin, at locus 12p11, represent 10–45% of the genotyped ACM patients (1).
In the present study, we performed molecular autopsy in a boy who died suddenly during physical exertion. In addition to post-mortem examination, a DNA sample was analyzed by NGS. The genetic analysis revealed a pathogenic heterozygous frameshift variant in the PKP2 gene (c.314del;p.Pro105Leufs*7). Cascade screening of 19 family members allowed us to identify 12 mutation carriers and to intervene on subjects at risk, many of whom were athletes.
A 13-year-old boy (Figure 1, IV:5) without prior medical history died suddenly during an hour of physical education following unsuccessful cardiopulmonary resuscitation, despite evidence of ventricular fibrillation treated by external defibrillation. Autopsy was requested and initial pathology report concluded for dilated cardiomyopathy with intramural fibrosis.
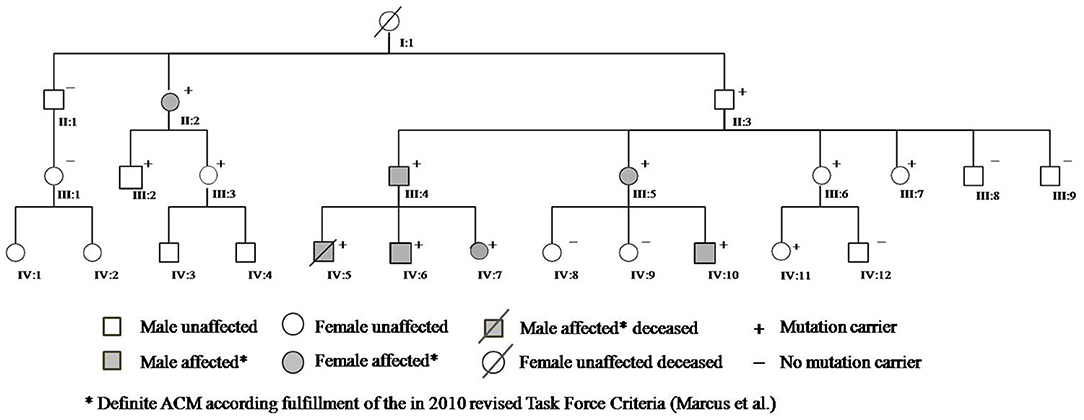
Figure 1. Family pedigree.
The parents, fearing for their other two children, underwent cardiologic evaluation at our institution. They reported that their son had complained of palpitations in the month before his death; after adequate information and psychological support for the bereavement process, they consented to genetic testing and molecular autopsy on their son's biological material. NGS analysis revealed a novel frameshift variant in the PKP2 gene (c.314del;p.Pro105Leufs*7). The PKP2 gene encodes plakophilin, which has been implicated in literature with arrhythmogenic cardiomyopathy (ACM). Therefore, we asked the family for permission to review the autoptic examination in light of the molecular data, a more thorough second analysis was consistent with ACM with biventricular involvement and fibrofatty replacement in both ventricles, supporting the diagnosis suggested by the genetic analysis. At this stage, adhering to the ACMG standards and guidelines and in the absence of segregation and functional data, this PKP2 variant was classified a class 4 “likely pathogenic” variant warranting further co-segregation analysis and predictive testing in the first-degree relatives (FDRs) (8).
