 Bo Bai
Bo Bai Yun Xu
Yun Xu- Department of Cardiology, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People's Hospital, Shenzhen, China
Aims: To evaluate the interrelation between neutrophil to lymphocyte ratio (NLR) coupled with gene signatures, inflammation, and diastolic dysfunction in patients with heart failure (HF) with preserved ejection fraction (HFpEF).
Methods: The clinical profile of 172 patients with HFpEF (EF ≥ 50%) and 173 non-HF control individuals was analyzed retrospectively. The association between NLR and HFpEF and the predictive performance of NLR for HFpEF were assessed by the binary logistic regression analysis and the receiver operating characteristic curve (ROC). Multivariate linear regression models further examined the associations between NLR and high-sensitivity C-reactive protein (hs-CRP), N-terminal prohormone of brain natriuretic peptide (NT-proBNP), and average septal-lateral E/e', respectively. The freshly isolated neutrophils from 30 HFpEF patients and 42 non-HF controls were subjected to transcriptomic profiling. The biomarkers related to neutrophil activation and inflammation were detected in serum samples.
Results: The HFpEF patients in Southeast China were lean and had comorbidity burden and worse cardiac structure/function. Compared with non-HF control individuals, HFpEF patients had a rise in NLR. NLR displayed an independent association with HFpEF [adjusted odds ratio, 2.351; 95% CI, 1.464–3.776; p < 0.001] and it predicted HFpEF with the area under the ROC 0.796 (95% CI, 0.748–0.845, p < 0.001). The positive associations between NLR and hs-CRP, NT-proBNP, and mitral E/e' were found in HFpEF patients. Moreover, patients had significantly elevated serum levels of neutrophil elastase and inflammatory biomarkers, both of which correlated with the mitral E/e' ratio. Finally, multiple molecules that drive neutrophil degranulation and inflammation, such as S100A8/A9/A12 and PADI4, were transcriptionally up-regulated in neutrophils of HFpEF patients.
Conclusions: The high NLR coupled with transcriptional activation of neutrophils correlates with systemic inflammation and functional impairment in HFpEF patients, which may suggest a causative role of neutrophils in the pathogenesis of the disease.
Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF) has conferred substantial morbidity and mortality on clinical patients. Its prevalence is increasing at an alarming rate, currently representing 50% of all HF worldwide (1). In contrast to positive outcomes in heart failure with reduced ejection fraction (HFrEF) treated by neurohumoral inhibition, most of the large-scale clinical trials to assess the efficiency of medical therapies for HFpEF have not shown positive results yet (2, 3). HFpEF represents a broad cohort of patients with a combination of multiple risk factors and comorbidities. As such, the failure of effective treatment for HFpEF is likely attributable to the heterogeneity in this clinical scenario (4). Despite the phenotypic diversity, an increasingly popular theory about HFpEF is that this syndrome reflects a pro-inflammatory state (5). By utilizing comprehensive proteomic approaches to analyze blood biomarkers of HFpEF patients, recent studies demonstrate that systemic inflammation is closely related to HFpEF symptomatology. Moreover, the inflammation appears to mediate the association between comorbidity burden, worse cardiac hemodynamic stress, and adverse outcomes (6, 7). The systemic inflammation is associated with increased cardiomyocyte passive tension and aberrant myocardial collagen deposition, both of which would result in impaired left ventricular (LV) compliance in HFpEF (5, 8). Intriguingly, the tissue or cellular source of these inflammatory biomarkers remains uncertain. Therefore, characterizing specific sources of inflammatory molecules involved in the pathogenesis of HFpEF is an essential issue to be clarified.
Neutrophils are the dominant type of leukocytes during acute inflammatory reactions. The emerging evidence that neutrophils contribute to the clinical manifestations of cardiovascular diseases has been well-discussed (9). In the context of congestive HF, the increased neutrophil lifespan positively correlates to the New York Heart Association (NYHA) class, plasma levels of C-reactive protein (CRP), and alkaline phosphatase (10). The I-PRESERVE trial (Irbesartan in Heart Failure with Preserved Ejection Fraction Study) demonstrates that high neutrophil counts serve as an independent risk factor associated with poor outcomes of HFpEF patients (11). The neutrophil to lymphocyte ratio (NLR) has been proposed as a valuable marker to stratify the risk of patients hospitalized with HFpEF (12). The high level of plasma myeloperoxidase secreted by neutrophils is thought to be suggestive footprints of microvascular endothelial inflammation in HFpEF patients (13). In the endomyocardial biopsy samples from HFpEF patients, a subset of inflammatory cells marked by CD11a and CD45 (pan-leukocyte markers) is increased, associated with the collagen accumulation and high tissue levels of transforming growth factor (TGF)-β (14). Our previous study reported a lean diabetic HFpEF mouse model. The HFpEF mice have diastolic dysfunction and LV stiffness, concurrent with apparent cardiac inflammation and interstitial fibrosis. Of note, these pathological alternations in mouse hearts are associated with massive neutrophil infiltration and neutrophil extracellular traps (NETs) formation (15).
It appears that neutrophils play significant roles in the pathogenic process of HFpEF. However, the pathological involvement of neutrophils in exacerbating the inflammation or functional impairment of HFpEF patients remains poorly understood. To this end, our study aimed to assess the interrelation between neutrophils coupled with transcriptomic profile, inflammatory biomarkers, and abnormal cardiac structure/function of clinical HFpEF patients.
Methods
Study Population
First, in retrospective analysis, the clinical data were obtained from 172 in-patients diagnosed with HFpEF (EF ≥50%) between January 2016 and December 2019 (16). Meanwhile, 173 gender and age-matched in-patients with mild to moderate hypertension but no HF symptoms were recruited as non-HF controls (27 patients had Grade1 hypertension, 144 patients had Grade 2 hypertension, and two patients had Grade 3 hypertension) (Table 1). Patients' clinical profile, including demographic variables, medical history, laboratory values, and echocardiographic variables, was well-documented after admission. Patients who had pulmonary infection, hematopoietic disease, and autoimmune disease or were undergoing antibiotic or immunosuppressive therapy were excluded from this study. Second, a total of 30 in-patients newly diagnosed with HFpEF and 42 non-HF control individuals in our hospital (from January to December 2020) were enrolled in this study (Supplementary Table 1). The circulating neutrophils were freshly isolated from blood samples of patients for transcriptomic analysis. The serum samples of patients were subjected to a biomarker assay.
