
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 03 February 2021
Sec. Cardiovascular Metabolism
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.629933
This article is part of the Research Topic COVID-19 Mechanisms on Cardio-Vascular Dysfunction: From membrane receptors to immune response View all 9 articles
 Jacob Roberts1
Jacob Roberts1 Antonia L. Pritchard1
Antonia L. Pritchard1 Andrew T. Treweeke1
Andrew T. Treweeke1 Adriano G. Rossi2Nicole Brace1
Adriano G. Rossi2Nicole Brace1 Paul Cahill3
Paul Cahill3 Sandra M. MacRury1Jun Wei1
Sandra M. MacRury1Jun Wei1 Ian L. Megson1*
Ian L. Megson1*Meta-analyses have indicated that individuals with type 1 or type 2 diabetes are at increased risk of suffering a severe form of COVID-19 and have a higher mortality rate than the non-diabetic population. Patients with diabetes have chronic, low-level systemic inflammation, which results in global cellular dysfunction underlying the wide variety of symptoms associated with the disease, including an increased risk of respiratory infection. While the increased severity of COVID-19 amongst patients with diabetes is not yet fully understood, the common features associated with both diseases are dysregulated immune and inflammatory responses. An additional key player in COVID-19 is the enzyme, angiotensin-converting enzyme 2 (ACE2), which is essential for adhesion and uptake of virus into cells prior to replication. Changes to the expression of ACE2 in diabetes have been documented, but they vary across different organs and the importance of such changes on COVID-19 severity are still under investigation. This review will examine and summarise existing data on how immune and inflammatory processes interplay with the pathogenesis of COVID-19, with a particular focus on the impacts that diabetes, endothelial dysfunction and the expression dynamics of ACE2 have on the disease severity.
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has been the focus of an unprecedented, dramatically augmented rate of scientific research since its emergence at the end of 2019. The direct and indirect health impacts of this disease have been felt by people all over the world: over 80 million people have been infected with the SARS-CoV-2 virus and the death toll has exceeded 1.75 million globally. Lockdown measures designed to curtail and contain outbreaks have led to rarely experienced increases in unemployment and many healthcare systems are being overwhelmed by severe coronavirus disease 2019 (COVID-19; the disease caused by the SARS-CoV-2 virus), lowering the rates of admission and treatment of many other health conditions. One of the most interesting and least well-understood aspects of the disease is the wide spectrum of disease severity experienced in those infected—from asymptomatic to fatal on account of respiratory or multi-organ failure. In addition, the long-term legacy of COVID-19 is, by definition, not yet fully appreciated, although worrying signs are emerging to suggest that some individuals who have survived the acute infection stage of the disease might be prone to a wide range of long-lasting health conditions (1).
A number of meta-analyses have been conducted in an effort to understand those factors that might affect the severity of COVID-19 (2–6). The outcome of these studies have identified a number of key factors that associate with critical/fatal events, most notably: gender (male, compound odds ratio (OR): 1.76), black, Asian or mixed ethnicity, BAME (OR: 1.9); age (OR: 6.06), smoking (OR: 2.04), cardiovascular disease (OR: 5.19), respiratory disease (OR: 5.15), malignancy (OR: 1.6) (2), hypertension (OR: 3.36) (7), severe mental illness (OR: 2.27) (8), and obesity (OR: 5.70) (9). Diabetes has been identified as a comorbidity associated with COVID-19 severity, with meta-analyses indicating that pre-existing diabetes is associated with a greater risk of severe COVID-19 and death (Table 1) with OR from 1.9 to 2.68 (4, 27, 32, 40, 55, 59, 64). Studies of COVID-19 patients in England found that the hazard risk for mortality was greater for patients with type 1 diabetes mellitus (T1DM: OR 2.23) than those with type 2 diabetes mellitus (T2DM: OR 1.61) (65). These findings were reinforced by a later study, which found similar mortality risks for T1DM (OR: 3.51) and T2DM (OR: 1.80) (66). In the former study, amongst patients with T2DM, hazard ratios were highest in those with poorest glycaemic control, but there was found to be an unusual U-shaped relationship with BMI and COVID-19 mortality in both types of diabetes. Association of this disease with severe COVID-19 symptoms and mortality is of particular concern on account of the spiralling prevalence of diabetes, with current estimates suggesting that >400 million people are living with diabetes worldwide and roughly half are undiagnosed (67). The rise in diabetes has been linked to rapidly escalating levels of obesity, particularly in highly populated regions of the world with known genetic pre-disposition to T2DM (e.g., South-East Asia). Until the COVID-19 pandemic, the autoimmune form of diabetes, T1DM, was considered to be relatively stable at ~1% of the population, but emerging evidence suggests that COVID-19 might be responsible for a recent rise in prevalence of T1DM (68).
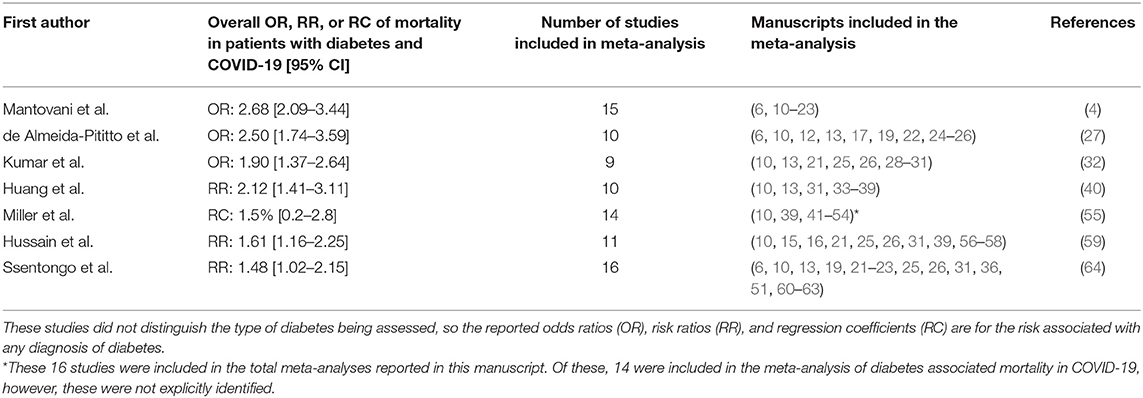
Table 1. Meta-analyses of the mortality risk associated with pre-existing diabetes in COVID-19 patients.
Given the novelty of COVID-19, our understanding of the reasons for the association between severity of COVID-19 symptoms and pre-existing diabetes is poorly developed and highly complex on account of the inter-relationship of T2DM in particular with many other risk factors for severe COVID-19 symptoms (age, ethnicity, obesity, cardiovascular disease and hypertension). The aim of this review is to bring together the early evidence emerging from COVID-19 studies with that from previous, related epidemics (e.g., SARS) in an effort to understand the mechanisms by which diabetes might prime individuals to suffer more severe symptoms than their healthy counterparts. In addition, the review will examine the evidence suggesting that COVID-19 might induce a diabetes-like syndrome in some patients and identify potential mechanisms behind this.
SARS-CoV-2 is transmitted primarily via respiratory droplets, with an average of 4–5 days before symptom onset (24, 69–71) and peak viral load within 5–6 days of symptom onset. COVID-19 symptoms include fever, cough, anosmia, ageusia, fatigue, myalgia, headache, sore throat, diarrhoea and dyspnoea (shortage of breath) (41–43, 72).
Severe COVID-19 cases progress to acute respiratory distress syndrome (ARDS) ~8–9 days after symptom onset (41, 42) and is characterised by pneumonia, pulmonary oedema, oxygen saturation (SpO2) <93%, respiratory failure requiring invasive ventilation, admission to the intensive care unit (ICU), coagulopathy, lymphopaenia, cytokine storm, viraemia, and multi-organ damage (6, 24, 42, 43, 72–74). Severe COVID-19 requires confirmation by physical and laboratory-based examinations, including SpO2, D-dimer assessment of fibrinolysis, inflammatory markers, leucocyte counts and computerised tomography (CT) scans (24, 75). Pulmonary oedema occurs due to excess fluid seeping out of the blood vessels in the lungs, affecting the exchange of gas (oxygen and carbon dioxide), leading to decreased SpO2, respiratory failure and ICU admission. Coagulopathy, including microvascular thrombosis and disseminated intravascular coagulopathy (DIC) (76, 77) and lymphopaenia could be a predictor of mortality in severe COVID-19 (78). Lung scarring and fibrosis, a hyper-inflammatory marker that suggests tissue damage due to overactive, dysregulated inflammation, occurs in survivors of severe disease. In a longitudinal study of 90 patients, CT scans showed lung abnormalities soon after the onset of symptoms. Ground-glass opacity was the most common sign, with an increase in a mixed pattern phenotype observed 12–17 days following the onset of symptoms (79). Another clinical report revealed that 58.3% of severe cases had bilateral patchy opacities compared to 30.4% of non-severe cases, suggesting the presence of a radiographic marker in severe COVID-19 (24). Lung autopsies of deceased COVID-19 patients demonstrated that neutrophil extracellular traps (NETs) were likely to be involved in inflammation-associated lung damage, thrombosis and fibrosis (80). Lung autopsies also showed type-2 pneumocyte hyperplasia in all cases examined (81), potentially suggesting a compensation for the loss of angiotensin converting enzyme 2 (ACE2) expressing cells in the lung. Severe infections have been associated with a sustained high viral load in the upper airways (82, 83) and patients who have died from severe COVID-19 showed SARS-CoV-2 virions can be detected in almost all tissues, including the brain (84, 85).
A substantial proportion of people infected with SARS-CoV-2 are asymptomatic. However, an accurate rate of asymptomatic infection of SARS-CoV-2 is difficult to ascertain due to the limitations of testing policies. Large-scale longitudinal testing of populations is required to provide a more precise picture of the number and demographics of asymptomatic carriers; computational modelling has predicted the number of infections to be 3 to 20 times higher than the number of confirmed cases (86). Asymptomatic infections could be due to the efficacy of the host immune responses, low viral load, cross-reactivity of existing immune effectors, or prior infection with/pre-existing immunity to other related human coronaviruses, conferring cross-immunity against SARS-CoV-2 (87–89) and resulting in a longer period of detection of viral RNA in the upper respiratory tracts of symptomatic patients (89).
To date, studies in COVID-19 hotspots have revealed that <20% of infections are classed as “severe,” while the remaining 80% are asymptomatic, mild, or moderate (24, 90). While there has been little evidence that demographic factors and co-morbidities impact on the risk of SARS-CoV-2 infection, it is clear they influence the severity of symptoms of COVID-19. That there is such a wide range of symptoms was initially perplexing; however, the associations with age, ethnicity and co-morbidities have provided vital clues as to why some individuals develop specific severe symptoms or succumb to their infection.
SARS-CoV-2 is a positive-sense, single-stranded RNA virus in the Nidovirales order and Betacoronavirinae genus (Wuhan-Hu-1 isolate reference genome NCBI ID: 86693; transcript ID: NC_045512.2; Figure 1) (92, 93). It is one of several coronaviruses that infect the human respiratory system, including SARS-CoV-1 (causing severe acute respiratory syndrome, SARS) (92, 94), MERS-CoV (causing Middle East respiratory syndrome, MERS) (92, 95), HCoV-229E, HCoV-HKU1, HCoV-NL63, and HCoV-OC43 (92). The virion encodes proteins required for viral replication (Figure 1), including the RNA-dependent RNA polymerase (RdRp) and structural proteins, including spike (S), membrane (M), nucleocapsid (N) and envelope (E) (96), which are likely to function similarly to those described in related coronaviruses.
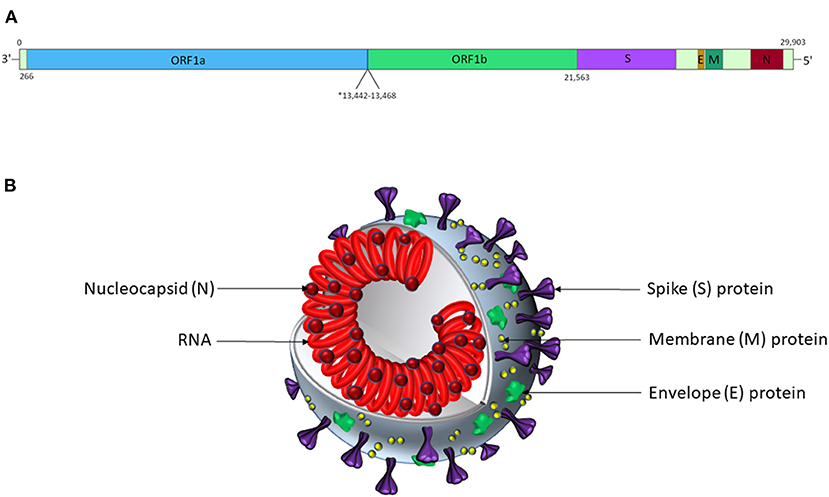
Figure 1. SARS-CoV-2 genomic and virion structures. (A) Genome schematic of SARS-CoV-2. The asterisk marks the overlapping reading frames of ORF1a and ORF1b, which encode a variety of non-structural proteins, including helicases, proteases, and an RNA-dependent RNA polymerase. The S protein is encoded by nt. 21,563-25,384; the E protein is encoded by nt. 26,245-26472; the M protein is encoded by nt. 26,523-27,191; and the N protein is encoded by nt. 28,274-29,533. Accessory proteins not shown. Adapted from Alanagreh et al. (91). (B) Schematic representation of the SARS-CoV-2 virion structure. This figure is not to scale, and relative abundances of the proteins shown are arbitrary.
The S protein of SARS-CoV-2 is a homotrimer and, like that of SARS-CoV-1, is a class I fusion protein [13] that recognises the host cell receptor, ACE2 as a key step in the viral internalisation process (Figure 2). ACE2 (Xp22.2) is a zinc metalloprotease with homology to both ACE and collectrin (CLTRN) (99, 100). X-ray crystallography has confirmed that the SARS-CoV-2 binding interface is similar to that of the SARS-CoV-1 S protein (97) and in vitro experiments have shown that SARS-CoV-2 internalisation requires ACE2 (101, 102).
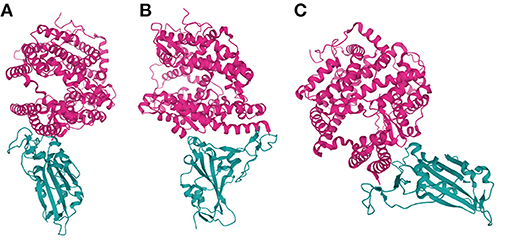
Figure 2. SARS-CoV-2 S interfacing with ACE2. Various orientations of an X-ray crystallography-derived cartoon representation of the Spike protein S1 subunit of SARS-CoV-2 (cyan) bound to an extracellular portion of ACE2 (residues 1-597; pink). (B) Is a 90° anticlockwise rotation, along the z-axis, of (A), while (C) is angled to highlight the shape of the binding interface between ACE2 and SARS-CoV-2 S, ~90° clockwise along the z-axis, and ~45° counter-clockwise along the x-axis, relative to A. Figure was created using *Mol. PDB ID: 6M0J, Crystal Structure of SARS-CoV-2 spike receptor-binding domain bound with ACE2 (97, 98).
Upon binding to ACE2, the conformation of the S protein changes to initiate the membrane fusion of virus particles and host cell, required for internalisation of the viral genome. The S1 subunit of the protein contains the receptor binding domain (RBD), which interfaces with the N-terminus of ACE2 on target host cells. The S2 subunit contains two distinct regions of heptad repeats, as well as a fusion domain, all of which are essential for viral entry into host cells (103). Following viral binding to ACE2, the heptad repeats of S2 associate to each other, forming a six-helical bundle (6HB) complex (104–106). In SARS-CoV-1, the formation of 6HB exposes the fusion domain of the S2 subunit, which is hydrophobic and embeds into the host cell membrane, initiating fusion (105).
The S protein exists in either “standing up” or “lying down” conformations prior to receptor binding. When standing up, the RBD of the S protein is more exposed, allowing for higher affinity binding to ACE2, but also presenting a highly immunogenic target (107–109). There is a 4 amino acid insertion seen in the SARS-CoV-2 S protein that is not present in SARS-CoV-1, which creates a furin cleavage site at the junction between the S1 and S2 subunits (103). In vitro, pre-treatment with furin enhances viral entry into ACE2 expressing cell lines, which is hypothesised to cause a transition from lying down to the standing up conformation (109).