Cascade screening of family members was subsequently initiated to evaluate possible carriers of the same mutation and identify other patients at risk with asymptomatic or unrecognized cardiomyopathy (Table 1). The first to be studied was his 42-year-old father (III:4) who reported palpitations. ECG evaluation revealed T wave inversion in V1–V3, minimal prolonged terminal activation duration (TAD) in V1, fragmented QRS in inferior leads, low QRS-voltage (Figure 2) and Holter recording showed frequent ventricular premature complexes (2388 PVCs/24 h) with two different morphologies (right bundle branch block and left bundle branch block pattern). Echocardiography showed enlargement of the right ventricular outflow tract (RVOT) (PLAX 41 mm and PSAX 36 mm, respectively). Cardiac magnetic resonance imaging highlighted the dilated right ventricle with contractile dysfunction (RVEF 35%, RVEDVi 105 mL/m2) and hypokinesia of the mid to apical free wall segments, fatty infiltration within the free wall of the right ventricle, and minimal adipose substitution in the epicardium of the apical left ventricle. A coronary angiography ruled out critical coronary stenosis. According to the 2010 Task Force Criteria, “definite” ACM was diagnosed in this patient for the presence of inverted T waves, prolonged TAD, and sudden death of 13-year-old son, and therapy with beta-blocker and ACE inhibitors was initiated. ACM mortality risk was estimated as 32.7% at 5 years, and an ICD was implanted for primary prevention (9–11).
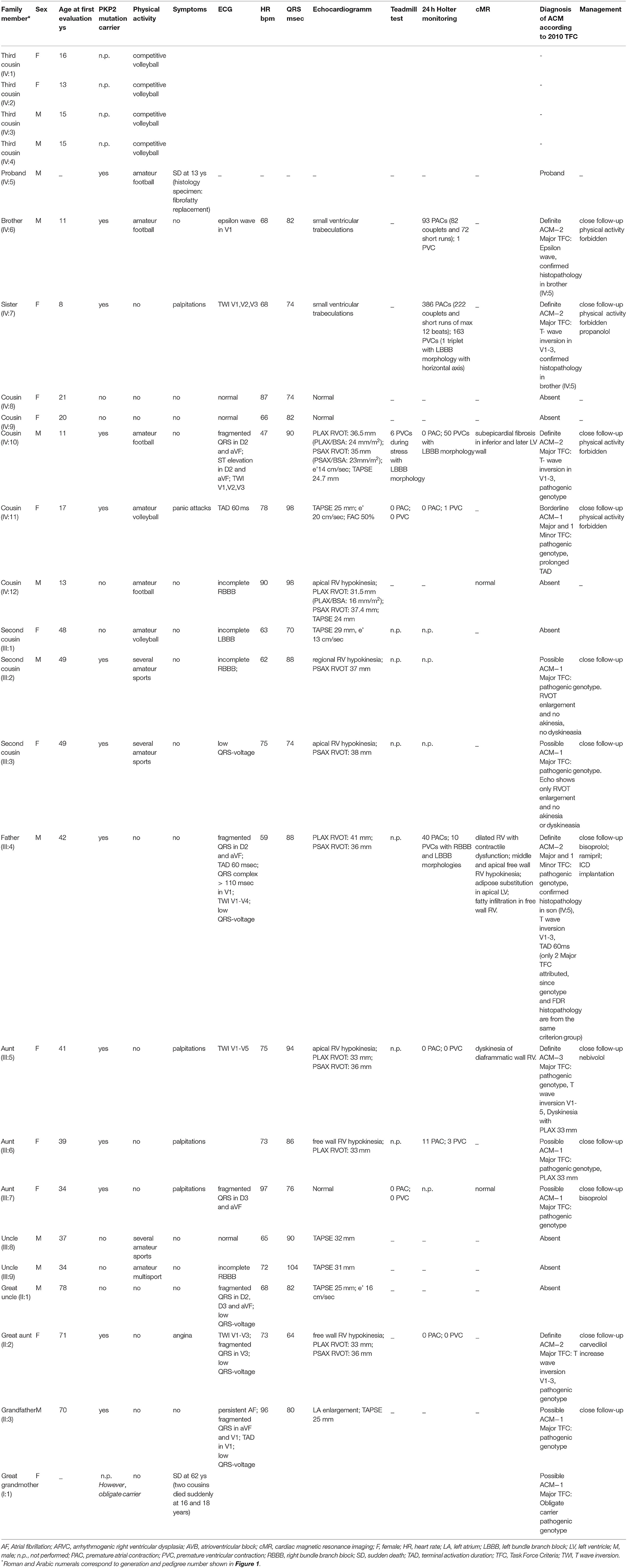
Table 1. Clinic and genetic features of family members.