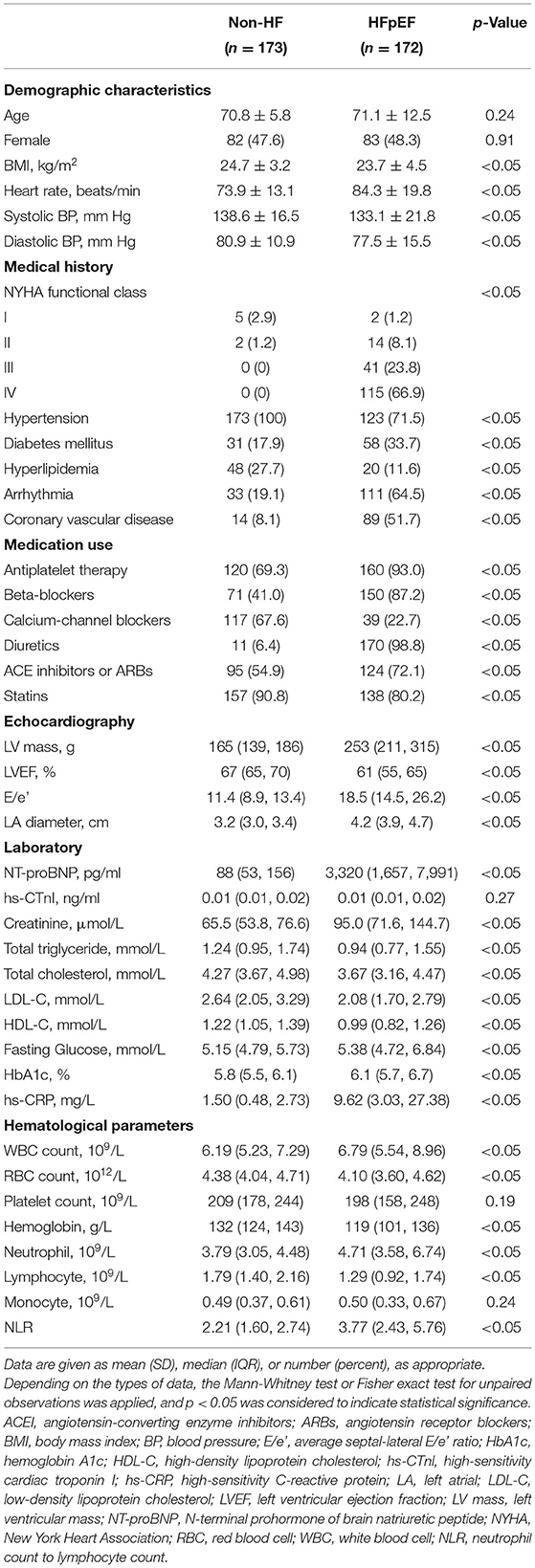
Table 1. The clinical characteristics of non-HF control individuals and HFpEF patients.
Assessment of Hematological Parameters
Laboratory variables, including complete blood cell counts, serum lipids, glucose, hemoglobin A1c (HbA1c), N-terminal prohormone of brain natriuretic peptide (NT-proBNP), were examined and documented. The NLR ratio was constructed as follows: NLR = neutrophil count to lymphocyte count.
Neutrophil Isolation
The circulating neutrophils were freshly isolated from eight HFpEF patients and 12 non-HF control individuals. The blood sample (2 mL) was carefully layered over Polymorphprep™ reagent (Axis-Shield, Scotland). After centrifuging at 500 g for 35 min at room temperature, the plasma and mononuclear cells (upper band of cells) were removed, and neutrophils were harvested. After washing with Hepes-buffered saline [0.85% (w/v) NaCl], cell pellet was resuspended in ammonium chloride lysis buffer [0.83% (w/v) NH4Cl, 10 mM Hepes-NaOH, pH 7.4] to remove any residual erythrocyte contamination. Then cells were harvested by centrifugation and stored in TRIzol™ reagent (Thermo Fisher Scientific, USA) for subsequent RNA extraction.
Transcriptome Sequencing of Neutrophils
RNA isolation and purification were performed using TRIzol-chloroform and RNeasy Mini Kit (Qiagen, Germany). The quality of RNA was checked with Agilent 2100 Bioanalyzer (Agilent, USA). The RNA-seq library was prepared by the Beijing Genomics Institute (Shenzhen, China). Sequence reads were obtained using BGIseq500 (Illumina) and successfully mapped to the human genome (Genome Reference Consortium Human Build 38 patch release 13, GRCh38.p13). Read counts were normalized based on reads per kilobase million (RPKM). The DEseq2 method was used to filter differential genes (17). The adjusted p-value (Q value) < = 0.05 was acceptable to indicate the gene expression with a significant difference. According to the results of differential gene detection, the R package heatmap was used to perform hierarchical clustering analysis on the union set differential genes. The Reactome enrichment was subsequently performed to investigate the molecular function and biological pathways that genes participate.
Determination of Biomarkers in Serum
The serum samples were collected from 30 HFpEF patients and 42 non-HF control individuals. Biomarkers related to systemic inflammation [interleukin (IL)-1β, IL-6, IL-10, tumor necrosis factor (TNFα), and soluble intercellular adhesion molecule-1 (sICAM-1)], tissue remodeling [matrix metalloproteinase 9 (MMP9)], as well as neutrophil activation [neutrophil elastase (NE)] were examined by the enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (NeoBioscience, China, and Abcam, UK).
Statistical Analysis
In the cohort study, data are given as means and standard deviations (SD), medians and interquartile 25th and 75th percentiles (IQRs), or numbers and percentages, as appropriate. The statistical calculations were performed using IBM SPSS statistics software. Depending on the types of data, the non-parametric Mann-Whitney test or Fisher exact test for unpaired observations was applied for statistical comparison. Binary logistic regression analysis was carried out using HFpEF as the dependent variable to analyze the association between NLR and HFpEF after adjusting for potential confounders, including age, gender, body mass index (BMI), high-sensitivity CRP (hs-CRP), and diabetes. The crude and adjusted odds ratios (OR) with 95% confidence intervals (CI) were calculated. Receiver operating characteristic (ROC) curves with area under the curve (AUC) were calculated to determine discriminative ability. The partial Pearson or the Spearman correlations were computed to describe the relationship between variables of interest after values were logarithmically transformed. Subsequently, the multivariate linear regression analysis was conducted to identify factors associated with hs-CRP, NT-proBNP, and average septal-lateral E/e', respectively. In the transcriptomic sequencing study, the fold changes of FPKM of interested genes were calculated and compared between the two groups. Independent samples were compared by a two-tailed unpaired t-test with Welch's correction. For all statistical analyses, p < 0.05 was considered to indicate statistical significance.
