 Ahmad Aljohmani
Ahmad Aljohmani Daniela Yildiz
Daniela Yildiz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 16 December 2020
Sec. Atherosclerosis and Vascular Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.608281
This article is part of the Research Topic Proteolysis of Membrane Proteins in Vascular Biology and Disease View all 7 articles
Despite recent advances in treatment strategies, infectious diseases are still under the leading causes of death worldwide. Although the activation of the inflammatory cascade is one prerequisite of defense, persistent and exuberant immune response, however, may lead to chronicity of inflammation predisposing to a temporal or permanent tissue damage not only of the site of infection but also among different body organs. The initial response to invading pathogens is mediated by the recognition through various pattern-recognition receptors along with cellular engulfment resulting in a coordinated release of soluble effector molecules and cytokines aiming to terminate the external stimuli. Members of the ‘a disintegrin and metalloproteinase’ (ADAM) family have the capability to proteolytically cleave transmembrane molecules close to the plasma membrane, a process called ectodomain shedding. In fact, in infectious diseases dysregulation of numerous ADAM substrates such as junction molecules (e.g., E-cadherin, VE-cadherin, JAM-A), adhesion molecules (e.g., ICAM-1, VCAM-1, L-selectin), and chemokines and cytokines (e.g., CXCL16, TNF-α) has been observed. The alpha-cleavage by ADAM proteases represents a rate limiting step for downstream regulated intramembrane proteolysis (RIPing) of several substrates, which influence cellular differentiation, cell signaling pathways and immune modulation. Both the substrates mentioned above and RIPing crucially contribute to a systematic damage in cardiovascular, endocrine, and/or gastrointestinal systems. This review will summarize the current knowledge of ADAM function and the subsequent RIPing in infectious diseases (e.g., pathogen recognition and clearance) and discuss the potential long-term effect on pathophysiological changes such as cardiovascular diseases.
Inflammation is a defensive process by which the immune system responds to an external or internal stimulus aiming to remove the injurious insult and initiate the healing process (1, 2). One major inflammatory insult is infection caused by viruses, prions, bacteria, fungi, and parasites. Besides the infectious particles themselves, their released toxins may cause severe disease (3).
The response to invading pathogens follows a well-defined cascade. The initial recognition of the pathogen occurs through germline-encoded pattern-recognition receptors (PRRs) such as the Toll-like receptors (TLRs), C-type lectin receptors, NOD-like receptors, retinoic acid-inducible gene I (RIG)-like receptors, cyclic GMP-AMP synthase and stimulator of interferon genes (cGAS_STING), and scavenger receptors which are not only expressed by immune cells but also non-immune/tissue cells such as endothelial and epithelial cells (4–6). The pathogens themselves provide pathogen-associated molecular pattern (PAMP) such as lipopolysaccharide, lipoteichoic acid, and flagellin as bacterial component as well as nucleic acids mostly associated with viral infection. Activation of the PRRs by PAMPs and the cellular uptake of the pathogens result in the release of danger molecules and cytokines such as interleukin-1β (IL-1β), IL-6, IL-18, and tumor necrosis factor-α (TNF-α) orchestrating the immune response and defense. Chemoattraction and activation of neutrophils, T cells, monocytes and dendritic cells promote antigen presentation, pathogen phagocytosis and immune modification (7, 8) allowing for pathogen clearance, resolution of inflammation, and initiation of tissue homeostasis restoration. However, uncontrolled inflammation and insufficient clearance may lead to chronic inflammation and permanent tissues damage in the worst case leading to organ failure and sepsis (9).
ADAMs belong to the class of metalloproteinases and consist of 34 members, in which 22 are well-described in human (10, 11). ADAMs are transmembrane molecules consisting of an N-terminal metalloproteinase pro-domain, followed by a disintegrin domain, an epidermal growth factor (EGF)-like domain, a cysteine-rich domain, a transmembrane domain and a cytoplasmic domain (12). As the EGF-like domain is missing in ADAM10 and ADAM17, the membrane-proximal domain is often referred to as stalk region (12, 13). Twelve of the human ADAM members (ADAM8, 9, 10, 12, 15, 17, 19, 20, 21, 28, 30, and 33) are proteolytically active upon removal of the pro-domain that blocks the zinc atom (14). The proteolytic cleavage of transmembrane substrates close to the cell surface is referred to as ectodomain shedding and results in the release of the soluble ectodomains. These ectodomains may act as agonists (e.g., TNF-α and EGF), driving the inflammatory surrogate, or antagonists (e.g., TNFR and IL-1R), decreasing the inflammatory responsiveness and driving the resolution of inflammation (for review see (15, 16). The transmembrane and the cytoplasmic domain remain as cell-associated fragment and can be further processed by regulated intramembrane proteolysis (RIPing) through, e.g., γ-secretases (17), as a downstream event. The resulting C-terminal intracellular domains (ICDs) fulfill divergent functions, e.g., acting as endogenous inhibitor [e.g., syndecans (18)] or transcription factor [e.g., Notch (19)]. Regulation of ADAMs occurs on the posttranslational rather than transcriptional level. Posttranslational regulation includes the cleavage of the inhibiting pro-domain by furin (e.g., ADAM9, 10, and 17) and autocatalytic activation (e.g., ADAM8), the change in cellular distribution, the release from intracellular stores, interaction with partners for cellular trafficking and signaling as well as conformational changes (15). Dysregulation of the proteolytic activity of ADAMs is a core factor in a number of pathologies such as cardiovascular disease, asthma, cancer, inflammation, and neurodegenerative disease (20–24). Although the general function of ADAMs in inflammatory diseases is well-described, their specific contribution in infectious diseases, especially in vivo (Table 2) is only partly understood. This review will summarize the current knowledge of ADAMs' function in infectious diseases addressing pathogen recognition and entrance, toxin handling, phagocytosis and clearance, cytokine release, leukocyte recruitment, resolution of inflammation and repair/regeneration, as well as local and systemic changes (Table 1). The RIPing may influence further signaling pathways leading to immune modulation. Both ADAM function and the subsequent RIPing may lead to long-term effects on pathophysiological changes causing, e.g., cardiovascular diseases.
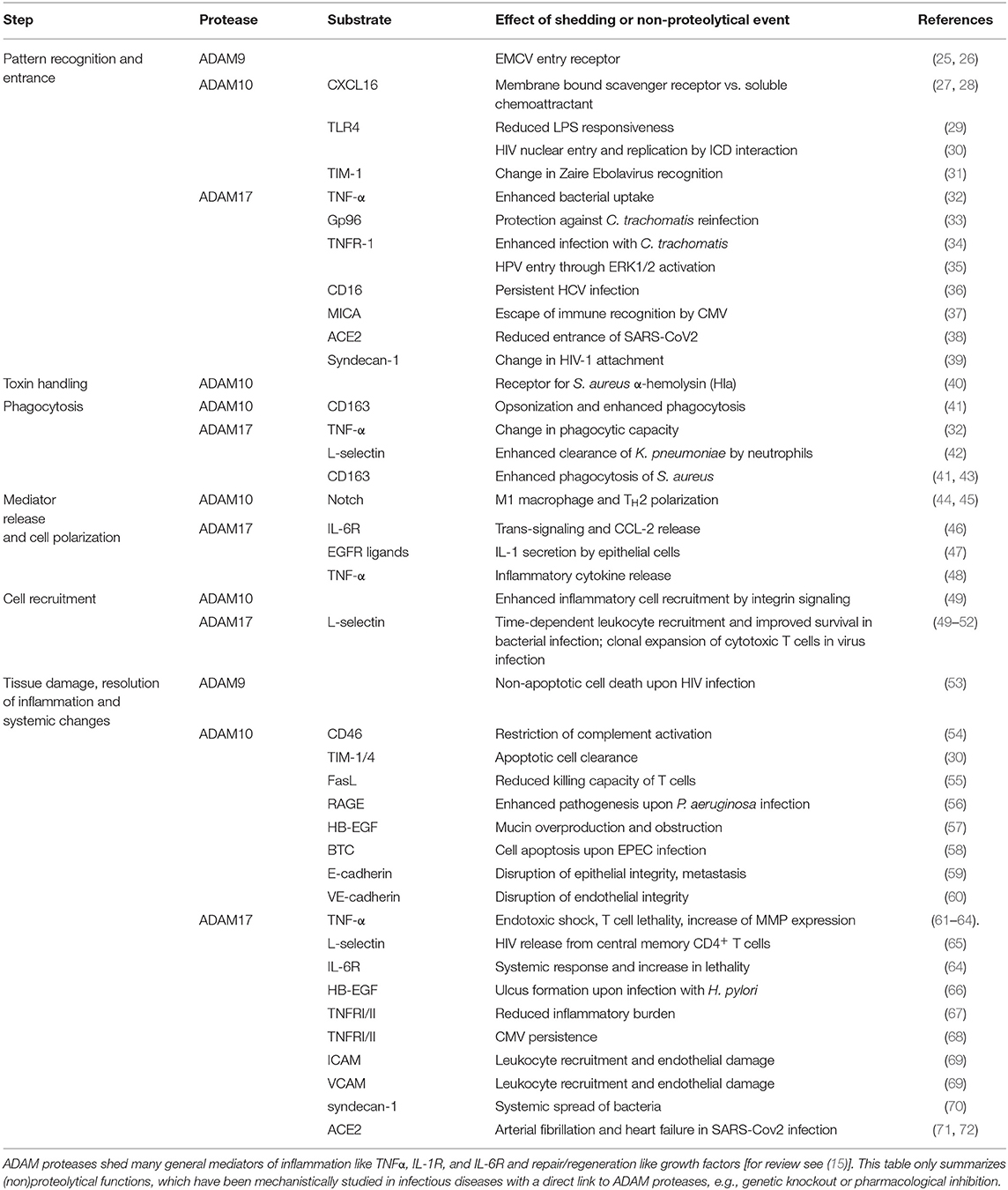
Table 1. Function of ADAM proteases in infection.
