 Jiayu Wu1
Jiayu Wu1 Johan W. M. Heemskerk
Johan W. M. Heemskerk Constance C. F. M. J. Baaten
Constance C. F. M. J. Baaten
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 08 January 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.608391
This article is part of the Research Topic Proteolysis of Membrane Proteins in Vascular Biology and Disease View all 7 articles
The activities of adhesion and signaling receptors in platelets are controlled by several mechanisms. An important way of regulation is provided by proteolytic cleavage of several of these receptors, leading to either a gain or a loss of platelet function. The proteases involved are of different origins and types: (i) present as precursor in plasma, (ii) secreted into the plasma by activated platelets or other blood cells, or (iii) intracellularly activated and cleaving cytosolic receptor domains. We provide a comprehensive overview of the proteases acting on the platelet membrane. We describe how these are activated, which are their target proteins, and how their proteolytic activity modulates platelet functions. The review focuses on coagulation-related proteases, plasmin, matrix metalloproteinases, ADAM(TS) isoforms, cathepsins, caspases, and calpains. We also describe how the proteolytic activities are determined by different platelet populations in a thrombus and conversely how proteolysis contributes to the formation of such populations.
According to a common concept of thrombosis and hemostasis, damage or injury of a vessel wall and ensuing exposure of extracellular matrix components to the blood stream triggers platelets from the circulation to become adherent and to assemble into a thrombus, thus limiting the extravasation of blood (1, 2). Within a thrombus, however, distinct types of activated platelets can be recognized, exhibiting different functions, although partial overlap between the populations exists (3).
The process of thrombus formation is considered to be initiated by von Willebrand factor (VWF) binding to exposed collagen or laminin in the damaged vessel wall, followed by shear-dependent platelet binding to VWF through the glycoprotein (GP)Ib-V–IX complex (1, 3, 4). The flow-dependent adhesion of platelets to those and other extracellular matrix components is stabilized by a panel of integrins, including integrin α2β1, αIIbβ3, and α6β1 (adhesive platelet population), while the initial activation of platelets is achieved by signaling via the collagen/laminin receptor, glycoprotein VI (GPVI). This provokes the release of thromboxane A2 and the secretion of granular contents including ADP and, via these autocrine agents, the subsequent recruitment of additional flowing platelets, which assemble into a growing thrombus via αIIbβ3-fibrinogen interactions (aggregating platelet population) (1). Upon prolonged high intracellular rises in Ca2+, platelets develop a procoagulant phenotype that is characterized by the surface membrane exposure of phosphatidylserine and by the inactivation of integrin αIIbβ3 (procoagulant platelet population) (5). Phosphatidylserine-exposing platelets, usually located around a thrombus, provide a negatively charged membrane surface, which supports coagulation factor binding and the formation of tenase and prothrombinase complexes (6, 7). Thrombin, which is generated at these phosphatidylserine sites, triggers the formation of fibrin fibers, which consolidate the platelet thrombus into a stable clot sealing the breach in a vessel wall (2). Independently of such activation processes, platelet heterogeneity can be achieved by aging and an accompanied inactivation (3). The latter changes are considered to have apoptosis-like properties (8).
Although these platelet activation processes are relatively well-studied as a function of the platelet environment, only since recently it is becoming clear that a multitude of proteases present in plasma or produced by platelets themselves are important for the distinct properties of platelet populations, often by cleaving specific receptors. Here, we review current knowledge how proteases act on platelet receptors and the platelet membrane surface. We describe how these are activated, their targets, their effect on platelet functions, and the consequences for platelet population formation. An overview of the discussed proteases, their known targets, and their effects on platelet function is given in Supplementary Table 1.
The coagulation process is accomplished by consecutive activity of several plasmatic serine proteases, which always circulate as zymogen precursors and, once activated, provide complex feedback loops to trigger other coagulation or anticoagulation factors (1, 9). Serine proteases (serine endopeptidases) are defined as proteolytic enzymes with a serine residue in their active site; they usually have a multidomain structure containing β-barrel regions and multiple surface loops surrounding the active site (10). Relevant coagulation serine proteases that have been reported to act on platelets are thrombin, factor Xa, and plasmin (11), as summarized in Supplementary Table 1. A structurally related serine protease is trypsin.
In the extrinsic coagulation process triggered by tissue factor, the protease factor Xa is produced from its zymogen factor X by factor VII/VIIa (1, 9). Cleavage into factor Xa is greatly enhanced in the presence of phosphatidylserine-exposing membranes, such as present on procoagulant platelets (12, 13). On such platelets, the so-called tenase complex, with as protease factor IXa and as cofactor VIIIa, catalyzes the conversion of factor X into factor Xa. Subsequently, the protease factor Xa along with cofactor Va (prothrombinase complex) catalyzes the conversion of prothrombin into α-thrombin (1, 14). Both factor Xa and thrombin cleave a range of substrates, including proteolytic-activated receptors (PARs), while thrombin also cleaves fibrinogen to form fibrin clots (Figure 1). A further substrate of thrombin is the serine protease plasmin, which is produced on endothelial cells from its precursor plasminogen, and acts as an essential factor in fibrinolysis (15). The activity of all three serine proteases—factor Xa, thrombin, plasmin—is tightly regulated by circulating serpins (serine protease inhibitors, such as antithrombin), which avidly bind to and inactivate the catalytic domains of the proteases (16). The binding of thrombin to endothelial-bound thrombomodulin, in addition, activates the anticoagulant pathway of protein C (17).
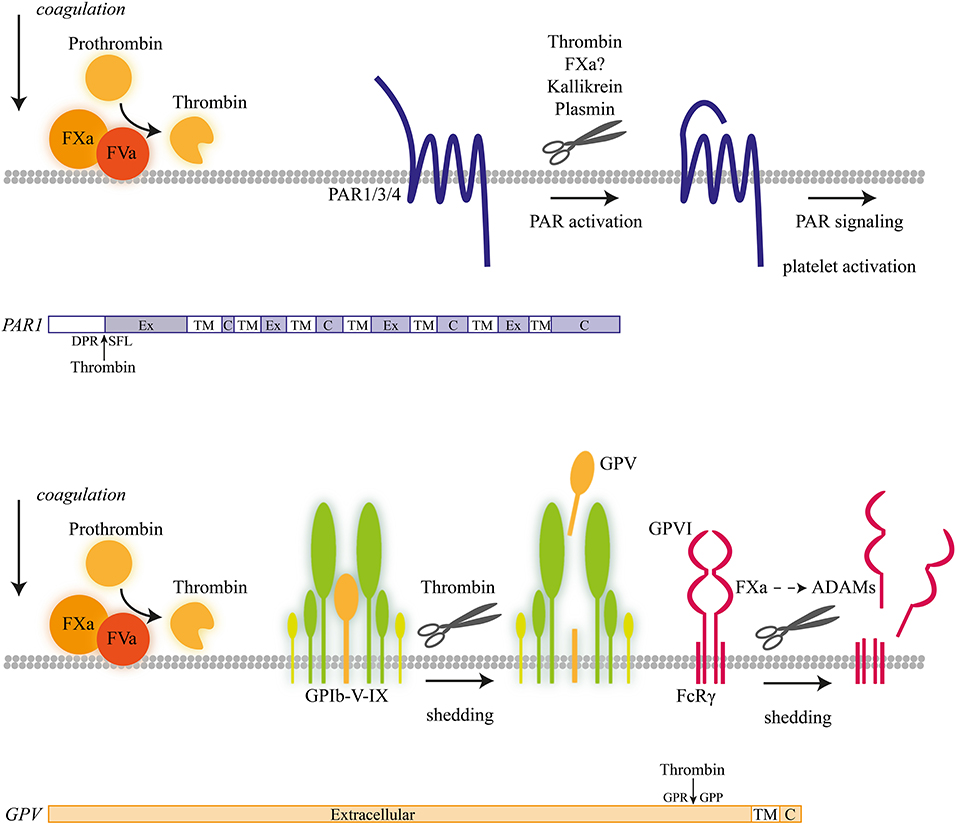
Figure 1. Coagulation-induced receptor proteolysis. Proteolytic activation mechanism of coagulation factors at the platelet membrane, progressing to thrombin-induced activation of proteolytic-activated receptor (PAR) isoforms and shedding of glycoprotein V (GPV) and GPVI with a schematic overview of proteolytic cleavage sites.
Other serine proteases such as trypsin (in the intestine) are also released as zymogens (trypsinogen) that require proteolytic cleavage (hetero- or autocatalytic) to give the active proteolytic enzyme. Trypsin (not present in blood plasma) has a strong preference for cleavage of the PAR2 receptors (18), although one report demonstrates trypsin IV-induced cleavage of PAR4 (19). Overall, it appears that the various serine proteases that affect platelets first need to be converted into an active form via proteolysis and that all are prone to inactivation by serpins. As a consequence, the platelet activation responses to these proteases are confined in space and time.
α-Thrombin is known as a potent platelet agonist. On platelets, it carries out the activating potential by inducing proteolytic events and furthermore by binding to the GPIb-V–IX complex (1, 20–22). Established thrombin substrates include PAR receptors, coagulation factors V, VIII, and XIII, and fibrinogen (23).
In human platelets, α-thrombin cleaves the N-terminal extracellular site of isoforms PAR1 and PAR4 (24, 25). The proteolysis liberates a new N-terminus, which autocatalytically activates the receptor molecule (26, 27). By ensuing signaling via G-proteins (in particular Gqα) (24, 28), thrombin induces a wide range of platelet responses, including shape change, Ca2+ fluxes, integrin activation, and secretion (23).