Results
The Clinical Characteristics of HFpEF Patients
Baseline demographic characteristics and laboratory variables of the entire study population were shown in Table 1. The age and gender distribution of HFpEF patients were comparable to that of the non-HF control population. Of note, both groups of cohorts were lean and with an average BMI below 30 kg/m2. The majority of HFpEF patients had severe cardiac function impairment. Approximately 90% of patients were classified in NYHA III to IV, together with a significant elevation of NT-proBNP in patients' blood. Besides hypertension, comorbid arrhythmia and coronary vascular disease were frequently present in HFpEF patients. Of note, we found diabetes was more common in the HFpEF group (30.8 vs.12.7% of cohorts who had HbA1c>6.5% for HFpEF vs. non-HF individuals). Compared with non-HF individuals, a greater proportion of HFpEF patients were on an antiplatelet, beta-blocker, diuretic, and angiotensin-converting enzyme inhibitors (ACEI)/angiotensin receptor blockers (ARBs) therapy. Echocardiography examination demonstrated that HFpEF patients had an overall prevalence of LV hypertrophy. Although HFpEF patients exhibited a preserved LV ejection fraction (LVEF, 55–65%), the significantly increased average septal-lateral E/e' ratio, one of the echocardiographic markers of LV filling pressure (18), was present among patients. Moreover, HFpEF patients exhibited an apparent left atrial (LA) dilatation compared to non-HF controls. In terms of laboratory variables, HFpEF patients had significantly elevated hs-CRP levels in circulation, underpinning a systemic inflammatory state in patients.
Correlations Between NLR, Inflammation, and Echo Characteristic
The total count of leukocyte, red blood cells, platelet, and monocytes of HFpEF patients was within normal range and was not distinctly different from that of non-HF controls. However, HFpEF patients had a higher neutrophil count but lower lymphocyte count than that of non-HF controls, which resulted in a significant rise in NLR of HFpEF patients (Table 1). The binary logistic regression analysis showed that NLR was significantly associated with HFpEF, independent of effects of age, gender, BMI, hs-CRP, and diabetes (adjusted OR, 2.351; 95% CI, 1.464–3.776; p < 0.001) (Table 2). Then, we calculated the AUC in the ROC curve to assess the predictive performance of NLR for HFpEF, which was 0.796 [95% CI (0.748–0.845), p < 0.001] (Figure 1).
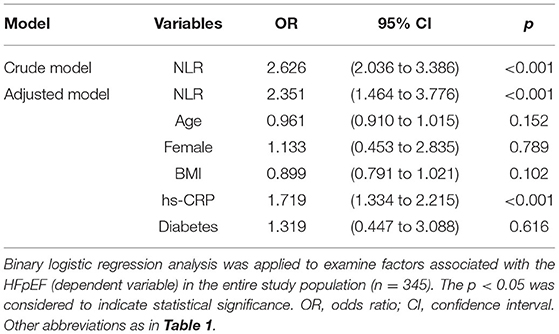
Table 2. Binary logistic regression analysis of factors associated with HFpEF.
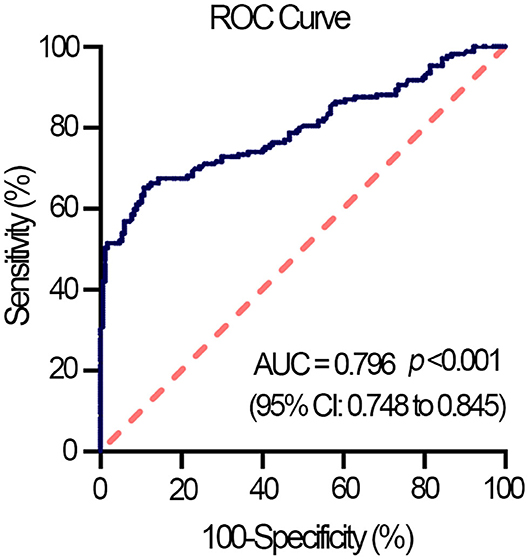
Figure 1. Receiver operating characteristic (ROC) curve displays NLR as a predictor of HFpEF in the entire study population (n = 345). AUC, the area under the curve; CI, confidence interval; HFpEF, heart failure with preserved ejection fraction.
The significant correlations between NLR and hs-CRP, NT-proBNP, and mitral E/e' ratio were found in HFpEF patients, which were not changed after adjusting age, gender, BMI, NYHA class, and diabetes (Supplementary Table 1). Furthermore, results of multivariate linear regression models suggested NLR was likely to be an independent predictor of hs-CRP (p < 0.001), NT-proBNP (p < 0.01), and mitral E/e' ratio (p < 0.05), respectively (Table 3). In contrast, NLR did not display any significant correlation with cardiac structural parameters in HFpEF patients (Supplementary Table 1).