The entry of SARS-CoV-2 into cells via ACE2 also relies on transmembrane serine protease 2 (TMPRSS2), which primes the S protein through proteolytic cleavage, altering its conformation in a manner that facilitates membrane fusion (109).
The main physiological role of ACE2 centres on its metalloprotease activity, processing multiple proteins involved in the renin-angiotensin-aldosterone-system (RAAS; Figure 3). Specifically, ACE2 converts the vasoconstrictor agents angiotensin-I into angiotensin-(1–9) and angiotensin-II into angiotensin-(1–7), both products being vasodilatory (110, 111). The action of ACE2 therefore shifts balance of vascular control in favour of vasodilation (112). A receptor for angiotensin-II, AT1R, is also present on macrophages, dendritic cells, T-cells, mesangial cells and vascular smooth muscle cells, meaning that ACE2 processing of angiotensin-II also influences function of these cells (113). Therefore, in addition to its role in SARS-CoV-2 cellular entry, ACE2 has normal physiological functions that directly link to COVID-19 features in the lung, including inflammation, oxidative stress and fibrosis (113).
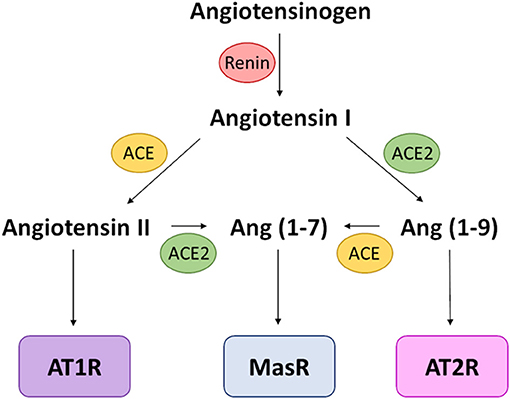
Figure 3. RAAS schematic. Simplified schematic focusing on the activity of ACE and ACE2 and the resulting receptor activation. The vasoconstrictive activity of this pathway is mediated by AT1R activation, which also causes increased oxidative stress; MasR and AT2R activation leads to vasodilation and a reduction in oxidative stress. Receptors are in boxes; enzymes that processes signalling peptides are in ovals.
In addition to its role in the RAAS, ACE2 inactivates the vasodilator, bradykinin. It is important to note that there is complex interplay between RAAS and the bradykinin system, with multiple feedback loops and intersections. The importance of bradykinin in the COVID-19 story has gained credence recently on account of an unbiased in silico approach to understanding the underlying reason for differential severity in COVID-19 symptoms (114). Bradykinin ligates specific G-protein-coupled receptors to mediate vasodilatation via endothelial nitric oxide (NO) and prostacyclin (PGI2), which drive vascular permeability and local oedema. Leaky capillaries in the lungs of COVID-19 patients would not only compromise breathing on account of oedema, but would also increase trafficking of inflammatory cells between the blood and lung tissue, together with the inflammatory mediators they convey.
Following the identification of ACE2 as the target of the S protein of SARS-CoV-1 [reviewed by (115)], ACE2 was shown to be expressed by lung alveolar epithelial cells, small intestine enterocytes, vascular endothelial cells and arterial smooth muscle cells derived from the brain, lungs, kidneys, large intestine and small intestine, using immunohistochemistry (116). ACE2 is also highly expressed by nasal epithelial cells (83), which is pertinent in the pathogenesis of cellular infection in the upper airways. Single-cell RNA sequencing has shown ACE2 mRNA expression in a range of cells: type II alveolar cells, myocardial cells, kidney proximal tubule cells, urothelial bladder cells, ileal epithelial cells and oesophageal epithelial cells, though this study did not examine protein levels (117).
ACE2 also exists in a soluble form (sACE2) in vivo, generated by the activity of TMPRSS2 and of a disintegrin and metalloproteinase domain 17 enzyme (ADAM-17; also known as tumour necrosis factor-alpha converting enzyme, TACE). These enzymes cleave ACE2 at p.R697 and p.K716 [Figure 4; (119, 120)]. Increased sACE2 activity has been proposed as a possible biomarker, predictive of poor patient outcome and heart failure following myocardial infarction or heart transplant (121). Whilst it has not yet been demonstrated that sACE2 is able to convert vasoconstrictor angiotensins to their vasodilator counterparts in vivo, assays using fluorogenic substrates show that sACE2 can hydrolyse angiotensin-I and angiotensin-II analogues (122). In addition, the initial characterisation of ACE2 that utilised a truncated, soluble version of the protein, indicated the production of angiotensin-(1–9) and angiotensin-(1–7), by mass spectrometry (123).

Figure 4. ACE2 protein schematic. An annotated schematic of the primary structure of ACE2. Arg273 is essential for substrate binding, by facilitating the formation of a salt-bridge. His345 and His505 assist with stabilising the substrates transition state during catalysis (118). The cleavage sites for the in vivo production of sACE2 are Arg697 and Lys716.
It has been suggested that cleavage of membrane-bound ACE2 to generate sACE2 may serve as a feedback mechanism in the RAAS. Evidence for this was presented in a mouse study, where continuous subcutaneous angiotensin-II release, via osmotic minipump, decreased myocardial membrane-bound ACE2 protein levels and increased the levels of sACE2 activity in plasma (122). This finding was reinforced in vitro in the human hepatocellular carcinoma cell line (HuH-7), with angiotensin-II exposure resulting in increased levels of ACE2 activity and sACE2 in supernatant (122).
The soluble portion of ACE2 comprises nearly the entirety of the protein's extracellular domain and could therefore plausibly act as a ligand for the SARS-CoV-2 S proteins. However, the conformation of the truncated protein has not been confirmed. While no evidence has been presented for how frequently an interaction between virions and endogenous sACE2 happens in COVID-19 patients, a case study showed that intravenous infusion of human recombinant soluble ACE2 (hrsACE2) could successfully treat severe COVID-19 (124); the putative mechanism behind this treatment may involve preventing SARS-CoV-2 from entering ACE2-expressing cells for replication and thereby-reducing angiotensin-II levels in the circulation. Moreover, the use of sACE2 as “bait” to sequester SARS-CoV-2 to potentially slow/halt the spread of infection in patients with viraemia is currently being investigated (122, 125).
SARS-CoV-2 initially infects cells of the upper airways. During the initial incubation phase, SARS-CoV-2 replicates in infected cells without detectably triggering the innate immune response, leading to initially high viral loads (126). During the robust phase of SARS-CoV-2 cellular infection, or uptake of virions by antigen-presenting cells (APC), toll-like receptors (TLR), including TLR3, TLR7, and TLR8 (127) are stimulated, producing a rapid innate immune response (128–130). This includes initiation of a signalling cascade that promotes the production of innate interferons (IFN-α, IFN-β, IFN-λ) via interferon regulatory factors (such as IRF1 and IRF7) (131) and pro-inflammatory mediators via NF-κB (132). Stimulator of interferon genes (STING) also performs a key role in the innate immune system's defence against viral infection [reviewed by (133, 134)]. The signalling of STING via NF-κB and IRF3 was initially shown to be stimulated by the presence of non-CpG intracellular DNA but there is evidence for its activity in response to viral RNA as well [reviewed by (135)]. Excessive STING activity induces pyroptosis in monocytes and macrophages, as well as elevated tissue factor (CD142) levels (136), both of which occur in severe COVID-19 patients (73, 137, 138). The overexpression of angiotensin-II activates the STING pathway in murine myocardial cells (139), which may indicate a route by which STING is overstimulated in COVID-19 patients with cardiovascular disease and T2DM. These rapid responses stimulate different effects, depending on the cell type in which they occur [reviewed in (140)].
Cross-reactive T-cell responses directed against either the SARS-CoV-2 spike or membrane proteins were present in healthy individuals who donated blood before the pandemic; in ~50% of CD8+ T-cells and ~30% of CD4+ T-cells (88, 141, 142). The majority of individuals who have had confirmed SARS-CoV-2 infection had a robust T-cell (88, 141, 142) and B-cell response, with detection of IgA, IgM, and IgG antibodies against the virus, which lasted at least 6 weeks (143–145). However, a long-lasting B-cell response was less common in those with mild COVID-19 (146). The adaptive immune response is initiated by intermediatory cells activated by innate effectors, such as dendritic cells that display antigen to CD8+, CD4+ helper T-cells (proinflammatory Th1 and Th17 cells and suppressive Th2 cells) and CD4+ Treg. B-cell responses can be either Th cell dependent or independent. Cytokines secreted during T-cell dependent activation encourage B-cell proliferation and isotype switching to maintain germinal centre size and longevity. Notable differences in these responses have been observed in patients with mild compared to severe COVID-19 [review by (147)].
A range of immune markers are markedly dysregulated in patients with severe complications from COVID-19 compared to those with mild/moderate infection, which provides clues as to the reason some people have a severe response to SARS-CoV-2 infection (Table 2) (10, 42, 126, 148–155).
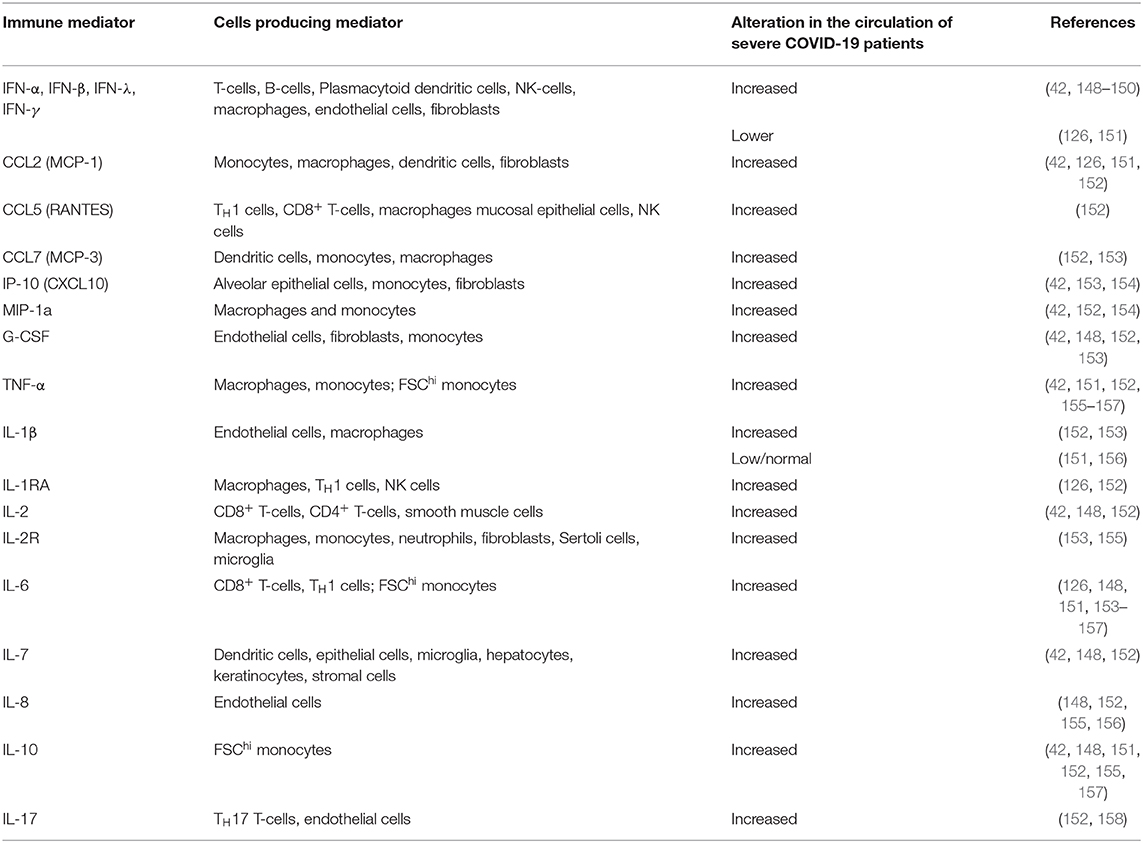
Table 2. Immune mediators dysregulated in severe COVID-19.
In patients with severe COVID-19, there is robust production of pro-inflammatory cytokines and chemokines, with a limited production of IFN-α, IFN-β, and IFN-λ (Table 2), suggesting an effective activation of NF-κB but not of IRF7 (126), proposed to result in an imbalance between the pro-inflammatory vs. pro-repair functions of airway macrophages (150). One of the features of severe COVID-19 that has been extensively described is a “cytokine storm,” which can lead to high risk of vascular hyperpermeability, multiorgan failure, and death [reviewed in (159–161)]. IL-6 is a key biomarker for COVID-19-related cytokine storms (162) and has shown an inverse correlation with impaired immunity (159, 163); T-cell numbers were negatively correlated to the concentrations of serum IL-6, IL-10, and TNF-α (164). Severe COVID-19 is consequently associated with impairment of T-cells, manifesting as lymphopenia and functional exhaustion of CD4+ (Th and Treg) and CD8+ T-cells; it is likely that the preceding innate immune dysregulation influences these observations (155, 163–165).
The B-cell response is also likely dysregulated, although reports are inconsistent, confounded by methodological issues surrounding the detection of antigen-specific immunoglobulins (166, 167) and whether systemic (circulatory) or local (particularly mucosal) responses were assessed (168). For example, enhanced secretory mucosal IgA responses, detected in the circulation in severe COVID-19, were hypothesised to confer damaging effects via induction of inflammatory cytokines (169–171); although IgA levels in saliva from COVID-19 patients showed only a moderate correlation with COVID-19 severity (172) and other studies found no difference with disease severity (155). Circulating IgG levels were higher in patients with severe COVID-19, which has been hypothesised to promote macrophage hyper-inflammatory responses (173); although the effect of viral load on the secretion of antibodies has not been ruled out.
There are chronic, persistent immune dysregulation features present in T1DM and T2DM, which are associated with disease-related pathology. In both T1DM and T2DM, there are notable changes in cytokine expression and phagocytic activity, such as suppressed chemokine responses, higher levels of pro-inflammatory mediators and lower rates of phagocytosis by neutrophils and macrophages necessary for uptake of infectious antigen (174–179).
The β-islet destruction that is a feature of T1DM is a consequence of the action of CD4+ and CD8+ T-cells with specificity for islet autoantigens (180), and local macrophages secreting pro-inflammatory cytokines (181); autoreactive antibodies are a secondary consequence to this. Individuals with T1DM do not have differences in the number of Treg cells compared to healthy individuals (180), but do have defects in Treg activation, which persist throughout their lifetime (182). Furthermore, the function of existing Treg is dysregulated, whereby their suppressive capabilities are reduced, leading to a sustained increase in pro-inflammatory cytokines (180).
In T2DM, the major immune feature is an imbalance in the Th17 and Treg cells, which reflects a loss in T-cell homeostasis and is a major contributor to inflammation and tissue specific immunity (183). Th17 cells have unique glycolysis and lipogenesis metabolic profiles that drives differentiation and cytokine production (184). The significant alterations in lipolysis and lipogenesis in patients with T2DM impacts Th17 function and suggests that despite the normalisation of blood glucose levels in T2DM, this might not be sufficient to reverse obesity-associated T-cell inflammation (185).
The association of diabetes with severe symptoms in COVID-19 is linked to glucose control, with little apparent dissociation of the effect between patients with T1DM and T2DM (65). Hyperglycaemia impairs host defences, including granulocyte and macrophage function and lymphopaenia, as further discussed below. People with diabetes are at increased risk of a wide range of infections, with death from infections substantially higher than in matched controls, particularly amongst patients with T1DM [OR 7.72; (186)]. HbA1C is a marker of glycaemic control that is closely associated with risk of infection, hospitalisation, and mortality (187). While these data provide interesting insights in terms of susceptibility to infection, severity, and mortality, they provide little information regarding the mechanism underpinning the association. An interesting additional observation, however, is that fasting blood glucose (i.e., acute, transient measure of glucose regulation) at hospital admission has been found to be an independent predictor of COVID-19 mortality in patients without pre-existing diabetes (188), which lends weight to the hypothesis that blood glucose itself is a key mediator of severe symptoms in COVID-19.