α-Thrombin easily activates and cleaves the PAR1 receptor (Arg41-Ser42) with an EC50 of around 0.1 nM, due to the presence of a second thrombin binding site on this receptor. The hexapeptide SFLLRN (TRAP6) acts as a specific agonist of PAR1, as it represents the neo-N-terminal site of the cleaved receptor. In addition, the α-thrombin precursor form meizothrombin (μ-thrombin), recognized as an obligatory intermediate during prothrombin conversion, contains proteolytic activity toward the platelet receptor but at a fairly reduced rate (29, 30). For the activation and cleavage of PAR4, higher thrombin concentrations are required, in the order of 5 nM. Hence, it is considered that PAR1 cleavage by thrombin precedes PAR4 cleavage (23).
While human platelets express only very low copy numbers of PAR3, this isoform is more abundant on rodent platelets, where it functionally replaces the PAR1 isoform. Thus, on human platelets, PAR3 cleavage hardly contributes to the thrombin-induced responses (23). The other receptor family member, PAR2, is not known as a thrombin receptor, but rather as a cleavable receptor for the coagulation factors VIIa and Xa (31, 32). On platelets, it is lacking.
Another high-affinity receptor for thrombin is the GPIbα chain of the GPIb-V–IX complex (33). Upon thrombin-induced platelet activation, GPIbα is considered to cooperate with the two PAR receptors (34). However, thrombin binding can also facilitate another, this time proteolytic, effect on the GPIb-V–IX complex, namely by cleaving the extracellular domain of GPV (35). Interestingly, mice lacking GPV showed an increased platelet activation and arterial thrombosis formation for reasons that are not well-understood (36). The cleaved, soluble GPV fragment has been used as a marker of platelet activation in vivo (37).
Next to binding to platelet receptors proteins, thrombin carries out additional proteolytic events at the platelet surface. On activated, procoagulant platelets, thrombin cleaves coagulation factor V (whether or not platelet-derived) into factor Va, thus enhancing the prothrombinase activity (38). Furthermore, thrombin cleaves platelet-bound fibrinogen to produce fibrin polymers that form the shield of so-called coated platelets (39, 40). The latter process preferentially occurs on procoagulant platelets, secondary to processes like ballooning and phosphatidylserine exposure (39).
A poorly understood finding is that platelet activation with the TRAP6 peptide, similarly to α-thrombin but at a lower extent, can induce cleavage or PAR1 (Arg41-Ser42) in a manner antagonized by serpin but not by metalloproteinase inhibition (41). This may imply the presence of another trypsin-like protease implicated in TRAP6-induced platelet activation.
Factor Xa itself is generated (from non-cleaved factor X) at the surface of procoagulant, phosphatidylserine-exposing platelets, where it serves to produce thrombin (42, 43). The combined activities of factor Xa and thrombin can be measured in thrombin generation assays under stasis and are also detectable under flow conditions in whole blood assays (12, 44). Intriguingly, the factor Xa receptor PAR2, present on endothelial cells (45), does not play a significant role in human platelet activation (46). While for endothelial cells a receptor cross-talk has been reported of factor Xa mediating PAR1 cleavage (47), this is not known for platelets or other PAR1-overexpressing cells (23). A recent report stipulates that in plasma- and blood-based systems, the blockage of factor Xa with rivaroxaban inhibits a factor Xa-driven platelet activation pathway via PAR1 (48). However, it cannot be ruled out that, in this setting, thrombin itself is the receptor-cleaving protease.
Another not well-understood effect of factor Xa on platelets is its reported ability to mediate coagulation-dependent shedding of the extracellular domain of GPVI (49). Evidence for this effect came from the finding that this shedding of GPVI was blocked by factor Xa inhibition (rivaroxaban), rather than by thrombin inhibition (dabigatran, hirudin). Markedly, the receptor shedding is also sensitive to metalloproteinase inhibition, which can point to an indirect rather than a direct role of the factor Xa enzyme (50).
Another plasma protease that is proposed to modulate PAR1 cleavage is kallikrein. It has been reported that a kallikrein preparation via proteolysis can disarm or inhibit PAR1 signaling (51). A different proposal is that kallikrein first binds to integrin αIIbβ3 and then potentiates ADP-induced platelet activation through prior cleavage of PAR1 (52). The evidence for this mechanism so far is pharmacological.
Recombinant factor VIIa (NovoSeven) is incidentally used to treat patients with coagulation of platelet defects (53), for instance in patients with Glanzmann's thrombasthenia (54). The mechanism of factor VII's proteolytic activity is peculiar, since the idea is that factor VIIa (in the apparent absence of tissue factor) enhances the generation of factor Xa and thrombin at the platelet surface and thereby promotes thrombin-induced thrombus formation under flow (55, 56). Factor VIIa hence can play a role in the serine protease cascade at the platelet membrane.
The receptor chain GPIbα, due to its large and charged extracellular domain and its abundance on platelets, has been considered as a “sink” of several coagulation factors, i.e., next to (pro)thrombin, also factors VII(a), VIII, and XI (11). Binding of factor XI to GPIbα was identified as a proteolytic feedback pathway for thrombin generation in the setting of angiotensin-II-induced hypertension (57). Earlier reports on GPIbα-dependent factor XI binding and activation on platelets (58) were later partly modified by proposing that also the platelet ApoER2 receptors act as factor XI ligands (59). During the intrinsic coagulation pathway, factor XIa cleaves factor IX, but so far, no proteolytic events of factor IXa or XIa on platelet membrane proteins have been reported.
To become an active serine protease, plasmin needs to be cleaved from its precursor plasminogen by the enzymes tissue or urokinase plasminogen activator. Several reports indicate that this proteolytic cleavage is promoted at the surface of activated (phosphatidylserine-exposing) platelets. One proposal is that polyphosphates released from platelets allow binding of factor XII(a) on platelets, which, in some way, enhances plasminogen cleavage into plasmin (60, 61). Plasmin is a key enzyme in the fibrinolytic process, causing proteolytic fibrin degradation, but in addition, plasmin is known to act on platelets. Reports indicate that plasmin cleaves and activates the receptors PAR1 and/or PAR4, however at a distinct site as thrombin, thereby suppressing thrombin-induced platelet activation (62, 63). Plasmin inactivation is accomplished by serpins such as the plasmin activator inhibitor-I (PAI-1).
Concerning cathepsins, another class of plasma proteases secreted by leukocytes and other cells, only little is known of proteolytic effects on platelets. Most of the cathepsins act as cysteine proteases. It is described that the neutrophil-derived cathepsin G evokes platelet activation via the proteolytic cleavage of PAR4 (64). Similarly, the mesenchymal-cell-derived cathepsin K can activate the PAR4 receptor (65). On the other hand, platelets produce a broad-range inhibitor of cathepsins, called cystatin A, which was proposed to interfere in thrombus formation in a matrix metalloproteinase (MMP)-dependent manner; the proteolytic events here are still unresolved (66).
MMPs belong to the metzincin superfamily of zinc-dependent metalloproteinases. In the human genome, 23 different members of MMPs have been identified. Conventionally, these are grouped into the following classes: collagenases, gelatinases, stromelysins, matrilysins, and membrane-bound MMPs. The classification is based on the enzymes' substrates, the structural domains, and their pericellular localization (67). As an overall structure, MMPs are composed of a propeptide sequence, a catalytic domain, a hinge region, and a hemopexin domain. Many of the MMPs are secreted from cells, after which they remain at the cell membrane to carry out proteolytic functions (68).
In the cell, MMPs are produced as inactive zymogens (pro-MMPs), which require proteolytic removal of a propeptide sequence to become active. A result of this activation step is exposure of the enzyme's catalytic domain containing a Zn2+ ion (67). However, protection against undesired activity is still provided by a range of abundantly expressed tissue inhibitors of metalloproteinases (TIMPs), which avidly bind to the MMP catalytic domains. With respect to platelets, specific MMPs preferentially interact with TIMP isoforms, although there is a certain degree of redundancy. Other than the name “matrix metalloproteinase” suggests, most MMPs cleave not only extracellular matrix components but also other pericellular proteins (67), several of which regulate platelet functionality.
Of the 23 known MMPs, expression in human megakaryocytes and platelets on the transcript and/or protein level is confirmed for 13 isoforms (i.e., MMP1, 2, 3, 7, 9, 11, 12, 14, 15, 17, 19, 24, 25) (69, 70). In addition, all four TIMP isoforms are expressed, and these appear to be secreted along with the MMPs upon platelet activation (70–72). How the 13 expressed MMP isoforms contribute to the overall high MMP activity of (activated) platelets and to platelet function is still mostly unclear. At present, a role in platelets has been elucidated for the isoforms MMP1, 2, 9, 12, 13, and 14 (Supplementary Table 1).
MMP1 belongs to the collagenase class of MMPs and has a granular distribution in resting platelets (73). The latent pro-MMP1 requires processing into active MMP1 upon secretion (74). Once at the plasma membrane, (pro)MMP1 was found to colocalize with integrins αIIbβ3 and α2β1 (74, 75).
Active MMP1 is recognized as one of the several proteases that can cleave the N-terminal domain of the thrombin receptor, PAR1; the resulting fragment can activate PAR1 and trigger downstream signaling events (75). In agreement with the role of MMP1 in platelet activation, it was shown in two different in vivo thrombosis models using guinea pigs that inhibition of MMP1 suppressed intravascular thrombus formation and prolonged the time to vessel occlusion (75). Here, coadministration of a PAR1 antagonist did not increase the effects of MMP1 inhibition, thus suggesting overlap of the roles of MMP1 and PAR1. Similarly, in a human whole blood model of thrombus formation, the inhibition of MMP1 activity led to a reduction in platelet adhesion, aggregate formation, and P-selectin expression (73). Conversely, the treatment of human platelets with recombinant MMP1 potentiated thrombin-induced aggregation (74). While these findings collectively point to a functional role of MMP1 in cleaving the PAR1 receptors (Figure 2), the metalloproteinase may also stimulate and cleave other proteins on platelets.