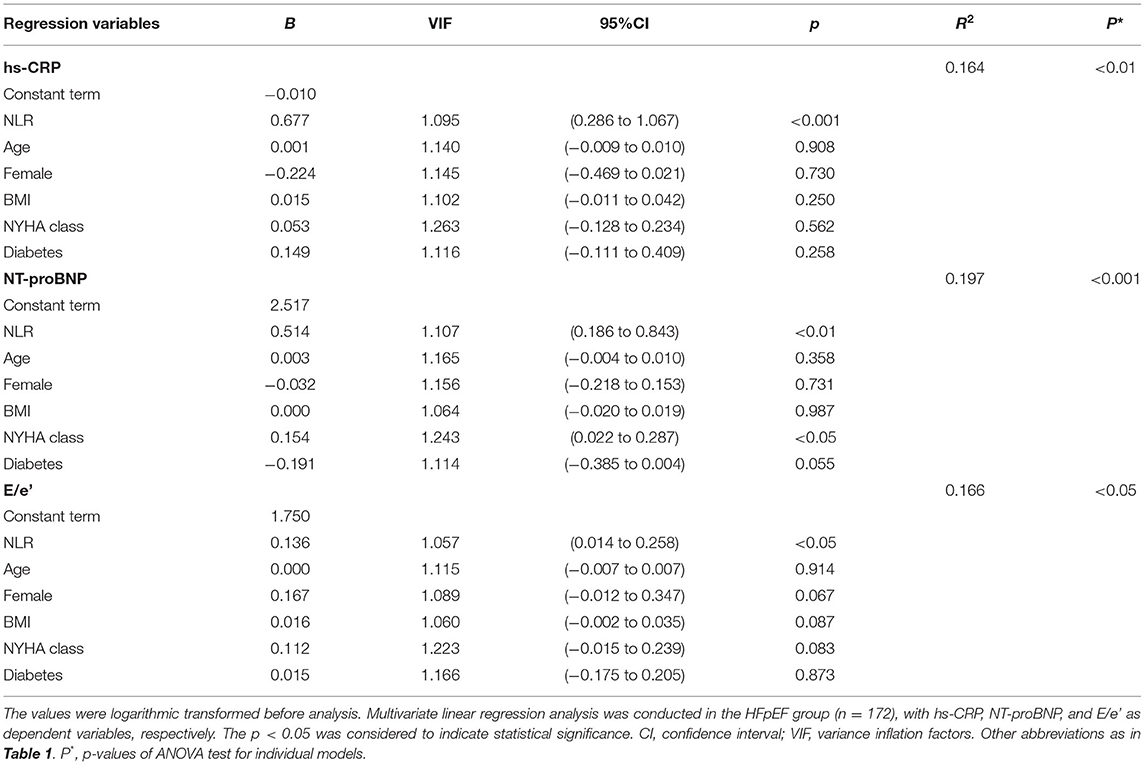
Table 3. Multivariate linear regression analysis with hs-CRP, NT-proBNP, and E/e' as dependent variables, respectively.
Correlations Between Neutrophil Activation and Systemic Inflammation
The circulating levels of pro-inflammatory biomarkers were further examined in HFpEF patients and non-HF control individuals (Table 4, Supplementary Table 2). Multiple pro-inflammatory biomarkers involved in systemic inflammation, such as TNFα, IL-1β, IL-6, IL-10, and sICAM-1 (6, 7), were substantially increased in HFpEF patients. Meanwhile, compared to non-HF controls, HFpEF patients had a higher level of MMP9, a serological marker of collagen turnover that predicts diastolic dysfunction and incidence of HFpEF (19). We also found serum level of NE, one of the neutrophil-derived serine proteases released upon neutrophil activation and degranulation (20), was significantly elevated in the HFpEF group (Table 4). The circulating level of NE correlated well with multiple inflammatory biomarkers, including TNFα, IL-1β, IL-6, and sICAM-1. Meanwhile, these inflammatory biomarkers had a significant correlation with the mitral E/e' ratio. Finally, a correlation between NE and the E/e' ratio (r = 0.562, p < 0.01) was observed in HFpEF patients. By contrast, although MMP9 showed a correlation trend with both NE and E/e' ratio, it did not reach a statistical significance (Table 5).
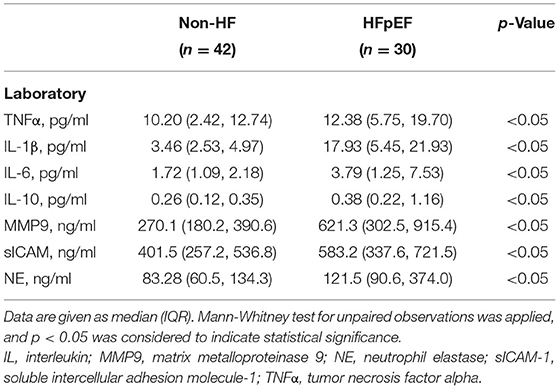
Table 4. The concentration of inflammatory biomarkers in the circulation of non-HF control individuals and HFpEF patients.
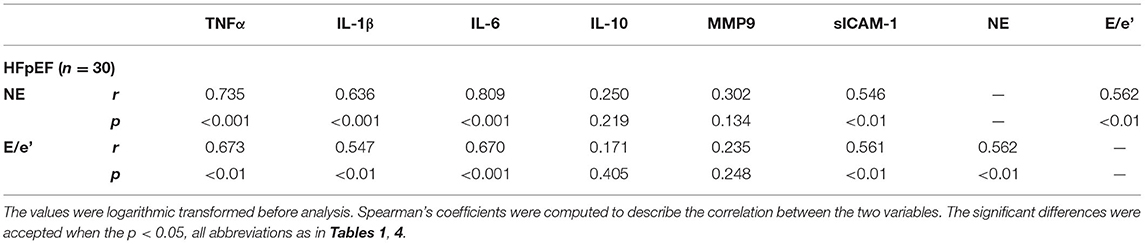
Table 5. The interrelation between neutrophil elastase, inflammatory biomarkers, and mitral E/e' ratio.
The Transcriptomic Characteristics of Neutrophils of HFpEF Patients
We further characterized the transcriptional plasticity of neutrophils collected from non-HF controls and HFpEF patients. A total of 19,813 genes were successfully identified by RNA-sequencing. Among them, 134 genes were filtered with a significantly differential expression between the two groups (Supplementary Table 3). Concretely, compared with neutrophils of the non-HF control group, there were 89 transcripts significantly increased, whereas 45 transcripts decreased in neutrophils of HFpEF patients. The representative gene expression profile in the form of a heatmap was generated (Figure 2A). The Reactome annotation classification was subsequently performed to enrich signaling pathways that genes participate. The most significant enrichments were found in signaling pathways relating to neutrophil degranulation (17 genes), immune system (33 genes), and innate immune system (20 genes) (Figure 2B). Importantly, we found all 17 genes involved in neutrophil degranulation were significantly up-regulated in the HFpEF group. In particular, the gene expression of S100A8 and S100A9, both of them encoding small calcium-binding protein S100A8/A9 complex, were significantly increased in neutrophils of HFpEF patients (Figure 2C). S100A8/A9 complex triggers leukocyte degranulation by promoting protein synthesis of leukotriene B4 (21) or by mechanisms dependent on p38 and JNK (22). In addition, HFpEF patients showed transcriptional up-regulation of S100A12 in circulating neutrophils. The S100A12 protein has been proved to mobilize neutrophils from bone marrow and activate the adhesion and migration of neutrophils toward inflammatory sites (23). Compared with the non-HF controls, neutrophils of the HFpEF patients also had much higher PADI4 gene expression that encodes the peptidyl arginine deiminase 4 (PAD4), a protein that critically regulates chromatin de-condensation and NETs formation (Figure 2C) (24). The gene-level of CD55, encoding a glycoprotein involved in the complement cascade regulation, was elevated in the HFpEF patients' neutrophils. On the resting neutrophil surface, the CD55 protein level is low, but that is highly expressed upon neutrophil activation (25). Finally, multiple transcripts (CDA, ALOX5AP, IL6R) with relatively high abundance in neutrophils were up-regulated in the HFpEF group as well. However, their pathophysiological relevance with cellular activation remains obscure yet.