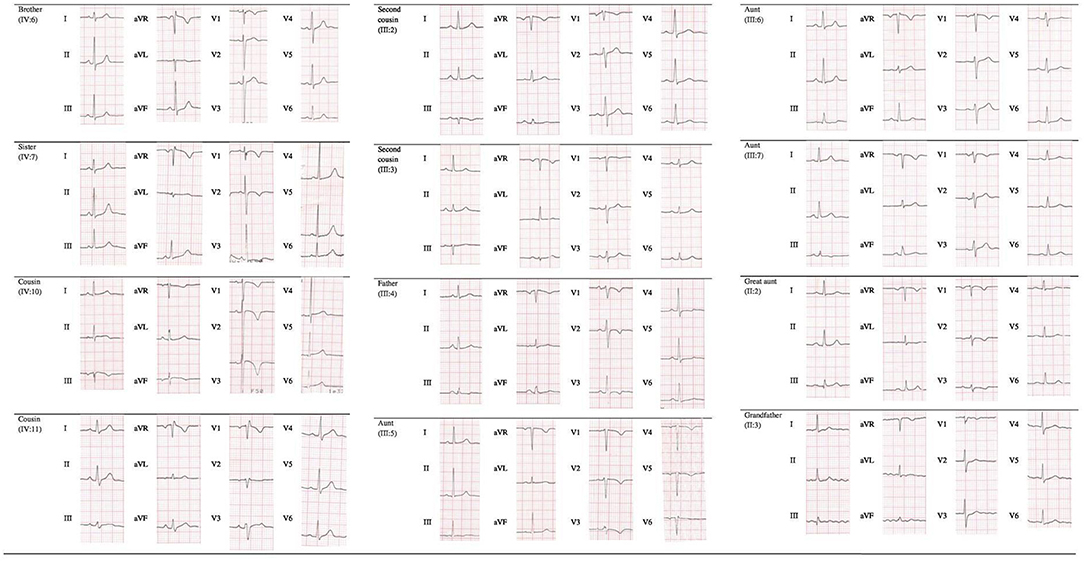
Figure 2. Electrocardiogram (ECG) finding in gene carriers.
Genetic evaluation revealed the presence of the same PKP2 variant that was deemed responsible for the sudden cardiac death of his son. This evidence of co-segregation of the genotype with the ACM phenotype allows re-classification of the PKP2 variant to a class 5 pathogenic mutation and further strengthening the need for predictive testing in other relatives of this family (8).
Cascade clinical and genetic screening was initiated with 19 relatives contacted in total; 12 additional subjects were found to be genotype positive and six of them diagnosed with two or more major TFC criteria; hence, definite ACM; five of mutation-positive individuals used to play high-intensity sports. Moreover, his paternal great-grandmother (I:1), who had died suddenly several years ago without apparent cause at the age of 62, and her two cousins known with sudden death at 16 and 18 years of age, involved in high-level sport activity (volleyball), were all likely affected.
Apart from genetic screening, the most prominent clinical traits found in the PKP2 mutation carriers were the ECG abnormalities, characterized by T wave inversion beyond V2 in the absence of intraventricular conduction disturbances, QRS fragmentation, epsilon wave, and TAD equal or longer than 55 ms, as shown in Table 1 and Figure 2. Echocardiographic abnormalities were less prevalent.
Depending on the individual findings, preventative measures were taken including medical therapy with beta blocker and lifestyle modification. According to current consensus, we forbade competitive sport in genotype-positive individuals and phenotypically affected relatives. However, to preserve psychological health and reduce the burden of intrusive lifestyle alterations, cardiogenetic counseling was done in conjunction with our psychologist team. In children, a gradual retreat from sport participation was imposed, and parents were advised to replace it with artistic activities, like theater and music performance.