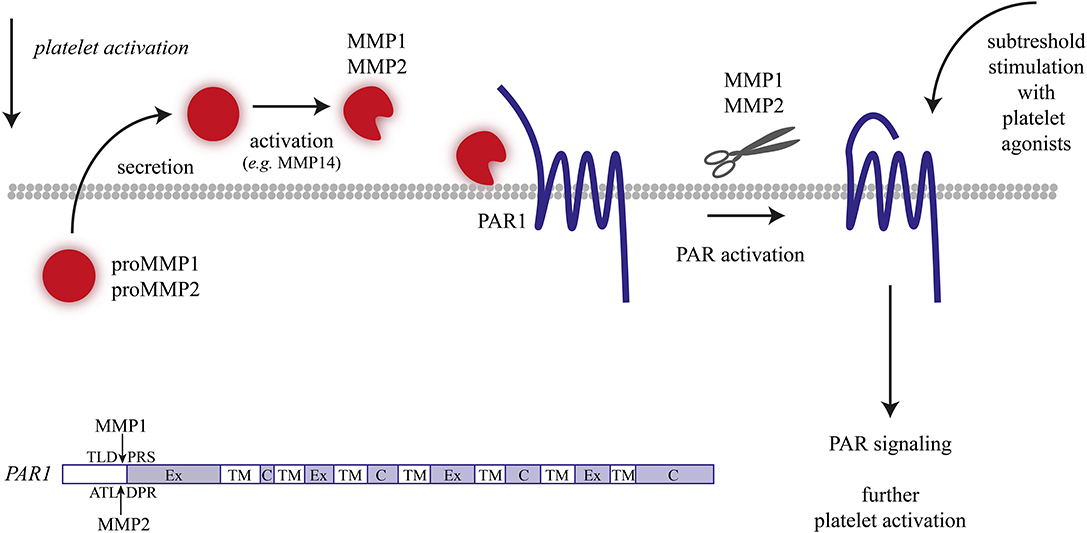
Figure 2. Matrix metalloproteinase (MMP)-1/2 induced platelet activation. Overview of mechanisms resulting in MMP-1/2 induced or enhanced platelet activation.
As a member of the gelatinase class, MMP2 is the most extensively studied MMP isoform in the context of platelet function. During platelet activation with collagen or thrombin, pro-MMP2 is released into the plasma and then translocates to the platelet surface (76, 77), where it binds to integrin αIIbβ3 (78). In vivo, the liberation of MMP2 has been detected at sites of vascular injury, in a manner relying on the extent of platelet activation (79). Along the same line, platelet inhibition by nitric oxide or prostacyclin appeared to inhibit the MMP2 release (76). While MMP2 itself is not considered as a platelet agonist, it enhances the activation process with subthreshold levels of agonists, as evidenced by an increased Ca2+ mobilization (76, 80). Evidence for such an enhancing role of MMP2 on platelet activation and (arterial) thrombus formation came from the use of MMP2 inhibitors and from mice with an MMP2 deficiency (76, 81). How these enhancing effects rely on proteolysis is still a matter of debate.
Several targets of MMP2 proteolysis have been identified, including the surface glycoproteins CD40L and CLEC2, of which extracellular soluble fragments are circulating in the plasma (82–84). Furthermore, MMP2 has been implicated in PAR1 activation by cleaving this receptor at a non-canonical site (i.e., different from the thrombin and MMP1 cleavage site), with integrin αIIbβ3 serving as a cofactor (Figure 2) (85). Cleavage generated a PAR1-activating peptide, which then stimulated signaling events via the G-proteins Gqα and G12/13α. However, the MMP2-generated peptide did not stimulate the Giα pathway, which was considered as an explanation why MMP2 did not elicit full platelet activation (85).
Whether or not the gelatinase MMP9 is expressed in platelets at relevant copy numbers is a matter of debate. Some research groups have detected measurable MMP9 activity in platelets (86, 87), while other groups could not confirm this finding (74, 88). Treatment of human platelets with recombinant MMP9 suppressed in a dose-dependent way thrombin- and collagen-induced aggregation, phosphoinositide breakdown, Ca2+ mobilization, and thromboxane A2 formation, which effects could be linked to cyclic guanosine monophosphate (cGMP) elevation and protein kinase C inactivation (86, 87). Similarly, platelet-inhibitory properties of (plasma-derived) MMP9 were seen in collagen-induced thrombus formation ex vivo (73). In addition, in mice, genetic deficiency of MMP9 resulted in the formation of larger thrombi under flow with higher levels of platelet activation in terms of P-selectin and phosphatidylserine exposure (73). If and how these effects rely on MMP9 proteolytic activity is unclear, and no definitive MMP9 targets are known so far. On the other hand, indirect evidence suggests a role of MMP9 in the extracellular cleavage of platelet CD40L (89).
Platelet-expressed MMP12 (a metalloelastase) has been reported to enhance collagen-induced aggregation and α-granule secretion but not aggregation by other agonists (90). Possibly linked to this finding, MMP12 can induce extracellular cleavage of the signaling receptor CAECAM1 at multiple sites, resulting in soluble fragments, of which one could reproduce the potentiating effect of MMP12 on collagen-induced activation (90). Other proteolytic activities of this protease on platelets are not reported.
There is limited evidence in the literature for a proteolytic role of the plasma-derived MMP13 (a collagenase) on platelet function (Supplementary Table 1). One group indicated that, at pathologically relevant concentrations, purified MMP13, added to whole blood, binds to integrin αIIbβ3 and GPVI and as such impairs human platelet aggregation and thrombus formation under flow (91). A distinct mechanism that was proposed is that MMP13-induced cleavage of VWF liberates a VWF product that more strongly interacts with platelets and partly loses its ability to adhere to collagen (92). In addition, MMP13 can (partially) degrade collagen, which may also alter the process of thrombus formation.
The isoform MMP14 is a membrane-type protease that remains tethered to the plasma membrane of resting and activated platelets. Its general function is supposed to be the breakdown of extracellular matrix components. Interestingly, upon GPVI-induced activation, caps of MMP14 were observed at the platelet surface, possibly indicating concentration of MMP activity (73). The MMP14 isoform likely cleaves a variety of protein substrates including pro-MMP2 and pro-MMP13 (93), which in turn alters platelet properties. Such a multisubstrate characteristic of MMP14 may also explain why inhibition of this isoform was found to increase collagen-dependent platelet activation and thrombus formation (73).
The ADAM proteases, as an abbreviation for “a disintegrin and metalloprotease,” belong to the adamalysin group of the metzincin metalloproteinases superfamily. Several of the ADAM isoforms have a profound influence on cellular functions, such as cell adhesion, migration, intracellular signaling, matrix degradation, and other proteolytic processes (94). As its name suggests, the ADAM molecular structure resembles that of other MMPs. Most ADAM proteins are made up of eight functional domains: a signal peptide domain; a prodomain present in the inactive form; a catalytic domain; disintegrin, cysteine-rich, and epidermal growth factor (EGF)-like domains (except for ADAM10 and ADAM17); a transmembrane domain; and a cytoplasmic tail (94, 95). Given the transmembrane localization with catalytic domain expressed extracellularly, ADAM-induced proteolysis occurs outside the cell surface near the plasma membrane.
Of the multiple ADAM domains, the catalytic domain—also called metalloprotease domain—is best known. It contains a highly conserved zinc-dependent endopeptidase motif with three histidine residues and a methionine turn in the active-site helix (94). Of the 21 presumed functional ADAMs, so far, 13 have been found to induce shedding of extracellular target (receptor) proteins. The other ADAM isoforms lack the endopeptidase motif and may be involved in transcellular cross-talk and cell adhesion via the disintegrin domain (94, 95). The common but not only mechanism of ADAM activation is the removal of the prodomain, thus liberating the Zn2+-dependent catalytic site.
In platelets, the isoforms ADAM10 and ADAM17 (also called TACE) are identified as major receptor sheddases of this family (96). Both ADAM10 and ADAM17 are single-pass transmembrane proteins that lack the EGF-like domain, although they only share 30% amino acid sequence identity. The list of recognized substrates for ADAM10 and ADAM17 is longer than that for other ADAM members (94), due to more extensive research.
Studies with cultured cells indicated that the expression of an active ADAM form at the cell surface relies on vesicular transport, cleavage of the prodomain by a convertase, and release by TIMPs blocking the ADAM active site (94). It is considered that ADAM10 is primarily blocked by TIMP1 and ADAM17 by TIMP3 (94).
In platelets, the isoforms ADAM10 and ADAM17 appeared to have a partly overlapping target spectrum (Figure 3). In vivo and in vitro studies using deficient mice revealed that ADAM10 preferentially cleaves the extracellular domain of GPVI on activated platelets (97), whereas ADAM17 rather targets GPIbα (97–99). On the other hand, in the GPIb-V–IX complex, the GPV chain can be cleaved by both enzymes. Other platelet receptors targeted by the ADAM isoforms are CD84, Sema7A, Sema4D, and JAM-A (99–103). In all these cases, cleavage will lead to an altered interaction of platelets with their environment. A related finding was the ability of ADAMs to act as a sheddase of platelet-bound amyloid precursor protein (APP) (104).