Pathogen recognition: Pathogen recognition is the first step of infection sensing. Cleavage of PRRs may cause [1] signaling initiation, [2] limitation of receptor-pathogen interaction, or [3] release of soluble scavenger receptor with opsonizing or antagonistic function. In the case of CD163, a scavenger receptor for both Gram-positive and Gram-negative bacteria (83), Staphylococcus aureus (S. aureus) leads to ADAM10-dependent release of soluble CD163, which binds to fibronectin on the bacterial surface resulting in further opsonization and higher clearance by phagocytes (41). Under certain conditions such as 1,25-dihydroxyvitamin D3 treatment, ADAM10 is able to shed TLR4 thereby limiting the inflammatory response to LPS (29). The third PRR is the receptor for advanced glycation end products (RAGE). Together with its secreted isoform (esRAGE), ADAM10-cleaved RAGE (cRAGE) builds the soluble fraction (sRAGE) (84), which has been shown to exert a detrimental action in Pseudomonas aeruginosa (P. aeruginosa) infection (56). However, a subsequent decrease of mature ADAM10 indicates a strict regulation and a time-dependent action of ADAM10 during P. aeruginosa infection (85). Besides its action in bacterial entry, ADAM10 might also be involved in viral recognition. T cell immunoglobulin and mucin domain (TIM)-1 and 4 are shed by ADAM10 (30), of which TIM-1 was shown to act as the receptor for the Zaire Ebola virus (31). Pathogen entrance/phagocytosis and toxin handling: ADAM10 may not only influence the initial pathogen interaction via cleavage of PRRs but also through its action as receptor itself. ADAM10 functions as a receptor for the S. aureus α-hemolysin (Hla) resulting in toxicity even at a low concentration (40). It was further shown that the intracellular domain of ADAM10 facilitates nuclear entry and replication of HIV-1 (86). CXCR6, the receptor for CXCL16 was previously identified as the HIV-1 coreceptor Bonzo (87). CXCL16, which is cleaved by ADAM10, exists as transmembrane form, mediating bacterial adhesion and phagocytosis, and soluble ectodomain with chemotactic function (27, 28, 88). Cell recruitment and polarization: ADAM10 is not only essential for the general recruitment of inflammatory cells (49), but also for the development of specific T cell responses. One crucial substrate of ADAM10, indicated by the lethality of ADAM10 knockout mice (81), is Notch which has divergent immune modulatory functions in infection. Upon infection with Listeria monocytogenes (L. monocytogenes), Notch downstream signaling results in pro-inflammatory polarization of macrophages to the classical M1 subtype (44). However, in allergic asthma induced by Alternaria alternata challenge, the T helper cell type 2 response was reduced upon Notch deficiency (45). Notch cleavage may also regulate invasiveness and epithelial to mesenchymal transition in epithelial cells infected with Epstein Barr virus (89). Tissue damage, resolution of inflammation, and systemic changes: There exist several evidences that ADAM10 does not only act in the initial phase of infection but is also involved in the development of tissue damage, the resolution of inflammation or development of systemic effects. E-cadherin and vascular endothelial (VE)-cadherin are junction molecules shed by ADAM10 in epithelial and endothelial cells, respectively (90, 91). Several pathogens, including P. aeruginosa, S. aureus and Helicobacter pylori (H. pylori) have been shown to induce cadherin shedding, thereby disrupting endothelial and epithelial barrier integrity leading to sepsis formation and persisting skin infection (59, 60, 73, 74, 92). Besides the mentioned molecules, shedding of growth factors such as heparin-binding EGF-like growth factor (HB-EGF) in epithelial cells may also contribute to persistent infection, e.g., through induction of mucin overproduction and obstruction (57). EGF receptor (EGFR) transactivation was further shown to induce epithelial cell apoptosis upon exposure to enteropathogenic E. coli (EPEC) (58). One essential step in the defense process and resolution of the concurrent infection is the removal of these apoptotic cells. Soluble TIM-1 and TIM-4 bind to phosphatidylserines on the surface of apoptotic cells, which could either prevent their engulfment or mediate phagocytosis by TIM-protein interaction (30). Furthermore, ADAM10 mediated shedding of Fas ligand (FasL) may reduce the killing capacity of T cells and their subsequent apoptosis, which could result in chronic inflammation (55). Additionally, ADAM10 sheds CD46 from apoptotic epithelial cells, which may reflect a strategy for restricted complement activation (54). As obvious, ADAM10 functions during different phases of infection (Tables 1, 2) and has to be tightly regulated via disease-specific mechanisms, which have to be taken into account when thinking about ADAM10 based treatment strategies.
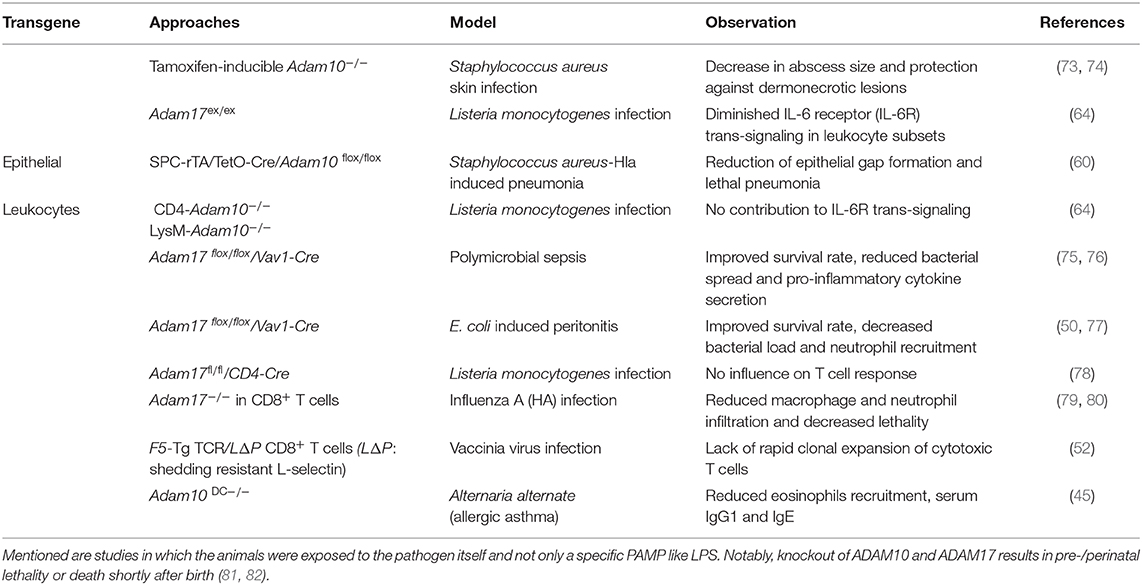
Table 2. Infectious diseases in ADAM knockout animals.
In addition to ADAM10, ADAM17 which is also known as tumor necrosis factor-α converting enzyme (TACE) (48) may play a divergent role during the different phases of bacterial, viral, and fungal infection. Pathogen recognition, entrance and phagocytosis: ADAM17 may influence the initial recognition and uptake of invading of pathogens by the cleavage of PRRs such as scavenger receptors and TLR2 and TLR4 (93, 94). However, it was recently shown that the limitation of bacterial uptake by ADAM17 in monocytes occurs in a cell-autonomous manner in parts dependent on TNF-α without significant changes in the surface expression of these receptors (32). Nevertheless, release of soluble CD163 from monocytes contributed to the recognition and phagocytosis of S. aureus (41, 43). In neutrophils, however, efficient bacterial clearance was dependent on ADAM17 activation and subsequent L-selectin shedding (42). Further, ADAM17 mediated shedding of the Chlamydia trachomatis receptor glucose regulated protein 96 (Gp96) protected against re-infection (33), whereas reduction of TNFRI expression on the cell surface facilitated the infection (34). With respect to viral infection ADAM17 facilitated the entry of the human papillomaviruses (HPV) (35), and led to downregulation of CD16 on natural killer (NK) cells during chronic hepatitis C virus (HCV) infection resulting in a lack of infection eradication (36). Furthermore, L-selectin was shown to support the preferential infection of central memory CD4+ T cells, with L-selectin shedding by ADAM17 being required for viral release (65). Cell recruitment and mediator release: ADAM17 sheds several junction and adhesion molecules like VCAM-1, ICAM-1 and JAM-A, which was shown to influence leukocyte recruitment and endothelial damage in both viral and sterile infection (69, 95, 96). Deficiency of ADAM17 in leukocytes resulted in enhanced recruitment of neutrophils to the site of infection, decreased bacterial load, and improved survival in both polymicrobial sepsis and peritonitis (50, 75, 77). Further investigations in models of sterile infection revealed a time-dependent effect of ADAM17 in neutrophil recruitment, which is dependent on L-selectin shedding at early time points (49–51). One essential step in the orchestration of the inflammatory response is the release of soluble mediators such as cytokines and growth factors. Stimulation of human airway epithelial cells with heat inactivated S. aureus and P. aeruginosa led to upregulation of ADAM17 surface expression and colocalization with IL-6R, initiating receptor trans-signaling and subsequent CCL-2 release (46). Similarly, ADAM17-mediated transactivation of EGFR was required for the upregulation of IL-1 in primary keratinocytes infected with S. aureus (47). The analysis of BAL samples from patients with community-acquired pneumonia showed that ADAM17 is involved in both the release of pro-inflammatory cytokines like TNF-α and IL-1β as well as their neutralizing soluble receptors (97). Furthermore, release of TNF-α by CD8+ T cells was essential for CXCL2 release and leukocyte recruitment upon influenza A infection (79). Additionally, L-selectin shedding promoted early clonal expansion of CD8+ T cell derived cytotoxic T cells in vaccinia virus infection (52). Tissue damage, resolution of inflammation, and systemic changes: Released mediators and their receptors do not only orchestrate the local immune response but are responsible for the systemic immune response resulting in either resolution of inflammation or in chronicity and damage of other organs. TNF-α shedding has been shown to be crucial for tissue damage and lethality in endotoxic shock, S. pneumoniae (serotype 3) caused meningitis, and L. monocytogenes infection, the latter one being further accompanied by IL-6R trans-signaling (61–64, 98). TNF-α is central during HIV infection and AIDS pathogenesis, in which a complex regulatory mechanism was observed. Nef induces both endocytosis of ADAM17, resulting in intracellular TNF-α cleavage, and ADAM17 release on extracellular vesicles, leading to release of TNF-α upon ingestion by PBMC, thereby decreasing the number of circulating CD4 lymphocytes (99, 100). Thus, the amount of circulating TNF-α has to be tightly regulated, e.g., through its soluble receptors TNFRI and TNFRII, which are shed by ADAM17 (67). On the one hand, enhanced shedding of TNFRs may reduce the inflammatory burden (80); on the other hand it may lead to reduced defense and persistent infection (68). A similar divergent function may apply for growth factor shedding. On the one hand, HB-EGF–mediated EGFR activation was shown to contribute to gastrointestinal ulcera and cancer formation upon H. pylori (66, 101, 102). On the other hand, TGF-α was required for β-defensins-3 (hBD-3) up-regulation upon Candida albicans infection (103). Escape mechanisms: ADAM17 does not only orchestrate the immune response but is also used by pathogens to escape the defense mechanisms. CMV promotes the shedding of MICA, an NK group 2D (NKG2D) receptor ligand, mimicking the tumor strategy in modulating immune recognition by NK cells and cytotoxic T cells (37). Furthermore, ADAM17 proteolytically cleaves the Ebola virus (EBOV) surface glycoprotein GP, which is known to block the antibodies responsible for virus neutralization (104). Thinking about treatment options, e.g., for polymicrobial sepsis (105), ADAM17 might be a suitable target in infectious diseases. However, due to its divergent functions (Tables 1, 2), as further addressed in the COVID-19 section, a substrate-specific, time-specific and site-specific therapeutic intervention has to be considered to achieve the best beneficial effect and reduction of severe side-effects.