A Cardiac Disease Sequencing Panel involving 76 genes (see Table 2 in the Supplements), including genes related to hypertrophic cardiomyopathy, dilated cardiomyopathy, and arrhythmogenic cardiomyopathy, was designed according to data obtained from scientific literature. Probes were designed using Agilent SureDesign Custom design tool (https://earray.chem.agilent.com/suredesign/:) the regions of interest (ROI) for this panel included all exons plus 25-bp flanking intron regions. The total amplicon number was 19.648, and the target size was 366.196 kb with a theoretical coverage of 99.32% for our targeted regions.
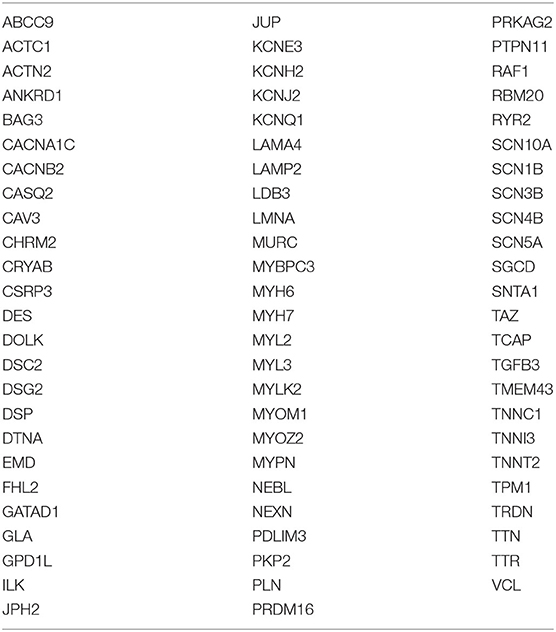
Table 2. List of analyzed genes.
The DNA of the patient was isolated from biological material. Peripheral blood samples were taken from his parents, and genomic DNA was isolated by using Bio Robot EZ1 (Quiagen, Solna, Sweden). A library of all coding regions of the 76 genes was obtained using the Haloplex target enrichment kit according to the manufacturer's instructions (Agilent Technologies, Santa Clara, CA, USA). At last, the enriched DNA fragments were sequenced on an Illumina MiSeq platform (Illumina, San Diego, CA, USA) using a MiSeq Reagent kit V3 300 cycles flow cell (Illumina, San Diego, CA, USA).
The produced raw paired-end reads underwent quality checking by using the FastQC tool (12) and then aligned to the hg19 reference genome sequence by means of Bowtie (13). Depth of coverage statistics for the target regions were calculated by TEQC ver. 3.47 (14).
Variants were called by means of the HaplotypeCaller tool of GATK ver. 3.8 (15), while functional annotation was carried out by ANNOVAR tool, using the NCBI RefSeq gene and transcript annotation system (updated to January 2018) (16). Variants were checked for their presence in public databases, such as dbSNP ver. 150 (17), ExAC ver. 0.3 (18), and Exome Variant Server (http://evs.gs.washington.edu/EVS, accessed at December 2017), HRC (19) Kaviar (20) and ClinVar (21). Predictions of functional consequences for missense variants were further collected by querying the dbNSFP ver. 3.5 resource and retrieving pre-computed pathogenicity predictions and evolutionary conservation measures (22).
The presence of the detected variant was confirmed by gold standard Sanger sequencing. A PCR was carried out to amplify the exon 2 of the PKP2 gene (NM_001005242) including the variant site, and the PCR product was purified by ExoSAP-IT PCR Product Cleanup Reagent (Thermo Fisher Scientific, Waltham, MA, USA) first, then sequenced on an ABI Prism 3100 Genetic Analyzer (Thermo Fisher Scientific Waltham, Massachusetts, USA) using BigDye Terminator v1.1 sequencing kit (Applied Biosystems, Foster City, CA, USA).
NGS analysis identified a heterozygous frameshift variant in the PKP2 gene (NM_001005242:exon2:c.314del;p.Pro105Leufs*7) in the patient (IV:5) and his father (III:4), detected with a depth of coverage of 127X.