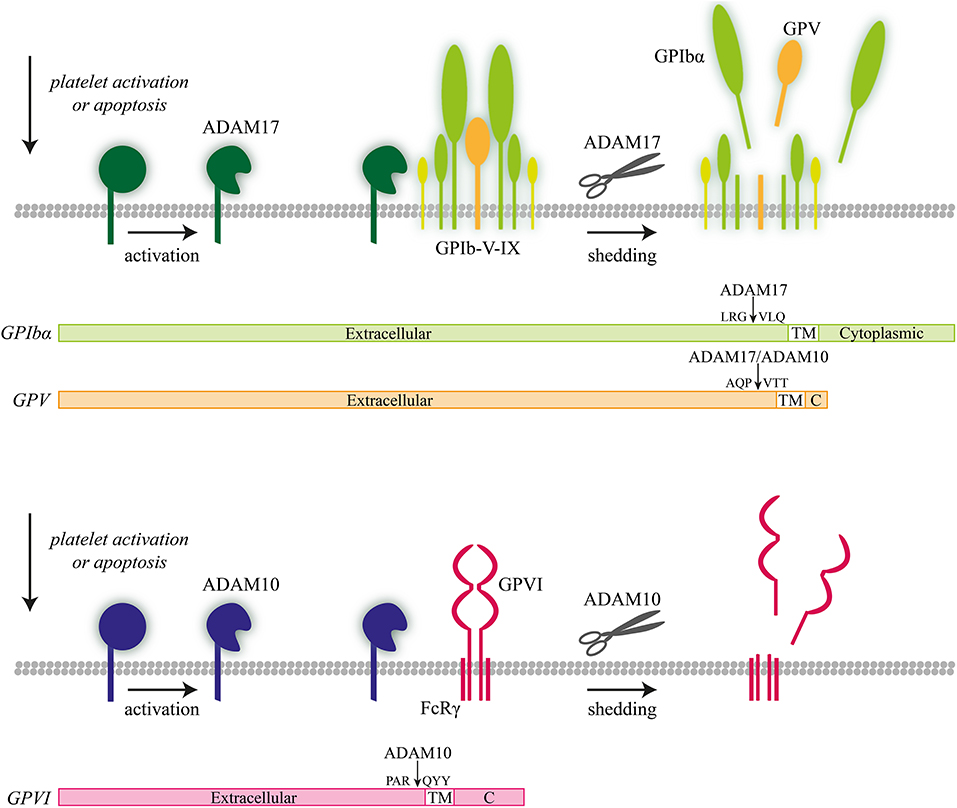
Figure 3. ADAM10- and ADAM 17-mediated proteolysis of platelet receptors. Illustration of ADAM10-mediated receptor shedding of glycoprotein V (GPV) and ADAM17-mediated receptor shedding of GPIbα with proteolytic cleavage sites indicated.
While constitutively active ADAM10 is already present on resting platelets, ADAM activity was found to drastically increase upon platelet activation (105). The signaling and activation mechanisms that lead to ADAM10- and ADAM17-mediated receptor shedding are unraveled only partly. Literature suggests that both extracellular and intracellular signals can enhance the proteolytic activity of ADAM forms (Supplementary Table 1). Recognized ADAM-activating agents that induce receptor shedding of GPIbα and/or GPVI include (i) inhibitors of the oxidative phosphorylation like CCCP (97, 98); (ii) inhibitors of protein kinase A (106); (iii) inhibitors of calmodulin (97, 107); (iv) reactive oxygen species (108); and (v) the coagulation product fibrin (109). Recently, our group has shown that ADAM-mediated receptor cleavage is confined to highly activated platelets (thrombin/CRP-XL), with, as a result, a defined population of phosphatidylserine-exposing platelets showing low GPIbα and GPVI expression (110). A further set of signaling processes that can trigger receptor shedding includes activation of protein kinase C (97, 110), p38 mitogen-activated protein kinase (108), and apoptosis (110, 111). Recently, it was shown that the Bruton's tyrosine kinase (Btk) inhibitor ibrutinib induced a time- and dose-dependent shedding of the GPIb-IX complex by increasing ADAM17 activation (112). Platelets from leukemia patients treated with ibrutinib showed reduced expression of the GPIb-IX complex, and also in mice, ibrutinib treatment resulted in elevated levels of soluble GPIbα (112).
In terms of function, it is assumed that ADAM10- and ADAM17-mediated receptor cleavage protects the platelets from a continued activation, by limiting receptor activity and the downstream signaling pathways (113). Alternatively, it can be stated that shedding of adhesive receptors such as GPIbα may steer removal from the circulation. However, in mice, platelet ADAM17 (TACE) deficiency reduced the levels of soluble GPIbα fragments in the plasma without affecting platelet count (98). Curiously, a decreased platelet ADAM10 level has been suggested as an early indicator for Alzheimer's disease. This relates to the fact that ADAM10, as an α-secretase, induces non-amyloidogenic proteolysis of the amyloid precursor protein, thus suppressing formation of the pathologic Aβ form (104, 114).
The isoforms of a disintegrin and metalloprotease with thrombospondin type 1 repeats (ADAMTS) are structurally highly similar to the ADAM proteases. While the enzymes of both families have in common a prodomain, catalytic, disintegrin, and cysteine-rich domains, the ADAMTS isoforms in addition contain one or more thrombospondin domains and a variable additional C-terminal domain (115). Since they lack a transmembrane domain, ADAMTS members do not function as integral membrane proteins. The most relevant isoform for platelet activation, ADAMTS13, contains seven thrombospondin domains and two CUB domains; it is secreted by liver cells into the circulation (115).
The proteolytic activity of ADAMTS13 affects platelet function in an indirect way, as it cleaves ultralarge multimers of VWF (residues Tyr1605 and Met1606), which are secreted by the endothelium (1). This cleavage weakens the interaction of VWF with platelets. For ADAMTS13 to become enzymatically active, vascular VWF multimers need to change into an elongated form, such as is triggered by high shear conditions (116). By implication, at sites of blood stasis, the ADAMTS13 activity is low. By shear-dependent cleavage of VWF multimers, ADAMTS13 has an important role in the hemostatic regulation. Thus, in patients with low ADAMTS13 activity, platelets become more easily activated by the ultralarge VWF multimers, which can lead to a condition of thrombotic thrombocytopenic purpura (TTP) (117). In patients with a congenital form of TTP (Upshaw–Schulman syndrome), recessive mutations of ADAMTS13 are present, while in patients with acquired TTP, autoantibodies are observed targeting ADAMTS13 (118). In addition, low ADAMTS13 activity has been associated with arterial thrombotic conditions such as stroke (119).
The calpain isoforms mu, 1 and 2, act as cytosolic proteases, which rely for their enzymatic activity on rises in Ca2+ (120). While studies in the early 1990s pointed to a role of calpains in (thrombin-induced) shape change, linked to actin cytoskeletal rearrangements (121), it later became clear that calpains regulate multiple signaling proteins including integrin αIIbβ3 (122–124). Calpains in particular appeared to play a role under conditions of prolonged rises in Ca2+ and contribute to agonist- or complement-induced shedding of microparticles (extracellular vesicles) (125, 126).
Regarding calpain receptor substrates in platelets, a marked finding was that calpains under certain, high-Ca2+ conditions can cleave the intracellular domain of the integrin β3 chain (Figure 4) (127). Markedly, this proteolysis was confined to β3 integrins of procoagulant, phosphatidylserine-exposing platelets (5). A recent proteomic neo N-terminal analysis of highly activated platelets (thrombin/convulxin or ionomycin) revealed up to 180 neo-N-terminal peptides sensitive to calpain inhibition, which corresponded to 106 proteins in particular cytoskeletal (filamin-A, talin-1, kindlin-3) and signaling proteins (128). Receptors cleaved by calpain included integrin β3, αIIb, and GPIbα, GPIbβ, GPIX, and CD226. Exactly which calpain isoform cleaves which intracellular proteins is still unclear.
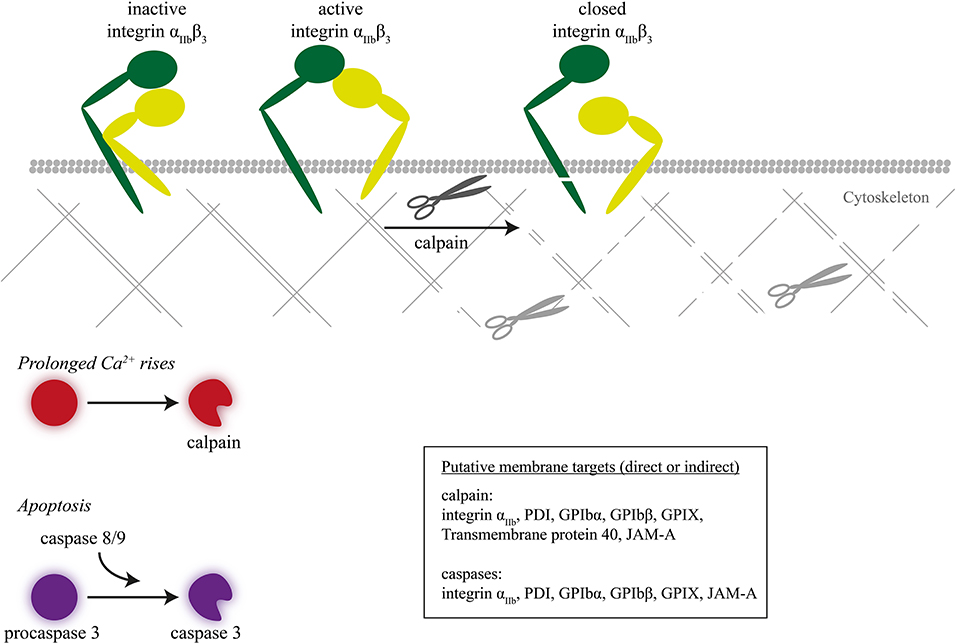
Figure 4. Inactivation of integrin β3 chain by intracellular proteolysis. Intracellular proteolysis mediated by calpains upon prolonged Ca2+ rise or by caspases during apoptosis leads to cleavage of the integrin β3 chain, next to cytoskeletal-linked signal proteins.