ADAM10 and ADAM17 as ubiquitously expressed proteases seem to play a central function in infectious diseases. Only a few studies have addressed the role of ADAM8. ADAM8 is less abundantly expressed with highest expression in leukocytes and carcinoma cells and seems to be dispensable for normal development and homeostasis (106, 107). In sterile inflammation caused by E. coli LPS, mainly leukocytic ADAM8 contributed to leukocyte recruitment in the acute phase of lung inflammation (108). ADAM8 protein expression was found to be upregulated during the formation of multinuclear giant cells by human salivary gland cells upon infection with human parainfluenza virus type 2 (HPIV2) (109). Further, macrophages of HIV-1 positive patients showed an upregulation of ADAM8 gene expression. In patients with periodontal diseases, which are mainly induced by bacteria and characterized by uncontrolled inflammation, a high protein expression ADAM8 was found in the gingival crevicular fluid, highly correlating with disease severity and destruction of the periodontal tissues (110). After non-surgical periodontal therapy, ADAM8 levels decreased and correlated with improvements in clinical parameters (111). Not only viral and bacterial infection may be influenced by ADAM8, but also infection by fungi. Transcriptional profiling of heart tissues mice infected with Candida albicans revealed an upregulation of ADAM8 gene expression, which was linked to cardiomyopathy and extracellular matrix remodeling (112). Thus, it seems feasible that ADAM8 plays a more important role in disease progression and chronicity vs. resolution of inflammation rather than in pathogen recognition and toxin handling.
ADAM9 expression was clearly shown in the brain, the lung, the developing heart, and the retina as well as several cell types such as fibroblasts, neutrophils, and platelets (15). Although ADAM9 is less abundantly expressed than ADAM10 and ADAM17, it is involved in several steps of inflammation, including cytokine release, neutrophil activation, endothelial transmigration, and growth factor signaling (15, 113–115). Enhanced mRNA expression in the blood, which was mostly restricted to monocytes and neutrophils, has been found in patients with influenza caused pneumonia, respiratory syncytial virus (RSV) infection, active tuberculosis, and several other bacterial infections, whereas stimulation with PAMPs only marginally increased ADAM9 transcript abundance compared to healthy volunteers [for review see Rinchai et al. (116)]. Further, ADAM9 mRNA expression increased in cystic fibrosis patients during acute exacerbation, with its decline being an independent predictor of the antibiotic treatment response (117).
Mechanistic studies in infection highlight the relevance of ADAM9 during viral infection from pathogen entrance to systemic changes. ADAM9 was shown to be an important and specific factor for encephalomyocarditis virus (EMCV) entry, which was inhibited upon ADAM9 knockout in myeloid leukemia derived cells and HEK293T cells (25, 26). Interestingly, pharmacological inhibition of ADAM9 did not show any effect, suggesting a non-proteolytical function of ADAM9 as EMCV receptor (25). Furthermore, ADAM9 mRNA was highly expressed in central memory CD4 T cells during chronic HIV infection, which associated with movement to cell cycle phases G1 and S leading to non-apoptotic cell death (53). ADAM9 knockout protected against remodeling processes like elastin degradation and subsequent reduction in lung compliance in LPS-induced ALI (118). Thus, it seems feasible that ADAM9 is not only involved in the acute phase of infection but also in the decision between resolution of inflammation, chronicity, and systemic changes. Further evidences for systemic changes upon upregulation of ADAM9 are given by enhanced development of metastases during hepatitis B virus-related hepatocellular carcinoma (119).
Although 22 ADAM proteases have been reported in humans, only 12 display catalytic activity based on the presence of the zinc-dependent metalloproteinase domain (14). ADAM10 and ADAM17 together with ADAM8 and ADAM9 seem to be the most relevant ADAM proteases in infection; however, some studies point toward an additional influence of ADAM12, ADAM15, and ADAM33. Two ADAM12 genetic mutations (rs11244787 and rs1871054) led to an increased risk to transfer an infection with Trypanosoma cruzi, a parasitic euglenoids, from the mother to the newborn (120). Single nucleotide polymorphism (SNP) analysis revealed that an ADAM33 SNP was associated with a higher risk to develop severe RSV bronchiolitis with airway remodeling in premature infants (121, 122). In addition, children with acute infectious pneumonia of viral etiology showed an increased protein expression of ADAM28 and ADAM33 in the lung, which was correlated with the severity of infection and is likely to be involved in the chronicity and fibrosis development (123). So far, the contribution of ADAM15 has been only investigated in models of sterile infection. In both, LPS-induced ALI and sepsis ADAM15 deficiency protected against vascular endothelial barrier dysfunction, resulting in reduced neutrophil transmigration (124, 125). Further, in vitro studies revealed a more complex action of ADAM15 in inflammation and infection. Whereas, negative regulation of ADAM15 mediated by microRNA-147b significantly improved endothelial barrier dysfunction (126), ADAM15 dampened the TLR3 and TLR4 stimulated pro-inflammatory cytokine production (127). Thus, further studies are required to specify the stimulus- and cell-type specific contribution of single ADAM proteases in infectious diseases.
Not only the released ectodomains but also the remaining transmembrane and intracellular fragments may regulate the infectious response. Intramembrane cleaving proteases, such as the γ-secretase presenilin 1 (PSEN1), liberate the ICD which might act as antagonist [e.g., syndecan-1 and 4 (18)] or translocate to the nucleus acting as transcription factor [e.g., Notch (128, 129). Prerequisite to γ-secretase action is the previous cleavage by α-secretases (e.g., ADAM9, ADAM10, and ADAM17 (130)] or β-secretases [e.g., BACE (131)]. The function of the Notch ICD (NICD) as transcriptional regulator is widely accepted (132). The NICD translocates to the nucleus, where it binds to the DNA-binding protein RBP-J with subsequent recruitment of superenhancers initiating transcription of Notch downstream targets like Hes5 (133). This does not only regulate transcriptional activity but may also regulate on the level of translation. It was shown that Notch1-RBP-J signaling is required for the activity of the MNK1/eIF4E axis, which controls the rapid induction of IRF8 upon TLR4 stimulation on the protein level. Thereby, Notch1 controls the IFR8 dependent expression of M1 macrophage-associated genes like Il12a and Nos2 (44). Syndecan-1 and 4 are subjected to constitutive and inflammation-induced shedding by ADAM17 (134). However, it seems that syndecan-1 plays a more prominent role in infectious diseases, which is already obvious from the huge variety of interaction partners including matrix proteins, proteases, cytokines, and growth factors [for review see (135)]. P. aeruginosa, S. aureus and S. pneumoniae were shown to induce syndecan-1 shedding correlating with susceptibility and systemic spread (70, 136, 137), and syndecan-1 serves as attachment receptor for HIV-1 (39). It has been reported that the SICD might acts inhibitory during tumor cell migration and that it might be essential for exosomal release (18, 135); however, a specific function in infectious disease has not been reported so far. Similarly, it is obvious from the mentioned studies that E-cadherin plays in important role in acute and chronic infectious diseases and has been recently reviewed (138). The EICD is associated with beta-catenin and α-catenin and thereby linked to the cytoskeleton. The disassembly of this complex upon ectodomain shedding and RIPing releases β-catenin, thereby orchestrating the canonical Wnt signaling pathways as integral component of the host response to infection (139, 140). However, it is important to note that E-cadherin is cleaved not only by ADAM10 and other metalloproteinase but also by bacterial proteases themselves. If these remaining transmembrane fragments are subjected to RIPing is still under investigation (138). Furthermore, the E-cadherin c-terminal fragment 2, produced by PSEN1, was shown to increase the degradation of transmembrane amyloid precursor protein (APP) derivatives, preventing the formation of the AICD and promoting the non-amyloidogenic degradation (141). Many in vitro and in vivo evidences including the analysis of patients' samples point toward a contribution of infection to the late-onset of Alzheimer disease. A strong association was shown for infection with Chlamydia pneumoniae (C. pneumoniae) [for review see (142)]. Infection of astrocytes with this intracellular pathogen showed an increase of BACE and PSEN1 expression, whereas the activity of ADAM10 was reduced, resulting in enhanced production of beta-amyloid (143). However, the overall mechanisms how infection contributes to late-onset of Alzheimer disease is still not clear. Apart from its sheddase activity, RIPing of ADAM10 itself has been reported. In macrophages infected with HIV-1, ADAM10, cleavage by ADAM15 resulted in the release of the intracellular domain by RIPing, mediating the above describe support of viral translocation (86, 144). Besides γ-secretase, which belongs to the aspartyl intramembrane proteases, rhomboids, site-2 protease, and Ras-converting enzyme 1 are intramembrane proteases. However, due to their subcellular localization these proteases are not involved in RIPing of ADAM substrates (145). Thus, there are clear evidences that RIPing by γ-secretase plays an important function in infectious diseases; however, little is known about the exact consequences of these processes.