The variant, reported as pathogenic in ClinVar (rs794729121), causes a shift in reading frame starting at codon proline 105 changing it to leucine, and creating a premature stop codon at position seven of the new reading frame, denoted p.Pro105Leufs*7.
The presence of the detected variant was assessed in the patients and in their parents by Sanger sequencing. Furthermore, after the genetic result of the patient (IV:5) and the father (III:4), we performed segregation analysis on other available relatives (II:1; II:2; II:3; III:1; III:2; III:3; III:5; III:6; III:7; III:8; III:9; IV:6; IV:7; IV:8; IV:9; IV:10; IV:11; IV:12). The sequencing analysis revealed further 11 carriers of the reported variant, nine (II:2; III:2; III:3; III:5; III:6; IV:6; IV:7; IV:10; IV:11) with electrocardiographic and/or echocardiographic signs of ACM, and two (II:3; III:7) without detectable ACM traits (Table 1 and Figure 2) conferring a clinical penetrance of 84% (11 with ACM expression out of 13 carriers) with variable expressivity. The estimated 5-year risk for lethal events among the mutation carriers ranges up to 32.7% (as calculated for III:4, according to ARVC calculator) justifying a holistic preventative approach adapted to the individual risk profile of each relative.
A single molecular autopsy in one proband resulted in the tailored improvement of the clinical management in 12 relatives ranging from close cardiac follow-up, lifestyle modification and medication, while immediate ICD implantation was warranted in one high-risk individual (III:4) to prevent a detrimental outcome.
Here we report a 13-year old boy who died suddenly during training despite the readily performed resuscitation maneuvers. NGS sequencing analysis helped physicians to establish the post-mortem diagnosis of arrhythmogenic cardiomyopathy by the finding of a pathogenic mutation in the PKP2 gene. Clinical diagnosis of ACM may be challenging, and therefore, predictive genetic testing may offer valid means to help and identify subjects at risk, before the presence of phenotypical traits, before the onset of early warning symptoms, and ultimately prior to malignant arrhythmic events (23–25). In this case, the discovery of a putative disease-causing variant in the PKP2 gene raised doubts about the initial cause of death reported in the first autopsy and helped us convince the family to consent to reassessment of the autopsy findings. Today, thanks to contemporary advances in DNA sequencing technologies, post-mortem genetic analysis can play an important role in the correct diagnosis of hereditary cardiac disease allowing critical review and interpretation of autopsy findings.
To date, the ClinVar database (26) provides a total of 1,005 annotated PKP2 variants (last accessed November 2020); roughly 65% are linked to cardiac conditions, the remaining 35% has been submitted without an associated clinical diagnosis (27). The variant reported here is a frameshift variant (NM_001005242:exon2:c.314del:p.Pro105Leufs*7) expected to result in either a truncated protein product or haplo-insufficiency through nonsense-mediated mRNA decay. Frameshift variants in PKP2 are strongly associated with ACM, and over the years, other frameshift mutations in the PKP2 gene have been reported in literature (28).
Lack of plakophilin-2 or incorporation of mutant plakophilin-2 in the cardiac desmosomes impairs cell–cell contacts and, as a consequence, disrupts adjacent cardiomyocytes, particularly in response to mechanical stress or stretch, providing a potential explanation for the high prevalence of the disorder in athletes, the frequent occurrence of ventricular tachyarrhythmias and sudden death during exercise, and the predominant affection of the right ventricle (29). In this family, the proband suffered SCD during physical education at school, and two more distant cousins died during a game of volleyball during adolescence underlining the importance of lifestyle modification as first-line preventative measure.
Beyond the NGS approach, segregation analysis represents a powerful methodology to further assess the putative correlation between genotype and phenotype, mainly for variants of uncertain significance (VUS), and to further strengthen the disease-causing probability of likely pathogenic variants. In this family, genetic screening detected the mutation in another 11 subjects following an autosomal dominant inheritance pattern, as reported in literature for other mutations in PKP2 (4).
This concept highlights the importance of performing family screening, beyond the index case.