Caspases (cysteine-aspartic proteases) are a family of proteolytic enzymes controlling programmed cell death or apoptosis. Presently, 12 caspase isoforms are known in man, which carry out the degradation of intracellular proteins without causing tissue necrosis. Caspases, once activated by proapoptotic stimuli, selectively cleave target proteins after an aspartic acid residue. In apoptotic platelets, the caspase pathways are also associated with phosphatidylserine exposure (129). When applied to platelets, the caspase-activating compound ABT-737 (an inhibitor of mitochondrial Bcl-2/Bcl-xL) appeared to act largely Ca2+ independently, implying caspase activities operating apart from calpains (130). In apoptotic platelets, especially the caspase-9 isoform plays a key role (131). Proteomic neo N-terminal analysis of ABT-737 stimulated platelets revealed 23 peptides and proteins (receptor, cytoskeletal, and signaling) sensitive to caspase inhibition, all with cleavage sites distinct from those of calpains (128). Next to integrin-associated proteins (filamin-A, kindlin-3), these were the integrin β3 and αIIb chains, GPIbα and GPIbβ, i.e., cleavage sites compatible with a defective adhesive phenotype.
In general, for the majority of proteases discussed, the proteolytic activity toward platelets increases upon activation. Platelets simultaneously exposed to collagen and thrombin or to a Ca2+ ionophore are characterized by prolonged cytosolic Ca2+ rises and ensuing exposure of phosphatidylserine (132). It is generally recognized now that these procoagulant platelets form a distinct population, morphologically characterized by swelling, ballooning, and microvesiculation (11, 133). Both phosphatidylserine exposure and ballooning are absent in activated platelets from Scott syndrome patients, lacking the Ca2+-activated channel protein anoctamin-6 (gene ANO6 or TMEM16F), as well as in activated platelets from Ano6 deficient mice (134).
Emerging evidence indicates that the high-Ca2+, phosphatidylserine-exposing platelets are focal sites of high protease activities on receptors and other membrane-bound proteins. For instance, a high activity of MMPs and ADAMs has been reported on platelets stimulated with collagen plus thrombin (73, 110). This type of stimulation appeared to increase the ADAM-mediated shedding of GPIbα and GPVI by 2.5-fold in comparison to single agonist stimulation (110). Markedly, shedding occurred normally in platelets from Scott patients, indicating that phosphatidylserine exposure is not a prerequisite for ADAM activity. As another example, the high Ca2+ rises in the phosphatidylserine-exposing platelet population potently stimulated intracellular receptor and actin cytoskeletal proteolysis by calpains (128). In addition, the coagulation-related proteolytic events are linked to platelet phosphatidylserine exposure. Accordingly, these platelets greatly promote coagulation factor proteolysis via tenase (factor Xa) and prothrombinase (thrombin) complexes (11). Herein, the binding of coagulation factors and the formation of fibrin can be further enhanced by ADAM-mediated glycoprotein shedding (110). In hemophilic (bleeding) conditions, these proteolytic events are greatly lowered (1). Along the same line, anticoagulant drugs suppress these platelet proteolytic activities, via inhibition of both coagulation factor proteolysis and metalloproteinase-mediated proteolysis (49, 135). In addition, phosphatidylserine-exposing platelets concentrate the protease plasmin, which stimulates platelet-dependent fibrinolysis (60).
A similar concentration of proteolytic events may occur in platelets that undergo apoptosis, e.g., induced by the drug ABT-737. Apoptotic platelets undergo caspase-dependent proteolytic events and phosphatidylserine exposure (130), although their procoagulant activity may be limited (129, 136). An apoptosis-like process is seen in (cold) stored platelets. Herein, sheddases like ADAM17 can induce the extracellular cleavage of GPIbα, GPV, and GPVI chains (137, 138), in a way that a lower GPVI expression correlated with a poorer platelet responsiveness (139). By storage with protease inhibitors or in mouse platelets lacking ADAM17, the hemostatic function of transfused platelets was improved (98, 140, 141). These findings provide novel options to improve platelet responsiveness upon transfusion.
Conclusively, on platelets, a range of proteases from various families—coagulation-related proteases, plasmin, MMPs, ADAM(TS) isoforms, cathepsins, caspases and calpains—appear to selectively activate or inactivate key adhesive and signaling receptors, thereby controlling platelet responses and functions. Furthermore, the high receptor-targeted proteolytic activity on phosphatidylserine-exposing (procoagulant) platelets implies a downregulation of the fractions of adhesive and aggregating platelets. Although relevant knowledge is gained on the regulation of these platelet- and plasma-derived proteases and their implications for platelet activation, still a better understanding of the spatiotemporal proteolytic activity within and in between platelets is needed to fully understand the complexity of receptor proteolysis upon thrombus formation in thrombosis and hemostasis.
JW, JH, and CB performed a literature search and wrote and revised the manuscript. All authors have seen and approved the final manuscript.
JW was supported by the China Scholarship Council (Grant No. 201908440512). CB was supported by the Alexander von Humboldt Foundation.
JH is a shareholder and cofounder of FlowChamber.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2020.608391/full#supplementary-material
1. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. (2013) 93:327–58. doi: 10.1152/physrev.00016.2011
2. Van der Meijden PE, Heemskerk JW. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. (2019) 16:166–79. doi: 10.1038/s41569-018-0110-0
3. Baaten CCFMJ, Ten Cate H, van der Meijden PE, Heemskerk JW. Platelet populations and priming in hematological diseases. Blood Rev. (2017) 31:389-399. doi: 10.1016/j.blre.2017.07.004
4. Ruggeri ZM, Mendolicchio GL. Interaction of von WIllebrand factor with platelets and the vessel wall. Haemostaseologie. (2015) 35:211–24. doi: 10.5482/HAMO-14-12-0081
5. Mattheij NJ, Gilio K, van Kruchten R, Jobe SM, Wieschhaus AJ, Chishti AH, et al. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J Biol Chem. (2013) 288:13325–36. doi: 10.1074/jbc.M112.428359
6. Krishnaswamy S, Church WR, Nesheim ME, Mann KG. Activation of human prothrombin by human prothrombinase. Influence of factor Va on the reaction mechanism. J Biol Chem. (1987) 262:3291–9.
7. Rosing J, Tans G, Govers-Riemslag JW, Zwaal RF, Hemker HC. The role of phospholipids and factor Va in the prothrombinase complex. J Biol Chem. (1980) 255:274–83. doi: 10.1055/s-0039-1684696
8. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. (2007) 128:1173–86. doi: 10.1016/j.cell.2007.01.037
9. Li X, Sim MMS, Wood JP. Recent insights Into the regulation of coagulation and thrombosis. Arterioscler. Thromb Vasc Biol. (2020) 40:e119–25. doi: 10.1161/ATVBAHA.120.312674
10. Goettig P, Brandstetter H, Magdolen V. Surface loops of trypsin-like serine proteases as determinants of function. Biochimie. (2019) 166:52–76. doi: 10.1016/j.biochi.2019.09.004
11. Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. (2013) 11:2–16. doi: 10.1111/jth.12045
12. Swieringa F, Kuijpers MJ, Lamers MM, van der Meijden PE, Heemskerk JW. Rate-limiting roles of the tenase complex of factors VIII and IX in platelet procoagulant activity and formation of platelet-fibrin thrombi under flow. Haematologica. (2015) 100:748–56. doi: 10.3324/haematol.2014.116863
13. Swords NA, Mann KG. The assembly of the prothrombinase complex on adherent platelets. Arterioscler Thromb. (1993) 13:1602–12. doi: 10.1161/01.ATV.13.11.1602
14. Bevers EM, Comfurius P, Zwaal RF. Platelet procoagulant activity: physiological significance and mechanisms of exposure. Blood Rev. (1991) 5:146–54. doi: 10.1016/0268-960X(91)90031-7
15. Hajjar KA, Nachman RL. Human Endothelial Cell Plasmin-Generating System (Colman, R. W, et al.), Hemostassis and Thrombosis. Philadelphia, PA, USA: Lippincott Williams and Wilkins (2001). p. 700–9.
16. Huntington JA Serpin structure function and dysfunction. J Thromb Haemost. (2011) 9(Suppl. 1):26–34. doi: 10.1111/j.1538-7836.2011.04360.x
17. Loghmani H, Conway EM. Exploring traditional and nontraditional roles for thrombomodulin. Blood. (2018) 132:148–58. doi: 10.1182/blood-2017-12-768994
18. Mackie EJ, Pagel CN, Smith R, de Niese MR, Song SJ, Pike RN. Protease-activated receptors: a means of converting extracellular proteolysis into intracellular signals. IUBMB Life. (2002) 53:277–81. doi: 10.1080/15216540213469
19. Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. (2004) 279:13532–9. doi: 10.1074/jbc.M312090200
20. Brass LF. Thrombin and platelet activation. Chest. (2003) 124:18s−25s. doi: 10.1378/chest.124.3_suppl.18S
21. Swieringa F, Spronk HM, Heemskerk JW, van der Meijden PE. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–60. doi: 10.1002/rth2.12107
22. De Candia E. Mechanisms of platelet activation by thrombin: a short history. Thromb Res. (2012) 129:250–6. doi: 10.1016/j.thromres.2011.11.001
23. Bahou WF. Thrombin receptors. In: Michelson AD, editor. Platelets. Amsterdam: Elsevier (2007). p. 179–220. doi: 10.1016/B978-012369367-9/50771-0
24. Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. (2006) 99:1293–304. doi: 10.1161/01.RES.0000251742.71301.16
25. Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. (2005) 3:1800–14. doi: 10.1111/j.1538-7836.2005.01377.x
26. Norton KJ, Scarborough RM, Kutok JL, Escobedo MA, Nannizzi L, Coller BS. Immunologic analysis of the cloned platelet thrombin receptor activation mechanism: evidence supporting receptor cleavage, release of the N-terminal peptide, and insertion of the tethered ligand into a protected environment. Blood. (1993) 82:2125–36. doi: 10.1182/blood.V82.7.2125.bloodjournal8272125
27. Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. (1991) 64:1057–68. doi: 10.1016/0092-8674(91)90261-V
28. Soto AG, Smith TH, Chen B, Bhattacharya S, Cordova IC, Kenakin T, et al. N-linked glycosylation of protease-activated receptor-1 at extracellular loop 2 regulates G-protein signaling bias. Proc Natl Acad Sci USA. (2015) 112:E3600–8. doi: 10.1073/pnas.1508838112
29. Doyle MF, Mann KG. Multiple active forms of thrombin. IV. Relative activities of meizothrombins. J Biol Chem. (1990) 265:10693–701.