There are only a few examples, in which a direct pathogen-ADAM protease-target molecule axis has been established. Besides ADAM10/S. aureus (40) and ADAM9/EMCV (25, 26), the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing COVID-19 is the most prominent example, showing the diverse protective and progressive action of ADAM proteases in infectious diseases. The extracellular domain of angiotensin converting enzyme II (ACE2), a crucial shedding substrate for ADAM17 (146), functions as cellular receptor for the virus' spike (S) protein (147). ACE2 is highly expressed in the lung and myocardium, and it has been speculated that ADAM17 overexpression could play a milestone protective role through mediating shedding of ACE2 leading to downregulation of its surface expression, inhibiting the key entrance of SARS-CoV-2 and further blocking circulation virus particles (71). After ligation of ACE2 with the spike protein, a proteolytic cut of the S glycoprotein mediated by furin is a crucial step for virus entry (148). One mediator regulating furin expression on the transcriptional level is Notch1 (149). Both, ADAM10 and ADAM17 are able to shed Notch1 and thereby activate down-stream signaling processes. However, cleavage occurs context dependent, with ADAM10 being responsible for ligand-induced signaling, whereas ligand-independent signaling requires ADAM17 (150). Ectodomain shedding of Notch1 is followed by γ-secretase cleavage leading to regulation of Notch targeting genes such as furin and enhancement of ADAM10 activity (149). On the other hand, Notch1 has been shown to function as negative regulator of ADAM17 through the transcription of miRNA-145 (151). Thus, inhibition of ADAM10/ADAM17 in intending to block Notch signaling resulting in downregulation of furin expression and its activation circle may serve as an important approach to prevent viral entry and SARS-CoV-2 infection (38). On the other hand, ADAM17 is the main sheddase of TNF-α and further involved in IL-6R shedding (48). Both, IL-6 and TNF-α are important components of the cytokine storm observed in COVID-19 patients (152). With respect to TNF-α, inhibition of ADAM17 as preventive mechanism could be desired. However, the effect on IL-6 signaling would be limited to ADAM17-dependent IL-6R trans-signaling but would not affect the binding of IL-6 to membrane-bound IL-6R or soluble IL-6R derived from alternative mRNA splicing (153, 154). Furthermore, ACE2 depletion on the cell surface is a key pathological feature of SARS-CoV-2 infection, disabling its protective role and consequently resulting in cardiovascular diseases such as atrial fibrillation and heart failure (72). Furthermore, down-regulation of ACE2 severely worsened the outcome in SARS-CoV infection (155), and pharmacological inhibition and gene silencing of ADAM17 showed a markedly decrease of infection in in vitro and in vivo models (156). Thus, the role of ADAM proteases during SARS-CoV-2 infection is controversy and has to be carefully evaluated for potential treatment options.
As mentioned above, local infection may cause severe side-effects in other organs or the whole body, such as sepsis formation upon infection with P. aeruginosa (157) and the development of hepatocellular carcinoma metastases upon hepatitis B virus infection (158). The most prominent example is in these days the development acute coronary syndromes, arrhythmias, exacerbation of heart failure, thromboembolism, and myocarditis upon SARS-CoV-2 infection, which may be related to the hyper-inflammatory response [for review see (159)]. TNF-α is one of the most essential cytokines that coordinates the host response against intracellular pathogens, including apoptosis for pathogen deposition, upregulation of adhesion molecules on endothelial cell and leukocytes for inflammatory cell recruitment and initiation of the systemic response and the action as negative regulator through the interaction with TNFRs (160, 161). Although this activity is tightly regulated, constantly elevated levels of TNF-α (e.g., chronic infection or cytokine storm) may cause detrimental vascular damage through the induction of endothelial apoptosis, vascular remodeling, pro-coagulatory effects such as enhanced expression of tissue factor, oxidative stress, and recruitment of inflammatory cells while reducing NO production and the number of regenerating stem cells [for review see (162, 163)]. Several studies have shown the direct link between infection and atherosclerosis development, e.g., with C. pneumonia and Pyrophyromonas gingivalis (164, 165). Based on autoimmune diseases, it is widely discussed if anti-TNF-α therapy could be used as treatment option in cardiovascular disease, but further studies are required for proof of concept (162). It is important to note that such a treatment would again enhance the susceptibility for infection (166). Besides TNF-α, ACE2 plays an essential role in SARS-CoV infection. As a consequence of infection, ACE2 is downregulated on the surface accompanied by enhanced levels of angiotensin II (Ang II) and activation of the renin-angiotensin system (167). It was shown that the conversion of Ang II to Ang 1-7 by ACE2 has cardioprotective function and that the described dysregulation of ACE2 is not unique for SARS-CoV-2, thus prompting toward administration of soluble ACE2 to prevent organ failure (168). Similar divergent functions of Notch activation in cardiovascular damage have been described. On the one hand, Notch exerts a pro-inflammatory action through the polarization of inflammatory cells and the production of IL-6 through the delta-like ligand 4/Notch1 axis (44, 45). On the other hand, it was shown that ADAM10-dependent signaling through Notch1 and Notch4 cleavage controls organ-specific vascular bed formation (169) and could be a therapeutic option to enhance the regenerative capacity of endothelial progenitor cells (170). Both Notch1 signaling and the shedding of E-cadherin were shown to contribute to endothelial malfunction upon S. aureus infection (40, 74, 171). Furthermore, infection with Neisseria meningitidis resulted in cleavage of the anticoagulant endothelial protein C receptor (EPCR) and impairment of protein C activation, thereby increasing the risk of thrombosis formation (172). Of course, a huge variety of ADAM substrates have been shown to be involved in vascular biology (15). The membrane-bound IL-6R was upregulated on SMCs of patients suffering from pulmonary arterial hypertension (173), and IL-6R trans-signaling critically contributed to pancreatitis-associated lung injury and in infection with L. monocytogenes (64, 174). Furthermore, ADAM8 is involved in the development of acute and chronic inflammatory lung diseases, and soluble ADAM8 was shown to be associated with atherosclerosis and myocardial infarction (175). Thus, it is obvious that both ectodomain shedding and RIPing in infection may contribute to the development of cardiovascular diseases. However, which substrates contribute in a time- and context-dependent manner has still to be investigated for the development of effective treatment options.
ADAM proteases fulfill different functions from pathogen recognition to resolution of the concurrent inflammation and the development of systemic effects (Tables 1, 2, Figure 1). Thereby, they are essential control elements of infectious diseases. However, there a still some limitations with respect to the development of effective ADAM based therapeutic intervention. First, a lot of evidences are based on in vitro studies not integrating the divergent functions of substrates in different cell types such as reported for Notch. Second, we did not discuss the complex regulation of ADAM proteases by interaction partners such as tetraspanins for ADAM10 [for review see (176)] and iRhoms for ADAM17 [for review see (177)] or the inhibition by endogenous inhibitors such as tissue inhibitor of metalloproteinase (TIMP) 1 or 3 (37, 178, 179). Of course, dysregulation of these factors may critically contribute to function of ADAM proteases in infectious diseases. Third, we focused on catalytic functions of ADAM proteases with direct evidences from infection studies. However, their general contribution by catalytic and non-catalytic functions to cell migration [for review see (180)] may further influence the defense against pathogens. Fourth, species specific differences in the use of cleavage events as has been reported for IL-6R (181) draw an even more complex view of the ADAM protease web in infection. Nevertheless, this web highlights a huge variety of molecular targets for the treatment of infectious diseases. Targeting ADAM proteases themselves could include inhibition of expression, maturation, or activation, inhibition of the active site by small molecules, application of the inhibitory pro-domain, or inhibition of substrate recognition [for review see (182)]. So far, clinical trials failed due to detrimental side-effects through global changes of ADAM activity and disturbance of tissue homeostasis and developmental processes or lack of efficacy [for review see (12, 13, 23, 183)]. However, several clinical trials are ongoing to address the activation of ADAM10 for the treatment of Alzheimer disease [for review see (184)]. Thus, future studies will have to address ADAM protease functions in a pathogen-, time-, and substrate-specific manner to evaluate specific and efficient treatment options with no or minimal side-effects.
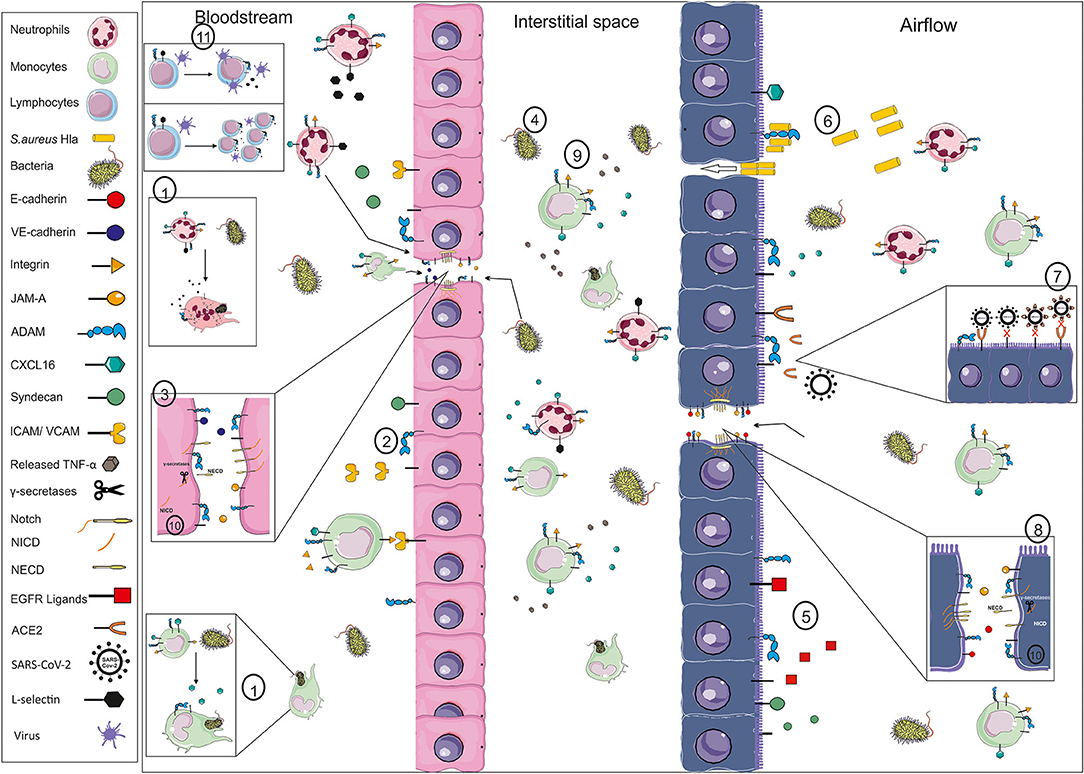
Figure 1. Exemplary illustration of ADAM function in infection. Pathogens like bacteria and their toxins may enter the body as exemplarily shown for the blood stream (left side) and the respiratory system (right side). The first line of defense for systemically entering pathogens is circulating monocytes and neutrophils and the endothelial barrier. ADAM proteases are involved in phagocytosis through, e.g., cleavage of scavenger receptors like CXCL16 or the cleavage of L-selectin (1), in the activation of the endothelium changing the recruitment of inflammatory cells through cleavage of adhesion and junction molecules like ICAM, VCAM, JAM-A and VE-cadherin (2), resulting in gap formation and disturbance of the barrier integrity (3). Thereby, not only inflammatory cells are able to enter the interstitial space but also pathogens (4). Mucus production and the ciliary movement are the natural barrier for pathogens entering via the airflow, which are influenced by the release of EGFR ligands (5). The epithelium expresses several receptors for pathogens, such as ADAM10 in the case of S. aureus HIa (6) or ACE2 in the case of SARS-CoV2 (7). Cleavage of ACE2 could limit the viral uptake through lack of receptor and the blocking of circulating virus particles. Pore formation and cleavage of junction molecules like E-cadherin (8) result in crossing of the pathogens and infection of the tissue. The infection is may be cleared by resident or newly recruited leukocytes and a beneficial systemic response, e.g., initiated by TNF-alpha release (9). However, systemic spread of the pathogens, e.g., amplified by the cleavage of syndecans and Notch-1, and enhanced release of pro-inflammatory mediators may cause systemic effects like sepsis or cardiovascular failure. These events are not only orchestrated by α-secretase cleavage but further regulated through RIPing processes mediated by γ-secretases (10). Furthermore, systemic effects may be based on changes in cytotoxic T cell expansion or viral release from central memory T cells (11), both involving L-selectin shedding. These are only a few examples of the ADAMs' impact in infection, which may differ in an organ- and site-specific manner. (VE-cadherin, vascular endothelial cadherin; JAM-A, junctional adhesion molecule A; ADAM, a disintegrin and metalloproteinase; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; TNF, tumor necrosis factor; NICD, Notch intracellular domain; NECD, Notch ectodomain; EGFR, epidermal growth factor; ACE2, angiotensin converting enzyme 2).