Perhaps the most controversial area in the COVID-19 story to date is the potential role of vasoconstrictor/pro-inflammatory angiotensin-II balance with vasodilator/anti-inflammatory angiotensin-(1–7) and -(1–9) in the pathophysiology of the disease and the development of severe and complex symptoms (Figure 3). Early reports during the pandemic suggested that angiotensin-II was found to be increased (3-fold) in patients with COVID-19 (189), although this finding has yet to be replicated (190). The initial advice around ACE inhibitors was mixed (191, 192) and in the face of this controversy, the early advice was not to prescribe ACE inhibitors or AT1R antagonists, but equally not to withdraw them from patients prescribed these drugs for pre-existing conditions (193–195). The latest study relating to ACE inhibitors suggest some benefit in terms of COVID-19 disease as a whole, if not reduced risk of ICU care (196) without increased risk of death (197, 198); however, there was also a complex interaction effect with ethnicity in that benefits were less apparent or absent in Black African groups in particular.
The primary reason for the conflicting suggestions and outcomes is the complex counter-regulatory mechanism that applies in this system. While loss of ACE2 to SARS-CoV-2 seems inevitable, expression of this enzyme is often found to be substantially up-regulated on account of a counter-regulatory mechanism mediated by ACE/angiotensin-II (199). Simultaneously, there appears to be down-regulation of ACE (200). In addition, an in silico study using worldwide databases suggested that there was a significant reduction in ACE2 expression in patients with diabetes (201). The ultimate impact of SARS-CoV-2 on RAAS mediators is likely to fluctuate with time and to be highly dependent on viral load. Because of the differential importance of ACE2 in metabolism of the various mediators, the response profiles will be distinct. Figure 5 provides a hypothetical response profile for some for the key mediators impacted by SARS-CoV-2, illustrating the importance of the time-course of the various responses on the pathophysiological outcome at any given time. In the event that a course of events similar to that illustrated is experienced, it is clear that both the amplitude of the impact (dictated by viral load) and the time-course and effectiveness of the various counter-regulatory events will have a substantial bearing on the nature and extent of the symptoms experienced. Superimposition of pre-existing ACE inhibition or AT1R antagonism on proceedings adds another layer of complexity, perhaps explaining the variety of outcomes and opinions relating to this particular topic. What is clear is that “one size fits all” does not apply in this regard.
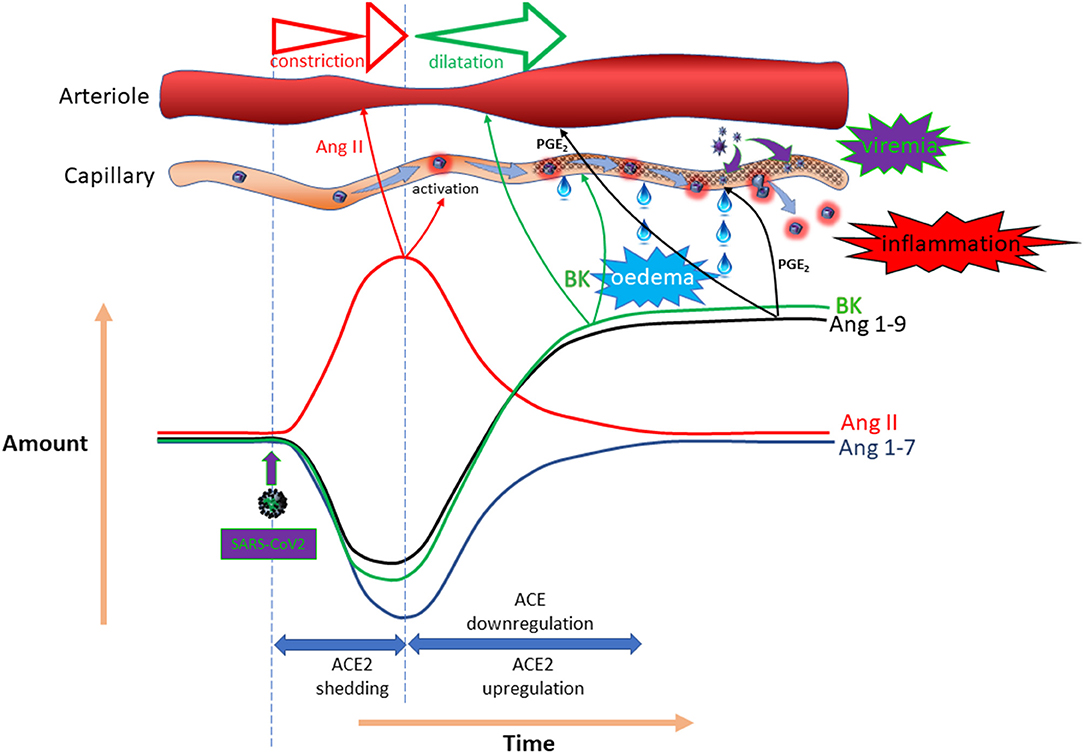
Figure 5. Potential temporal impact of COVID-19 on the RAAS. Hypothetical impact of SARS-CoV-2 on local expression of ACE/ACE-2 enzymes and the possible outcome in terms of angiotensin-II, angiotensin 1–7, angiotensin 1–9 and bradykinin (BK) concentrations, as well as the vascular impact.
There is consensus that dysregulation of the RAAS is a feature of diabetes and represents a valid target for drug intervention, particularly with respect to renal complications associated with the disease [e.g., nephropathy; reviewed here: (202)]. The precise nature of the dysregulation, however, is much less clear, with claims and counter-claims as to the relative expression of ACE and ACE2 in diabetes; certainly evidence from animal models appears to point to down-regulation of ACE2 in the kidney (203). Perhaps the most relevant evidence, however, is derived from a genome wide association study (GWAS) on markers associated with increased pulmonary ACE2 expression (204). This study notably identified association with polymorphisms previously linked to T2DM; although the association did not withstand correction for multiple testing (at a false discovery rate of <0.05), so should be viewed with caution. The association was weaker for T1DM (204). Taking all these data together, it is conceivable that ACE2 expression is differentially affected in different tissues within an individual with diabetes and that the pattern of effect on ACE2 varies from person to person. This concept is supported by a study from an animal model in which the expression and activity of ACE and ACE2 were measured in various tissues from a non-obese, diabetic mouse model; the pattern of expression of ACE and ACE2 varied dramatically across serum and various tissues, including lung, where ACE2 expression was increased, but to a lesser extent than ACE, resulting in an overall decrease in ACE2:ACE in this tissue. The reverse was true in serum (205). This variance is critical in understanding the impact of COVID-19 at tissue/organ level with respect to susceptibility to COVID-19: higher ACE2 expression in the lungs of patients with diabetes might correspond to increased susceptibility to binding and subsequent replication of SARS-CoV-2, which in turn could drive a further up-regulation of ACE2, resulting in bradykinin and angiotensin 1–9-mediated pulmonary oedema [hypothesised here: (206)]. Decreased renal ACE2 in the same individual might, however, offer some protection from viral infection associated with viraemia. Figure 6 is a hypothetical illustration of the complex interactions that might accrue in diabetes on account of modulation of ACE/ACE2 expression by COVID-19 and diabetes.
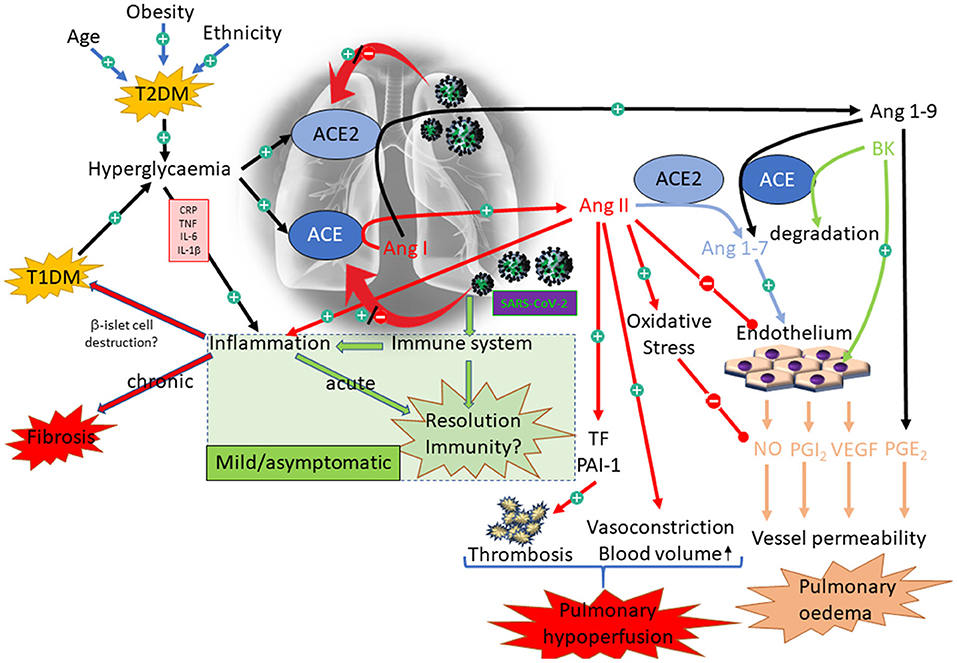
Figure 6. Interaction between diabetes and COVID-19 with respect to the RAAS. Illustration of the complex interactions associated with diabetes and COVID-19. The green box illustrates the appropriate response to SARS-CoV-2, resulting in only mild symptoms. The alternative pathways illustrate some of the dysregulation that might be primed by diabetes, resulting in acute (pulmonary hypoperfusion and oedema) and chronic (fibrosis) outcomes.
While the RAAS has been the focus of much attention in COVID-19 on account of the key role of the ACE2 receptor, there are a number of other critical elements associated with viral infection and host response that may be influenced by, but are not entirely dependent upon, changes in RAAS activity. Each of these elements are also affected by both types of diabetes, with the potential to increase susceptibility to severe symptoms in COVID-19.
As previously described, T1DM and T2DM are associated with distinct, chronic immune profiles. Pre-pandemic, it was already established that patients with diabetes had a greater risk of lower respiratory tract infections (207) and more severe outcomes to respiratory infections [e.g., MERS (208), influenza (209), and bacterial (207)]. There are notable confounders to the poor outcomes to respiratory viruses (including COVID-19) associated with diabetes, particularly increased age and male sex, which are also factors previously linked to poor immune response to other respiratory viruses (210–213), risk of T2DM (214) and poorer outcomes to COVID-19 (215). Lower respiratory tract infections occur when the immune response in the upper airways fails to contain the viral spread, which is a key feature of severe COVID-19. Therefore, it is plausible the immune dysregulation in T1DM and T2DM is a contributor to the less favourable outcomes with SARS-CoV-2 infection. Of course, it is an over-simplification to limit diabetes to T1DM and T2DM: there are significant patient subsets within these populations, at least some of which display significant differences in B-lymphocyte populations (216). It is not yet known whether susceptibility to severe COVID-19 symptoms align with certain diabetes sub-populations on account of their B-cell function or whether these subtleties are overwhelmed by other contributory factors like age, glycaemic control, ethnic origin or BMI.
Patients with T1DM have a dysregulated Treg response and patients with T2DM have an aberrantly active Th17 response, both of which lead to a sustained increase in pro-inflammatory cytokines (180, 182, 183, 185). The increased pro-inflammatory cytokine profile already present in T1DM and T2DM could therefore skew the immune response to SARS-CoV-2 infection toward an inflammatory response, increasing the likelihood of severe COVID-19, with associated cytokine storm, tissue damage, and respiratory failure. Given the propensity for SARS-CoV-2 to require a higher viral load to trigger detection by the immune system even in healthy individuals, the capacity to raise an acute immune response might be further compromised in patients with diabetes, resulting in a viral load that rapidly overwhelms the immune response capacity. Patients hospitalised with diabetes and COVID-19 who had a more controlled blood glucose concentration had lower incidences of lymphopaenia (30.5 vs. 49.6%) and neutrophilia (10.7 vs. 19.4%) than patients with a high (>7.5 mmol/L) blood glucose concentration, indicating that good glycaemic control is also important in maintaining a balanced immune system (11, 215).
Several reports demonstrate that persistent inflammation is associated with a compensatory anti-inflammatory response that could prevent excessive tissue damage by increasing immunosuppressive activity (217–219). The anti-inflammatory responses include the induction of myeloid-biassed haematopoietic stem cell differentiation, increased expansion of Treg cells and increased expression of anti-inflammatory cytokines such as IL-10 and TGF-β (219). The activation of immunosuppressive cells suppresses the functions of both innate and adaptive immunities. Accordingly, T2DM-related immunosuppression may facilitate SARS-CoV-2 replication in ACE2-expressing cells and the development of damage-associated molecular patterns (DAMP)-promoted cytokine storms in these patients.
In summary, from the gathered evidence of altered immune mediators in patients with T1DM and T2DM without SARS-CoV-2 infection (174–180, 182, 183, 185) and more severe outcomes during respiratory infections, including SARS-CoV-2, (6, 10, 41–43, 72, 73, 155, 186, 187, 207–213), it is highly likely that at least part of the poor outcome in patients with diabetes to COVID-19 is due to dysregulated, abnormal immune responses.
People over 60 years of age have a weaker immune response to respiratory infection compared to younger individuals (210–213) and coordinated immune responses specifically to SARS-CoV-2 are disrupted in individuals aged over 65 years old (220), indicating a reason why elderly people have high risk of developing severe COVID-19 after infection with SARS-CoV-2 virus (221). Individuals with impaired immune function cannot effectively clear SARS-CoV-2 from the upper respiratory tract and as a result, the virus can quickly disseminate to the lung with rapid replication in ACE2-expressing cells. Older patients with COVID-19 can reduce their viral titres, only to rapidly descend into a state of shock due to hyperactivation of the immune system, known as a cytokine storm. One mechanism by which this might occur is via virus-injured cells releasing the DAMPs that can promote hyperinflammation (222, 223).
Another key aspect associated with age that might influence severity of COVID-19 symptoms is ACE2 receptor expression. It is known that ACE2 receptor expression in epithelial and endothelial cells in rat lungs fall substantially in older individuals and is lower overall in male compared to female animals. This concept is interrogated in a recent viewpoint article, which postulates that a reduced ACE2 expression profile at onset of COVID-19 results in an exaggerated inflammatory response on account of the acute additional loss of ACE2 receptors in the face of the virus (224).
While the mechanism behind severe COVID-19 is unknown, individuals with conditions associated with metabolic disorders such as obesity, diabetes and cardiovascular disease have a higher risk of developing the cytokine storm and coagulopathy that play crucial roles in progression to a critical clinical condition (225, 226). The risk of in-hospital deaths with COVID-19 was significantly higher in patients with diabetes than those without diabetes, with hazard ratio of 2.36 (227). Because metabolic disorders are associated with NET dysfunction (222, 228, 229), an increase in activated neutrophils and NET formation may contribute to severe COVID-19 in patients with T2DM and obesity. Increased neutrophil-to-lymphocyte ratios have been identified as an early indicator of SARS-CoV-2 infection, predicting severe COVID-19, cytokine storm and poor clinical outcomes (41, 230). A recent study revealed that NETs triggered by SARS-CoV-2 were likely to depend on ACE2, virus replication, serine protease, and protein arginine deiminase 4 (231). Furthermore, it has been reported that patients with T2DM had an elevated NET release after infection, not only with SARS-CoV-2 (232), but also with other invading microbes (233). This suggests that hyperglycaemia may be a key contributor to the formation of NETs in COVID-19 patients with diabetes, since high glucose levels would increase microvascular permeability and oedema, and facilitate the transendothelial migration of capillary neutrophils to alveolar air spaces (225) (Figure 7). It is not yet unclear, however, whether it is the hyperglycaemia per se, or other features associated with T2DM, that induces this response. Pulmonary NETs may also promote the oxidation of SARS-CoV-2 due to the generation of reactive oxygen species by NAD(P)H oxidases during NET formation (228). Oxidised viruses could activate TLR4 that is highly expressed in inflammatory immune cells, and subsequently enhance the TLR4-mediated inflammatory response and cytokine storm (234).