30. Smith RD, Owen WG. Platelet responses to compound interactions with thrombin. Biochemistry. (1999) 38:8936–47. doi: 10.1021/bi9827518
31. Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA. (2001) 98:7742–7. doi: 10.1073/pnas.141126698
32. Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X- dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci USA. (2000) 97:5255–60. doi: 10.1073/pnas.97.10.5255
33. Zarpellon A, Celikel R, Roberts JR, McClintock RA, Mendolicchio GL, Moore KL, et al. Binding of alpha-thrombin to surface-anchored platelet glycoprotein Ibα sulfotyrosines through a two-site mechanism involving exosite I. Proc Natl Acad Sci USA. (2011) 108:8628–33. doi: 10.1073/pnas.1017042108
34. Estevez B, Kim K, Delaney MK, Stojanovic-Terpo A, Shen B, Ruan C, et al. Signaling-mediated cooperativity between glycoprotein Ib-IX and protease-activated receptors in thrombin-induced platelet activation. Blood. (2016) 127:626–36. doi: 10.1182/blood-2015-04-638387
35. Ravanat C, Morales M, Azorsa DO, Moog S, Schuhler S, Grunert P, et al. Gene cloning of rat and mouse platelet glycoprotein V: identification of megakaryocyte-specific promoters and demonstration of functional thrombin cleavage. Blood. (1997) 89:3253–62. doi: 10.1182/blood.V89.9.3253
36. Ramakrishnan V, DeGuzman F, Bao M, Hall SW, Leung LL, Phillips DR. A thrombin receptor function for platelet glycoprotein Ib-IX unmasked by cleavage of glycoprotein V. Proc Natl Acad Sci USA. (2001) 98:1823–8. doi: 10.1073/pnas.98.4.1823
37. Ravanat C, Freund M, Mangin P, Azorsa DO, Schwartz C, Moog S, et al. GPV is a marker of in vivo platelet activation–study in a rat thrombosis model. Thromb Haemost. (2000) 83:327–33. doi: 10.1055/s-0037-1613807
38. Baruch D, Hemker HC, Lindhout T. Kinetics of thrombin-induced release and activation of platelet factor V. Eur J Biochem. (1986) 154:213–8. doi: 10.1111/j.1432-1033.1986.tb09381.x
39. Mattheij NJ, Swieringa F, Mastenbroek TG, Berny-Lang MA, May F, Baaten CC, et al. Coated platelets function in platelet-dependent fibrin formation via integrin αIIbβ3 and transglutaminase factor XIII. Haematologica. (2016) 101:427–36. doi: 10.3324/haematol.2015.131441
40. Berny MA, Munnix IC, Auger JM, Schols SE, Cosemans JM, Panizzi P, et al. Spatial distribution of factor Xa, thrombin, and fibrin(ogen) on thrombi at venous shear. PLoS ONE. (2010) 5:e10415. doi: 10.1371/journal.pone.0010415
41. Ofosu FA, Freedman J, Dewar L, Song Y, Fenton JW. A trypsin-like platelet protease propagates protease-activated receptor-1 cleavage and platelet activation. Biochem J. (1998) 336:283–5. doi: 10.1042/bj3360283
42. Haynes LM, Bouchard BA, Tracy PB, Mann KG. Prothrombin activation by platelet-associated prothrombinase proceeds through the prethrombin-2 pathway via a concerted mechanism. J Biol Chem. (2012) 287:38647–55. doi: 10.1074/jbc.M112.407791
43. Vanschoonbeek K, Feijge MA, Van Kampen RJ, Kenis H, Hemker HC, Giesen PL, et al. Initiating and potentiating role of platelets in tissue factor-induced thrombin generation in the presence of plasma: subject-dependent variation in thrombogram characteristics. J Thromb Haemost. (2004) 2:476–84. doi: 10.1111/j.1538-7933.2004.00618.x
44. Brouns SLN, van Geffen JP, Campello E, Swieringa F, Spiezia L, van Oerle R, et al. Platelet-primed interactions of coagulation and anticoagulation pathways in flow-dependent thrombus formation. Sci Rep. (2020) 10:11910. doi: 10.1038/s41598-020-68438-9
45. Senden NH, Jeunhomme TM, Heemskerk JW, Wagenvoord R, van't Veer C, Hemker HC, et al. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J Immunol. (1998) 161:4318–24.
46. Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA. (1998) 95:6642–6. doi: 10.1073/pnas.95.12.6642
47. Schuepbach RA, Riewald M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C receptor. J Thromb Haemost. (2010) 8:379–88. doi: 10.1111/j.1538-7836.2009.03682.x
48. Petzold T, Thienel M, Dannenberg L, Mourikis P, Helten C, Ayhan A, et al. Rivaroxaban reduces arterial thrombosis by inhibition of FXa-driven platelet activation via protease activated receptor-1. Circ Res. (2020) 126:486–500. doi: 10.1161/CIRCRESAHA.119.315099
49. Al-Tamimi M, Grigoriadis G, Tran H, Paul E, Servadei P, Berndt MC, et al. Coagulation-induced shedding of platelet glycoprotein VI mediated by factor Xa. Blood. (2011) 117:3912–20. doi: 10.1182/blood-2010-08-301523
50. Nieswandt B, Heemskerk JW. Dividing VI by X(a). Blood. (2011) 117:3704–5. doi: 10.1182/blood-2011-02-332742
51. Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I, Blaber M, Blaber SI, et al. Proteinase-activated receptors, targets for kallikrein signaling. J Biol Chem. (2006) 281:32095–112. doi: 10.1074/jbc.M513138200
52. Ottaiano TF, Andrade SS, de Oliveira C, Silva MCC, Buri MV, Juliano MA, et al. Plasma kallikrein enhances platelet aggregation response by subthreshold doses of ADP. Biochimie. (2017) 135:72–81. doi: 10.1016/j.biochi.2017.01.010
53. Ghorashian S, Hunt BJ. “Off-license” use of recombinant activated factor VII. Blood Rev. (2004) 18:245–59. doi: 10.1016/j.blre.2003.12.003
54. Poon MC, Di Minno G, d'Oiron R, Zotz R. New insights into the treatment of Glanzmann thrombasthenia. Transfus Med Rev. (2016) 30:92–9. doi: 10.1016/j.tmrv.2016.01.001
55. Hoffman M, Monroe DM, Roberts HR. Platelet-dependent action of high-dose factor VIIa. Blood. (2002) 100:364. doi: 10.1182/blood-2002-03-0736
56. Lisman T, Adelmeijer J, Cauwenberghs S, Van Pampus EC, Heemskerk JW, De Groot PG Recombinant factor VIIa enhances platelet adhesion and activation under flow conditions at normal and reduced platelet count. J Thromb Haemost. (2005) 3:742–51. doi: 10.1111/j.1538-7836.2005.01227.x
57. Kossmann S, Lagrange J, Jäckel S, Jurk K, Ehlken M, Schönfelder T, et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med. (2017) 9:eaah4923. doi: 10.1126/scitranslmed.aah4923
58. Yun TH, Baglia FA, Myles T, Navaneetham D, López JA, Walsh PN, et al. Thrombin activation of factor XI on activated platelets requires the interaction of factor XI and platelet glycoprotein Ib alpha with thrombin anion-binding exosites I and II, respectively. J Biol Chem. (2007) 282:29067. doi: 10.1074/jbc.M306925200
59. White-Adams TC, Berny MA, Tucker EI, Gertz JM, Gailani D, Urbanus RT, et al. Identification of coagulation factor XI as a ligand for platelet apolipoprotein E receptor 2 (ApoER2). Arterioscler Thromb Vasc Biol. (2009) 29:1602–7. doi: 10.1161/ATVBAHA.109.187393
60. Whyte CS, Swieringa F, Mastenbroek TG, Lionikiene AS, Lancé MD, van der Meijden PE, et al. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood. (2015) 125:2568–78. doi: 10.1182/blood-2014-09-599480
61. Mitchell JL, Lionikiene AS, Georgiev G, Klemmer A, Brain C, Kim PY, et al. Polyphosphate colocalizes with factor XII on platelet-bound fibrin and augments its plasminogen activator activity. Blood. (2016) 128:2834–45. doi: 10.1182/blood-2015-10-673285
62. Kimura M, Andersen TT, Fenton JW, Bahou WF, Aviv A. Plasmin-platelet interaction involves cleavage of functional thrombin receptor. Am J Physiol. (1996) 271:C54–60. doi: 10.1152/ajpcell.1996.271.1.C54
63. Quinton TM, Kim S, Derian CK, Jin J, Kunapuli SP. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J Biol Chem. (2004) 279:18434–9. doi: 10.1074/jbc.M401431200
64. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. (2000) 275:6819–23. doi: 10.1074/jbc.275.10.6819
65. Andrade SS, Gouvea IE, Silva MCC, Castro ED, de Paula CAA, Okamoto D, et al. Cathepsin K induces platelte dysfunction and affects cell signaling in breast cancer - molecularly distinct behavior of cathepsin K in breast cancer. BMC Cancer. (2016) 16:173. doi: 10.1186/s12885-016-2203-7
66. Mezzapesa A, Bastelica D, Crescence L, Poggi M, Grino M, Peiretti F, et al. Increased levels of the megakaryocyte and platelet expressed cysteine proteases stefin A and cystatin A prevent thrombosis. Sci Rep. (2019) 9:9631. doi: 10.1038/s41598-019-45805-9
67. Nigase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. (2006) 69:562–73. doi: 10.1016/j.cardiores.2005.12.002