AA and DY viewed the literature and wrote the review.
This work was supported by the DFG grant DR1013/1-1 (DY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the HIPS-UdS TANDEM initiative for the support.
1. Ferrero-Miliani L, Nielsen OH, Andersen PS, Girardin SE. Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1beta generation. Clin Exp Immunol. (2007) 147:227–35. doi: 10.1111/j.1365-2249.2006.03261.x
2. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. (2010) 140:771–6. doi: 10.1016/j.cell.2010.03.006
3. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208
4. Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. (2000) 97:13766–71. doi: 10.1073/pnas.250476497
5. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
6. Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
7. Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflamm Allergy. (2005) 4:281–6. doi: 10.2174/1568010054022024
8. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
9. Zhou Y, Hong Y, Huang H. Triptolide attenuates inflammatory response in membranous glomerulo-nephritis rat via downregulation of NF-kappaB signaling pathway. Kidney Blood Press Res. (2016) 41:901–10. doi: 10.1159/000452591
10. Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. (2003) 17:7–30. doi: 10.1101/gad.1039703
11. Andreini C, Banci L, Bertini I, Elmi S, Rosato A. Comparative analysis of the ADAM and ADAMTS families. J Proteome Res. (2005) 4:881–8. doi: 10.1021/pr0500096
12. Giebeler N, Zigrino P. A disintegrin and metalloprotease (ADAM): historical overview of their functions. Toxins. (2016) 8:122. doi: 10.3390/toxins8040122
13. Lambrecht BN, Vanderkerken M, Hammad H. The emerging role of ADAM metalloproteinases in immunity. Nat Rev Immunol. (2018) 18:745–758. doi: 10.1038/s41577-018-0068-5
14. Takeda S. ADAM and ADAMTS family proteins and snake venom metalloproteinases: a structural overview. Toxins. (2016) 8:155. doi: 10.3390/toxins8050155
15. Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM-mediated shedding in vascular biology. Eur J Cell Biol. (2012) 91:472–485. doi: 10.1016/j.ejcb.2011.09.003
16. Dreymueller D, Uhlig S, Ludwig A. ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L325–43. doi: 10.1152/ajplung.00294.2014
17. Jurisch-Yaksi N, Sannerud R, Annaert W. A fast growing spectrum of biological functions of gamma-secretase in development and disease. Biochim Biophys Acta. (2013) 1828:2815–27. doi: 10.1016/j.bbamem.2013.04.016
18. Pasqualon T, Pruessmeyer J, Jankowski V, Babendreyer A, Groth E, Schumacher J, et al. A cytoplasmic C-terminal fragment of Syndecan-1 is generated by sequential proteolysis and antagonizes Syndecan-1 dependent lung tumor cell migration. Oncotarget. (2015) 6:31295–312. doi: 10.18632/oncotarget.5174
19. Groot AJ, Vooijs MA. The role of adams in notch signaling. Adv Exp Med Biol. (2012) 727:15–36. doi: 10.1007/978-1-4614-0899-4_2
20. van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. (2002) 418:426–30. doi: 10.1038/nature00878
21. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. (2008) 29:258–89. doi: 10.1016/j.mam.2008.08.001
22. Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. (2008) 8:929–41. doi: 10.1038/nrc2459
23. Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. (2012) 139:3693–709. doi: 10.1242/dev.076398
24. Zhang P, Shen M, Fernandez-Patron C, Kassiri Z. ADAMs family and relatives in cardiovascular physiology and pathology. J Mol Cell Cardiol. (2016) 93:186–99. doi: 10.1016/j.yjmcc.2015.10.031
25. Baggen J, Thibaut HJ, Hurdiss DL, Wahedi M, Marceau CD, van Vliet ALW, et al. Identification of the cell-surface protease ADAM9 as an entry factor for encephalomyocarditis virus. mBio. (2019) 10:e01780-19. doi: 10.1128/mBio.01780-19
26. Bazzone LE, King M, Mackay CR, Kyawe PP, Meraner P, Lindstrom D, et al. A disintegrin and metalloproteinase 9 domain (ADAM9) is a major susceptibility factor in the early stages of encephalomyocarditis virus infection. mBio. (2019) 10:e02734-18. doi: 10.1128/mBio.02734-18
27. Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, et al. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol. (2004) 172:6362–72. doi: 10.4049/jimmunol.172.10.6362
28. Gough PJ, Garton KJ, Wille PT, Rychlewski M, Dempsey PJ, Raines EW. A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16. J Immunol. (2004) 172:3678–85. doi: 10.4049/jimmunol.172.6.3678
29. Yang WS, Kim JJ, Han NJ, Lee EK, Park SK. 1,25-dihydroxyvitamin D3 attenuates the effects of lipopolysaccharide by causing ADAM10-dependent ectodomain shedding of toll-like receptor 4. Cell Physiol Biochem. (2017) 41:2104–16. doi: 10.1159/000475449
30. Schweigert O, Dewitz C, Moller-Hackbarth K, Trad A, Garbers C, Rose-John S, et al. Soluble T cell immunoglobulin and mucin domain (TIM)-1 and−4 generated by A disintegrin and metalloprotease (ADAM)-10 and−17 bind to phosphatidylserine. Biochim Biophys Acta. (2014) 1843:275–87. doi: 10.1016/j.bbamcr.2013.11.014
31. Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire ebolavirus and lake victoria marburgvirus. Proc Natl Acad Sci USA. (2011) 108:8426–31. doi: 10.1073/pnas.1019030108
32. Seifert A, Wozniak J, Dusterhoft S, Kasparek P, Sedlacek R, Dreschers S, et al. The iRhom2/ADAM17 axis attenuates bacterial uptake by phagocytes in a cell autonomous manner. Int J Mol Sci. (2020) 21:5978. doi: 10.3390/ijms21175978
33. Karunakaran K, Subbarayal P, Vollmuth N, Rudel T. Chlamydia-infected cells shed Gp96 to prevent chlamydial re-infection. Mol Microbiol. (2015) 98:694–711. doi: 10.1111/mmi.13151
34. Paland N, Bohme L, Gurumurthy RK, Maurer A, Szczepek AJ, Rudel T. Reduced display of tumor necrosis factor receptor I at the host cell surface supports infection with Chlamydia trachomatis. J Biol Chem. (2008) 283:6438–48. doi: 10.1074/jbc.M708422200
35. Mikulicic S, Finke J, Boukhallouk F, Wustenhagen E, Sons D, Homsi Y, et al. ADAM17-dependent signaling is required for oncogenic human papillomavirus entry platform assembly. Elife. (2019) 8:e44345. doi: 10.7554/eLife.44345
36. Oliviero B, Mantovani S, Varchetta S, Mele D, Grossi G, Ludovisi S, et al. Hepatitis C virus-induced NK cell activation causes metzincin-mediated CD16 cleavage and impaired antibody-dependent cytotoxicity. J Hepatol. (2017) 66:1130–7. doi: 10.1016/j.jhep.2017.01.032
37. Esteso G, Luzon E, Sarmiento E, Gomez-Caro R, Steinle A, Murphy G, et al. Altered microRNA expression after infection with human cytomegalovirus leads to TIMP3 downregulation and increased shedding of metalloprotease substrates, including MICA. J Immunol. (2014) 193:1344–52. doi: 10.4049/jimmunol.1303441
38. Rizzo P, Vieceli Dalla Sega F, Fortini F, Marracino L, Rapezzi C, Ferrari R. COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res Cardiol. (2020) 115:31. doi: 10.1007/s00395-020-0791-5
39. Saphire AC, Bobardt MD, Zhang Z, David G, Gallay PA. Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J Virol. (2001) 75:9187–200. doi: 10.1128/JVI.75.19.9187-9200.2001
40. Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci USA. (2010) 107:13473–13478. doi: 10.1073/pnas.1001815107
41. Kneidl J, Loffler B, Erat MC, Kalinka J, Peters G, Roth J, et al. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell Microbiol. (2012) 14:914–36. doi: 10.1111/j.1462-5822.2012.01766.x
42. Cappenberg A, Margraf A, Thomas K, Bardel B, Mccreedy DA, Van Marck V, et al. L-selectin shedding affects bacterial clearance in the lung: a new regulatory pathway for integrin outside-in signaling. Blood. (2019) 134:1445–57. doi: 10.1182/blood.2019000685
43. Etzerodt A, Maniecki MB, Moller K, Moller HJ, Moestrup SK. Tumor necrosis factor alpha-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol. (2010) 88:1201–5. doi: 10.1189/jlb.0410235
44. Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol. (2012) 13:642–50. doi: 10.1038/ni.2304
45. Damle SR, Martin RK, Cockburn CL, Lownik JC, Carlyon JA, Smith AD, et al. ADAM10 and Notch1 on murine dendritic cells control the development of type 2 immunity and IgE production. Allergy. (2018) 73:125–36. doi: 10.1111/all.13261
46. Gomez MI, Sokol SH, Muir AB, Soong G, Bastien J, Prince AS. Bacterial induction of TNF-alpha converting enzyme expression and IL-6 receptor alpha shedding regulates airway inflammatory signaling. J Immunol. (2005) 175:1930–36. doi: 10.4049/jimmunol.175.3.1930
47. Simanski M, Rademacher F, Schroder L, Glaser R, Harder J. The inflammasome and the epidermal growth factor receptor (EGFR) are involved in the Staphylococcus aureus-mediated induction of IL-1alpha and IL-1beta in human keratinocytes. PLoS ONE. (2016) 11:e0147118. doi: 10.1371/journal.pone.0147118
48. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. (1997) 385:729–33. doi: 10.1038/385729a0
49. Pruessmeyer J, Hess FM, Alert H, Groth E, Pasqualon T, Schwarz N, et al. Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood. (2014) 123:4077–88. doi: 10.1182/blood-2013-09-511543
50. Long C, Hosseinkhani MR, Wang Y, Sriramarao P, Walcheck B. ADAM17 activation in circulating neutrophils following bacterial challenge impairs their recruitment. J Leukoc Biol. (2012) 92:667–72. doi: 10.1189/jlb.0312112