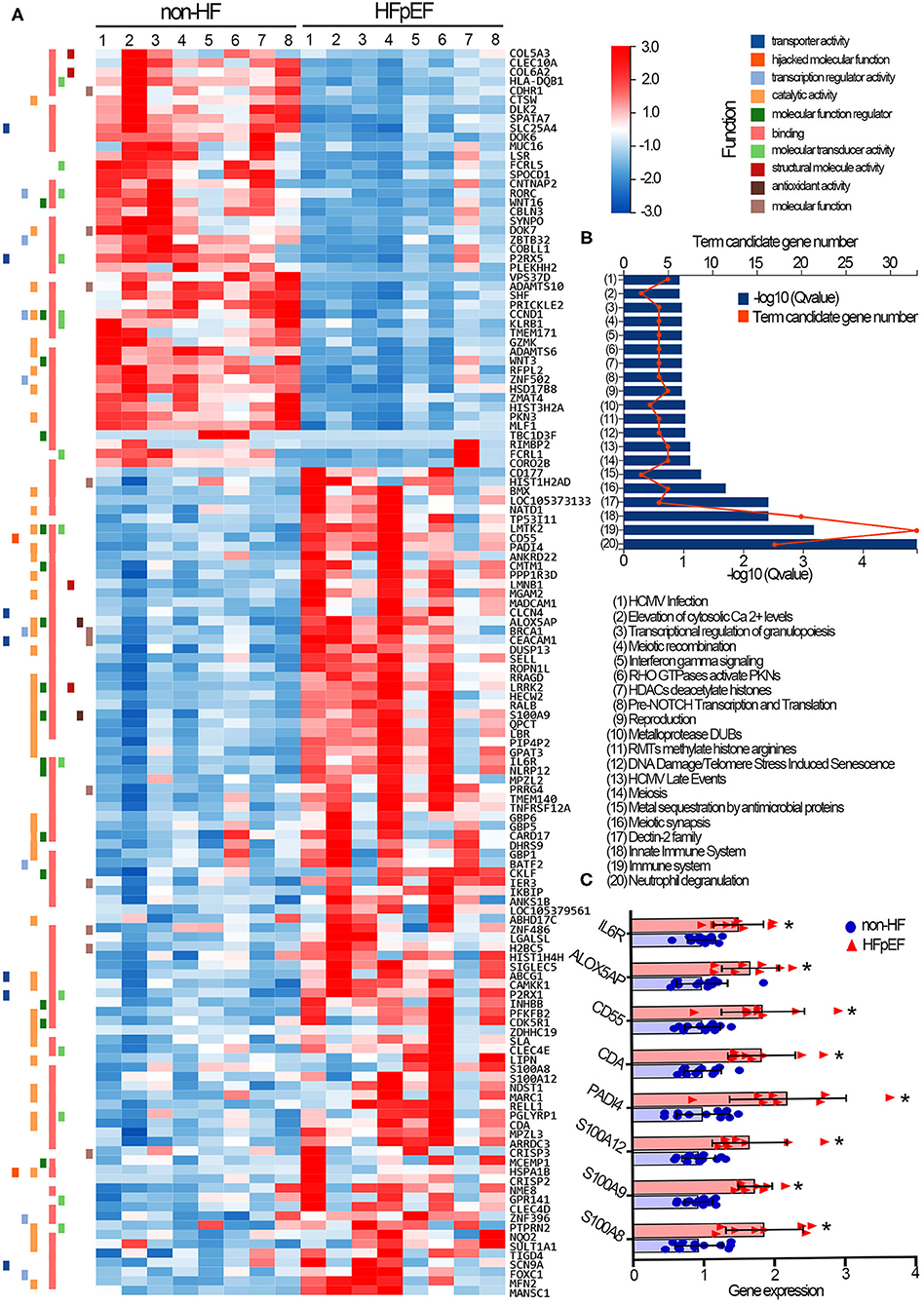
Figure 2. Transcriptomic profile of circulating neutrophils collected from non-HF control individuals and HFpEF patients. (A) The representative heatmap of the 134 genes that were differentially expressed in neutrophils of non-HF control individuals and HFpEF patients. Red indicates relative gene up-regulation, and blue indicates relative gene down-regulation. (B) The Reactome enrichment analysis was performed to characterize signaling pathways that genes participate. (C) Gene expression levels of S100A8, S100A9, S100A12, PADI4, CDA, CD55, ALOX5AP, and IL6R were compared in neutrophils of two groups. Fold change was calculated for comparison and was presented with means and SD. *p < 0.05, compared with non-HF control individuals. Non-HF, non-heart failure controls (n = 12); HFpEF, heart failure with preserved ejection fraction (n = 8).
Discussion
The main findings from the present HFpEF study were as follows: (1) the patients in Southeast China were lean and who had comorbidity burden and worse cardiac structure and function; (2) the high NLR was predictive to HFpEF and independently associated with hs-CRP, NT-proBNP, and mitral E/e' ratio; (3) the heightened serum NE levels correlated with the systemic inflammation and mitral E/e' ratio in HFpEF patients; (4) multiple molecules that drive neutrophil degranulation and inflammation were transcriptionally up-regulated in neutrophils of HFpEF patients.