68. Murphy G, Nagase H. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. (2011) 278:2–15. doi: 10.1111/j.1742-4658.2010.07918.x
69. Malara A, Ligi D, Di Buduo CA, Mannello F, Balduini A. Sub-cellular localization of metalloproteinases in megakaryocytes. Cells. (2018) 7:80. doi: 10.3390/cells7070080
70. Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood. (2011) 118:1903–11. doi: 10.1182/blood-2010-12-324517
71. Villeneuve J, Block A, Le Bousse-Kerdilès MC, Lepreux S, Nurden P, Ripoche J, et al. Tissue inhibitors of matrix metalloproteinases in platelets and megakaryocytes: a novel organization for these secreted proteins. Exp Hematol. (2009) 37:849–56. doi: 10.1016/j.exphem.2009.03.009
72. Radomski A, Jurasz P, Sanders EJ, Overall CM, Bigg HF, Edwards DR, et al. Identification, regulation and role of tissue inhibitor of metalloproteinases-4 (TIMP-4) in human platelets. Br J Pharmacol. (2002) 137:1330–8. doi: 10.1038/sj.bjp.0704936
73. Mastenbroek TG, Feijge MA, Kremers RM, van den Bosch MT, Swieringa F, De Groef L, et al. Platelet-associated matrix metalloproteinases regulate thrombus formation and exert local collagenolytic activity. Arterioscler Thromb Vasc Biol. (2015) 35:2554–61. doi: 10.1161/ATVBAHA.115.306153
74. Galt SW, Lindemann S, Allen L, Medd DJ, Falk JM, McIntyre TM, et al. Outside-in signals delivered by matrix metalloproteinase-1 regulate platelet function. Circ Res. (2002) 90:1093–9. doi: 10.1161/01.RES.0000019241.12929.EB
75. Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, et al. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. (2009) 137:332–43. doi: 10.1016/j.cell.2009.02.018
76. Sawicki G, Salas E, Murat J, Miszta-Lane H, Radomski MW. Release of gelatinase A during platelet activation mediates aggregation. Nature. (1997) 386:616–9. doi: 10.1038/386616a0
77. Kazes I, Elalamy I, Sraer JD, Hatmi M, Nguyen G. Platelet release of trimolecular complex components MT1-MMP/TIMP2/MMP2: involvement in MMP2 activation and platelet aggregation. Blood. (2000) 96:3064–9. doi: 10.1182/blood.V96.9.3064.h8003064_3064_3069
78. Choi WS, Jeon OH, Kim HH, Kim DS. MMP-2 regulates human platelet activation by interacting with integrin alphaIIbbeta3. J Thromb Haemost. (2008) 6:517–23. doi: 10.1111/j.1538-7836.2007.02871.x
79. Falcinelli E, Giannini S, Boschetti E, Gresele P. Platelets release active matrix metalloproteinase-2 in vivo in humans at a site of vascular injury: lack of inhibition by aspirin. Br J Haematol. (2007) 138:221–30. doi: 10.1111/j.1365-2141.2007.06632.x
80. Falcinelli E, Guglielmini G, Torti M, Gresele P. Intraplatelet signaling mechanisms of the priming effect of matrix metalloproteinase-2 on platelet aggregation. J Thromb Haemost. (2005) 3:2526–35. doi: 10.1111/j.1538-7836.2005.01614.x
81. Momi S, Falcinelli E, Giannini S, Ruggeri L, Cecchetti L, Corazzi T, et al. Loss of matrix metalloproteinase 2 in platelets reduces arterial thrombosis in vivo. J Exp Med. (2009) 206:2365–79. doi: 10.1084/jem.20090687
82. Reinboldt S, Wenzel F, Rauch BH, Hohlfeld T, Grandoch M, Fischer JW, et al. Preliminary evidence for a matrix metalloproteinase-2 (MMP-2)-dependent shedding of soluble CD40 ligand (sCD40L) from activated platelets. Platelets. (2009) 20:441–4. doi: 10.1080/09537100903096684
83. Inoue O, Osada M, Nakamura J, Kazama F, Shirai T, Tsukiji N, et al. Soluble CLEC-2 is generated independently of ADAM10 and is increased in plasma in acute coronary syndrome: comparison with soluble GPVI. Int J Hematol. (2019) 110:285–94. doi: 10.1007/s12185-019-02680-4
84. Choi WS, Jeon OH, Kim DS. CD40 ligand shedding is regulated by interaction between matrix metalloproteinase-2 and platelet integrin alpha(IIb)beta(3). J Thromb Haemost. (2010) 8:1364–71. doi: 10.1111/j.1538-7836.2010.03837.x
85. Sebastiano M, Momi S, Falcinelli E, Bury L, Hoylaerts MF, Gresele P. A novel mechanism regulating human platelet activation by MMP-2-mediated PAR1 biased signaling. Blood. (2017) 129:883–95. doi: 10.1182/blood-2016-06-724245
86. Sheu JR, Fong TH, Liu CM, Shen MY, Chen TL, Chang Y, et al. Expression of matrix metalloproteinase-9 in human platelets: regulation of platelet activation in in vitro and in vivo studies. Br J Pharmacol. (2004) 143:193–201. doi: 10.1038/sj.bjp.0705917
87. Fernandez-Patron C, Martinez-Cuesta MA, Salas E, Sawicki G, Wozniak M, Radomski MW, et al. Differential regulation of platelet aggregation by matrix metalloproteinases-9 and−2. Thromb Haemost. (1999) 82:1730–5. doi: 10.1055/s-0037-1614906
88. Kälvegren H, Jönsson S, Jonasson L. Release of matrix metalloproteinases-1 and -2, but not -9, from activated platelets measured by enzyme-linked immunosorbent assay. Platelets. (2011) 22:572–8. doi: 10.3109/09537104.2011.583300
89. Rahman M, Zhang S, Chew M, Syk I, Jeppsson B, Thorlacius H. Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J Thromb Haemost. (2013) 11:1385–98. doi: 10.1111/jth.12273
90. Wang J, Ye Y, Wei G, Hu W, Li L, Lu S, et al. Matrix metalloproteinase 12 facilitated platelet activation by shedding carcinoembryonic antigen related cell adhesion molecule 1. Biochem Biophys Res Commun. (2017) 486:1103–9. doi: 10.1016/j.bbrc.2017.04.001
91. Howes JM, Pugh N, Hamaia SW, Jung SM, Knäuper V, Malcor JD, et al. MMP-13 binds to platelet receptors αIIbβ3 and GPVI and impairs aggregation and thrombus formation. Res Pract Thromb Haemost. (2018) 2:370–9. doi: 10.1002/rth2.12088
92. Howes JM, Knäuper V, Malcor JD, Farndale RW. Cleavage by MMP-13 renders VWF unable to bind to collagen but increases its platelet reactivity. J Thromb Haemost. (2020) 18:942–54. doi: 10.1111/jth.14729
93. Itoh Y Membrane-type matrix metalloproteinases: their functions and regulations. Matrix Biol. (2015) 46:207–23. doi: 10.1016/j.matbio.2015.03.004
94. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. (2008) 29:258–89. doi: 10.1016/j.mam.2008.08.001
95. Benarroch EE. ADAM proteins, their ligands, clinical implications. Neurology. (2012) 78:914–20. doi: 10.1212/WNL.0b013e31824c4728
96. Matthews AL, Noy PJ, Reyat JS, Tomlinson MG. Regulation of a disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: the emerging role of tetraspanins and rhomboids. Platelets. (2017) 28:333–41. doi: 10.1080/09537104.2016.1184751
97. Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J Thromb Haemost. (2007) 5:1530–7. doi: 10.1111/j.1538-7836.2007.02590.x
98. Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, et al. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates GPIbα shedding from platelets in vitro and in vivo. Circ Res. (2004) 95:677–83. doi: 10.1161/01.RES.0000143899.73453.11
99. Montague SJ, Andrews RK, Gardiner EE. Mechanisms of receptor shedding in platelets. Blood. (2018) 132:2535–45. doi: 10.1182/blood-2018-03-742668
100. Hofmann S, Vögtle T, Bender M, Rose-John S, Nieswandt B. The SLAM family member CD84 is regulated by ADAM10 and calpain in platelets. J Thromb Haemost. (2012) 10:2581–92. doi: 10.1111/jth.12013
101. Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, et al. Deciphering the human platelet sheddome. Blood. (2011) 117:e15–26. doi: 10.1182/blood-2010-05-283838
102. Zhu L, Bergmeier W, Wu J, Jiang H, Stalker TJ, Cieslak M, et al. Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc Natl Acad Sci USA. (2007) 104:1621–6. doi: 10.1073/pnas.0606344104
103. Chen T, Xu DZ, Li Q, Mou P, Zeng Z, Brass LF, et al. The regulation of Sema4D exodomain shedding by protein kinase A in platelets. Platelets. (2016) 27:673–9. doi: 10.3109/09537104.2016.1154141
104. Canobbio I, Catricalà S, Balduini C, Torti M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim Biophys Acta. (2011) 1813:500–6. doi: 10.1016/j.bbamcr.2010.12.002
105. Facey A, Pinar I, Arthur JF, Qiao J, Jing J, Mado B, et al. A-disintegrin-and-metalloproteinase (ADAM) 10 activity on resting and activated platelets. Biochemistry. (2016) 55:1187–94. doi: 10.1021/acs.biochem.5b01102
106. Dai K, Yan R, Li S, Fan Y, Zhuang F, Ruan C. Prolonged inhibition of protein kinase A results in metalloproteinase-dependent platelet GPIbα shedding. Thromb Res. (2009) 124:101–9. doi: 10.1016/j.thromres.2008.12.044