51. Arndt PG, Strahan B, Wang Y, Long C, Horiuchi K, Walcheck B. Leukocyte ADAM17 regulates acute pulmonary inflammation. PLoS ONE. (2011) 6:e19938. doi: 10.1371/journal.pone.0019938
52. Mohammed RN, Wehenkel SC, Galkina EV, Yates EK, Preece G, Newman A, et al. ADAM17-dependent proteolysis of L-selectin promotes early clonal expansion of cytotoxic T cells. Sci Rep. (2019) 9:5487. doi: 10.1038/s41598-019-41811-z
53. Olvera-Garcia G, Aguilar-Garcia T, Gutierrez-Jasso F, Imaz-Rosshandler I, Rangel-Escareno C, Orozco L, et al. A transcriptome-based model of central memory CD4 T cell death in HIV infection. BMC Genomics. (2016) 17:956. doi: 10.1186/s12864-016-3308-8
54. Hakulinen J, Keski-Oja J. ADAM10-mediated release of complement membrane cofactor protein during apoptosis of epithelial cells. J Biol Chem. (2006) 281:21369–76. doi: 10.1074/jbc.M602053200
55. Schulte M, Reiss K, Lettau M, Maretzky T, Ludwig A, Hartmann D, et al. ADAM10 regulates FasL cell surface expression and modulates FasL-induced cytotoxicity and activation-induced cell death. Cell Death Differ. (2007) 14:1040–9. doi: 10.1038/sj.cdd.4402101
56. Antonelli A, Di Maggio S, Rejman J, Sanvito F, Rossi A, Catucci A, et al. The shedding-derived soluble receptor for advanced glycation endproducts sustains inflammation during acute Pseudomonas aeruginosa lung infection. Biochim Biophys Acta Gen Subj. (2017) 1861:354–64. doi: 10.1016/j.bbagen.2016.11.040
57. Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. Mechanisms by which gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR). Novartis Found Symp. (2002) 248:171–6. doi: 10.1002/0470860790.ch11
58. Ramachandran RP, Spiegel C, Keren Y, Danieli T, Melamed-Book N, Pal RR, et al. Mitochondrial targeting of the enteropathogenic escherichia coli map triggers calcium mobilization, ADAM10-MAP kinase signaling, and host cell apoptosis. mBio. (2020)11:e01397-20. doi: 10.1128/mBio.01397-20
59. Schirrmeister W, Gnad T, Wex T, Higashiyama S, Wolke C, Naumann M, et al. Ectodomain shedding of E-cadherin and c-Met is induced by helicobacter pylori infection. Exp Cell Res. (2009) 315:3500–8. doi: 10.1016/j.yexcr.2009.07.029
60. Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. (2011) 17:1310–4. doi: 10.1038/nm.2451
61. Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, et al. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. (2007) 179:2686–9. doi: 10.4049/jimmunol.179.5.2686
62. Mcilwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science. (2012) 335:229–32. doi: 10.1126/science.1214448
63. Ali M, Saroha A, Pewzner-Jung Y, Futerman AH. LPS-mediated septic shock is augmented in ceramide synthase 2 null mice due to elevated activity of TNFalpha-converting enzyme. FEBS Lett. (2015) 589:2213–7. doi: 10.1016/j.febslet.2015.06.045
64. Yan I, Schwarz J, Lucke K, Schumacher N, Schumacher V, Schmidt S, et al. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Ralpha from the cell surface of leukocytes during inflammatory responses. J Leukoc Biol. (2016) 99:749–60. doi: 10.1189/jlb.3A0515-207R
65. Kononchik J, Ireland J, Zou Z, Segura J, Holzapfel G, Chastain A, et al. HIV-1 targets L-selectin for adhesion and induces its shedding for viral release. Nat Commun. (2018) 9:2825. doi: 10.1038/s41467-018-05197-2
66. Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology. (2010) 139:239–48. doi: 10.1053/j.gastro.2010.03.036
67. Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J Leukoc Biol. (2007) 82:173–6. doi: 10.1189/jlb.0307193
68. Jakobsen PH, Dodt KK, Meyer CN, Katzenstein T, Gerstoft J, Skinhoj P. Increased levels of soluble tumour necrosis factor receptor-I (P55) and decreased IgG1 reactivities in HIV-1 patients with cytomegalovirus disease. Scand J Immunol. (1998) 47:591–5. doi: 10.1046/j.1365-3083.1998.00338.x
69. Nordoy I, Muller F, Nordal KP, Rollag H, Aukrust P, Froland SS. Chemokines and soluble adhesion molecules in renal transplant recipients with cytomegalovirus infection. Clin Exp Immunol. (2000) 120:333–7. doi: 10.1046/j.1365-2249.2000.01221.x
70. Park PW, Pier GB, Hinkes MT, Bernfield M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature. (2001) 411:98–102. doi: 10.1038/35075100
71. Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. (2014) 66:167–76. doi: 10.1016/j.yjmcc.2013.11.017
72. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th Anniversary of the discovery of ACE2. Circ Res. (2020) 126:1456–74. doi: 10.1161/CIRCRESAHA.120.317015
73. Inoshima N, Wang Y, Bubeck Wardenburg J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J Invest Dermatol. (2012) 132:1513–6. doi: 10.1038/jid.2011.462
74. Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. ADAM10 mediates vascular injury induced by Staphylococcus aureus alpha-hemolysin. J Infect Dis. (2012) 206:352–6. doi: 10.1093/infdis/jis192
75. Mishra HK, Johnson TJ, Seelig DM, Walcheck B. Targeting ADAM17 in leukocytes increases neutrophil recruitment and reduces bacterial spread during polymicrobial sepsis. J Leukoc Biol. (2016) 100:999–1004. doi: 10.1189/jlb.3VMAB1115-496RR
76. Mishra HK, Ma J, Walcheck B. Ectodomain shedding by ADAM17: its role in neutrophil recruitment and the impairment of this process during sepsis. Front Cell Infect Microbiol. (2017) 7:138. doi: 10.3389/fcimb.2017.00138
77. Long C, Wang Y, Herrera AH, Horiuchi K, Walcheck B. In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J Leukoc Biol. (2010) 87:1097–101. doi: 10.1189/jlb.1109763
78. Link MA, Lucke K, Schmid J, Schumacher V, Eden T, Rose-John S, et al. The role of ADAM17 in the T-cell response against bacterial pathogens. PLoS ONE. (2017) 12:e0184320. doi: 10.1371/journal.pone.0184320
79. Deberge MP, Ely KH, Cheng GS, Enelow RI. ADAM17-mediated processing of TNF-alpha expressed by antiviral effector CD8+ T cells is required for severe T-cell-mediated lung injury. PLoS ONE. (2013) 8:e79340. doi: 10.1371/journal.pone.0079340
80. Deberge MP, Ely KH, Wright PF, Thorp EB, Enelow RI. Shedding of TNF receptor 2 by effector CD8(+) T cells by ADAM17 is important for regulating TNF-alpha availability during influenza infection. J Leukoc Biol. (2015) 98:423–34. doi: 10.1189/jlb.3A0914-432RR
81. Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. (2002) 11:2615–24. doi: 10.1093/hmg/11.21.2615
82. Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science. (1998) 282:1281–4. doi: 10.1126/science.282.5392.1281
83. Fabriek BO, van Bruggen R, Deng DM, Ligtenberg AJ, Nazmi K, Schornagel K, et al. The macrophage scavenger receptor CD163 functions as an innate immune sensor for bacteria. Blood. (2009) 113:887–92. doi: 10.1182/blood-2008-07-167064
84. Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. (2008) 22:3716–27. doi: 10.1096/fj.08-109033
85. Gopal P, Gosker HR, Theije CC, Eurlings IM, Sell DR, Monnier VM, et al. Effect of chronic hypoxia on RAGE and its soluble forms in lungs and plasma of mice. Biochim Biophys Acta. (2015) 1852:992–1000. doi: 10.1016/j.bbadis.2015.02.003
86. Friedrich BM, Murray JL, Li G, Sheng J, Hodge TW, Rubin DH, et al. A functional role for ADAM10 in human immunodeficiency virus type-1 replication. Retrovirology. (2011) 8:32. doi: 10.1186/1742-4690-8-32
87. Loetscher M, Amara A, Oberlin E, Brass N, Legler D, Loetscher P, et al. TYMSTR, a putative chemokine receptor selectively expressed in activated T cells, exhibits HIV-1 coreceptor function. Curr Biol. (1997) 7:652–60. doi: 10.1016/S0960-9822(06)00292-2
88. Shimaoka T, Nakayama T, Kume N, Takahashi S, Yamaguchi J, Minami M, et al. Cutting edge: SR-PSOX/CXC chemokine ligand 16 mediates bacterial phagocytosis by APCs through its chemokine domain. J Immunol. (2003) 171:1647–51. doi: 10.4049/jimmunol.171.4.1647
89. Park GB, Kim D, Kim YS, Kim JW, Sun H, Roh KH, et al. Regulation of ADAM10 and ADAM17 by sorafenib inhibits epithelial-to-mesenchymal transition in epstein-barr virus-infected retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2015) 56:5162–73. doi: 10.1167/iovs.14-16058
90. Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, et al. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, beta-catenin translocation. Proc Natl Acad Sci USA. (2005) 102:9182–7. doi: 10.1073/pnas.0500918102
91. Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, et al. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. (2008) 102:1192–201. doi: 10.1161/CIRCRESAHA.107.169805
92. Reboud E, Bouillot S, Patot S, Beganton B, Attree I, Huber P. Pseudomonas aeruginosa ExlA and Serratia marcescens ShlA trigger cadherin cleavage by promoting calcium influx and ADAM10 activation. PLoS Pathog. (2017) 13:e1006579. doi: 10.1371/journal.ppat.1006579
93. Zager RA, Johnson AC, Lund S, Randolph-Habecker J. Toll-like receptor (TLR4) shedding and depletion: acute proximal tubular cell responses to hypoxic and toxic injury. Am J Physiol Renal Physiol. (2007) 292:F304–12. doi: 10.1152/ajprenal.00237.2006
94. Langjahr P, Diaz-Jimenez D, de La Fuente M, Rubio E, Golenbock D, Bronfman FC, et al. Metalloproteinase-dependent TLR2 ectodomain shedding is involved in soluble toll-like receptor 2 (sTLR2) production. PLoS ONE. (2014) 9:e104624. doi: 10.1371/journal.pone.0104624