The clinical HFpEF is frequently bound with a broad of comorbidities (1, 4, 26). Among those comorbidities, obesity is highly prevalent in Western patients. The obese patients (BMI>30 Kg/m2) exhibit comorbidity-driven microvascular inflammation, HF severity, and fibrosis (27). By comparison, recent epidemiologic studies suggest a unique lean phenotype of HFpEF in Asia. The lean HFpEF patients have a high prevalence of diabetes and worse quality of life (28). Our HFpEF cohorts were lean, with an average BMI below 30 Kg/m2, in line with findings from the China HF registry (29). Besides the high prevalence of comorbidities, such as hypertension (71.5%), arrhythmia (64.5%), and coronary artery diseases (51.7%), diabetes was also found to be relatively common in HFpEF patients (33.7%) than that in non-HF cohorts (17.9%). In terms of pharmacological therapies, to date, the evidence-based therapies for HFpEF are scant. As such, current management of HFpEF is primarily directed toward associated cardiovascular comorbidities and control of hypervolemia (26). We found most HFpEF patients were on diuretics, ACEI/ARBs, and beta-blocker therapies. Although the evidence that beta-blockers improve symptoms in HFpEF patients is lacking, these medications were frequently prescribed to our HFpEF patients (87.2%) in order to lower cardiac oxygen demand and prolong diastolic filling time. The high rate of beta-blocker use is also found in HFpEF patients from the Asian-HF registry (78.9%), CHECK-HF registry (78%), and EMPEROR-Preserved trial (86%) (28, 30, 31). There has been compelling evidence to support the prominent role of inflammation in the pathogenesis and progression of HFpEF (5–7, 32, 33). Mechanically, pro-inflammatory molecules augment oxidative stress, impair nitric oxide bioavailability, reduce cyclic guanosine monophosphate activity but raise cardiomyocyte hypertrophy and passive stiffness. Microvascular inflammation drives the proliferation and activation of myofibroblasts. Abnormal extracellular matrix turnover triggered by pro-inflammatory molecules contributes to cardiac interstitial fibrosis (5, 8, 34). Our lean HFpEF patients had high levels of inflammatory biomarkers in circulation, including hs-CRP, TNFα, IL-1β, IL-6, IL-10, and sICAM-10. At present, identifying which organ(s) or cells are inflamed in patients is still tricky.
Systemic inflammation and immune cell homeostasis are two interlinked processes that constantly emphasize each other. The important role of monocytes and macrophages in cardiovascular inflammation has been historically appreciated (35). By contrast, neutrophils have been neglected in the context of cardiovascular research for a long time. Currently, our understanding of the pleiotropic roles of neutrophils in chronic inflammation has been advanced (9, 36). Aberrant neutrophils in circulation can stratify the risk of patients hospitalized with HFpEF or predict the poor prognosis of patients (11–13). In cardiac specimens of both HFpEF patients and animals, the neutrophil infiltration is found to be associated with inflammatory and fibrotic damages that result in LV stiffness (14, 15). In lean HFpEF patients, we observed an apparent rise in NLR ratio and serum levels of NE. Multivariate regression analysis revealed a clear association between the NLR and hs-CRP, NT-proBNP, and mitral E/e' ratio. Moreover, NLR was likely to be predictive of the increased risk of HFpEF. Of note, such associations were independent of the effect of age, gender, BMI, NYHA class, and diabetes. Meanwhile, the elevated serum NE levels in HFpEF patients significantly correlated with multiple pro-inflammatory biomarkers. Both of them also displayed a close correlation with the mitral E/e' ratio of patients. These interrelations collectively indicated the pathological potential of activated neutrophils in aggravating systemic inflammation and diastolic dysfunction of HFpEF patients. To date, the pathophysiological mechanisms responsible for neutrophils' detrimental effects on heart tissues remain to be elucidated yet. In the future study, it is of significant interest to resolve this doubt by investigating the cardiac phenotypes and systemic inflammation levels in our lean HFpEF mice after the genetic depletion of neutrophils (15).
So far, a number of risk factors have been proposed to drive granulopoiesis, including metabolic alternations (hypercholesterolemia and hyperglycemia), inflammasome pathways, aging, stress, and disturbed lifestyle (9). We noted that the distribution of age and gender was comparable between HFpEF patients and non-HF control individuals. The comorbid hyperlipidemia was neither prevalent in HFpEF patients (11.6%). By comparison, diabetes was found to be relatively common in HFpEF patients. Hyperglycemia directly induces proliferation and expansion of bone marrow myeloid progenitors (37, 38). Under chronic inflammation, some cytokines function as critical pro-inflammatory “emergency” signals to drive myeloid differentiation. IL-1β directly accelerates myeloid differentiation of hematopoietic stem cells via precocious activation of a PU.1-dependent gene program (39). Myocardial infarction results in rapid recruitment of neutrophils to the infarct. The infiltrated neutrophils release IL-1β, which may contribute to the cytokine pool. As a consequence, IL-1β acts with hematopoietic progenitor cells in the bone marrow and further stimulates granulopoiesis in a cell-autonomous manner (40). Alternatively, other inflamed tissues or cells may produce cytokine that accelerates myelopoiesis and neutrophil production, leading to neutrophil recruitment in heart tissues. However, the risk factors that drive neutrophilia in lean HFpEF patients remain unknown yet.
Although neutrophils are traditionally considered to be transcriptionally silent, the transcriptional plasticity of neutrophils upon sterile stimulation and microbial insults has been unraveled (41). We further found the transcriptional signatures of neutrophils of HFpEF patients were distinctive to that of non-HF control individuals. Beyond our expectation, circulating neutrophils from HFpEF patients do not show robust transcriptomic changes of the classical pro-inflammatory cytokine found in primed neutrophils in vitro (42). However, we noted multiple molecules that drive neutrophil degranulation and inflammation were transcriptionally up-regulated in neutrophils of HFpEF patients. Of note, among 134 genes with differential expression, all genes enriched in the neutrophil degranulation pathway were up-regulated in HFpEF patients' neutrophils, consistent with an increased level of neutrophil-derived NE in patients' blood. Moreover, genes encoding a small calcium-binding protein family (S100A8/A9/A12) were transcriptionally up-regulated in neutrophils of HFpEF patients. S100A8/A9 functions as neutrophil-derived alarmins that can activate CD11b and induce neutrophil adhesion to fibrinogen, leading to neutrophil migration to inflammatory sites (43). Hyperglycemia can increase the release of S100A8/S100A9 from neutrophils, and this protein complex interacts with the receptor for advanced glycation end products on myeloid progenitor cells and enhance myelopoiesis (37, 38). In infarct myocardial tissues, S100A8/S100A9 released from neutrophils can bind to Toll-like receptor (TLR) 4 and prime the nod-like receptor family pyrin domain-containing 3 inflammasome in naive neutrophils, resulting in IL-1β-driven granulopoiesis (40). It is particularly worth noting that PADI4 is up-regulated substantially in neutrophils of HFpEF patients. PAD4 critically regulates chromatin de-condensation and NETs formation (24). The pathogenic potential of NETs in cardiovascular inflammation has so far been well-documented (44). NETs license macrophages to turn on transcriptional regulation of IL-6 and pro-IL-1β via TLR2/4 in atherosclerosis (45). NETs stimulate human lung fibroblasts to a myofibroblast with elevated α-smooth muscle actin expression (46) and mediates extracellular matrix remodeling (47). The cytotoxic histone and deoxyribonucleic acid bound to NETs induces organ fibrosis in aged mice (48). However, in clinical HFpEF patients, it is still difficult to determine whether neutrophils with high expression of PADI4 are prone to form NETs in failing heart tissues. In lean HFpEF mouse hearts, we observed the presence of NETs and increased PAD4 protein levels, which was paralleled with cardiac inflammation and fibrosis (15). Our ongoing study further demonstrated neutrophils from lean HFpEF mice were prone to form NETs. The NETs-containing media significantly enhanced alpha-smooth muscle actin expression in co-cultured myocardial fibroblasts, suggesting a pro-fibrotic action of NETs (unpublished data).