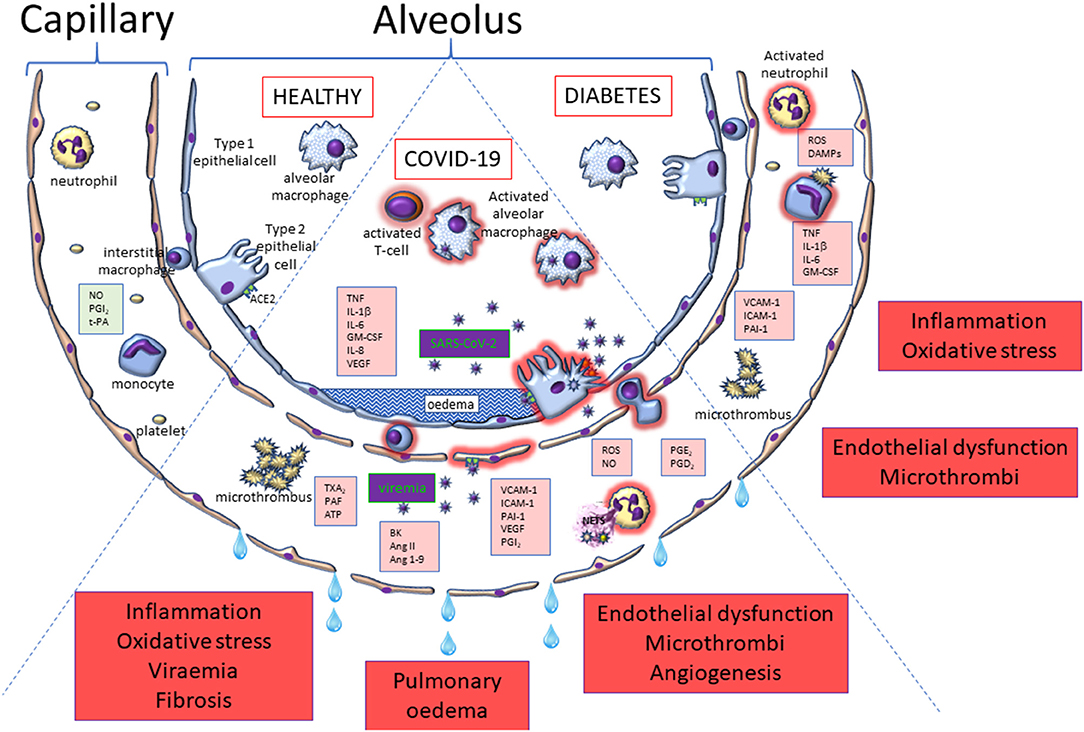
Figure 7. Alveolar and capillary pathophysiology relevant to COVID-19 and diabetes compared to the healthy scenario. This figure highlights some of the key cells that become activated in COVID-19 (middle segment) and diabetes (right segment) that have the potential to drive dysfunction in endothelial (diabetes and COVID-19) and alveolar epithelial (COVID-19 only) cells. The pathophysiological consequences are highlighted in the red boxes.
Given the route of entry of the SARS-CoV-2 virus into the body via the lungs, together with the primary symptoms that can accrue in terms of pulmonary oedema, inflammation and pneumonia, it is clear that the first-line cellular contact for the virus constitutes alveolar type 1 and type 2 epithelial cells that line the respiratory tract from nose to alveoli, together with the resident macrophages in the lungs, which help to raise the alarm for other inflammatory and immune cells to drive a defensive reaction (Figure 7). However, early in the pandemic, it was noted that cardiovascular complications were also widely experienced in severe cases of COVID-19, with symptoms associated with thrombosis and coagulation most prominent, increasing the risk of deep vein thrombosis, heart attack, stroke and peripheral ischaemia, often presenting in the form of “COVID toe” (235–237).
The endothelium was quickly identified as central to the vascular effects associated with COVID-19, not only because of the intimate proximity of alveolar epithelial and endothelial cells to facilitate efficient gas exchange, but also because endothelial cells express ACE2 and are highly regulated by many of the mediators that are modulated by changes in ACE2 presence or activity. An early case study involving post-mortem tissue suggested that SARS-CoV-2 can be found in endothelial cells, not only in the lungs (238) but also in other organs, and that inflammation and occlusion of small blood vessels was also evident in these patients (239).
Endothelial dysfunction is a feature of diabetes, driven by a range of mediating processes, including oxidative stress and inflammation (Figure 7). Indeed, endothelial dysfunction is considered to be the prime cause of accelerated atherogenesis in diabetes, leading to many of the secondary conditions, including coronary artery disease, ischaemic stroke, peripheral vascular disease (contributing to neuropathy and foot ulcers) and nephropathy (240). Hyperglycaemia-induced oxidative stress is a likely mechanism underpinning much of the endothelial dysfunction associated with diabetes (241), but elevated inflammation, resulting in enhanced expression of adhesion molecules [VCAM-1, ICAM-1, P-selectin (242–245)], is also considered to be key. Given that the endothelium in patients with diabetes is already compromised via these processes, it is perhaps inevitable that the impact of further endothelial dysfunction driven by COVID-19 might be more profound in this patient group, particularly if compounded by the effects of old age. What is not yet clear is the relative importance of changes in endothelial function in the lung to drive systemic inflammation, either through ensuing viraemia, extensive inflammation, or both.
Endothelial cells are uniquely positioned at the interface between circulating blood and underlying tissues. The key role of the endothelium as the local modulator of vascular tone in response to a wide range of local physical, chemical and inflammatory mediators, necessitates a vast array of sensors and receptors which combine to generate an integrated, outgoing range of signals that in turn influence vascular smooth muscle contraction, proliferation, permeability, angiogenesis, thrombosis and inflammation (246, 247). Prime amongst these mediators is NO (248), but PGI2 is another important protective agent (249, 250), acting in opposition to the contracting factors that include angiotensin-II (251), thromboxane A2 (TXA2) (252) and endothelin-1 (ET-1) (253). These mediators have influence over other processes that could be important in either moderating or driving severe symptoms associated with COVID-19, both directly related to cardiovascular function and indirectly associated with acute and chronic respiratory symptoms. Some of the key mediators will be considered in turn, with a particular focus on possible interplay between diabetes and COVID-19.
NO is a key indicator of endothelial function and its wide-ranging actions ensure that it is critical in determining vascular health. NO is synthesised de novo on demand and its free radical characteristics ensure that its effects are confined to the locality of its generation and are short-lived [reviewed by (254)]. The level of stimulation is determined by an integrated Ca2+ signal derived from multiple inputs, including sheer stress (255), hypoxia (256), and several endothelium-dependent vasodilators [e.g., bradykinin, angiotensin-(1–7), reviewed in (257)]. The reaction with superoxide (.), is particularly important because it not only inactivates NO, but also generates a highly oxidising, cytotoxic product in the form of peroxynitrite (ONOO−) [reviewed by (258)].
A healthy endothelium promotes vasodilatation, vascular permeability and controlled angiogenesis, while at the same time inhibiting vascular smooth muscle cell proliferation, platelet adhesion and aggregation, and inflammatory cell adhesion (240). The cardiovascular impact of diabetes is at least partly due to loss of NO on account of both reduced synthesis and increased inactivation by superoxide (259, 260). The role of NO in COVID-19-induced changes in vascular function that could contribute to severe symptoms in the acute phase of COVID-19 are currently unknown. Certainly, reduced capacity for pulmonary dilatation, together with increased adhesion and infiltration of inflammatory cells and the additional risk of microthrombi precipitated by reduced NO, would all contribute to poor lung function and exacerbated inflammation. However, reduced NO production would also lead to reduced risk of vascular endothelial growth factor (VEGF)-induced hyperpermeability and angiogenesis, which is counter to the clinical findings (42, 238). Likewise, VEGF is increased in diabetes (261), although the profile of expression varies between T1DM and T2DM (262).
It is worth noting, however, that the contribution of eNOS to overall NO can be relatively small under pathological conditions, with high concentrations generated from the inducible isoform of the enzyme (iNOS) in activated macrophages and activated neutrophils (263), both of which are associated with COVID-19 (42). NO generated from inflammatory cells can act to destroy invading pathogens and indeed, iNOS-derived NO is considered critical in driving vascular collapse in animal models of sepsis, although the role in complications associated with sepsis in humans is less clear-cut (264). Until we fully understand the impact of COVID-19 on NO, it is difficult to determine whether inhibition of iNOS to reduce NO or delivery of inhaled NO (265) to alleviate symptoms driven by loss of eNOS-derived NO is advantageous. Clearly, if endothelium-associated microvascular dysfunction is critical in COVID-19, pre-existing diabetes will constitute a risk factor.
Though VEGF-A is predominantly associated with angiogenesis, it also plays a significant role in the regulation of endothelial function [reviewed in: (266)] through, for example, the regulation of vascular permeability, or facilitating diapedesis of leukocytes (267). Expression of VEGF is controlled by hypoxia-induced expression of the transcription factor, HIF-1 (268), and it is not implausible to presume that both localised and systemic hypoxia caused by COVID-19 would lead to the overexpression or dysregulation of VEGF. The consequences of this would likely be an increase in vascular permeability and leakage, leading to increased oedema, as well as increased infiltration of leukocytes into surrounding tissues, further exacerbating the severe inflammatory symptoms of the infection. Angiogenesis is another symptom that has been found in patients who have died from COVID-19 (238) that is almost certain to involve VEGF.
The dual effect of loss of vasoprotective NO and the generation of cytotoxic, oxidising ONOO− is considered to be important in the initiation and progression of atherogenesis (240, 269), not least in patients with diabetes (241). NO, together with PGI2 is not only a vital local mediator of vasodilatation, but also of inhibition of platelet aggregation. Diminution of this antithrombotic effect is exacerbated by increased oxidative stress in platelets of individuals with diabetes, which further inactivates NO in the target platelets themselves. Furthermore, evidence is accumulating to suggest that an imbalance in pro- (tissue plasminogen activator; t-PA) and anti- (plasminogen activator inhibitor-1; PAI-1) fibrinolytic factors in favour of PAI-1 in diabetes (270), could depress the fibrinolytic process that helps reverse coagulation associated with thrombus. Oxidative stress is also understood to be a driver of inflammation in endothelial cells, mediated by the expression of adhesion molecules on the surface, such as VCAM-1, ICAM-1 (240) and P-selectin [reviewed by (271–273)].
Eicosanoids are powerful lipid mediators of inflammation that are implicated in COVID-19. The role of eicosanoids in COVID-19 patients with diabetes is complex, as an imbalance in lipid mediator production is a feature of diabetes, contributing to the pathogenesis of the disease and associated complications [reviewed by (274, 275)]. Eicosanoids are lipid mediators synthesised by cyclo-oxygenase (COX), lipoxygenase (LOX) and P450 enzymes. While their impact in COVID-19 is currently poorly understood, SARS-CoV-1 can bind directly to the COX-2 promotor, increasing the expression of this inducible iso-enzyme, which is central to synthesis of prostanoids (276). In addition, the endoplasmic reticulum stress response, which can promote a cytokine storm, activates inositol-requiring enzyme 1[α] to upregulate COX-2 and prostaglandin E synthase, triggering the production of PGE2 amongst other prostaglandins (277). These findings suggest that an “eicosanoid storm” [reviewed by (278)] could contribute to the intense inflammatory response that accompanies severe COVID-19.
There have been contradictory reports regarding the use of non-steroidal anti-inflammatory drugs (NSAIDs) to treat patients with COVID-19. NSAIDs include selective and non-selective inhibitors of the COX enzymes that catalyse the first committed step in synthesis of prostanoids, which contribute to pain and fever [reviewed in: (279)]. COX-1 is constitutive and central to synthesis TxA2 (amongst other prostanoid mediators) in platelets and endothelial cells, resulting in platelet aggregation and vasoconstriction. Aspirin is a weakly COX-1 selective inhibitor which is routinely used in primary prevention of myocardial infarction, except in patients with diabetes, where there is controversy over its benefits (280–284), perhaps suggesting eicosanoid imbalance in this patient group. By contrast, inducible COX-2 primarily synthesises PGI2, especially in microvascular endothelial cells, which opposes the actions of TxA2, resulting in inhibition of platelet aggregation and vasodilation (285, 286). The balance between PGI2 and TXA2 is critical for cardiovascular homeostasis (287, 288) and upsetting the balance can be detrimental. In addition, NSAIDs have differential and multifactorial effects on growth factor–induced angiogenesis and vascular permeability (289, 290) through inhibition of prostaglandins, particularly PGE2, which regulates vascular permeability (291) via the induction of VEGF and basic fibroblast growth factor (292). The anti-inflammatory effects of NSAIDs are broad: alongside the inhibition of prostaglandin synthesis, NSAIDs inhibit cytokine production, including IL-12 (293), IFN-γ (294), and IL-4 (295), thereby preventing Th1 and Th2-mediated responses. The use of NSAID may oppose the anti-inflammatory and pro-resolving roles of COX and ultimately lead to counter-intuitive prolongation of inflammation.
Advice early in the pandemic was to favour paracetamol over ibuprofen for COVID-19-related pain relief (296), due in part, to the observation that ibuprofen upregulates ACE2 in diabetic rats (297). However, this finding was not replicated in human clinical studies (298, 299) and this advice has since been withdrawn (300).
The corticosteroid, dexamethasone, promotes the production of pro-resolving mediators (301) and is now recommended for the treatment of the most severe symptoms in COVID-19 on account of reduced 28-day mortality amongst the treatment group of patients receiving invasive mechanical intervention or oxygen (302). However, the same trial found that there was a modest detrimental effect in patients not receiving respiratory support, again highlighting the importance of appropriate balance in the inflammatory response to COVID-19. In patients with diabetes, corticosteroids can have an impact on glycaemic control and other metabolic parameters and their use should be under careful clinical review (303).
An early clinical symptom identified in severe cases of COVID-19 was coagulopathy, with anywhere between 16–49% of patients admitted to ICU suffering thrombotic complications (304). The root cause(s) of thrombotic complications are not yet fully understood, but inflammatory cytokines have been implicated, along with endothelial dysfunction and stasis on account of immobility; this is not unique amongst severe infections. Post-mortem data from early casualties of COVID-19 indicated diffuse thrombosis throughout the lung microvasculature (305). Interestingly, while D-dimer levels are frequently raised in COVID-19 related coagulopathy, other coagulation cascade markers are not apparently consumed, as is seen with disseminated intravascular coagulation, perhaps pointing away from coagulation as the root cause and implicating platelet activation or dysfunctional fibrinolysis instead (306). Both of these possible causes would lead back to endothelial dysfunction as a crucial player in determining thrombotic potential, both through loss of anti-thrombotic NO and PGI2, and through a potential imbalance between endothelium-derived, fibrinolytic t-PA, and its countermeasure, PAI-1, in favour of the latter. With diabetes in mind as a potential primer for thrombosis in COVID-19, not only is diabetes associated with increased circulating inflammatory cytokines, but also with endothelial dysfunction, platelets in a hyperactive state, and an imbalance of t-PA and PAI-1 – all exacerbated by oxidative stress (307). The drive toward coagulopathy in COVID-19 is also likely to be boosted through enhanced generation of NETS, a feature that is common to diabetes (308) and infection (309).
Prophylactic and therapeutic uses of anticoagulant treatments are being investigated for COVID-19 patients (310). A retrospective study of 449 COVID-19 patients in China found no significant effect of heparin on 28-day mortality. However, there was a significant effect when only patients with a sepsis-induced coagulopathy score of >4 were analysed (311). Direct oral anticoagulants (DOACs) are also being investigated as potential COVID-19 treatments. There is evidence, from a study in ApoE−/− mice, that DOACs may help to alleviate the endothelial dysfunction associated with diabetes (312), a potential confounding factor of COVID-19 coagulopathy in patients with diabetes. However, combining DOAC and antiviral treatments can lead to a sharp increase in plasma DOAC concentrations, which may increase the risk of haemorrhage in patients (313).
In children, the manifestation of β-islet autoimmunity is known to correlate with recent respiratory illness (314), including SARS-CoV-1 infection (315). In the case of SARS-CoV-2, there have been reports of COVID-19 patients exhibiting T1DM-like symptoms, despite no history of diabetes prior to hospitalisation. The first case study described a patient who presented with diabetes-like symptoms 1 month after they had been diagnosed with COVID-19, despite being normoglycaemic during their initial hospitalisation (316). Autoantibody production against β-islets was assessed, with the patient testing positive for glutamic acid decarboxylase-65 and negative for tyrosine phosphatase IA2 antibodies and zinc transporter 8 antibodies. As there was evidence of autoimmunity driving the development of diabetes, the patient was classified as having T1DM (316). Following this initial report, an increase in new-onset T1DM cases amongst children admitted to hospital with COVID-19 has been reported (68).