95. Eriksson BM, Sjolin J, Claesson K, Wirgart BZ, Grillner L, Totterman TH. Circulating soluble vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in immunocompetent and renal transplant patients: correlation with cytomegalovirus disease and renal function. Scand J Infect Dis. (2001) 33:350–4. doi: 10.1080/003655401750173968
96. Dreymueller D, Martin C, Kogel T, Pruessmeyer J, Hess FM, Horiuchi K, et al. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol Med. (2012) 4:412–23. doi: 10.1002/emmm.201200217
97. Greene C, Lowe G, Taggart C, Gallagher P, Mcelvaney N, O'neill S. Tumor necrosis factor-alpha-converting enzyme: its role in community-acquired pneumonia. J Infect Dis. (2002) 186:1790–6. doi: 10.1086/345799
98. Leib SL, Clements JM, Lindberg RL, Heimgartner C, Loeffler JM, Pfister LA, et al. Inhibition of matrix metalloproteinases and tumour necrosis factor alpha converting enzyme as adjuvant therapy in pneumococcal meningitis. Brain. (2001) 124:1734–42. doi: 10.1093/brain/124.9.1734
99. Lee JH, Schierer S, Blume K, Dindorf J, Wittki S, Xiang W, et al. HIV-Nef and ADAM17-containing plasma extracellular vesicles induce and correlate with immune pathogenesis in chronic HIV infection. EBioMedicine. (2016) 6:103–13. doi: 10.1016/j.ebiom.2016.03.004
100. Ostalecki C, Wittki S, Lee JH, Geist MM, Tibroni N, Harrer T, et al. HIV Nef- and Notch1-dependent Endocytosis of ADAM17 induces vesicular TNF secretion in chronic HIV infection. EBioMedicine. (2016) 13:294–304. doi: 10.1016/j.ebiom.2016.10.027
101. Mcclurg UL, Danjo K, King HO, Scott GB, Robinson PA, Crabtree JE. Epithelial cell ADAM17 activation by helicobacter pylori: role of ADAM17 C-terminus and Threonine-735 phosphorylation. Microbes Infect. (2015) 17:205–14. doi: 10.1016/j.micinf.2014.11.011
102. Slomiany BL, Slomiany A. Helicobacter pylori-induced gastric mucosal TGF-alpha ectodomain shedding and EGFR transactivation involves Rac1/p38 MAPK-dependent TACE activation. Inflammopharmacology. (2016) 24:23–31. doi: 10.1007/s10787-015-0254-z
103. Pahl R, Brunke G, Steubesand N, Schubert S, Bottner M, Wedel T, et al. IL-1beta and ADAM17 are central regulators of beta-defensin expression in Candida esophagitis. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G547–53. doi: 10.1152/ajpgi.00251.2010
104. Dolnik O, Volchkova V, Garten W, Carbonnelle C, Becker S, Kahnt J, et al. Ectodomain shedding of the glycoprotein GP of Ebola virus. EMBO J. (2004) 23:2175–84. doi: 10.1038/sj.emboj.7600219
105. Mishra HK, Ma J, Mendez D, Hullsiek R, Pore N, Walcheck B. Blocking ADAM17 function with a monoclonal antibody improves sepsis survival in a murine model of polymicrobial sepsis. Int J Mol Sci. (2020) 21:6688. doi: 10.3390/ijms21186688
106. Kelly K, Hutchinson G, Nebenius-Oosthuizen D, Smith AJ, Bartsch JW, Horiuchi K, et al. Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn. (2005) 232:221–31. doi: 10.1002/dvdy.20221
107. Schlomann U, Koller G, Conrad C, Ferdous T, Golfi P, Garcia AM, et al. ADAM8 as a drug target in pancreatic cancer. Nat Commun. (2015) 6:6175. doi: 10.1038/ncomms7175
108. Dreymueller D, Pruessmeyer J, Schumacher J, Fellendorf S, Hess FM, Seifert A, et al. The metalloproteinase ADAM8 promotes leukocyte recruitment in vitro and in acute lung inflammation. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L602–14. doi: 10.1152/ajplung.00444.2016
109. Ma GF, Miettinen S, Porola P, Hedman K, Salo J, Konttinen YT. Human parainfluenza virus type 2 (HPIV2) induced host ADAM8 expression in human salivary adenocarcinoma cell line (HSY) during cell fusion. BMC Microbiol. (2009) 9:55. doi: 10.1186/1471-2180-9-55
110. Khongkhunthian S, Techasatian P, Supanchart C, Bandhaya P, Montreekachon P, Thawanaphong S, et al. Elevated levels of a disintegrin and metalloproteinase 8 in gingival crevicular fluid of patients with periodontal diseases. J Periodontol. (2013) 84:520–8. doi: 10.1902/jop.2012.120262
111. Nimcharoen T, Aung WPP, Makeudom A, Sastraruji T, Khongkhunthian S, Sirinirund B, et al. Reduced ADAM8 levels upon non-surgical periodontal therapy in patients with chronic periodontitis. Arch Oral Biol. (2019) 97:137–43. doi: 10.1016/j.archoralbio.2018.10.021
112. Mullick A, Leon Z, Min-Oo G, Berghout J, Lo R, Daniels E, et al. Cardiac failure in C5-deficient A/J mice after Candida albicans infection. Infect Immun. (2006) 74:4439–51. doi: 10.1128/IAI.00159-06
113. Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. (2005) 6:32–43. doi: 10.1038/nrm1548
114. Amendola RS, Martin AC, Selistre-De-Araujo HS, Paula-Neto HA, Saldanha-Gama R, Barja-Fidalgo C. ADAM9 disintegrin domain activates human neutrophils through an autocrine circuit involving integrins and CXCR2. J Leukoc Biol. (2015) 97:951–62. doi: 10.1189/jlb.3A0914-455R
115. English WR, Siviter RJ, Hansen M, Murphy G. ADAM9 is present at endothelial cell - cell junctions and regulates monocyte - endothelial transmigration. Biochem Biophys Res Commun. (2017) 493:1057–62. doi: 10.1016/j.bbrc.2017.09.089
116. Rinchai D, Kewcharoenwong C, Kessler B, Lertmemongkolchai G, Chaussabel D. Increased abundance of ADAM9 transcripts in the blood is associated with tissue damage. F1000Res. (2015) 4:89. doi: 10.12688/f1000research.6241.1
117. Saavedra MT, Hughes GJ, Sanders LA, Carr M, Rodman DM, Coldren CD, et al. Circulating RNA transcripts identify therapeutic response in cystic fibrosis lung disease. Am J Respir Crit Care Med. (2008) 178:929–38. doi: 10.1164/rccm.200803-387OC
118. Roychaudhuri R, Hergrueter AH, Polverino F, Laucho-Contreras ME, Gupta K, Borregaard N, et al. ADAM9 is a novel product of polymorphonuclear neutrophils: regulation of expression and contributions to extracellular matrix protein degradation during acute lung injury. J Immunol. (2014) 193:2469–82. doi: 10.4049/jimmunol.1303370
119. Xiang LY, Ou HH, Liu XC, Chen ZJ, Li XH, Huang Y, et al. Loss of tumor suppressor miR-126 contributes to the development of hepatitis B virus-related hepatocellular carcinoma metastasis through the upregulation of ADAM9. Tumour Biol. (2017) 39:1010428317709128. doi: 10.1177/1010428317709128
120. Juiz NA, Cayo NM, Burgos M, Salvo ME, Nasser JR, Bua J, et al. Human polymorphisms in placentally expressed genes and their association with susceptibility to congenital Trypanosoma cruzi infection. J Infect Dis. (2016) 213:1299–306. doi: 10.1093/infdis/jiv561
121. Siezen CL, Bont L, Hodemaekers HM, Ermers MJ, Doornbos G, Van't Slot R, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis in preterm children is associated with airway remodeling genes and innate immune genes. Pediatr Infect Dis J. (2009) 28:333–5. doi: 10.1097/INF.0b013e31818e2aa9
122. Drysdale SB, Prendergast M, Alcazar M, Wilson T, Smith M, Zuckerman M, et al. Genetic predisposition of RSV infection-related respiratory morbidity in preterm infants. Eur J Pediatr. (2014) 173:905–12. doi: 10.1007/s00431-014-2263-0
123. Baurakiades E, Costa VH, Raboni SM, De Almeida VR, Larsen KS, Kohler JN, et al. The roles of ADAM33, ADAM28, IL-13 and IL-4 in the development of lung injuries in children with lethal non-pandemic acute infectious pneumonia. J Clin Virol. (2014) 61:585–9. doi: 10.1016/j.jcv.2014.10.004
124. Sun C, Beard RS, Mclean DL, Rigor RR, Konia T, Wu MH, et al. ADAM15 deficiency attenuates pulmonary hyperpermeability and acute lung injury in lipopolysaccharide-treated mice. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L135–42. doi: 10.1152/ajplung.00133.2012
125. Yang X, Meegan JE, Jannaway M, Coleman DC, Yuan SY. A disintegrin and metalloproteinase 15-mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc Res. (2018) 114:1752–63. doi: 10.1093/cvr/cvy167
126. Chatterjee V, Beard RS, Reynolds JJ, Haines R, Guo M, Rubin M, et al. MicroRNA-147b regulates vascular endothelial barrier function by targeting ADAM15 expression. PLoS ONE. (2014) 9:e110286. doi: 10.1371/journal.pone.0110286
127. Ahmed S, Maratha A, Butt AQ, Shevlin E, Miggin SM. TRIF-mediated TLR3 and TLR4 signaling is negatively regulated by ADAM15. J Immunol. (2013) 190:2217–28. doi: 10.4049/jimmunol.1201630
128. Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. (2000) 228:151–65. doi: 10.1006/dbio.2000.9960
129. Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. (2004) 5:499–504. doi: 10.1038/nrm1406
130. Deuss M, Reiss K, Hartmann D. Part-time alpha-secretases: the functional biology of ADAM 9, 10 and 17. Curr Alzheimer Res. (2008) 5:187–201. doi: 10.2174/156720508783954686
131. Venugopal C, Demos CM, Rao KS, Pappolla MA, Sambamurti K. Beta-secretase: structure, function, and evolution. CNS Neurol Disord Drug Targets. (2008) 7:278–94. doi: 10.2174/187152708784936626
132. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. (2012) 13:654–66. doi: 10.1038/nrg3272
133. Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci USA. (2014) 111:705–10. doi: 10.1073/pnas.1315023111
134. Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, et al. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and−4 by lung epithelial cells. J Biol Chem. (2010) 285:555–64. doi: 10.1074/jbc.M109.059394
135. Stepp MA, Pal-Ghosh S, Tadvalkar G, Pajoohesh-Ganji A. Syndecan-1 and its expanding list of contacts. Adv Wound Care. (2015) 4:235–49. doi: 10.1089/wound.2014.0555