Here, we were able to establish a genotype–phenotype correlation in 12 additional informative individuals, which is a very strong evidence that this PKP2 variant is the predominant cause for the ACM phenotype displayed in this family.
Furthermore, cascade screening allows the identification of other ACM patients at risk and enables individualized preventative measures. Clinical examination showed heterogeneous phenotypic expression, mainly represented by typical electrocardiographic alterations.
Although echocardiographic findings and cardiac MRI examination were not diagnostic in most cases, ECG abnormalities should raise suspicion for the presence of arrhythmogenic cardiomyopathy, in particular, in the precordial leads, characterized by T wave inversion beyond V2, epsilon wave, prolonged TAD (≥55 ms), and QRS fragmentation. In our patients, prevalence of akinesia/dyskinesia was rare; it should be hypothesized that early carrier identification is allowed in the initial ACM stage, without overt phenotype, permitting a better primary care prevention.
Based on the individual clinical characteristics, close follow-up, tailored lifestyle modifications, and medical treatments were advocated. Our young proband carried out physical activity regularly, playing soccer three times a week, a condition that may promote disease expression and progression. In contrast, none of the affected relatives reported being aware of a pre-existing cardiac condition that can progress and exacerbate lethal arrhythmias due to strenuous exercise as many of them were participating regularly in sport activities without any screening. This finding underlines the importance of pre-participation evaluation by electrocardiographic screening integrated with clinical and familiar history for sport-related sudden death prevention (30).
Moreover, according to recent recommendations for participation in competitive and leisure time sports in athletes with cardiomyopathies, athletes who have unequivocal or probable diagnosis of ACM or are genetic carriers of pathogenic desmosomal mutations (even in the absence of phenotypic expression of the disease), should not participate in competitive sports (Class IIa, Level C); these athletes should be advised to limit their exercise programs to leisure-time activities and remain under close clinical surveillance (31). In this perspective, as several family members performed both amateur and competitive sport, in many of them, the diagnosis of ACM represented a reason to suspend these activities.
The psychological reaction to cardiomyopathy diagnosis, in particular, during childhood or adolescence, could have important implications when advocating lifestyle modifications according to the disease state and when confronted with the latent risk to develop disease progression and sudden cardiac death. It is important to identify and address potential psychological suffering early and to incorporate psychological support into the cardiogenetics care team. Patients may experience feelings of anxiety and depression following the diagnosis of an inherited cardiomyopathy that is deemed responsible for the loss of a close relative. The initial proposal for treatment and lifestyle modification can be negotiated and adapted based on the disease stage and patient belief and level of acceptance, allowing intense cooperation between the patient, family, and the healthcare professionals (32).
Our study shows that molecular autopsy is an essential tool in autopsy-negative cases to help establish the cause of death and enable familial screening for other individuals at risk. Such analysis requires highly specialized techniques and trained specialists including geneticists, cardiologists, counselors and psychologists, and should therefore be performed in multi-disciplinary-dedicated cardiogenetics clinics that are experienced in managing hereditary cardiac diseases and cases of sudden death in the young (33).
Healthcare policy makers and cardiovascular societies should support the notion of specialized cardiogenetic hubs that operate in a wider network and be accessed more easily by peripheral centers to permit the extensive cardiogenetic evaluation of these particular SCD cases that may have far-reaching implications for often unknowing relatives and for better understanding of inherited cardiac disorders (34).
We aim to highlight the importance of molecular autopsy in unsolved sudden cardiac death cases as it may help establish the correct diagnosis, allow the identification of other relatives at risk, and start preventative measures where appropriate. In this case, a single molecular autopsy revealed a highly penetrant PKP2 frameshift mutation underlying ACM and having far-reaching implications on cardiac management of the family members including follow-up, lifestyle modification, medical treatment, and outcome. Such analysis requires highly specialized and highly trained multi-disciplinary care teams and should therefore be performed in dedicated cardiogenetics clinics.