The induction of diabetes-like symptoms by SARS-CoV-1 infection was found in 39/520 SARS patients, who had no history of diabetes; only 2 patients were classified as having diabetes at 3 years post-hospitalisation (315). Diabetes was only assessed by glucose tolerance tests, with no measurement of autoantibodies; these type(s) of diabetes therefore cannot be classified. This study concluded that SARS-CoV-1 entered β-islets via ACE2 and the resultant damage caused an acute diabetes-like syndrome (315). Indeed, ACE2 is expressed throughout the pancreas, including in endocrine tissues and SARS-CoV-2 can infect both adult human pancreatic β cells and human pluripotent stem cell-derived pancreatic β cells (317).
Furthermore, a study in rats showed that an increase in angiotensin-II levels can result in an acute reduction in β-islet blood flow (318). This leads to the hypothesis that diabetes-like symptoms are caused by SARS-CoV-2 infection drive decreased ACE2 activity, which increases angiotensin-II and decreases β-islet function.
Therefore, the presence of diabetes-like symptoms in some patients after COVID-19 could be due to (i) induction of autoantibodies; (ii) direct infection of β-islets by SARS-CoV-2; or (iii) temporary β-islet function loss due to increased angiotensin-II. This is an evolving area of research and larger studies of “long-COVID,” the long-term effects of COVID-19, are now underway. We hypothesise a potential underlying mechanism of some of the long-COVID symptoms, particularly those of fatigue and general malaise, might be underlined by the dysregulation of metabolism and hyperglycaemia associated with a SARS-CoV-2 induced diabetes-like syndrome. There is some early evidence emerging to suggest that glucose control suffers perpetuated dysregulation at least in the acute phase after hospitalisation in hyperglycaemic patients with severe COVID-19 symptoms (319), although whether this is cause or effect is still to be deduced.
COVID-19 is a disease with a wide array of possible outcomes, ranging from the benign right through to death. It soon became clear that age and pre-existing disease were two major risk factors for severe symptoms, and that diabetes was one of several disease profiles that was implicated as a risk factor. On the face of it, the potential links between diabetes and severe COVID-19 symptoms seem obscure, given that COVID-19 drives respiratory collapse, whereas diabetes is related to outcomes that are mediated by cardiovascular dysfunction, leading to retinopathy, neuropathy, peripheral vascular disease, stroke and myocardial infarction. Dig deeper, however, and parallels appear, not least with respect to dysfunction in various interconnecting systems and processes that could come together to drive a more severe package of symptoms in response to SARS-CoV-2 infection. Without doubt, subtle changes in the immune system and RAAS, together with inflammation, oxidative stress and endothelial dysfunction in diabetes have the potential to exaggerate the response triggered by SARS-CoV-2, ultimately driving one or more of the cellular processes that result in pulmonary thrombosis, increased vascular permeability and/or cytokine storm, resulting in respiratory failure.
Clearly, the story is a highly complex one: it must be remembered that the diabetes population is highly heterogeneous, with extremely diverse disease aetiology and severity, just as COVID-19 shows extreme diversity. Nevertheless, it is apparent that at least a sub-population of those with diabetes are less equipped to affect a quiet and efficient resolution of COVID-19, resulting instead in a monumental counter-regulatory failure with potentially fatal consequences. Additionally, evidence indicates that some individuals with COVID-19 develop a diabetes-like syndrome and it will be important to understand the mechanism by which this may occur. Current plausible explanations range through RAAS dysregulation, temporary β-islet destruction and induction of β-islet autoantibodies. More data on testing for β-islet autoantibodies and tracking the long-COVID symptoms are needed to begin to identify the underlying mechanisms behind these observations.
There is still a great deal to learn about COVID-19 before firm conclusions can be drawn as to the importance of the various potential players in determining the severity of the disease. The promise shown by the powerful anti-inflammatory agent, dexamethasone, which inhibits a multitude of pro-inflammatory pathways as a prophylactic agent or treatment for such symptoms, presents strong evidence for a central role for inflammation in symptom severity (302), but offers little insight to help predict what specific elements of the highly complex inflammatory system predispose to a poor outcome.
JR, AP, AT, AR, NB, PC, SM, JW, and IM were all involved in writing and editing the manuscript through several iterations. All authors contributed to the article and approved the submitted version.
JR was supported by the European Union's INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB; IVA5034). NB is funded through the CoS: IGN Programme at the University of the Highlands and Islands. AP was funded by a grant from Highlands & Islands Enterprise (HMS 9353763).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Yelin D, Wirtheim E, Vetter P, Kalil AC, Bruchfeld J, Runold M, et al. Long-term consequences of COVID-19: research needs. Lancet Infect Dis. (2020) 20:1115–7. doi: 10.1016/S1473-3099(20)30701-5
2. Zheng Z, Peng F, Xu B, Zhao J, Liu H, Peng J, et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J Infect. (2020) 81:e16–25. doi: 10.1016/j.jinf.2020.04.021
3. Gold MS, Sehayek D, Gabrielli S, Zhang X, McCusker C, Ben-Shoshan M. COVID-19 and comorbidities: a systematic review and meta-analysis. Postgrad Med. (2020) 132:1–7. doi: 10.1080/00325481.2020.1786964
4. Mantovani A, Byrne CD, Zheng MH, Targher G. Diabetes as a risk factor for greater COVID-19 severity and in-hospital death: a meta-analysis of observational studies. Nutr Metab Cardiovasc Dis. (2020) 30:1236–48. doi: 10.1016/j.numecd.2020.05.014
5. Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. (2020) 94:91–5. doi: 10.1016/j.ijid.2020.03.017
6. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
7. Zuin M, Rigatelli G, Zuliani G, Rigatelli A, Mazza A, Roncon L. Arterial hypertension and risk of death in patients with COVID-19 infection: systematic review and meta-analysis. J Infect. (2020) 81:e84–6. doi: 10.1016/j.jinf.2020.03.059
8. Lee SW, Yang JM, Moon SY, Yoo IK, Ha EK, Kim SY, et al. Association between mental illness and COVID-19 susceptibility and clinical outcomes in South Korea: a nationwide cohort study. Lancet Psychiatry. (2020) 7:P1025–31. doi: 10.1016/S2215-0366(20)30421-1
9. Dyett J. Possible link between obesity and severe COVID-19. Med J Aust. (2020). 213:380–380.e1 doi: 10.5694/mja2.50793
10. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
11. Zhu L, She ZG, Cheng X, Qin JJ, Zhang XJ, Cai J, et al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing Type 2 diabetes. Cell Metab. (2020) 31:1068–77 e3. doi: 10.1016/j.cmet.2020.04.021
12. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
13. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. (2020) 368:m1091. doi: 10.1136/bmj.m1091
14. Tu WJ, Cao J, Yu L, Hu X, Liu Q. Clinicolaboratory study of 25 fatal cases of COVID-19 in Wuhan. Intensive Care Med. (2020) 46:1117–20. doi: 10.1007/s00134-020-06023-4
15. Chen T, Dai Z, Mo P, Li X, Ma Z, Song S, et al. Clinical characteristics and outcomes of older patients with coronavirus disease 2019 (COVID-19) in Wuhan, China: a single-centered, retrospective study. J Gerontol A Biol Sci Med Sci. (2020) 75:1788–95. doi: 10.1093/gerona/glaa089
16. Yao Q, Wang P, Wang X, Qie G, Meng M, Tong X, et al. A retrospective study of risk factors for severe acute respiratory syndrome coronavirus 2 infections in hospitalized adult patients. Pol Arch Intern Med. (2020) 130:390–9. doi: 10.20452/pamw.15312
17. Zhang J, Liu P, Wang M, Wang J, Chen J, Yuan W, et al. The clinical data from 19 critically ill patients with coronavirus disease 2019: a single-centered, retrospective, observational study. Z Gesundh Wiss. (2020). doi: 10.21203/rs.3.rs-20800/v1. [Epub ahead of print].
18. Wang K, Zhang Z, Yu M, Tao Y, Xie M. 15-day mortality and associated risk factors for hospitalized patients with COVID-19 in Wuhan, China: an ambispective observational cohort study. Intensive Care Med. (2020) 46:1472–4. doi: 10.1007/s00134-020-06047-w
19. Li J, Wang X, Chen J, Zhang H, Deng A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) Infection in Wuhan, China. JAMA Cardiol. (2020) 5:825–30. doi: 10.1001/jamacardio.2020.1624
20. Inciardi RM, Adamo M, Lupi L, Cani DS, Di Pasquale M, Tomasoni D, et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur Heart J. (2020) 41:1821–9. doi: 10.1093/eurheartj/ehaa388
21. Du RH, Liang LR, Yang CQ, Wang W, Cao TZ, Li M, et al. Predictors of mortality for patients with COVID-19 pneumonia caused by SARS-CoV-2: a prospective cohort study. Eur Respir J. (2020) 55:2000524. doi: 10.1183/13993003.00524-2020
22. Guan WJ, Liang WH, Zhao Y, Liang HR, Chen ZS, Li YM, et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur Respir J. (2020) 55:2001227. doi: 10.1183/13993003.01227-2020
23. Epidemiology Working Group for Ncip Epidemic Response CCfDC, Prevention. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Zhonghua Liu Xing Bing Xue Za Zhi. (2020) 41:145–51. doi: 10.3760/cma.j.issn.0254-6450.2020.02.003
24. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of Coronavirus Disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
25. Deng Y, Liu W, Liu K, Fang YY, Shang J, Zhou L, et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 in Wuhan, China: a retrospective study. Chin Med J. (2020) 133:1261–7. doi: 10.1097/CM9.0000000000000824
26. Wang L, He W, Yu X, Hu D, Bao M, Liu H, et al. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J Infect. (2020) 80:639–45. doi: 10.1016/j.jinf.2020.03.019
27. de Almeida-Pititto B, Dualib PM, Zajdenverg L, Dantas JR, de Souza FD, Rodacki M, et al. Severity and mortality of COVID 19 in patients with diabetes, hypertension and cardiovascular disease: a meta-analysis. Diabetol Metab Syndr. (2020) 12:75. doi: 10.1186/s13098-020-00586-4
28. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
29. Wang Y, Lu X, Li Y, Chen H, Chen T, Su N, et al. Clinical Course and Outcomes of 344 Intensive Care Patients with COVID-19. Am J Respir Crit Care Med. (2020) 201:1430–4. doi: 10.1164/rccm.202003-0736LE
30. Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HHX, et al. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J Infect. (2020) 81:e6–12. doi: 10.1016/j.jinf.2020.04.002
31. Cao J, Tu WJ, Cheng W, Yu L, Liu YK, Hu X, et al. Clinical features and short-term outcomes of 102 patients with Coronavirus Disease 2019 in Wuhan, China. Clin Infect Dis. (2020) 71:748–55. doi: 10.1093/cid/ciaa243
32. Kumar A, Arora A, Sharma P, Anikhindi SA, Bansal N, Singla V, et al. Is diabetes mellitus associated with mortality and severity of COVID-19? A meta-analysis. Diabetes Metab Syndr. (2020) 14:535–45. doi: 10.1016/j.dsx.2020.04.044
33. Akbari A, Emami A, Javanmardi F, Pirbonyeh N, Fadakar N. Early epidemiological analysis of COVID-19: first report from South of Iran. Res Square. (2020). doi: 10.21203/rs.3.rs-19915/v1
34. Bai T, Tu S, Wei Y, Xiao L, Jin Y, Zhang L, et al. Clinical and laboratory factors predicting the prognosis of patients with COVID-19: an analysis of 127 patients in Wuhan, China. Lancet Respir Med. (2020). doi: 10.2139/ssrn.3546118
35. Lu Z, Chen M, Fan Y, Wu X, Zhang L, Guo T, et al. Clinical characteristics and risk factors for fatal outcome in patients with 2019-Coronavirus Infected Disease (COVID-19) in Wuhan. Lancet. (2020). doi: 10.2139/ssrn.3546069. [Epub ahead of print].