Given the significant roles of neutrophils in cardiovascular inflammation, the specific intervention of neutrophils may open the door for the development of a novel therapeutic strategy. Interestingly, metformin, a drug representing a worldwide cornerstone in anti-diabetes therapy, can exert inflammation-inhibitory effects independently from glucose control (49). Metformin can inhibit NETs in vitro (50), decrease NLR in the diabetic population, and suppress plasma cytokine levels in the non-diabetic heart failure cohort (51). It is recently reported that, in patients who are infected with coronavirus disease 2019, metformin users have a lower level of neutrophil counts but a higher level of lymphocyte counts in the blood. Meanwhile, serum inflammatory factors (CRP, IL-6, TNF-α) and cardiac injury indicators (NT-proBNP) are marked lower in the metformin group (52). Therefore, we think repurposing metformin to inflammation-driven chronic HFpEF would be an active field investigated in the future.
Study Limitations
Several limitations should be considered when interpreting the results of the present study. This was a small single cohort study. There was, therefore, a potential lack of power. The roles of neutrophils contributing to systemic or myocardial inflammation should be investigated in larger HFpEF cohorts. In addition, our investigation was not exploratory but based on the published hypothesis that inflammation is a critical pathogenic stimulus in HFpEF. We observed several correlations between neutrophil activation, systemic inflammation, and ventricular functional impairment in HFpEF patients. However, the tissue or cellular source of inflammatory molecules and their interrelation with neutrophilia in patients remain uncertain yet. Given the limitation to obtain heart specimens from clinical patients, the mechanisms by which neutrophils and NETs impair cardiac function need to be addressed by more intensive animal and in vitro studies.
Conclusion
The high NLR coupled with transcriptional activation of neutrophils correlates with systemic inflammation and functional impairment in HFpEF patients, which may suggest a causative role of neutrophils in the pathogenesis of the disease.
Data Availability Statement
The data used for the transcriptomic analysis were deposited in the NCBI Sequence Read Archive (SRA) database. The data are accessible via the SRA accession: PRJNA717666.
Ethics Statement
The studies involving human participants were reviewed and approved by Research Ethics Committees of Shenzhen Second People's Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
BB, HC, and YX were responsible for the study design and manuscript writing. BB, MC, LJ, and JX contributed to the acquisition and analysis of the data. All authors gave final approval and agreed to be accountable for all aspects of work to ensure integrity and accuracy.
Funding
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2021A1515010696) and Seed Funding for Young Individual Research of Shenzhen Second People's Hospital (4001019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.614757/full#supplementary-material
References
1. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2017) 14:591–602. doi: 10.1038/nrcardio.2017.65
2. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. (2019) 381:1609–20. doi: 10.1056/NEJMoa1908655
3. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. (2014) 370:1383–92. doi: 10.1056/NEJMoa1313731
4. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, et al. Phenotype-specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation. (2016) 134:73–90. doi: 10.1161/CIRCULATIONAHA.116.021884
5. Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2021) 18:400–23. doi: 10.1038/s41569-021-00516-5
6. Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, et al. Proteomic evaluation of the comorbidity-inflammation paradigm in heart failure with preserved ejection fraction: results from the PROMIS-HFpEF study. Circulation. (2020) 142:2029–44. doi: 10.1161/CIRCULATIONAHA.120.045810
7. Chirinos JA, Orlenko A, Zhao L, Basso MD, Cvijic ME, Li Z, et al. Multiple plasma biomarkers for risk stratification in patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. (2020) 75:1281–95. doi: 10.1016/j.jacc.2019.12.069
8. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
9. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17:327–40. doi: 10.1038/s41569-019-0326-7
10. Tracchi I, Ghigliotti G, Mura M, Garibaldi S, Spallarossa P, Barisione C, et al. Increased neutrophil lifespan in patients with congestive heart failure. Eur J Heart Fail. (2009) 11:378–85. doi: 10.1093/eurjhf/hfp031
11. Komajda M, Carson PE, Hetzel S, McKelvie R, McMurray J, Ptaszynska A, et al. Factors associated with outcome in heart failure with preserved ejection fraction: findings from the Irbesartan in heart failure with preserved ejection fraction study (I-PRESERVE). Circ Heart Fail. (2011) 4:27–35. doi: 10.1161/CIRCHEARTFAILURE.109.932996
12. Boralkar KA, Kobayashi Y, Amsallem M, Ataam JA, Moneghetti KJ, Cauwenberghs N, et al. Value of neutrophil to lymphocyte ratio and its trajectory in patients hospitalized with acute heart failure and preserved ejection fraction. Am J Cardiol. (2020) 125:229–35. doi: 10.1016/j.amjcard.2019.10.020
13. Hage C, Michaelsson E, Kull B, Miliotis T, Svedlund S, Linde C, et al. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. (2020) 7:1534–46. doi: 10.1002/ehf2.12700
14. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. (2011) 4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451
15. Bai B, Yang W, Fu Y, Foon HL, Tay WT, Yang K, et al. Seipin knockout mice develop heart failure with preserved ejection fraction. JACC Basic Transl Sci. (2019) 4:924–37. doi: 10.1016/j.jacbts.2019.07.008
16. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. (2018) 39:3021–104. doi: 10.1093/eurheartj/ehy339
17. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
18. Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quinones MA. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. (1997) 30:1527–33. doi: 10.1016/S0735-1097(97)00344-6