The data presented in this study are available on request from the corresponding author. Data can also be found here https://databases.lovd.nl/shared/variants/0000708305#00001012.
The studies involving human participants were reviewed and approved by IRCCS Fondazione Casa Sollievo della Sofferenza ethics committee. The patients/participants provided their written informed consent to participate in this study.
ML and PP contributed to conception of the manuscript, data collection and interpretation, drafting the article. JS contributed to critical revision and final approval of the version to be published. SM and GDS contributed to conception of the manuscript, data collection and interpretation, drafting the article, critical revision and final approval of the version to be published. SC and TM contributed to data collection, interpretation, and drafting the article. CA and DP contributed to drafting the article and critical revision. AP contributed to drafting the article. MCas contributed to conception of the manuscript. MCar contributed to conception of the manuscript, critical revision and final approval of the version to be published. All authors contributed to the article and approved the submitted version.
https://databases.lovd.nl/shared/variants/0000708305#00001012
This work was supported by the Italian Ministry of Health for research grant RC 2001CA08.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACM, arrhythmogenic cardiomyopathy; ECG, electrocardiogram; FDRs, first degree relatives; NGS, next-generation sequencing; SCD, sudden cardiac death.
1. Pilichou K, Thiene G, Bauce B, Rigato I, Lazzarini E, Migliore F, et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis. (2016) 11:1–17. doi: 10.1186/s13023-016-0407-1
2. Akdis D, Brunckhorst C, Duru F, Saguner AM. Arrhythmogenic cardiomyopathy: electrical and structural phenotypes. Arrhythmia Electrophysiol Rev. (2016) 5:90–101. doi: 10.15420/AER.2016.4.3
3. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D). Circulation. (2010) 121:1533–41. doi: 10.1161/CIRCULATIONAHA.108.840827
4. Pinamonti B, Brun F, Mestroni L, Sinagra G. Arrhythmogenic right ventricular cardiomyopathy: From genetics to diagnostic and therapeutic challenges. World J Cardiol. (2014) 6:1234–44. doi: 10.4330/wjc.v6.i12.1234
5. Vimalanathan AK, Ehler E, Gehmlich K. Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardiomyopathy. Biophys Rev. (2018) 10:973–82. doi: 10.1007/s12551-018-0437-0
6. Bonné S, van Hengel J, van Roy F. Assignment of the plakophilin-2 gene (PKP2) and a plakophilin-2 pseudogene (PKP2P1) to human chromosome bands 12p11 and 12p13, respectively, by in situ hybridization. Cytogenet Cell Genet. (2000) 88:286–7. doi: 10.1159/000015540
7. Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. (2004) 167:149–60. doi: 10.1083/jcb.200402096
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. ARVC Risk Calculator, n.d. Available online at: https://arvcrisk.com/ (accessed November 7, 2020).
10. Cadrin-Tourigny J, Bosman LP, Nozza A, Wang W, Tadros R, Bhonsale A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. (2019) 40:1850–8. doi: 10.1093/eurheartj/ehz103
11. Sweeting J, Semsarian C. Sudden cardiac death in athletes. Heart Lung Circ. (2018) 27:1072–7. doi: 10.1016/j.hlc.2018.03.026
12. Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data, n.d. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed December 29, 2019).
13. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
14. Hummel M, Bonnin S, Lowy E, Roma G. TEQC: an R package for quality control in target capture experiments. Bioinforma Oxf Engl. (2011) 27:1316–7. doi: 10.1093/bioinformatics/btr122
15. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
16. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
17. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. (2001) 29:308–11. doi: 10.1093/nar/29.1.308
18. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
19. McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. (2016) 48:1279–83. doi: 10.1038/ng.3643
20. Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC. Kaviar: an accessible system for testing SNV novelty. Bioinformatics. (2011) 27:3216–7. doi: 10.1093/bioinformatics/btr540
21. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
22. Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. (2011) 32:894–9. doi: 10.1002/humu.21517
23. Poloni G, De Bortoli M, Calore M, Rampazzo A, Lorenzon A. Arrhythmogenic right-ventricular cardiomyopathy: molecular genetics into clinical practice in the era of next generation sequencing. J Cardiovasc Med Hagerstown Md. (2016) 17:399–407. doi: 10.2459/JCM.0000000000000385
24. Cho Y. Arrhythmogenic right ventricular cardiomyopathy. J Arrhythmia. (2018) 34:356–68. doi: 10.1002/joa3.12012
25. Ingles J, Bagnall RD, Yeates L, McGrady M, Berman Y, Whalley D, et al. Concealed arrhythmogenic right ventricular cardiomyopathy in sudden unexplained cardiac death events. Circ Genomic Precis Med. (2018) 11:e002355. doi: 10.1161/CIRCGEN.118.002355
26. ClinVar, n.d. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed January 6, 2021).
27. Novelli V, Malkani K, Cerrone M, Pleiotropic phenotypes associated with PKP2 variants. Front Cardiovasc Med. (2018) 5. doi: 10.3389/fcvm.2018.00184
28. van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld ACP, Wilde AAM, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. (2006) 113:1650–8. doi: 10.1161/CIRCULATIONAHA.105.609719
29. Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. (2004) 36:1162–4. doi: 10.1038/ng1461
30. Heidbuchel H, Arbelo E, D'Ascenzi F, Borjesson M, Boveda S, Castelletti S, et al. Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions. Part 2: ventricular arrhythmias, channelopathies, and implantable defibrillators. Europace. (2021) 23:147–8. doi: 10.1093/europace/euaa106
31. Pelliccia A, Solberg EE, Papadakis M, Adami PE, Biffi A, Caselli S, et al. Recommendations for participation in competitive and leisure time sport in athletes with cardiomyopathies, myocarditis, and pericarditis: position statement of the Sport Cardiology Section of the European Association of Preventive Cardiology (EAPC). Eur Heart J. (2019) 40:19–33. doi: 10.1093/eurheartj/ehy730
32. Bregman L. Kübler-ross and the re-visioning of death as loss: religious appropriation and responses. J Pastor Care Couns JPCC. (2019) 73:4–8. doi: 10.1177/1542305019831943
33. Wiley KA, Demo EM, Walker P, Shuler CO. Exploring the discussion of risk of sudden cardiac death. Pediatr Cardiol. (2016) 37:262–70. doi: 10.1007/s00246-015-1272-8
34. Hodgkinson K, Dicks E, Connors S, Young T-L, Parfrey P, Pullman D. Translation of research discoveries to clinical care in arrhythmogenic right ventricular cardiomyopathy in Newfoundland and Labrador: lessons for health policy in genetic disease. Genet Med Off J Am Coll Med Genet. (2009) 11:859–65. doi: 10.1097/GIM.0b013e3181c20bb3
Keywords: novel PKP2 mutation, arrhythmogenic cardiomyopathy, juvenile sudden death, molecular autopsy, sport restriction
Citation: Leone MP, Palumbo P, Saenen J, Mastroianno S, Castellana S, Amico C, Mazza T, Potenza DR, Petracca A, Castori M, Carella M and Di Stolfo G (2021) Phenotypic Variability of a Pathogenic PKP2 Mutation in an Italian Family Affected by Arrhythmogenic Cardiomyopathy and Juvenile Sudden Death: Considerations From Molecular Autopsy to Sport Restriction. Front. Cardiovasc. Med. 8:635141. doi: 10.3389/fcvm.2021.635141
Received: 29 November 2020; Accepted: 01 March 2021;
Published: 20 May 2021.
Edited by:
Julia Ramírez, Queen Mary University of London, United KingdomReviewed by:
Oscar Campuzano, University of Girona, SpainCopyright © 2021 Leone, Palumbo, Saenen, Mastroianno, Castellana, Amico, Mazza, Potenza, Petracca, Castori, Carella and Di Stolfo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Di Stolfo, Z2l1c2VwcGVkaXN0b2xmb0B5YWhvby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.