36. Fu L, Fei J, Xiang H-X, Xiang Y, Tan Z-X, Li M-D, et al. Influence factors of death risk among COVID-19 patients in Wuhan, China: a hospital-based case-cohort study. medRxiv. (2020). doi: 10.1101/2020.03.13.20035329
37. Li K, Chen D, Chen S, Feng Y, Chang C, Wang Z, et al. Predictors of fatality including radiographic findings in adults with COVID-19. Respir Res.(2020) 21:146. doi: 10.1186/s12931-020-01411-2
38. Luo X, Xia H, Yang W, Wang B, Guo T, Xiong J, et al. Characteristics of patients with COVID-19 during epidemic ongoing outbreak in Wuhan, China. medRxiv. (2020). doi: 10.1101/2020.03.19.20033175
39. Yuan M, Yin W, Tao Z, Tan W, Hu Y. Association of radiologic findings with mortality of patients infected with 2019 novel coronavirus in Wuhan, China. PLoS ONE. (2020) 15:e0230548. doi: 10.1371/journal.pone.0230548
40. Huang I, Lim MA, Pranata R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia - A systematic review, meta-analysis, and meta-regression. Diabetes Metab Syndr. (2020) 14:395–403. doi: 10.1016/j.dsx.2020.04.018
41. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
42. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
43. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
44. Chen L, Liu HG, Liu W, Liu J, Liu K, Shang J, et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. (2020) 43:E005. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
45. Fu H, Li H, Tang X, Li X, Shen J, Zhou Y, et al. Analysis on the clinical characteristics of 36 cases of novel coronavirus pneumonia in Kunming. medRxiv. (2020). doi: 10.1101/2020.02.28.20029173
46. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:811–8. doi: 10.1001/jamacardio.2020.1017
47. Liu K, Fang YY, Deng Y, Liu W, Wang MF, Ma JP, et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin Med J. (2020) 133:1025–31. doi: 10.1097/CM9.0000000000000744
48. Liu L, Zhang D, Tang S, Chen H, Chen L, He X, et al. The epidemiological and clinical characteristics of 2019 novel coronalvirus infection in Changsha, China. Lancet Infect Dis. (2020). doi: 10.2139/ssrn.3537093
49. Peng YD, Meng K, Guan HQ, Leng L, Zhu RR, Wang BY, et al. Clinical characteristics and outcomes of 112 cardiovascular disease patients infected by 2019-nCoV. Zhonghua Xin Xue Guan Bing Za Zhi. (2020) 48:450–5. doi: 10.3760/cma.j.cn112148-20200220-00105
50. Shi H, Han X, Jiang N, Cao Y, Alwalid O, Gu J, et al. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. (2020) 20:425–34. doi: 10.1016/S1473-3099(20)30086-4
51. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. (2020) 5:802–10. doi: 10.1001/jamacardio.2020.0950
52. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. (2020) 18:844–7. doi: 10.1111/jth.14768
53. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical Features of 69 Cases With Coronavirus Disease 2019 in Wuhan, China. Clin Infect Dis. (2020) 71:769–77. doi: 10.1093/cid/ciaa272
54. Xu XW, Wu XX, Jiang XG, Xu KJ, Ying LJ, Ma CL, et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ. (2020) 368:m606. doi: 10.1136/bmj.m606
55. Miller LE, Bhattacharyya R, Miller AL. Diabetes mellitus increases the risk of hospital mortality in patients with Covid-19: systematic review with meta-analysis. Medicine. (2020) 99:e22439. doi: 10.1097/MD.0000000000022439
56. Du Y, Tu L, Zhu P, Mu M, Wang R, Yang P, et al. Clinical Features of 85 Fatal Cases of COVID-19 from Wuhan. A Retrospective Observational Study. Am J Respir Crit Care Med. (2020) 201:1372–9. doi: 10.1164/rccm.202003-0543OC
57. Li X, Wang L, Yan S, Yang F, Xiang L, Zhu J, et al. Clinical characteristics of 25 death cases with COVID-19: A retrospective review of medical records in a single medical center, Wuhan, China. Int J Infect Dis. (2020) 94:128–32. doi: 10.1016/j.ijid.2020.03.053
58. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City Area. JAMA. (2020) 323:2052–9. doi: 10.1001/jama.2020.6775
59. Hussain S, Baxi H, Chand Jamali M, Nisar N, Hussain MS. Burden of diabetes mellitus and its impact on COVID-19 patients: a meta-analysis of real-world evidence. Diabetes Metab Syndr. (2020) 14:1595–602. doi: 10.1016/j.dsx.2020.08.014
60. Gu T, Chu Q, Yu Z, Fa B, Li A, Xu L, et al. History of coronary heart disease increases the mortality rate of COVID-19 patients: a nested case-control study. BMJ Open. (2020) 10:e038976. doi: 10.1136/bmjopen-2020-038976
61. COVIDSurg Collaborative. Mortality and pulmonary complications in patients undergoing surgery with perioperative SARS-CoV-2 infection: an international cohort study. Lancet. (2020) 396:27–38. doi: 10.1016/S0140-6736(20)31182-X
62. Chen Y, Yang D, Cheng B, Chen J, Peng A, Yang C, et al. Clinical characteristics and outcomes of patients with diabetes and COVID-19 in association with glucose-lowering medication. Diabetes Care. (2020) 43:1399–407. doi: 10.2337/dc20-0660
63. Zhang F, Yang D, Li J, Gao P, Chen T, Cheng Z, et al. Myocardial injury is associated with in-hospital mortality of confirmed or suspected COVID-19 in Wuhan, China: a single center retrospective cohort study. medRxiv. (2020). doi: 10.1101/2020.03.21.20040121
64. Ssentongo P, Ssentongo AE, Heilbrunn ES, Ba DM, Chinchilli VM. Association of cardiovascular disease and 10 other pre-existing comorbidities with COVID-19 mortality: a systematic review and meta-analysis. PLoS ONE. (2020) 15:e0238215. doi: 10.1371/journal.pone.0238215
65. Holman N, Knighton P, Kar P, O'Keefe J, Curley M, Weaver A, et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: a population-based cohort study. Lancet Diabetes Endocrinol. (2020) 8:823–33. doi: 10.1016/S2213-8587(20)30271-0
66. Barron E, Bakhai C, Kar P, Weaver A, Bradley D, Ismail H, et al. Associations of type 1 and type 2 diabetes with COVID-19-related mortality in England: a whole-population study. Lancet Diabetes Endocrinol. (2020) 8:813–22. doi: 10.1016/S2213-8587(20)30272-2
67. Beagley J, Guariguata L, Weil C, Motala AA. Global estimates of undiagnosed diabetes in adults. Diabetes Res Clin Pract. (2014) 103:150–60. doi: 10.1016/j.diabres.2013.11.001
68. Unsworth R, Wallace S, Oliver NS, Yeung S, Kshirsagar A, Naidu H, et al. New-onset type 1 diabetes in children during COVID-19: multicenter regional findings in the U.K. Diabetes Care. (2020) 43:e170–1. doi: 10.2337/dc20-1551
69. Pung R, Chiew CJ, Young BE, Chin S, Chen MI, Clapham HE, et al. Investigation of three clusters of COVID-19 in Singapore: implications for surveillance and response measures. Lancet. (2020) 395:1039–46. doi: 10.1016/S0140-6736(20)30528-6
70. Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of Coronavirus Disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med. (2020) 172:577–82. doi: 10.7326/M20-0504
71. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early Transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. (2020) 382:1199–207. doi: 10.1056/NEJMoa2001316
72. Song JW, Zhang C, Fan X, Meng FP, Xu Z, Xia P, et al. Immunological and inflammatory profiles in mild and severe cases of COVID-19. Nat Commun. (2020) 11:3410. doi: 10.1038/s41467-020-17240-2
73. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1172/JCI137244
74. Keeley P, Buchanan D, Carolan C, Pivodic L, Tavabie S, Noble S. Symptom burden and clinical profile of COVID-19 deaths: a rapid systematic review and evidence summary. BMJ Support Palliat Care. (2020) 10:381–4. doi: 10.1136/bmjspcare-2020-002368
75. Zhou S, Zhu T, Wang Y, Xia L. Imaging features and evolution on CT in 100 COVID-19 pneumonia patients in Wuhan, China. Eur Radiol. (2020) 30:5446–54. doi: 10.1007/s00330-020-06879-6
76. Fei Y, Tang N, Liu H, Cao W. Coagulation Dysfunction. Arch Pathol Lab Med. (2020) 144:1223–9. doi: 10.5858/arpa.2020-0324-SA
77. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. (2020) 135:2033–40. doi: 10.1182/blood.2020006000
78. Tavakolpour S, Rakhshandehroo T, Wei EX, Rashidian M. Lymphopenia during the COVID-19 infection: what it shows and what can be learned. Immunol Lett. (2020) 225:31–2. doi: 10.1016/j.imlet.2020.06.013
79. Wang Y, Dong C, Hu Y, Li C, Ren Q, Zhang X, et al. Temporal changes of CT findings in 90 patients with COVID-19 Pneumonia: a Longitudinal Study. Radiology. (2020) 296:E55–64. doi: 10.1148/radiol.2020200843
80. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d'Emal C, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med. (2020) 217:e20201012. doi: 10.1084/jem.20201012
81. Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. (2020) 20:1135–40. doi: 10.1016/S1473-3099(20)30434-5
82. Liu Y, Yan LM, Wan L, Xiang TX, Le A, Liu JM, et al. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis. (2020) 20:656–7. doi: 10.1016/S1473-3099(20)30232-2
83. Sungnak W, Huang N, Becavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
84. Al-Sarraj S, Troakes C, Hanley B, Osborn M, Richardson MP, Hotopf M, et al. The spectrum of neuropathology in COVID-19. Neuropathol Appl Neurobiol. (2020). doi: 10.1111/nan.12667
85. Vasquez-Bonilla WO, Orozco R, Argueta V, Sierra M, Zambrano LI, Munoz-Lara F, et al. A review of the main histopathological findings in coronavirus disease 2019. Hum Pathol. (2020) 105:74–83. doi: 10.1016/j.humpath.2020.07.023
86. Wu SL, Mertens AN, Crider YS, Nguyen A, Pokpongkiat NN, Djajadi S, et al. Substantial underestimation of SARS-CoV-2 infection in the United States. Nat Commun. (2020) 11:4507. doi: 10.1038/s41467-020-18272-4
87. Sette A, Crotty S. Pre-existing immunity to SARS-CoV-2: the knowns and unknowns. Nat Rev Immunol. (2020) 20:457–8. doi: 10.1038/s41577-020-0389-z
88. Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. (2020) 584:457–62. doi: 10.1038/s41586-020-2550-z
89. Lee S, Kim T, Lee E, Lee C, Kim H, Rhee H, et al. Clinical course and molecular viral shedding among asymptomatic and symptomatic patients with SARS-CoV-2 infection in a Community Treatment Center in the Republic of Korea. JAMA Intern Med. (2020) 180:1447–52. doi: 10.1001/jamainternmed.2020.3862
90. Eckerle I, Meyer B. SARS-CoV-2 seroprevalence in COVID-19 hotspots. Lancet. (2020) 396:514–5. doi: 10.1016/S0140-6736(20)31482-3
91. Alanagreh L, Alzoughool F, Atoum M. The Human Coronavirus Disease COVID-19: its origin, characteristics, and insights into potential drugs and its mechanisms. Pathogens. (2020) 9:331. doi: 10.3390/pathogens9050331
92. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. (2015) 1282:1–23. doi: 10.1007/978-1-4939-2438-7_1
93. Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell. (2020) 183:730–8.e13. doi: 10.1101/2020.07.08.192104
94. Skowronski DM, Astell C, Brunham RC, Low DE, Petric M, Roper RL, et al. Severe acute respiratory syndrome (SARS): a year in review. Annu Rev Med. (2005) 56:357–81. doi: 10.1146/annurev.med.56.091103.134135
95. Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet. (2015) 386:995–1007. doi: 10.1016/S0140-6736(15)60454-8
96. Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. (2020) 19:100682. doi: 10.1016/j.genrep.2020.100682
97. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. (2020) 581:215–20. doi: 10.1038/s41586-020-2180-5
98. Sehnal D, Rose AS, Kovca J, Burley SK, Velankar S. Mol*: Towards a common library and tools for web molecular graphics. In: MolVA/EuroVis Proceedings (Graz) (2018).
99. Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. (2010) 128:119–28. doi: 10.1016/j.pharmthera.2010.06.003
100. Zhang Y, Wada J. Collectrin, a homologue of ACE2, its transcriptional control and functional perspectives. Biochem Biophys Res Commun. (2007) 363:1–5. doi: 10.1016/j.bbrc.2007.08.136
101. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
102. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80 e8. doi: 10.1016/j.cell.2020.02.052
103. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92 e6. doi: 10.1016/j.cell.2020.02.058
104. Xia S, Zhu Y, Liu M, Lan Q, Xu W, Wu Y, et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. (2020) 17:765–7. doi: 10.1038/s41423-020-0374-2
105. Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. (2004) 363:938–47. doi: 10.1016/S0140-6736(04)15788-7
106. Xu Y, Zhu J, Liu Y, Lou Z, Yuan F, Liu Y, et al. Characterization of the heptad repeat regions, HR1 and HR2, and design of a fusion core structure model of the spike protein from severe acute respiratory syndrome (SARS) coronavirus. Biochemistry. (2004) 43:14064–71. doi: 10.1021/bi049101q
107. Gui M, Song W, Zhou H, Xu J, Chen S, Xiang Y, et al. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. (2017) 27:119–29. doi: 10.1038/cr.2016.152
108. Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun. (2017) 8:15092. doi: 10.1038/ncomms15092
109. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. (2020) 117:11727–34. doi: 10.1073/pnas.2003138117
110. McKinney CA, Fattah C, Loughrey CM, Milligan G, Nicklin SA. Angiotensin-(1–7) and angiotensin-(1–9): function in cardiac and vascular remodelling. Clin Sci. (2014) 126:815–27. doi: 10.1042/CS20130436
111. Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. (2003) 24:261–71. doi: 10.1210/er.2003-0001
112. Hayashi N, Yamamoto K, Ohishi M, Tatara Y, Takeya Y, Shiota A, et al. The counterregulating role of ACE2 and ACE2-mediated angiotensin 1–7 signaling against angiotensin II stimulation in vascular cells. Hypertens Res. (2010) 33:1182–5. doi: 10.1038/hr.2010.147
113. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. (2010) 2:247–57. doi: 10.1002/emmm.201000080
114. Garvin MR, Alvarez C, Miller JI, Prates ET, Walker AM, Amos BK, et al. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. Elife. (2020) 9:e59177. doi: 10.7554/eLife.59177
115. Turner AJ, Hiscox JA, Hooper NM. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci. (2004) 25:291–4. doi: 10.1016/j.tips.2004.04.001
116. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203:631–7. doi: 10.1002/path.1570
117. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. (2020) 14:185–92. doi: 10.1007/s11684-020-0754-0
118. Guy JL, Jackson RM, Jensen HA, Hooper NM, Turner AJ. Identification of critical active-site residues in angiotensin-converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J. (2005) 272:3512–20. doi: 10.1111/j.1742-4658.2005.04756.x
119. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
120. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. (2014) 88:1293–307. doi: 10.1128/JVI.02202-13
121. Epelman S, Tang WH, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol. (2008) 52:750–4. doi: 10.1016/j.jacc.2008.02.088
122. Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. (2014) 66:167–76. doi: 10.1016/j.yjmcc.2013.11.017
123. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. (2000) 275:33238–43. doi: 10.1074/jbc.M002615200
124. Zoufaly A, Poglitsch M, Aberle JH, Hoepler W, Seitz T, Traugott M, et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir Med. (2020) 8:1154–8. doi: 10.1016/S2213-2600(20)30418-5
125. Batlle D, Wysocki J, Satchell K. Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci. (2020) 134:543–5. doi: 10.1042/CS20200163
126. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45 e9. doi: 10.1016/j.cell.2020.04.026
127. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
128. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. (2004) 303:1526–9. doi: 10.1126/science.1093620
129. Zhang Z, Ohto U, Shibata T, Taoka M, Yamauchi Y, Sato R, et al. Structural analyses of toll-like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. (2018) 25:3371–81 e5. doi: 10.1016/j.celrep.2018.11.081
130. Tanji H, Ohto U, Shibata T, Taoka M, Yamauchi Y, Isobe T, et al. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat Struct Mol Biol. (2015) 22:109–15. doi: 10.1038/nsmb.2943
131. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
132. Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. (2007) 315:1274–8. doi: 10.1126/science.1136567
133. Ran Y, Shu H-B, Wang Y-Y. MITA/STING: a central and multifaceted mediator in innate immune response. Cytokine Growth Factor Rev. (2014) 25:631–9. doi: 10.1016/j.cytogfr.2014.05.003
134. Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. (2011) 23:10–20. doi: 10.1016/j.coi.2010.12.015
135. Maringer K, Fernandez-Sesma A. Message in a bottle: lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev. (2014) 25:669–79. doi: 10.1016/j.cytogfr.2014.08.004
136. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. (2019) 50:1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
137. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J Immunol. (2020) 205:307. doi: 10.4049/jimmunol.2000513
138. Berthelot J-M, Drouet L, Lioté F. Kawasaki-like diseases and thrombotic coagulopathy in COVID-19: delayed over-activation of the STING pathway? Emerg Microbes Infect. (2020) 9:1514–22. doi: 10.1080/22221751.2020.1785336
139. Zhang Y, Chen W, Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomedicine & Pharmacotherapy. (2020) 125:110022. doi: 10.1016/j.biopha.2020.110022
140. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discovery. (2007) 6:975–90. doi: 10.1038/nrd2422
141. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. (2020) 181:1489–501 e15. doi: 10.1016/j.cell.2020.05.015
142. Sekine T, Perez-Potti A, Rivera-Ballesteros O, Stralin K, Gorin JB, Olsson A, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. (2020) 183:158–68 e14. doi: 10.1101/2020.06.29.174888
143. Wang X, Guo X, Xin Q, Pan Y, Hu Y, Li J, et al. Neutralizing antibodies responses to SARS-CoV-2 in COVID-19 inpatients and convalescent patients. Clin Infect Dis. (2020). doi: 10.1101/2020.04.15.20065623
144. Ng K, Faulkner N, Cornish G, Rosa A, Harvey R, Hussain S, et al. Pre-existing and de novo humoral immunity to SARS-CoV-2 in humans. Science. (2020) 370:1339–43. doi: 10.1126/science.abe1107
145. Wu F, Wang A, Liu M, Wang Q, Chen J, Xia S, et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv. (2020). doi: 10.2139/ssrn.3566211
146. Muecksch F, Wise H, Batchelor B, Squires M, Semple E, Richardson C, et al. Longitudinal analysis of serology and neutralizing antibody levels in COVID19 convalescents. J Infect Dis. (2020) jiaa659. doi: 10.1093/infdis/jiaa659
147. Chen Z, John Wherry E. T cell responses in patients with COVID-19. Nat Rev Immunol. (2020) 20:529–36. doi: 10.1038/s41577-020-0402-6
148. Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. (2020) 26:1200–4. doi: 10.1038/s41591-020-0965-6
149. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. (2020) 5:abd1554. doi: 10.1126/sciimmunol.abd1554
150. Acharya D, Liu G, Gack MU. Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol. (2020) 20:397–8. doi: 10.1038/s41577-020-0346-x
151. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
152. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
153. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. (2020) 146:119–27 e4. doi: 10.1016/j.jaci.2020.04.027
154. Mann ER, Menon M, Knight SB, Konkel JE, Jagger C, Shaw TN, et al. Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci Immunol. (2020) 5:abd6197. doi: 10.1126/sciimmunol.abd6197
155. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. (2020) 71:762–8. doi: 10.1093/cid/ciaa248
156. Bryce C, Grimes Z, Pujadas E, Ahuja S, Beasley MB, Albrecht R, et al. Pathophysiology of SARS-CoV-2: targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. medRxiv. (2020). doi: 10.1101/2020.05.18.20099960
157. Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, et al. COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J Leukoc Biol. (2020) 109:13–22. doi: 10.1002/JLB.4HI0720-470R
158. Muir R, Osbourn M, Dubois AV, Doran E, Small DM, Monahan A, et al. Innate lymphoid cells are the predominant source of il-17a during the early pathogenesis of acute respiratory distress syndrome. Am J Respir Crit Care Med. (2016) 193:407–16. doi: 10.1164/rccm.201410-1782OC
159. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine Storm in COVID-19: the current evidence and treatment strategies. Front Immunol. (2020) 11:1708. doi: 10.3389/fimmu.2020.01708
160. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
161. Gao YM, Xu G, Wang B, Liu BC. Cytokine storm syndrome in coronavirus disease 2019: a narrative review. J Intern Med. (2020). doi: 10.1111/joim.13144
162. Leisman DE, Ronner L, Pinotti R, Taylor MD, Sinha P, Calfee CS, et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir Med. (2020) 8:1233–44. doi: 10.1016/S2213-2600(20)30404-5
163. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest. (2020) 130:4694–703. doi: 10.1172/JCI138554
164. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol. (2020) 11:827. doi: 10.3389/fimmu.2020.00827
165. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
166. The National SARS-CoV-2 Serology Assay Evaluation Group. Performance characteristics of five immunoassays for SARS-CoV-2: a head-to-head benchmark comparison. Lancet Infect Dis. (2020) 20:1390–400. doi: 10.1016/S1473-3099(20)30634-4
167. Whitman JD, Hiatt J, Mowery CT, Shy BR, Yu R, Yamamoto TN, et al. Evaluation of SARS-CoV-2 serology assays reveals a range of test performance. Nat Biotechnol. (2020) 38:1174–83. doi: 10.1038/s41587-020-0659-0
168. Faustini SE, Jossi SE, Perez-Toledo M, Shields A, Allen JD, Watanabe Y, et al. Detection of antibodies to the SARS-CoV-2 spike glycoprotein in both serum and saliva enhances detection of infection. medRxiv. (2020). doi: 10.1101/2020.06.16.20133025
169. Yu HQ, Sun BQ, Fang ZF, Zhao JC, Liu XY, Li YM, et al. Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. Eur Respir J. (2020) 56:2020. doi: 10.1183/13993003.01526-2020
170. Ma H, Zeng W, He H, Zhao D, Jiang D, Zhou P, et al. Serum IgA, IgM, and IgG responses in COVID-19. Cell Mol Immunol. (2020) 17:773–5. doi: 10.1038/s41423-020-0474-z
171. Hasan Ali O, Bomze D, Risch L, Brugger S, Paprotny M, Weber M, et al. Severe COVID-19 is associated with elevated serum IgA and antiphospholipid IgA-antibodies. Clin Infect Dis. (2020) ciaa1496. doi: 10.1093/cid/ciaa1496
172. Varadhachary A, Chatterjee D, Garza J, Garr RP, Foley C, Letkeman AF, et al. Salivary anti-SARS-CoV-2 IgA as an :accessible biomarker of mucosal immunity against COVID-19. medRxiv. (2020). doi: 10.1101/2020.08.07.20170258
173. Zhang B, Zhou X, Zhu C, Song Y, Feng F, Qiu Y, et al. Immune phenotyping based on the neutrophil-to-lymphocyte ratio and IgG level predicts disease severity and outcome for patients with COVID-19. Front Mol Biosci. (2020) 7:157. doi: 10.3389/fmolb.2020.00157
174. Alexandraki KI, Piperi C, Ziakas PD, Apostolopoulos NV, Makrilakis K, Syriou V, et al. Cytokine secretion in long-standing diabetes mellitus type 1 and 2: associations with low-grade systemic inflammation. J Clin Immunol. (2008) 28:314–21. doi: 10.1007/s10875-007-9164-1
175. Davidson NJ, Sowden JM, Fletcher J. Defective phagocytosis in insulin controlled diabetics: evidence for a reaction between glucose and opsonising proteins. J Clin Pathol. (1984) 37:783–6. doi: 10.1136/jcp.37.7.783
176. Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, Genetet B. Impaired leucocyte functions in diabetic patients. Diabet Med. (1997) 14:29–34.