19. Martos R, Baugh J, Ledwidge M, O'Loughlin C, Murphy NF, Conlon C, et al. Diagnosis of heart failure with preserved ejection fraction: improved accuracy with the use of markers of collagen turnover. Eur J Heart Fail. (2009) 11:191–7. doi: 10.1093/eurjhf/hfn036
20. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. (2006) 6:541–50. doi: 10.1038/nri1841
21. Kannan S. Inflammation: a novel mechanism for the transport of extracellular nucleotide-induced arachidonic acid by S100A8/A9 for transcellular metabolism. Cell Biol Int. (2003) 27:593–5. doi: 10.1016/S1065-6995(03)00082-9
22. Simard JC, Girard D, Tessier PA. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J Leukoc Biol. (2010) 87:905–14. doi: 10.1189/jlb.1009676
23. Rouleau P, Vandal K, Ryckman C, Poubelle PE, Boivin A, Talbot M, et al. The calcium-binding protein S100A12 induces neutrophil adhesion, migration, and release from bone marrow in mouse at concentrations similar to those found in human inflammatory arthritis. Clin Immunol. (2003) 107:46–54. doi: 10.1016/S1521-6616(02)00043-8
24. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. (2015) 21:815–9. doi: 10.1038/nm.3887
25. Berger M, Medof ME. Increased expression of complement decay-accelerating factor during activation of human neutrophils. J Clin Invest. (1987) 79:214–20. doi: 10.1172/JCI112786
26. Borlaug BA. Evaluation and management of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2020) 17:559–73. doi: 10.1038/s41569-020-0363-2
27. Sabbah MS, Fayyaz AU, de Denus S, Felker GM, Borlaug BA, Dasari S, et al. Obese-inflammatory phenotypes in heart failure with preserved ejection fraction. Circ Heart Fail. (2020) 13:e006414. doi: 10.1161/CIRCHEARTFAILURE.119.006414
28. MacDonald MR, Tay WT, Teng TK, Anand I, Ling LH, Yap J, et al. Regional variation of mortality in heart failure with reduced and preserved ejection fraction across asia: outcomes in the ASIAN-HF registry. J Am Heart Assoc. (2020) 9:e012199. doi: 10.1161/JAHA.119.012199
29. Zhang Y, Zhang J, Butler J, Yang X, Xie P, Guo D, et al. Contemporary epidemiology, management, and outcomes of patients hospitalized for heart failure in china: results from the china heart failure (China-HF) registry. J Card Fail. (2017) 23:868–75. doi: 10.1016/j.cardfail.2017.09.014
30. Uijl A, Veenis JF, Brunner-La Rocca HP, van Empel V, Linssen GCM, Asselbergs FW, et al. Clinical profile and contemporary management of patients with heart failure with preserved ejection fraction: results from the CHECK-HF registry. Neth Heart J. (2021). doi: 10.1007/s12471-020-01534-7. [Epub ahead of print].
31. Anker SD, Butler J, Filippatos G, Shahzeb Khan M, Ferreira JP, Bocchi E, et al. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur J Heart Fail. (2020) 22:2383–92. doi: 10.1002/ejhf.2064
32. Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, et al. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. (2018) 72:1081–90. doi: 10.1016/j.jacc.2018.06.050
33. Glezeva N, Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev. (2014) 19:681–94. doi: 10.1007/s10741-013-9405-8
34. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. (2021) 117:1450–88. doi: 10.1093/cvr/cvaa324
35. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. (2018) 18:733–44. doi: 10.1038/s41577-018-0065-8
36. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. (2017) 17:248–61. doi: 10.1038/nri.2017.10
37. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. (2013) 17:695–708. doi: 10.1016/j.cmet.2013.04.001
38. Flynn MC, Kraakman MJ, Tikellis C, Lee MKS, Hanssen NMJ, Kammoun HL, et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ Res. (2020) 127:877–92. doi: 10.1161/CIRCRESAHA.120.316653
39. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. (2016) 18:607–18. doi: 10.1038/ncb3346
40. Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation. (2020) 141:1080–94. doi: 10.1161/CIRCULATIONAHA.119.043833
41. Naranbhai V, Fairfax BP, Makino S, Humburg P, Wong D, Ng E, et al. Genomic modulators of gene expression in human neutrophils. Nat Commun. (2015) 6:7545. doi: 10.1038/ncomms8545
42. Wright HL, Thomas HB, Moots RJ, Edwards SW. RNA-seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS ONE. (2013) 8:e58598. doi: 10.1371/journal.pone.0058598
43. Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Pro-inflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. (2003) 170:3233–42. doi: 10.4049/jimmunol.170.6.3233
44. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
45. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
46. Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol. (2014) 233:294–307. doi: 10.1002/path.4359
47. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaa04227. doi: 10.1126/science.aao4227
48. Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med. (2017) 214:439–58. doi: 10.1084/jem.20160530
49. Bai B, Chen H. Metformin: a novel weapon against inflammation. Front Pharmacol. (2021) 12:622262. doi: 10.3389/fphar.2021.622262
50. Menegazzo L, Scattolini V, Cappellari R, Bonora BM, Albiero M, Bortolozzi M, et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. (2018) 55:593–601. doi: 10.1007/s00592-018-1129-8
51. Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, et al. Anti-Inflammatory effects of metformin irrespective of diabetes status. Circ Res. (2016) 119:652–65. doi: 10.1161/CIRCRESAHA.116.308445
Keywords: heart failure with preserved ejection fraction, neutrophil to lymphocyte ratio, inflammation, diastolic dysfunction, gene signature
Citation: Bai B, Cheng M, Jiang L, Xu J, Chen H and Xu Y (2021) High Neutrophil to Lymphocyte Ratio and Its Gene Signatures Correlate With Diastolic Dysfunction in Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 8:614757. doi: 10.3389/fcvm.2021.614757
Received: 07 October 2020; Accepted: 18 May 2021;
Published: 24 June 2021.
Edited by:
Jian Zhang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Eisuke Amiya, The University of Tokyo Hospital, JapanRisto Kerkela, University of Oulu, Finland
Copyright © 2021 Bai, Cheng, Jiang, Xu, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Xu, eXgxNDEySEZAeWVhaC5uZXQ=