107. Bender M, Hofmann S, Stegner D, Chalaris A, Bösl M, Braun A, et al. Differentially regulated GPVI ectodomain shedding by multiple platelet-expressed proteinases. Blood. (2010) 116:3347–55. doi: 10.1182/blood-2010-06-289108
108. Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res. (2009) 84:137–44. doi: 10.1093/cvr/cvp176
109. Montague SJ, Hicks SM, Lee CSM, Coupland LA, Parish CR, Lee WM, et al. Fibrin exposure triggers αIIbβ3-independent platelet aggregate formation, ADAM10 activity and glycoprotein VI shedding in a charge-dependent manner. J Thromb Haemost. (2020) 18:1447–58. doi: 10.1111/jth.14797
110. Baaten CC, Swieringa F, Misztal T, Mastenbroek TG, Feijge MA, Bock PE, et al. Platelet heterogeneity in activation-induced glycoprotein shedding: functional effects. Blood Adv. (2018) 2:2320–31. doi: 10.1182/bloodadvances.2017011544
111. Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. (2011) 118:1663–74. doi: 10.1182/blood-2011-04-347849
112. Dobie G, Kuriri FA, Omar MMA, Alanazi F, Gazwani AM, Tang CPS, et al. Ibrutinib, but not zanubrutinib, induces platelet receptor shedding of GPIb-IX-V complex and integrin αIIbβ3 in mice and humans. Blood Adv. (2019) 3:4298–311. doi: 10.1182/bloodadvances.2019000640
113. Berndt MC, Karunakaran D, Gardiner EE, Andrews RK. Programmed autologous cleavage of platelet receptors. J Thromb Haemost. (2007) 5(Suppl. 1):212–9. doi: 10.1111/j.1538-7836.2007.02484.x
114. Tang K, Hynan LS, Baskin F, Rosenberg RN. Platelet amyloid precursos protein processing: a biomarker for Alzheimer's disease. J Neurol Sci. (2006) 240:53–8. doi: 10.1016/j.jns.2005.09.002
115. Zhong S, Khalil RA. A disintegrin and metalloproteinase (ADAM) and ADAM with thrombospondin motifs (ADAMTS) family in vascular biology and disease. Biochem Pharmacol. (2019) 164:188–204. doi: 10.1016/j.bcp.2019.03.033
116. Crawley JT, de Groot R, Xiang Y, Luken BM, Lane DA. Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood. (2011) 118:3212–21. doi: 10.1182/blood-2011-02-306597
117. Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. (2001) 413:488–94. doi: 10.1038/35097008
118. Verbij FC, Fijnheer R, Voorberg J, Sorvillo N. Acquired TTP: ADAMTS13 meets the immune system. Blood Rev. (2014) 28:227–34. doi: 10.1016/j.blre.2014.07.004
119. Sonneveld MA, de Maat MP, Portegies ML, Kavousi M, Hofman A, Turecek PL, et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood. (2015) 126:2739–46. doi: 10.1182/blood-2015-05-643338
120. Kuchay SM, Chishti AH. Calpain-mediated regulation of platelet signaling pathways. Curr Opin Hematol. (2007) 14:249–54. doi: 10.1097/MOH.0b013e3280ef68f8
121. Ishii H, Suzuki Y, Kuboki M, Morikawa M, Inoue M, Kazama M. Activation of calpain I in thrombin-stimulated platelets is regulated by the initial elevation of the cytosolic Ca2+ concentration. Biochem J. (1992) 284:755–60. doi: 10.1042/bj2840755
122. Schoenwaelder SM, Yuan Y, Cooray P, Salem HH, Jackson SP. Calpain cleavage of focal adhesion proteins regulates the cytoskeletal attachment of integrin αIIbβ3 (platelet glycoprotein IIb/IIIa) and the cellular retraction of fibrin clots. J Biol Chem. (1997) 272:1694–702. doi: 10.1074/jbc.272.3.1694
123. Montsarrat N, Racaud-Sultan C, Mauco G, Plantavid M, Payrastre B, Breton-Douillon M, et al. Calpains are involved in phosphatidylinositol 3',4'-bisphosphate synthesis dependent on the aIIbb3 integrin engagement in thrombin-stimulated platelets. FEBS Lett. (1997) 404:23–6. doi: 10.1016/S0014-5793(97)00079-3
124. Yan B, Calderwood DA, Yaspan B, Ginsberg MH. Calpain cleavage promotes talin binding to the β3 integrin cytoplasmic domain. J Biol Chem. (2001) 276:28164–70. doi: 10.1074/jbc.M104161200
125. Wiedmer T, Shattil SJ, Cunningham M, Sims PJ. Role of calcium and calpain in complement-induced vesiculation of the platelet plasma membrane and in the exposure of the platelet factor Va receptor. Biochemistry. (1990) 29:623–32. doi: 10.1021/bi00455a005
126. Pasquet JM, Dachary-Prigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem. (1996) 239:647–54. doi: 10.1111/j.1432-1033.1996.0647u.x
127. Pfaff M, Du X, Ginsberg MH. Calpain cleavage of integrin beta cytoplasmic domains. FEBS Lett. (1999) 460:17–22. doi: 10.1016/S0014-5793(99)01250-8
128. Solari FA, Mattheij NJ, Burkhart JM, Swieringa F, Collins PW, Cosemans JM, et al. Combined quantification of the global proteome, phosphoproteome, and proteolytic cleavage to characterize altered platelet functions in the human Scott syndrome. Mol Cell Proteomics. (2016) 15:3154–9. doi: 10.1074/mcp.M116.060368
129. Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Yao Y, Mason KD, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. (2009) 114:663–6. doi: 10.1182/blood-2009-01-200345
130. Van Kruchten R, Mattheij NJ, Saunders C, Feijge MA, Swieringa F, Wolfs JL, et al. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood. (2013) 121:1850–7. doi: 10.1182/blood-2012-09-454314
131. White MJ, Schoenwaelder SM, Josefsson EC, Jarman KE, Henley KJ, James C, et al. Caspase-9 mediates the apoptotic death of megakaryocytes and platelets, but is dispensable for their generation and function. Blood. (2012) 119:4283–90. doi: 10.1182/blood-2011-11-394858
132. Siljander P, Farndale RW, Feijge MA, Comfurius P, Kos S, Bevers EM, et al. Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: involvement of p38 MAP kinase and calpain. Arterioscler Thromb Vasc Biol. (2001) 21:618–27. doi: 10.1161/01.ATV.21.4.618
133. Agbani EO, van den Bosch MTJ, Brown E, Williams CM, Mattheij NJA, Cosemans JMEM, et al. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation. (2015) 132:1414–24. doi: 10.1161/CIRCULATIONAHA.114.015036
134. Mattheij NJ, Braun A, van Kruchten R, Castoldi E, Pircher J, Baaten CC, et al. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling and protein cleavage. FASEB J. (2016) 30:727–37. doi: 10.1096/fj.15-280446
135. Ringwala SM, Dibattiste PM, Schneider DJ. Effects on platelet function of a direct acting antagonist of coagulation factor Xa. J Thromb Thrombolysis. (2012) 34:291–6. doi: 10.1007/s11239-012-0727-5
136. Kholmukhamedov A, Jobe SM. Necrotic but not apoptotic platelets are functionally procoagulant. Blood. (2018) 132:2420. doi: 10.1182/blood-2018-99-116972
137. Hosseini E, Ghasemzadeh M, Nassaji F, Jamaat ZP. GPVI modulation during platelet activation and storage: its expression levels and ectodomain shedding compared to markers of platelet storage lesion. Platelets. (2017) 28:498–508. doi: 10.1080/09537104.2016.1235692
138. Prudova A, Serrano K, Eckhard U, Fortelny N, Devine DV, Overall CM. TAILS N-terminomics of human platelets reveals pervasive metalloproteinase-dependent proteolytic processing in storage. Blood. (2014) 124:e49–60. doi: 10.1182/blood-2014-04-569640
139. Hosseini E, Beshkar P, Ghasemzadeh M. Reverse correlations of collagen-dependent platelet aggregation and adhesion with GPVI shedding during storage. J Thromb Thrombolysis. (2018) 46:534–40. doi: 10.1007/s11239-018-1739-6
140. Chen W, Liang X, Syed AK, Jessup P, Church WR, Ware J, et al. Inhibiting GPIbα shedding preserves post-transfusion recovery and hemostatic function of platelets after prolonged storage. Arterioscler Thromb Vasc Biol. (2016) 36:1821–8. doi: 10.1161/ATVBAHA.116.307639
Keywords: ADAM, calpain, caspase, coagulation factors, MMP, platelets, receptor proteolysis
Citation: Wu J, Heemskerk JWM and Baaten CCFMJ (2021) Platelet Membrane Receptor Proteolysis: Implications for Platelet Function. Front. Cardiovasc. Med. 7:608391. doi: 10.3389/fcvm.2020.608391
Received: 20 September 2020; Accepted: 24 November 2020;
Published: 08 January 2021.
Edited by:
Andreas Ludwig, RWTH Aachen University, GermanyReviewed by:
Marvin T. Nieman, Case Western Reserve University, United StatesCopyright © 2021 Wu, Heemskerk and Baaten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan W. M. Heemskerk, andtLmhlZW1za2Vya0BtYWFzdHJpY2h0dW5pdmVyc2l0eS5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.