177. Huang J, Xiao Y, Zheng P, Zhou W, Wang Y, Huang G, et al. Distinct neutrophil counts and functions in newly diagnosed type 1 diabetes, latent autoimmune diabetes in adults, and type 2 diabetes. Diabetes Metab Res Rev. (2019) 35:e3064. doi: 10.1002/dmrr.3064
178. Lecube A, Pachon G, Petriz J, Hernandez C, Simo R. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS ONE. (2011) 6:e23366. doi: 10.1371/journal.pone.0023366
179. Marhoffer W, Stein M, Maeser E, Federlin K. Impairment of polymorphonuclear leukocyte function and metabolic control of diabetes. Diabetes Care. (1992) 15:256–60. doi: 10.2337/diacare.15.2.256
180. Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what's broken and how can we fix it? Diabetologia. (2017) 60:1839–50. doi: 10.1007/s00125-017-4377-1
181. Blandino-Rosano M, Perez-Arana G, Mellado-Gil JM, Segundo C, Aguilar-Diosdado M. Anti-proliferative effect of pro-inflammatory cytokines in cultured beta cells is associated with extracellular signal-regulated kinase 1/2 pathway inhibition: protective role of glucagon-like peptide−1. J Mol Endocrinol. (2008) 41:35–44. doi: 10.1677/JME-07-0154
182. Okubo Y, Torrey H, Butterworth J, Zheng H, Faustman DL. Treg activation defect in type 1 diabetes: correction with TNFR2 agonism. Clin Transl Immunol. (2016) 5:e56. doi: 10.1038/cti.2015.43
183. Ip B, Cilfone NA, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M, et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity. (2016) 24:102–12. doi: 10.1002/oby.21243
184. Shen H, Shi LZ. Metabolic regulation of TH17 cells. Mol Immunol. (2019) 109:81–7. doi: 10.1016/j.molimm.2019.03.005
185. Nicholas DA, Proctor EA, Agrawal M, Belkina AC, Van Nostrand SC, Panneerseelan-Bharath L, et al. Fatty acid metabolites combine with reduced beta oxidation to activate Th17 inflammation in human type 2 diabetes. Cell Metab. (2019) 30:447–61 e5. doi: 10.1016/j.cmet.2019.07.004
186. Carey IM, Critchley JA, DeWilde S, Harris T, Hosking FJ, Cook DG. Risk of infection in Type 1 and Type 2 diabetes compared with the general population: a matched cohort study. Diabetes Care. (2018) 41:513–21. doi: 10.2337/dc17-2131
187. Critchley JA, Carey IM, Harris T, DeWilde S, Hosking FJ, Cook DG. Glycemic control and risk of infections among people with type 1 or Type 2 diabetes in a large primary care Cohort Study. Diabetes Care. (2018) 41:2127–35. doi: 10.2337/dc18-0287
188. Wang S, Ma P, Zhang S, Song S, Wang Z, Ma Y, et al. Fasting blood glucose at admission is an independent predictor for 28-day mortality in patients with COVID-19 without previous diagnosis of diabetes: a multi-centre retrospective study. Diabetologia. (2020) 63:2102–11. doi: 10.1007/s00125-020-05209-1
189. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. (2020) 63:364–74. doi: 10.1007/s11427-020-1643-8
190. Henry BM, Benoit S, Lippi G, Benoit J. Circulating plasma levels of angiotensin II and aldosterone in patients with coronavirus disease 2019 (COVID-19): a preliminary report. Prog Cardiovasc Dis. (2020) 63:702–3. doi: 10.1016/j.pcad.2020.07.006
191. Jarcho JA, Ingelfinger JR, Hamel MB, D'Agostino RB Sr., Harrington DP. Inhibitors of the renin-angiotensin-aldosterone system and Covid-19. N Engl J Med. (2020) 382:2462–4. doi: 10.1056/NEJMe2012924
192. Tralhao A, Moita LF, Povoa P. Potential benefit of angiotensin II in COVID-19 patients: beyond reasonable doubt? Crit Care. (2020) 24:324. doi: 10.1186/s13054-020-03027-w
193. Flacco ME, Acuti Martellucci C, Bravi F, Parruti G, Cappadona R, Mascitelli A, et al. Treatment with ACE inhibitors or ARBs and risk of severe/lethal COVID-19: a meta-analysis. Heart. (2020) 106:1519–24. doi: 10.1136/heartjnl-2020-317336
194. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. (2020) 382:1653–9. doi: 10.1056/NEJMsr2005760
195. Aronson JK, Ferner RE. Drugs and the renin-angiotensin system in covid-19. BMJ. (2020) 369:m1313. doi: 10.1136/bmj.m1313
196. Hippisley-Cox J, Young D, Coupland C, Channon KM, Tan PS, Harrison DA, et al. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: cohort study including 8.3 million people. Heart. (2020) 106:1503–11. doi: 10.1136/heartjnl-2020-317393
197. Braude P, Carter B, Short R, Vilches-Moraga A, Verduri A, Pearce L, et al. The influence of ACE inhibitors and ARBs on hospital length of stay and survival in people with COVID-19. IJC Heart Vasculature. (2020) 31:100660. doi: 10.1016/j.ijcha.2020.100660
198. Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN. Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19. New England J Med. (2020) 382:e102. doi: 10.1056/NEJMoa2007621
199. Koka V, Huang XR, Chung AC, Wang W, Truong LD, Lan HY. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol. (2008) 172:1174–83. doi: 10.2353/ajpath.2008.070762
200. Soler MJ, Wysocki J, Ye M, Lloveras J, Kanwar Y, Batlle D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. (2007) 72:614–23. doi: 10.1038/sj.ki.5002373
201. Chen J, Jiang Q, Xia X, Liu K, Yu Z, Tao W, et al. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell. (2020) 19:e13168. doi: 10.1111/acel.13168
202. Anderson S, Jung FF, Ingelfinger JR. Renal renin-angiotensin system in diabetes: functional, immunohistochemical, and molecular biological correlations. Am J Physiol. (1993) 265(4 Pt 2):F477–86. doi: 10.1152/ajprenal.1993.265.4.F477
203. Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, et al. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. (2003) 41:392–7. doi: 10.1161/01.HYP.0000060689.38912.CB
204. Rao S, Lau A, So HC. Exploring Diseases/traits and blood proteins causally related to expression of ACE2, the Putative Receptor of SARS-CoV-2: a mendelian randomization analysis highlights tentative relevance of diabetes-related traits. Diabetes Care. (2020) 43:1416–26. doi: 10.2337/dc20-0643
205. Roca-Ho H, Riera M, Palau V, Pascual J, Soler MJ. Characterization of ACE and ACE2 expression within different organs of the NOD mouse. Int J Mol Sci. (2017) 18:563. doi: 10.3390/ijms18030563
206. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. (2020) 8:e21. doi: 10.1016/S2213-2600(20)30116-8
207. Muller LM, Gorter KJ, Hak E, Goudzwaard WL, Schellevis FG, Hoepelman AI, et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis. (2005) 41:281–8. doi: 10.1086/431587
208. Kulcsar KA, Coleman CM, Beck SE, Frieman MB. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight. (2019) 4:e131774. doi: 10.1172/jci.insight.131774
209. Hulme KD, Gallo LA, Short KR. Influenza virus and glycemic variability in diabetes: a killer combination? Front Microbiol. (2017) 8:861. doi: 10.3389/fmicb.2017.00861
210. Carroll ML, Yerkovich ST, Pritchard AL, Davies JM, Upham JW. Adaptive immunity to rhinoviruses: sex and age matter. Respir Res. (2010) 11:184. doi: 10.1186/1465-9921-11-184
211. Giefing-Kroll C, Berger P, Lepperdinger G, Grubeck-Loebenstein B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell. (2015) 14:309–21. doi: 10.1111/acel.12326
212. Klein SL. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays. (2012) 34:1050–9. doi: 10.1002/bies.201200099
213. Oh SJ, Lee JK, Shin OS. Aging and the Immune System: the Impact of Immunosenescence on viral infection, immunity and vaccine immunogenicity. Immune Netw. (2019) 19:e37. doi: 10.4110/in.2019.19.e37
214. Kautzky-Willer A, Harreiter J, Pacini G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev. (2016) 37:278–316. doi: 10.1210/er.2015-1137
215. Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A, Del Prato S. COVID-19 in people with diabetes: understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol. (2020) 8:782–92. doi: 10.1016/S2213-8587(20)30238-2
216. Deng C, Xiang Y, Tan T, Ren Z, Cao C, Huang G, et al. Altered peripheral B-lymphocyte subsets in type 1 diabetes and latent autoimmune diabetes in adults. Diabetes Care. (2016) 39:434–40. doi: 10.2337/dc15-1765
217. Mira JC, Brakenridge SC, Moldawer LL, Moore FA. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin. (2017) 33:245–58. doi: 10.1016/j.ccc.2016.12.001
218. Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, et al. Chronic critical illness and the persistent inflammation, immunosuppression, and catabolism syndrome. Front Immunol. (2018) 9:1511. doi: 10.3389/fimmu.2018.01511
219. Salminen A. Increased immunosuppression impairs tissue homeostasis with aging and age-related diseases. J Mol Med. (2021) 99:1–20. doi: 10.1007/s00109-020-01988-7
220. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in Acute COVID-19 and associations with age and disease severity. Cell. (2020) 183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038
221. Daoust JF. Elderly people and responses to COVID-19 in 27 Countries. PLoS ONE. (2020) 15:e0235590. doi: 10.1371/journal.pone.0235590
222. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two Potential Targets for COVID-19 Treatment. Mediators Inflamm. (2020) 2020:7527953. doi: 10.1155/2020/7527953
223. Land WG. Role of damage-associated molecular patterns in light of modern environmental research: a tautological approach. Int J Environ Res. (2020) 14:583–604. doi: 10.1007/s41742-020-00276-z
224. AlGhatrif M, Cingolani O, Lakatta EG. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights From Cardiovascular Aging Science. JAMA Cardiol. (2020) 5:747–8. doi: 10.1001/jamacardio.2020.1329
225. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells. (2020) 9:1383. doi: 10.3390/cells9061383
226. Webb BJ, Peltan ID, Jensen P, Hoda D, Hunter B, Silver A, et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: a cohort study. Lancet Rheumatol. (2020) 2:e754–63. doi: 10.1016/S2665-9913(20)30343-X
227. The Open SC, Williamson E, Walker A, Bhaskaran K, Bacon S, Bates C, etal. OpenSAFELY: factors associated with COVID-19-related hospital death in the linked electronic health records of 17 million adult NHS patients. Nature. (2020) 584:430–6. doi: 10.1038/s41586-020-2521-4
228. Thierry AR, Roch B. Neutrophil extracellular traps and by-products play a key role in COVID-19: pathogenesis, risk factors, and therapy. J Clin Med. (2020) 9:2942. doi: 10.3390/jcm9092942
229. Kalyanaraman B. Do free radical NETwork and oxidative stress disparities in African Americans enhance their vulnerability to SARS-CoV-2 infection and COVID-19 severity? Redox Biol. (2020) 37:101721. doi: 10.1016/j.redox.2020.101721
230. Wang S, Fu L, Huang K, Han J, Zhang R, Fu Z. Neutrophil-to-lymphocyte ratio at admission is an independent risk factors for the severity and mortality in patients with coronavirus disease 2019. J Infect. (2020). doi: 10.1016/j.jinf.2020.09.022
231. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217:e20201129. doi: 10.1084/jem.20201129
232. de Vries JJ, Hoppenbrouwers T, Martinez-Torres C, Majied R, Ozcan B, van Hoek M, et al. Effects of diabetes mellitus on fibrin clot structure and mechanics in a model of acute Neutrophil Extracellular Traps (NETs) Formation. Int J Mol Sci. (2020) 21:7107. doi: 10.3390/ijms21197107
233. Jin L, Liu Y, Jing C, Wang R, Wang Q, Wang H. Neutrophil extracellular traps (NETs)-mediated killing of carbapenem-resistant hypervirulent Klebsiella pneumoniae (CR-hvKP) are impaired in patients with diabetes mellitus. Virulence. (2020) 11:1122–30. doi: 10.1080/21505594.2020.1809325
234. Birra D, Benucci M, Landolfi L, Merchionda A, Loi G, Amato P, et al. COVID 19: a clue from innate immunity. Immunol Res. (2020) 68:161–8. doi: 10.1007/s12026-020-09137-5
235. Bandyopadhyay D, Akhtar T, Hajra A, Gupta M, Das A, Chakraborty S, et al. COVID-19 pandemic: cardiovascular complications and future implications. Am J Cardiovasc Drugs. (2020) 20:311–24. doi: 10.1007/s40256-020-00420-2
236. Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID-19 and cardiovascular disease. Circulation. (2020) 141:1648–55. doi: 10.1161/CIRCULATIONAHA.120.046941
237. Seirafianpour F, Sodagar S, Pour Mohammad A, Panahi P, Mozafarpoor S, Almasi S, et al. Cutaneous manifestations and considerations in COVID-19 pandemic: a systematic review. Dermatol Ther. (2020) 33:e13986. doi: 10.1111/dth.13986
238. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
239. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
240. Le Brocq M, Leslie SJ, Milliken P, Megson IL. Endothelial dysfunction: from molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid Redox Signal. (2008) 10:1631–74. doi: 10.1089/ars.2007.2013
241. Shaw A, Doherty MK, Mutch NJ, MacRury SM, Megson IL. Endothelial cell oxidative stress in diabetes: a key driver of cardiovascular complications? Biochem Soc Trans. (2014) 42:928–33. doi: 10.1042/BST20140113
242. Gu HF, Ma J, Gu KT, Brismar K. Association of intercellular adhesion molecule 1 (ICAM1) with diabetes and diabetic nephropathy. Front Endocrinol. (2012) 3:179. doi: 10.3389/fendo.2012.00179
243. Ribau JC, Hadcock SJ, Teoh K, DeReske M, Richardson M. Endothelial adhesion molecule expression is enhanced in the aorta and internal mammary artery of diabetic patients. J Surg Res. (1999) 85:225–33. doi: 10.1006/jsre.1999.5682
244. McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. (1995) 147:642–53.
245. Lim HS, Blann AD, Lip GY. Soluble CD40 ligand, soluble P-selectin, interleukin-6, and tissue factor in diabetes mellitus: relationships to cardiovascular disease and risk factor intervention. Circulation. (2004) 109:2524–8. doi: 10.1161/01.CIR.0000129773.70647.94
246. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. (2010) 4:302–12. doi: 10.2174/1874192401004010302
247. Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
248. Moncada S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann N Y Acad Sci. (1997) 811:60–7; discussion 7–9. doi: 10.1111/j.1749-6632.1997.tb51989.x
249. Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. (1987) 92:639–46. doi: 10.1111/j.1476-5381.1987.tb11367.x
250. Moncada S. Adventures in vascular biology: a tale of two mediators. Philos Trans R Soc Lond B Biol Sci. (2006) 361:735–59. doi: 10.1098/rstb.2005.1775
251. Darimont C, Vassaux G, Gaillard D, Ailhaud G, Negrel R. In situ microdialysis of prostaglandins in adipose tissue: stimulation of prostacyclin release by angiotensin II. Int J Obes Relat Metab Disord. (1994) 18:783–8.
252. Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. (2002) 296:539–41. doi: 10.1126/science.1068711
253. Zellers TM, McCormick J, Wu Y. Interaction among ET-1, endothelium-derived nitric oxide, and prostacyclin in pulmonary arteries and veins. Am J Physiol. (1994) 267(1 Pt 2):H139–47. doi: 10.1152/ajpheart.1994.267.1.H139
254. Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. (2012) 10:4–18. doi: 10.2174/157016112798829760
255. Noris M, Morigi M, Donadelli R, Aiello S, Foppolo M, Todeschini M, et al. Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ Res. (1995) 76:536–43. doi: 10.1161/01.RES.76.4.536
256. Arnet UA, McMillan A, Dinerman JL, Ballermann B, Lowenstein CJ. Regulation of endothelial nitric-oxide synthase during hypoxia. J Biol Chem. (1996) 271:15069–73. doi: 10.1074/jbc.271.25.15069
257. Godo S, Shimokawa H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic Biol Med. (2017) 109:4–10. doi: 10.1016/j.freeradbiomed.2016.12.019
258. Salvemini D, Doyle TM, Cuzzocrea S. Superoxide, peroxynitrite and oxidative/nitrative stress in inflammation. Biochem Soc Trans. (2006) 34(Pt 5):965–70. doi: 10.1042/BST0340965
259. Stockklauser-Farber K, Ballhausen T, Laufer A, Rosen P. Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim Biophys Acta. (2000) 1535:10–20. doi: 10.1016/S0925-4439(00)00078-8
260. Endemann DH, Schiffrin EL. Nitric oxide, oxidative excess, and vascular complications of diabetes mellitus. Curr Hypertens Rep. (2004) 6:85–9. doi: 10.1007/s11906-004-0081-x
261. Treweeke A, Hall J, Lambie S, Leslie SJ, Megson IL, MacRury SM. Preliminary study of hypoxia-related cardiovascular mediator-markers in patients with end-stage renal disease with and without diabetes and the effects of haemodialysis. PLoS ONE. (2017) 12:e0178171. doi: 10.1371/journal.pone.0178171
262. Zafar MI, Mills K, Ye X, Blakely B, Min J, Kong W, et al. Association between the expression of vascular endothelial growth factors and metabolic syndrome or its components: a systematic review and meta-analysis. Diabetol Metab Syndr. (2018) 10:62. doi: 10.1186/s13098-018-0363-0
263. Wright CD, Mulsch A, Busse R, Osswald H. Generation of nitric oxide by human neutrophils. Biochem Biophys Res Commun. (1989) 160:813–9. doi: 10.1016/0006-291X(89)92506-0
264. Lambden S. Bench to bedside review: therapeutic modulation of nitric oxide in sepsis-an update. Intensive Care Med Exp. (2019) 7:64. doi: 10.1186/s40635-019-0274-x
265. Alvarez RA, Berra L, Gladwin MT. Home Nitric Oxide Therapy for COVID-19. Am J Respir Crit Care Med. (2020) 202:16–20. doi: 10.1164/rccm.202005-1906ED
266. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
267. Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. (2011) 30:4157–70. doi: 10.1038/emboj.2011.304
268. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. (1996) 16:4604–13. doi: 10.1128/MCB.16.9.4604
269. Burtenshaw D, Kitching M, Redmond EM, Megson IL, Cahill PA. Reactive Oxygen Species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front Cardiovasc Med. (2019) 6:89. doi: 10.3389/fcvm.2019.00089
270. Fiorello ML, Treweeke AT, Macfarlane DP, Megson IL. The impact of glucose exposure on bioenergetics and function in a cultured endothelial cell model and the implications for cardiovascular health in diabetes. Sci Rep. (2020) 10:19547. doi: 10.1038/s41598-020-76505-4
271. Andre P. P-selectin in haemostasis. Br J Haematol. (2004) 126:298–306. doi: 10.1111/j.1365-2141.2004.05032.x
272. Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost. (2005) 3:1590–6. doi: 10.1111/j.1538-7836.2005.01373.x
273. Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. (2003) 24:2166–79. doi: 10.1016/j.ehj.2003.08.021
274. Tessaro FH, Ayala TS, Martins JO. Lipid mediators are critical in resolving inflammation: a review of the emerging roles of eicosanoids in diabetes mellitus. BioMed Research International. (2015) 2015:568408. doi: 10.1155/2015/568408
275. Luo P, Wang M-H. Eicosanoids, β-cell function, and diabetes. Prostagl Other Lipid Mediators. (2011) 95:1–10. doi: 10.1016/j.prostaglandins.2011.06.001
276. Yan X, Hao Q, Mu Y, Timani KA, Ye L, Zhu Y, et al. Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-kappa B and CCAAT/enhancer binding protein. Int J Biochem Cell Biol. (2006) 38:1417–28. doi: 10.1016/j.biocel.2006.02.003
277. Chopra S, Giovanelli P, Alvarado-Vazquez PA, Alonso S, Song M, Sandoval TA, et al. IRE1alpha-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science. (2019) 365:eaau6499. doi: 10.1126/science.aau6499
278. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. (2015) 15:511–23. doi: 10.1038/nri3859
279. Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. (1998) 104:2S-22S. doi: 10.1016/S0002-9343(97)00203-9
280. Ogawa H, Nakayama M, Morimoto T, Uemura S, Kanauchi M, Doi N, et al. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. (2008) 300:2134–41. doi: 10.1001/jama.2008.623
281. Belch J, MacCuish A, Campbell I, Cobbe S, Taylor R, Prescott R, et al. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ. (2008) 337:a1840. doi: 10.1136/bmj.a1840
282. Pignone M, Alberts MJ, Colwell JA, Cushman M, Inzucchi SE, Mukherjee D, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Diabetes care. (2010) 33:1395–402. doi: 10.2337/dc10-0555
283. Group ASC, Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med. (2018) 379:1529–39. doi: 10.1056/NEJMoa1804988
284. Xing Y, Tan KC. Aspirin for primary prevention of cardiovascular disease in diabetes. J Diabetes Investig. (2019) 10:899–901. doi: 10.1111/jdi.13006
285. FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. (2001) 345:433–42. doi: 10.1056/NEJM200108093450607
286. Howard PA, Delafontaine P. Nonsteroidal anti-inflammatory drugs and cardiovascular risk. J Am College Cardiol. (2004) 43:519–25. doi: 10.1016/j.jacc.2003.09.043
287. Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and−2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. (2000) 102:840–5. doi: 10.1161/01.CIR.102.8.840
288. Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. (2001) 345:1809–17. doi: 10.1056/NEJMoa003199
289. Reichman HR, Farrell CL, Del Maestro RF. Effects of steroids and nonsteroid anti-inflammatory agents on vascular permeability in a rat glioma model. J Neurosurg. (1986) 65:233–7. doi: 10.3171/jns.1986.65.2.0233
290. Tarnawski AS, Jones MK. Inhibition of angiogenesis by NSAIDs: molecular mechanisms and clinical implications. J Mol Med. (2003) 81:627–36. doi: 10.1007/s00109-003-0479-y
291. Williams TJ, Morley J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. (1973) 246:215–7. doi: 10.1038/246215a0
292. Pai R, Szabo IL, Soreghan BA, Atay S, Kawanaka H, Tarnawski AS. PGE(2) stimulates VEGF expression in endothelial cells via ERK2/JNK1 signaling pathways. Biochem Biophys Res Commun. (2001) 286:923–8. doi: 10.1006/bbrc.2001.5494
293. Mazzeo D, Panina-Bordignon P, Recalde H, Sinigaglia F, D'Ambrosio D. Decreased IL-12 production and Th1 cell development by acetyl salicylic acid-mediated inhibition of NF-kappaB. Eur J Immunol. (1998) 28:3205–13.
294. Inaoka M, Kimishima M, Takahashi R, Shiohara T. Non-steroidal anti-inflammatory drugs selectively inhibit cytokine production by NK cells and gamma delta T cells. Exp Dermatol. (2006) 15:981–90. doi: 10.1111/j.1600-0625.2006.00505.x
295. Cianferoni A, Schroeder JT, Kim J, Schmidt JW, Lichtenstein LM, Georas SN, et al. Selective inhibition of interleukin-4 gene expression in human T cells by aspirin. Blood. (2001) 97:1742–9. doi: 10.1182/blood.V97.6.1742
296. Torjesen I. Covid-19: NICE advises against using NSAIDs for fever in patients with suspected cases. BMJ. (2020) 369:m1409. doi: 10.1136/bmj.m1409
297. Qiao W, Wang C, Chen B, Zhang F, Liu Y, Lu Q, et al. Ibuprofen attenuates cardiac fibrosis in streptozotocin-induced diabetic rats. Cardiology. (2015) 131:97–106. doi: 10.1159/000375362
298. Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med. (2020) 382:2431–40. doi: 10.1056/NEJMoa2006923
299. Chen JS, Alfajaro MM, Wei J, Chow RD, Filler RB, Eisenbarth SC, et al. Cyclooxgenase-2 is induced by SARS-CoV-2 infection but does not affect viral entry or replication. bioRxiv. (2020). doi: 10.1101/2020.09.24.312769
300. Torjesen I. Covid-19: ibuprofen can be used for symptoms, says UK agency, but reasons for change in advice are unclear. BMJ. (2020) 369:m1555. doi: 10.1136/bmj.m1555
301. Pyrillou K, Chairakaki AD, Tamvakopoulos C, Andreakos E. Dexamethasone induces omega3-derived immunoresolvents driving resolution of allergic airway inflammation. J Allergy Clin Immunol. (2018) 142:691–5.e4. doi: 10.1016/j.jaci.2018.04.004
302. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with Covid-19 - preliminary report. N Engl J Med. (2020). doi: 10.1056/NEJMoa2021436
303. Alessi J, de Oliveira GB, Schaan BD, Telo GH. Dexamethasone in the era of COVID-19: friend or foe? An essay on the effects of dexamethasone and the potential risks of its inadvertent use in patients with diabetes. Diabetol Metab Syndr. (2020) 12:80–91. doi: 10.1186/s13098-020-00583-7
304. The Lancet Haematology. COVID-19 coagulopathy: an evolving story. Lancet Haematol. (2020) 7:e425. doi: 10.1016/S2352-3026(20)30151-4
305. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
306. Martin-Rojas RM, Perez-Rus G, Delgado-Pinos VE, Domingo-Gonzalez A, Regalado-Artamendi I, Alba-Urdiales N, et al. COVID-19 coagulopathy: An in-depth analysis of the coagulation system. Eur J Haematol. (2020) 105:741–50. doi: 10.1111/ejh.13501
307. Pretorius L, Thomson GJA, Adams RCM, Nell TA, Laubscher WA, Pretorius E. Platelet activity and hypercoagulation in type 2 diabetes. Cardiovasc Diabetol. (2018) 17:141. doi: 10.1186/s12933-018-0783-z
308. Wang L, Zhou X, Yin Y, Mai Y, Wang D, Zhang X. Hyperglycemia induces neutrophil extracellular traps formation through an NADPH oxidase-dependent pathway in diabetic retinopathy. Front Immunol. (2018) 9:3076. doi: 10.3389/fimmu.2018.03076
309. Zucoloto AZ, Jenne CN. Platelet-neutrophil interplay: insights into Neutrophil Extracellular Trap (NET)-driven coagulation in infection. Front Cardiovasc Med. (2019) 6:85. doi: 10.3389/fcvm.2019.00085
310. Abou-Ismail MY, Diamond A, Kapoor S, Arafah Y, Nayak L. The hypercoagulable state in COVID-19: incidence, pathophysiology, and management. Thrombosis Res. (2020) 194:101–15. doi: 10.1016/j.thromres.2020.06.029
311. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thrombosis Haemostasis. (2020) 18:1094–9. doi: 10.1111/jth.14817
312. Rahadian A, Fukuda D, Salim HM, Yagi S, Kusunose K, Yamada H, et al. Canagliflozin Prevents Diabetes-Induced Vascular Dysfunction in ApoE-Deficient Mice. Journal of Atherosclerosis and Thrombosis. (2020) 27:1141–51. doi: 10.5551/jat.52100
313. Testa S, Prandoni P, Paoletti O, Morandini R, Tala M, Dellanoce C, et al. Direct oral anticoagulant plasma levels' striking increase in severe COVID-19 respiratory syndrome patients treated with antiviral agents: the Cremona experience. J Thromb Haemost. (2020) 18:1320–3. doi: 10.1111/jth.14871
314. Lonnrot M, Lynch KF, Elding Larsson H, Lernmark A, Rewers MJ, Torn C, et al. Respiratory infections are temporally associated with initiation of type 1 diabetes autoimmunity: the TEDDY study. Diabetologia. (2017) 60:1931–40. doi: 10.1007/s00125-017-4365-5
315. Yang JK, Lin SS, Ji XJ, Guo LM. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. (2010) 47:193–9. doi: 10.1007/s00592-009-0109-4
316. Marchand L, Pecquet M, Luyton C. Type 1 diabetes onset triggered by COVID-19. Acta Diabetol. (2020) 57:1265–6. doi: 10.1007/s00592-020-01570-0
317. Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, et al. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell. (2020) 27:125–36 e7. doi: 10.1016/j.stem.2020.06.015
318. Carlsson PO, Berne C, Jansson L. Angiotensin II and the endocrine pancreas: effects on islet blood flow and insulin secretion in rats. Diabetologia. (1998) 41:127–33. doi: 10.1007/s001250050880
Keywords: COVID-19, SARS– CoV– 2, diabetes, endothelium, oxidative stress, angiotensin converting enzyme-2, inflammation, immune response
Citation: Roberts J, Pritchard AL, Treweeke AT, Rossi AG, Brace N, Cahill P, MacRury SM, Wei J and Megson IL (2021) Why Is COVID-19 More Severe in Patients With Diabetes? The Role of Angiotensin-Converting Enzyme 2, Endothelial Dysfunction and the Immunoinflammatory System. Front. Cardiovasc. Med. 7:629933. doi: 10.3389/fcvm.2020.629933
Received: 16 November 2020; Accepted: 15 December 2020;
Published: 03 February 2021.
Edited by:
Amarylis Wanschel, University of Miami Hospital, United StatesReviewed by:
Nayara Leite, Harvard University, United StatesCopyright © 2021 Roberts, Pritchard, Treweeke, Rossi, Brace, Cahill, MacRury, Wei and Megson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian L. Megson, aWFuLm1lZ3NvbkB1aGkuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.