136. Park PW, Foster TJ, Nishi E, Duncan SJ, Klagsbrun M, Chen Y. Activation of syndecan-1 ectodomain shedding by Staphylococcus aureus alpha-toxin and beta-toxin. J Biol Chem. (2004) 279:251–8. doi: 10.1074/jbc.M308537200
137. Haynes A, Ruda F, Oliver J, Hamood AN, Griswold JA, Park PW, et al. Syndecan 1 shedding contributes to Pseudomonas aeruginosa sepsis. Infect Immun. (2005) 73:7914–21. doi: 10.1128/IAI.73.12.7914-7921.2005
138. Devaux CA, Mezouar S, Mege JL. The E-cadherin cleavage associated to pathogenic bacteria infections can favor bacterial invasion and transmigration, dysregulation of the immune response and cancer induction in humans. Front Microbiol. (2019) 10:2598. doi: 10.3389/fmicb.2019.02598
139. Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. (2002) 21:1948–56. doi: 10.1093/emboj/21.8.1948
140. Ljungberg JK, Kling JC, Tran TT, Blumenthal A. Functions of the WNT signaling network in shaping host responses to infection. Front Immunol. (2019) 10:2521. doi: 10.3389/fimmu.2019.02521
141. Agiostratidou G, Muros RM, Shioi J, Marambaud P, Robakis NK. The cytoplasmic sequence of E-cadherin promotes non-amyloidogenic degradation of A beta precursors. J Neurochem. (2006) 96:1182–8. doi: 10.1111/j.1471-4159.2005.03616.x
142. Balin BJ, Hammond CJ, Little CS, Hingley ST, Al-Atrache Z, Appelt DM, et al. Chlamydia pneumoniae: an etiologic agent for late-onset dementia. Front Aging Neurosci. (2018) 10:302. doi: 10.3389/fnagi.2018.00302
143. Al-Atrache Z, Lopez DB, Hingley ST, Appelt DM. Astrocytes infected with Chlamydia pneumoniae demonstrate altered expression and activity of secretases involved in the generation of beta-amyloid found in Alzheimer disease. BMC Neurosci. (2019) 20:6. doi: 10.1186/s12868-019-0489-5
144. Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, et al. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, the gamma-secretase. J Biol Chem. (2009) 284:11738–47. doi: 10.1074/jbc.M805894200
145. Kuhnle N, Dederer V, Lemberg MK. Intramembrane proteolysis at a glance: from signalling to protein degradation. J Cell Sci. (2019)132:jsc.217745. doi: 10.1242/jcs.217745
146. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
147. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. (2020) 367:1444–8. doi: 10.1126/science.abb2762
148. Walls AC, Park YJ, Tortorici MA, Wall A, Mcguire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92.e286. doi: 10.1016/j.cell.2020.02.058
149. Qiu H, Tang X, Ma J, Shaverdashvili K, Zhang K, Bedogni B. Notch1 autoactivation via transcriptional regulation of furin, which sustains notch1 signaling by processing notch1-activating proteases ADAM10 and membrane Type 1 matrix metalloproteinase. Mol Cell Biol. (2015) 35:3622–32. doi: 10.1128/MCB.00116-15
150. Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. (2009) 29:5679–95. doi: 10.1128/MCB.00406-09
151. Doberstein K, Steinmeyer N, Hartmetz AK, Eberhardt W, Mittelbronn M, Harter PN, et al. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia. (2013) 15:218–30. doi: 10.1593/neo.121222
152. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. (2020) 26:1636–4. doi: 10.1038/s41591-020-1051-9
153. Horiuchi S, Koyanagi Y, Zhou Y, Miyamoto H, Tanaka Y, Waki M, et al. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur J Immunol. (1994) 24:1945–8. doi: 10.1002/eji.1830240837
154. Schumacher N, Rose-John S. ADAM17 Activity and IL-6 trans-signaling in inflammation and cancer. Cancers. (2019) 11:1736. doi: 10.3390/cancers11111736
155. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. (2005) 11:875–9. doi: 10.1038/nm1267
156. Haga S, Nagata N, Okamura T, Yamamoto N, Sata T, Yamamoto N, et al. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antiviral Res. (2010) 85:551–5. doi: 10.1016/j.antiviral.2009.12.001
157. Kurahashi K, Kajikawa O, Sawa T, Ohara M, Gropper MA, Frank DW, et al. Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J Clin Invest. (1999) 104:743–50. doi: 10.1172/JCI7124
158. Xie Y. Hepatitis B virus-associated hepatocellular carcinoma. Adv Exp Med Biol. (2017) 1018:11–21. doi: 10.1007/978-981-10-5765-6_2
159. Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. (2020) 5:831–40. doi: 10.1001/jamacardio.2020.1286
161. Zganiacz A, Santosuosso M, Wang J, Yang T, Chen L, Anzulovic M, et al. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J Clin Invest. (2004) 113:401–13. doi: 10.1172/JCI18991
162. Mckellar GE, Mccarey DW, Sattar N, Mcinnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. (2009) 6:410–7. doi: 10.1038/nrcardio.2009.57
163. Zhang H, Park Y, Wu J, Chen X, Lee S, Yang J, et al. Role of TNF-alpha in vascular dysfunction. Clin Sci. (2009) 116:219–30. doi: 10.1042/CS20080196
164. Campbell LA, Rosenfeld ME. Infection and atherosclerosis development. Arch Med Res. (2015) 46:339–50. doi: 10.1016/j.arcmed.2015.05.006
165. Kim HJ, Cha GS, Kim HJ, Kwon EY, Lee JY, Choi J, et al. Porphyromonas gingivalis accelerates atherosclerosis through oxidation of high-density lipoprotein. J Periodontal Implant Sci. (2018) 48:60–8. doi: 10.5051/jpis.2018.48.1.60
166. Crawford M, Curtis JR. Tumor necrosis factor inhibitors and infection complications. Curr Rheumatol Rep. (2008) 10:383–9. doi: 10.1007/s11926-008-0062-1
167. Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. (2020) 24:422. doi: 10.1186/s13054-020-03120-0
168. Vaduganathan M, Vardeny O, Michel T, Mcmurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with covid-19. N Engl J Med. (2020) 382:1653–9. doi: 10.1056/NEJMsr2005760
169. Alabi RO, Glomski K, Haxaire C, Weskamp G, Monette S, Blobel CP. ADAM10-dependent signaling through Notch1 and Notch4 controls development of organ-specific vascular beds. Circ Res. (2016) 119:519–31. doi: 10.1161/CIRCRESAHA.115.307738
170. Rizzo P, Miele L, Ferrari R. The Notch pathway: a crossroad between the life and death of the endothelium. Eur Heart J. (2013) 34:2504–9. doi: 10.1093/eurheartj/ehs141
171. Hernandez SL, Nelson M, Sampedro GR, Bagrodia N, Defnet AM, Lec B, et al. Staphylococcus aureus alpha toxin activates Notch in vascular cells. Angiogenesis. (2019) 22:197–209. doi: 10.1007/s10456-018-9650-5
172. Lecuyer H, Virion Z, Barnier JP, Matczak S, Bourdoulous S, Bianchini E, et al. An ADAM-10 dependent EPCR shedding links meningococcal interaction with endothelial cells to purpura fulminans. PLoS Pathog. (2018) 14:e1006981. doi: 10.1371/journal.ppat.1006981
173. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. (2018) 128:1956–970. doi: 10.1172/JCI96462
174. Zhang H, Neuhofer P, Song L, Rabe B, Lesina M, Kurkowski MU, et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J Clin Invest. (2013) 123:1019–31. doi: 10.1172/JCI64931
175. Schick D, Babendreyer A, Wozniak J, Awan T, Noels H, Liehn E, et al. Elevated expression of the metalloproteinase ADAM8 associates with vascular diseases in mice and humans. Atherosclerosis. (2019) 286:163–71. doi: 10.1016/j.atherosclerosis.2019.03.008
176. Matthews AL, Koo CZ, Szyroka J, Harrison N, Kanhere A, Tomlinson MG. Regulation of leukocytes by TspanC8 tetraspanins and the “Molecular scissor” ADAM10. Front Immunol. (2018) 9:1451. doi: 10.3389/fimmu.2018.01451
177. Dulloo I, Muliyil S, Freeman M. The molecular, cellular and pathophysiological roles of iRhom pseudoproteases. Open Biol. (2019) 9:190003. doi: 10.1098/rsob.190003
178. Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, et al. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. (2000) 473:275–279. doi: 10.1016/S0014-5793(00)01528-3
179. Grotzinger J, Lorenzen I, Dusterhoft S. Molecular insights into the multilayered regulation of ADAM17: the role of the extracellular region. Biochim Biophys Acta Mol Cell. (2017) 1864:2088–95. doi: 10.1016/j.bbamcr.2017.05.024
180. Dreymueller D, Theodorou K, Donners M, Ludwig A. Fine tuning cell migration by a disintegrin and metalloproteinases. Mediators Inflamm. (2017) 2017:9621724. doi: 10.1155/2017/9621724
181. Garbers C, Janner N, Chalaris A, Moss ML, Floss DM, Meyer D, et al. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J Biol Chem. (2011) 286:14804–11. doi: 10.1074/jbc.M111.229393
182. Dreymueller D, Ludwig A. Considerations on inhibition approaches for proinflammatory functions of ADAM proteases. Platelets. (2017) 28:354–61. doi: 10.1080/09537104.2016.1203396
183. Zunke F, Rose-John S. The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res. (2017) 1864:2059–70. doi: 10.1016/j.bbamcr.2017.07.001
Keywords: metalloproteinase, infection, regulated intramembrane proteolysis, SARS – CoV, cardiovascular
Citation: Aljohmani A and Yildiz D (2020) A Disintegrin and Metalloproteinase—Control Elements in Infectious Diseases. Front. Cardiovasc. Med. 7:608281. doi: 10.3389/fcvm.2020.608281
Received: 19 September 2020; Accepted: 09 November 2020;
Published: 16 December 2020.
Edited by:
Michael Graham Tomlinson, University of Birmingham, United KingdomReviewed by:
Christoph Garbers, University Hospital Magdeburg, GermanyCopyright © 2020 Aljohmani and Yildiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Yildiz, ZGFuaWVsYS55aWxkaXpAdWtzLmV1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.